a single heterologously expressed plant cellulose synthase ... · a single heterologously expressed...
TRANSCRIPT

A single heterologously expressed plant cellulosesynthase isoform is sufficient for cellulosemicrofibril formation in vitroPallinti Purushothama, Sung Hyun Chob, Sara M. Díaz-Morenoc, Manish Kumard, B. Tracy Nixonb, Vincent Bulonec,e,and Jochen Zimmera,1
aDepartment of Molecular Physiology and Biological Physics, University of Virginia School of Medicine, Charlottesville, VA 22908; bDepartment ofBiochemistry and Molecular Biology, The Pennsylvania State University, University Park, PA 16802; cDivision of Glycoscience, Royal Institute of Technology,Stockholm, SE-10691, Sweden; dDepartment of Chemical Engineering, The Pennsylvania State University, University Park, PA 16802; and eAustralianResearch Council Centre of Excellence in Plant Cell Walls, School of Agriculture, Food and Wine, University of Adelaide, Waite Campus, Urrbrae, SA,5064, Australia
Edited by Chris R. Somerville, University of California, Berkeley, CA, and approved August 9, 2016 (received for review April 18, 2016)
Plant cell walls are a composite material of polysaccharides, proteins,and other noncarbohydrate polymers. In the majority of plant tissues,the most abundant polysaccharide is cellulose, a linear polymer ofglucose molecules. As the load-bearing component of the cell wall,individual cellulose chains are frequently bundled into micro andmacrofibrils and are wrapped around the cell. Cellulose is synthe-sized by membrane-integrated and processive glycosyltransferasesthat polymerize UDP-activated glucose and secrete the nascentpolymer through a channel formed by their own transmembraneregions. Plants express several different cellulose synthase isoformsduring primary and secondary cell wall formation; however, so far,none has been functionally reconstituted in vitro for detailedbiochemical analyses. Here we report the heterologous expression,purification, and functional reconstitution of Populus tremula xtremuloides CesA8 (PttCesA8), implicated in secondary cell wallformation. The recombinant enzyme polymerizes UDP-activatedglucose to cellulose, as determined by enzyme degradation, perme-thylation glycosyl linkage analysis, electron microscopy, and muta-genesis studies. Catalytic activity is dependent on the presence of alipid bilayer environment and divalent manganese cations. Further,electron microscopy analyses reveal that PttCesA8 produces cellulosefibers several micrometers long that occasionally are capped by glob-ular particles, likely representing PttCesA8 complexes. Deletion ofthe enzyme’s N-terminal RING-finger domain almost completelyabolishes fiber formation but not cellulose biosynthetic activity. Ourresults demonstrate that reconstituted PttCesA8 is not only sufficientfor cellulose biosynthesis in vitro but also suffices to bundle individualglucan chains into cellulose microfibrils.
biopolymer | cellulose | glycosyltransferase | plant cell wall |membrane transport
Cellulose is an abundant biopolymer primarily produced byvascular plants and also by some bacteria, algae, and tunicates
(1). It is a polymer of glucose molecules connected between theirC1 and C4 carbons via β-glycosidic linkages. The unbranchedpolymer is further stabilized by intramolecular hydrogen bondsbetween the ring oxygen and C2 hydroxyl of one sugar and theC3 and C6 hydroxyl groups, respectively, of a neighboringsugar unit.Cellulose is synthesized by cellulose synthase, CesA (1), a
membrane-integrated processive family-2 glycosyltransferase thatshares significant similarity with chitin, hyaluronan, and alginatesynthases (2). CesA combines two functions: the enzyme poly-merizes UDP-activated glucose (UDP-Glc) in an SN2-like nu-cleophilic displacement reaction (3) and translocates the growingcellulose polymer across the plasma membrane through a poreformed by its own transmembrane (TM) region. Depending onthe tissue, the synthesized polymers can be of astonishing lengths,some containing tens of thousands of sugar units (1).
In most plant tissues, cellulose polymers are bundled intocable-like structures that are wrapped around the cell to form theload-bearing component of the cell wall (1). The smallest repeatingunit, the cellulose microfibril, consists of a currently unknown [butmost likely 18–24 (4, 5)] number of cellulose polymers. The mi-crofibrils are stabilized by van der Waals interactions between thepolymer’s glucopyranose rings and by hydrophilic intermolecularcontacts mediated by the sugars’ equatorial hydroxyl groups.The mechanism by which CesA synthesizes and secretes individual
glucan chains has been delineated recently for bacterial cellu-lose synthase (6–8); however, the process by which plant CesAsbundle these glucan chains into cellulose microfibrils is currentlyunknown. An intriguing hypothesis is that the oligomeric stateof CesAs in the plasma membrane dictates the spontaneousassociation of the extruded glucan chains into microfibrils(9, 10). Indeed, plant cellulose synthases have been shown toform large membrane-embedded complexes displaying pseudo-sixfoldsymmetry referred to as “CesA rosettes” (11, 12). In some speciesthese rosettes are organized further into 2D arrays from which cel-lulose fibrils originate (13). Thus it seems likely that close proximityof CesAs during cellulose biosynthesis is sufficient for microfibrilformation. This notion is supported by the observation thatcellulose produced by monomeric bacterial cellulose synthase isamorphous, with no detectable higher-order structure (14).
