alkenes, di-and polyenes bond system and restricted rotation
TRANSCRIPT

Alkenes, di-and polyenes bond system and restricted rotation. Physical and chemical properties of alkenes. Electrophilic and
radical addition reactions. Markovnikov's rule and interpretation. Anti-Markovnikov orientation. Polymerization
and types. Allylic substitution.
-bond cleavage or oxidation reactions with chain cleavage. Addition reactions of conjugated dienes, 1,2 - and 1,4-addition and interpretation. Diels-Alder cycloaddition. Preparation of
Alkenes and dienes.

The simplest: + model Base: sp2 hybridized pillar carbon atoms-skeleton from the hybrid sp2 AO-s, the -bond from side overlapping of pz AO's (maximum overlap = minimum energy requirements: parallel pz AO's)
Consequence: stronger, shorter bond than single bonddC=C = 0.134 nm ↔ dC-C = 0.154 nmEC=C = 610-630 kJ/mole ↔ EC-C = 345-355 kJ/mole
DE! E+ < E2 (690-710 kJ/mól) - bond is weaker: reasons: smaller overlap
The result of the appearance of the double bond: restricted or hindered rotation (rotation makes the bond weaker or cases bond cleavage). The bond cleavage (isomerization) can be induced by heat or UV light.The -skeleton geometry basically planar trigonal (bond angle = 120 °), substituents may distort this geometry.
Open-chain or cyclic hydrocarbons containing one or more double bonds. General formula: CnH2n, CnH2n-2 (alkene or diene)Based on the number of double bonds: mono-olefin (alkene), diene, triene, polyene
Classification of dienes
Alkenes (olefins) and their classification
Bonding system of alkenes

The existence of diastereomers ("geometric isomers") is a consequence of restricted rotation
Configuration: cis / trans (up to 1,2-disubstituted systems!)In general, E (entgegen (Ger.)= against) / Z (Zusammen (Ger.) = together) - it
always can be usedBase: ranking of substituents on a specific carbon according to CIP convention
Physical Properties of Alkenes
Melting point, boiling point: - strong similarity to the alkanes, weak induced dipole-induced dipole or dipole-dipole interactions (Low mp, bp)
Appearance of dipole moment - depending on the geometry!
Solubility in water and highly polar solvents is weak, in non-polar solvents unlimited or good.

Stability of alkenes
Compared to alkanes their thermodynamic stability is lower (see energy data) - reason: destabilizing effect of high electron density between the two carbon atoms
Comparison of stability of alkenes with different structure1. Stability: cis < trans
Reason: van de Waals repulsion between the alkyl groups
positioned on the same side
2. Alkyl groups have stabilizing effectMono< di< tri< tetra subs.Reason: hyperconjugation
Hhydrogenation
KJ/mol Kcal/mol
Ethene CH2=CH2 -137 -32,8
Propene CH3CH=CH2 -126 -30,1
Z-but-2-ene
E-but-2-ene
2-methylpropene
-120
-116
-119
-28,6
-27,6
-28,4
2-methylbut-2-ene -113 -26,9
2,3-dimethylbut-2-ene -111 -26,6
the heat evolved on hydrogenation of one mole of an alkene is its heat of hydrogenation

Degree of substitution. We classify double bonds as monosubstituted, disubstituted,trisubstituted, or tetrasubstituted according to the number of carbon atoms that are directly attached to the C=C structural unit.
In general, alkenes with more highly substituted double bonds are more stable than isomerswith less substituted double bonds.

Like the sp2-hybridized carbons of carbocations and free radicals, the sp2- hybridized carbons of double bonds are electron attracting, and alkenes are stabilized by substituents that release electrons to these carbons. Alkyl groups are better electron-releasing substituents than hydrogen and are, therefore, better able to stabilize an alkene.An effect that results when two or more atoms or groups interact so as to alter the electron distribution in a system is called an electronic effect. The greater stability of more highly substituted alkenes is an example of an electronic effect.
van der Waals strain. Alkenes are more stable when large substituents are trans to each other than when they are cis.The difference in stability between stereoisomeric alkenes is even more pronounced with larger alkyl groups on the double bond.

