advanced crystal structure analysis - shelx...
TRANSCRIPT

ACSA
Advanced Crystal Structure AnalysisMrTailor: PDB file Preparation to generate External Restraints
Tim GrüneGeorg-August-UniversitätInstitut für Strukturchemie
http://shelx.uni-ac.gwdg.de/~tg
Tim Grüne mrtailor 1/25

ACSA
Refinement at low Resolution
Resolution[Å] refined parameters data/parameters ratio3.0 x,y,z 0.9:12.3 x,y,z; B 1.5:11.8 x,y,z; B 3.1:11.5 x,y,z; B 5.4:11.5 x,y,z; U11U12U13U23U22U33 2.4:11.1 x,y,z; U11U12U13U23U22U33 6.1:10.8 x,y,z; U11U12U13U23U22U33 16:1
G. Sheldrick
Tim Grüne mrtailor 2/25

ACSA
Restraints and Constraints
Improvement of Data:Parameters by
Restraints
• Increase data points
• e.g. Engh-Huber parameters (Engh & Huber Parameters, Acta Cryst. (1991), A47, 392–400), 1,2- and1,3-distance restraints
• shelxl: DFIX, DANG, CHIV, FLAT, . . .
Constraints
• Decrease number of parameters
• e.g. −5 < Ueq(H) < −0.5; Invariom database.
Tim Grüne mrtailor 3/25

ACSA
Refinement at d > 3 Å
• Cα trace for 2Y9Y “Chromatin Remodelling FactorISW1A”• Data resolution: 30 Å– 3.25 Å• Cell: 206.97 206.97 215.31 90 90 120 (H32)
1. How much model bias, how much data?2. Interpretation requires experience (no peak picking)3. Removing loops without density = removing phasing
power
Tim Grüne mrtailor 4/25

ACSA
Refinement at d > 3 Å
1. Data:Parameters very low
2. 1,2- and 1,3-distances (Engh-Huber-Restraints) not shape determining
3. Molecular Replacement: heavy model bias towards template
Tim Grüne mrtailor 5/25

ACSA
Improvement of Data:Parameters
• NCS
• knowledge from deposited structures
Tim Grüne mrtailor 6/25