Significance
Cellulose is an abundant natural polymer synthesized primarilyby vascular plants in which it forms the load-bearing compo-nent of the cell wall. It is a linear polymer of glucose moleculessynthesized by membrane-embedded cellulose synthases thatcouple polymer synthesis with its secretion across the plasmamembrane. Plants express multiple cellulose synthase isoformsthat are organized into large macromolecular assemblies ofvarying composition that are likely responsible for aligning thecellulose strands into microfibrils. Here we show that recom-binantly expressed and purified Populus tremula x tremuloides(hybrid aspen) cellulose synthase-8 is sufficient for cellulosebiosynthesis and produces cellulose microfibrils in vitro. Ourresults demonstrate that no other plant-derived factors arerequired for cellulose microfibril biosynthesis.
Author contributions: P.P. and J.Z. designed all biochemical experiments; S.H.C., M.K., andB.T.N. designed all EM experiments; S.M.D.-M. and V.B. designed the linkage analysis exper-iments; P.P., S.H.C., and S.M.D.-M. performed all experiments; P.P., S.H.C., S.M.D.-M., M.K.,B.T.N., V.B., and J.Z. contributed to analyzing and writing the manuscript.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.1To whom correspondence should be addressed. Email: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1606210113/-/DCSupplemental.
11360–11365 | PNAS | October 4, 2016 | vol. 113 | no. 40 www.pnas.org/cgi/doi/10.1073/pnas.1606210113
Despite many years of study, cellulose biosynthesis has beenreconstituted in vitro only recently from purified componentsfrom Rhodobacter sphaeroides (14). In these studies, the cellulosesynthase was BcsA, which requires a membrane-anchored, periplasmicsubunit (BcsB) for catalytic activity. Plant cellulose biosynthetic activityhas been demonstrated in membrane extracts from several planttissues (15–17), but none of the catalytically active synthases hasbeen purified to date. Because of these difficulties, it was spec-ulated that the plant enzymes also might require an additional,weakly associated factor for catalytic activity, similar to theBcsA–B complex in bacteria.Plants produce several CesA isoforms; some are required for
primary and others for secondary cell wall formation (1). To de-termine whether a single CesA isoform is catalytically active, weheterologously expressed and purified Populus tremula × tremuloides(hybrid aspen) CesA isoform 8 (PttCesA8), which is implicated insecondary cell wall formation, in Pichia pastoris. The enzyme wasreconstituted into proteoliposomes where it synthesizes cellulosemicrofibrils in an UDP-Glc– and manganese-dependent manner,thereby demonstrating that a single CesA isoform suffices forcellulose biosynthesis and microfibril formation.
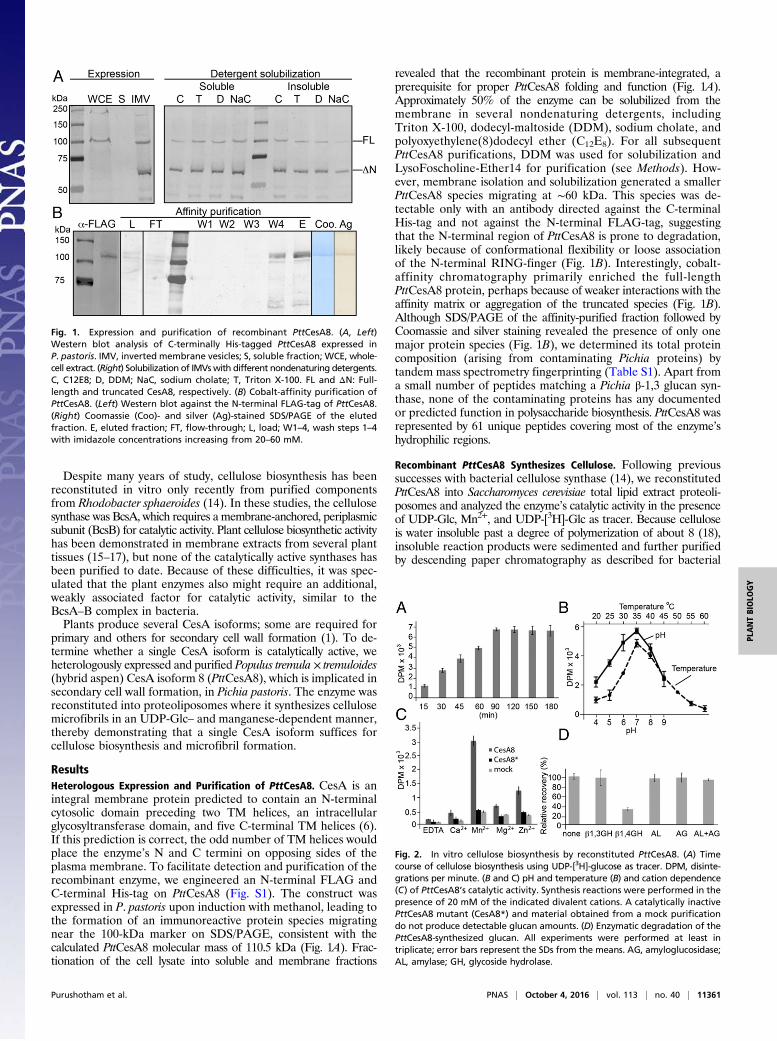
ResultsHeterologous Expression and Purification of PttCesA8. CesA is anintegral membrane protein predicted to contain an N-terminalcytosolic domain preceding two TM helices, an intracellularglycosyltransferase domain, and five C-terminal TM helices (6).If this prediction is correct, the odd number of TM helices wouldplace the enzyme’s N and C termini on opposing sides of theplasma membrane. To facilitate detection and purification of therecombinant enzyme, we engineered an N-terminal FLAG andC-terminal His-tag on PttCesA8 (Fig. S1). The construct wasexpressed in P. pastoris upon induction with methanol, leading tothe formation of an immunoreactive protein species migratingnear the 100-kDa marker on SDS/PAGE, consistent with thecalculated PttCesA8 molecular mass of 110.5 kDa (Fig. 1A). Frac-tionation of the cell lysate into soluble and membrane fractions