HyperconjugationThe interaction of the electron pair of σ-bond of C-H bond with an adjacentπ-electron pair or empty pz orbitals to give an extended MO that increasesthe stability of the system (electron donating effects of alkyl groups) – stabilising effect!!!
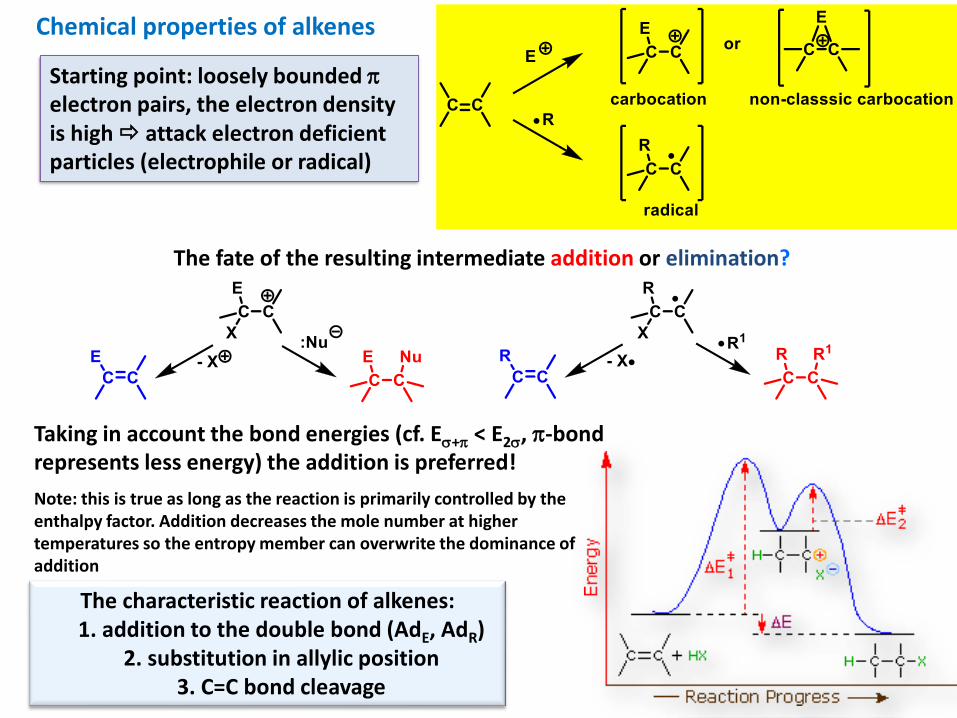
Chemical properties of alkenes
Starting point: loosely bounded electron pairs, the electron density is high attack electron deficient particles (electrophile or radical)
The fate of the resulting intermediate addition or elimination?
Taking in account the bond energies (cf. E+ < E2, -bond represents less energy) the addition is preferred!
Note: this is true as long as the reaction is primarily controlled by the enthalpy factor. Addition decreases the mole number at higher temperatures so the entropy member can overwrite the dominance of addition
The characteristic reaction of alkenes:1. addition to the double bond (AdE, AdR)
2. substitution in allylic position3. C=C bond cleavage

1. Electrophylic addition of alkenes (AdE)
1.1. Halogenation –in solution (without radical initiation)
Due to spatial reasons the nucleophile can attack only back side stereoselective reaction, the two halogen is in antiperiplanar position („trans addition”)
Note: This type of addition is observed in cyclic compounds (or in specific structural conditions), in non-cyclic compounds due to the free rotation of C-C bond systems is not observable
The intermediate character is controlled by the size of
electrophile

1.2. Further additions taking place through the "non-classical cation" intermediate Oxymercuration - demercuration
Applicable in the presence of acid sensitive R groups
Hypohalous acid addition
These reactions can be regioselective. In the case of alkenes containing different-orderpillar carbon atoms, hydroxyl group is attached to that C atom which is higher-order carbon