ACSA
Multi-subunit structures
• 70S ribosome with mRNA (PDB-ID 2wdk)• 3.5 Å• PDB: 24 chains!
• Nucleosome Core Particle (PDB-ID 3mvd)• 2.9 Å• 8 Histones + double stranded DNA (190 bp)
• “hot” structures often multi-subunit structures
• subunits often known, sometimes at better resolution than complex
Tim Grüne mrtailor 7/25
ACSA
External Restraints
Idea: Additional restraints beyond 1,3-restraints from same or similar structure
Automated in most MX refinement programs:
• Buster-TNT (http://www.globalphasing.com/buster/wiki/index.cgi?AutoBusterExample2e4ytarget)
• phenix.refine (http://www.phenix-online.org/documentation/refinement.htm#anch163)
• CNS (http://cns-online.org/v1.3/)
• refmac5 (http://www2.mrc-lmb.cam.ac.uk/groups/murshudov/) via prosmart
Tim Grüne mrtailor 8/25
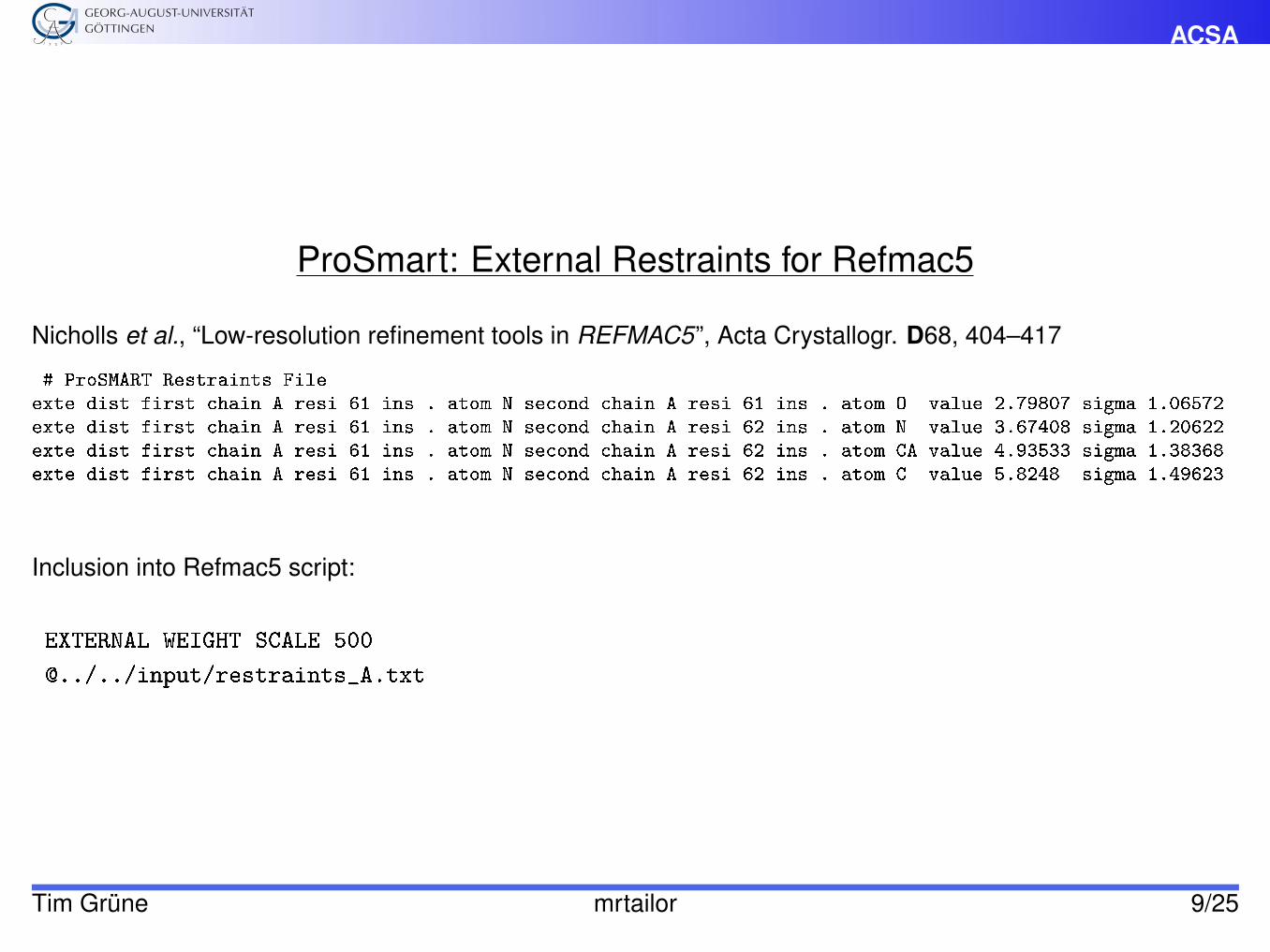
ACSA
ProSmart: External Restraints for Refmac5
Nicholls et al., “Low-resolution refinement tools in REFMAC5”, Acta Crystallogr. D68, 404–417
# ProSMART Restraints Fileexte dist first chain A resi 61 ins . atom N second chain A resi 61 ins . atom O value 2.79807 sigma 1.06572exte dist first chain A resi 61 ins . atom N second chain A resi 62 ins . atom N value 3.67408 sigma 1.20622exte dist first chain A resi 61 ins . atom N second chain A resi 62 ins . atom CA value 4.93533 sigma 1.38368exte dist first chain A resi 61 ins . atom N second chain A resi 62 ins . atom C value 5.8248 sigma 1.49623
Inclusion into Refmac5 script:
EXTERNAL WEIGHT SCALE 500
@../../input/restraints_A.txt
Tim Grüne mrtailor 9/25

ACSA
Model Bias
From Nicholls et al., “Low-resolution refinement tools in REFMAC5”, Acta Crystallogr. D68, 404–417:
“It is, however, necessary for local structure to be sufficiently well conserved along the chain so thatthe effect on refinement is positive.”
Tim Grüne mrtailor 10/25