revealed that the recombinant protein is membrane-integrated, aprerequisite for proper PttCesA8 folding and function (Fig. 1A).Approximately 50% of the enzyme can be solubilized from themembrane in several nondenaturing detergents, includingTriton X-100, dodecyl-maltoside (DDM), sodium cholate, andpolyoxyethylene(8)dodecyl ether (C12E8). For all subsequentPttCesA8 purifications, DDM was used for solubilization andLysoFoscholine-Ether14 for purification (see Methods). How-ever, membrane isolation and solubilization generated a smallerPttCesA8 species migrating at ∼60 kDa. This species was de-tectable only with an antibody directed against the C-terminalHis-tag and not against the N-terminal FLAG-tag, suggestingthat the N-terminal region of PttCesA8 is prone to degradation,likely because of conformational flexibility or loose associationof the N-terminal RING-finger (Fig. 1B). Interestingly, cobalt-affinity chromatography primarily enriched the full-lengthPttCesA8 protein, perhaps because of weaker interactions with theaffinity matrix or aggregation of the truncated species (Fig. 1B).Although SDS/PAGE of the affinity-purified fraction followed byCoomassie and silver staining revealed the presence of only onemajor protein species (Fig. 1B), we determined its total proteincomposition (arising from contaminating Pichia proteins) bytandem mass spectrometry fingerprinting (Table S1). Apart froma small number of peptides matching a Pichia β-1,3 glucan syn-thase, none of the contaminating proteins has any documentedor predicted function in polysaccharide biosynthesis. PttCesA8 wasrepresented by 61 unique peptides covering most of the enzyme’shydrophilic regions.
Recombinant PttCesA8 Synthesizes Cellulose. Following previoussuccesses with bacterial cellulose synthase (14), we reconstitutedPttCesA8 into Saccharomyces cerevisiae total lipid extract proteoli-posomes and analyzed the enzyme’s catalytic activity in the presenceof UDP-Glc, Mn2+, and UDP-[3H]-Glc as tracer. Because celluloseis water insoluble past a degree of polymerization of about 8 (18),insoluble reaction products were sedimented and further purifiedby descending paper chromatography as described for bacterial
Fig. 1. Expression and purification of recombinant PttCesA8. (A, Left)Western blot analysis of C-terminally His-tagged PttCesA8 expressed inP. pastoris. IMV, inverted membrane vesicles; S, soluble fraction; WCE, whole-cell extract. (Right) Solubilization of IMVswith different nondenaturing detergents.C, C12E8; D, DDM; NaC, sodium cholate; T, Triton X-100. FL and ΔN: Full-length and truncated CesA8, respectively. (B) Cobalt-affinity purification ofPttCesA8. (Left) Western blot against the N-terminal FLAG-tag of PttCesA8.(Right) Coomassie (Coo)- and silver (Ag)-stained SDS/PAGE of the elutedfraction. E, eluted fraction; FT, flow-through; L, load; W1–4, wash steps 1–4with imidazole concentrations increasing from 20–60 mM.
Fig. 2. In vitro cellulose biosynthesis by reconstituted PttCesA8. (A) Timecourse of cellulose biosynthesis using UDP-[3H]-glucose as tracer. DPM, disinte-grations per minute. (B and C) pH and temperature (B) and cation dependence(C) of PttCesA8’s catalytic activity. Synthesis reactions were performed in thepresence of 20 mM of the indicated divalent cations. A catalytically inactivePttCesA8 mutant (CesA8*) and material obtained from a mock purificationdo not produce detectable glucan amounts. (D) Enzymatic degradation of thePttCesA8-synthesized glucan. All experiments were performed at least intriplicate; error bars represent the SDs from the means. AG, amyloglucosidase;AL, amylase; GH, glycoside hydrolase.
