alkyl amine bevirimat derivatives are potent and broadly...
TRANSCRIPT

Alkyl Amine Bevirimat Derivatives Are Potent and Broadly ActiveHIV-1 Maturation Inhibitors
Emiko Urano,a Sherimay D. Ablan,a Rebecca Mandt,a Gary T. Pauly,b Dina M. Sigano,b Joel P. Schneider,b David E. Martin,c
Theodore J. Nitz,c Carl T. Wild,c Eric O. Freeda
Virus-Cell Interaction Section, HIV Dynamics and Replication Program, Center for Cancer Research, National Cancer Institute, Frederick, Maryland, USAa; ChemicalSynthesis Group, Chemical Biology Laboratory, Center for Cancer Research, National Cancer Institute, Frederick, Maryland, USAb; DFH Pharma, Gaithersburg, Maryland,USAc
Concomitant with the release of human immunodeficiency virus type 1 (HIV-1) particles from the infected cell, the viral pro-tease cleaves the Gag polyprotein precursor at a number of sites to trigger virus maturation. We previously reported that a bet-ulinic acid-derived compound, bevirimat (BVM), blocks HIV-1 maturation by disrupting a late step in protease-mediated Gagprocessing: the cleavage of the capsid-spacer peptide 1 (CA-SP1) intermediate to mature CA. BVM was shown in multiple clinicaltrials to be safe and effective in reducing viral loads in HIV-1-infected patients. However, naturally occurring polymorphisms inthe SP1 region of Gag (e.g., SP1-V7A) led to a variable response in some BVM-treated patients. The reduced susceptibility of SP1-polymorphic HIV-1 to BVM resulted in the discontinuation of its clinical development. To overcome the loss of BVM activityinduced by polymorphisms in SP1, we carried out an extensive medicinal chemistry campaign to develop novel maturation in-hibitors. In this study, we focused on alkyl amine derivatives modified at the C-28 position of the BVM scaffold. We identified aset of derivatives that are markedly more potent than BVM against an HIV-1 clade B clone (NL4-3) and show robust antiviralactivity against a variant of NL4-3 containing the V7A polymorphism in SP1. One of the most potent of these compounds alsostrongly inhibited a multiclade panel of primary HIV-1 isolates. These data demonstrate that C-28 alkyl amine derivatives ofBVM can, to a large extent, overcome the loss of susceptibility imposed by polymorphisms in SP1.
Human immunodeficiency virus type 1 (HIV-1), the primarycausative agent of AIDS, is currently estimated to infect �33
million people worldwide (http://www.healthline.com/health/hiv-aids/facts-statistics-infographic). A number of inhibitors havebeen developed that suppress HIV-1 replication in infected pa-tients, and there are currently more than two dozen anti-HIV-1drugs approved for clinical use (1). These inhibitors, which areadministered in combination (combination antiretroviral therapy[cART]), fall into several major classes. Inhibitors of the viral en-zymes reverse transcriptase (RT), protease (PR), and integrase(IN) form the backbone of current cART regimens. Inhibitors thattarget fusion and entry are also available (1).
Although current cART is able to suppress viral loads to belowthe level of detection of standard commercial assays in the major-ity of treatment-compliant individuals, available therapies are notcurative and thus require lifelong drug adherence. Long-termtreatment is associated with a variety of issues related to drugtoxicity, unfavorable drug-drug interactions, and patient non-compliance. Multidrug resistance is likely to limit treatment op-tions in an increasing number of patients over time, particularly inresource-limited settings, where viral load testing is not widelyavailable (2–5). Thus, it is imperative that continued efforts bemade to develop novel drugs targeting steps in the viral replicationcycle not affected by current therapies. As an added benefit, devel-oping inhibitors against novel targets provides a wealth of basicmechanistic information about fundamental aspects of viral rep-lication.
Maturation of HIV-1 particles, which is triggered by the actionof the viral PR, occurs concomitantly with virion release from theinfected cell (6–8). PR cleaves a number of sites in the Gag poly-protein precursor, Pr55Gag, the major structural protein responsi-ble for the formation of virus particles. PR-mediated Gag cleavage
gives rise to the matrix (MA), capsid (CA), nucleocapsid (NC),and p6 proteins and to two small spacer peptides, SP1 and SP2,located between CA and NC and between NC and p6, respectively.PR also cleaves the Gag-Pol polyprotein precursor to generate themature viral enzymes, i.e., PR, RT, and IN. Cleavage of the Gagand Gag-Pol polyproteins results in a marked change in virionmorphology. In the immature particle, the Gag precursor proteinsare arranged radially around the outer edge of the virus particle,whereas in the mature virion the CA proteins assemble into acentrally located, conical core (referred to as the capsid) in whichthe viral RNA genome and the viral enzymes RT and IN reside.Both Pr55Gag and mature CA assemble into a largely hexamericlattice, though the unit-to-unit spacing of the lattice and the in-tersubunit contacts differ between the immature and mature lat-tices (9). The strain of curvature is accommodated in the imma-ture Gag lattice by the presence of gaps, whereas in the maturecapsid the inclusion of a total of 12 pentamers in the otherwisehexameric capsid lattice allows the capsid to close off at both ends(10–12). Maturation is critical to particle infectivity (7).
Received 1 September 2015 Returned for modification 30 September 2015Accepted 12 October 2015
Accepted manuscript posted online 19 October 2015
Citation Urano E, Ablan SD, Mandt R, Pauly GT, Sigano DM, Schneider JP, MartinDE, Nitz TJ, Wild CT, Freed EO. 2016. Alkyl amine bevirimat derivatives are potentand broadly active HIV-1 maturation inhibitors. Antimicrob Agents Chemother60:190 –197. doi:10.1128/AAC.02121-15.
Address correspondence to Eric O. Freed, [email protected].
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02121-15.
Copyright © 2015, American Society for Microbiology. All Rights Reserved.
crossmark
190 aac.asm.org January 2016 Volume 60 Number 1Antimicrobial Agents and Chemotherapy
on Novem
ber 11, 2018 by guesthttp://aac.asm
.org/D
ownloaded from

Each processing site within the Gag and Gag-Pol polyproteinprecursors is cleaved by PR with distinct kinetics, largely due to theunique primary amino acid sequence at each site (13–19). Theconsequence of the differential rates of cleavage is that Gag andGag-Pol processing occurs as a highly ordered cascade of cleavageevents. This highly ordered processing is required for proper mat-uration. Defects in maturation can affect both virus entry (20, 21)and subsequent postentry events. Even partial disruption of pro-cessing at several sites in Gag leads to severely impaired virus in-fectivity (22–24), highlighting the utility of Gag processing as atarget for antiretrovirals.
