activation of mitogen-activated protein kinase pathways by cyclic gmp and cyclic gmp-dependent...
TRANSCRIPT

Hanjoong Jo and Thomas M. LincolnPadmini Komalavilas, Paras K. Shah, Contractile Vascular Smooth Muscle CellsCyclic GMP-dependent Protein Kinase inKinase Pathways by Cyclic GMP and Activation of Mitogen-activated ProteinCELL BIOLOGY AND METABOLISM:
doi: 10.1074/jbc.274.48.343011999, 274:34301-34309.J. Biol. Chem.
http://www.jbc.org/content/274/48/34301Access the most updated version of this article at
.JBC Affinity SitesFind articles, minireviews, Reflections and Classics on similar topics on the
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/274/48/34301.full.html#ref-list-1
This article cites 66 references, 36 of which can be accessed free at
at Kansas State U
niversity Libraries on July 19, 2014
http://ww
w.jbc.org/
Dow
nloaded from
at Kansas State U
niversity Libraries on July 19, 2014
http://ww
w.jbc.org/
Dow
nloaded from

Activation of Mitogen-activated Protein Kinase Pathways by CyclicGMP and Cyclic GMP-dependent Protein Kinase in ContractileVascular Smooth Muscle Cells*
(Received for publication, June 7, 1999, and in revised form, September 1, 1999)
Padmini Komalavilas‡, Paras K. Shah, Hanjoong Jo, and Thomas M. Lincoln
From the Department of Pathology, Division of Molecular and Cellular Pathology, University of Alabama at Birmingham,Birmingham, Alabama 35294-0019
Vascular smooth muscle cells (VSMC) exist in either acontractile or a synthetic phenotype in vitro and in vivo.The molecular mechanisms regulating phenotypic mod-ulation are unknown. Previous studies have suggestedthat the serine/threonine protein kinase mediator of ni-tric oxide (NO) and cyclic GMP (cGMP) signaling, thecGMP-dependent protein kinase (PKG) promotes modu-lation to the contractile phenotype in cultured rat aorticsmooth muscle cells (RASMC). Because of the potentialimportance of the mitogen-activated protein kinase(MAP kinase) pathways in VSMC proliferation and phe-notypic modulation, the effects of PKG expression inPKG-deficient and PKG-expressing adult RASMC onMAP kinases were examined. In PKG-expressing adultRASMC, 8-para-chlorophenylthio-cGMP activated ex-tracellular signal- regulated kinases (ERK1/2) and c-JunN-terminal kinase (JNK). The major effect of PKG acti-vation was increased activation by MAP kinase kinase(MEK). The cAMP analog, 8-Br-cAMP inhibited ERK1/2activation in PKG-deficient and PKG-expressingRASMC but had no effect on JNK activity. The effects ofPKG on ERK and JNK activity were additive with thoseof platelet-derived growth factor (PDGF), suggestingthat PKG activates MEK through a pathway not used byPDGF. The stimulatory effects of cGMP on ERK and JNKactivation were also observed in low-passaged, contract-ile RASMC still expressing endogenous PKG, suggestingthat the effects of PKG expression were not artifacts ofcell transfections. These results suggest that in contract-ile adult RASMC, NO-cGMP signaling increases MAPkinase activity. Increased activation of these MAP ki-nase pathways may be one mechanism by which cGMPand PKG activation mediate c-fos induction and in-creased proliferation of contractile adult RASMC.
Vascular smooth muscle cell (VSMC)1 proliferation and mi-gration are associated with several vascular diseases such as
atherosclerosis and restenosis following vascular injury (1–5).VSMC from several species when cultured in vitro undergo achange in phenotype from a contractile state to a syntheticstate similar to the changes that occur with VSMC in vivo inresponse to vascular injury (6–8). Several lines of evidencesuggest that nitric oxide (NO) inhibits VSMC proliferation(9–13), suggesting that signal transduction pathways regu-lated by NO may be important in VSMC phenotypic modula-tion. NO stimulates the production of cyclic GMP (14), which,in turn, regulates several functions of VSMC such as smoothmuscle relaxation (15). The major receptor protein for cGMP inVSMC is cGMP-dependent protein kinase (PKG), a serine/threonine kinase that catalyzes the phosphorylation of impor-tant proteins that regulate intracellular Ca21 levels and relax-ation of vascular smooth muscle (16). Our laboratory hasrecently demonstrated that PKG also plays a major role in theregulation of the phenotype and morphology of VSMC (17, 18).
The expression of PKG is highly variable in VSMC. Whenadult rat aortic SMC are subcultured in vitro, PKG expressionis reduced to nearly undetectable levels (16, 19). Coincidentwith the loss of expression of PKG, VSMC assume the moresynthetic phenotype. Transfection of the PKG-deficient VSMCwith cDNAs encoding either PKG Ia or the catalytic domain oftype I PKG reverted the morphology from synthetic to theoriginal contractile morphology (17, 18).
Mitogen-activated protein kinases are important mediatorsof signal transduction from the cell surface to the nucleus,thereby regulating several cellular responses such as growthand differentiation (20–22). The MAP kinase pathways areactivated during proliferation (23–25) and differentiation (26,27), and it has been proposed that it is the duration of the MAPkinase activation that determines whether a stimulus evokesproliferation or differentiation (28). Three distinct MAP kinasepathways have been identified in mammalian cells: extracellu-lar signal-regulated kinases (ERK), c-Jun N-terminal kinas-e(JNK)/stress-activated protein kinase, and p38 kinase. MAPkinases are activated upon phosphorylation on both threonineand tyrosine residues by the dual function kinase MAP kinasekinase or MEK (mitogen-activated extracellular-regulated pro-tein kinase kinase). The various MEKs are themselves acti-vated by serine phosphorylation. In the case of the MEK thatactivates ERKs, phosphorylation on serine by Raf kinase is thefirst step in ERK activation. Using constitutively active andnonactive mutants of MEK, it was demonstrated that activa-tion of MEK was necessary and sufficient for PC12 differenti-ation and transformation of NIH 3T3 cells (27).
The two isoforms of ERK, p42 and p44 (ERK2 and ERK1,respectively), are present in adult contractile mammalianVSMC at relatively high concentrations (24, 29–31). Addition-ally, it has been demonstrated that ERK1 and -2 are partiallyactivated in fully differentiated contractile smooth muscle by
* This work was supported by grants from the National Institutes ofHealth (HL34646 and HL53426) and the American Heart Association,Southeastern Affiliate (ALG970010). The costs of publication of thisarticle were defrayed in part by the payment of page charges. Thisarticle must therefore be hereby marked “advertisement” in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.
‡ To whom correspondence should be addressed. Fax: 205-934-1775;E-mail: [email protected].
1 The abbreviations used are: VSMC, vascular smooth muscle cell;PKG, cGMP-dependent protein kinase; PKA, cAMP-dependent proteinkinase; RASMC, rat aortic smooth muscle cell; MAP, mitogen-activatedprotein; ERK, extracellular signal-regulated kinase; MEK, MAP kinasekinase; JNK, c-Jun N-terminal kinase; GST, glutathione S-transferase;PDGF, platelet-derived growth factor; TNF-a, tumor necrosis factor-a;PAGE, polyacrylamide gel electrophoresis; Br-cAMP, bromo-cAMP;DMEM, Dulbecco’s modified Eagle’s medium; CPT, chlorophenylthio-.
THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 274, No. 48, Issue of November 26, pp. 34301–34309, 1999© 1999 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A.
