acute antiinflammatory properties of statins involve...
TRANSCRIPT

Acute Antiinflammatory Properties of Statins InvolvePeroxisome Proliferator–Activated Receptor-� via Inhibition
of the Protein Kinase C Signaling PathwayRejane Paumelle, Christophe Blanquart, Olivier Briand, Olivier Barbier, Christian Duhem,
Gaetane Woerly, Frederic Percevault, Jean-Charles Fruchart, David Dombrowicz,Corine Glineur, Bart Staels
Abstract—Statins are inhibitors of 3-hydroxy-3-methylglutaryl–coenzyme A (HMG-CoA) reductase used in the preventionof cardiovascular disease (CVD). In addition to their cholesterol-lowering activities, statins exert pleiotropicantiinflammatory effects, which might contribute to their beneficial effects not only on CVD but also on lipid-unrelatedimmune and inflammatory diseases, such as rheumatoid arthritis, asthma, stroke, and transplant rejection. However, themolecular mechanisms involved in these antiinflammatory properties of statins are unresolved. Here we show that theperoxisome proliferator–activated receptor (PPAR) � mediates antiinflammatory effects of simvastatin in vivo inmodels of acute inflammation. The inhibitory effects of statins on lipopolysaccharide-induced inflammatory responsegenes were abolished in PPAR�-deficient macrophages and neutrophils. Moreover, simvastatin inhibited PPAR�phosphorylation by lipopolysaccharide-activated protein kinase C (PKC) �. A constitutive active form of PKC�inhibited nuclear factor �B transrepression by PPAR� whereas simvastatin enhanced transrepression activity ofwild-type PPAR�, but not of PPAR� mutated in its PKC phosphorylation sites. These data indicate that the acuteantiinflammatory effect of simvastatin occurs via PPAR� by a mechanism involving inhibition of PKC� inactivationof PPAR� transrepression activity. (Circ Res. 2006;98:361-369.)
Key Words: inflammation � macrophages � neutrophils � nuclear receptors � statins � PKC
Statins, competitive inhibitors of 3-hydroxy-3-methylglu-taryl–coenzyme A (HMG-CoA) reductase, the rate-
limiting enzyme in cholesterol synthesis, are widely pre-scribed for the treatment of hypercholesterolemia.1 Inaddition to plasma lipid-modulating action, statins exertpleiotropic antiinflammatory effects, which might contributeto their beneficial effects on cardiovascular disease (CVD).2
Emerging evidences also suggest beneficial therapeutic ac-tivities of statins in immune and inflammatory diseases suchas multiple sclerosis, Alzheimer’s disease, ischemic stroke,transplant rejection, rheumatoid arthritis, and asthma.3–6 Severalclinical observations indicate that these effects cannot beattributed to their cholesterol-lowering activities only.7 Statintherapy decreases plasma concentrations of inflammatorymarkers, such as C-reactive protein (CRP), within 1 weekafter treatment initiation, before any lipid changes are ob-served.8 Statin treatment reduces the incidence of ischemicstroke for which plasma cholesterol levels are not considereda risk factor.9 Moreover, statins also exert antiinflammatoryactions in animal models, which are resistant to their hypo-
lipidemic actions.10 In models of acute and chronic inflam-mation, statins inhibit endothelial adhesion and transendothe-lial migration of leukocytes to sites of inflammation,10 actingboth on endothelial cells and leukocytes. Statins modulatemacrophage functions by inhibiting the activation of inflam-matory response genes, such as interleukin (IL)-1b and IL-6,tumor necrosis factor (TNF) �, metalloproteinase (MMP)-2,and MMP-9, and inducible nitric oxide synthase (iNOS).11
These antiinflammatory actions of statins are attributed totheir ability to modulate signal transduction pathways acti-vating proinflammatory transcription factors, such as nuclearfactor (NF) �B.12
PPAR� is a nuclear receptor that regulates gene expressionby binding with its heterodimeric partner the retinoid-X-receptor (RXR) to PPAR-responsive elements (PPREs).PPAR� not only regulates lipid metabolism13 but also exertspronounced antiinflammatory activities.14 Clinical trials haveshown that fibrates decrease inflammation and have benefi-cial effects on CVD and stroke.14,15 In animals, PPAR�
deficiency induces a prolonged inflammatory response in a
Original received September 12, 2005; revision received December 15, 2005; accepted December 20, 2005.From the Institut Pasteur de Lille (R.P., C.B., O. Briand, O. Barbier, C.D., F.P., J.-C.F., C.G., B.S.), Departement d’Atherosclerose; INSERM, U545
(R.P., C.B., O. Briand, O. Barbier, C.D., F.P., J.-C.F., C.G., B.S.); Universite de Lille 2 (R.P., C.B., O. Briand, O. Barbier, C.D., F.P., J.-C.F., C.G., B.S.);and INSERM, U547 (G.W., D.D.), Institut Pasteur de Lille, Lille, France.
Correspondence to Bart Staels, INSERM U545, Institut Pasteur de Lille, 1 rue Calmette, BP 245, 59019 Lille, France. E-mail [email protected]
© 2006 American Heart Association, Inc.
Circulation Research is available at http://circres.ahajournals.org DOI: 10.1161/01.RES.0000202706.70992.95
361
by guest on June 15, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on June 15, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on June 15, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on June 15, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on June 15, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on June 15, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on June 15, 2018http://circres.ahajournals.org/
Dow
nloaded from

mouse ear-swelling model. PPAR� exerts antiinflammatoryactivities by negatively interfering with proinflammatorysignaling pathways including NF�B. This molecular action isexemplified by the inhibition of inflammatory induction ofgenes, such as vascular cell adhesion molecule-1, MMP-9,IL-6, and TNF�.14
These similarities between the antiinflammatory effects ofstatins and PPAR� led us to investigate whether PPAR�
could mediate antiinflammatory effects of statins in vivo inmodels of acute inflammation and in vitro in macrophagesand neutrophils.
Materials and Methods
Inflammation TestsSubcutaneous dorsal pouches and carrageenan footpad edema wereinduced in C57BL6 wild-type and PPAR�-null mice as described.16,17
Simvastatin at indicated doses or vehicle (CMC 0.5%) was given bygavage to mice 1 hour before inflammatory challenges (see the onlinedata supplement available at http://circres.ahajournals.org).
Cell CultureLipopolysaccharide (LPS)-elicited neutrophils from air pouches andthioglycollate-elicited peritoneal macrophages were isolated as de-scribed.18 Cells were treated with the indicated reagents (see theonline data supplement).
RNA AnalysisRNA extraction was performed using TRIzol reagent followed byreverse transcription (Invitrogen Life Technologies, Cergy-Pontoise,France). cDNA was quantified by real-time PCR on a MX4000apparatus (Stratagene) using specific primers (see the online datasupplement).
Kinase Assays and ImmunoblotAfter treatment, cells were washed with PBS and suspended inprotein kinase C (PKC) lysis buffer, sonicated (Vibracell Hiddock72442), and centrifuged at 4°C (3000 rpm, 15 minutes). Cell extracts(10 �g) or cell extract–immunoprecipitated PKC� (200 �g) wereincubated in kinase reaction buffer, histone H1 (1 �g), or purifiedPPAR� protein (400 ng) as substrates and (�-32P)ATP (5 �Ci) (2000Ci/mmol). Kinase reactions were performed as described previous-ly.19 Immunoblots were performed using the Aurora detectionsystem (ICN Pharmaceuticals, Orsay, France) (see the online datasupplement).
Transient Transfections and Metabolic LabelingCOS-7 cells were transfected by lipofection with reporter andexpression plasmids as indicated and incubated overnight withDMEM supplemented with 2% Ultroser. Cells were collected andluciferase and �-galactosidase assays performed. For 35S-methioninelabeling, cells were cultured in methionine-free minimum essentialmedium for 1 hour before supplementation with 35S-methionine (100�Ci) for an additional 3 hours. For 33P-phosphate labeling, cells weredeprived in phosphate-free minimum essential medium for 2 hoursbefore supplementing the medium with 33P-phosphate (500 �Ci) for5 hours, followed by PPAR� immunoprecipitation (see the onlinedata supplement).
Statistical AnalysisStatistical significance was determined using nonparametric Mann–Whitney or multivariate ANOVA tests followed by Scheffe post hocor the unpaired t tests (transient transfections). Values of P�0.05were considered as significant.