We and others previously reported that the first-in-class HIV-1maturation inhibitor 3-O-(3=-3=-dimethylsuccinyl) betulinic acid(bevirimat [BVM]) (25) acts by blocking cleavage at the CA-SP1junction (26, 27). We selected a panel of resistant mutants bypropagating HIV-1 in the presence of BVM; the resistance muta-tions mapped to the CA-SP1 boundary region (28). Clinical trialsshowed that BVM was safe and efficacious. However, polymor-phisms present in circulating strains of HIV-1 diminished the ef-ficacy of BVM. These polymorphisms cluster in a Gln-Val-Thr(QVT) sequence spanning SP1 residues 6 to 8. In particular, weobserved that, in culture, a Val-to-Ala change at SP1 residue 7(V7A) almost completely abrogated the ability of BVM to blockCA-SP1 processing and virus replication (29). Our analysis of thestructurally distinct maturation inhibitor PF-46396 (30) demon-strated that this compound retained activity against a derivative ofclade B NL4-3 bearing the V7A change (31). This result, togetherwith recent reports describing chemically modified BVM deriva-tives (32–34), suggested that the insensitivity of SP1-polymorphicHIV-1 to maturation inhibitors could be overcome.
In this study, we synthesized a collection of novel C-28 alkylamine derivatives of BVM and tested their ability to block CA-SP1processing and virus replication by using wild-type (WT) NL4-3and a V7A variant of NL4-3 that is resistant to BVM. The activitiesof these compounds against a multiclade panel of HIV-1 isolateswere also determined. The results demonstrated that several ofthese derivatives were markedly more potent and more broadlyactive than the parental BVM compound, with low-nanomolarantiviral activity against both WT NL4-3 and the V7A derivative.
MATERIALS AND METHODSChemical synthesis. Scaffold 7 compounds (Table 1) were synthesized bymodification of C-3 and C-28 of betulin. Oxidation of C-28 allowed acy-lation followed by deprotection at C-3 to provide the left-hand side of themolecule. Further homologation following by oxidation and reductiveanimation at C-28 furnished the right-hand side of the molecule, resultingin compounds 7a to 7t (Table 1). See the supplemental material for addi-tional details.
Cell culture, plasmids, and transfections. The MT-4 T-cell line wasmaintained in RPMI 1640 medium supplemented with 10% (vol/vol)fetal bovine serum (FBS), 2 mM glutamine, 100 U/ml penicillin, and 100�g/ml streptomycin at 37°C in a humidified 5% CO2 atmosphere. HeLaand 293T cells were maintained in Dulbecco’s modified Eagle’s medium(DMEM) supplemented with 10% (vol/vol) FBS, 2 mM glutamine, 100U/ml penicillin, and 100 �g/ml streptomycin. The molecular clones usedin this study were WT pNL4-3 (35) and a derivative encoding a Val-to-Alamutation at SP1 residue 7, referred to as the V7A variant (29). HeLa and293T cells were transfected by use of linear polyethylenimine (L-PEI) orLipofectamine 2000 (Invitrogen) (36, 37).
CA-SP1 accumulation assay. CA-SP1 accumulation assays were per-formed as described previously (28, 31, 37), with some modification.Briefly, HeLa cells were transfected with WT pNL4-3 or the V7A variant.
At 24 h posttransfection, cells were starved in Met/Cys-free medium for 30min and then metabolically labeled with [35S]Met/Cys-Pro mix(PerkinElmer) for 2 to 3 h. Maturation inhibitors were maintained in thecultures throughout the transfection and labeling period. Viruses werecollected by ultracentrifugation at 75,000 � g for 45 to 60 min. Viruspellets were resuspended in Triton X-100 lysis buffer (300 mM NaCl, 50mM Tris-HCl [pH 7.5], 0.5% Triton X-100, 10 mM iodoacetamide, andprotease inhibitor cocktail tablets [Roche]). CA and CA-SP1 proteinswere separated in gels containing between 13.5 and 15% polyacrylamideby SDS-polyacrylamide gel electrophoresis, exposed to a phosphorimagerplate (Fuji), and quantified by use of Quantity One software (Bio-Rad).
Antiviral assays. To produce HIV-1 stocks, 293T cells were trans-fected with WT pNL4-3 or the V7A variant, and culture supernatants werecollected at 24 h posttransfection. RT-normalized virus supernatants wereused to infect MT-4 cells at room temperature for 30 min. Cells were thencultured in the presence of serial dilutions of each compound. At 4 dayspostinfection, the culture supernatants were subjected to RT assay tomonitor viral replication as previously described (37). The 50% inhibitoryconcentrations (IC50s) were defined as compound concentrations thatreduced RT levels to 50% of those measured in the absence of inhibitor(dimethyl sulfoxide [DMSO]-only controls) and were determined usingGraphPad Prism 6 software (38). The IC50s in primary peripheral bloodmononuclear cells (PBMCs) were determined by Southern Research In-stitute (Frederick, MD) for a multiclade panel of HIV-1 isolates, as re-ported previously (38). PBMCs were purified from anonymized, healthyblood donors by Ficoll-Hypaque gradient centrifugation. Cells were stim-ulated for 3 days with phytohemagglutinin and interleukin-2 prior toinfection with HIV-1. IC50s were calculated as described above, based onreductions in RT activity on day 6 postinfection.
Cytotoxicity assays. Cytotoxicity was measured with the CellTiter-Blue cell viability assay according to the manufacturer’s protocol (Pro-mega). Two cell lines, MT-4 and HeLa, were used in this assay. Cells wereincubated with serial dilutions of compounds at 37°C for 4 or 8 days andthen treated with CellTiter-Blue reagent. The fluorescence signal wasmeasured at 560 nm (excitation)/590 nm (emission) by use of a platereader (Infinite M1000 Pro; Tecan). The 50% cytotoxic concentrations(CC50s) were defined as compound concentrations at which the fluores-cence signals were reduced by 50% relative to that of the no-inhibitor(DMSO only) controls and were determined using GraphPad Prism 6software. Cytotoxicity in PBMCs was determined as described previ-ously (38).