ACSA
Model Bias
Original data with 1OFC, no exter-nal restraints
with 1OFC, prosmartrestraints
with 1OFC, mrtailorrestraints
2Y9Y, chain A replaced with 1OFC
Tim Grüne mrtailor 11/25

ACSA
How to avoid model bias before knowing the structure?
PDB 2Y9Y (3.25 Å); chain A replaced with 1OFC (1.9 Å)
PDB 3ee6 (orange; PhD thesis A. Pal) with 1dbi
(from the mrtailor tutorial at http://shelx.uni-ac.gwdg.de/~tg/research/programs/mrtailor/tutorial/)
Tim Grüne mrtailor 12/25

ACSA
Sequence Alignment with BLAST
• Proteins: sequence⇒ structure
• “Blast” search against various data bases http://blast.ncbi.nlm.nih.gov:
– non-redundant data base (24584348 sequences): multiple sequence alignment, conservation of residuesor outliers
– PDB (63301 sequences): search models for molecular replacement
Tim Grüne mrtailor 13/25

ACSA
Sequence Alignment with BLAST
. . . : : .. :. . * : : :. : .gi|329665881 MLLNPTKRERKENYSIDNYYKDVLNTGRSSTPSHPRMPKPHVFHSHQLQPPQLKVLYEKERMWTAKKTGYVPTMDDVKAA 80gi|329665879 MLLNPTKRERKENYSIDNYYKDVLNTGRSSTPSHPRMPKPHVFHSHQLQPPQLQVLYEKERMWTAKKTGYVPTMDDVKAA 80gi|34809857 -----GAMERKANYAVDAYFREALRVSEPKAPKAPRPPKQPIVQDFQFFPPRLFELLDQEIYYFRKTVGYKVPKN----T 71gi|158429118 ------------------------MVSEPKVPKAPRPPKQPNVQDFQFFPPRLFELLEKEILYYRKTIGYKVPRN----P 52gi|18655633 -----------------MGHLGKTRWTREEDEKLKKLVEQNGTDDWKVIANYLPNRTDVQCQHRWQKV---LNPELIKGP 60
. . : :: : .: . : : .. * *.: : . : . : * : **: :gi|329665881 YGDISDEEEKK--QKLELLKLSVNNSQPLTEEEEKMKADWESE-----GFTNWNKLEFRKFITVSGKYGRNSIQAIAREL 153gi|329665879 YGKISDQEEQK--EKLELLKLSVNNSQPLTEEEEKMKADWESE-----GFTNWNKLEFRKFITVSGKYGRNSIQAIAREL 153gi|34809857 ELGSDATKVQR--EE----QRKIDEAEPLTEEEIQEKENLLSQ-----GFTAWTKRDFNQFIKANEKYGRDDIDNIAKDV 140gi|158429118 DLPNSA-QVQK--EE----QLKIDEAEPLNDEELEEKEKLLTQ-----GFTNWNKRDFNQFIKANEKWGRDDIENIAREV 120gi|18655633 WTKEEDQRVIKLVQKYGPKRWSVIAKHLKGRIGKQCRERWHNHLNPEVKKTSWTEEEDRIIYQAHKRLG-NRWAEIAKLL 139
*:* : : : . gi|329665881 APGKTLEEVRAYAKAFWSNIERIEDYEKYLKIIENEEEKIKRVKMQQEALRRKLSEYKNPFFDLKLKHPPSSNNKRTYSE 233gi|329665879 APGKTLEEVRAYAKAFWSNIERIEDYEKYLKIIENEEEKIKRVKMQQEALRRKLSEYKNPFFDLKLKHPPSSNNKRTYSE 233gi|34809857 -EGKTPEEVIEYNAVFWERCTELQDIERIMGQIERGEGKIQRRLSIKKALDQKMSRYRAPFHQLRLQYGN--NKGKNYTE 217gi|158429118 -EGKTPEEVIEYSAVFWERCNELQDIEKTMAQIERGEARIQRRISI---LEHHHHHH----------------------- 173gi|18655633 -PGRTDNAIKNHWNSTMRRKV----------------------------------------------------------- 159
gi|329665881 EEDRFILLMLFKYGLDRDDVYELVRDEIRDCPLFELDFYFRSRTPVELARRGNTLLQCLEKEFNAGIVLDDATKDRMKKE 313gi|329665879 EEDRFILLMLFKYGLDRDDVYELVRDEIRDCPLFELDFYFRSRTPVELARRGNTLLQCLEKEFNAGIVLDDATKDRMKKE 313gi|34809857 IEDRFLVCMLHKLGFDKENVYEELRAAIRASPQFRFDWFIKSRTALELQRRCNTLITLIEREN---IELEEKERAEKKKK 294gi|158429118 -------------------------------------------------------------------------------- 173gi|18655633 -------------------------------------------------------------------------------- 159
Tim Grüne mrtailor 14/25

