alkaptonuric ochronosis with aortic valve and joint
TRANSCRIPT

ABSTRACT
Alkaptonuria is a rare autosomal recessive disorder of metabolism caused bydeficiency of homogentisic acid oxidase and resulting in accumulation ofhomogentisic acid in collagenous structures. It is characterized by homogentisicaciduria, bluish-black discoloration of connective tissues (ochronosis) andarthropathy of large joints. Less common manifestations include cardiovascularabnormalities, renal, urethral and prostate calculi. Bone fractures are unusual inochronosis.
In this report, we describe a woman, 69 years of age, with a history of dark urinesince childhood and progressive pigmentation of the skin, sclera, and auricularcartilages. She had severe arthropathy requiring total joint replacement in both ofher knees and right hip. She also had severe aortic stenosis requiring valvereplacement, and asymptomatic nephrolithiasis. She presented with a low traumafracture of the distal femur despite two years of alendroate therapy.
We review the etiology, pathogenesis, clinical presentation, diagnosis andtreatment of alkaptonuric ochronosis. Early detection is important for preventionand treatment of multiple systems. Nitisinone, a potent inhibitor of 4-hydroxyphenylpyruvate dioxygenase, dramatically reduces production andurinary excretion of homogentisic acid; however, the long-term efficacy and sideeffects of such therapy are unknown. Identifying the gene for alkaptonuria offersthe potential for a new therapeutic approach (replacement therapy with arecombinant enzyme) in the treatment of alkaptonuric ochronosis.
INTRODUCTION
Alkaptonuria, a rare autosomal recessive disorder of phenylalanine and tyrosinemetabolism caused by deficiency in homogentisate 1,2-dioxygenase activity,leads to accumulation of large amounts of homogentisic acid. Homogentisic acidis excreted in urine, turning dark brown or black upon oxygenation andalkalinization. Homogentisic acid is deposited as an oxidized and polymerisedpigment (ochronotic pigment) in various tissues and organs binding irreversiblyto collagen and causing bluish-black pigmentation (ochronosis).
Alkaptonuria affects between 1 in 250,000 to 1 in 1,000,000 people,1-3 althoughin some areas such as Slovakia and the Dominican Republic, the incidence ismuch higher (e.g., up to 1 in 19,000 in Slovakia4).
209
Clinical Medicine & ResearchVolume 2, Number 4: 209-215©2004 Clinical Medicine & Research http://www.mfldclin.edu/clinmedres
AUGUST 18, 2004RECEIVED:
SEPTEMBER 14, 2004REVISED:
ACCEPTED:SEPTEMBER 28, 2004
REPRINT REQUESTS:Michael W. Davis, MBBS, FRACPDepartment of Geriatric MedicineThe Canberra HospitalPO Box 11, WODEN ACT 2606AUSTRALIATelephone: +61-2-62442926Fax: +61-2-62444036
Alkaptonuric Ochronosis with Aortic Valve and Joint Replacements and Femoral Fracture
A Case Report and Literature Review
Alexander A. Fisher, MD, FRACP, PhD, Department of Geriatric Medicine, The Canberra Hospital, Canberra, Australia.Michael W. Davis, MBBS, FRACP, Director of the Department of Geriatric Medicine, The Canberra Hospital, Canberra, Australia.
KEYWORDS:Alkaptonuria; Ochronosis; Arthroplasty, replacement;Aortic valve/surgery; Femoral fractures
CLINICAL OVERVIEWCLINICAL OVERVIEW
November 2004 Issue.qxd 11/8/04 1:52 PM Page 209

The most common clinical features are discoloration of theurine; pigmentation of the skin, sclerae, and ear cartilage;and ochronotic arthropathy affecting mainly the vertebraldiscs and large joints. Less common manifestations includerenal, urethral and prostate calculi and cardiovascularabnormalities, especially valvular disease. Few reports ofbone fracture (mainly vertebral) have been published,5-8 andnone have documented distal femur fractures.
We report a case history of a patient with severe multiplesystem ochronosis who underwent three joint replacements,an aortic valve replacement, and presented with a lowtrauma fracture of the distal femur. The disease history,etiology, pathogenesis, clinical presentation and treatmentare reviewed.
CASE REPORT
A woman, 69 years of age, was transferred from a districthospital with a low trauma fracture of her left distal femurthat she reported occurred as she walked past a tennis courtand “twisted her body” during an attempt to pick up andthrow a stray tennis ball over the fence surrounding thecourt. She stated that she heard a crack in her left leg andthen the knee gave way. She developed severe pain in the legand was unable to bear any weight on it. X-ray examinationshowed a fracture of the left distal femur.