RESULTSC-28 BVM analogs inhibit CA-SP1 processing of both WTHIV-1 strain NL4-3 and the SP1-V7A derivative. Previous resultsfrom this laboratory indicated that compounds containing scaf-fold 7 possessed some activity against V7A variants (data notshown). To identify BVM analogs with an increased potency andbreadth of activity, we synthesized a panel of related C-28-homol-ogated amines (Table 1; see Materials and Methods and the sup-plemental information). We then tested the ability of these deriv-atives to inhibit CA-SP1 processing of a clade B viral clone, NL4-3,and the BVM-resistant V7A variant (29). HeLa cells transfectedwith pNL4-3 or the V7A variant were left untreated or weretreated with 100 nM (each) compounds 7a to 7j and then meta-bolically radiolabeled with [35S]Met/Cys. Released virus particleswere concentrated by ultracentrifugation, and lysates were sub-jected to SDS-PAGE and fluorography (see Materials and Meth-ods). This approach evaluates the ability of a maturation inhibitorto block CA-SP1 processing in a quantitative manner by measur-ing the percentage of CA-SP1 accumulation. As indicated in Fig.1A, treatment with BVM or with analogs 7a to 7j led to a markedaccumulation (�50 to 80%) of CA-SP1 for WT NL4-3. In con-trast, and consistent with our previous report (29), for samples
Potent HIV-1 Maturation Inhibitors
January 2016 Volume 60 Number 1 aac.asm.org 191Antimicrobial Agents and Chemotherapy
on Novem
ber 11, 2018 by guesthttp://aac.asm
.org/D
ownloaded from
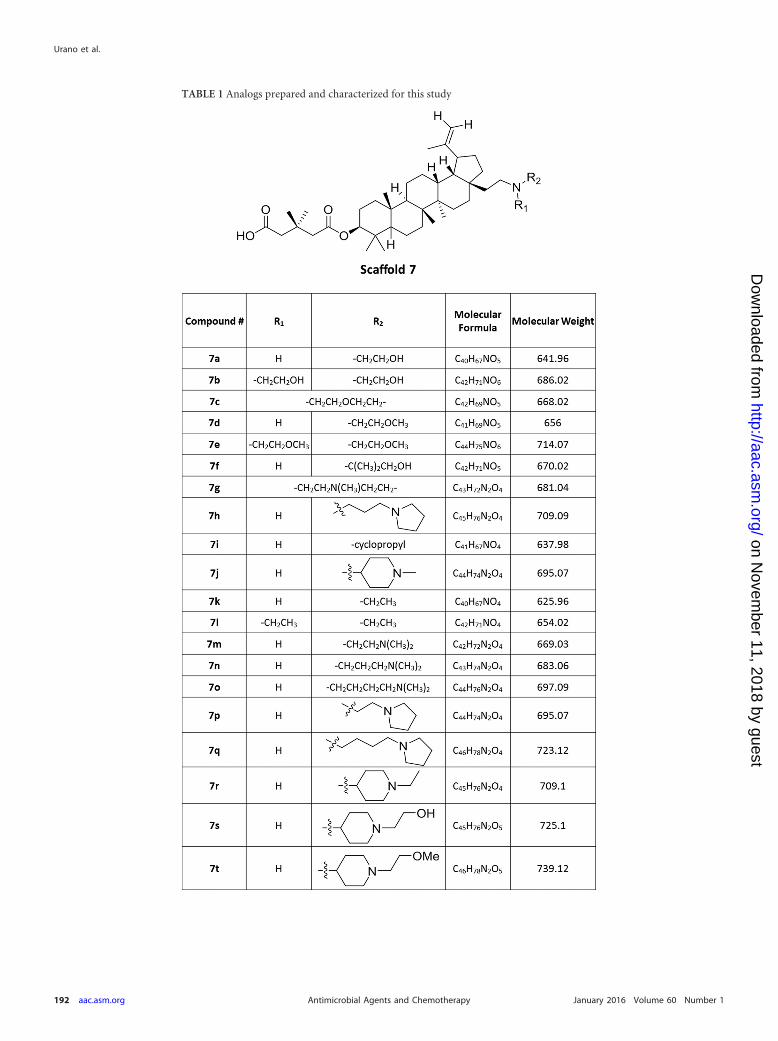
TABLE 1 Analogs prepared and characterized for this study
Urano et al.
192 aac.asm.org January 2016 Volume 60 Number 1Antimicrobial Agents and Chemotherapy
on Novem
ber 11, 2018 by guesthttp://aac.asm
.org/D
ownloaded from

transfected with the V7A mutant the BVM treatment resulted inonly �10% accumulation of CA-SP1, which is comparable to thelevel for the DMSO control (Fig. 1B). For the remainingcompounds in this set, levels of CA-SP1 accumulation rangedfrom �15 to 55%, with compounds 7h and 7j, which incorporatean additional amino grouping into the C-28 side chain, showingthe greatest potencies in disrupting CA-SP1 processing. Based onthese results, we synthesized a new set of compounds (7k to 7t)and evaluated their potency in the CA-SP1 accumulation assay.Compared to BVM, compounds 7k to 7t were considerably moreeffective against WT NL4-3 (Fig. 1C) and showed a high degree ofpotency against the V7A mutant (Fig. 1D), with V7A mutant CA-SP1 accumulation in the 60% range for many of these compounds(Fig. 1D).