1.3. HX addition
1st step: attack by small electrophile classical carbenium ion is formed. Depending on the nucleophile different products can be formed
Acid catalysed water addition, a classical alcohol synthesis starting from alkenes
Regioselectivity (orientation) in addition reaction of unsymmetrical alkenes containing different order pillar carbon
Theoretically two products, in practice one of them dominates
alkyl halides
alkyl hydrogen sulfate
alcohol
ether
The intermediates of the acid catalysed water or alcohol addition
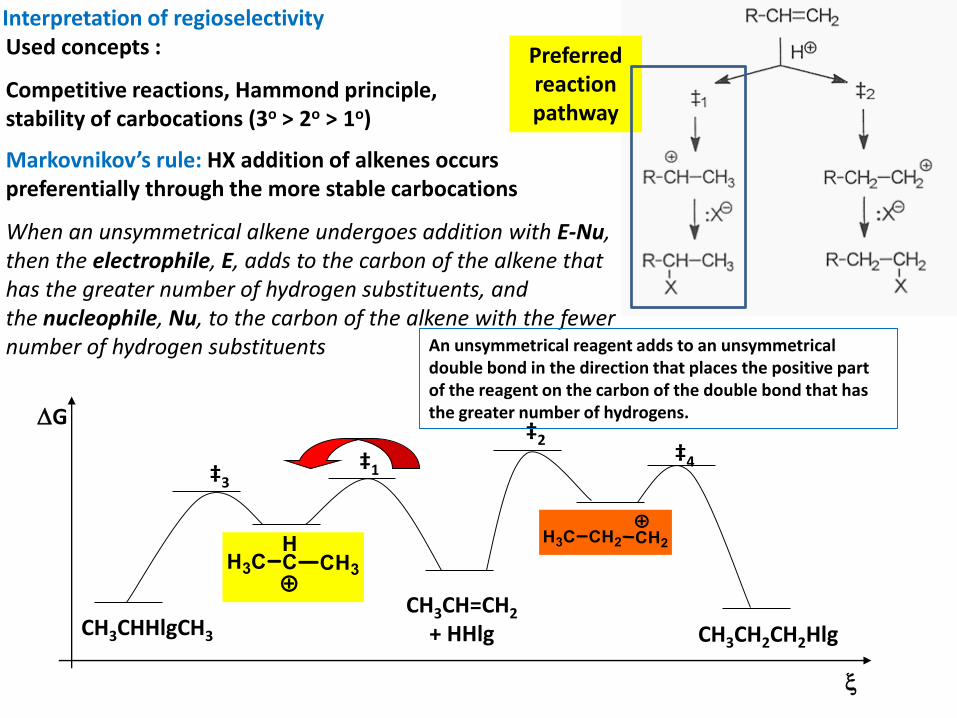
Interpretation of regioselectivityUsed concepts :
Competitive reactions, Hammond principle, stability of carbocations (3o > 2o > 1o)
Markovnikov’s rule: HX addition of alkenes occurs preferentially through the more stable carbocations
When an unsymmetrical alkene undergoes addition with E-Nu, then the electrophile, E, adds to the carbon of the alkene that has the greater number of hydrogen substituents, and the nucleophile, Nu, to the carbon of the alkene with the fewer number of hydrogen substituents
G
‡1
‡2
‡3
‡4
CH3CH=CH2
+ HHlg CH3CH2CH2HlgCH3CHHlgCH3
Preferredreactionpathway
An unsymmetrical reagent adds to an unsymmetrical double bond in the direction that places the positive part of the reagent on the carbon of the double bond that has the greater number of hydrogens.

1.4. Cationic polymerization
At the beginning HX (or Lewis acid) only a catalytic amount because low X
concentration the -electron pair of the alkene attacks as a nucleophile ("escape route") chain growth, chain reaction
In case of ethene it has little practical significance, it is frequently if R group is electron donating!1.5. Hydroboration-oxidation
Alkenes containing different ordered pillar carbons, the lower-order alcohols formed! It is a synthetic pathway for the formation of isomers of previously synthetized alcohols.
The Markovnikov orientation only apparently is compromised, in the 1st step the electrophilic species is BH3 (boron hydride) not H+
Acid-catalyzed hydration converts alkenes to alcohols with Markovnikov rule regioselectivity.Frequently, however, one needs an alcohol having a structure that corresponds to hydration of an alkene with a regioselectivity apparently opposite to that of Markovnikov’s rule.