Purushotham et al. PNAS | October 4, 2016 | vol. 113 | no. 40 | 11361
PLANTBIOLO
GY

cellulose synthase (14). As shown in Fig. 2 A and B, PttCesA8continued to form a water-insoluble glucan over 90 min at an opti-mal pH and temperature of 7.0 and 35 °C, respectively. The catalyticactivity stalls after 90 min of incubation, likely because of productinhibition or cosedimentation of the synthase and polymeric product.Processive and nonprocessive inverting glycosyltransferases re-
quire a divalent cation, often a magnesium or manganese ion, forcatalytic activity (3). Accordingly, PttCesA8’s catalytic activitydepends on the presence of Mn2+; no significant enzymatic activitywas observed in the presence of Ca2+ or Mg2+, and only modestactivity was seen in the presence of Zn2+ (Fig. 2C).To confirm that the observed in vitro catalytic activity is indeed
caused by recombinant PttCesA8 and not by a minor contamina-tion with a Pichia protein, we generated a catalytically inactivePttCesA8 mutant. Cellulose biosynthesis requires a general base todeprotonate the acceptor’s hydroxyl group during glycosyl transfer(3, 7, 19). This base is formed by the Asp residue of an invariantTED motif, which we replaced in PttCesA8 with an Asn (D676N)to render the enzyme catalytically inactive. The PttCesA8 mutant(PttCesA8*) showed no significant cellulose biosynthetic activity,comparable to material obtained from a mock purification in whichPichia had been transformed with an empty pPICZA vector (Fig.2C), confirming that the observed biosynthetic activity results fromPttCesA8. Further, to establish that the in vitro-synthesized mate-rial indeed represents a β-1,4 glucan, the reaction product washydrolyzed with glucanases specifically degrading β-1,3–, β-1,4–, orα-1,4–linked glucans. Consistent with the formation of authenticcellulose, the PttCesA8-produced material is degraded only by aβ-1,4–specific glucanase, a cellulase (Fig. 2D).In addition to testing the polymer’s sensitivity to enzymatic
degradation, we also characterized it by permethylation glycosyllinkage analysis. The product obtained from a 60-min synthesisreaction contained primarily 1,4-linked glucose together with asmall amount of terminal glucose (Fig. 3 and Fig. S2). The identityof the derivatives was confirmed by electron-impact mass spec-trometry (Fig. S2). However, the peak corresponding to terminalglucose also contained another unidentified compound that was nota sugar derivative. This compound prevents the calculation of anaccurate degree of polymerization, because the fraction of the peakarising from terminal glucose cannot be determined accurately.Some PttCesA8 preparations also showed minor product accu-
mulation in the presence of Mg2+ in addition to the Mn2+-stimulated
PttCesA8 activity (Fig. S3). In this case, glycosyl linkage analysisof the product also revealed the presence of a 1,3-linked glucan,most likely arising from a contaminating Mg2+-dependent β-1,3-glucan synthase and not from PttCesA8. As expected for a con-tamination, the presence of the β-1,3-glucan synthase activityvaried among purifications, and we used only enzyme prepara-tions exhibiting negligible catalytic activity in the presence ofmagnesium (Fig. 2C) for further analyses.
Kinetic Characterization, Product Inhibition, and Lipid Dependence.Cellulose synthase binds the substrate UDP-Glc via its cytosolicglycosyltransferase domain and transfers the activated glucose tothe C4 hydroxyl of the nascent cellulose chain, thereby extendingthe cellulose polymer and forming UDP as a second reactionproduct (8, 14, 20). Monitoring PttCesA8’s catalytic activity un-der increasing UDP-Glc concentrations and at a constant Mn2+
concentration suggests monophasic Michaelis–Menten kineticswith an apparent Km of 30 μM (Fig. 4A). If we assume that k−1is negligible compared with k2, Km can be assimilated to an affinityconstant. This affinity compares to an apparent Km for UDP-Glcof about 500 μM for R. sphaeroides BcsA (14), perhaps reflectingdifferent cytosolic UDP-Glc levels in plant and bacteria. Per-forming cellulose synthesis reactions at a constant UDP-Glc butincreasing UDP concentrations reveals a profound inhibitory effectof UDP, with an apparent inhibitory constant (Ki) of 27 μM,matching the enzyme’s affinity (Km) for the substrate UDP-Glc (Fig.4B). Other nucleoside diphosphates, such as ADP and GDP, do notinhibit PttCesA8, suggesting that UDP, a reaction product ofPttCesA8, rebinds to the active site and inhibits the enzyme com-petitively. Indeed, UDP inhibition is overcome at a UDP-Glc con-centration about 30 times above the Km, as expected for competitiveinhibition (Fig. 4C). A similar product inhibition has been observedfor bacterial cellulose and hyaluronan synthases and is in accordancewith a conserved reaction mechanism of these processive family-2glycosyltransferases (14, 21).
Fig. 3. Glycosyl linkage analysis of PttCesA8-synthesized cellulose. Gaschromatogram corresponding to the separated permethylated alditol ace-tates. The identity of the derivatives corresponding to 4-linked and terminalglucose was confirmed by electron-impact mass spectrometry (Fig. S2). Allother peaks of the chromatogram, except for the peak marked with an as-terisk, correspond to unidentified derivatives that are not alditol acetates, asevidenced by the corresponding fragmentation data. The peak marked withan asterisk corresponds to 4-linked xylose, a known instrument contamina-tion at the time of analysis.