ACSA
MrTailor
Blast search against PDBwith target sequence
multiple alignment
prosmart
modifiedPDB
clustal
externalrestraints
refmac5
qscores
model PDB
mrtailor
pdb/mtz
Tim Grüne mrtailor 15/25

ACSA
MrTailor Applications
1. Sequence Mapping after Molecular Replacement
2. Model Preparation for Prosmart
Tim Grüne mrtailor 16/25
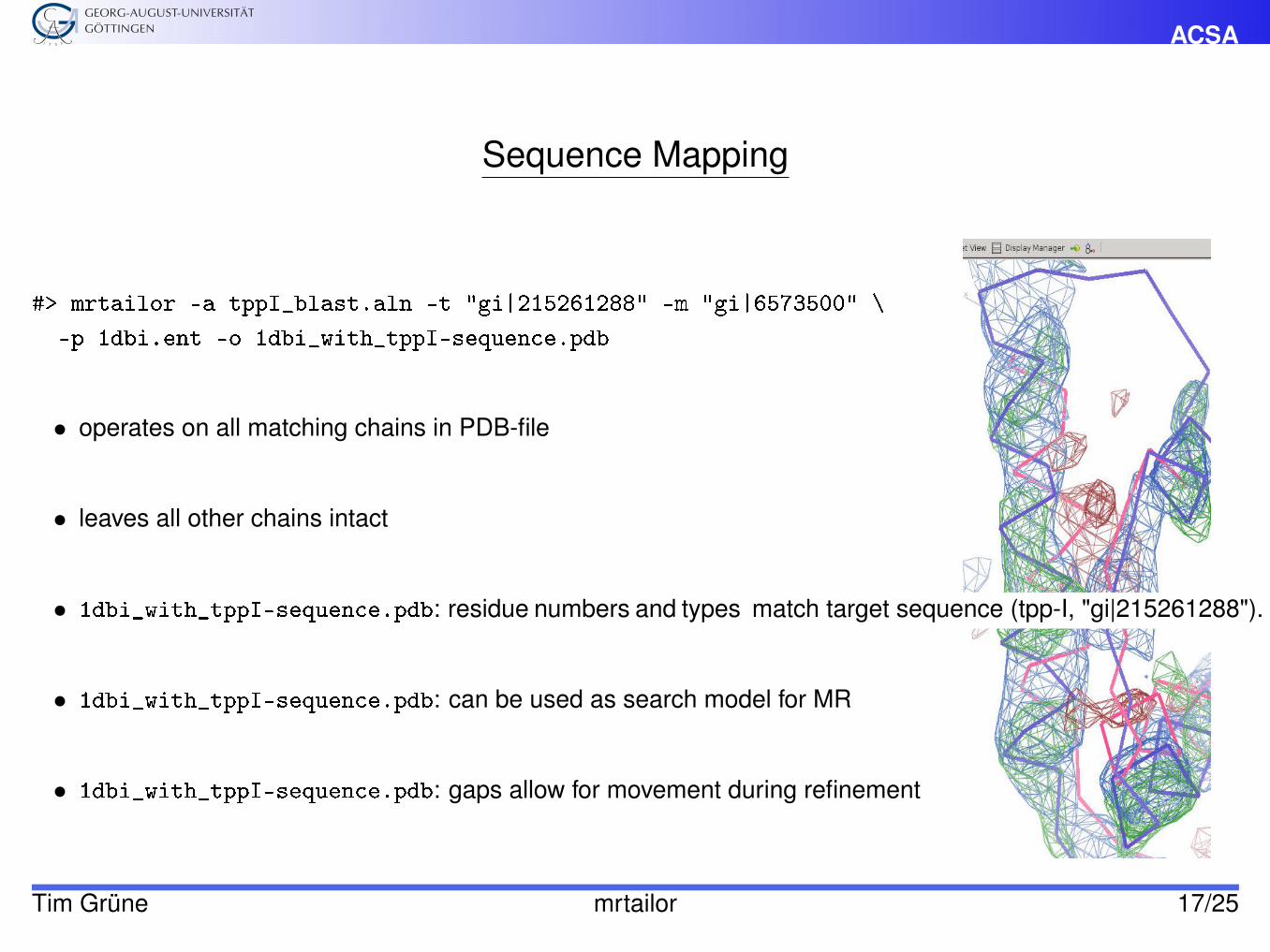
ACSA
Sequence Mapping
#> mrtailor -a tppI_blast.aln -t "gi|215261288" -m "gi|6573500" \
-p 1dbi.ent -o 1dbi_with_tppI-sequence.pdb
• operates on all matching chains in PDB-file