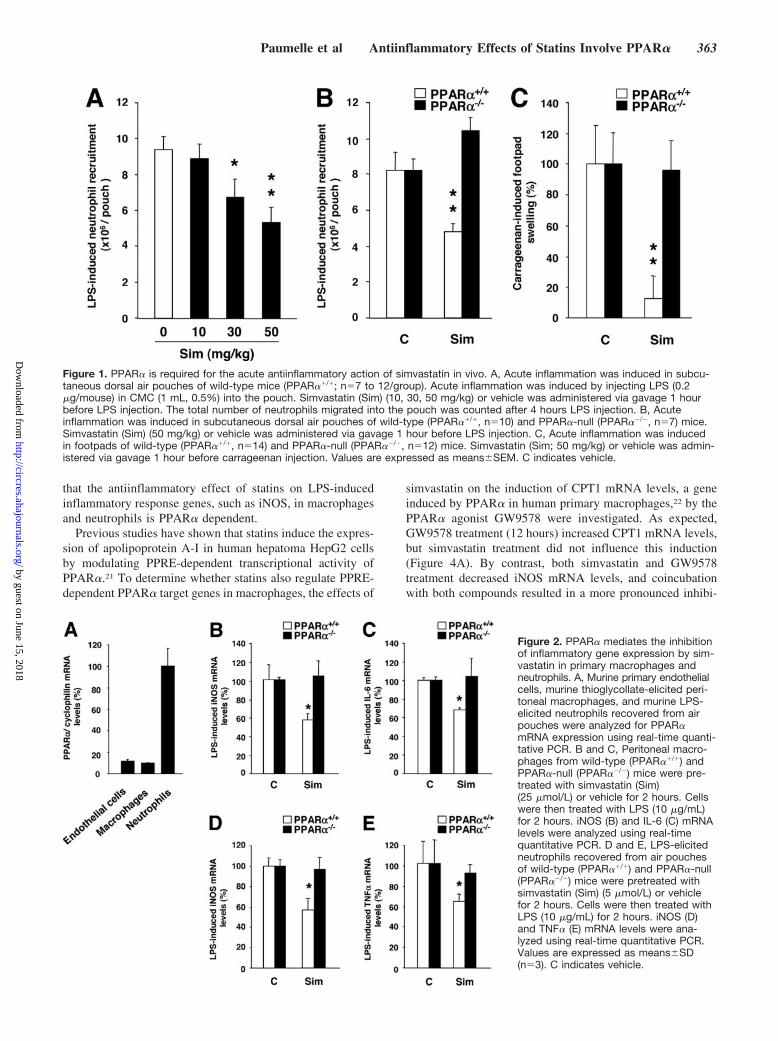
ResultsPPAR� Mediates the Acute AntiinflammatoryAction of Simvastatin In VivoTo investigate whether PPAR� plays a role in inflammatoryresponse modulation by statins in vivo, the influence ofsimvastatin was tested in wild-type and PPAR�-null miceusing 2 models of acute inflammation in which statins displayantiinflammatory activity.16,17 Doses were chosen in accor-dance with these previous studies.16,17 The acute antiinflam-matory action of simvastatin (10 to 50 mg/kg) administeredorally 1 hour before LPS was first measured by the number ofneutrophils recruited in air pouches by LPS.16 Simvastatintreatment decreased neutrophil recruitment in a dose-dependent manner (Figure 1A). Administration of a singledose of atorvastatin (30 mg/kg) exerted similar effects onneutrophil recruitment (not shown). Interestingly, the de-crease of LPS-induced neutrophil recruitment by simvastatinwas only observed in wild-type, but not in PPAR�-null mice(Figure 1B). Similarly, in the carrageenan-induced footpadinflammation mouse model,17 a single dose of simvastatingiven 1 hour before carrageenan injection blocked swellingonly in wild-type, but not in PPAR�-null mice (Figure 1C).These effects occurred independently of alterations in plasmalipid levels, because plasma cholesterol levels did not changeafter simvastatin treatment in either model (not shown). Thus,PPAR� mediates the lipid-independent acute antiinflamma-tory activity of simvastatin in mice.
PPAR� Mediates the Inhibition of LPS-InducedInflammatory Response Genes by Simvastatin inPrimary Macrophages and NeutrophilsBecause neutrophils are a major cell type mediating theinflammatory response in these in vivo models, the expres-sion of PPAR� was analyzed in LPS-elicited neutrophilsrecovered from air pouches and compared with other celltypes. PPAR� mRNA levels were highest in neutrophils,whereas macrophages express similar levels as primary en-dothelial cells, a cell type in which the antiinflammatoryeffects of PPAR� have been well documented20 (Figure 2A).
Subsequently, the role of PPAR� in the modulation ofinflammatory response gene expression (iNOS, TNF�, orIL-6) by statins was investigated in macrophages and neutro-phils isolated from wild-type and PPAR�-null mice. Pretreat-ment (2 hours) of macrophages isolated from wild-type micewith a single dose of simvastatin, sufficient to inhibit HMG-CoA reductase activity,18 significantly decreased LPS-induced iNOS and IL-6 mRNA levels (Figure 2B and 2C). Bycontrast, simvastatin was without effect in macrophagesisolated from PPAR�-null mice. Similarly, pretreatment (2hours) with simvastatin also significantly decreased LPS-induced iNOS and TNF� mRNA levels in neutrophils iso-lated from wild-type but not from PPAR�-null mice (Figure2D and 2E). Simvastatin induced a dose-dependent decreasein iNOS mRNA and protein levels after LPS induction onlyin wild-type but not in PPAR�-null macrophages (Figure 3Aand 3B). Atorvastatin and fluvastatin pretreatment also de-creased LPS-induced iNOS expression in a PPAR�-dependent manner (Figure 3C and 3D). These data indicate
362 Circulation Research February 17, 2006
by guest on June 15, 2018http://circres.ahajournals.org/
Dow
nloaded from
that the antiinflammatory effect of statins on LPS-inducedinflammatory response genes, such as iNOS, in macrophagesand neutrophils is PPAR� dependent.
Previous studies have shown that statins induce the expres-sion of apolipoprotein A-I in human hepatoma HepG2 cellsby modulating PPRE-dependent transcriptional activity ofPPAR�.21 To determine whether statins also regulate PPRE-dependent PPAR� target genes in macrophages, the effects of
simvastatin on the induction of CPT1 mRNA levels, a geneinduced by PPAR� in human primary macrophages,22 by thePPAR� agonist GW9578 were investigated. As expected,GW9578 treatment (12 hours) increased CPT1 mRNA levels,but simvastatin treatment did not influence this induction(Figure 4A). By contrast, both simvastatin and GW9578treatment decreased iNOS mRNA levels, and coincubationwith both compounds resulted in a more pronounced inhibi-
Figure 1. PPAR� is required for the acute antiinflammatory action of simvastatin in vivo. A, Acute inflammation was induced in subcu-taneous dorsal air pouches of wild-type mice (PPAR��/�; n�7 to 12/group). Acute inflammation was induced by injecting LPS (0.2�g/mouse) in CMC (1 mL, 0.5%) into the pouch. Simvastatin (Sim) (10, 30, 50 mg/kg) or vehicle was administered via gavage 1 hourbefore LPS injection. The total number of neutrophils migrated into the pouch was counted after 4 hours LPS injection. B, Acuteinflammation was induced in subcutaneous dorsal air pouches of wild-type (PPAR��/�, n�10) and PPAR�-null (PPAR��/�, n�7) mice.Simvastatin (Sim) (50 mg/kg) or vehicle was administered via gavage 1 hour before LPS injection. C, Acute inflammation was inducedin footpads of wild-type (PPAR��/�, n�14) and PPAR�-null (PPAR��/�, n�12) mice. Simvastatin (Sim; 50 mg/kg) or vehicle was admin-istered via gavage 1 hour before carrageenan injection. Values are expressed as means�SEM. C indicates vehicle.
Figure 2. PPAR� mediates the inhibitionof inflammatory gene expression by sim-vastatin in primary macrophages andneutrophils. A, Murine primary endothelialcells, murine thioglycollate-elicited peri-toneal macrophages, and murine LPS-elicited neutrophils recovered from airpouches were analyzed for PPAR�mRNA expression using real-time quanti-tative PCR. B and C, Peritoneal macro-phages from wild-type (PPAR��/�) andPPAR�-null (PPAR��/�) mice were pre-treated with simvastatin (Sim)(25 �mol/L) or vehicle for 2 hours. Cellswere then treated with LPS (10 �g/mL)for 2 hours. iNOS (B) and IL-6 (C) mRNAlevels were analyzed using real-timequantitative PCR. D and E, LPS-elicitedneutrophils recovered from air pouchesof wild-type (PPAR��/�) and PPAR�-null(PPAR��/�) mice were pretreated withsimvastatin (Sim) (5 �mol/L) or vehiclefor 2 hours. Cells were then treated withLPS (10 �g/mL) for 2 hours. iNOS (D)and TNF� (E) mRNA levels were ana-lyzed using real-time quantitative PCR.Values are expressed as means�SD(n�3). C indicates vehicle.
