akt activation mediates acquired resistance to fibroblast ...small molecule therapeutics akt...
TRANSCRIPT

Small Molecule Therapeutics
Akt Activation Mediates Acquired Resistanceto Fibroblast Growth Factor ReceptorInhibitor BGJ398Jharna Datta1, Senthilkumar Damodaran1,2, Hannah Parks1, Cristina Ocrainiciuc1,Jharna Miya1, Lianbo Yu3, Elijah P. Gardner1, Eric Samorodnitsky1, Michele R.Wing1,Darshna Bhatt1, John Hays1,2, Julie W. Reeser1, and Sameek Roychowdhury1,2,4
Abstract
Activation of FGFR signaling through mutations, amplifica-tions, or fusions involving FGFR1, 2, 3, or 4 is seen in multipletumors, including lung, bladder, and cholangiocarcinoma.Currently, several clinical trials are evaluating the role of novelFGFR inhibitors in solid tumors. As we move forward withFGFR inhibitors clinically, we anticipate the emergence ofresistance with treatment. Consequently, we sought to studythe mechanism(s) of acquired resistance to FGFR inhibitorsusing annotated cancer cell lines. We identified cancer cell linesthat have activating mutations in FGFR1, 2, or 3 and treatedthem chronically with the selective FGFR inhibitor, BGJ398.We observed resistance to chronic BGJ398 exposure in
DMS114 (small-cell lung cancer, FGFR1 amplification) andRT112 (urothelial carcinoma, FGFR3 fusion/amplification) celllines based on viability assays. Reverse-phase protein array(RPPA) analysis showed increased phosphorylation of Akt(T308 and S473) and its downstream target GSK3 (S9 andS21) in both the resistant cell lines when compared withmatching controls. Results of RPPA were confirmed usingimmunoblots. Consequently, the addition of an Akt inhibitor(GSK2141795) or siRNA was able to restore sensitivityto BGJ398 in resistant cell lines. These data suggest a role for Aktpathway in mediating acquired resistance to FGFR inhibition.Mol Cancer Ther; 16(4); 614–24. �2017 AACR.
IntroductionFGFRs play essential roles in mediating cell proliferation,
migration, and survival (1). FGFR belongs to the receptor tyrosinekinase (RTK) family of proteins that also includes EGFR andVEGFR family. The FGFR family is comprised of four receptors,FGFR1, FGFR2, FGFR3, and FGFR4 with 18 known ligands.Ligand binding leads to dimerization and conformational changein the receptor, resulting in phosphorylation of tyrosine kinasedomains. The phosphorylated tyrosine residues in turn act as adocking site for FGFR substrate 2 (FRS2), a vital adapter protein inthe FGFR signaling cascade. Phosphorylation of FRS2 leads to
recruitment of adapter proteins, such as SOS, GRB2, and GAB1,resulting in the activation of downstream MAPK or PI3K–Aktpathways.
Deregulation and activation of FGFR signaling has been iden-tified in multiple cancers that include bladder, lung, biliary,prostate, and breast (1, 2). FGFR1 amplification has been reportedin 20% of squamous cell carcinoma of the lung (3). FGFR3mutations are reported in approximately 70% of low-gradeurothelial carcinomas (4). In addition to oncogenic fusionsinvolving FGFR3 in bladder cancers, FGFR3 fusions have alsobeen reported in breast, biliary, and prostate cancers (5, 6).
Preclinical studies have shown that FGFR alterations predictsensitivity to FGFR inhibitors (7, 8). Multiple clinical trials toevaluate FGFR inhibitors, both selective and nonselective, arecurrently in progress. A phase II trial of dovitinib (TKI258), anFGFR1, FGFR2, and FGFR3 inhibitor, showed activity in estro-gen receptor–positive (ERþ), FGFR1-amplified breast cancers(9). However, second-line treatment of dovitinib in FGFR2-mutated advanced endometrial cancers did not show anysignificant activity (10). Lucitanib, an oral, FGFR1/2/3 andVEGFR inhibitor, has shown activity in FGFR1 and FGF3/4/19-amplified tumors (11). Also, JNJ-42756493, an oral pan-FGFR inhibitor, showed significant activity in advanced tumorswith FGFR alterations in a phase I dose escalation study (12). Asstudies with other small-molecule inhibitors have illustrated,despite initial response rates, tumor cells acquire resistancemechanisms with chronic exposure. Therefore, it is importantto be cognizant of emerging resistance with treatment.Although FGFR alterations appear to predict sensitivity tomatching inhibitors, little is known about acquired resistancemechanisms to FGFR inhibition. Consequently, we sought to
1Comprehensive Cancer Center, The Ohio State University, Columbus, Ohio.2Division of Medical Oncology, Department of Internal Medicine, The Ohio StateUniversity, Columbus, Ohio. 3Center for Biostatistics, Department of BiomedicalInformatics, The Ohio State University, Columbus, Ohio. 4Department of Phar-macology, The Ohio State University, Columbus, Ohio.
Note: Supplementary data for this article are available at Molecular CancerTherapeutics Online (http://mct.aacrjournals.org/).
J. Datta and S. Damodaran contributed equally to this article.
Current address for S. Damodaran: Departments of BreastMedicalOncology andInvestigational Cancer Therapeutics, UT MD Anderson Cancer Center, Houston,TX.
Corresponding Author: Sameek Roychowdhury, The Ohio State UniversityComprehensive Cancer Center, 460 West 12th Avenue, Columbus, OH 43210.Phone: 614-685-5842; Fax: 614-292-6356; E-mail:[email protected]
doi: 10.1158/1535-7163.MCT-15-1010
�2017 American Association for Cancer Research.
MolecularCancerTherapeutics
Mol Cancer Ther; 16(4) April 2017614
on April 11, 2017. © 2017 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst March 2, 2017; DOI: 10.1158/1535-7163.MCT-15-1010

identify mechanisms of acquired resistance to FGFR inhibitorsutilizing a high-throughput proteomic approach. In this article,we characterize Akt pathway activation in annotated cancer celllines with activating FGFR alterations as a mechanism ofacquired resistance to BGJ398, a selective, oral, pan-FGFRinhibitor.
Materials and MethodsCell lines, reagents, and antibodies
Human cancer cell lines DMS114 (small-cell lung cancer;FGFR1 amplification) and RT112 (bladder cancer; FGFR3 fusionand amplification) were obtained from ATCC and CLS, respec-tively, during December 2012. All the cell lines (control andresistant) were authenticated at the University of Arizona GeneticCore facility (STR profiling) in February 2016. BGJ398 waspurchased from Selleck Chemicals. Akt inhibitor, GSK2141795,was purchased fromMedChem Express. Inhibitors were preparedas 10mmol/L stock solutions inDMSO.CellTiter-Glo reagentwaspurchased from Promega Corporation. Lipofectamine 2000 wasobtained from Invitrogen. Control and Akt siRNA were obtainedfrom Cell Signaling Technology. Antibodies to phospho-FRS2a(y196), FGFR1,GSK3a, GSK3b, p-GSK3a/b S9/S21, p-GSK3b S9,p-Akt S473, p-Akt T308, pan-Akt, pMEK1/2, MEK1/2, pERK1/2,ERK1/2, pYAP-S127, YAP, TSC1, Cyclin B1, and FOXM1 anti-bodieswere obtained fromCell Signaling Technology. Antibodiesagainst FGFR3, FRS2a, and GAPDH were obtained from SantaCruz Biotechnology. Antibodies to p-FGFR1 (y653/y654) fromEMD Millipore and p-FGFR3 (y724) from Abcam were alsoutilized.