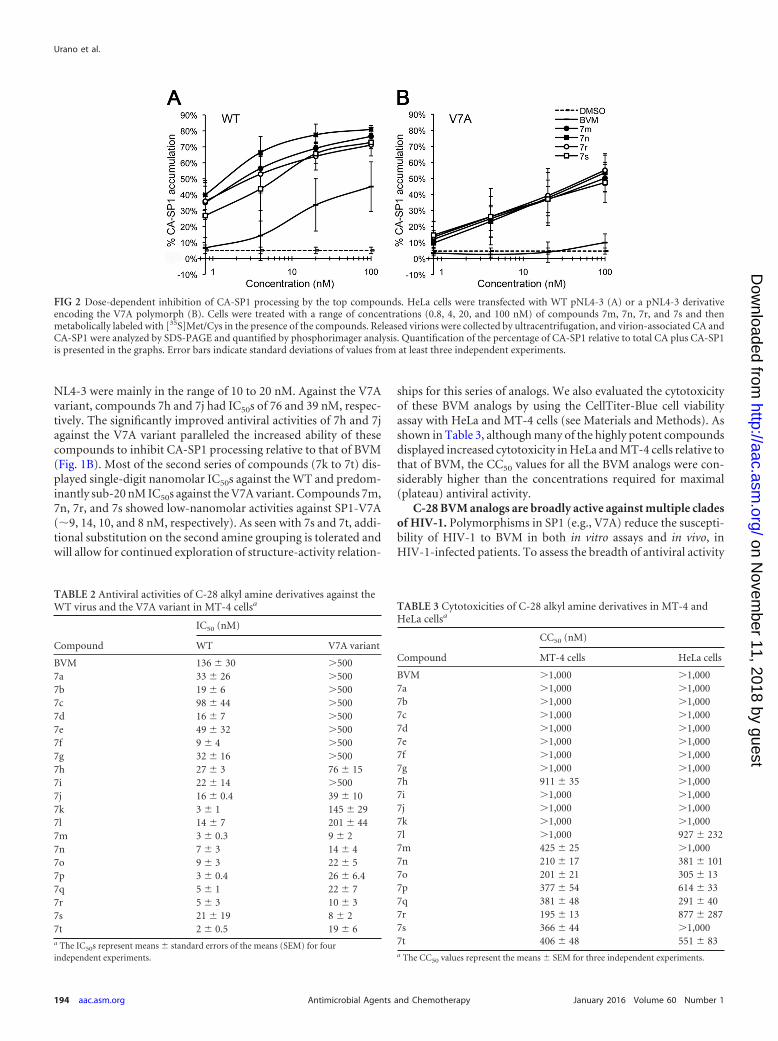
To refine our analysis of CA-SP1 accumulation, we focused oncompounds with the lowest IC50s against the V7A variant, namely,7m, 7n, 7r, and 7s, and performed a dose-response analysis. Virus-producing cells were treated with increasing concentrations of
compound (0.8 to 100 nM). WT NL4-3 showed a dose-dependentaccumulation of CA-SP1 when cells were treated with BVM, whileSP1-V7A was resistant to all concentrations used (Fig. 2A and B).In contrast, compounds 7m, 7n, 7r, and 7s showed strong dose-responsive accumulations of CA-SP1 for both WT NL4-3 (Fig.2A) and the V7A mutant (Fig. 2B).
C-28 BVM analogs display low-nanomolar antiviral activityagainst WT NL4-3 and the SP1-V7A derivative. Our previousstudies established a close correlation between the inhibition ofCA-SP1 processing imposed by BVM and the compound’s antivi-ral activity (28, 29, 31). To determine whether this correlationextends to the BVM analogs being studied here, we used a virusreplication assay based on spreading HIV-1 infection in the MT-4T-cell line to measure the IC50s of the compounds. HIV-1-in-fected MT-4 cells were cultured for 4 days in the presence of serialdilutions of each compound. Antiviral activity was monitored byreductions in RT activity in culture supernatants. As shown inTable 2, the IC50s of the first series of compounds against WT
FIG 1 C-28 alkyl amine BVM derivatives potently disrupt CA-SP1 processing. HeLa cells were transfected with WT pNL4-3 (A and C) or a pNL4-3 derivativeencoding the V7A polymorph (B and D). Cells were treated with 100 nM (each) compounds 7a to 7j (A and B) or 7k to 7t (C and D) and then metabolically labeledwith [35S]Met/Cys in the presence of the compounds. Released virions were collected by ultracentrifugation, and virion-associated CA and CA-SP1 were analyzedby SDS-PAGE and quantified by phosphorimager analysis. A representative gel image is shown at the top of each panel, and quantification of the percentage ofCA-SP1 relative to total CA plus CA-SP1 is presented in the graphs. Error bars indicate standard deviations of values from three independent experiments.
Potent HIV-1 Maturation Inhibitors
January 2016 Volume 60 Number 1 aac.asm.org 193Antimicrobial Agents and Chemotherapy
on Novem
ber 11, 2018 by guesthttp://aac.asm
.org/D
ownloaded from
NL4-3 were mainly in the range of 10 to 20 nM. Against the V7Avariant, compounds 7h and 7j had IC50s of 76 and 39 nM, respec-tively. The significantly improved antiviral activities of 7h and 7jagainst the V7A variant paralleled the increased ability of thesecompounds to inhibit CA-SP1 processing relative to that of BVM(Fig. 1B). Most of the second series of compounds (7k to 7t) dis-played single-digit nanomolar IC50s against the WT and predom-inantly sub-20 nM IC50s against the V7A variant. Compounds 7m,7n, 7r, and 7s showed low-nanomolar activities against SP1-V7A(�9, 14, 10, and 8 nM, respectively). As seen with 7s and 7t, addi-tional substitution on the second amine grouping is tolerated andwill allow for continued exploration of structure-activity relation-
ships for this series of analogs. We also evaluated the cytotoxicityof these BVM analogs by using the CellTiter-Blue cell viabilityassay with HeLa and MT-4 cells (see Materials and Methods). Asshown in Table 3, although many of the highly potent compoundsdisplayed increased cytotoxicity in HeLa and MT-4 cells relative tothat of BVM, the CC50 values for all the BVM analogs were con-siderably higher than the concentrations required for maximal(plateau) antiviral activity.
C-28 BVM analogs are broadly active against multiple cladesof HIV-1. Polymorphisms in SP1 (e.g., V7A) reduce the suscepti-bility of HIV-1 to BVM in both in vitro assays and in vivo, inHIV-1-infected patients. To assess the breadth of antiviral activity
FIG 2 Dose-dependent inhibition of CA-SP1 processing by the top compounds. HeLa cells were transfected with WT pNL4-3 (A) or a pNL4-3 derivativeencoding the V7A polymorph (B). Cells were treated with a range of concentrations (0.8, 4, 20, and 100 nM) of compounds 7m, 7n, 7r, and 7s and thenmetabolically labeled with [35S]Met/Cys in the presence of the compounds. Released virions were collected by ultracentrifugation, and virion-associated CA andCA-SP1 were analyzed by SDS-PAGE and quantified by phosphorimager analysis. Quantification of the percentage of CA-SP1 relative to total CA plus CA-SP1is presented in the graphs. Error bars indicate standard deviations of values from at least three independent experiments.