Paumelle et al Antiinflammatory Effects of Statins Involve PPAR� 363
by guest on June 15, 2018http://circres.ahajournals.org/
Dow
nloaded from
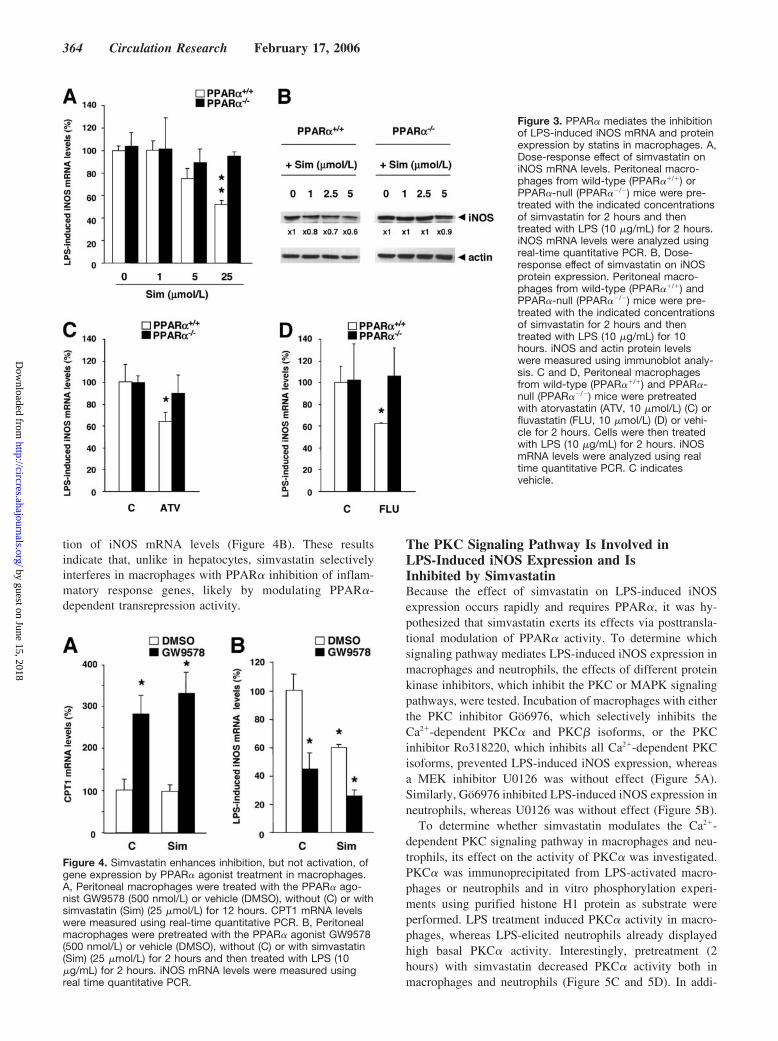
tion of iNOS mRNA levels (Figure 4B). These resultsindicate that, unlike in hepatocytes, simvastatin selectivelyinterferes in macrophages with PPAR� inhibition of inflam-matory response genes, likely by modulating PPAR�-dependent transrepression activity.
The PKC Signaling Pathway Is Involved inLPS-Induced iNOS Expression and IsInhibited by SimvastatinBecause the effect of simvastatin on LPS-induced iNOSexpression occurs rapidly and requires PPAR�, it was hy-pothesized that simvastatin exerts its effects via posttransla-tional modulation of PPAR� activity. To determine whichsignaling pathway mediates LPS-induced iNOS expression inmacrophages and neutrophils, the effects of different proteinkinase inhibitors, which inhibit the PKC or MAPK signalingpathways, were tested. Incubation of macrophages with eitherthe PKC inhibitor Go6976, which selectively inhibits theCa2�-dependent PKC� and PKC� isoforms, or the PKCinhibitor Ro318220, which inhibits all Ca2�-dependent PKCisoforms, prevented LPS-induced iNOS expression, whereasa MEK inhibitor U0126 was without effect (Figure 5A).Similarly, Go6976 inhibited LPS-induced iNOS expression inneutrophils, whereas U0126 was without effect (Figure 5B).
To determine whether simvastatin modulates the Ca2�-dependent PKC signaling pathway in macrophages and neu-trophils, its effect on the activity of PKC� was investigated.PKC� was immunoprecipitated from LPS-activated macro-phages or neutrophils and in vitro phosphorylation experi-ments using purified histone H1 protein as substrate wereperformed. LPS treatment induced PKC� activity in macro-phages, whereas LPS-elicited neutrophils already displayedhigh basal PKC� activity. Interestingly, pretreatment (2hours) with simvastatin decreased PKC� activity both inmacrophages and neutrophils (Figure 5C and 5D). In addi-
Figure 3. PPAR� mediates the inhibitionof LPS-induced iNOS mRNA and proteinexpression by statins in macrophages. A,Dose-response effect of simvastatin oniNOS mRNA levels. Peritoneal macro-phages from wild-type (PPAR��/�) orPPAR�-null (PPAR��/�) mice were pre-treated with the indicated concentrationsof simvastatin for 2 hours and thentreated with LPS (10 �g/mL) for 2 hours.iNOS mRNA levels were analyzed usingreal-time quantitative PCR. B, Dose-response effect of simvastatin on iNOSprotein expression. Peritoneal macro-phages from wild-type (PPAR��/�) andPPAR�-null (PPAR��/�) mice were pre-treated with the indicated concentrationsof simvastatin for 2 hours and thentreated with LPS (10 �g/mL) for 10hours. iNOS and actin protein levelswere measured using immunoblot analy-sis. C and D, Peritoneal macrophagesfrom wild-type (PPAR��/�) and PPAR�-null (PPAR��/�) mice were pretreatedwith atorvastatin (ATV, 10 �mol/L) (C) orfluvastatin (FLU, 10 �mol/L) (D) or vehi-cle for 2 hours. Cells were then treatedwith LPS (10 �g/mL) for 2 hours. iNOSmRNA levels were analyzed using realtime quantitative PCR. C indicatesvehicle.
Figure 4. Simvastatin enhances inhibition, but not activation, ofgene expression by PPAR� agonist treatment in macrophages.A, Peritoneal macrophages were treated with the PPAR� ago-nist GW9578 (500 nmol/L) or vehicle (DMSO), without (C) or withsimvastatin (Sim) (25 �mol/L) for 12 hours. CPT1 mRNA levelswere measured using real-time quantitative PCR. B, Peritonealmacrophages were pretreated with the PPAR� agonist GW9578(500 nmol/L) or vehicle (DMSO), without (C) or with simvastatin(Sim) (25 �mol/L) for 2 hours and then treated with LPS (10�g/mL) for 2 hours. iNOS mRNA levels were measured usingreal time quantitative PCR.
364 Circulation Research February 17, 2006
by guest on June 15, 2018http://circres.ahajournals.org/
Dow
nloaded from
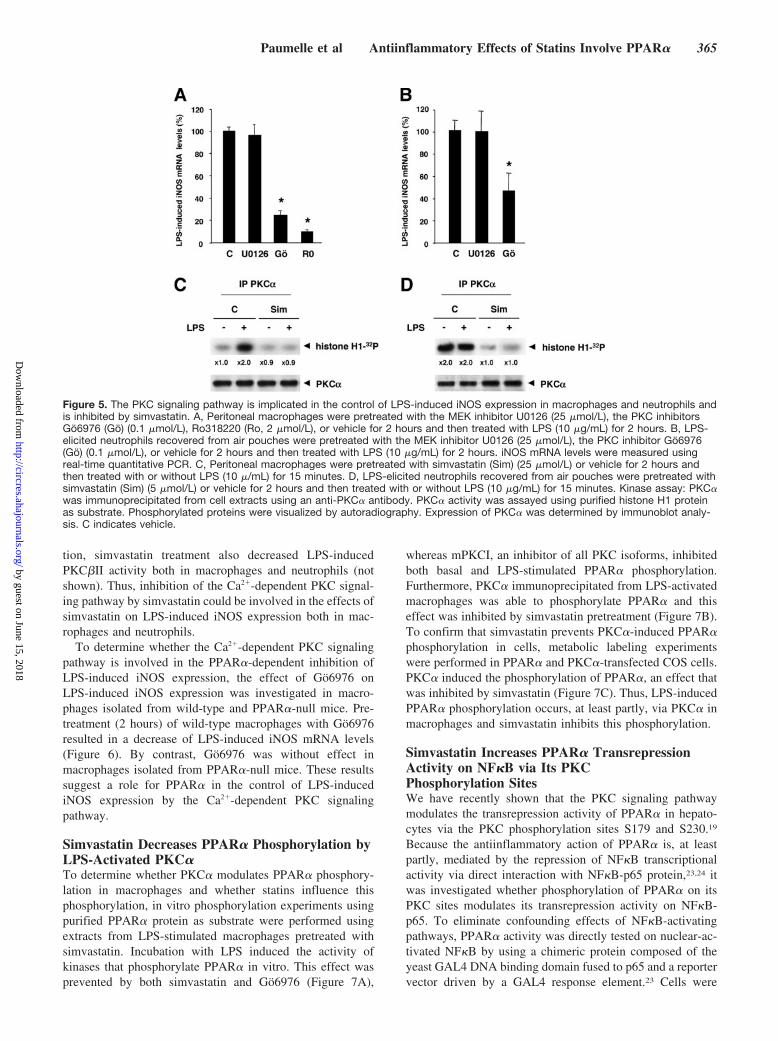
tion, simvastatin treatment also decreased LPS-inducedPKC�II activity both in macrophages and neutrophils (notshown). Thus, inhibition of the Ca2�-dependent PKC signal-ing pathway by simvastatin could be involved in the effects ofsimvastatin on LPS-induced iNOS expression both in mac-rophages and neutrophils.