Cell cultureDMS114 cell line was cultured in Waymouth's medium
supplemented with 10% heat-inactivated FBS, 1% penicillin(100 U/mL), and streptomycin (100 mg/mL). Similarly, RT112cells were grown in RPMI1640 and 2 mmol/L L-glutamine. Cellswere grown as monolayer cultures and maintained in a humid-ified atmosphere of 5% CO2 in air at 37�C. Cell morphologywas monitored using an EVOS XL Core Cell Imaging Systemunder a phase-contrast microscope (Life Technologies). Cellswere routinely tested for mycoplasma contamination monthlyusing the MycoAlert Plus Mycoplasma Detection Kit (Lonza)following the manufacturer's protocol. Cells were last tested inMay 2016.
Cell viability and generation of resistanceCell lines were treated chronically at a fixed concentration
(DMS114; 3 mmol/L) or gradually at increasing concentrations(RT112) with BGJ398. Control cells were treated with vehicleDMSO. To assess the development of resistance, cell cultures weresampled every 4 to 6 weeks and assayed for viability with theCellTiter-Glo assay (Promega). For viability assays, cells wereseeded (in quadruplicates) in 96-well plates at a density of2,000 to 3,000 cells per well in a volume of 100 mL. Twenty-fourhours later, media containing BGJ398 or GSK2141795 at variousdilutions or DMSO were added. After 72 hours, CellTiter-Gloreagent was added and luminescence wasmeasured following themanufacturer's protocol. Cell viability was evaluated as percent-age relative to vehicle controls (100%). Viability curves wereplotted using GraphPad Prism software. Upon manifesting resis-tance, cell lines weremaintained with continued drug exposure atconcentrations that displayed resistance.
Western blotsFor immunoblots, cell lysates were isolated and homogenized
in RIPA buffer (10mmol/L Tris-HCl, pH7.5, 150mmol/LNaCl, 5mmol/L EDTA, 0.5% SDS, 1.0% NP40, 1.0% sodium deoxycho-late, containing protease and phosphatase inhibitors). Total cel-lular protein was prepared, mixed with 4� Laemmli's buffer,boiled at 97�C for 5 minutes, and then separated on SDS-poly-acrylamide gels. Proteins were transferred onto nitrocellulosemembranes and probed with antibodies described above. Afterovernight incubation at 4�C, membranes were blotted for onehour with HRP-conjugated secondary anti-rabbit/mouse antibo-dies (1:2,500). Next, bound antibody complexes were detectedand visualized using enhanced chemiluminescence substratedetection system (Bio-Rad). Blots were stripped and reprobedwith GAPDH antibody to control for loading. The intensity of theprotein bands was quantified by ImageJ software (https://imagej.nih.gov/ij/).
Reverse-phase protein arrayControl and resistant cell lines were plated in 60-mm culture
plates. After 24hours, cellswere lysedusing lysis buffer (1%TritonX-100, 50mmol/L HEPES pH 7.4, 150 nmol/L NaCl, 1.5mmol/LMgCl2, 1 mmol/L EGTA, 100 mmol/L sodium pyrophosphate, 1mmol/L Na3VO4, 10% glycerol, phosphatase and protease inhi-bitors) and centrifuged at 4�C for 10 minutes at 12,700� g. Afterprotein concentrations were quantified, the lysate wasmixedwith4� SDS sample buffer (40% glycerol, 8% SDS, 0.25 mol/L Tris-HCl pH 6.8), boiled for 5 minutes, and stored at �80�C. Subse-quently, samples were sent to the Functional Proteomics CoreFacility at MD Anderson for reverse-phase protein array (RPPA)analysis.
Briefly, cellular proteins were denatured by 1% SDS (withb-mercaptoethanol) and diluted in five 2-fold serial dilutions(from undiluted to 1:16 dilution) in dilution buffer (lysisbuffer containing 1% SDS). Serial diluted lysates were arrayedon nitrocellulose-coated slides (Grace Bio-Labs) by Aushon2470 Arrayer (Aushon BioSystems). Each slide was probedwith a validated primary antibody plus a biotin-conjugatedsecondary antibody. Only antibodies with a Pearson correla-tion coefficient between RPPA and Western blotting of greaterthan 0.7 were used for analysis. The signal obtained wasamplified using a DakoCytomation Catalyzed Signal Amplifi-cation System (Dako) and visualized by diaminobenzidinecolorimetric reaction. The slides were scanned, analyzed, andquantified using a customized software MicroVigene (Vigene-Tech Inc.) to generate spot intensity. Spots from TIFF imageswere identified, and the density was quantified by Array-ProAnalyzer software. Each dilution curve was fitted with a logisticmodel (supercurve fitting) developed by the Department ofBioinformatics and Computational Biology in MD AndersonCancer Center (Houston, TX). All the data points were nor-malized for protein loading and transformed to linear valuesfor analysis.
RNA sequencingWe used 4 mg of each cell line's total RNA and prepared the
libraries following Illumina's TruSeq Stranded mRNA SamplePreparation HS (high sample) protocol without modification.We performed 15 cycles of PCR for amplification of the adapter-ligated DNA library and assessed the quality of the final DNAlibrary using the High Sensitivity D1K ScreenTape (TapeStation
Akt Activation Confers Acquired Resistance to FGFR Inhibitor
www.aacrjournals.org Mol Cancer Ther; 16(4) April 2017 615
on April 11, 2017. © 2017 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst March 2, 2017; DOI: 10.1158/1535-7163.MCT-15-1010

2200; Agilent Technologies). Per manufacturer's protocol, librarypeak size was approximately at 260 bp. We prepared indexedlibraries and sent them for 100-bp paired-end sequencing (2 �100 bp) on an Illumina HiSeq 2000 at Beijing Genomics Institute(China). All four (DMS114, RT112 controls and respective resis-tant cells) libraries were pooled and sequenced in one lane. Geoaccession number is GSE92651.
RNAi assayResistant DMS114 and RT112 cells were plated for 24 hours
andwere transfectedwith control or Akt-siRNA targeting Akt1 andAkt2 (60–100 nmol/L) using lipofectamine 2000 following themanufacturer's protocol (Invitrogen). Cells were harvested after48hours for protein immunoblot analysis, colony formation, andinvasion assays.
Clonogenic survival assayClonogenic survival assay was performed as described previ-
ously with minor modifications (13). Briefly, equal number ofcontrol siRNA- or Akt siRNA–treated cells were allowed to grow incomplete growth media on 60-mm culture plates until visiblecolonies were formed (12–14 days). Next, cell colonies wererinsed with PBS, fixed with 4% paraformaldehyde, stained withcrystal violet solution, washed with water, and air-dried. Visiblecolonies were counted manually.
Cell proliferation assayFor cell proliferation assay, resistant cell lines plated on 96
strip–well plate were transfected as described above. Cell viabilitywas monitored every 24 hours for 5 days using the CellTiter-Gloassay. Ratios of cell viability for each day relative to day 1 (24hours after transfection) were plotted.
Cell motility assayCell motility assays were carried out with the resistant cells
transfected in 6-well culture plates (14). After 24 hours oftransfection, at approximately 90% confluence, cells werewashed with PBS and a fine scratch in the form of a groovewas made with the help of a sterile pipette tip and immediatelyphotographed (time 0 hour). Next, cells were supplementedwith complete growth medium and allowed to grow. Migrationof cells from the edge of the groove toward the center wasmonitored at 24 hours (�100 magnification). The width of thescratch was measured at 0 hour and at 24 hours to calculate thepercentage of the gap covered by the cells in a 24-hour timeperiod.