This paper is available on line at http://www.jbc.org 34301
at Kansas State U
niversity Libraries on July 19, 2014
http://ww
w.jbc.org/
Dow
nloaded from

contractile agonists (29, 32–34). Several reports suggest thatERK activation may be involved in regulating contraction ofdifferentiated smooth muscle (35), but others have found onlysmall effects of ERK activation on contraction (34). Neverthe-less, these findings suggest that the MAP kinase pathway maybe involved in other functions besides the regulation of prolif-eration in fully contractile smooth muscle cells. Hence, it ispossible that MAP kinase activation is regulatory in pheno-typic modulation. Because PKG itself appears to be importantfor the expression of the VSMC contractile phenotype, it was ofinterest to examine the effects of PKG expression on MAPkinase pathways in VSMC compared with VSMC that do notexpress PKG. In addition, accumulating evidence indicatesthat PKG activation increases the expression of a number ofprotooncogene transcription factors in a variety of cell types(36–40). In particular, increased expression of c-fos and Jun-Bin response to PKG activation may underlie the increases inVSMC proliferation observed by some investigators (41, 42)and endothelial cells (43). In the case of endothelial cells, Hoodand Granger (43) have shown that cGMP analogs activateRaf-1, an upstream regulator of ERK1/2. The results reportedhere are the first to examine the role of PKG on MAP kinaseactivation in contractile VSMC.
EXPERIMENTAL PROCEDURES
Materials—Collagenase (CLS III) and elastase were from Worthing-ton. Fetal bovine and calf serum, Dulbecco’s modified Eagle’s medium(DMEM) and platelet-derived growth factor BB (PDGF) were purchasedfrom Life Technologies, Inc. Transfectam reagent was from Promega(Madison, WI). 8-(para-chlorophenylthio)-guanosine-39,59-monophos-phate (8-CPT-cGMP) was from Biolog (La Jolla, CA). Geneticin waspurchased from Sigma. Antibodies specific to the phosphorylated formsof ERK1/2, total ERK1/2, phospho-p38, total p38, phospho-MEK, andtotal MEK were purchased from New England Biolabs, Inc. (Beverly,MA). Anti-human JNK-1 antibodies were from PharMingen (San Diego,CA), and the anti-phospho JNK-1 antibodies were from Santa CruzBiotechnology Inc. (Santa Cruz, CA). Purified antibodies to PKG-I wereproduced in our own laboratory or were purchased from StressGenBiotechnologies Corp. (Victoria, British Columbia, Canada). All otherreagents were purchased from either Sigma or Fisher.
Culture of Rat Aortic Smooth Muscle Cells (RASMC)—RASMC wereisolated from the thoracic and abdominal aortas of adult Harlan Spra-gue-Dawley rats (150–200 g, Harlan) as described previously (16). Ratswere sacrificed by CO2 inhalation, and aortae were excised and placedin a wash medium of DMEM containing 20 mM HEPES, 1 mg/ml bovineserum albumin, 5 mg/ml amphotericin B, and 50 mg/ml gentamicin. Theaortae were cleaned and placed in digestion medium (wash mediumcontaining 1 mg/ml elastase and 130 units/ml collagenase) for 8 min.The tunicae adventitiae were removed, and the medial layers wereminced and further digested for 1–2 h in digestion medium containing200 units/ml collagenase until a single-cell suspension was obtained.Cells were washed twice with the wash medium and plated in culturedishes. Cells were maintained in DMEM containing 10% fetal bovineserum and 50 mg/ml gentamicin and grown under 10% CO2 and sub-cultured every 6 days.
Transfection of RASMC—The transfection of RASMC with the bo-vine PKG Ia cDNA was performed as described previously (17, 18).Cells at passage 3 were transfected with 5 mg of recombinantpcDNA1-neo/PKG Ia vector or empty (control) pcDNA1-neo vector us-ing 10 ml of Transfectam reagent with precipitation of the DNA-lipo-some complex for 15 min at room temperature. The precipitate wasadded to the cell monolayer, and the cells were incubated for 6 h at37 °C and 10% CO2. The transfection was terminated by adding DMEMwith 20% fetal bovine serum. Stably transfected cells were selectedusing 500 mg/ml Geneticin (G418). After isolating the colonies, thetransfected cell lines were maintained in 250 mg/ml G418. The trans-fected cells from passage 6–12 were used for the experiments. For theexperiments reported here, at least three different clones of cells ex-pressing physiological levels of PKG and three different clones of cellstransfected with empty vector and not expressing PKG were examined,as reported previously (17, 18).
Cell Treatments and Preparation of Cell Lysates—Cells plated in 60mm culture dishes and grown to confluence were serum-deprived inDMEM containing 1 mg/ml bovine serum albumin for 24 h, treated with
various agents in serum-free medium for different time intervals,quick-frozen in liquid nitrogen, and stored at 280 °C. Cells were har-vested by scraping in 0.25–0.5 ml of ice-cold extraction buffer (50 mM
HEPES, pH 7.4, 150 mM NaCl, 1% Triton X-100, 10% glycerol, 1 mM
sodium orthovanadate, 0.1 mM phenylmethylsulfonyl fluoride, 10 mg/mleach leupeptin and pepstatin A, 5 mg/ml aprotinin A, and 10 nM calicu-lyn A), rotated for 15 min at 4 °C, and centrifuged at 20,000 3 g for 10min. The supernatants were separated, and protein contents wereestimated by using the Coomassie Plus protein assay reagent (Pierce)using bovine serum albumin as standard.
JNK Activation Assay—The activity of JNK was measured by animmunocomplex kinase assay using an antibody specific for JNK-1(PharMingen, clone G 151–333) and c-Jun (amino acids 5–89) fused toglutathione S-transferase (GST c-Jun) as the substrate as describedpreviously (44). 200 mg of cell lysate protein was incubated with antiJNK-1 antibodies for 1 h at 4 °C, followed by an additional 1-h incuba-tion with Protein G-agarose beads. The immunocomplexes were washedfour times with the extraction buffer followed by two washes with JNKassay buffer (20 mM HEPES, pH 7.6, 20 mM MgCl2, 20 mM b-glycero-phosphate, 20 mM p-nitrophenyl phosphate, 0.1 mM sodium orthovana-date, 2 mM dithiothreitol). The immunocomplexes were further incu-bated in 20 ml of JNK assay buffer containing GST-c-Jun (5 mg/sample),50 mCi of [g-32P]ATP for 8 min at 30 °C. The reaction was terminated bythe addition of 5 ml of a 53 electrophoresis stop buffer (312.5 mM
Tris-Cl, pH 6.9, 10 mM EDTA, 0.5 M sucrose, 15% sodium dodecyl sulfate(SDS), 0.8 M 2-mercaptoethanol, and 0.002% bromphenol blue). Thesamples were heated for 10 min at 100 °C, and the denatured proteinswere resolved by 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE). The proteins were transferred to a polyvinylidene difluoridemembrane (Millipore) and subjected to autoradiography. The radioac-tivity incorporated into the phosphorylated GST-c-Jun protein bandswas quantitated by scintillation counting and by densitometry of theautoradiogram.
Western Blot Analysis for the Activation of ERK1/2, MEK1/2, JNK1,and p38 Kinases—Cell lysates (10–50 mg of protein) from the treatedsamples were resolved by 10% SDS-PAGE, protein transferred to nitro-cellulose membranes (Bio-Rad), and probed with antibodies specific tothe phosphorylated forms of ERK1/2, p38, JNK-1, and MEK1/2. As acontrol, the amount of total ERK1/2 protein, p38 protein, JNK-1 pro-tein, and MEK1/2 protein was determined by using antibodies that arespecific to total ERK1/2, p38, JNK-1, and MEK1/2, respectively. PKGwas detected using goat anti-bovine type I PKG prepared in our labo-ratory or purchased from StressGen. Proteins were visualized using theenhanced chemiluminescence (ECL) detection system (Pierce). The in-tensity of the bands was quantitated by densitometry.
Statistical Analysis—Experiments were performed a minimum ofthree times, and the data are presented as the mean 6 S.E. andanalyzed by Student’s t test. A p value of less than 0.05 was used as thecriterion of significance.