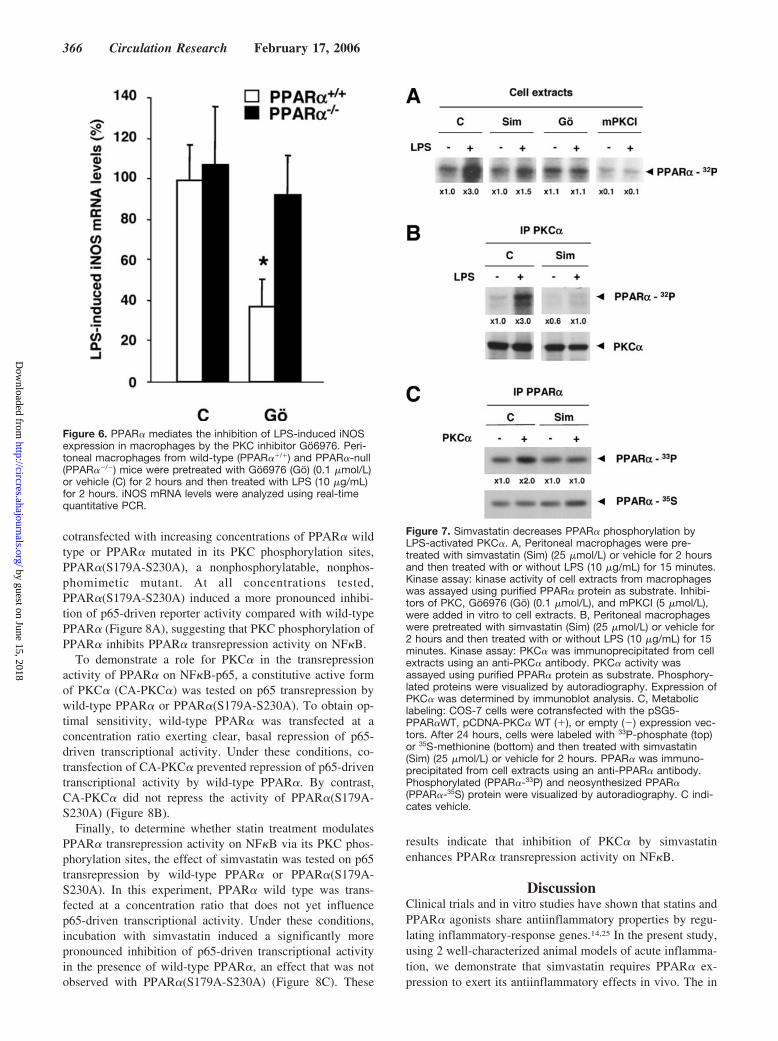
To determine whether the Ca2�-dependent PKC signalingpathway is involved in the PPAR�-dependent inhibition ofLPS-induced iNOS expression, the effect of Go6976 onLPS-induced iNOS expression was investigated in macro-phages isolated from wild-type and PPAR�-null mice. Pre-treatment (2 hours) of wild-type macrophages with Go6976resulted in a decrease of LPS-induced iNOS mRNA levels(Figure 6). By contrast, Go6976 was without effect inmacrophages isolated from PPAR�-null mice. These resultssuggest a role for PPAR� in the control of LPS-inducediNOS expression by the Ca2�-dependent PKC signalingpathway.
Simvastatin Decreases PPAR� Phosphorylation byLPS-Activated PKC�To determine whether PKC� modulates PPAR� phosphory-lation in macrophages and whether statins influence thisphosphorylation, in vitro phosphorylation experiments usingpurified PPAR� protein as substrate were performed usingextracts from LPS-stimulated macrophages pretreated withsimvastatin. Incubation with LPS induced the activity ofkinases that phosphorylate PPAR� in vitro. This effect wasprevented by both simvastatin and Go6976 (Figure 7A),
whereas mPKCI, an inhibitor of all PKC isoforms, inhibitedboth basal and LPS-stimulated PPAR� phosphorylation.Furthermore, PKC� immunoprecipitated from LPS-activatedmacrophages was able to phosphorylate PPAR� and thiseffect was inhibited by simvastatin pretreatment (Figure 7B).To confirm that simvastatin prevents PKC�-induced PPAR�phosphorylation in cells, metabolic labeling experimentswere performed in PPAR� and PKC�-transfected COS cells.PKC� induced the phosphorylation of PPAR�, an effect thatwas inhibited by simvastatin (Figure 7C). Thus, LPS-inducedPPAR� phosphorylation occurs, at least partly, via PKC� inmacrophages and simvastatin inhibits this phosphorylation.
Simvastatin Increases PPAR� TransrepressionActivity on NF�B via Its PKCPhosphorylation SitesWe have recently shown that the PKC signaling pathwaymodulates the transrepression activity of PPAR� in hepato-cytes via the PKC phosphorylation sites S179 and S230.19
Because the antiinflammatory action of PPAR� is, at leastpartly, mediated by the repression of NF�B transcriptionalactivity via direct interaction with NF�B-p65 protein,23,24 itwas investigated whether phosphorylation of PPAR� on itsPKC sites modulates its transrepression activity on NF�B-p65. To eliminate confounding effects of NF�B-activatingpathways, PPAR� activity was directly tested on nuclear-ac-tivated NF�B by using a chimeric protein composed of theyeast GAL4 DNA binding domain fused to p65 and a reportervector driven by a GAL4 response element.23 Cells were
Figure 5. The PKC signaling pathway is implicated in the control of LPS-induced iNOS expression in macrophages and neutrophils andis inhibited by simvastatin. A, Peritoneal macrophages were pretreated with the MEK inhibitor U0126 (25 �mol/L), the PKC inhibitorsGo6976 (Go) (0.1 �mol/L), Ro318220 (Ro, 2 �mol/L), or vehicle for 2 hours and then treated with LPS (10 �g/mL) for 2 hours. B, LPS-elicited neutrophils recovered from air pouches were pretreated with the MEK inhibitor U0126 (25 �mol/L), the PKC inhibitor Go6976(Go) (0.1 �mol/L), or vehicle for 2 hours and then treated with LPS (10 �g/mL) for 2 hours. iNOS mRNA levels were measured usingreal-time quantitative PCR. C, Peritoneal macrophages were pretreated with simvastatin (Sim) (25 �mol/L) or vehicle for 2 hours andthen treated with or without LPS (10 �/mL) for 15 minutes. D, LPS-elicited neutrophils recovered from air pouches were pretreated withsimvastatin (Sim) (5 �mol/L) or vehicle for 2 hours and then treated with or without LPS (10 �g/mL) for 15 minutes. Kinase assay: PKC�was immunoprecipitated from cell extracts using an anti-PKC� antibody. PKC� activity was assayed using purified histone H1 proteinas substrate. Phosphorylated proteins were visualized by autoradiography. Expression of PKC� was determined by immunoblot analy-sis. C indicates vehicle.
Paumelle et al Antiinflammatory Effects of Statins Involve PPAR� 365
by guest on June 15, 2018http://circres.ahajournals.org/
Dow
nloaded from
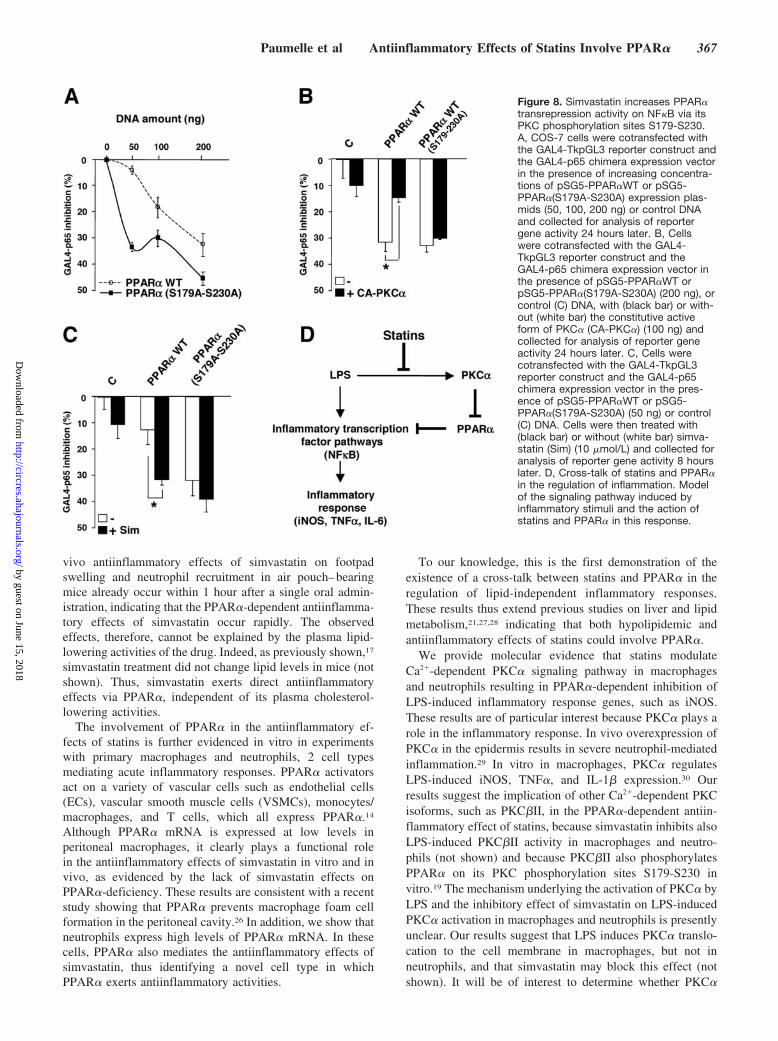
cotransfected with increasing concentrations of PPAR� wildtype or PPAR� mutated in its PKC phosphorylation sites,PPAR�(S179A-S230A), a nonphosphorylatable, nonphos-phomimetic mutant. At all concentrations tested,PPAR�(S179A-S230A) induced a more pronounced inhibi-tion of p65-driven reporter activity compared with wild-typePPAR� (Figure 8A), suggesting that PKC phosphorylation ofPPAR� inhibits PPAR� transrepression activity on NF�B.