Fig. 4. Kinetic analyses of PttCesA8. (A) Titration of UDP-Glc and quantifi-cation of the synthesized cellulose. The data were fit to monophasicMichaelis–Menten kinetics, yielding a Km of 30 μM. (B) Cellulose biosynthesisin the presence of 30 μM UDP-Glc and increasing concentrations of UDP(circles), ADP (triangles), and GDP (squares). UDP inhibits cellulose synthesiswith a Ki of about 27 μM. (C) UDP competitively inhibits PttCesA8. Thecatalytic activity of PttCesA8 in the presence of 27 μM UDP and increasingUDP-Glc concentrations is shown relative to its activity in the absence ofUDP. (Inset) Lineweaver Burk plot of data shown in A and C. (D) Detergentinhibition of PttCesA8’s catalytic activity. Comparison of PttCesA8’s catalyticactivity in intact (light gray bars) and solubilized (dark gray bars) proteoli-posomes formed from S. cerevisiae or E. coli total lipid extracts. (Inset) Westernblot of S. cerevisiae (Sc) and E. coli (Ec) proteoliposomes. All experiments wereperformed at least in triplicate; error bars represent SDs from the means.
11362 | www.pnas.org/cgi/doi/10.1073/pnas.1606210113 Purushotham et al.

The heterologously expressed PttCesA8 was solubilized fromPichia membranes and purified in the detergent DDM. Severalattempts failed to enrich the enzyme in a detergent-solubilizedstate by the product entrapment method used previously toisolate for example chitin synthases (22). The method relies onthe catalytic activity of the synthase in a detergent-solubilizedstate, so that it cosediments with the synthesized water-insolublepolysaccharide. To test whether PttCesA8 is catalytically active ina detergent micelle, we solubilized PttCesA8-containing pro-teoliposomes with DDM and analyzed the enzyme’s catalyticactivity according to our established protocol. As shown in Fig.4D, PttCesA8 almost completely loses its catalytic activity upondetergent solubilization of the vesicles, suggesting that enzymaticactivity requires an intact lipid bilayer. This loss of catalytic activityis in contrast to bacterial BcsA, which is catalytically active inseveral nondenaturing detergents (14). Lack of catalytic activity indetergents could be caused either by a particular sensitivity of theenzyme toward solubilizing detergents and/or by the disruption ofquaternary structures required for proper function. Despite thisdependence, the enzyme exhibits comparable activity in proteoli-posomes formed from either S. cerevisiae or E. coli total lipid ex-tracts (Fig. 4D), indicating that no eukaryote-specific lipid, unlesscopurifying with the enzyme, is required for catalytic activity.
PttCesA8’s N-Terminal Cytosolic Domain Is Dispensable for CatalyticActivity. Primarily plant CesAs contain extended cytosolic Ntermini of about 170 residues that precede the first TM helix(Fig. 5A). The first ∼65 residues share significant homology withRING-finger domains, which are specialized Zn-binding domainscoordinating two Zn2+ ions via a Cys-rich motif. The following ∼100residues are less well conserved and have no known function (Fig. S4).Because RING-fingers are often implicated in mediating protein–protein interactions, in particular dimerization (23), and isolated CesARING-fingers have been shown to form homo- and heterodimersin vitro (24), it is likely that these domains stabilize membrane-embedded CesA oligomers.To test whether PttCesA8’s N terminus is required for catalytic
activity, we generated two N-terminally truncated mutants, onedevoid of only the RING-finger (CesA8N60, starting with Glu60)and the other missing the entire N-terminal cytosolic domain(CesA8N168, starting with Asn168) (Fig. 5A and Fig. S4). Bothmutants exhibit significant catalytic activity relative to the wild-typeenzyme, albeit reduced by about 25 and 50% for CesA8N60 andCesA8N168, respectively (Fig. 5B). The lower activity of CesA8N168likely results from improper folding and/or membrane integra-tion, because this mutant expresses at a reduced level and isprone to proteolytic degradation. No significant change in catalyticactivity was observed for any of the enzymes in the presence ofboth Mn2+ and Zn2+, as compared with Mn2+ only, suggestingthat an intact and Zn2+-coordinating RING-finger does not directly
affect CesAs catalytic activity. Further, permethylation linkageanalysis of cellulose produced by PttCesA8N60 shows a ratio ofterminal to internal glucose similar to that observed for the wild-type product (Fig. 3), indicating that the two enzymes producecellulose polymers of similar lengths (Fig. S5). Although in bothanalyses the exact amount of terminal glucose is obscured by thepresence of either unidentified or 1,3-glucan contaminations, theresults suggest a degree of polymerization of at least 25–35 andmost likely significantly higher (Fig. 3 and Fig. S5).