Transwell invasion assayDMS114 and RT112 resistant cells were treated with control
and Akt-specific siRNA as described above. Matrigel invasionassay was performed as described previously (15). Briefly, 48hours after transfection, equal number of control and AktsiRNA–treated resistant cells (30,000) were seeded per well inthe Matrigel invasion chambers (Becton Dickinson) andallowed to invade toward 10% FBS containing media (in thebottom of the well). Following incubation of the plate for 24hours, the noninvaded cells were removed, and the invadedcells at the bottom of the chambers were fixed, stained withviolet, washed, and then counted using an inverted microscope(�100 magnification).
RNA sequencing analysisTo calculate gene expression, TopHat2 (version 2.0.10) was
used for aligning the FASTQ files to the human reference genomeUCSC build hg19 assembly (16). A UCSC gene annotation file inGTF format was also used during the alignment. The aligned BAMfile fromTopHat2was fed intoRNASeqQC (version 1.1.7; https://www.broadinstitute.org/cancer/cga/rnaseqc_download) to gen-erate alignment metrics. Gene expression for known genes wascalculated as FPKM (fragments per kilobase per million mappedreads) using CuffLinks (version 2.1.1) from the Tuxedo suite,whereas the differentially significant genes from a parent andresistant cell line comparison were found using Cuffdiff (version2.1.1; ref. 17). The gene annotation file from UCSC was providedto keep the gene format consistent throughout the pipeline.
Statistical analysisDescriptive statistics (mean, SD) were used to describe parental
and resistant cell lines in viability curves. To compare differencesin protein phosphorylation between the parent and resistant celllines in RPPA, two-sample t test (with equal variances) wasutilized. To adjust P values for multiple comparisons, FDR usingthe Benjamini–Hochbergmethodwas employed (18). A cutoff ofFDR <0.05 (q value) was used as a threshold for statisticalsignificance. Microsoft Excel and R (www.r-project.org; v 2.5.0)were used for FDR analysis.
Principal component analysis (PCA) was used to explore thestructure of the samples based on protein profiling data in RPPA(19). The samples were projected onto the principle components(i.e., PC1, PC2) for each sample, where PC1 and PC2 are the firsttwo dimensions with the largest variation in the expression dataand are the linear combinations of all proteins' expression. Topproteins with largest factor loadings on PC1 and PC2 are iden-tified. Protein expression heatmaps with the imposed two-wayhierarchical clustering (based on average linkage and Euclideandistance) were generated for each cell line separately. Heatmapswere generated using MeV 4.9. Statistical analyses for PCA wereperformed in R 3.2.
ResultsBGJ398 inhibits FGFR signaling pathway
We assessed the levels of FGFR 1 and 3 in DMS114 and RT112cell lines by RNA sequencing (RNA-Seq) analysis (SupplementaryTable S1).Western blot analysis confirmed that RT112 has FGFR3expression and no FGFR1, whereas DMS114 has mainly FGFR1with a small amount of FGFR3 expression (Supplementary Fig.S1). Next, we assessed the effect of BGJ398 on total and phos-phorylated forms of FGFR (FGFR1 for DMS114 and FGFR3 forRT112) and its substrate FRS2 (Fig. 1). Western blots showeddose-dependent decreases in phosphorylation of FGFR and FRS2in both the cell lines, demonstrating that BGJ398 (SupplementaryFig. S2) inhibits FGFR signaling. The levels of total FGFR1/FGFR3and FRS2 were also significantly decreased in a dose-dependentmanner. On the other hand, TotalMEK and ERKwere not affectedeven at higher doses indicating FGFR specificity (Fig. 1).
Development of resistance to BGJ398We cultured DMS114 and RT112 cell lines, in the presence of
DMSO (vehicle control) or BGJ398 at fixed concentration orgradually increasing concentrations, chronically to stimulateacquired resistance. With prolonged exposure, both resistant cell
Datta et al.
Mol Cancer Ther; 16(4) April 2017 Molecular Cancer Therapeutics616
on April 11, 2017. © 2017 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst March 2, 2017; DOI: 10.1158/1535-7163.MCT-15-1010

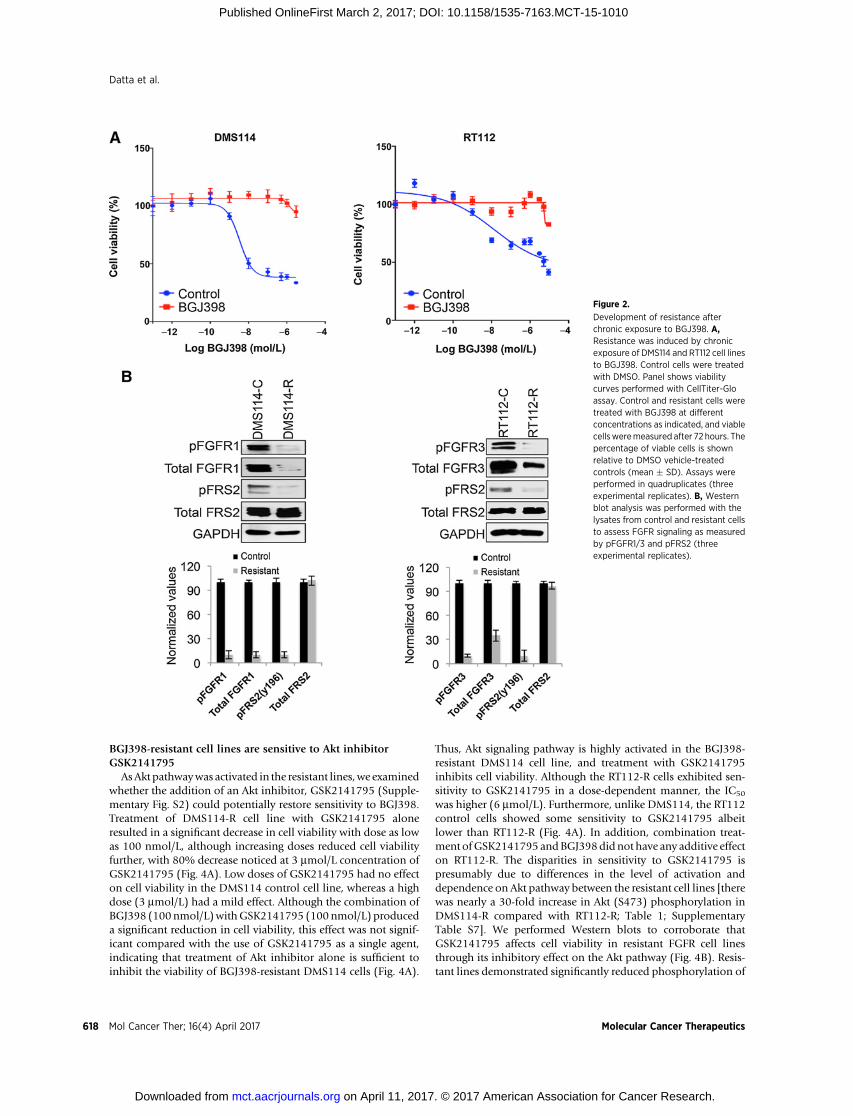
lines displayed marked differences in the sensitivity to BGJ398compared with the corresponding controls (Fig. 2A). Morpho-logically, resistant cell lines were irregular, elongated with protru-sions, whereas the control cells were regular and remained inclusters, as described previously (Supplementary Fig. S3; ref. 20).Next, we performed Western blots to evaluate the total andphosphorylated forms of FGFR1/FGFR3 (DMS114/RT112) andFRS2 in the control and resistant cell lines.Weobserved significantdecreases in pFGFR1or 3, total FGFR1or 3, andpFRS2 inDMS114and RT112 cell lines compared with controls (Fig. 2B) However,there was no significant change in total FRS2. Western blottingdata corroborated that BGJ398 continued to block FGFR activityin both the resistant cell lines (Fig. 2B) without any appreciableeffect on cell viability (Fig. 2A).