The patient’s medical history was remarkable for progressivedegenerative arthritis affecting the hips, knees, shoulders,and spine beginning in her late 40s. A diagnosis ofochronosis was made by radiological exam, and alkaptonuriawas confirmed 10 years after the first presentation. At theage of 62 years she underwent total replacement of the righthip because of intractable pain, no improvement with non-steroidal anti-inflammatory drugs and physiotherapy,and restricted mobility and sitting tolerance of less that halfan hour. Two years later she underwent bilateral kneereplacement. Following these operations she was able towalk without walking aids. At 66 years of age, she receivedan aortic valve replacement (pericardial prosthesis) forsevere calcified aortic valvular stenosis with a peak trans-valvular gradient of 90 mm Hg. At surgery, severeannular calcification extending into the left ventricularoutflow tract and dark blue discoloration of the valve cuspsand aortic wall were noted. Microscopic examination of theaortic valve leaflets revealed nodular calcification, andochronotic dark pigment within the areas of calcificationand focally in the non-calcified valvular tissue (figure 1).Angiography demonstrated minor coronary artery disease;the left ventricle was hypertrophied but functioning well(ejection fraction 60%). A year later, at 67 years of age, shewas diagnosed with low-grade papillary transitional cellcarcinoma of the bladder. No invasion was identified onrepeated biopsies (stage pTa). The same year she sustained alow trauma fracture of the right distal radius. Treatment withalendronate and calcium was initiated at this time. Thepatient complained of chronic dull pain and stiffness in herlower back and shoulders, and previously noted brown black
Figure 2. Ochronotic pigmentation of the ear cartilage (A)and the sclera of both eyes (B).
Figure 3. A urine sample before (A) and after (B) sodiumhydroxide addition.
Figure 1. Photomicrograph showing ochronotic pigmentdeposition in aortic valve leaflet (haematoxylin and eosinstained, magnification = 100X)
210 CM&R 2004 : 4 (November) Fisher and Davis
November 2004 Issue.qxd 11/5/04 3:07 PM Page 210
pigmentation in the sclera of the eyes, face, ears and palms.Black discoloration of her undergarments was noticed sincechildhood.
The patient’s family history was unremarkable for geneticdisorders, including alkaptonuria. Her parents were notconsanguineous.
Her medications included aspirin, celecoxib, atorvastatin,calcium and alendronate for the previous 2 years. She hadnever used antimalarial, dopamine or phenolic drugs. Beforestarting the anti-osteoporotic therapy, bone mineral density T score at the femoral neck measured by dual-energy x-rayabsorptiometry was – 3.21, and after 21 months of treatmentwas – 2.60. Lumbar spine bone mineral density was elevatedon both occasions with T score values +1.92 and +1.74,respectively.
General examination revealed brown pigmentation of thesclera of both eyes (in the nasal and temporal regions), thecartilage of the external ears, the face, thenar, hypothenar,fingertips, and dorsum of both hands and the soles of thefeet (figure 2). The pigment deposits were not tender. Thevisible mucous membranes were normal. Her weight was 49 kg with a body mass index of 21.7 kg/m2. She hadmoderate thoracic kyphosis, loss of lumbar lordosis withlimited range of motion and tenderness in the mid-lowerthoracic and lumbar spine. Range of motion in bothshoulders was also reduced and more painful on the rightside. The joints of the hands and feet were not affected.
Cardiovascular examination and echocardiogram wereconsistent with an artificial aortic valve and left ventricularhypertrophy. The electrocardiogram showed sinus rhythm at75 beats/min and bifascicular block (right and left bundle
branch block). The respiratory, neurological, and abdominalexaminations were normal.
Laboratory analyses included complete blood count,erythrocyte sedimentation rate, C-reactive protein, urea,creatinine, uric acid, liver and thyroid function tests,calcium, phosphate, magnesium, parathyroid hormone (5.6 pmol/l), 25-OH vitamin D (61 nmol/l), vitamin B12,folate, and ferritin, as well as urinalysis. All tests werewithin normal ranges. Rheumatoid factor and HLA-B27were negative. The patient’s freshly passed urine turnedblack immediately after sodium hydroxide was added (figure 3).