To demonstrate a role for PKC� in the transrepressionactivity of PPAR� on NF�B-p65, a constitutive active formof PKC� (CA-PKC�) was tested on p65 transrepression bywild-type PPAR� or PPAR�(S179A-S230A). To obtain op-timal sensitivity, wild-type PPAR� was transfected at aconcentration ratio exerting clear, basal repression of p65-driven transcriptional activity. Under these conditions, co-transfection of CA-PKC� prevented repression of p65-driventranscriptional activity by wild-type PPAR�. By contrast,CA-PKC� did not repress the activity of PPAR�(S179A-S230A) (Figure 8B).
Finally, to determine whether statin treatment modulatesPPAR� transrepression activity on NF�B via its PKC phos-phorylation sites, the effect of simvastatin was tested on p65transrepression by wild-type PPAR� or PPAR�(S179A-S230A). In this experiment, PPAR� wild type was trans-fected at a concentration ratio that does not yet influencep65-driven transcriptional activity. Under these conditions,incubation with simvastatin induced a significantly morepronounced inhibition of p65-driven transcriptional activityin the presence of wild-type PPAR�, an effect that was notobserved with PPAR�(S179A-S230A) (Figure 8C). These
results indicate that inhibition of PKC� by simvastatinenhances PPAR� transrepression activity on NF�B.
DiscussionClinical trials and in vitro studies have shown that statins andPPAR� agonists share antiinflammatory properties by regu-lating inflammatory-response genes.14,25 In the present study,using 2 well-characterized animal models of acute inflamma-tion, we demonstrate that simvastatin requires PPAR� ex-pression to exert its antiinflammatory effects in vivo. The in
Figure 6. PPAR� mediates the inhibition of LPS-induced iNOSexpression in macrophages by the PKC inhibitor Go6976. Peri-toneal macrophages from wild-type (PPAR��/�) and PPAR�-null(PPAR��/�) mice were pretreated with Go6976 (Go) (0.1 �mol/L)or vehicle (C) for 2 hours and then treated with LPS (10 �g/mL)for 2 hours. iNOS mRNA levels were analyzed using real-timequantitative PCR.
Figure 7. Simvastatin decreases PPAR� phosphorylation byLPS-activated PKC�. A, Peritoneal macrophages were pre-treated with simvastatin (Sim) (25 �mol/L) or vehicle for 2 hoursand then treated with or without LPS (10 �g/mL) for 15 minutes.Kinase assay: kinase activity of cell extracts from macrophageswas assayed using purified PPAR� protein as substrate. Inhibi-tors of PKC, Go6976 (Go) (0.1 �mol/L), and mPKCI (5 �mol/L),were added in vitro to cell extracts. B, Peritoneal macrophageswere pretreated with simvastatin (Sim) (25 �mol/L) or vehicle for2 hours and then treated with or without LPS (10 �g/mL) for 15minutes. Kinase assay: PKC� was immunoprecipitated from cellextracts using an anti-PKC� antibody. PKC� activity wasassayed using purified PPAR� protein as substrate. Phosphory-lated proteins were visualized by autoradiography. Expression ofPKC� was determined by immunoblot analysis. C, Metaboliclabeling: COS-7 cells were cotransfected with the pSG5-PPAR�WT, pCDNA-PKC� WT (�), or empty (�) expression vec-tors. After 24 hours, cells were labeled with 33P-phosphate (top)or 35S-methionine (bottom) and then treated with simvastatin(Sim) (25 �mol/L) or vehicle for 2 hours. PPAR� was immuno-precipitated from cell extracts using an anti-PPAR� antibody.Phosphorylated (PPAR�-33P) and neosynthesized PPAR�(PPAR�-35S) protein were visualized by autoradiography. C indi-cates vehicle.
366 Circulation Research February 17, 2006
by guest on June 15, 2018http://circres.ahajournals.org/
Dow
nloaded from
vivo antiinflammatory effects of simvastatin on footpadswelling and neutrophil recruitment in air pouch–bearingmice already occur within 1 hour after a single oral admin-istration, indicating that the PPAR�-dependent antiinflamma-tory effects of simvastatin occur rapidly. The observedeffects, therefore, cannot be explained by the plasma lipid-lowering activities of the drug. Indeed, as previously shown,17
simvastatin treatment did not change lipid levels in mice (notshown). Thus, simvastatin exerts direct antiinflammatoryeffects via PPAR�, independent of its plasma cholesterol-lowering activities.
The involvement of PPAR� in the antiinflammatory ef-fects of statins is further evidenced in vitro in experimentswith primary macrophages and neutrophils, 2 cell typesmediating acute inflammatory responses. PPAR� activatorsact on a variety of vascular cells such as endothelial cells(ECs), vascular smooth muscle cells (VSMCs), monocytes/macrophages, and T cells, which all express PPAR�.14
Although PPAR� mRNA is expressed at low levels inperitoneal macrophages, it clearly plays a functional rolein the antiinflammatory effects of simvastatin in vitro and invivo, as evidenced by the lack of simvastatin effects onPPAR�-deficiency. These results are consistent with a recentstudy showing that PPAR� prevents macrophage foam cellformation in the peritoneal cavity.26 In addition, we show thatneutrophils express high levels of PPAR� mRNA. In thesecells, PPAR� also mediates the antiinflammatory effects ofsimvastatin, thus identifying a novel cell type in whichPPAR� exerts antiinflammatory activities.
To our knowledge, this is the first demonstration of theexistence of a cross-talk between statins and PPAR� in theregulation of lipid-independent inflammatory responses.These results thus extend previous studies on liver and lipidmetabolism,21,27,28 indicating that both hypolipidemic andantiinflammatory effects of statins could involve PPAR�.
We provide molecular evidence that statins modulateCa2�-dependent PKC� signaling pathway in macrophagesand neutrophils resulting in PPAR�-dependent inhibition ofLPS-induced inflammatory response genes, such as iNOS.These results are of particular interest because PKC� plays arole in the inflammatory response. In vivo overexpression ofPKC� in the epidermis results in severe neutrophil-mediatedinflammation.29 In vitro in macrophages, PKC� regulatesLPS-induced iNOS, TNF�, and IL-1� expression.30 Ourresults suggest the implication of other Ca2�-dependent PKCisoforms, such as PKC�II, in the PPAR�-dependent antiin-flammatory effect of statins, because simvastatin inhibits alsoLPS-induced PKC�II activity in macrophages and neutro-phils (not shown) and because PKC�II also phosphorylatesPPAR� on its PKC phosphorylation sites S179-S230 invitro.19 The mechanism underlying the activation of PKC� byLPS and the inhibitory effect of simvastatin on LPS-inducedPKC� activation in macrophages and neutrophils is presentlyunclear. Our results suggest that LPS induces PKC� translo-cation to the cell membrane in macrophages, but not inneutrophils, and that simvastatin may block this effect (notshown). It will be of interest to determine whether PKC�
Figure 8. Simvastatin increases PPAR�transrepression activity on NF�B via itsPKC phosphorylation sites S179-S230.A, COS-7 cells were cotransfected withthe GAL4-TkpGL3 reporter construct andthe GAL4-p65 chimera expression vectorin the presence of increasing concentra-tions of pSG5-PPAR�WT or pSG5-PPAR�(S179A-S230A) expression plas-mids (50, 100, 200 ng) or control DNAand collected for analysis of reportergene activity 24 hours later. B, Cellswere cotransfected with the GAL4-TkpGL3 reporter construct and theGAL4-p65 chimera expression vector inthe presence of pSG5-PPAR�WT orpSG5-PPAR�(S179A-S230A) (200 ng), orcontrol (C) DNA, with (black bar) or with-out (white bar) the constitutive activeform of PKC� (CA-PKC�) (100 ng) andcollected for analysis of reporter geneactivity 24 hours later. C, Cells werecotransfected with the GAL4-TkpGL3reporter construct and the GAL4-p65chimera expression vector in the pres-ence of pSG5-PPAR�WT or pSG5-PPAR�(S179A-S230A) (50 ng) or control(C) DNA. Cells were then treated with(black bar) or without (white bar) simva-statin (Sim) (10 �mol/L) and collected foranalysis of reporter gene activity 8 hourslater. D, Cross-talk of statins and PPAR�in the regulation of inflammation. Modelof the signaling pathway induced byinflammatory stimuli and the action ofstatins and PPAR� in this response.
Paumelle et al Antiinflammatory Effects of Statins Involve PPAR� 367
by guest on June 15, 2018http://circres.ahajournals.org/
Dow
nloaded from

activators (PLC, PDK1) or repressors (DAGK or PP1 phos-phatases)31 are regulated by simvastatin in these cells.