• leaves all other chains intact
• 1dbi_with_tppI-sequence.pdb: residue numbers and types match target sequence (tpp-I, "gi|215261288").
• 1dbi_with_tppI-sequence.pdb: can be used as search model for MR
• 1dbi_with_tppI-sequence.pdb: gaps allow for movement during refinement
Tim Grüne mrtailor 17/25
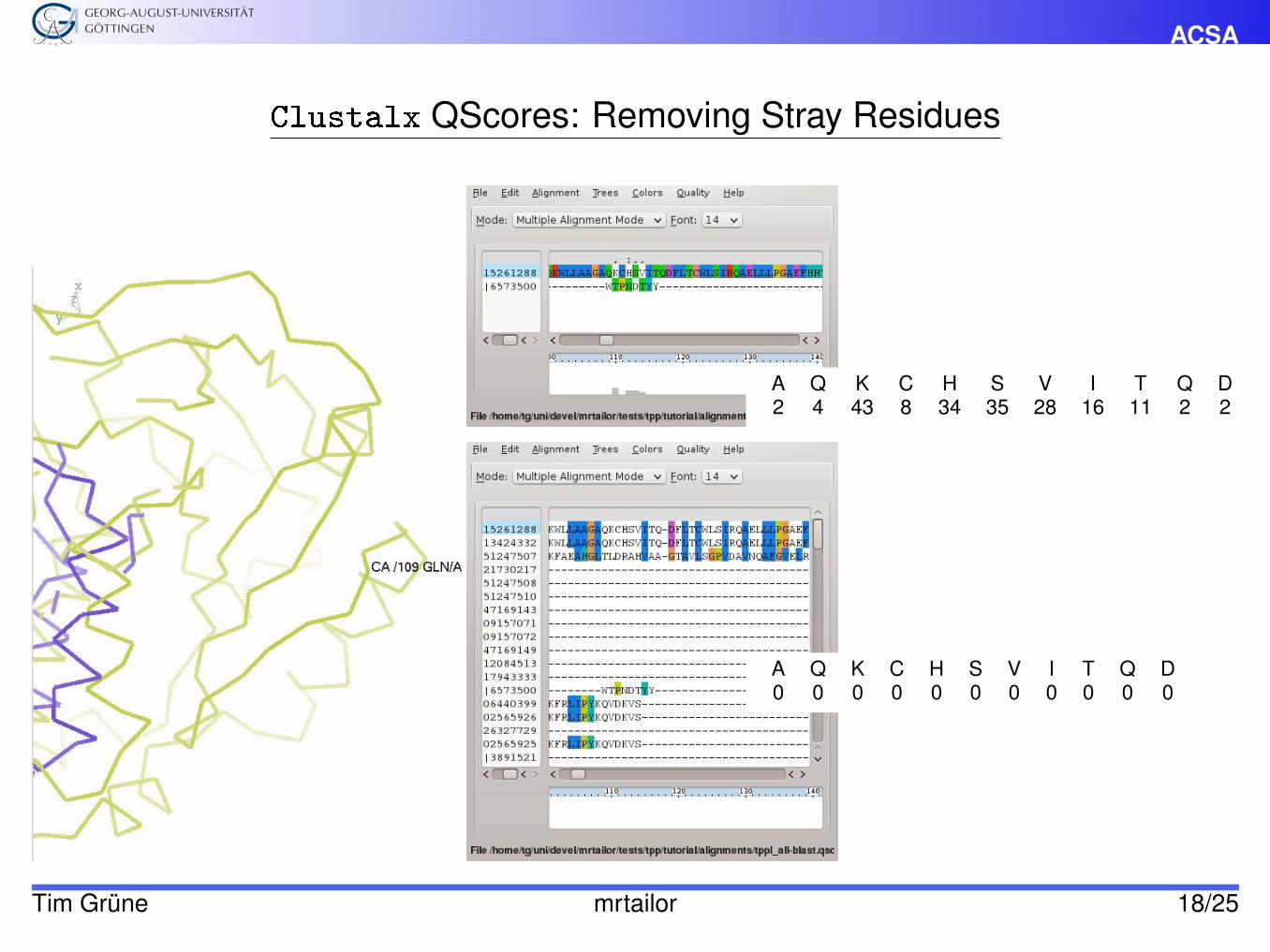
ACSA
Clustalx QScores: Removing Stray Residues
A Q K C H S V I T Q D2 4 43 8 34 35 28 16 11 2 2
A Q K C H S V I T Q D0 0 0 0 0 0 0 0 0 0 0
Tim Grüne mrtailor 18/25