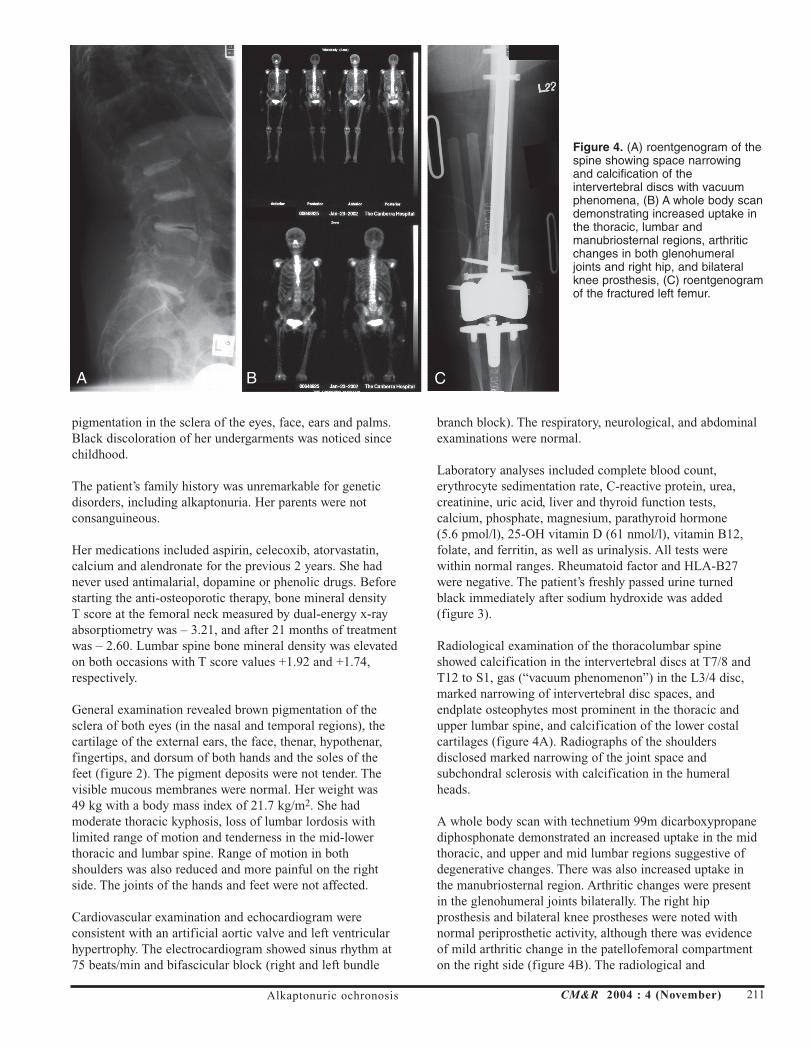
Radiological examination of the thoracolumbar spineshowed calcification in the intervertebral discs at T7/8 andT12 to S1, gas (“vacuum phenomenon”) in the L3/4 disc,marked narrowing of intervertebral disc spaces, andendplate osteophytes most prominent in the thoracic andupper lumbar spine, and calcification of the lower costalcartilages (figure 4A). Radiographs of the shouldersdisclosed marked narrowing of the joint space andsubchondral sclerosis with calcification in the humeralheads.
A whole body scan with technetium 99m dicarboxypropanediphosphonate demonstrated an increased uptake in the midthoracic, and upper and mid lumbar regions suggestive ofdegenerative changes. There was also increased uptake inthe manubriosternal region. Arthritic changes were presentin the glenohumeral joints bilaterally. The right hipprosthesis and bilateral knee prostheses were noted withnormal periprosthetic activity, although there was evidenceof mild arthritic change in the patellofemoral compartmenton the right side (figure 4B). The radiological and
Alkaptonuric ochronosis CM&R 2004 : 4 (November) 211
Figure 4. (A) roentgenogram of thespine showing space narrowingand calcification of theintervertebral discs with vacuumphenomena, (B) A whole body scandemonstrating increased uptake inthe thoracic, lumbar andmanubriosternal regions, arthriticchanges in both glenohumeraljoints and right hip, and bilateralknee prosthesis, (C) roentgenogramof the fractured left femur.
A B C
November 2004 Issue.qxd 11/5/04 3:07 PM Page 211

scintigraphic findings were confirmed by computedtomography that also showed a calcific density in the upperpole of the left kidney (a small calculus), but no renalobstruction was noted.
The patient had an open reduction with internal fixation(retrograde nail insertion) of the fractured left femur (figure 4C).Her operation and hospital stay were uneventful, and shewas discharged to her home.