PttCesA8-Synthesized Cellulose Is Partially Resistant to Acid Hydrolysis.Because of its organization into cellulose micro- and macrofibrils,native plant cellulose exhibits a remarkable resistance to acid hydro-lysis. In particular, treatment of cellulosic tissues with Updegraff re-agent [80% acetic acid and concentrated nitric acid at a 10:1 (vol:vol)ratio] (25) has been shown to hydrolyze noncellulosic cell wallmaterials efficiently as well as isolated glucan chains not organizedinto fibrils (16, 26). Hence, resistance to acid hydrolysis correlateswith a higher-order organization of cellulose.We compared the resistance to acid hydrolysis of PttCesA8
and bacterial BcsA-synthesized cellulose, the latter formingnoncrystalline cellulose expected to be susceptible to acid hydro-lysis. To this end, cellulose synthesized in vitro by PttCesA8 andBcsA was resuspended in Updegraff reagent and incubated for20 min at 40 °C before quantification. Although the reagentcompletely hydrolyzed bacterial cellulose, ∼25% of the PttCesA8-synthesized cellulose was resistant to hydrolysis, suggesting a differentorganization, at least in part, for BcsA- and PttCesA8-producedcellulose (Fig. 6A). To confirm that the PttCesA8-produced mate-rial resistant to acid hydrolysis indeed represents cellulose, theremaining material was washed several times in deionized waterand subsequently incubated with β-1,3– and β-1,4–specific glucanasesof which only the cellulase was able to degrade the product (Fig. 6B).Interestingly, cellulose produced by PttCesA8 lacking either
the N-terminal RING-finger (CesA8N60) or the entire N-terminalcytosolic domain (CesA8N168) was completely hydrolyzed by theUpdegraff reagent, perhaps because of the formation of primarilynoncrystalline cellulose (Fig. 6A).
PttCesA8 Forms Fiber-Like Cellulosic Structures. The partial resistanceof PttCesA8-synthesized cellulose to acid hydrolysis (Fig. 6A), notobserved for noncrystalline bacterial cellulose, is consistent with ahigher-order organization of the glucan chains. To analyze theorganization of the PttCesA8 cellulose product, we visualized thepolymers by transmission electron microscopy (TEM) after nega-tive staining. Cellulose was synthesized from proteoliposome-reconstituted PttCesA8, and the vesicles were solubilized in thedetergent Triton X-100, after which the sample was diluted at least100-fold before preparing EM grids. Proteoliposomes containing
Fig. 5. PttCesA8’s N terminus is dispensable for catalytic activity. (A) TMtopology diagram of PttCesA8 as predicted by TOPCONS (41). The N terminiof the generated PttCesA8 truncations are indicated. GT, glycosyltransferase.(B) Comparison of the catalytic activities of all PttCesA8 variants in thepresence of 20 mM of the indicated divalent cations or EDTA. ∅ denotesreactions in the absence of additional cations or EDTA. (Inset) Western blotanalysis of proteoliposomes containing the indicated PttCesA8 variants.
Fig. 6. PttCesA8-synthesized cellulose is partially acid resistant. (A) Quan-tification of cellulose resistant to acid hydrolysis. The amount of celluloseresistant to hydrolysis by 80% acetic acid and concentrated nitric acid [10:1(vol:vol)] is shown relative to the initial cellulosic material produced by eachcellulose synthase variant (dark and light gray bars, respectively). (B) Enzy-matic degradation of the acid-resistant cellulose produced by PttCesA8. GH,glycoside hydrolase.
Purushotham et al. PNAS | October 4, 2016 | vol. 113 | no. 40 | 11363
PLANTBIOLO
GY

wild-type PttCesA8 produced fiber-like polymers several microme-ters long only when incubated in the presence of substrate. Controlreactions in the absence of UDP-Glc did not contain any polymericmaterial (Fig. 7). Some samples contained apparently incompletelysolubilized membrane fragments, clearly visible by negative-stainEM, from which cellulose fibrils originated, suggesting that mi-crofibril formation occurs directly after the release of the nascentglucans from their site of synthesis.The apparent cellulose microfibrils were produced only by
wild-type PttCesA8 and not by the catalytically inactive PttCesA8*mutant, confirming that fiber formation indeed is a result ofPttCesA8’s catalytic activity (Fig. 7). Additionally, the synthesized fiberscould be degraded with cellulase, resulting in almost complete lossof visible fibers within 10 min of incubation (Fig. 7 and Fig. S6).Analysis of frozen hydrated fibers by cryo-electron microscopy
(cryoEM) suggests a diameter of 48 ± 10Å (Fig. S7), which is inagreement with diameter estimates for cellulose microfibrilsobtained from spruce wood and celery collenchyma (27, 28). Assuminga stacking distance of about 5 Å between glucan chains within acellulose microfibril, the estimated width of the PttCesA8-producedfiber is sufficient to accommodate at least 18–24 glucan chains. Indeed,comparable cellulose fibers have been synthesized from solubilizedPhyscomitrella patens CesA5 (17), with a similar estimated diameter of20–30 Å, as well as from blackberry membrane extracts (16).Some fibers are capped at one end by globular structures ∼250 Å
in diameter (Fig. 7). Similar structures have been observed oncellulose fibers produced by CesAs solubilized from blackberryand P. patens membranes and most likely represent polymer-attached CesAs (16, 17). The strong interaction of cellulose synthasewith the nascent cellulose polymer is exemplified by RhodobacterBcsA, which, upon heterologous expression in E. coli and detergentsolubilization, copurifies with a cellulose chain (7). The width of thefibril-attached particle is consistent with size estimates for plantCesA oligomers, referred to as “rosettes,” observed by freeze-frac-ture EM (11, 29, 30). CesA rosettes have recently been interpretedas hexamers of CesA trimers, yielding a total of 18 CesA moleculesper complex and, accordingly, a maximum of 18 glucan chains percellulose microfibril (31, 32).Interestingly, cellulose produced by the N-terminally truncated
PttCesA8s contained very little to no fibrous material (Fig. 7), de-spite significant in vitro catalytic activity (Fig. 5B) and an apparentpolymer length similar to that obtained from wild-type PttCesA8
(Fig. 3 and Fig. S5). Given also that the mutant’s cellulosic productis particularly sensitive to acid hydrolysis (Fig. 6A), our data suggestthat N-terminally truncated PttCesA8 cannot form cellulose mi-crofibrils. Of note, our cellulose biosynthesis assay reports onlyon the formation of water-insoluble glucans, irrespective of theirsupramolecular organization. Negative-stain EM, as used in thiswork, is able to visualize only fibrous material and not noncrystallinecellulose, as confirmed by the observation that BcsA-producednoncrystalline cellulose is not detected (Fig. 7). Taken together, ourdata suggest that cellulose microfibril formation is not an intrinsicproperty of CesA and more likely is a secondary effect perhapsarising from a particular CesA quaternary structure supported bythe enzyme’s N-terminal cytosolic domain.