RPPAdemonstrates Akt activation in BGJ398-resistant cell linesTo identify differentially phosphorylated proteins in the
BGJ398-resistant cells, we performed RPPA analysis for bothresistant cell lines and their respective DMSO-treated controls.Heatmaps showed differential expression of proteins betweenthe resistant and control cell lines (Supplementary Fig. S4;Supplementary Tables S2 and S3). Using FDR <0.05, we iden-tified proteins differentially expressed for each of the resistantcell lines compared with controls (Fig. 3A; SupplementaryTables S4 and S5). Next, we investigated protein changes thatwere common in both DMS114 and RT112 resistant cell lines(differentially up- or downregulated). Increased levels of pAkt(T308 and S473), pGSK3b (S9), and pGSK3a/b (S21/S9) wereobserved in resistant cell lines compared with the controls (Fig.3B). However, the levels of phosphorylation significantly var-ied between the two cell lines, with DMS114 showing markedlyhigher levels of Akt and GSK3 phosphorylation compared withthe RT112 cell line (Table 1). Phosphorylated YAP (pYAP-S127) and TSC1 levels were also significantly higher in theresistant lines compared with their respective controls (Table1). Conversely, Cyclin B1 and FOXM1 were downregulated inthe resistant cell lines. PCA revealed that the resistant cell linesclustered separately from their corresponding control cell lines(Supplementary Fig. S5). Factor-loading plots for each princi-pal component (PC) were created to identify the proteins
responsible for group clustering. Data for all proteins in RPPAwere projected in the first two dimensions (PC1 and PC2). AsPC1 seemed to best associate with group clustering, the top 20proteins for PC1 were determined (Supplementary Fig. S5).PCA showed Akt phosphorylation (S473 and T308) as PC1 inboth the resistant cell lines.
Validation of RPPA protein changes by Western blot analysisNext, we performedWestern blots to corroborate pAkt (S473,
T308), pGSK3b (S9), pGSK3a/b (S21/S9), pYAP (S127), andTSC1 proteins that were upregulated in resistant cell linescompared with the controls. These were performed with thesame protein extracts used for RPPA. There were increased levelsof phosphorylation of Akt (both S473 and T308), pGSK3b(S9), and pGSK3a/b (S21/S9) observed in both the resistantcell lines compared with their matching control cell lines (Fig.3C). Although there were no appreciable changes in total levelsof these proteins in DMS114, lower levels of total Akt wereobserved in resistant RT112 cells. Furthermore, phosphorylatedYAP (S127) and TSC1 protein expression were significantlyhigher compared with the corresponding controls. Thesechanges were concordant with the changes that were observedin RPPA (Table 1; Fig. 3; Supplementary Tables S4 and S5). Incontrast, Western blot data demonstrated upregulation ofCyclin B1 and FOXM1 (Fig. 3C) in the resistant cell lines.Furthermore, decreases in phosphorylation of MEK and ERKwere observed in resistant DMS114 and RT112 cells (Supple-mentary Fig. S6). Taken together, data from RPPA and Westernblots demonstrated Akt pathway activation in both the resistantFGFR cell lines as evidenced by increased phosphorylation ofAkt and its downstream substrate GSK3. We reviewed RPPAdata to identify upstream RTKs that could potentially activateAkt. No significant changes in ERBB2, EGFR, and MET wereobserved in DMS114-R (Supplementary Table S4). On theother hand, lower levels of EGFR and ERBB2 were observedin RT112-R (Supplementary Table S5). However, RNA-Seqanalysis did not reveal any significant changes in the expressionof EGFR, ERBB2, or MET (Supplementary Table S6). AlthoughRNA-Seq showed increased EGFR expression in DMS114-R, thiswas not statistically significant (q ¼ 0.91).
Figure 1.
BGJ398 inhibits signaling in cell lineswith activating FGFR alterations.Immunoblot demonstrates theexpression of total/pFGFRs and total/pFRS2 in DMSO and BGJ398 in treatedcell lines (A) DMS114. B, RT112. Lysateswere prepared from cells exposed toBGJ398 (at the indicatedconcentrations) for 24 hours andimmunoblots performed with therespective antibodies. Representativedata are shown from threeexperimental replicates. Bar graphsdisplay densitometric analysis ofprotein bands using GAPDH as acontrol. BGJ398 treatment results indecreased levels of pFGFR1/pFGFR3and pFRS2/total FRS2, respectively.
Akt Activation Confers Acquired Resistance to FGFR Inhibitor
www.aacrjournals.org Mol Cancer Ther; 16(4) April 2017 617
on April 11, 2017. © 2017 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst March 2, 2017; DOI: 10.1158/1535-7163.MCT-15-1010
BGJ398-resistant cell lines are sensitive to Akt inhibitorGSK2141795
AsAkt pathwaywas activated in the resistant lines,we examinedwhether the addition of an Akt inhibitor, GSK2141795 (Supple-mentary Fig. S2) could potentially restore sensitivity to BGJ398.Treatment of DMS114-R cell line with GSK2141795 aloneresulted in a significant decrease in cell viability with dose as lowas 100 nmol/L, although increasing doses reduced cell viabilityfurther, with 80% decrease noticed at 3 mmol/L concentration ofGSK2141795 (Fig. 4A). Low doses of GSK2141795 had no effecton cell viability in the DMS114 control cell line, whereas a highdose (3 mmol/L) had a mild effect. Although the combination ofBGJ398 (100nmol/L)withGSK2141795 (100nmol/L) produceda significant reduction in cell viability, this effect was not signif-icant compared with the use of GSK2141795 as a single agent,indicating that treatment of Akt inhibitor alone is sufficient toinhibit the viability of BGJ398-resistant DMS114 cells (Fig. 4A).
Thus, Akt signaling pathway is highly activated in the BGJ398-resistant DMS114 cell line, and treatment with GSK2141795inhibits cell viability. Although the RT112-R cells exhibited sen-sitivity to GSK2141795 in a dose-dependent manner, the IC50
was higher (6 mmol/L). Furthermore, unlike DMS114, the RT112control cells showed some sensitivity to GSK2141795 albeitlower than RT112-R (Fig. 4A). In addition, combination treat-ment ofGSK2141795 andBGJ398didnot have any additive effecton RT112-R. The disparities in sensitivity to GSK2141795 ispresumably due to differences in the level of activation anddependence on Akt pathway between the resistant cell lines [therewas nearly a 30-fold increase in Akt (S473) phosphorylation inDMS114-R compared with RT112-R; Table 1; SupplementaryTable S7]. We performed Western blots to corroborate thatGSK2141795 affects cell viability in resistant FGFR cell linesthrough its inhibitory effect on the Akt pathway (Fig. 4B). Resis-tant lines demonstrated significantly reduced phosphorylation of
Figure 2.