TABLE 2 Antiviral activities of C-28 alkyl amine derivatives against theWT virus and the V7A variant in MT-4 cellsa
Compound
IC50 (nM)
WT V7A variant
BVM 136 � 30 �5007a 33 � 26 �5007b 19 � 6 �5007c 98 � 44 �5007d 16 � 7 �5007e 49 � 32 �5007f 9 � 4 �5007g 32 � 16 �5007h 27 � 3 76 � 157i 22 � 14 �5007j 16 � 0.4 39 � 107k 3 � 1 145 � 297l 14 � 7 201 � 447m 3 � 0.3 9 � 27n 7 � 3 14 � 47o 9 � 3 22 � 57p 3 � 0.4 26 � 6.47q 5 � 1 22 � 77r 5 � 3 10 � 37s 21 � 19 8 � 27t 2 � 0.5 19 � 6a The IC50s represent means � standard errors of the means (SEM) for fourindependent experiments.
TABLE 3 Cytotoxicities of C-28 alkyl amine derivatives in MT-4 andHeLa cellsa
Compound
CC50 (nM)
MT-4 cells HeLa cells
BVM �1,000 �1,0007a �1,000 �1,0007b �1,000 �1,0007c �1,000 �1,0007d �1,000 �1,0007e �1,000 �1,0007f �1,000 �1,0007g �1,000 �1,0007h 911 � 35 �1,0007i �1,000 �1,0007j �1,000 �1,0007k �1,000 �1,0007l �1,000 927 � 2327m 425 � 25 �1,0007n 210 � 17 381 � 1017o 201 � 21 305 � 137p 377 � 54 614 � 337q 381 � 48 291 � 407r 195 � 13 877 � 2877s 366 � 44 �1,0007t 406 � 48 551 � 83a The CC50 values represent the means � SEM for three independent experiments.
Urano et al.
194 aac.asm.org January 2016 Volume 60 Number 1Antimicrobial Agents and Chemotherapy
on Novem
ber 11, 2018 by guesthttp://aac.asm
.org/D
ownloaded from

displayed by our novel panel of BVM analogs, we extended ouranalyses to a panel of primary HIV-1 isolates that includes repre-sentative viruses from HIV-1 subtypes A, B, C, D, E, F, and G. Thispanel of isolates was selected in part because of their highly diverseSP1 sequences (Fig. 3). Compound 7r was selected for this analysisbecause of its high level of potency against the V7A variant. Anti-viral activity in PBMCs was measured by quantifying the reduc-tion in RT activity in the culture supernatant at 6 days postinfec-tion. BVM displayed low-nanomolar IC50s against only some ofthe isolates (Table 4). In contrast, compound 7r was active acrossall the primary isolates, except the subtype A isolate 92UG031.This virus contains both CA-V230I and SP1-V7A polymorphisms(Fig. 3). Our preliminary resistance data suggest that the combi-nation of the V230I and V7A polymorphisms confers resistance toBVM analogs (data not shown). However, the frequency of bothof these polymorphisms occurring together is very low, i.e., �1%of �30,000 viral sequences compiled in the Los Alamos HIV-1sequence database (http://www.hiv.lanl.gov/content/index). Cy-totoxicity was limited in PBMCs, with a BVM CC50 of �10,000nM and no toxicity observed for compound 7r at concentrationsof up to 1,000 nM (data not shown). These data clearly demon-strate that relative to parental BVM, the C-28 alkyl amine deriva-tives of BVM display marked improvements in potency andbreadth of activity.
DISCUSSION
In this study, we conducted an extensive medicinal chemistrycampaign to develop novel maturation inhibitors that not onlydemonstrate increased potency against WT HIV-1 but also areactive, in the low-nanomolar concentration range, against SP1-polymorphic HIV-1 isolates. The lack of antiviral activity against
HIV-1 isolates with polymorphisms near the CA-SP1 cleavagesite, particularly in SP1 residues 6 to 8, was a major reason for thediscontinuation of BVM clinical development. In the presentstudy, we synthesized a series of BVM analogs with C-28 modifi-cations, with the top candidates potently inhibiting the processingof CA-SP1 and the viral replication of a representative polymor-phic virus, the V7A variant. These compounds were also activeagainst a multiclade panel of HIV-1 isolates in PBMCs. Whileseveral of the most potent compounds exhibited increased cyto-toxicity in laboratory cell lines relative to that of BVM, cytotoxicitywas not observed in PBMCs for concentrations of up to 1,000 nM.
Recently, several other groups reported the synthesis and anti-viral activity of BVM analogs. Most of these analogs have C-28modifications and demonstrated slightly improved antiviral activ-ity compared to that of BVM against prototypic clade B HIV-1strains (34, 39, 40). Interestingly, some of these BVM analogs re-portedly inhibited both viral maturation and viral entry (32, 41).In our study, the antiviral activity correlated well with CA-SP1accumulation, and infection with a vesicular stomatitis virus gly-coprotein G (VSV-G)-pseudotyped virus was potently inhibitedby the BVM derivatives in single-cycle assays. In addition, theinfectivity of virions bearing the HIV-1 Env glycoprotein was notaffected by the inhibitors when they were present at the time ofinfection (data not shown). Thus, the compounds synthesizedhere act as maturation inhibitors, with no evidence of an entry-based inhibitory activity. In addition to C-28 modifications, it wasreported that position C-3 can be derivatized without a loss ofantiviral activity (39). More recently, Dang et al. identified a C-28BVM derivative that exhibited a 20-fold increased activity againsta virus bearing the V7A polymorphism (32). A series of macrocy-clized betulinic acid derivatives was also reported; however, thesecompounds were not able to overcome the resistance conferred bythe V7A polymorphism (40). The most potent compounds inour study demonstrated IC50s against the V7A polymorph thatwere �50-fold lower than that of parental BVM. Thus, thesecompounds significantly contribute to the available informa-tion concerning the structure-activity relationships of HIV-1maturation inhibitors.