DISCUSSION
The earliest verified case of ochronosis was described in anEgyptian mummy dating to 1500 B.C. Radiological andbiochemical examination of the intervertebral discs, hipsand knees was used to confirm the diagnosis.9,10 In 1584,Scribonius reported a boy who passed urine as black asink.11 The term alkaptonuria was first used in 1859 byBoedeker to describe urine discoloration due to a reducingcompound. The compound was identified as homogentisicacid in 1891 by Wolkow and Baumann.3 In 1866, Virchowcalled the condition ochronosis (meaning “yellow disease”in Greek) because the accumulated pigment in theconnective tissues appears as ochre (yellow) when examinedmicroscopically.12
At the beginning of the last century alkaptonuria was thefirst disorder to be found to conform to the applicability ofthe rediscovered Mendelian laws of autosomal recessiveinheritance13 and became a cornerstone of the fundamentalconcept of “inborn errors of metabolism” (MendelianInheritance in Man number [McKusick] 203500). A halfcentury later, the specific enzyme defect in the liver of apatient with alkaptonuria was demonstrated to be adeficiency of homogentisic acid 1,2-dioxygenase (HGO)activity, one of six enzymes required for catabolism of thearomatic amino acids phenylalanine and tyrosine.14 Thehuman HGO gene locus has been mapped to chromosome3q21-q23.2,15,16 In the last decade the HGO gene has beencloned, characterized, and its promoter region identified.17,18
A total of 84 mutations impairing this enzyme have beenfound in the HGO gene from humans and model organisms,and 43 of these mutations result in single amino acidsubstitutions.19 More than 40 different mutations have beenidentified in approximately 100 unrelated patients withalkaptonuria from many different countries.20 The mostwidespread HGO mutations are probably old mutations thathave spread throughout Europe and Asia during humanmigration.21,22
Alkaptonuria is characterized by a remarkable allelicheterogeneity. Affected persons are either homozygous orcompound heterozygous for loss of function mutation(s) inHGO. In a recent series, 46 out of 58 patients werecompound heterozygotes and no correlation between thepresence or absence of any type of HGO mutation and eitherlevel of urinary homogentisic acid excretion or severity ofdisease was found.3
Inability to convert homogentisic acid to maleylacetoaceticacid results in accumulation of homogentisic acid and aproduct of its oxidation, benzoquinone, which induces tissueinjury. This accumulation causes the classic clinical triad:(1) homogentisic aciduria which presents at birth(pathognomonic sign: urine blackens on standing whenoxidized or alkalinized); (2) gradual development ofochronosis after 20 to 30 years of age (deposition ofpolymers of oxidized homogentisic acid in connectivetissues leads to intensive eumelanin-like pigmentation ofskin, sclera, cartilages, etc); and (3) degenerative ochronicarthropathies usually in the fourth decade of life. Otherimportant but more rare consequences of alkaptonuricochronosis are cardiovascular and urinary tract involvement.
Our patient had dark urine and demonstrated extensive,progressive pigmentation of the skin, sclera, and auricularcartilages; advanced spondyloarthropathy necessitating threejoint replacements; severe aortic stenosis requiring valvereplacement; asymptomatic nephrolithiasis; and low traumafractures of the radius and distal femur.
It has been suggested that clinical manifestations ofalkaptonuric ochronosis are usually delayed, not appearinguntil the fourth decade of life because with ageing the renalclearance of homogentisic acid decreases.23 Case reports ofochronotic nephropathy and renal failure further emphasizedthe role of renal tubular secretion in eliminatinghomogentisic acid from the body.24-26 However, there wereno signs of renal impairment in our patient indicating therole of other mechanisms contributing to the severity of thedisease. Indeed, of 58 subjects with alkaptonuric ochronosis,reduced creatinine clearance was documented only in onepatient with diabetic nephropathy.3
Ochronotic spondyloarthropathy is the most commoncomplication of alkaptonuria affecting large weight bearingjoints and later the shoulders. Typically, involvement of thelarge peripheral joints usually occurs several years afterspinal involvement. It is suggested that the characteristicextensive calcification and vacuum phenomena inochronotic intervertebral discs are pathologically differentfrom degenerative disc disease and are related to cartilagebrittleness and fragmentation similar to that in the affectedperipheral joints.27 In contrast to rheumatoid arthritis, thesmall joints of the hands and feet are usually not affected,and in contrast to ankylosing spondylitis, bamboo spine,annular ossification, syndesmophytes, erosion, and fusion ofsacroiliac joints do not occur.
It was claimed that arthropathy, especially axialinvolvement, is more severe in HLA-B27-positiveindividuals.27,28 Coexistence of ochronosis and rheumatoidarthritis,29,30 ankylosing spondylitis,31,33 or chondrocalcinosis34
has also been reported.