RESULTS
Expression of PKG Ia in Transfected RASMC—It was dem-onstrated earlier that the level of PKG decreases with eachpassage when adult RASMC are cultured in vitro (16, 19).Similar results have been reported by other laboratories foradult rat aortic and pulmonary arterial smooth muscle cells(13, 45).2 On the other hand, RASMC isolated from juvenileanimals have been reported to maintain PKG expression undercertain plating and growth conditions (46). Therefore, in orderto ensure that PKG expression was suppressed for the studiesdescribed below, cells were isolated from adult animals andpassaged several times. The lack of PKG expression was alsoexamined by Western blot analysis, Northern blot analysis,and enzymatic assay. Any non-transfected cell lines still ex-pressing PKG were excluded from these studies. Transfectionof RASMC with either the cDNA encoding PKG Ia or thecatalytic domain of PKG-I in passages 3–5 reversed the mor-phology of the cells to a more contractile phenotype (17, 18).Fig. 1 shows the morphology of RASMC stably expressing phys-iological levels of PKG Ia compared with control transfectednot expressing PKG. The RASMC transfected with the emptyvector had a synthetic morphology as demonstrated by the
2 C. M. Brophy, unpublished observations.
Activation of MAP Kinases by PKG34302
at Kansas State U
niversity Libraries on July 19, 2014
http://ww
w.jbc.org/
Dow
nloaded from

flattened appearance and the irregular growth pattern (panelA), whereas the RASMC transfected with pcDNA1-neo/PKG Iahad a contractile morphology as demonstrated by the spindle-shaped appearance and “hill and valley” growth pattern (panelB). Similar results were obtained by transfection of the cata-lytic domain (data not shown). To determine whether the phe-notypic changes were associated with alterations in MAP ki-nase expression, RASMC deficient in PKG and thoseexpressing PKG Ia were used to study the activation ofERK1/2, JNK, and p38. Fig. 2 demonstrates the levels of pro-tein expression for these MAP kinases along with PKG expres-sion in transfected RASMC. It is important to note the absenceof detectable PKG expression in RASMC transfected with theempty vector. No PKG was detected by Western blot analysiswhen up to 50 mg of soluble protein were probed using either apolyclonal, immunopurified anti-PKG generated in our ownlaboratory or an antibody purchased from StressGen. It is alsoimportant to note that the loss of PKG expression in passagedadult rat VSMC has been found by at least three other labora-tories independent of our own (13, 45).2 RASMC expressingPKG Ia (Ia) have somewhat higher endogenous levels of ERK1and -2, especially with respect to the 42-kDa protein (2.9 60.87-fold) compared with RASMC deficient in PKG expression.There were no significant differences in the levels of JNK andp38 kinases in these two cell types.
Time Course of Activation of ERK1/2, JNK, and p38 Kinasesby 8-CPT-cGMP—Empty vector- and PKG Ia-transfected cellswere treated with an analog of cGMP, 8-CPT-cGMP, for differ-ent time intervals. Fig. 3 is the time course of activation ofERK1/2 by cGMP in vector only (lanes 1–4) and PKG Ia-transfected RASMC (lanes 5–8). Compared with empty vector-transfected cells, the untreated (time 0) RASMC transfectedwith PKG Ia demonstrated increased activation of ERK1/2(phospho-ERK1/2, lane 5 compared with lane 1). Greater levelsof ERK in the PKG-expressing cells did not appear to accountfor the increased levels of phospho-ERK, suggesting that thebasal activation state of ERK is greater in PKG-expressingcells (panel B). 8-CPT-cGMP increased ERK1/2 activity at 15,30, and 60 min with peak increases by cGMP observed at 15min. On the other hand, no significant increases in ERK1/2activation by 8-CPT-cGMP were observed in the vector alone-transfected cells. The statistical analysis of ERK1/2 activationin PKG-deficient and PKG-expressing cells is shown in panel Dof Fig. 3. The effects of PKG Ia transfection and 8-CPT-cGMPon ERK1/2 activity were reproducibly observed in three differ-ent clonally isolated cells expressing PKG compared with threedifferent clonally isolated cells transfected with empty vector.We had previously described the controls performed to ensure
that the results observed with PKG expression were not arti-facts associated with the transfection and isolation procedureitself (17, 18). Therefore, the results observed were not due tounique characteristics of a particular cell type derived from alone colony of RASMC, but rather reflected the consequences ofPKG expression in the RASMC.
Fig. 3 (panel E) is a time course of activation of JNK inPKG-deficient and PKG-expressing cells. As shown in thepanel, cGMP also activates JNK kinase as represented byGST-cJun phosphorylation of the immunoprecipitated JNK(vector only cells, lanes 1–4 and PKG Ia cells, lanes 5–8).Compared with empty vector-transfected cells, the untreatedcontrol (time 0) from the PKG-Ia RASMC had an increasedactivity of JNK. 8-CPT-cGMP increased JNK activation at 15min in the PKG-expressing cells but had no significant effectson JNK activation in the PKG-deficient cells. The total level ofJNK protein in these two cell lines was not significantly differ-ent (panel F), and the statistical analysis of the data for JNKactivation by 8-CPT-cGMP is shown in panel G. PKG transfec-tion and 8-CPT-cGMP had no significant stimulatory effects onp38 kinase in the PKG-expressing cells compared with thePKG-deficient cells (data not shown) and was not studied fur-ther. Panel C in Fig. 3 illustrates the level of PKG in the vectoralone-transfected cells (lanes 1-4) and the PKG Ia-transfectedcells (lanes 5–8). Again, it is important to note the lack ofdetectable expression of PKG-I in vector alone-transfectedadult RASMC.
Sorbitol is an osmotically stressful agent to cultured cellsthat is known to activate MAP kinase pathways. Sorbitol sig-nificantly activated both ERK1/2 and JNK in vector alone-transfected RASMC (panels A and C and the statistical analy-sis in panel D in Fig. 4). In PKG-expressing RASMC, sorbitolincreased JNK activation more potently than did 8-CPT-cGMP.In these same cells, however, sorbitol was not more potent inincreasing ERK1/2 than was 8-CPT-cGMP. The total increasein JNK activation by sorbitol in PKG-deficient and PKG-ex-pressing cells was similar.
Activation of ERK1/2 and JNK by PDGF—Growth factorssuch as PDGF activate MAP kinases through both Ras-depend-ent pathways and G-protein coupled pathways in smooth mus-cle cells (47). To gain further insight into possible mechanismsby which PKG activation increases MAP kinase activity inRASMC, the effects of PDGF on ERK and JNK activation weremonitored in PKG-expressing and PKG-deficient cells. Maxi-mal concentrations of PDGF (10 ng/ml) increased ERK1/2 ac-tivity in both PKG-deficient and PKG-expressing cells (Fig. 5,panels A and C). ERK1/2 activity at 10 min induced by PDGFwas significantly greater in the PKG-expressing cells comparedwith the PKG-deficient cells. Because total ERK1/2 was notsignificantly different between RASMC deficient in PKG and
FIG. 1. Effect of PKG expression on the morphology of RASMC.Stably transfected cell lines expressing PKG Ia or control cell linestransfected with empty vector were made as described under “Experi-mental Procedures.” Panel A is a representative photomicrograph ofstably transfected RASMC with empty vector deficient in PKG Ia (con-trol). Panel B is a representative photomicrograph of cells stably ex-pressing PKG Ia.
FIG. 2. Expression of MAP kinases and PKG Ia in vector-trans-fected (C) and PKG-transfected (1a) RASMC. Shown is a repre-sentative Western blot for p38, ERK1/2, JNK-1, and PKG Ia in extractsfrom vector alone-transfected RASMC and PKG Ia-transfected RASMC(1a). 10–50 mg of total protein were prepared and analyzed by Westernblot as described under “Experimental Procedures” using antibodiesspecific for these kinases. The arrows indicate the kinases at theirrespective molecular sizes. The band above the 46-kDa JNK1 is arelated 54–55-kDa JNK isoform.