2. Radical addition of alkenes (AdR)
General schemeThe initiator •X is re-formed Chain reaction
The formation of •X depends on reaction condition
2.1. Halogenation (X2) – in gas phase (high T or UV light)
In practice: Hlg = Cl, Br Hlg F Cl Br I
H (kJ/mól) -544 -178 -95 +16
2.2. Radical HX addition (X = Br, I, in apolar solvents)!
or
Initiation
Chain propagation

Regioselectivity (orientation) in radical addition reaction of unsymmetrical alkenes containing different order pillar carbon; theoretically two products can be formed, in practice one of them dominates
Predominant orientation anti-Markovnikovproduct (exclusively)
G
‡1
‡2
‡3
‡4
CH3CH=CH2
+ HHlgCH3CH2CH2Hlg CH3CHHlgCH3
Used concepts : Competitive reactions, Hammond principle, stability of carbon radicals (3o > 2o > 1o)
The reason of the reverse regioselectivity: the attacking electron-deficient particle is different now!·Br attacks then ·H is involved
Preferred reaction pathway

2.3. Radical polymerisation
Explanation
Typical initiators - peroxidesSignificance: most commonly used polymerization method in the industry R = H (polyethylene), R = Cl (PVC), R = OAc (polyvinyl acetate, starting materials of polyvinyl alcohols, varnish and paint industry), R = COOQ (polyacrylates) Anionic polymerization also exists (if R strongly electron-withdrawing)
Elemental steps:
low X-Y concentration chain growth, chain reaction ("escape route")
high X-Y concentration simple addition reaction
Termination by recombination of radicals or by disproportion

3. Catalytic hydrogenation
Chemisorption on the surface of the catalyst -bond and H-H bond get weakerGenerally cis addition – explanation: „four-center” model
4. Oxidation4.1 -bond cleavage by oxidation4.1.1. Hydroxylation
Stereoselective cis additionIt is used for the identification of double bond as a qualitative test (Baeyer’s test)
Surface of the catalyst

4.1.2. Epoxidation
Electron rich nature of -bond opportunity to attack the electron-poor oxygen.
Typical epoxidizing agents: percarboxylic acids Industry: O2/Ag
Practical significance: epoxides are valuable intermediates, possible ring opening by nucleophiles
4.2. Oxidation reactions with + bond cleavage (chain cleavage)
Practical importance: in case of special alkenes or the manufacture of mixtures of longer fatty acid
5. Substitution in allylic position
At high T (> 350 oC), in gas phase in the presence of Cl2(Br2) addition does NOT take place
Radical substitution reaction (SR)
percarboxylic acids

Radical chain reaction (SR) - substitution
Addition Substitution
T (oC) Go (kJ/mól) Termékarány Go (kJ/mól)
227 -118 74 26 -116
327 -105 22 78 -116
527 -79 1 99 -118
Mole number is increased it is favored
from entropy side!
The privileged position of allylic position is explained by the structure of allyl radical that has extra stability
Substitution is preferred to addition → at high temperature the entropy factor becomesimportant, it overcompensate the enthalpy factor
The radical centre is sp2 hybridised 3 pz orbital can overlap side-by-side, tricentered MO can be formed
electron delocalization (energy gain) see resonance structures:
Product ratio

Isolated diene units are those in which two carbon–carbon double bond units are separated from each other by one or more sp3-hybridized carbon atoms.
CLASSES OF DIENES
A hydrocarbon that contains two double bonds is called an alkadiene, and the relationshipbetween the double bonds may be described as isolated, conjugated, or cumulated.
Conjugated dienes are those in which two carbon–carbon double bond units are directlyconnected to each other by a single bond.
Cumulated dienes are those in which one carbon atom is common to two carbon–carbondouble bonds. The simplest cumulated diene is 1,2-propadiene, also called allene,and compounds of this class are generally referred to as allenes.