The mechanism by which C-28 BVM derivatives potently andbroadly inhibit HIV-1 replication relative to the action of the pa-rental compound remains to be determined. The modificationsmay allow the C-28 side chain of the compound to interact moretightly with the putative Gag substrate. Although the binding sitefor BVM and its analogs has not been characterized structurally,several lines of evidence support the hypothesis that contacts aremade between these compounds and two regions of Gag: the CA-SP1 boundary region and the major homology region (MHR) ofCA. An early study demonstrated that BVM is specifically incor-porated into immature but not mature particles (42). Associationof 3H-labeled BVM with immature particles was reduced by resis-tance-conferring mutations at the CA-SP1 cleavage site (43).Nguyen et al. demonstrated that a photoaffinity BVM analog in-teracted with residues around the CA-SP1 cleavage site and the CAMHR (44). In a previous study (31), we demonstrated that resis-tance to a structurally distinct maturation inhibitor, PF-46396,mapped not only to the vicinity of the CA-SP1 cleavage site (whereBVM resistance mutations map) but also to the MHR. The MHRmutants were strongly compound dependent and could revert inculture by acquiring a mutation at residue 8 of SP1 (SP1-T8I). Theability of an SP1 mutation to rescue the replication defect con-
FIG 3 Amino acid sequences of the CA-SP1 boundary region for the multi-clade panel of HIV-1 isolates shown in Table 4. Asterisks indicate amino acidsequence identity with WT NL4-3 (top line); gaps are indicated by hyphens.The QVT motif spanning SP1 residues 6 to 8 is in bold.
TABLE 4 Antiviral activities of C-28 alkyl amine derivatives against amulticlade panel of HIV-1 isolates in PBMCsa
Isolate Subtype
IC50 (nM)
Compound 7r BVM Zidovudine
92UG031 A �1,000 2,829 1192BR030 B 199 5,235 1293IN101 C 67 2,651 1999UGA07412MI D 15 15 6CMU02 E 96 3,327 893BR029 F 13 7 3JV1083 G 221 4,188 13a Data are representative of two independent experiments.
Potent HIV-1 Maturation Inhibitors
January 2016 Volume 60 Number 1 aac.asm.org 195Antimicrobial Agents and Chemotherapy
on Novem
ber 11, 2018 by guesthttp://aac.asm
.org/D
ownloaded from

ferred by an MHR substitution suggested functional cross talkbetween the MHR and SP1. It is interesting that residue 8 of SP1falls within the so-called QVT motif (SP1 residues 6 to 8) previ-ously shown to influence BVM activity (29). The poor solubility ofBVM and its analogs has hampered structural characterization ofBVM binding, although a BVM derivative with improved solubil-ity has been reported and shown by nuclear magnetic resonance(NMR) to bind a CA-SP1-NC peptide (45). Adding to the chal-lenging nature of defining the maturation inhibitor-binding site isthe fact that the structure of the CA-SP1 boundary region of Gag ispoorly defined. Although recent progress has been made in delin-eating the structure of the CA domain in the immature Gag lattice,the high level of flexibility of SP1 has thus far precluded an atomic-resolution structure of SP1 in the immature particle (9). Bindingof maturation inhibitors to Gag may stabilize SP1 (46, 47), thuspotentially facilitating structural studies of this region. Mutationsin SP1 may also limit the conformational flexibility of this peptide,providing another useful tool for structural studies.
The novel HIV-1 maturation inhibitors developed in this studyprovide insights into the structure-activity relationships of thisclass of compounds. Ongoing studies are aimed at further opti-mizing the potency and breadth of activity of these inhibitors.
ACKNOWLEDGMENTS
We thank members of the Freed laboratory for helpful discussions andcritical reviews of the manuscript and Wei Shau (National Cancer Insti-tute) for HIV-1 sequence analyses. We gratefully acknowledge SouthernResearch Institute, Frederick, MD, for the multiclade analysis shown inTable 4 and Ioannis Kagiampakis (National Cancer Institute) for assis-tance with GraphPad statistical analyses.
FUNDING INFORMATIONHHS | National Institutes of Health (NIH) provided funding to Eric O.Freed and Joel P. Schneider and under grant numbers BC 010778 and BC011515 from the Intramural Research Program. HHS | NIH providedfunding to Carl Wild under grant number 1R41AI108457-1 and by con-tract N01-AI-70041. This work was also supported by the IntramuralAIDS Targeted Antiviral Program and was conducted in part by SouthernResearch Institute, using federal funds from the Division of AIDS, NIH,under contract HHSN272200700041C, entitled “Confirmatory In VitroEvaluations of HIV Therapeutics.”
REFERENCES1. Kinch MS, Patridge E. 2014. An analysis of FDA-approved drugs for
infectious disease: HIV/AIDS drugs. Drug Discov Today 19:1510 –1513.http://dx.doi.org/10.1016/j.drudis.2014.05.012.
2. Bertagnolio S, Perno CF, Vella S, Pillay D. 2013. The impact of HIV drugresistance on the selection of first- and second-line ART in resource-limited settings. J Infect Dis 207(Suppl 2):S45–S48. http://dx.doi.org/10.1093/infdis/jit121.
3. Hosseinipour MC, Gupta RK, Van Zyl G, Eron JJ, Nachega JB. 2013.Emergence of HIV drug resistance during first- and second-line antiret-roviral therapy in resource-limited settings. J Infect Dis 207(Suppl 2):S49 –S56. http://dx.doi.org/10.1093/infdis/jit107.
4. Iyidogan P, Anderson KS. 2014. Current perspectives on HIV-1 antiret-roviral drug resistance. Viruses 6:4095– 4139. http://dx.doi.org/10.3390/v6104095.
5. Stadeli KM, Richman DD. 2013. Rates of emergence of HIV drug resis-tance in resource-limited settings: a systematic review. Antivir Ther 18:115–123. http://dx.doi.org/10.3851/IMP2437.
6. Freed EO. 2015. HIV-1 assembly, release and maturation. Nat Rev Mi-crobiol 13:484 – 496. http://dx.doi.org/10.1038/nrmicro3490.
7. Konvalinka J, Krausslich HG, Muller B. 2015. Retroviral proteases andtheir roles in virion maturation. Virology 479 – 480:403– 417. http://dx.doi.org/10.1016/j.virol.2015.03.021.
8. Lee SK, Potempa M, Swanstrom R. 2012. The choreography of HIV-1proteolytic processing and virion assembly. J Biol Chem 287:40867–40874. http://dx.doi.org/10.1074/jbc.R112.399444.
9. Schur FK, Hagen WJ, Rumlova M, Ruml T, Muller B, Krausslich HG,Briggs JA. 2015. Structure of the immature HIV-1 capsid in intact virusparticles at 8.8 A resolution. Nature 517:505–508. http://dx.doi.org/10.1038/nature13838.