Our patient lacked rheumatoid factor and the HLA-B27antigen. She did not have signs of rheumatoid arthritis,
212 CM&R 2004 : 4 (November) Fisher and Davis
November 2004 Issue.qxd 11/5/04 3:07 PM Page 212

ankylosing spondylitis or chondrocalcinosis. Shedemonstrated the spectrum of clinical and radiological signstypical of severe ochronotic arthropathy with involvement ofthe spine, knees, hips, and shoulders and had under wenttotal joint replacement of both knees and the right hip. In aseries of 58 ochronotic patients, 8 (13.8%) had three ormore joints replaced.3 There are two other reports of fourtotal joint replacement arthroplasties in ochronosis,35,36 andin one case seven joints had been replaced.25
To our knowledge, the case we present here is the firstreport of low trauma distal femur fracture in an ochronoticpatient. While degenerative changes of the spine and majorjoints have been frequently reported, there are only threereports of spine fractures,5,7,8 one associated with cortisonetherapy,5 and only one report of femoral neck fracture.6
In ochronosis, the changes in the bone are thought to be lesssevere than those in cartilage.37 The accumulation ofoxidized and polymerized products of homogentisic acidreduces the cross-linkage of collagen fibers leading toconnective tissue failure, cartilage erosion, and progressivedegenerative changes.23 Although ochronosis in boneinduces the same changes as in other connective tissues, theseverity appears to be limited by calcification and boneremodelling.37 It is suggested that the detrimental effects ofochronotic pigment on the fibrils of soft connective tissuesare avoided by the collagen fibrils of the bones because theyare encrusted by a mineral substance and because the newlyformed osteoid matrix remains uncalcified for too short of atime to be modified by the pigment. In an ochronoticfemoral head, the pigment was not found in osteoblasts butwas present in the calcified matrix as well as in thecytoplasmic vacuoles of osteoclasts and in osteocytes, someof which were degenerate or dead.37 In a series ofochronotic patients, the biochemical markers of boneturnover showed increased bone resorption (high urinaryexcretion of crosslinked N-telopeptides of type I collagen)with an almost normal bone formation in 6 out of 7 patientsindicating accelerated bone loss.38 Importantly, thesechanges were associated with reduced femoral bone mineraldensity. Moreover, in organ cultures of embryonic chickcalvaria it was shown that homogentisic acid inhibitsintracellular hydroxylysine formation diminishingintermolecular cross-links that are critically important forthe structural function of the newly synthesized collagen.39
It should be mentioned that in our patient, as in otherreported cases,38 while femoral neck bone mineral densitywas markedly reduced, the lumbar spine bone mineraldensity was normal or increased. This seeming paradoxmight be due to extensive intervertebral disc calcification.
The case we present demonstrates that in ochronosis bonystructures may be severely affected. Our patient had twonon-vertebral fractures (distal radium and distal femur)within two years time, but did not have vitamin D deficiencyor secondary hyperparathyroidism. She has no other riskfactors for osteoporosis such as malnutrition, immobility,smoking, medications (corticosteroids, anticonvulsants), or
family history of osteoporotic fractures. Moreover, she hasreceived alendronate therapy for the last two years withimprovement in bone mineral density. It now seems practicalthat adequate antiresorptive therapy to prevent bonefractures in ochronotic patients should be considered muchearlier in the course of treatment.
Our patient has also had a severe aortic stenosis withcalcified valves and gross pigmentation of the aorta but nocoronary artery involvement. Some case series have showedno increase in frequency of calcification and stenosis ofaortic valves or coronary artery disease,40,41 but numerousother observations suggest that ochronosis may beassociated with pigment deposition in aortic and mitralvalves, endocardium, pericardium, aortic intima, coronaryarteries and especially with valvular dystrophic calcification,aortic stenosis, and coronary disease.42-49 In a series of 58ochronotic patients, 3 (5.2%) had aortic valve replacement,and 50% had computed tomographic evidence of coronaryartery calcification by 59 years of age.3
Patients with alkaptonuria are known to be at increased riskof nephrolithiasis. Kidney stones caused by ochronosis werereported in 16 out of 58 patients (27.6%), and more often inmales.3 An asymptomatic small calculus in the left kidneywas documented in our patient.