Development of resistance afterchronic exposure to BGJ398. A,Resistance was induced by chronicexposure of DMS114 and RT112 cell linesto BGJ398. Control cells were treatedwith DMSO. Panel shows viabilitycurves performed with CellTiter-Gloassay. Control and resistant cells weretreated with BGJ398 at differentconcentrations as indicated, and viablecellsweremeasured after 72 hours. Thepercentage of viable cells is shownrelative to DMSO vehicle-treatedcontrols (mean � SD). Assays wereperformed in quadruplicates (threeexperimental replicates). B, Westernblot analysis was performed with thelysates from control and resistant cellsto assess FGFR signaling as measuredby pFGFR1/3 and pFRS2 (threeexperimental replicates).
Datta et al.
Mol Cancer Ther; 16(4) April 2017 Molecular Cancer Therapeutics618
on April 11, 2017. © 2017 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst March 2, 2017; DOI: 10.1158/1535-7163.MCT-15-1010

GSK3b (S9) and GSK3a/b (S21/S9), the downstream targets ofAkt. Interestingly, increased phosphorylation of Akt (S473 andT308) was observed in both the resistant cell lines withGSK2141795 treatment, likely due to feedback increase in Aktphosphorylation as described previously (21, 22). Overall, theseresults suggest that Akt pathway activation can mediate acquiredresistance to FGFR targeting, and Akt inhibitor treatment canrestore sensitivity.
siRNA-mediated Akt knockdown inhibits growth andmigration of resistant FGFR cancer cells
RNA-Seq analysis showed that Akt1 and Akt2 are the com-monest isoforms in DMS114 and RT112 cell lines (Supple-mentary Table S7). Next, we used an Akt siRNA that specificallyinhibited Akt1 and Akt2 expression. We evaluated the effect ofsiRNA treatment on the levels of Akt phosphorylation and itsdownstream substrates, GSK3b and GSK3a/b. Treatment of
Figure 3.
RPPA reveals Akt pathway activation in BGJ398-resistant cell lines. A, Heatmaps show RPPA proteins that were differentially phosphorylated in resistant andcontrol (q < 0.05) DMS114 and RT112 cell lines. B, Venn diagram shows proteins that were commonly upregulated (left) and downregulated (right) in bothresistant cell lines C. Western blot analysis to confirm protein changes that were upregulated (left) and downregulated (3) in resistant cell lines (representativedata from three experimental replicates are shown).
Table 1. Protein changes in resistant FGFR cell lines
Cell line Protein Percent change (up/down) P q-value
DMS114 Akt_pS473 3622.68645 1.54E�06 0.00016487Akt_pT308 2252.07737 6.47E�05 0.00125812GSK3-alpha-beta_pS21_S9 284.091792 2.50E�07 5.35E�05GSK3_pS9 187.53704 4.94E�06 0.00026516YAP_pS127 32.06119 0.01389659 0.04373338TSC1 31.3146193 0.00084919 0.00586215Cyclin_B1 �24.410473 0.00499741 0.02088713FoxM1 �56.218837 0.00027636 0.00295707
RT112 Akt_pS473 123.622596 0.00038911 0.01055472Akt_pT308 203.350928 0.00126369 0.01890811GSK3-alpha-beta_pS21_S9 41.1993588 0.00134332 0.01890811GSK3_pS9 64.2870708 0.00011599 0.00629257YAP_pS127 76.7742588 0.00493268 0.04129296TSC1 24.6593776 0.0027762 0.02868744Cyclin_B1 �28.205219 0.0065222 0.04424411FoxM1 �36.057898 0.0022811 0.0261498
Akt Activation Confers Acquired Resistance to FGFR Inhibitor
www.aacrjournals.org Mol Cancer Ther; 16(4) April 2017 619
on April 11, 2017. © 2017 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst March 2, 2017; DOI: 10.1158/1535-7163.MCT-15-1010

each resistant cell line with Akt siRNA resulted in significantdecreases in both the total Akt and pAkt (S473 and T308)expression compared with their respective siRNA control–trea-ted cells (P < 0.001; Fig. 5A). Differential levels of total Aktdepletion was observed in DMS114-R (�80%) and RT112-R(�70%) cell lines. This could potentially be due to variationin transfection efficiency between these cell lines. No effectwas observed on MEK1/2 phosphorylation, confirming thespecificity of Akt siRNA. Although the levels of both totalGSK3a/b and their phosphorylated forms (S21/S9) weredecreased significantly with Akt siRNA–treated DMS114-R, thelevels of pGSK3a/b (S21/S9) were only moderately decreasedwith Akt siRNA in RT112-R compared with control siRNAcells. Furthermore, we tested whether Akt siRNA treatmentaffected cell proliferation, colony formation, cell migration,and cell invasion in resistant lines (Fig. 5B–D). Cell prolifer-ation was reduced in resistant cell lines following Akt siRNAtreatment. Consistent with Akt inhibitor treatment, theobserved effect of siRNA was lower in RT112-R in comparison
with DMS114-R cell line. Akt siRNA treatment also significantlyinhibited colony formation, cell migration, and cell invasion inboth resistant cell lines compared with controls (Fig. 5C and D,P < 0.001).
DiscussionActivation of FGFR signaling pathway due to FGFR gene
amplifications, mutations, or fusions has been identified invarious cancers, such as, lung, bladder, biliary, and breast (1).While therapeutic strategies targeting FGFR are being explored inclinical trials, we sought to study acquired drug resistance inannotated cancer cell lines (23–25). As observed with inhibitionof other oncogenic kinases, such as ALK and BRAF, we hypoth-esized that oncogenic addiction could lead to reactivation ofdownstreamelements of FGFR signaling (26, 27). Also, secondaryresistance can be mediated due to clonal selection of resistantcells, often present prior to treatment, with observed clonaldivergence across various metastatic sites (28, 29). We applied
Figure 4.
BGJ398-resistant cell lines aresensitive to Akt inhibitor. A, Viabilitycurves for control and resistant cellstreated with DMSO vehicle, BGJ398(varying concentrations as indicated),and GSK2141795 (at a fixedconcentration: DMS114 0.1, mmol/L;RT112, 1 mmol/L) for 72 hours. Gray andblack vertical dotted lines denote3 mmol/L and 0.1 mmol/L (DMS114),5 mmol/L and 1 mmol/L (RT112) drugconcentrations, respectively, that areshown in the bar graph below. Bargraphs show differences in cellviability in control and resistant celllines to BGJ398 and GSK2141795.Assays performed in quadruplicatesand data are presented as mean � SD(three experimental replicates).��, Statistical significant difference,P <0.001.B,Western blot analysiswasperformed with lysates from resistantcell lines treated either with vehicle(DMSO) or GSK2141795 (1 mmol/L) for24 hours to assess inhibition of Akt(pAKT) and downstream (pGSK3)signaling (three experimentalreplicates).
Datta et al.
Mol Cancer Ther; 16(4) April 2017 Molecular Cancer Therapeutics620
on April 11, 2017. © 2017 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst March 2, 2017; DOI: 10.1158/1535-7163.MCT-15-1010

ahigh-throughput proteomics approach to identify candidates foracquired resistance. Early preclinical work may guide evaluationof precious clinical tumor specimens from patients participatingin ongoing therapeutic trials.