Activation of MAP Kinases by PKG 34303
at Kansas State U
niversity Libraries on July 19, 2014
http://ww
w.jbc.org/
Dow
nloaded from

those expressing PKG (Fig. 5, panel B), these results suggestthat PKG expression appeared to augment ERK activation byPDGF. Similarly, PDGF activated JNK in both PKG-deficientand PKG-expressing cells (Fig. 5, panels D and F). PKG-trans-fected cells showed increased basal activity of JNK, which wasfurther stimulated by PDGF treatment for 10 min when com-pared with vector alone-transfected cells. Thus, JNK activationwas augmented by the presence of PKG. Basal or PDGF-stim-ulated activation of p38 kinase was not significantly differentin the PKG-deficient and PKG Ia-expressing RASMC (data notshown).
Activation of MAP Kinase Pathways by TNF-a—Inflamma-tory cytokines such as TNF-a activate MAP kinase pathways in
smooth muscle cells predominantly through STAT proteinphosphorylation (48). To gain some insight into whether theactivation of MAP kinases by PKG shares a similar pathway,we examined the effects of PKG expression on ERK1/2 andJNK activation in TNF-a-treated cells. As shown in Fig. 6(panels A and C), the time course of activation of ERK1/2 byTNF-a was different in the two RASMC cell types with thePKG Ia-expressing RASMC having a more prolonged activa-tion of ERK1/2 compared with PKG-deficient cells. However,the peak level of ERK1/2 activation by TNF-a was similar.Likewise, panels D and F in Fig. 6 show that TNF-a activatedJNK to a similar extent at 15 min in both the PKG-deficientand PKG-expressing cells. There were no differences in TNF-astimulated p38 kinase activation in the PKG-expressing andPKG-deficient cells (data not shown).
Activation of MEK by 8-CPT-cGMP—Since MEK is the up-stream kinase that catalyzes the phosphorylation and activa-tion of ERK1/2, the effects of cGMP on the activity of MEK wasstudied in the PKG-expressing and PKG-deficient cells. Asshown in Fig. 7, treatment of cells with 8-CPT-cGMP for 10 minonly stimulated the activity of MEK in the PKG Ia-expressingcells as determined by immunoblotting for phospho-MEK1/2.There were no significant differences in the level of total MEKin the PKG-expressing and PKG-deficient RASMC.
Activation of ERK1/2, JNK, and MEK in Primary Culture ofRASMC—We have monitored the activation of ERK, JNK, andMEK in several colonies of cells expressing PKG through stabletransfection and compared these results with colonies of cellstransfected with empty vector and deficient in the expression ofthis kinase. All colonies expressing PKG demonstrated activa-tion of MAP kinase pathways (i.e. ERK1/2, JNK, MEK) com-
FIG. 3. Time course of activation of MAP kinases by 8-CPT-cGMP in transfected RASMC. The cells were grown as describedunder “Experimental Procedures,” and the confluent cells were serum-deprived for 24 h and treated with 100 mM 8-CPT-cGMP. At the varioustimes indicated, vector alone-transfected RASMC (lanes 1–4) and PKG-Ia-transfected RASMC (lanes 5–8) were analyzed for activated (phos-pho-)ERK1/2 (panel A) and total ERK1/2 (panel B), PKG Ia levels (panelC), activity of JNK (GST-cJun, panel E), and total JNK (panel F).Panels D and G represent averaged data quantified by densitometry ofimmunoblots (D) or autoradiographs (G), expressed as -fold increases inactivation in which the activation observed in unstimulated vectoralone-transfected cells was defined as 1.0. The -fold activations (themeans 6 S.E. for three independent experiments, *, p , 0.05 forincrease in -fold activation with 8-CPT-cGMP at 15 min compared tobasal activation in PKG-expressing cells) for the various different celllines examined are presented as bar graphs for ERK1/2 (panel D) andJNK (panel G). In panels D and G, the open bars are vector alone-transfected cells and the filled bars are PKG Ia-transfected cells. Thepreparation of cell extracts, Western blots, and JNK kinase assays wereperformed as described under “Experimental Procedures.”
FIG. 4. Effects of sorbitol and 8-CPT-cGMP on the activation ofMAP kinases in transfected RASMC. The cells were grown as de-scribed under “Experimental Procedures” and the confluent cells wereserum-deprived for 24 h and treated with 100 mM 8-CPT-cGMP orsorbitol for 30 min. Panel A, Western blot for ERK1/2 activation; panelB, total ERK; panel C, GST-c-Jun phosphorylation. Panels D representaveraged data quantified by densitometry of immunoblots (ERK1/2) orautoradiographs (GST c-Jun), expressed as relative intensity of thebands (means 6 S.E. for three independent experiments) for the variousdifferent cell lines. The open bars are vector alone-transfected cells, andthe filled bars are PKG Ia-transfected cells. *, p , 0.05 for increase in-fold activation with 8-CPT-cGMP compared with activation in vector-transfected cells.
Activation of MAP Kinases by PKG34304
at Kansas State U
niversity Libraries on July 19, 2014
http://ww
w.jbc.org/
Dow
nloaded from

pared with colonies of cells deficient in PKG expression. Be-cause PKG expression promotes modulation of synthetic adultRASMC to contractile RASMC, these results imply that thephenotypic state of the cells may underlie the greater levels ofMAP kinase activity in the transfected cell lines. To examinethis possibility further, and to rule out the possibility that theactivation of MAP kinases is unique to only transfectedRASMC, we examined the activation state of MAP kinases infreshly isolated, primary cultures of adult RASMC. Primarycultures of adult RASMC in the first passage still express acontractile-like phenotype and still express physiological levelsof PKG, as shown in Fig. 8 (panel E). 8-CPT-cGMP activatedERK1/2 (panel A), MEK (panel C), and JNK (panel D) in theprimary cultures of RASMC but not in the passaged adultRASMC devoid of PKG. Panel F is the summary of the datafrom three separate experiments utilizing primary cultures ofnon-transfected RASMC. It is important to note the absence ofexpression of PKG-I in the passaged adult RASMC in panel E.
Effects of 8-Br-cAMP on ERK Activity in RASMC—It is well
established that PKG and the cAMP-dependent protein kinase(PKA) share several signaling functions in smooth muscle suchas the mediation of relaxation. Furthermore, it has been sug-gested that cAMP is involved in phenotypic modulation ofVSMC (49), and it is well known that cAMP is a physiologicalinhibitor of VSMC proliferation (50–53). Therefore, it was ofinterest to examine the effects of cAMP on MAP kinase activ-ities in adult RASMC transfected with PKG. This was madepossible from the standpoint that PKA expression, unlike thatof PKG, is not reduced during culture of RASMC (54).3 Vectoralone- and PKG Ia-transfected RASMC were treated with 100mM 8-Br-cAMP for different time intervals and the activation ofERK and JNK was monitored. In contrast to the effects of8-CPT-cGMP, 8-Br-cAMP markedly inhibited the activation ofERK1/2 in a time-dependent manner in both PKG-deficient
3 P. Komalavilas, P. K. Shah, H. Jo, and T. M. Lincoln, unpublishedobservations.
FIG. 5. Time course of activation of MAP kinases by PDGF intransfected RASMC. The cells were grown as described under “Ex-perimental Procedures,” and the confluent cells were serum-deprivedfor 24 h and treated with PDGF (10 ng/ml) for the times indicated. Thecell lysates were prepared, and activation of ERK and JNK was meas-ured as described under “Experimental Procedures.” Lanes 1–4 areprotein extracts prepared from vector alone-transfected RASMC, andlanes 5–8 are protein extracts prepared from PKG-Ia-transfectedRASMC. Panel A is a immunoblot for ERK1/2 activation; panel B, totalERK protein; panel D, GST-c-Jun phosphorylation; panel E, total JNKenzyme. Panels C and F represent averaged data quantified by densi-tometry of immunoblots (C) or autoradiographs (F), expressed as -foldincreases in activation in which the activation observed in unstimu-lated vector alone-transfected cells was defined as 1.0. The -fold acti-vation (the means 6 S.E. for three independent experiments; *, p , 0.05for increase in -fold activation with PDGF compared with basal activa-tion in PKG-deficient and PKG-expressing cells) for the various differ-ent cell lines examined are presented as bar graphs for ERK1/2 (panelC) and JNK (panel F). The open bars are vector alone-transfected cells,and the filled bars are PKG Ia-transfected cells.