10. Briggs JA, Riches JD, Glass B, Bartonova V, Zanetti G, Krausslich HG.2009. Structure and assembly of immature HIV. Proc Natl Acad Sci U S A106:11090 –11095. http://dx.doi.org/10.1073/pnas.0903535106.
11. Fuller SD, Wilk T, Gowen BE, Krausslich HG, Vogt VM. 1997.Cryo-electron microscopy reveals ordered domains in the immatureHIV-1 particle. Curr Biol 7:729 –738. http://dx.doi.org/10.1016/S0960-9822(06)00331-9.
12. Li S, Hill CP, Sundquist WI, Finch JT. 2000. Image reconstructions ofhelical assemblies of the HIV-1 CA protein. Nature 407:409 – 413. http://dx.doi.org/10.1038/35030177.
13. Erickson-Viitanen S, Manfredi J, Viitanen P, Tribe DE, Tritch R,Hutchison CA, III, Loeb DD, Swanstrom R. 1989. Cleavage of HIV-1 gagpolyprotein synthesized in vitro: sequential cleavage by the viral protease.AIDS Res Hum Retroviruses 5:577–591. http://dx.doi.org/10.1089/aid.1989.5.577.
14. Krausslich HG, Schneider H, Zybarth G, Carter CA, Wimmer E. 1988.Processing of in vitro-synthesized Gag precursor proteins of human im-munodeficiency virus (HIV) type 1 by HIV proteinase generated in Esch-erichia coli. J Virol 62:4393– 4397.
15. Mervis RJ, Ahmad N, Lillehoj EP, Raum MG, Salazar FH, Chan HW,Venkatesan S. 1988. The gag gene products of human immunodeficiencyvirus type 1: alignment within the gag open reading frame, identificationof posttranslational modifications, and evidence for alternative gag pre-cursors. J Virol 62:3993– 4002.
16. Pettit SC, Moody MD, Wehbie RS, Kaplan AH, Nantermet PV, KleinCA, Swanstrom R. 1994. The p2 domain of human immunodeficiencyvirus type 1 Gag regulates sequential proteolytic processing and is requiredto produce fully infectious virions. J Virol 68:8017– 8027.
17. Tritch RJ, Cheng YE, Yin FH, Erickson-Viitanen S. 1991. Mutagenesis ofprotease cleavage sites in the human immunodeficiency virus type 1 Gagpolyprotein. J Virol 65:922–930.
18. Vogt VM. 1996. Proteolytic processing and particle maturation. Curr TopMicrobiol Immunol 214:95–131.
19. Wiegers K, Rutter G, Kottler H, Tessmer U, Hohenberg H, KrausslichHG. 1998. Sequential steps in human immunodeficiency virus particlematuration revealed by alterations of individual Gag polyprotein cleavagesites. J Virol 72:2846 –2854.
20. Murakami T, Ablan S, Freed EO, Tanaka Y. 2004. Regulation of humanimmunodeficiency virus type 1 Env-mediated membrane fusion by viralprotease activity. J Virol 78:1026 –1031. http://dx.doi.org/10.1128/JVI.78.2.1026-1031.2004.
21. Wyma DJ, Jiang J, Shi J, Zhou J, Lineberger JE, Miller MD, Aiken C.2004. Coupling of human immunodeficiency virus type 1 fusion to virionmaturation: a novel role of the gp41 cytoplasmic tail. J Virol 78:3429 –3435. http://dx.doi.org/10.1128/JVI.78.7.3429-3435.2004.
22. Checkley MA, Luttge BG, Soheilian F, Nagashima K, Freed EO. 2010.The capsid-spacer peptide 1 Gag processing intermediate is a dominant-negative inhibitor of HIV-1 maturation. Virology 400:137–144. http://dx.doi.org/10.1016/j.virol.2010.01.028.
23. Lee SK, Harris J, Swanstrom R. 2009. A strongly transdominant muta-tion in the human immunodeficiency virus type 1 gag gene defines anAchilles heel in the virus life cycle. J Virol 83:8536 – 8543. http://dx.doi.org/10.1128/JVI.00317-09.
24. Muller B, Anders M, Akiyama H, Welsch S, Glass B, Nikovics K, ClavelF, Tervo HM, Keppler OT, Krausslich HG. 2009. HIV-1 Gag processingintermediates trans-dominantly interfere with HIV-1 infectivity. J BiolChem 284:29692–29703. http://dx.doi.org/10.1074/jbc.M109.027144.
25. Kanamoto T, Kashiwada Y, Kanbara K, Gotoh K, Yoshimori M, GotoT, Sano K, Nakashima H. 2001. Anti-human immunodeficiency virusactivity of YK-FH312 (a betulinic acid derivative), a novel compoundblocking viral maturation. Antimicrob Agents Chemother 45:1225–1230.http://dx.doi.org/10.1128/AAC.45.4.1225-1230.2001.
26. Li F, Goila-Gaur R, Salzwedel K, Kilgore NR, Reddick M, Matallana C,Castillo A, Zoumplis D, Martin DE, Orenstein JM, Allaway GP, FreedEO, Wild CT. 2003. PA-457: a potent HIV inhibitor that disrupts core
Urano et al.
196 aac.asm.org January 2016 Volume 60 Number 1Antimicrobial Agents and Chemotherapy
on Novem
ber 11, 2018 by guesthttp://aac.asm
.org/D
ownloaded from

condensation by targeting a late step in Gag processing. Proc Natl Acad SciU S A 100:13555–13560. http://dx.doi.org/10.1073/pnas.2234683100.
27. Zhou J, Yuan X, Dismuke D, Forshey BM, Lundquist C, Lee KH, AikenC, Chen CH. 2004. Small-molecule inhibition of human immunodefi-ciency virus type 1 replication by specific targeting of the final step ofvirion maturation. J Virol 78:922–929. http://dx.doi.org/10.1128/JVI.78.2.922-929.2004.
28. Adamson CS, Ablan SD, Boeras I, Goila-Gaur R, Soheilian F, Na-gashima K, Li F, Salzwedel K, Sakalian M, Wild CT, Freed EO. 2006. Invitro resistance to the human immunodeficiency virus type 1 maturationinhibitor PA-457 (bevirimat). J Virol 80:10957–10971. http://dx.doi.org/10.1128/JVI.01369-06.
29. Adamson CS, Sakalian M, Salzwedel K, Freed EO. 2010. Polymorphismsin Gag spacer peptide 1 confer varying levels of resistance to the HIV-1maturation inhibitor bevirimat. Retrovirology 7:36. http://dx.doi.org/10.1186/1742-4690-7-36.