In this study, we generated two cell lines, DMS114 (small-celllung cancer line with FGFR1 amplification) and RT112 (bladdercarcinoma with FGFR3-TACC3 fusion and FGFR3 amplification)asmanifesting acquired resistance after chronic BGJ398 exposure,based on viability assays (Fig. 2). To assess the relative activity ofproteins involved in FGFR signal transduction, we performedproteomic analysis with RPPA. RPPA demonstrated Akt pathwayactivation in both the resistant cell lines despite ongoing FGFRblockade in the presence of BGJ398 (Fig. 3). These results werecorroborated with Western blots. Treatment of the resistant cells
with Akt siRNA or GSK2141795, an oral, competitive, pan-iso-form Akt inhibitor, currently under evaluation in clinical trials(30, 31), was able to restore sensitivity of resistant cell lines (Figs.4 and 5).
Activated FGFR signals primarily through one of three path-ways: MAPK, PI3K/Akt/mTOR, and phospholipase C-gamma(PLCg). FGFR preferentially signals through FRS2 to the MAPKpathway, although depending on the cellular context, other path-ways, such as p38 MAPK and STAT, could also be activated (1).ActivatedAkt,mediated byphosphorylation at T308byPDK1andphosphorylation at S473 by PDK2, regulates multiple down-stream substrates, including GSK3 a/b, FOXO, PRAS40, andTSC1/2 (32). Phosphorylation of S9 in GSK3b or S21 in GSK3amediated by Akt can potentially lead to reactivation of b-catenin,
Figure 5.
BGJ398-resistant cell lines are sensitive to Akt siRNA knockdown. A, Resistant cells were treated with control siRNA or Akt siRNA (60 nmol/L for 48 hours). Whole-cell protein lysates were extracted, and immunoblots were performed to assess Akt and downstream proteins. Representative data have been shown (threeexperimental replicates). B, Resistant cells were transfected with control or Akt-specific siRNA, and cell proliferation was measured using the CellTiter-Glo assay(mean � SD; � , P < 0.005). Assays were performed in quadruplicates with four experimental replicates. C, Colonies were stained with crystal violet and countedafter transfection (control or Akt siRNA) of resistant cell lines. Bar graph shows colony counts in control and Akt siRNA–treated resistant cell lines (mean � SD;� , P � 0.005). Each assay was performed in triplicate with three experimental replicates. D, Left, motility assay was performed after treatment with controlor Akt siRNA (magnification, �100). Bar graphs depict the surface area covered by the cells at 24 hours (mean � SD; � , P � 0.005). Right, invasion assay wasperformed using Boyden chamber after treatment with control or Akt siRNA. Bar graphs depict the number of cells that migrated through the Matrigel at 24 hours(mean � SD; � , P � 0.005). Representative data from three experimental replicates are shown.
Akt Activation Confers Acquired Resistance to FGFR Inhibitor
www.aacrjournals.org Mol Cancer Ther; 16(4) April 2017 621
on April 11, 2017. © 2017 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst March 2, 2017; DOI: 10.1158/1535-7163.MCT-15-1010

which in turn can increase cyclin D1 and transcription factoractivity leading to cell-cycle progression (33–35). Increased phos-phorylation of GSK3a at S21 and GSK3b at S9 was observed inboth the resistant cell lines consistent with Akt activation. Fur-thermore, an increase in phosphorylation of proline-rich Aktsubstrate of 40 kDa (PRAS40) at T246 was observed inDMS114-R. PRAS40 is a crucial mediator of Akt signaling andits phosphorylation (T246), mediated by Akt, facilitates thebinding of 14-3-3 protein, which disrupts the inhibitory actionof PRAS40 on mTORC1 leading to phosphorylation of down-stream substrates (e.g., S6 kinase, 4EBP1) and cell growth (36,37). Accordingly, increased expression of 14-3-3 (z) was observedin DMS114-R. Overall, data from RPPA substantiated activationof Akt pathway in cell lines resistant to BGJ398.
Several studies have suggested a reciprocal relationshipbetween FGFR and othermembers of the RTK family inmediatingacquired resistance to FGFR inhibition. Herrera-Abreu and col-leagues used parallel RNA interference genetic screens to showthat EGFR activation limits sensitivity to FGFR inhibition in celllineswith FGFR3mutations (38). Conversely, activation of FGF2–FGFR1 pathway has been shown tomediate acquired resistance toEGFR inhibition in lung cancer (39). Ligand-mediated activationof ERBB2/3 was shown tomediate resistance to FGFR3 inhibition(40). Similarly, HGF-mediated ligand activation of MET wasshown to rescue RT112 cells from the effect of FGFR inhibitionby BGJ398, suggesting that compensatory activation of othermembers of the RTK family can mediate resistance to FGFRtargeting (41). In contrast, Chell and colleagues identified asecondary gatekeeper mutation in FGFR3 (V555M) as a mecha-nism of acquired resistance to ATP-competitive FGFR inhibitors,AZD4547 andPD173074 (42). In our study, despite differences inthe tissue of origin and the genomic alteration in FGFR, theresistant cell lines showed activation of Akt pathway, suggestingsecondary activationof bypass signaling as apotentialmechanismof resistance to FGFR targeting.
Our observation is consistent with what has been previouslydescribed with respect to a prominent role for Akt in FGFRsignaling (43). Hu and colleagues observed that FGFR inhibitionwith BGJ398 led to transient inhibition of ERK1/2 phosphoryla-tion due to negative feedback, whereas the effect on Akt phos-phorylation was sustained in RT112 and KATOIII cell lines. Inaddition, they showed that a constitutively activated form of Akt(myristoylated Akt) effectively rescued cells from BGJ398 growthinhibition. Grygielewicz and colleagues evaluated resistance inSNU-16, an FGFR2-amplified gastric cancer cell line, to FGFRinhibitors AZD4547, BGJ398, and PD173074. While epithelial–mesenchymal transition was primarily implicated as mediatingresistance to the FGFR inhibitors, increased levels of pAkt (alongwithpSTAT andpERK)were observed in the resistant cell lines andtreatment with pictilisib, a PI3K inhibitor, was able to restoresensitivity (44). Furthermore, acquired resistance to cetuximab,an mAb for EGFR, was associated with Akt activation in lungcancer cell lines, and pharmacologic inhibition of Akt withthe PI3K inhibitor LY294002 enhanced the inhibitory effect ofcetuximab (45). Thus, there is interest in whether Akt orPI3K inhibitors could overcome resistance in cancers driven byRTKs. Although there is a breadth of candidate mechanisms ofresistance in preclinical models suggesting a potential role forcombination therapies with FGFR inhibitors, this will depend onthe evaluation of resistance in patient tumor samples in ongoingclinical trials.
To characterize resistance mechanisms in patients, pretreat-ment and postprogression tumor biopsies will be needed tosupport assays such as RPPA, DNA/RNA-Seq, or patient-derivedxenograft studies. However, tumor specimens are finite and maynot be able to practically support all of these assays. Consequent-ly, preclinical models are often utilized to provide preliminaryevidence of resistance mechanisms in humans and guide evalu-ation in precious tumor specimens. In addition to tumor biopsies,circulating tumor cells or circulating tumor DNA (ctDNA) mayprovide a rapid and noninvasive strategy to assess resistance incases where tumors cannot be readily biopsied. For example,using ctDNA and xenograft, secondary NTRK1 mutations wereidentified as a resistance mechanism to entrectinib, a pan-TRKinhibitor, in an advanced colorectal cancer patientwith anNTRK1fusion (46). Similarly, ctDNA has been successfully utilized totrack clonal evolution and resistance to EGFR inhibition (47).Identification of resistance mechanisms can potentially revealrational targeted therapy combinations. The understanding thatreactivation of MAPK pathway mediates resistance to BRAF inhi-bition led to the use of combination of BRAF andMEK inhibitorsin melanoma patients harboring BRAF V600 mutations, leadingto improved survival (48). In another example, identification ofPI3K/Akt/mTOR pathway activation as amechanism of resistanceto endocrine therapy led to the evaluation and approval ofcombination of everolimus, an mTOR inhibitor, and exemestanein hormone receptor–positive breast cancer patients (49). Ourstudy provides preclinical evidence that activation of Aktmediatesresistance to FGFR inhibition, supports the need for furtherevaluation of this pathway in patients, and underscores a poten-tial opportunity for combination therapy.