DiscussionCesA functions as polymer synthase and translocase because theenzyme synthesizes glucan chains several hundred to thousandsugar units long and translocates the polymer across the plasmamembrane during its synthesis. In addition, cellulose polymerssynthesized by vascular plants and many other eukaryotic speciesare assembled into fibrillar structures, thereby establishing theirphysical properties as the load-bearing component of the cell wall.Plant CesAs have been shown to interact with several com-
ponents, including cytosolic microtubule-binding proteins, andalso with membrane-integrated or -attached factors, such as theβ-1,4 glucanase KORRIGAN (33). However, the cellulose bio-synthetic activity of recombinantly expressed and reconstitutedCesA8 from hybrid aspen demonstrates that no other plant-specificproteinaceous or lipidic factors are required for its catalytic activity.Recent advances in bacterial cellulose biosynthesis provide
detailed insights into how the synthase elongates the nascentcellulose polymer one glucose unit at a time with translocationsteps between each round of polymer extension (8). However, itis currently unknown how cellulose biosynthesis is initiated andin particular whether a specific primer is required to form thefirst acceptor of glycosyl transfer. Several proposed models forbacterial and plant cellulose biosynthesis include the in-volvement of lipid-linked oligosaccharides or glycolipids, such assitosterol-glucoside, which has been found only in plants so far(34–36). However, the catalytic activity of Pichia-expressedPttCesA8 suggests that the enzyme either initiates cellulosebiosynthesis in vivo using an expression host-provided factor [asobserved for Rhodobacter BcsA upon expression in E. coli (14)] orthat cellulose biosynthesis initiates in vitro upon incubation withUDP-Glc and Mn2+. Based on the Rhodobacter BcsA structure,priming of cellulose biosynthesis with monomeric glucose (derivedfromUDP-Glc hydrolysis) seems likely, as recently discussed (6, 37).In contrast to BcsA, PttCesA8-synthesized cellulose appears as
long fibers that are partially resistant to acid hydrolysis. It istempting to speculate that in vivo these microfibrils could co-alesce to form macrofibrils, similar to those isolated from nativetissues and giving rise to X-ray powder diffraction (28). The lim-ited amount and random orientation of the PttCesA8-producedmicrofibrils currently preclude a similar analysis of our product.However, the observations that recombinant PttCesA8 formscellulose microfibrils and BcsA does not suggest that microfibrilformation is caused by CesA-specific features. An attractivemodel is that CesA oligomerization suffices to drive glucan chainalignment, consistent with the large globular densities cappingsome of the cellulose microfibrils (Fig. 7). The observations thatN-terminally truncated PttCesA8s are catalytically active but failto form cellulose microfibrils further supports this hypothesis.CesA’s N-terminal Zn2+-binding region has strong similarity toRING-fingers and likely mediates CesA–CesA interactions.Further, plant CesAs contain two prominent insertions withinthe evolutionarily conserved glycosyltransferase domain, referredto as “plant-conserved” and “class-specific” regions (38), which havebeen proposed to be involved in CesA oligomerization as well (9).
Fig. 7. PttCesA8 produces cellulose microfibrils in vitro. Cellulose synthesizedby reconstituted PttCesA8 was visualized by negative-stain EM. Shown arerepresentative images of cellulosic material formed by the indicated PttCesA8constructs. Cellulose microfibrils are seen and in some cases are associated withglobular particles that may be cellulose synthase complexes. Cellulose micro-fibrils originating from a membrane sheet are indicated by a white arrow.Negative controls in the absence of UDP-Glc or with the inactive PttCesA8*mutant result in no fiber formation. Infrequent fiber formation is seen with theN-terminal truncation mutants. Treatment of the sample with cellulase for10 min before grid preparation results in the loss of cellulose fibers. BcsA-produced cellulose was not detectable.
11364 | www.pnas.org/cgi/doi/10.1073/pnas.1606210113 Purushotham et al.