DienesIsolated dienes – separated + bonds, no interaction between bonds
Cumulated dienes – 2 C(sp2) + C(sp) special geometry, perpendicular -bonds no interaction between -bonds BUT e-density is high around the C strong and short bonds, BUT its E is high and the molecule is destabilized
Consequence: high reactivity, increased readiness of addition and
tendency to rearrange
Conjugated dienes – side overlapping of 4 pz is possible, four centred MO conjugated -bonds, e- delocalisation (E gain)
Longer and weaker double bonds (bond order < 2), stronger, shorter single bonds (bond order > 1)
Illustration resonance structural forms :
Decreasing heat of hydroge-nation increasing stability of the double bond

The most important reactions of conjugated dienes
Compared to simple alkenes it has increased reactivity, easier (faster) additions. Cause: at electrophilic attack stable allyl-type cations are formed.
Total addition2 equiv. reagent is added
Partial addition –1 equiv. reagent is added
Interpretation of appearances of 1,2 - and 1,4-products

The ratio of 1,2- and 1,4-addition
By rising the reaction temperature
1,4-addition becomes more dominant
Interpretation:
kinetic versus thermodynamic control
the 1,4-product is a disubstituted ethene more stable thermodynamic control
but the intermediate cation of 1,2-adduct has lower energy kinetic control
G
‡1
‡2
kinetic control
thermodynamic control
kinetic thermodynamic
product

When the major product of a reaction is the one that is formed at the fastest rate, we say that the reaction is governed by kinetic control. Most organic reactions fall into this category, and the electrophilic addition of hydrogen bromide to 1,3-butadiene at lowtemperature is a kinetically controlled reaction.
The product of 1,4 addition, 1-bromo-2-butene, contains an internal double bond and so is more stable than the product of 1,2 addition, 3-bromo-1-butene, which has a terminaldouble bond.
When addition occurs under conditions in which the products can equilibrate, thecomposition of the reaction mixture no longer reflects the relative rates of formation ofthe products but tends to reflect their relative stabilities. Reactions of this type are saidto be governed by thermodynamic control.

Polymerization reactions of conjugated dienes
Practical significance: rubber and synthetic rubber production - usually radical mechanism
Buta-1,3-diene and 2-chloro-1 ,3-diene similarly polymerisable
Natural rubber
2-methyl-1,3-butadienIsoprene
Vulcanization of raw rubber
Latex: polyisoprene(n= 20-40.000) all-cis
Another synthetic rubber: polychloroprene 2-Chloro-1,3-butadien
= chloroprene

Diels-Alder-reactions of conjugated dienes (1928, Nobel prize: 1950)
AlderDiels
Known as a pericyclic reaction (one TS, simultaneous migration of electrons in a closed ring) fast reaction if X, Y are electron withdrawing groups
Some common dienophile
Stereo chemically bound reaction:
simultaneous bond formation
out of exo- and endo-products the formation
of endo is favoured

Preparation of alkenes
1. 1,2-elimination (-elimination)
Terms: X = good leaving group (LG) e.g. Hlg, OH + H, NR3
1.1 Base-induced elimination of alkyl halides
The most common bases: alkali hydroxides (NaOH/ KOH), salts of alcohols (tBuOK), tertiary amines (Et3N)
In case of elimination reaction of non-symmetrical starting compounds the formation of themost highly substituted alkene (thermodynamically more stable) is the major product of the βelimination.
Zaitsev's rule
1.2 Acid-catalyzed dehydration of alcohols
Dehydrating reagents: cc. H2SO4, KHSO4, H3PO4, AlCl3, ZnCl2, Al2O3/Typically, Zaitsev products are formed
2. Dehalogenation of 1,2-dihalo alkanes
Reagents: • Zn(Mg)/ROH/• „soft” nucleophiles (I, SH, S2, etc.)

3. Partial reduction of alkynes
4. Wittig reaction
Nobel-prize: 1979ylide = Wittig reagentgeneration:
RR’CHHlg + PPh3 then baseThe most efficient synthesis of alkenes,
aliphatic and aromatic aldehydes and ketones react well.
Preparation of industrially important dienes
1. Catalytic dehydrogenation
2. Dehydration of ethanol and its dimerization
Starting material: ethanol derived from fermentation or from petrochemical way
liq.

3. Acetylene-based isoprene production
Previously a process for the synthesis of butadiene has been developed that operates in a similar way but it is uneconomic today
Note: In the first half of the XX. century the acetylene-based procedures were widespread
C → CaC2 → HC≡CH