30. Blair WS, Cao J, Fok-Seang J, Griffin P, Isaacson J, Jackson RL, MurrayE, Patick AK, Peng Q, Perros M, Pickford C, Wu H, Butler SL. 2009.New small-molecule inhibitor class targeting human immunodeficiencyvirus type 1 virion maturation. Antimicrob Agents Chemother 53:5080 –5087. http://dx.doi.org/10.1128/AAC.00759-09.
31. Waki K, Durell SR, Soheilian F, Nagashima K, Butler SL, Freed EO.2012. Structural and functional insights into the HIV-1 maturation inhib-itor binding pocket. PLoS Pathog 8:e1002997. http://dx.doi.org/10.1371/journal.ppat.1002997.
32. Dang Z, Ho P, Zhu L, Qian K, Lee KH, Huang L, Chen CH. 2013. Newbetulinic acid derivatives for bevirimat-resistant human immunodefi-ciency virus type-1. J Med Chem 56:2029 –2037. http://dx.doi.org/10.1021/jm3016969.
33. Dang Z, Qian K, Ho P, Zhu L, Lee KH, Huang L, Chen CH. 2012.Synthesis of betulinic acid derivatives as entry inhibitors against HIV-1and bevirimat-resistant HIV-1 variants. Bioorg Med Chem Lett 22:5190 –5194. http://dx.doi.org/10.1016/j.bmcl.2012.06.080.
34. Qian K, Bori ID, Chen CH, Huang L, Lee KH. 2012. Anti-AIDS agents90. Novel C-28 modified bevirimat analogues as potent HIV maturationinhibitors. J Med Chem 55:8128 – 8136.
35. Adachi A, Koenig S, Gendelman HE, Daugherty D, Gattoni-Celli S,Fauci AS, Martin MA. 1987. Productive, persistent infection of hu-man colorectal cell lines with human immunodeficiency virus. J Virol61:209 –213.
36. Brissault B, Kichler A, Guis C, Leborgne C, Danos O, Cheradame H.2003. Synthesis of linear polyethylenimine derivatives for DNA transfec-tion. Bioconjug Chem 14:581–587. http://dx.doi.org/10.1021/bc0200529.
37. Waheed AA, Ono A, Freed EO. 2009. Methods for the study of HIV-1assembly. Methods Mol Biol 485:163–184. http://dx.doi.org/10.1007/978-1-59745-170-3_12.
38. Ptak RG, Gentry BG, Hartman TL, Watson KM, Osterling MC, Buck-heit RW, Jr, Townsend LB, Drach JC. 2010. Inhibition of human im-munodeficiency virus type 1 by triciribine involves the accessory proteinNef. Antimicrob Agents Chemother 54:1512–1519. http://dx.doi.org/10.1128/AAC.01443-09.
39. Qian K, Kuo RY, Chen CH, Huang L, Morris-Natschke SL, Lee KH.2010. Anti-AIDS agents 81. Design, synthesis, and structure-activity rela-tionship study of betulinic acid and moronic acid derivatives as potentHIV maturation inhibitors. J Med Chem 53:3133–3141. http://dx.doi.org/10.1021/jm901782m.
40. Tang J, Jones SA, Jeffery JL, Miranda SR, Galardi CM, Irlbeck DM,Brown KW, McDanal CB, Han N, Gao D, Wu Y, Shen B, Liu C, Xi C,Yang H, Li R, Yu Y, Sun Y, Jin Z, Wang E, Johns BA. 2014. Synthesis andbiological evaluation of macrocyclized betulin derivatives as a novel classof anti-HIV-1 maturation inhibitors. Open Med Chem J 8:23–27. http://dx.doi.org/10.2174/1874104501408010023.
41. Huang L, Ho P, Lee KH, Chen CH. 2006. Synthesis and anti-HIV activityof bi-functional betulinic acid derivatives. Bioorg Med Chem 14:2279 –2289. http://dx.doi.org/10.1016/j.bmc.2005.11.016.
42. Zhou J, Huang L, Hachey DL, Chen CH, Aiken C. 2005. Inhibition ofHIV-1 maturation via drug association with the viral Gag protein in im-mature HIV-1 particles. J Biol Chem 280:42149 – 42155. http://dx.doi.org/10.1074/jbc.M508951200.
43. Zhou J, Chen CH, Aiken C. 2006. Human immunodeficiency virus type1 resistance to the small molecule maturation inhibitor 3-O-(3=,3=-dimethylsuccinyl)-betulinic acid is conferred by a variety of single aminoacid substitutions at the CA-SP1 cleavage site in Gag. J Virol 80:12095–12101. http://dx.doi.org/10.1128/JVI.01626-06.
44. Nguyen AT, Feasley CL, Jackson KW, Nitz TJ, Salzwedel K, Air GM,Sakalian M. 2011. The prototype HIV-1 maturation inhibitor, bevirimat,binds to the CA-SP1 cleavage site in immature Gag particles. Retrovirol-ogy 8:101. http://dx.doi.org/10.1186/1742-4690-8-101.
45. Coric P, Turcaud S, Souquet F, Briant L, Gay B, Royer J, Chazal N,Bouaziz S. 2013. Synthesis and biological evaluation of a new derivative ofbevirimat that targets the Gag CA-SP1 cleavage site. Eur J Med Chem62:453– 465. http://dx.doi.org/10.1016/j.ejmech.2013.01.013.
46. Keller PW, Adamson CS, Heymann JB, Freed EO, Steven AC. 2011.HIV-1 maturation inhibitor bevirimat stabilizes the immature Gag lattice.J Virol 85:1420 –1428. http://dx.doi.org/10.1128/JVI.01926-10.
47. Keller PW, Huang RK, England MR, Waki K, Cheng N, Heymann JB,Craven RC, Freed EO, Steven AC. 2013. A two-pronged structural anal-ysis of retroviral maturation indicates that core formation proceeds by adisassembly-reassembly pathway rather than a displacive transition. J Vi-rol 87:13655–13664. http://dx.doi.org/10.1128/JVI.01408-13.
Potent HIV-1 Maturation Inhibitors
January 2016 Volume 60 Number 1 aac.asm.org 197Antimicrobial Agents and Chemotherapy
on Novem
ber 11, 2018 by guesthttp://aac.asm
.org/D
ownloaded from