FIG. 6. Time course of activation of MAP kinases by TNF-a intransfected RASMC. The cells were serum-deprived for 24 h andtreated with 50 ng/ml TNF-a for the times indicated as described in Fig.5 legend. Lanes 1–4 are protein extracts prepared from vector alone-transfected RASMC, and lanes 5–8 are extracts prepared from PKGIa-transfected RASMC. Panel A is a Western blot for ERK1/2 activa-tion, panel B is total ERK, panel D is the autoradiograph for GST-c-Junphosphorylation, and panel E is the immunoblot for total JNK. PanelsC and F represent averaged data quantified by densitometry of immu-noblots (C) or autoradiographs (F), expressed as -fold increases inactivation in which the activation observed in unstimulated vectoralone-transfected cells was defined as 1.0. The -fold activation (themeans 6 S.E. for three independent experiments; *, p , 0.05 forincrease in -fold activation with TNF-a compared with basal activationin PKG-deficient or PKG-expressing cells) for the various different celllines examined are presented as bar graphs for ERK1/2 (panel C) andJNK (panel F). The open bars are vector alone-transfected cells, and thefilled bars are PKG Ia-transfected cells.
Activation of MAP Kinases by PKG 34305
at Kansas State U
niversity Libraries on July 19, 2014
http://ww
w.jbc.org/
Dow
nloaded from

cells and PKG-expressing cells (Fig. 9, panels A and B). Theactivation of ERK1/2 produced as a result of PKG Ia expressionwas reduced by 50%, the same percentage of inhibition ob-served in the PKG-deficient cells. The inhibitory effects of8-Br-cAMP on ERK1/2 were likely due to the inhibition of MEKactivation as illustrated in panels C and D of Fig. 9. On theother hand, there were no significant or consistent changes inthe activation of JNK in the PKG-deficient and PKG-express-ing cells in response to 8-Br-cAMP. These results are consistentwith previous reports that PKA activation only inhibits ERK(55, 56), and suggests that PKA and PKG regulate the ERKpathway in opposing directions.
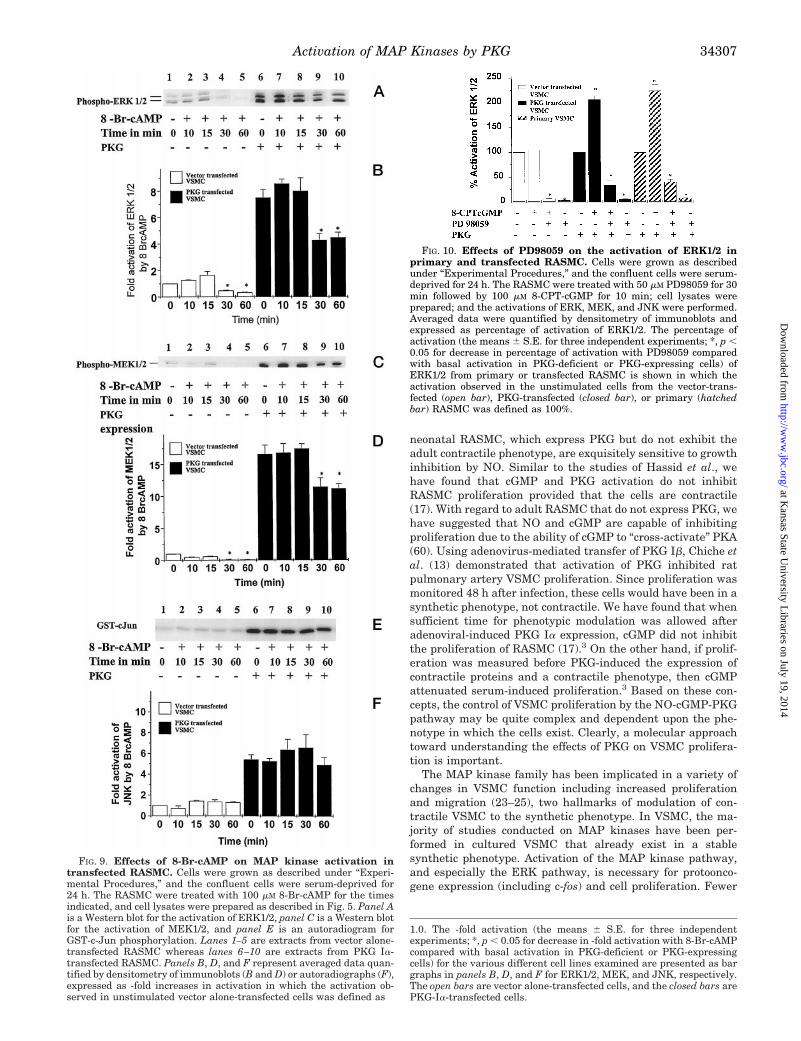
Inhibition of ERK Activation by Selective Kinase Inhibitors—Previous studies have examined the effects of protein kinaseinhibitors on MAP kinase activation in various cell types (12,57). Although it is evident that compounds previously believedto be selective for inhibiting PKA and PKG are not selectivewhen used as described in intact cells (58, 59), we were inter-ested in determining whether or not there were any effects ofthese compounds on MAP kinase activity in the PKG-deficientand PKG-expressing adult RASMC. Incubation of PKG Ia-transfected RASMC with 1 mM KT 5823 (a compound reportedto be a selective inhibitor of PKG) for 30 min partially inhibitedthe stimulation of ERK activity (13.6 6 2.1%, p , 0.0067)produced by 8-CPT-cGMP in PKG-transfected cells. Incubationwith 1 mM KT 5720 (reportedly a selective inhibitor of PKA) didnot significantly inhibit the stimulation of ERK activity (0.99 61.2%, p . 0.474) in these same cells treated with 8-CPT-cGMP(data not shown). Neither KT 5823 nor KT 5720 inhibited thebasal activity of ERK in either vector alone- or PKG-trans-fected RASMC. In primary cultures of RASMC expressingPKG, incubation of the cells for 30 min with 1 mM KT 5823partially inhibited ERK activation produced by 8-CPT-cGMPby 27.5 6 7.5%. In contrast, the selective MEK inhibitor, PD98059, potently inhibited the effects of 8-CPT-cGMP on ERK1/2
activation in both PKG Ia-transfected RASMC and primarycultures of RASMC, as well as in vector alone-transfectedRASMC (Fig. 10).