Disclosure of Potential Conflicts of InterestS. Roychowdhury receives funding fromNovartis and Ariad Pharmaceuticals
for conducting clinical trials. S.Roychowdhury's immediate family membersown stock in Johnson and Johnson. No potential conflicts of interest weredisclosed by the other authors.
Authors' ContributionsConception and design: S. Damodaran, J. Datta, H. Parks, J. Hays,S. RoychowdhuryDevelopment of methodology: S. Damodaran, J. Datta, H. Parks, J. HaysAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): S. Damodaran, J. Datta, C. Ocrainiciuc, E.P. GardnerAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): S. Damodaran, J. Datta, H. Parks, C. Ocrainiciuc,J. Miya, L. Yu, E.P. Gardner, E. Samorodnitsky, M.R. Wing, J. HaysWriting, review, and/or revision of the manuscript: S. Damodaran, J. Datta,J. Miya, L. Yu, J. Hays, J.W. Reeser, S. RoychowdhuryAdministrative, technical, or material support (i.e., reporting or organizingdata, constructing databases): J. Datta, D. BhattStudy supervision: J. Datta, J. Hays, S. Roychowdhury
AcknowledgmentsWe would like to acknowledge the Comprehensive Cancer Center (CCC),
Arthur G. James Cancer Hospital and Richard A. Solove Research Instituteat The Ohio State University Wexner Medical Center, and The Ohio Super-computer Center for supporting this study. We also would like to thank EskoKautto and Mikayla Dantuono for technical help and Jenny Badillo foradministrative support.
Grant SupportS. Damodaran was supported by the American Society of Clinical Oncology
Young Investigator Award. E. Samorodnitsky was supported by a PelotoniaPostdoctoral Fellowship. S. Roychowdhury was supported by the American
Datta et al.
Mol Cancer Ther; 16(4) April 2017 Molecular Cancer Therapeutics622
on April 11, 2017. © 2017 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst March 2, 2017; DOI: 10.1158/1535-7163.MCT-15-1010

Cancer Society (MRSG-12-194-01-TBG), the Prostate Cancer Foundation,NHGRIUM1HG006508-01A1, Fore Cancer Research, American Lung Associa-tion, and Pelotonia.
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby marked
advertisement in accordance with 18 U.S.C. Section 1734 solely to indicatethis fact.
Received December 28, 2015; revised December 21, 2016; accepted Decem-ber 25, 2016; published OnlineFirst March 2, 2017.
References1. Turner N, Grose R. Fibroblast growth factor signalling: from development
to cancer. Nat Rev Cancer 2010;10:116–29.2. Yang F, ZhangY, Ressler SJ, IttmannMM,AyalaGE,Dang TD, et al. FGFR1 is
essential for prostate cancer progression and metastasis. Cancer Res2013;73:3716–24.
3. Weiss J, Sos ML, Seidel D, Peifer M, Zander T, Heuckmann JM, et al.Frequent and focal FGFR1 amplification associates with therapeuticallytractable FGFR1 dependency in squamous cell lung cancer. Sci Transl Med2010;2:62ra93.
4. Wesche J, Haglund K, Haugsten EM. Fibroblast growth factors and theirreceptors in cancer. Biochem J 2011;437:199–213.
5. Williams SV, Hurst CD, Knowles MA. Oncogenic FGFR3 gene fusions inbladder cancer. Hum Mol Genet 2013;22:795–803.
6. Wu YM, Su F, Kalyana-Sundaram S, Khazanov N, Ateeq B, Cao X, et al.Identification of targetable FGFR gene fusions in diverse cancers. CancerDiscov 2013;3:636–47.
7. Guagnano V, Kauffmann A, Wohrle S, StammC, Ito M, Barys L, et al. FGFRgenetic alterations predict for sensitivity to NVP-BGJ398, a selective pan-FGFR inhibitor. Cancer Discov 2012;2:1118–33.
8. Gozgit JM, Wong MJ, Moran L, Wardwell S, Mohemmad QK, NarasimhanNI, et al. Ponatinib (AP24534), a multitargeted pan-FGFR inhibitor withactivity inmultiple FGFR-amplified ormutated cancermodels. Mol CancerTher 2012;11:690–9.
9. Andre F, Bachelot T, Campone M, Dalenc F, Perez-Garcia JM, Hurvitz SA,et al. Targeting FGFR with dovitinib (TKI258): preclinical and clinical datain breast cancer. Clin Cancer Res 2013;19:3693–702.
10. Konecny GE, Finkler N, Garcia AA, Lorusso D, Lee PS, Rocconi RP, et al.Second-linedovitinib (TKI258) inpatientswith FGFR2-mutated or FGFR2-non-mutated advanced or metastatic endometrial cancer: a non-rando-mised, open-label, two-group, two-stage, phase 2 study. Lancet Oncol2015;16:686–94.
11. Soria JC, DeBraud F, Bahleda R, Adamo B, Andre F, Dienstmann R, et al.Phase I/IIa study evaluating the safety, efficacy, pharmacokinetics, andpharmacodynamics of lucitanib in advanced solid tumors. Ann Oncol2014;25:2244–51.
12. Tabernero J, Bahleda R, Dienstmann R, Infante JR, Mita A, Italiano A, et al.Phase I dose-escalation study of JNJ-42756493, an oral pan-fibroblastgrowth factor receptor inhibitor, in patients with advanced solid tumors.J Clin Oncol 2015;33:3401–8.
13. Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenicassay of cells in vitro. Nat Protoc 2006;1:2315–9.
14. Islam M, Datta J, Lang JC, Teknos TN. Down regulation of RhoC bymicroRNA-138 results in de-activation of FAK, Src and Erk1/2 signalingpathway in head and neck squamous cell carcinoma. Oral Oncol2014;50:448–56.
15. Valster A, Tran NL, Nakada M, Berens ME, Chan AY, Symons M. Cellmigration and invasion assays. Methods 2005;37:208–15.
16. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2:accurate alignment of transcriptomes in the presence of insertions, dele-tions and gene fusions. Genome Biol 2013;14:R36.
17. Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ,et al. Transcript assembly and quantification by RNA-Seq reveals unan-notated transcripts and isoform switching during cell differentiation. NatBiotechnol 2010;28:511–5.
18. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practicaland powerful approach to multiple testing. J Royal Stat Soc Ser B1995;57:289–300.
19. Abdi H, Williams LJ. Principal component analysis. Wiley Interdiscip RevComput Stat 2010;2:433–59.
20. Du F, Wu X, Liu Y, Wang T, Qi X, Mao Y, et al. Acquisition of paclitaxelresistance via PI3Kdependent epithelialmesenchymal transition in A2780human ovarian cancer cells. Oncol Rep 2013;30:1113–8.