We previously demonstrated that classical PKCs phosphor-ylate PPAR� in vitro.19 Here, we show that LPS induction ofPKC� in macrophages results in increased PPAR� phosphor-ylation in vitro and that PKC� overexpression increasedPPAR� phosphorylation in cells. Moreover, simvastatin in-hibited PKC�-induced PPAR� phosphorylation. PPAR� is aphosphoprotein phosphorylated by different kinases, such asextracellular signal-regulated kinase,32 p38,33 and PKA.34
Previously identified PPAR�-phosphorylating kinases all en-hanced PPAR� transcriptional activity. In our report, weshow, by using PPAR� mutated on its PKC phosphorylationsites (S179-S230) as well as a CA-PKC�, that activatedPKC� inhibits the transrepression properties of PPAR� onNF�B-p65. By contrast, simvastatin enhances PPAR� trans-repression activity acting via its PKC phosphorylation sites(S179-S230), suggesting that simvastatin stimulates PPAR�transrepression activity via inhibition of PPAR� inactivationby PKC�. Whereas in liver cells, the PKC signaling pathwayalso regulates the ligand-dependent PPAR� transactivationproperties, as demonstrated by enhanced CPT1 induction,19 inmacrophages, simvastatin treatment did not modify PPAR�-induced CPT1 expression, even in the presence of a PPAR�agonist. Inhibition of the Ca2�-dependent PKC signalingpathway by simvastatin thus only influences the transrepres-sion properties of PPAR� in macrophages. We propose thatactivation of PKC� by inflammatory stimuli, such as LPS,leads to the phosphorylation and subsequent deactivation ofPPAR�. Statins prevent PKC� activation by LPS and, as aconsequence, inhibit PPAR� phosphorylation by PKC�,leading to enhanced PPAR� transrepressive activity onNF�B (Figure 8D).
The effects of statins on inflammation could also involveNF�B-independent mechanisms, eg, via modulation ofCD62L and CD11b adhesion molecule expression in mono-cytes.35 However, we did not observe any effect of statins onthe expression of these adhesion molecules in neutrophils(not shown). Nonetheless, our results do not exclude thatother PPAR�- and PKC-independent mechanisms contributealso to the antiinflammatory effects of statins because statinsregulate other signaling pathways such as phosphatidylinosi-tol 3-kinase and mitogen-activated protein kinase.36,37
Macrophages and neutrophils are mediators of the earlyinflammatory response that play a major role in the inflam-mation and tissue damage associated with both infectious andnoninfectious diseases, such as sepsis, acute coronary syn-drome, rheumatoid arthritis, and ischemic stroke.38–41 Resultsfrom basic research and clinical trials indicate that thepleiotropic antiinflammatory effects of statins may result inclinical benefit in such inflammatory diseases.42–44 Our re-sults demonstrating that statins exert their antiinflammatoryeffects through PPAR� provide further evidence for theimportance of such pleiotropic activities. Clinical studies withPPAR� agonists have shown significant protective effectsagainst CVD and stroke, effects that cannot be attributed totheir cholesterol-lowering activities alone.14,15 Our resultsthus provide a potential clinically relevant mechanism for thepleiotropic effects of statins through PPAR�.
AcknowledgmentsThis work was supported by grants from the Fondation pour laRecherche Medicale (to R.P. and C.B.), Institut de France (O.B.),Fondation Leducq, and European community grant QLRT-1999-01007. We thank Drs Parker and Haegeman for providingexpression plasmids.
References1. Evans M, Roberts A, Davies S, Rees A. Medical lipid-regulating therapy:
current evidence, ongoing trials and future developments. Drugs. 2004;64:1181–1196.
2. Landmesser U, Bahlmann F, Mueller M, Spiekermann S, Kirchhoff N,Schulz S, Manes C, Fischer D, de Groot K, Fliser D, Fauler G, Marz W,Drexler H. Simvastatin versus ezetimibe: pleiotropic and lipid-loweringeffects on endothelial function in humans. Circulation. 2005;111:2356–2363.
3. Stuve O, Youssef S, Steinman L, Zamvil SS. Statins as potential thera-peutic agents in neuroinflammatory disorders. Curr Opin Neurol. 2003;16:393–401.
4. McKay A, Leung BP, McInnes IB, Thomson NC, Liew FY. A novelanti-inflammatory role of simvastatin in a murine model of allergicasthma. J Immunol. 2004;172:2903–2908.
5. McCarey DW, McInnes IB, Madhok R, Hampson R, Scherbakov O, FordI, Capell HA, Sattar N. Trial of Atorvastatin in Rheumatoid Arthritis(TARA): double-blind, randomised placebo-controlled trial. Lancet.2004;363:2015–2021.
6. Maggard MA, Ke B, Wang T, Kaldas F, Seu P, Busuttil RW, ImagawaDK. Effects of pravastatin on chronic rejection of rat cardiac allografts.Transplantation. 1998;65:149–155.
7. Crisby M. Modulation of the inflammatory process by statins. DrugsToday (Barc). 2003;39:137–143.
8. Ansell BJ, Watson KE, Weiss RE, Fonarow GC. hsCRP and HDL Effectsof Statins Trial (CHEST): rapid effect of statin therapy on C-reactiveprotein and high-density lipoprotein levels. A clinical investigation. HeartDis. 2003;5:2–7.
9. Plutzky J, Ridker PM. Statins for stroke: the second story? Circulation.2001;103:348–350.
10. Weitz-Schmidt G. Statins as anti-inflammatory agents. Trends PharmacolSci. 2002;23:482–486.
11. Takemoto M, Liao JK. Pleiotropic effects of 3-hydroxy-3-methylglutarylcoenzyme A reductase inhibitors. Arterioscler Thromb Vasc Biol. 2001;21:1712–1719.
12. Dichtl W, Dulak J, Frick M, Alber HF, Schwarzacher SP, Ares MP,Nilsson J, Pachinger O, Weidinger F. HMG-CoA reductase inhibitorsregulate inflammatory transcription factors in human endothelial andvascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2003;23:58–63.
13. Staels B, Dallongeville J, Auwerx J, Schoonjans K, Leitersdorf E,Fruchart JC. Mechanism of action of fibrates on lipid and lipoproteinmetabolism. Circulation. 1998;98:2088–2093.
14. Marx N, Duez H, Fruchart JC, Staels B. Peroxisome proliferator-activatedreceptors and atherogenesis: regulators of gene expression in vascularcells. Circ Res. 2004;94:1168–1178.
15. Bloomfield Rubins H, Davenport J, Babikian V, Brass LM, Collins D,Wexler L, Wagner S, Papademetriou V, Rutan G, Robins SJ. Reductionin stroke with gemfibrozil in men with coronary heart disease and lowHDL cholesterol: The Veterans Affairs HDL Intervention Trial(VA-HIT). Circulation. 2001;103:2828–2833.
16. Diomede L, Albani D, Sottocorno M, Donati MB, Bianchi M, FruscellaP, Salmona M. In vivo anti-inflammatory effect of statins is mediated bynonsterol mevalonate products. Arterioscler Thromb Vasc Biol. 2001;21:1327–1332.
17. Sparrow CP, Burton CA, Hernandez M, Mundt S, Hassing H, Patel S,Rosa R, Hermanowski-Vosatka A, Wang PR, Zhang D, Peterson L,Detmers PA, Chao YS, Wright SD. Simvastatin has anti-inflammatoryand antiatherosclerotic activities independent of plasma cholesterollowering. Arterioscler Thromb Vasc Biol. 2001;21:115–121.
18. Bellosta S, Via D, Canavesi M, Pfister P, Fumagalli R, Paoletti R, BerniniF. HMG-CoA reductase inhibitors reduce MMP-9 secretion by macro-phages. Arterioscler Thromb Vasc Biol. 1998;18:1671–1678.
19. Blanquart C, Mansouri R, Paumelle R, Fruchart JC, Staels B, Glineur C.The protein kinase C signaling pathway regulates a molecular switchbetween transactivation and transrepression activity of the peroxisome
368 Circulation Research February 17, 2006
by guest on June 15, 2018http://circres.ahajournals.org/
Dow
nloaded from

proliferator-activated receptor alpha. Mol Endocrinol. 2004;18:1906–1918.
20. Delerive P, Martin-Nizard F, Chinetti G, Trottein F, Fruchart JC, Najib J,Duriez P, Staels B. Peroxisome proliferator-activated receptor activatorsinhibit thrombin- induced endothelin-1 production in human vascularendothelial cells by inhibiting the activator protein-1 signaling pathway.Circ Res. 1999;85:394–402.
21. Martin G, Duez H, Blanquart C, Berezowski V, Poulain P, Fruchart JC,Najib-Fruchart J, Glineur C, Staels B. Statin-induced inhibition of theRho-signaling pathway activates PPARalpha and induces HDL apoA-I.J Clin Invest. 2001;107:1423–1432.
22. Chinetti G, Lestavel S, Fruchart JC, Clavey V, Staels B. Peroxisomeproliferator-activated receptor alpha reduces cholesterol esterification inmacrophages. Circ Res. 2003;92:212–217.
23. Delerive P, De Bosscher K, Besnard S, Vanden Berghe W, Peters JM,Gonzalez FJ, Fruchart JC, Tedgui A, Haegeman G, Staels B. Peroxisomeproliferator-activated receptor alpha negatively regulates the vascularinflammatory gene response by negative cross-talk with transcriptionfactors NF-kappaB and AP-1. J Biol Chem. 1999;274:32048–32054.