DISCUSSION
One of the hallmarks of cGMP and PKG action in a variety ofcultured cells is activation of c-fos gene expression (36–40). Inturn, c-fos expression promotes increased cell proliferation.Currently there is much debate and controversy surroundingthe role of cGMP and PKG in the regulation of cell prolifera-tion. There are several studies suggesting that cGMP inhibitsthe proliferation of VSMC. Most of these studies were per-formed using adult VSMC that had been passaged severaltimes and that exist in the synthetic phenotype. Except forneonatal or juvenile VSMC (or cell lines derived from embry-onic VSMC such as the A7r5), synthetic phenotype VSMC donot express appreciable levels PKG. Thus, the role of the NO-cGMP-PKG signal transduction pathway in regulating adultVSMC proliferation is still unknown. In one report, Hassid andcolleagues (41) demonstrated that NO and cGMP do not inhibitthe proliferation of freshly isolated adult RASMC, which ex-press PKG and exhibit the contractile phenotype. In contrast,
FIG. 7. Activation of MEK1/2 by 8-CPT-cGMP in transfectedRASMC. The cells were serum-deprived for 24 h at confluence andtreated with 100 mM 8-CPT-cGMP for 10 min. 20 mg of total cell proteinwere analyzed as described under “Experimental Procedures” by SDS-PAGE. Lanes 1 and 2 of panel A are Western blots for the activation ofMEK from vector alone-transfected RASMC, while lanes 3 and 4 areWestern blots for activation of MEK from PKG Ia-transfected RASMC.Panel B is the immunoblot for total MEK protein. Panel C representsaveraged data quantified by densitometry of immunoblots, expressed as-fold increases in activation in which the activation observed in un-stimulated vector alone-transfected cells was defined as 1.0. The -foldactivation (the means 6 S.E. for three independent experiments; *, p ,0.05 for increase in -fold activation with 8-CPT-cGMP compared withbasal activation in PKG deficient cells) for the various different celllines examined are presented as bar graphs for MEK. The open bars arevector alone-transfected cells and the filled bars PKG Ia-transfectedcells.
FIG. 8. Activation of ERK, MEK, and JNK in low-passaged,primary cultures of RASMC. RASMC were isolated from rat aortaand grown as described under “Experimental Procedures.” After pas-sage 1 (PKG-positive, lanes 1 and 2) or passage 6 (PKG-negative cells,lanes 3 and 4), the cells were serum-deprived for 24 h and treated with100 mM 8-CPT-cGMP for 10 min. Cell lysates were prepared, and theactivations of ERK, MEK, and JNK were performed as described under“Experimental Procedures.” Panel A is a Western blot for the activationof ERK1/2, panel B is for total ERK protein, panel C is a Western blotfor activation of MEK1/2 (phospho MEK), panel D is an autoradiographfor GST-c-Jun phosphorylation, and panel E is PKG expression inpassage 1 and 6 cells. Lanes 1 and 2 are cells examined in the firstpassage (PKG-positive) whereas lanes 3 and 4 are cells in passage 6(PKG-negative). Cells were treated in the absence (lanes 1 and 3) orpresence (lanes 2 and 4) of 8-CPT-cGMP for 10 min. Panel F representaveraged data quantified by densitometry of immunoblots or autora-diographs, expressed as -fold activation from Primary or passagedRASMC in which the activation observed in the unstimulated cells wasdefined as 1.0. The -fold activation of ERK, MEK and JNK (the means 6S.E. for three independent experiments; *, p , 0.05 for increase in -foldactivation with 8-CPT-cGMP compared with basal activation in PKG-expressing cells) for the various different cell lines is shown as bargraphs. The hatched bars are passage 1 primary cultured VSMC, andthe open bars are passage 6 VSMC.
Activation of MAP Kinases by PKG34306
at Kansas State U
niversity Libraries on July 19, 2014
http://ww
w.jbc.org/
Dow
nloaded from
neonatal RASMC, which express PKG but do not exhibit theadult contractile phenotype, are exquisitely sensitive to growthinhibition by NO. Similar to the studies of Hassid et al., wehave found that cGMP and PKG activation do not inhibitRASMC proliferation provided that the cells are contractile(17). With regard to adult RASMC that do not express PKG, wehave suggested that NO and cGMP are capable of inhibitingproliferation due to the ability of cGMP to “cross-activate” PKA(60). Using adenovirus-mediated transfer of PKG Ib, Chiche etal. (13) demonstrated that activation of PKG inhibited ratpulmonary artery VSMC proliferation. Since proliferation wasmonitored 48 h after infection, these cells would have been in asynthetic phenotype, not contractile. We have found that whensufficient time for phenotypic modulation was allowed afteradenoviral-induced PKG Ia expression, cGMP did not inhibitthe proliferation of RASMC (17).3 On the other hand, if prolif-eration was measured before PKG-induced the expression ofcontractile proteins and a contractile phenotype, then cGMPattenuated serum-induced proliferation.3 Based on these con-cepts, the control of VSMC proliferation by the NO-cGMP-PKGpathway may be quite complex and dependent upon the phe-notype in which the cells exist. Clearly, a molecular approachtoward understanding the effects of PKG on VSMC prolifera-tion is important.
The MAP kinase family has been implicated in a variety ofchanges in VSMC function including increased proliferationand migration (23–25), two hallmarks of modulation of con-tractile VSMC to the synthetic phenotype. In VSMC, the ma-jority of studies conducted on MAP kinases have been per-formed in cultured VSMC that already exist in a stablesynthetic phenotype. Activation of the MAP kinase pathway,and especially the ERK pathway, is necessary for protoonco-gene expression (including c-fos) and cell proliferation. Fewer
1.0. The -fold activation (the means 6 S.E. for three independentexperiments; *, p , 0.05 for decrease in -fold activation with 8-Br-cAMPcompared with basal activation in PKG-deficient or PKG-expressingcells) for the various different cell lines examined are presented as bargraphs in panels B, D, and F for ERK1/2, MEK, and JNK, respectively.The open bars are vector alone-transfected cells, and the closed bars arePKG-Ia-transfected cells.
FIG. 9. Effects of 8-Br-cAMP on MAP kinase activation intransfected RASMC. Cells were grown as described under “Experi-mental Procedures,” and the confluent cells were serum-deprived for24 h. The RASMC were treated with 100 mM 8-Br-cAMP for the timesindicated, and cell lysates were prepared as described in Fig. 5. Panel Ais a Western blot for the activation of ERK1/2, panel C is a Western blotfor the activation of MEK1/2, and panel E is an autoradiogram forGST-c-Jun phosphorylation. Lanes 1–5 are extracts from vector alone-transfected RASMC whereas lanes 6–10 are extracts from PKG Ia-transfected RASMC. Panels B, D, and F represent averaged data quan-tified by densitometry of immunoblots (B and D) or autoradiographs (F),expressed as -fold increases in activation in which the activation ob-served in unstimulated vector alone-transfected cells was defined as
FIG. 10. Effects of PD98059 on the activation of ERK1/2 inprimary and transfected RASMC. Cells were grown as describedunder “Experimental Procedures,” and the confluent cells were serum-deprived for 24 h. The RASMC were treated with 50 mM PD98059 for 30min followed by 100 mM 8-CPT-cGMP for 10 min; cell lysates wereprepared; and the activations of ERK, MEK, and JNK were performed.Averaged data were quantified by densitometry of immunoblots andexpressed as percentage of activation of ERK1/2. The percentage ofactivation (the means 6 S.E. for three independent experiments; *, p ,0.05 for decrease in percentage of activation with PD98059 comparedwith basal activation in PKG-deficient or PKG-expressing cells) ofERK1/2 from primary or transfected RASMC is shown in which theactivation observed in the unstimulated cells from the vector-trans-fected (open bar), PKG-transfected (closed bar), or primary (hatchedbar) RASMC was defined as 100%.
Activation of MAP Kinases by PKG 34307
at Kansas State U
niversity Libraries on July 19, 2014
http://ww
w.jbc.org/
Dow
nloaded from

studies have been performed on fully contractile smooth musclecells, and those studies that have been done, a role for the MAPkinases in regulating contractile behavior has been suggested(32–35). Therefore, it was of great interest to us to examine therole of PKG on MAP kinase activity in RASMC whose contract-ile phenotype had been restored.