It is worth mentioning that clinical variability ofalkaptonuric ochronosis, which reflects the spectrum ofHGO mutations, may delay the diagnosis and lead tomismanagement including unnecessary biopsies and surgery.In one case ocular ochronotic pigmentation wasmisdiagnosed as melanosarcoma and one eye wasmistakenly enucleated.50
Although the diagnostic confirmation of alkaptonuria iseasily made by alkanization of urine with quantitativedetermination of homogentisic acid in urine available, only21% of patients are diagnosed before 1 year of age.3 In arecently reported case,51 despite marked mucocutaneouspigmentation, advanced spondyloarthropathy, unilateral hipreplacement, and blackened urine on standing, the diagnosiswas not established until the patient was 82 years of age.Moreover, of 755 respondents to a “medical mystery”published in the New England Journal of Medicine51 only43% correctly diagnosed alkaptonuric ochronosis, while23% thought of melanoma and 23% of porphyria orporphyria cutanea tarda. Paroxysmal nocturnalhaemoglobinuria and ingestion of arsenic or silver wereamong other responses. Pseudo-ochronosis, which is not aninherited disorder, has been described as a result of argyria52
and long term use of levodopa, methyldopa,53 antimalarials,or products containing hydroquinone, phenol, resorcinol,mercury or picric acid.54
Currently there is no specific and effective treatment foralkaptonuria. Although some advocate dietary proteinrestriction (mainly phenylalanine and tyrosine), and ascorbicacid to reduce urinary homogentisic acid excretion and
Alkaptonuric ochronosis CM&R 2004 : 4 (November) 213
November 2004 Issue.qxd 11/5/04 3:07 PM Page 213

possibly reverse bone abnormalities,55 these observationshave not been confirmed in other studies.3,56
A direct pharmacologic reduction of homogentisic acidproduction could be achieved with nitisinone therapy.Nitisinone is a triketone herbicide and potent inhibitor of 4-hydroxy-phenylpyruvate dioxygenase which is responsiblefor catalyzing the formation of homogentisic acid fromhydroxyphenylpyruvic acid. Nitisinone reduced urinaryhomogentisic acid excretion by approximately 70% in twopatients with alkaptonuria. Long-term side effects ofnitisinone therapy are under consideration.3
Understanding the genetic and molecular basis ofalkaptonuria has the potential to offer a new therapeuticapproach, enzyme replacement therapy with recombinantHGO. However, despite the theoretical advantage, such astrategy may be difficult to employ. Moreover, it is notknown whether accumulation of toxic metabolites oftyrosine will occur, thus excluding this as an acceptablealternative therapy. Before human trials can be undertaken,therapies would need to be carefully tested in animalmodels.
In advanced cases, such as the one we present here, surgicalreplacement of joints and aortic valves result in significantimprovement. Usually the disorder does not affect life span.Physiotherapy, analgesia, and adequate anti-osteoporotictherapy will be continued in our patient to prevent furtherdisability.
In conclusion, diagnosis and management of patients withalkaptonuric ochronosis, a rare inherited disorder, iscomplex. Advances in orthopaedic and cardiac surgery haveenabled many patients to overcome progressive disability.Physicians and surgeons should be aware of multiple systeminvolvement in this disorder, as early recognition andappropriate treatment may significantly improve the qualityof life in these patients.
ACKNOWLEDGEMENTS
The authors wish to thank Lyn Cridland and Kim Forsythfor technical assistance in the preparation of this manuscript,and James Davison for his help in preparation of the figures.
REFERENCES1. Kottinen YT, Hoikka V, Landtman M, Saari H, Santavirta S,
Metsarinne K, Seegmiller JE. Ochronosis: a report of a caseand a review of literature. Clin Exp Rheumatol 1989;7:435-444.
2. Janocha S, Wolz W, Srsen S, Srsnova K, Montagutelli X, GuenetJL, Grimm T, Kress W, Mueller CR. The human gene foralkaptonuria (AKU) maps to chromosome 3q. Genomics1994;19:5-8.
3. Phornphutkul C, Introne WJ, Perry MB, Bernardini I, MurpheyMD, Fitzpatrick DL, Anderson PD, Huizing M, Anikster Y,Gerber LH, Gahl WA. Natural history of alkaptonuria. NEngl J Med 2002;347:2111-2121.
4. Zatkova A, Chmelikova A, Polakova H, Ferakova E, Kadasi L.Rapid detection methods for five HGO gene mutationscausing alkaptonuria. Clin Genet 2003;63:145-149.
5. Savastano AA, Quigley DG, Scala ME. Spontaneous fracturesassociated with cortisone therapy in a patient withochronosis. Am J Orthop Surg 1969;11:116-120.
6. Waschulewski H. [Medial femoral neck fracture in ochronoticalkaptonuria]. Zentralbl Chir 1969;94:806-810.
7. Millea TP, Segal LS, Liss RG, Stauffer ES. Spine fracture inochronosis. Report of a case. Clin Orthop 1992:208-211.
8. Marsile C, Menozzi C, Menozzi C. [A rare case of high dorsalradicular-medullary compression in a patient with ochronoticarthropathy. Clinico-radiological features]. Minerva Med1995;86:61-66.
9. Stenn FF, Milgram JW, Lee SL, Weigand RJ, Veis A.Biochemical identification of homogentisic acid pigment inan ochronotic egyptian mummy. Science 1977;197:566-568.
10. Lee SL, Stenn FF. Characterization of mummy bone ochronoticpigment. JAMA 1978;240:136-138.
11. Gaines JJ, Jr. The pathology of alkaptonuric ochronosis. HumPathol 1989;20:40-46.
12. Vijaikumar M, Thappa DM, Srikanth S, Sethuraman G,Nadarajan S. Alkaptonuric ochronosis presenting aspalmoplantar pigmentation. Clin Exp Dermatol 2000;25:305-307.