24. Staels B, Koenig W, Habib A, Merval R, Lebret M, Torra IP, Delerive P,Fadel A, Chinetti G, Fruchart JC, Najib J, Maclouf J, Tedgui A. Acti-vation of human aortic smooth-muscle cells is inhibited by PPARalphabut not by PPARgamma activators. Nature. 1998;393:790–793.
25. Delerive P, Fruchart JC, Staels B. Peroxisome proliferator-activatedreceptors in inflammation control. J Endocrinol. 2001;169:453–459.
26. Li AC, Binder CJ, Gutierrez A, Brown KK, Plotkin CR, Pattison JW,Valledor AF, Davis RA, Willson TM, Witztum JL, Palinski W, Glass CK.Differential inhibition of macrophage foam-cell formation and atheroscle-rosis in mice by PPARalpha, beta/delta, and gamma. J Clin Invest.2004;114:1564–1576.
27. Kleemann R, Verschuren L, de Rooij BJ, Lindeman J, de Maat MM,Szalai AJ, Princen HM, Kooistra T. Evidence for anti-inflammatoryactivity of statins and PPARalpha activators in human C-reactive proteintransgenic mice in vivo and in cultured human hepatocytes in vitro.Blood. 2004;103:4188–4194.
28. Landrier JF, Thomas C, Grober J, Duez H, Percevault F, Souidi M, LinardC, Staels B, Besnard P. Statin induction of liver fatty acid-binding protein(L-FABP) gene expression is peroxisome proliferator-activated receptor-alpha-dependent. J Biol Chem. 2004;279:45512–45518.
29. Cataisson C, Joseloff E, Murillas R, Wang A, Atwell C, Torgerson S,Gerdes M, Subleski J, Gao JL, Murphy PM, Wiltrout RH, Vinson C,Yuspa SH. Activation of cutaneous protein kinase C alpha induces kera-tinocyte apoptosis and intraepidermal inflammation by independent sig-naling pathways. J Immunol. 2003;171:2703–2713.
30. Chen CC, Wang JK, Lin SB. Antisense oligonucleotides targeting proteinkinase C-alpha, -beta I, or -delta but not -eta inhibit lipopolysaccharide-
induced nitric oxide synthase expression in RAW 264.7 macrophages:involvement of a nuclear factor kappa B-dependent mechanism.J Immunol. 1998;161:6206–6214.
31. Parekh DB, Ziegler W, Parker PJ. Multiple pathways control proteinkinase C phosphorylation. EMBO J. 2000;19:496–503.
32. Juge-Aubry CE, Hammar E, Siegrist-Kaiser C, Pernin A, Takeshita A,Chin WW, Burger AG, Meier CA. Regulation of the transcriptionalactivity of the peroxisome proliferator-activated receptor alpha by phos-phorylation of a ligand- independent trans-activating domain. J BiolChem. 1999;274:10505–10510.
33. Barger PM, Browning AC, Garner AN, Kelly DP. p38 mitogen-activatedprotein kinase activates peroxisome proliferator-activated receptor alpha:a potential role in the cardiac metabolic stress response. J Biol Chem.2001;276:44495–44501.
34. Lazennec G, Canaple L, Saugy D, Wahli W. Activation of peroxisomeproliferator-activated receptors (PPARs) by their ligands and proteinkinase A activators. Mol Endocrinol. 2000;14:1962–1975.
35. Serrano CV Jr, Yoshida VM, Venturinelli ML, D’Amico E, Monteiro HP,Ramires JA, da Luz PL. Effect of simvastatin on monocyte adhesionmolecule expression in patients with hypercholesterolemia. Atheroscle-rosis. 2001;157:505–512.
36. Liao JK. Statin therapy for cardiac hypertrophy and heart failure.J Investig Med. 2004;52:248–253.
37. Hillyard DZ, Jardine AG, McDonald KJ, Cameron AJ. Fluvastatininhibits raft dependent Fcgamma receptor signalling in humanmonocytes. Atherosclerosis. 2004;172:219–228.
38. Strassheim D, Park JS, Abraham E. Sepsis: current concepts in intra-cellular signaling. Int J Biochem Cell Biol. 2002;34:1527–1533.
39. Naruko T, Ueda M, Haze K, van der Wal AC, van der Loos CM, ItohA, Komatsu R, Ikura Y, Ogami M, Shimada Y, Ehara S, YoshiyamaM, Takeuchi K, Yoshikawa J, Becker AE. Neutrophil infiltration ofculprit lesions in acute coronary syndromes. Circulation. 2002;106:2894 –2900.
40. Kato H, Kogure K. Biochemical and molecular characteristics of the brainwith developing cerebral infarction. Cell Mol Neurobiol. 1999;19:93–108.
41. Pettipher ER. Pathogenesis and treatment of chronic arthritis. Sci Prog.1989;73:521–534.
42. Almog Y. Statins, inflammation, and sepsis: hypothesis. Chest. 2003;124:740–743.
43. Leung BP, Sattar N, Crilly A, Prach M, McCarey DW, Payne H, MadhokR, Campbell C, Gracie JA, Liew FY, McInnes IB. A novel anti-inflammatory role for simvastatin in inflammatory arthritis. J Immunol.2003;170:1524–1530.
44. Vaughan CJ. Prevention of stroke and dementia with statins: effectsbeyond lipid lowering. Am J Cardiol. 2003;91:23B–29B.
Paumelle et al Antiinflammatory Effects of Statins Involve PPAR� 369
by guest on June 15, 2018http://circres.ahajournals.org/
Dow
nloaded from

Glineur and Bart StaelsGaëtane Woerly, Frédéric Percevault, Jean-Charles Fruchart, David Dombrowicz, Corine
Réjane Paumelle, Christophe Blanquart, Olivier Briand, Olivier Barbier, Christian Duhem, via Inhibition of the Protein Kinase C Signaling PathwayαReceptor-
Activated−Acute Antiinflammatory Properties of Statins Involve Peroxisome Proliferator
Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 2006 American Heart Association, Inc. All rights reserved.is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation Research
doi: 10.1161/01.RES.0000202706.70992.952006;98:361-369; originally published online January 5, 2006;Circ Res.
http://circres.ahajournals.org/content/98/3/361World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circres.ahajournals.org/content/suppl/2006/01/05/01.RES.0000202706.70992.95.DC1Data Supplement (unedited) at:
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further informationEditorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Researchin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on June 15, 2018http://circres.ahajournals.org/
Dow
nloaded from

Online Data Supplement Paumelle et al.
Material and methods
Inflammation tests
Air-pouch model: Subcutaneous dorsal pouches were induced in female C57BL6 wild-type and
PPARα-null mice, 8 weeks of age (7-12/group) was induced by injection of 5 ml of steril air
followed 3 days later by reinjection of 3 ml of steril air 1. On day 6, LPS (0.2µg/mouse) in 1 ml of
CMC (0.5%) was injected into the air-pouches. Simvastatin at indicated doses in CMC (0.5%) or
vehicle (CMC) was given by oral administration to air pouch-bearing mice 1h before LPS
injection for an additional 4h. The animals were then killed by cervical dislocation and the
pouches were flushed with cold PBS (2 ml). The lavage fluid was immediately cooled on ice, its
volume determined, and neutrophils counted using a haemocytometer. Viability, as determined
by trypan blue exclusion, was consistently greater than 95%. Neutrophil purity, as determined
by Wright's-stained cytospin preparations, was greater than 90-95%.
Carrageenan footpad edema model: female C57BL6 wild-type and PPARα-null mice between 8
and 12 weeks of age were used (12-14/group). Footpad swelling was induced by a single
subplantar injection of 0,05 ml of a sterile 1% solution of carrageenan in water 2. Simvastatin at
indicated doses in CMC (0.5%) or vehicle (CMC) was given by oral administration to mice 1h
before carrageenan injection for an additional 3h. Footpad swelling was then measured using a
micrometer and compared with the preinjection volume of the same paw. Swelling was then
expressed in percent relative to the level of carrageenan-treated controls (CMC) group.
Cell culture and treatments
Murine endothelial cells from adipose tissue were isolated as described 3. Murine thioglycollate-
elicited peritoneal macrophages were isolated as described 4. Briefly, mouse peritoneal
macrophages were collected by peritoneal lavage with saccharose (0.34 mol/L) from mice given
a 3 ml intraperitoneal injection of 4% thioglycollate (Sigma Aldrich, St Quentin, France) in water
for 3 days. Cells were then washed twice with serum-free RPMI (GIBCO BRL), plated and
allowed to adhere to dishes for 2h. Plates were then washed 3 times with RPMI to remove
nonadherent cells and incubated in RPMI containing fetal calf serum (FCS, 10%) over night.

Online Data Supplement Paumelle et al.