In this study, we demonstrated that restoration of PKGexpression to PKG-deficient adult RASMC mediates cGMP ac-tivation of MEK, ERK1/2, and JNK. These effects were ob-served in all clonally derived cell lines expressing PKG. Thecellular phenotype is clearly contractile in the PKG-expressingcell lines as judged by morphological, biochemical, and func-tional criteria. In PKG-deficient cell lines that are synthetic inphenotype, 8-CPT-cGMP has little or no effect on MAP kinaseactivities. In addition to the findings using stably transfectedcells, we also observed that cGMP activates the same MAPkinases in freshly isolated, primary RASMC that still expressPKG and exist in the contractile phenotype (Fig. 8). Therefore,the stimulatory effects of cGMP and PKG on MAP kinaseactivities are not due to prolonged culturing of the cells or dueto artifacts associated with transfection. These results are sim-ilar to those of Hood and Granger (43) for the effects of PKG incultured endothelial cells. To our knowledge, this is the firstreport demonstrating the PKG-mediated activation of MAPkinases in contractile smooth muscle cells. Hence, the findingthat PKG activates MAP kinase pathways provides a plausiblemechanism underlying NO- and cGMP-induced increases inc-fos expression and proliferation of adult, contractile VSMCand other cell types.
As mentioned above, every clonally derived RASMC lineexpressing PKG demonstrated higher levels of ERK and JNKactivity compared with clonally derived RASMC lines trans-fected with empty vector and deficient in PKG expression. Weobserved significant variability in the basal ERK and JNKactivity in PKG-expressing cells in these experiments. Thevariable levels of phospho-ERK and JNK were likely due to therapidly fluctuating levels of intracellular cGMP, a phenomenonthat is known to occur in cultured cells (61). During the growthphase of RASMC in the presence of serum, the intracellularlevels of cGMP are some 10–100-fold greater when comparedwith cells approaching confluence (62).3 These high levels ofcGMP activate PKG Ia during growth, thus accounting for thehigher basal activity of ERK and JNK and in the stably trans-fected cell lines.
At first, we were somewhat surprised by these effects of PKGgiven previous reports that NO and cGMP inhibit VSMC pro-liferation (9, 10, 13, 24, 57). Yu et al. (12) reported that cGMP-elevating agents suppress proliferation of VSMC, presumablythrough the activation of PKG and the phosphorylation ofRaf-1. The major evidence presented that suggested the in-volvement of PKG was the sensitivity of the effects to thepurported PKG inhibitor, KT 5823. In another study usingBHK cells, Suhasini et al. (57) reported that overexpression ofPKG inhibits ERK activation via the phosphorylation of Raf-1kinase on serine 43. In neither of these studies were the effectsof cAMP and PKA activation on MAP kinase activity and Raf-1phosphorylation determined. A well known effect of cAMP andPKA activation is the phosphorylation of Raf-1 on serine 43,leading to the inability of ras to activate the pathway (55, 56).Conceivably, activation of the PKA pathway could have beenresponsible for the effects of cGMP elevating agents, inasmuchas high concentrations of cGMP are known to activate PKA(60). It is also clear that PKA and PKG share overlappingsubstrate specificities in vitro and in the intact cell (15). Hence,overexpression of PKG in cells may result in the phosphoryla-tion of substrate proteins normally selective for PKA. In either
case, our results clearly demonstrate a specific effect of PKG toactivate MEK and the ERK pathway. Indeed, the exact oppo-site effects of 8-Br-cAMP were observed in the current studywhere there was a clear inhibition by 8-Br-cAMP of ERK acti-vation in both PKG-expressing and -deficient RASMC. Thus, itis possible that the inhibitory effects of cGMP on MAP kinaseactivity observed by other investigators using PKG-deficientVSMC is due to PKA-dependent phosphorylation of Raf-1 onserine-43.
Our finding that MAP kinases are also activated in differen-tiated contractile smooth muscle is consistent with these re-sults and those obtained earlier by several investigators whostudy contractile function of differentiated VSMC. For exam-ple, the role of MAP kinase in the contractile response ofsmooth muscle may involve the phosphorylation of caldesmon(35). Although these results are somewhat controversial (34),the function of MAP kinase in the contractile phenotype ofvascular smooth muscle is likely to be very different from itsrole in the synthetic phenotype.
Because JNK is also activated in the PKG Ia-transfectedRASMC, the possibility exists that NO and cGMP may regulatestress pathway outcomes such as apoptosis. Induction of apop-tosis by PKG in VSMC has been documented by Pollman et al.(63) and Chiche et al. (13). JNK has been implicated in theapoptotic pathway in at least some cell types (22). Thus, JNKactivation in the PKG-expressing contractile VSMC may ex-plain the increased sensitivity of these cells for apoptosis upongrowth factor withdrawal, compared with the synthetic PKG-deficient VSMC.
The mechanism by which PKG leads to MAP kinase acti-vation in the contractile phenotype is unclear. The resultsshown in Fig. 5 indicate that PKG-mediated activation ofERK is at least partially additive with PDGF, suggesting analternative mechanism to the pathways utilized by thesebiological modulators. Since MEK is also activated in thesecells, PKG may be acting upstream to MEK. Potential candi-dates for regulation by PKG include p21 Ras-GTPase-activat-ing protein since Ras-GTPase-activating protein is phospho-rylated in vitro using PKG.4 Other possible candidatesinclude the MAP kinase phosphatase 1, since it has beendemonstrated that PKG regulates the expression of at leastone protein-tyrosine phosphatase in VSMC (64–67). Morerecently, phenotypic modulation of vascular and non-vascu-lar smooth muscle cells has been linked to the balance be-tween phosphoinositide 3-kinase and Akt with MAP kinases(68). Perhaps an upstream point of divergence between PKG-regulated MAP kinases and the PDGF and TNF-a signalingpathways is at the level of phosphoinositide 3-kinase. On theother hand, the results in Fig. 6 demonstrating that PKG andTNF-a may utilize similar pathways in the activation of ERKand JNK which suggest a common target for these two sig-naling pathways. Clearly, more studies are needed to identifythe steps regulated by PKG in VSMC.
REFERENCES
1. Ross, R. (1995) Ann. N. Y. Acad. Sci. 748, 1–42. Moss, P., Campbell, G., Wang, Z., and Campbell, J. (1985) Lab. Invest. 53,
556–5623. Schwartz, S., Heimar, R., and Majesky, M. (1990) Physiol. Rev. 70, 1177–12094. Ross, R. (1993) Nature 362, 801–8095. Fingerle, J., Johnson, R., Clowes, A., Majesky, M., and Reidy, M. (1989) Proc.
Natl. Acad. Sci. U. S. A. 86, 8412–84166. Campbell, J. H., and Campbell, G. R. (1985) Exp. Mol. Pathol. 42, 136–1627. Gown, A. M., Tsukada, T., and Ross, R. (1986) Am. J. Pathol. 125, 191–2078. Majesky, M. W., Giachelli, C. M., Reidy, M. A., and Schwartz, S. M. (1992) Circ.
Res. 71, 759–7689. Garg, U. C., and Hassid, A. (1989) J. Clin. Invest. 83, 1774–1777
10. Kariya, K., Kawahara, Y., Araki, S., Fukuzaki, H., and Takai, Y. (1989)
4 P. Komalavilas and T. M. Lincoln, unpublished results.
Activation of MAP Kinases by PKG34308
at Kansas State U
niversity Libraries on July 19, 2014
http://ww
w.jbc.org/
Dow
nloaded from

Atherosclerosis 80, 143–14711. Nakaki, T. M., Nakayama, M., and Kato, R. (1990) Eur. J. Pharmacol. 189,
347–35312. Yu, S. M., Hung, L. M., and Lin, C. C. (1997) Circulation 95, 1269–127713. Chiche, J.-D., Schlutsmeyer, S. M., Bloch, D. B., de la Monte, S. M., Roberts,
J. D., Jr., Fillippov, G., Janssens, S. P., Rosenzweig, A., and Bloch, K. D.(1998) J. Biol. Chem. 273, 34263–24271
14. Gruetter, C. A., Barry, B. K., McNamara, D. B., Kadowitz, P. J., and Ignarro,L. (1980) J. Pharmacol. Exp. Ther. 214, 9–15
15. Lincoln, T. M. (1994) Cyclic GMP: Biochemistry, Physiology, and Pathophysi-ology, Landes Inc., Austin, TX
16. Cornwell, T. L., and Lincoln, T. M. (1989) J. Biol. Chem. 264, 1146–115517. Boerth, N. J., Dey, N. B., Cornwell, T. L., and Lincoln, T. M. (1997) J. Vasc.