ACSA
Clustalx QScores: Removing Stray Residues
Example TPP-I (PDB 3ee6 vs. 1dbi) Input PDB-file for ProSmart after using mrtailor with qscore cut-off:
Tim Grüne mrtailor 19/25

ACSA
Number of External Restraints
Refmac5 with unmodified 1dbi + Prosmart
Number of atoms : 4062Number of residues : 542Number of chains : 2
Standard External AllBonds: 7862 23442 31304
Refmac5 and Prosmart with mrtailored 1dbi
Number of atoms : 2334Number of residues : 464Number of chains : 2
Standard External AllBonds: 3784 14290 18074
Tim Grüne mrtailor 20/25

ACSA
Algorithm Details
1 2 3 4 4
3EE6 VTTVGGTSFQ---EPFLITNEIVDYISGG <-- Target
1DBI VIAVGAVDQYDRLASF---------SNYG <-- Reference
1. Identical residues (number & type): reference PDB unmodified
2. Different residue types: reduced to Alanine in reference PDB
3. gap in target sequence: residue removed from reference PDB
4. gap in reference sequence: renumbering and renaming (F194 S195→ F397 I407)
Tim Grüne mrtailor 21/25

ACSA
GUI
Tim Grüne mrtailor 22/25
ACSA
Tutorial
http://shelx.uni-ac.gwdg.de/~tg/research/programs/mrtailor/tutorial
• Based on TPP-I "tripeptidyl-peptidase I" (TPP), 2.35 Å
• Pal, A. et al. "Structure of Tripeptidyl-peptidase I Provides Insight into the Molecular Basis of Late InfantileNeuronal Ceroid Lipofuscinosis" J. Biol. Chem. 2009, 284: 3976-3984
Tim Grüne mrtailor 23/25

ACSA
Conclusions
• mrtailor can remove model bias from a molecular replacement solution
• mrtailor can improve the effect of external restraints
• At low resolution the answer is not unique: try several options, e.g.
1. no external restraints (freedom to move)
2. external restraints from original PDB file (more restraints)
3. external restraints after using mrtailor (with and without alignment scoring) (compromise between 1+2)
Tim Grüne mrtailor 24/25