Then, cells were incubated in RPMI containing 1% Nutridoma for 24h before treatment. Murine
LPS-elicited neutrophils were isolated from air-pouches after 4h LPS treatment and incubated in
RPMI containing FCS (0.5%) and treated with the indicated reagents. COS-7 cells were cultured
in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with FCS (10%) and antibiotics
at 37°C. Cells were treated with the indicated reagents (GW9578 (GlaxoSmithKline), U0126 and
mPKCI (Promega, Madison, USA), Gö6976 and Ro318220 (Calbiochem, San Diego,
California), E.coli 026:B6 LPS (Sigma Aldrich, St Quentin, France)). For in vitro studies,
simvastatin (Zocor, Merck laboratories) was converted to the active compound 5. Atorvastatin
(Pfizer) and fluvastatin (Novartis pharma) were diluted in DMSO
RNA analysis
RNA extraction was performed using TRIzol reagent and reverse transcription was performed
according to the manufacturer's protocole (Invitrogen Life technologies, Cergy-Pontoise,
France). RNA levels were measured by quantitative PCR using brilliant SYBR Green QPCR
Master Mix on the MX4000 detection system (Stratagene). The amplifying primers were: murine
PPARα (FOR: 5'-AGGCGGTTGCCACTGTTCAG-3' and REV: 5'-
AGCCCTCTTCATCCCCAAGC-3'), murine iNOS (FOR: 5'-TTGCCCCTGGAAGTTTCTCTTC-3’
and REV 5'-GGAGCCATTTTGGTGACTCTTAGT-3'), murine TNFα (FOR, 5'-
ATCCAGTTTGGTGTCGCGGAGC-3' and REV, 5'-CGTCGTCGTCGAAATGGGCATC-3'),
murine IL-6 (FOR, 5'-CCAGTTGCCTTCTTGGGACTG-3' and REV, 5'-
CAGGTCTGTTGGGAGTGGTATCC-3') and murine CPT1 (FOR: 5'-
CATCATGACTATGCGCTACTC-3' and REV: 5'-CAGTGCTGTCATGCGTTGG-3') and as
control, murine cyclophilin (FOR, 5'-GCATACGGGTCCTGGCATCTTGTCC-3' and REV,
5'ATGGTGATCTTCTTGCTGGTCTTGC-3'). Crossing threshold (Ct) values were determined for
target genes and normalysed to the Ct of cyclophilin using the following equation: relative
values= 2 –(Ct target gene- Ct cyclophilin) . Results are expressed as means -/+ SD (n=3) relative to the
level of LPS-treated controls. All experiments were repeated at least 3 times.
Kinase assays and immunoprecipitation

Online Data Supplement Paumelle et al.
After treatment, cells were washed with PBS and suspended in PKC lysis buffer (50 mmol/L Tris
pH 7.5, 3 mmol/L DDT, 5 mmol/L EDTA and 10 mmol/L EGTA) containing freshly added
protease and phosphatase inhibitors (1 mmol/L β-Glycerophosphate, 1 mmol/L Na3VO4, 1
mmol/L PMSF, 10 µg/ml aprotinin). Lysates were sonicated using a Vibracell hiddock 72442
(70%, 80 J/sec, 5 s) and then clarified by centrifugation at 4°C (3000 rpm, 15 min). Cell extracts
(10 µg) or immunoprecipitated PKCα from cell extract (200 µg) were incubated for 30 min at 30
°C in 20 µl of kinase reaction buffer containing 1/3 Calcium Buffer (12 mmol/L CaCl2, 50 mmol/L
Tris), 1/3 Lipid activator (Sigma Aldrich, St Quentin, France), 1/3 DTT buffer (30 mmol/L DTT,
50 mmol/L Tris), 1 µg of Histone H1 or 400 ng of purified PPARα protein as substrates and 5
µCi of (γ-32P) ATP (2000 Ci/mmol). For histone H1 protein substrate, kinase reaction was
stopped by addition of 10 µl of 3X Laemmli buffer and electrophoresed on 12.5% SDS-PAGE.
For the purified PPARα protein substrate, kinase reaction was stopped by addition of ice cold
RIPA with inhibitors and PPARα was immunoprecipitated overnight at 4°C using anti-PPARα
polyclonal antibody as described previously 6. Immune complexes were incubated with 30 µl of
protein-A sepharose beads for 1h at 4 °C and then washed once with RIPA, RIPA/NaCl 1 mol/L,
RIPA/TNE and TNE. Immunoprecipitates were resuspended in 10 µl of 3X Laemmli buffer and
electrophoresed on 10% SDS-polyacrylamide gel electrophoresis. For equal loading of purified
poteins, gels electrophoresis were stained with coomassie blue (data not shown). Gels were
then dried and incorporation of (γ-32P) ATP was visualised by autoradiography. Signal intensity
corresponding to the various bands was quantified using the Image 1D software.
Immunoblot
After treatment, cells were washed with PBS and suspended in PKC lysis buffer for PKCα
detection or lysis buffer (25 mmol/L Hepes pH 7.5, 100 mmol/L NaCl, 1.5 mmol/L MgCl2, 0.25
mmol/L EDTA, 0.5 mmol/L EGTA, 10 mmol/L NaF and 0.1% NP40) for other protein detection
containing freshly added protease and phosphatase inhibitors (1 mmol/L β-Glycerophosphate, 1
mmol/L Na3VO4, 1 mmol/L PMSF, 10 µg/ml aprotinin). Lysates were clarified by centrifugation at
4°C (14000 rpm, 30 min). Cell extracts were resolved on 10% SDS-polyacrylamide gel and

Online Data Supplement Paumelle et al.
transferred onto PVDF membrane. Immunoblots were performed using the Aurora detection
system (ICN pharmaceuticals, Orsay, France) as previously described 7 using anti-iNOS
polyclonal antibody (BD Biosciences, Le Pont de Claix, France), anti-PKCα C20 and anti-actin
I19 polyclonal antibodies (Santa-Cruz Biotechnology, Le Perray en Yvelines, France).
Transient transfection assay
COS-7 cells, grown to 50%-60% confluence in DMEM supplemented with 10% FCS, were
transiently transfected by lipofection with reporter and expression plasmids as indicated in the
figure legends. The GAL4-TkpGL3, pSG5-PPARαWT pSG5-PPARα(S179A-S230A) 8, GAL4-
p65 9, CA-PKCα 10 plasmids were obtained as described. The pCDNA-PKCα WT was a kind
gift of Laurence Suaud. A β-galactosidase expression plasmid was co-transfected as a control
for transfection efficiency. The total amount of transfected DNA was kept constant by adding
empty vector. After 2h (COS-7 cells), cells were refed with DMEM supplemented with 2%
Ultroser and incubated overnight. Cells were then treated as indicated in the figure legends.
After the indicated time, cells were collected and subjected to luciferase and β-galactosidase
assays. All experiments were repeated at least 3 times.
References
1. Diomede L, Albani D, Sottocorno M, Donati MB, Bianchi M, Fruscella P, Salmona M. In
vivo anti-inflammatory effect of statins is mediated by nonsterol mevalonate products.
Arterioscler Thromb Vasc Biol. 2001;21:1327-32.
2. Sparrow CP, Burton CA, Hernandez M, Mundt S, Hassing H, Patel S, Rosa R,
Hermanowski-Vosatka A, Wang PR, Zhang D, Peterson L, Detmers PA, Chao YS,
Wright SD. Simvastatin has anti-inflammatory and antiatherosclerotic activities
independent of plasma cholesterol lowering. Arterioscler Thromb Vasc Biol.
2001;21:115-21.
3. Hewett PW, Murray JC, Price EA, Watts ME, Woodcock M. Isolation and
characterization of microvessel endothelial cells from human mammary adipose tissue.
In Vitro Cell Dev Biol Anim. 1993;29A:325-31.

Online Data Supplement Paumelle et al.
4. Bellosta S, Via D, Canavesi M, Pfister P, Fumagalli R, Paoletti R, Bernini F. HMG-CoA
reductase inhibitors reduce MMP-9 secretion by macrophages. Arterioscler Thromb
Vasc Biol. 1998;18:1671-8.
5. Kita T, Brown MS, Goldstein JL. Feedback regulation of 3-hydroxy-3-methylglutaryl
coenzyme A reductase in livers of mice treated with mevinolin, a competitive inhibitor of
the reductase. J Clin Invest. 1980;66:1094-100.
6. Blanquart C, Barbier O, Fruchart JC, Staels B, Glineur C. Peroxisome proliferator-
activated receptor alpha (PPARalpha ) turnover by the ubiquitin-proteasome system
controls the ligand-induced expression level of its target genes. J Biol Chem.
2002;277:37254-9.
7. Paumelle R, Tulasne D, Leroy C, Coll J, Vandenbunder B, Fafeur V. Sequential
activation of ERK and repression of JNK by scatter factor/hepatocyte growth factor in
madin-darby canine kidney epithelial cells. Mol Biol Cell. 2000;11:3751-63.
8. Blanquart C, Mansouri R, Paumelle R, Fruchart JC, Staels B, Glineur C. The protein
kinase C signaling pathway regulates a molecular switch between transactivation and
transrepression activity of the peroxisome proliferator-activated receptor alpha. Mol
Endocrinol. 2004;18:1906-18.
9. Delerive P, De Bosscher K, Besnard S, Vanden Berghe W, Peters JM, Gonzalez FJ,
Fruchart JC, Tedgui A, Haegeman G, Staels B. Peroxisome proliferator-activated
receptor alpha negatively regulates the vascular inflammatory gene response by
negative cross-talk with transcription factors NF-kappaB and AP-1. J Biol Chem.
1999;274:32048-54.
10. Schonwasser DC, Marais RM, Marshall CJ, Parker PJ. Activation of the mitogen-
activated protein kinase/extracellular signal- regulated kinase pathway by conventional,
novel, and atypical protein kinase C isotypes. Mol Cell Biol. 1998;18:790-8.