21. Dumble M, Crouthamel MC, Zhang SY, Schaber M, Levy D, Robell K,et al. Discovery of novel AKT inhibitors with enhanced anti-tumoreffects in combination with the MEK inhibitor. PLoS One 2014;9:e100880.
22. Rhodes N, Heerding DA, Duckett DR, Eberwein DJ, Knick VB,Lansing TJ, et al. Characterization of an Akt kinase inhibitor withpotent pharmacodynamic and antitumor activity. Cancer Res 2008;68:2366–74.
23. Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA,et al. COT drives resistance to RAF inhibition throughMAP kinase pathwayreactivation. Nature 2010;468:968–72.
24. Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, et al. Melanomasacquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregula-tion. Nature 2010;468:973–7.
25. Poulikakos PI, Persaud Y, JanakiramanM, Kong X, Ng C,Moriceau G, et al.RAF inhibitor resistance is mediated by dimerization of aberrantly splicedBRAF(V600E). Nature 2011;480:387–90.
26. Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAFinhibitors. Nat Med 2013;19:1401–9.
27. Wilson FH, Johannessen CM, Piccioni F, Tamayo P, Kim JW, Van Allen EM,et al. A functional landscape of resistance to ALK inhibition in lung cancer.Cancer Cell 2015;27:397–408.
28. Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E, et al.Preexistence and clonal selection of MET amplification in EGFR mutantNSCLC. Cancer Cell 2010;17:77–88.
29. Morrissy AS, Garzia L, Shih DJ, Zuyderduyn S, Huang X, Skowron P, et al.Divergent clonal selection dominates medulloblastoma at recurrence.Nature 2016;529:351–7.
30. ClinicalTrials.gov. Trametinib and Akt inhibitor GSK2141795 in treat-ing patients with metastatic triple-negative breast cancer. Bethesda, MD:NIH; 2017. Available from: https://www.clinicaltrials.gov/ct2/show/NCT01964924.
31. Burris HA, Siu LL, Infante JR, Wheler JJ, Kurkjian C, Opalinska J, SmithDA, et al. Safety, pharmacokinetics (PK), pharmacodynamics (PD),and clinical activity of the oral AKT inhibitor GSK2141795 (GSK795)in a phase I first-in-human study. J Clin Oncol 29: 2011(suppl; abstr3003).
32. Franke TF. PI3K/Akt: getting it right matters. Oncogene 2008;27:6473–88.
33. Luo J. Glycogen synthase kinase 3beta (GSK3beta) in tumorigenesis andcancer chemotherapy. Cancer Lett 2009;273:194–200.
34. Medina M, Wandosell F. Deconstructing GSK-3: The Fine Regulation of ItsActivity. Int J Alzheimer's Dis 2011;2011:479249.
35. Mavila N, James D, Utley S, Cu N, Coblens O, Mak K, et al.Fibroblast growth factor receptor-mediated activation of AKT-beta-catenin-CBP pathway regulates survival and proliferation of murinehepatoblasts and hepatic tumor initiating stem cells. PLoS One2012;7:e50401.
36. Wiza C, Nascimento EB, Ouwens DM. Role of PRAS40 in Akt and mTORsignaling in health and disease. Am J Physiol Endocrinol Metab 2012;302:E1453–60.
37. Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, KimDH. Insulin signallingto mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol2007;9:316–23.
38. Herrera-Abreu MT, Pearson A, Campbell J, Shnyder SD, Knowles MA,Ashworth A, et al. Parallel RNA interference screens identify EGFR activa-tion as an escape mechanism in FGFR3-mutant cancer. Cancer Discov2013;3:1058–71.
39. Terai H, Soejima K, Yasuda H, Nakayama S, Hamamoto J, Arai D, et al.Activation of the FGF2-FGFR1 autocrine pathway: a novel mechanismof acquired resistance to gefitinib in NSCLC. Mol Cancer Res 2013;11:759–67.
Akt Activation Confers Acquired Resistance to FGFR Inhibitor
www.aacrjournals.org Mol Cancer Ther; 16(4) April 2017 623
on April 11, 2017. © 2017 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst March 2, 2017; DOI: 10.1158/1535-7163.MCT-15-1010

40. Wang J, Mikse O, Liao RG, Li Y, Tan L, Janne PA, et al. Ligand-associatedERBB2/3 activation confers acquired resistance to FGFR inhibition inFGFR3-dependent cancer cells. Oncogene 2015;34:2167–77.
41. Harbinski F,CraigVJ, Sanghavi S, JefferyD, Liu L, SheppardKA, et al. Rescuescreens with secreted proteins reveal compensatory potential of receptortyrosine kinases in driving cancer growth. Cancer Discov 2012;2:948–59.
42. Chell V, Balmanno K, Little AS, Wilson M, Andrews S, Blockley L, et al.Tumour cell responses to new fibroblast growth factor receptor tyrosinekinase inhibitors and identification of a gatekeepermutation in FGFR3 as amechanism of acquired resistance. Oncogene 2013;32:3059–70.
43. Hu Y, LuH, Zhang J, Chen J, Chai Z, Zhang J. Essential role of AKT in tumorcells addicted to FGFR. Anticancer Drugs 2014;25:183–8.
44. Grygielewicz P, Dymek B, Bujak A, Gunerka P, Stanczak A, Lamparska-Przybysz M, et al. Epithelial-mesenchymal transition confers resistance toselective FGFR inhibitors in SNU-16 gastric cancer cells. Gastric Cancer2016;19:53–62.
45. Takata M, Chikumi H, Miyake N, Adachi K, Kanamori Y, Yamasaki A,et al. Lack of AKT activation in lung cancer cells with EGFR mutation isa novel marker of cetuximab sensitivity. Cancer Biol Ther 2012;13:369–78.
46. Russo M, Misale S, Wei G, Siravegna G, Crisafulli G, Lazzari L, et al.Acquired resistance to the TRK inhibitor entrectinib in colorectal cancer.Cancer Discov 2016;6:36–44.
47. Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G,et al. Clonal evolution and resistance to EGFR blockade in the blood ofcolorectal cancer patients. Nat Med 2015;21:795–801.
48. Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al.Combined BRAF and MEK inhibition in melanoma with BRAF V600mutations. N Engl J Med 2012;367:1694–703.
49. Baselga J, Campone M, Piccart M, Burris HA 3rd, Rugo HS, Sahmoud T,et al. Everolimus in postmenopausal hormone-receptor-positive advancedbreast cancer. N Engl J Med 2012;366:520–9.
Mol Cancer Ther; 16(4) April 2017 Molecular Cancer Therapeutics624
Datta et al.
on April 11, 2017. © 2017 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst March 2, 2017; DOI: 10.1158/1535-7163.MCT-15-1010

2017;16:614-624. Published OnlineFirst March 2, 2017.Mol Cancer Ther Jharna Datta, Senthilkumar Damodaran, Hannah Parks, et al. Factor Receptor Inhibitor BGJ398Akt Activation Mediates Acquired Resistance to Fibroblast Growth
Updated version
10.1158/1535-7163.MCT-15-1010doi:
Access the most recent version of this article at:
Cited articles
http://mct.aacrjournals.org/content/16/4/614.full.html#ref-list-1
This article cites 47 articles, 16 of which you can access for free at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
To request permission to re-use all or part of this article, contact the AACR Publications Department at
on April 11, 2017. © 2017 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst March 2, 2017; DOI: 10.1158/1535-7163.MCT-15-1010