Res. 34, 245–25918. Dey, N. B., Boerth, N. J., Murphy-Ullrich, J. E., Chang, P. L., Prince, C. W.,
and Lincoln, T. M. (1998) Circ. Res. 82, 139–14619. Cornwell, T. L., Soff, G. A., Traynor, A. E., and Lincoln, T. M. (1994) J. Vasc.
Res. 31, 330–33720. Karin, M. (1994) Curr. Opin. Cell. Biol. 6, 415–42421. Robinson, M. J., and Cobb, M. H. (1997) Curr. Opin. Cell. Biol. 9, 180–18622. Lewis, T. S., Shapiro, P. S., and Ahn, N. G. (1998) Adv. Cancer Res. 74, 49–5723. Pages, G., Lenormand, P., L’Allemain, G., Chambard, J. C., Meloche, S., and
Pouyssegur, J. (1993) Proc. Natl. Acad. Sci. U. S. A. 90, 8319–832324. Watson, M. H., Venance, S. L., Pang, S. P., and Mak, A. S. (1993) Circ. Res. 73,
109–11725. Mansour, S. J., Matten, W. T., Hermann, A. S., Candia, J. M., Rong, S.,
Fukasawa, K., Vande Woude, G. F., and Ahn, N. G. (1994) Science 265,966–970
26. Qui, M. S., and Green, S. H. (1992) Neuron 9, 705–71727. Cowley, S., Paterson, H., Kemp, P., and Marshall, C. J. (1994) Cell 77, 841–85228. Traverse, S., Gomez, N., Paterson, H., Marshall, C., and Cohen, P. (1992)
Biochem. J. 288, 351–35529. Adam, L. P., Gapinski, C. J., and Hathaway, D. R. (1992) FEBS Lett. 302,
223–22630. Childs, T. J., Watson, M. H., Sanghera, J. S., Campbell, D. L., Pelech, S. L., and
Mak, A. S. (1992) J. Biol. Chem. 267, 22853–2285931. Ishida, Y., Kawahara, Y., Tsuda, T., Koide, M., and Yokoyama, M. C. S. (1992)
FEBS Lett. 310, 41–4532. Khalil, R. A., and Morgan, K. G. (1993) Am. J. Physiol. 265, C406–C41133. Katoch, S. S., and Moreland, R. S. (1995) Am. J. Physiol. 269, H222–H22934. Gorenne, I., Su, X., and Moreland, R. S. (1998) Am. J. Physiol. 275,
H131–H13835. Adam, L. P. (1996) in Biochemistry of Smooth Muscle Contraction (Barany, M.,
ed) pp. 167–177, Academic Press, Inc., San Diego36. Dhaunsi, G. S., and Hassid, A. (1996) Cardiovasc Res 31, 37–4737. Gudi, T., Huvar, I., Meinecke, M., Lohmann, S. M., Boss, G. R., and Pilz, R. B.
(1996) J. Biol. Chem. 271, 4597–460038. Peunova, N., and Enikolopov, G. (1993) Nature 364, 450–45339. Haby, C., LIsovoski, F., Aunis, D., and Zwiller, J. (1994) J. Neurochem. 63,
496–50140. Pilz, R. B., Suhasini, M., Idriss, S., Meinkoth, J. L., and Boss, G. R. (1995)
FASEB J. 9, 552–558
41. Hassid, A., Arabshahi, H., Bourcier, T., Dhaunsi, G. S., and Matthews, C.(1994) Am. J. Physiol. 267, H1040–H1048
42. Sciorati, C., Nisitco, G., Meldolesi, J., and Clementi, E. (1997) Br. J. Pharma-col. 122, 687–697
43. Hood, J., and Granger, H. J. (1998) J. Biol. Chem. 273, 23504–2350844. Jo, H., Sipos, K., Go, Y.-M., Law, R., Rong, J., and McDonald, J. M. (1997)
J. Biol. Chem. 272, 1395–140145. Wyatt, T. A., Naftilan, A. J., Francis, S. H., and Corbin, J. C. (1998) Am. J.
Physiol. 274, H448–H45546. Brown, C., Pan, X., and Hassid, A. (1999) Circ. Res. 84, 655–66747. Berk, B. C., and Corson, M. A. (1997) Circ. Res. 80, 607–61648. Ihle, J. N. (1995) Nature 377, 591–59449. Drab, M., Haller, H., Bychkov, R., Erdmann, B., Lindschau, C., Hasse, H.,
Morano, I., Luft, F. C., and Wobus, A. M. (1997) FASEB J. 11, 905–91550. Jonzon, B., Nilsson, J., and Fredholm, B. B. (1985) J. Cell. Physiol. 124,
451–45651. Morisaki, N., Kanzaki, T., Motoyama, N., Saito, Y., and Yoshida, S. (1988)
Atherosclerosis 71, 165–17152. Southgate, J. E., and Newby, A. C. (1990) Atherosclerosis 82, 113–12353. Souness, J. E., Hassall, G. A., and Parrott, D. P. (1992) Biochem. Pharmacol.
44, 857–86654. Lincoln, T. M., Cornwell, T. L., and Taylor, A. E. (1990) Am. J. Physiol. 258,
C399–C40755. Hafner, S., Adler, H. S., Mischak, H., Janosch, P., Heidecder, G., Wolfman, A.,
Fippig, S., Lohse, M., Ueffing, M., and Kolch, W. (1994) Mol. Cell. Biol. 14,6696–6703
56. Wu, J., Dent, P., Jelinek, T., Wolfman, A., Weber, M. J., and Sturgill, T. W.(1993) Science 262, 1065–1068
57. Suhasini, M., Li, H., Lohmann, S. M., Boss, G. R., and Pilz, R. B. (1998) Mol.Cell. Biol. 18, 6983–6994
58. Komalavilas, P., and Lincoln, T. M. (1996) J. Biol. Chem. 271, 21933–2193859. Smolenski, A., Burkhardt, A. M., Eigenthaler, M., Butt, E., Gambaryan, S.,
Lohmann, S. M., and Walter, U. (1998) Naunyn-Schmiedeberg’s Arch. Phar-macol. 358, 134–139
60. Cornwell, T. L., Arnold, E., Boerth, N. J., and Lincoln, T. M. (1994) Am. J.Physiol. 267, C1405–C1413
61. Zeilig, C., and Goldberg, N. (1977) Proc. Natl. Acad. Sci. U. S. A. 74,1052–1056
62. Lermioglu, F., Goyal, J., and Hassid, A. (1991) Biochem. J. 274, 323–32863. Pollman, M. J., Yamada, T., Horiuchi, M., and Gibbons, G. H. (1996) Circ. Res.
79, 748–75664. Duff, J. L., Monia, B. P., and Berk, B. C. (1995) J. Biol. Chem. 270, 7161–716665. Dhaunsi, G. S., Matthews, C., Kaur, K., and Hassid, A. (1997) Am. J. Physiol.
272, H1613–H161966. Sugimoto, T., Haneda, M., Togawa, M., Isono, M., Shikano, T., Araki, S.,
Nakagawa, T., Kashiwagi, A., Guan, K.-L., and Kikkawa R. (1996) J. Biol.Chem. 271, 544–547
67. Begum, N., Ragolia, L., Rienzie, J., McCarthy, M., and Duddy, N. (1998)J. Biol. Chem. 273, 25164–25170
68. Hayashi, K., Takashashi, M., Kumura, K., Nishida, W., and Saga, H. (1999)J. Cell Biol. 145, 727–740
Activation of MAP Kinases by PKG 34309
at Kansas State U
niversity Libraries on July 19, 2014
http://ww
w.jbc.org/
Dow
nloaded from