13. Garrod E. The incidence of alkaptonuria: a study in chemicalindividuality. Lancet 1902;2:1616-1620.
14. La Du BN, Seegmiller JE, Laster L, Zannoni V. Alcaptonuriaand ochronotic arthritis. Bull Rheum Dis 1958;8:163-164.
15. Pollak MR, Chou YH, Cerda JJ, Steinmann B, La Du BN,Seidman JG, Seidman CD. Homozygosity mapping of thegene for alkaptonuria to chromosome 3q2. Nat Genet1993;5:201-204.
16. Fernandez-Canon JM, Granadino B, Beltran-Valero de BernabeD, Renedo M, Fernandez-Ruiz E, Penalva MA, Rodriguez deCordoba S. The molecular basis of alkaptonuria. Nat Genet1996;14:19-24.
17. Fernandez-Canon JM, Penalva MA. Molecular characterizationof a gene encoding a homogentisate dioxygenase fromAspergillus nidulans and identification of its human andplant homologues. J Biol Chem 1995;270:21199-21205.
18. Granadino B, Beltran-Valero de Bernabe D, Fernandez-CanonJM, Penalva MA, Rodriguez de Cordoba S. The humanhomogentisate 1,2-dioxygenase (HGO) gene. Genomics1997;43:115-122.
19. Rodriguez JM, Timm DE, Titus GP, Beltran-Valero De BernabeD, Criado O, Mueller HA, Rodriguez De Cordoba S, PenalvaMA. Structural and functional analysis of mutations inalkaptonuria. Hum Mol Genet 2000;9:2341-2350.
20. Goicoechea De Jorge E, Lorda I, Gallardo ME, Perez B, PerezDe Ferran C, Mendoza H, Rodriguez De Cordoba S.Alkaptonuria in the Dominican Republic: identification ofthe founder AKU mutation and further evidence of mutationhot spots in the HGO gene. J Med Genet 2002;39:E40.
21. Uyguner O, Goicoechea de Jorge E, Cefle A, Baykal T,Kayserili H, Cefle K, Demirkol M, Yuksel-Apak M,Rodriguez de Cordoba S, Wollnik B. Molecular analyses ofthe HGO gene mutations in Turkish alkaptonuria patientssuggest that the R58fs mutation originated from central Asiaand was spread throughout Europe and Anatolia by humanmigrations. J Inherit Metab Dis 2003;26:17-23.
22. Beltran-Valero de Bernabe D, Granadino B, Chiarelli I, PorfirioB, Mayatepek E, Aquaron R, Moore MM, Festen JJ, SanmartiR, Penalva MA, de Cordoba SR. Mutation and polymorphismanalysis of the human homogentisate 1,2-dioxygenase genein alkaptonuria patients. Am J Hum Genet 1998;62:776-784.
23. Hamdi N, Cooke TD, Hassan B. Ochronotic arthropathy: casereport and review of the literature. Int Orthop 1999;23:122-125.
24. Venkataseshan VS, Chandra B, Graziano V, Steinlauf P,Marquet E, Irmiere V, Needle MA. Alkaptonuria and renalfailure: a case report and review of the literature. Mod Pathol1992;5:464-471.
214 CM&R 2004 : 4 (November) Fisher and Davis
November 2004 Issue.qxd 11/5/04 3:07 PM Page 214

25. Jagose JT, Bailey RR, Rothwell AG. Alkaptonuria withochronotic nephropathy and multiple joint replacement forochronotic arthropathy. N Z Med J 1997;110:235-236.
26. Introne WJ, Phornphutkul C, Bernardini I, McLaughlin K,Fitzpatrick D, Gahl WA. Exacerbation of the ochronosis ofalkaptonuria due to renal insufficiency and improvementafter renal transplantation. Mol Genet Metab 2002;77:136-142.
27. Borman P, Bodur H, Ciliz D. Ochronotic arthropathy.Rheumatol Int 2002;21:205-209.
28. Kabasakal Y, Kiyici I, Ozmen D, Yagci A, Gumusdis G. Spinalabnormalities similar to ankylosing spondylitis in a 58-year-old woman with ochronosis. Clin Rheumatol 1995;14:355-357.
29. Kihara T, Yasuda M, Watanabe H, Suenaga Y, Shiokawa S,Wada T, Nonaka S, Suzuki T, Nobunaga M. Coexistence ofochronosis and rheumatoid arthritis. Clin Rheumatol1994;13:135-138.
30. Simianer S, Krause D, Rau R. [Concomitant manifestation ofochronosis and chronic polyarthritis in a patient]. ZRheumatol 1998;57:50-52.
31. Gemignani G, Olivieri I, Semeria R, Giustarini S, Pasero G.Coexistence of ochronosis and ankylosing spondylitis. JRheumatol 1990;17:1707-1709.
32. Yagan R, Khan MA. The coexistence of ochronosis andankylosing spondylitis. J Rheumatol 1991;18:1639-1640.
33. Weinberger KA. The coexistence of ochronosis and ankylosingspondylitis. J Rheumatol 1991;18:1948-1949.
34. Roth A, Schmidt K, Muller KM, Haaker R. [A case report:ochronosis in combination with chondrocalcinosis]. Z OrthopIhre Grenzgeb 1999;137:76-78.
35. Carrier DA, Harris CM. Bilateral hip and bilateral kneearthroplasties in a patient with ochronotic arthropathy.Orthop Rev 1990;19:1005-1009.
36. Demir S. Alkaptonuric ochronosis: a case with multiple jointreplacement arthroplasties. Clin Rheumatol 2003;22:437-439.
37. Di Franco M, Coari G, Bonucci E. A morphological study ofbone and articular cartilage in ochronosis. Virchows Arch2000;436:74-81.
38. Aliberti G, Pulignano I, Schiappoli A, Minisola S, RomagnoliE, Proietta M. Bone metabolism in ochronotic patients. JIntern Med 2003;254:296-300.
39. Murray JC, Lindberg KA, Pinnell SR. In vitro inhibition ofchick embryo lysyl hydroxylase by homogentisic acid. Aproposed connective tissue defect in alkaptonuria. J ClinInvest 1977;59:1071-1079.
40. O’Brien WM, La Du BN, Bunim JJ. Biochemical, pathologicand clinical aspects of alcaptonuria, ochronosis andochotronotic arthropathy. Review of world literature (1584-1962). Am J Med 1963;34:813-838.
41. Cortina R, Moris C, Astudillo A, Gosalbez F, Cortina A.Familial ochronosis. Eur Heart J 1995;16:285-286.
42. Vlay SC, Hartman AR, Culliford AT. Alkaptonuria and aorticstenosis. Ann Intern Med 1986;104:446.
43. Gaines JJ Jr, Pai GM. Cardiovascular ochronosis. Arch PatholLab Med 1987;111:991-994.
44. Dereymaeker L, Van Parijs G, Bayart M, Daenen W,Lauwerijns J, De Geest H. Ochronosis and alkaptonuria:report of a new case with calcified aortic valve stenosis. ActaCardiol 1990;45:87-92.
45. Kragel AH, Lapa JA, Roberts WC. Cardiovascular findings inalkaptonuric ochronosis. Am Heart J 1990;120(6 Pt 1):1460-1463.
46. Kenny D, Ptacin MJ, Bamrah VS, Almagro U. Cardiovascularochronosis: a case report and review of the medical literature.Cardiology 1990;77:477-483.
47. Helou J, Masters RG, Keon WJ, Veinot JP. Ochronosis: anunusual finding at aortic valve replacement. Can J Cardiol1999;15:1013-1015.
48. Zund G, Schmid AC, Vogt PR, Grunenfelder J, Turina MI.Green aortic valve: alcaptonuria (ochronosis) with severeaortic stenosis. Ann Thorac Surg 1999;67:1805.
49. Cercek M, Prokselj K, Kozelj M. Aortic valve stenosis inalkaptonuric ochronosis. J Heart Valve Dis 2002;11:386-388.
50. Soker Cakmak S, Cevik R, Aksunger A, Unlu K, Ava S. Ocularochronosis: A case report and clinical findings. ActaOphthalmol Scand 2002;80:340-342.
51. Nikkels AF, Pierard GE. Medical mystery: the answer. N Engl JMed 2001;344:1642-1643.
52. Robinson-Bostom L, Pomerantz D, Wilkel C, Mader R, LernerL, Dufresne R, Flotte T. Localized argyria with pseudo-ochronosis. J Am Acad Dermatol 2002;46:222-227.
53. Rausing A, Rosen U. Black cartilage after therapy withlevodopa and methyldopa. Arch Pathol Lab Med1994;118:531-535.
54. Levin CY, Maibach H. Exogenous ochronosis. An update onclinical features, causative agents and treatment options. AmJ Clin Dermatol 2001;2:213-217.
55. Morava E, Kosztolanyi G, Engelke UF, Wevers RA. Reversal ofclinical symptoms and radiographic abnormalities withprotein restriction and ascorbic acid in alkaptonuria. AnnClin Biochem 2003;40:108-111.
56. Forslind K, Wollheim FA, Akesson B, Rydholm U.Alkaptonuria and ochronosis in three siblings. Ascorbic acidtreatment monitored by urinary HGA excretion. Clin ExpRheumatol 1988;6:289-292.
Alkaptonuric ochronosis CM&R 2004 : 4 (November) 215
November 2004 Issue.qxd 11/5/04 3:07 PM Page 215