CesA oligomers (rosettes) contain different isoforms; for example,in Arabidopsis and Populus CesA1, 3, and 6 are involved in primaryand CesA4, 7, and 8 are involved in secondary cell wall biosynthesis(1). Stoichiometry estimates of CesA rosettes formed during primaryor secondary cell wall formation suggest an equal number of isoformsper complex (39, 40). Although the arrangements and functionsof the individual CesAs in a rosette are currently unknown, ourdata demonstrate that a single isoform suffices to form a cellulosemicrofibril. This finding raises the possibility that CesA rosettesrepresent higher-order complexes of catalytically active CesAhomooligomers, perhaps preformed dimers or trimers, as recentlyobserved for an isolated glycosyltransferase domain from ArabidopsisCesA1 (31). Rosette formation then could be driven by interactionsbetween preformed CesA homooligomers of different isoforms.The reconstitution of cellulose biosynthesis from heterolo-
gously expressed and purified PttCesA8 provides a powerful toolto delineate many crucial steps during cellulose microfibril for-mation. These steps include interaction studies of PttCesA8 andits cellulosic product with other proteins and complex carbohy-drates, thereby providing an in vitro platform for investigatingthe many steps implicated in plant cell wall formation.
MethodsPopulus tremula × tremuloides CesA8 was expressed as a C-terminally His-taggedspecies in P. pastoris and was purified in the detergent LysoFoscholine-Ether14 bymetal-affinity chromatography. The purified protein was reconstituted into pro-teoliposomes, in which it synthesized cellulose upon incubation with the substrateUDP-glucose and Mn2+ cations. The synthesized polymer was quantified by scin-tillation counting upon incorporation of trace amounts of 3H-labeled glucoseas described (14). The synthesis of authentic cellulose was confirmed by en-zymatic degradation and permethylation glycosyl linkage analysis as describedfor bacterial cellulose synthase (14). Uranyl formate-stained electron micro-graphs were obtained from in vitro-synthesized cellulose after the proteoli-posomes were solubilized in the detergent Triton X-100. Detailed methods areprovided in SI Methods.
ACKNOWLEDGMENTS. We thank Chao Fang and Caitlin Hubbard for assis-tance with negative-stain EM and total internal reflection fluorescencemicroscopy, respectively. Protein MS-MS fingerprinting was performedby the Taplin Mass Spectrometry Facility, Harvard Medical School. P.P.,S.H.C., M.K., B.T.N., and J.Z. are supported by the Center for LignocelluloseStructure and Formation, Energy Frontier Research Center, US Departmentof Energy, Office of Science Award DE-SC0001090. S.M.D.-M. and V.B.are supported by the Australian Research Council Centre of Excellencein Plant Cell Walls and matching funding from the Royal Institute ofTechnology, Stockholm.
1. McFarlane HE, Döring A, Persson S (2014) The cell biology of cellulose synthesis. AnnuRev Plant Biol 65:69–94.
2. Bi Y, Hubbard C, Purushotham P, Zimmer J (2015) Insights into the structure andfunction of membrane-integrated processive glycosyltransferases. Curr Opin StructBiol 34:78–86.
3. Lairson LL, Henrissat B, Davies GJ, Withers SG (2008) Glycosyltransferases: Structures,functions, and mechanisms. Annu Rev Biochem 77:521–555.
4. Newman RH, Hill SJ, Harris PJ (2013) Wide-angle x-ray scattering and solid-state nu-clear magnetic resonance data combined to test models for cellulose microfibrils inmung bean cell walls. Plant Physiol 163(4):1558–1567.
5. Jarvis MC (2013) Cellulose biosynthesis: Counting the chains. Plant Physiol 163(4):1485–1486.
6. McNamara J, Morgan JLW, Zimmer J (2015) A molecular description of cellulosebiosynthesis. Annu Rev Biochem 84:17.11–17.27.
7. Morgan JL, Strumillo J, Zimmer J (2013) Crystallographic snapshot of cellulose syn-thesis and membrane translocation. Nature 493(7431):181–186.
8. Morgan JL, et al. (2016) Observing cellulose biosynthesis and membrane translocationin crystallo. Nature 531(7594):329–334.
9. Sethaphong L, et al. (2013) Tertiary model of a plant cellulose synthase. Proc NatlAcad Sci USA 110(18):7512–7517.
10. Slabaugh E, Davis JK, Haigler CH, Yingling YG, Zimmer J (2014) Cellulose synthases:New insights from crystallography and modeling. Trends Plant Sci 19(2):99–106.
11. Kimura S, et al. (1999) Immunogold labeling of rosette terminal cellulose-synthesizingcomplexes in the vascular plant Vigna angularis. Plant Cell 11(11):2075–2086.
12. Brown RM (2003) Cellulose structure and biosynthesis: What is in store for the 21stcentury? J Polym Sci A Polym Chem 42(3):487–495.
13. Giddings TH, Jr, Brower DL, Staehelin LA (1980) Visualization of particle complexes inthe plasma membrane of Micrasterias denticulata associated with the formation ofcellulose fibrils in primary and secondary cell walls. J Cell Biol 84(2):327–339.
14. Omadjela O, et al. (2013) BcsA and BcsB form the catalytically active core of bacterialcellulose synthase sufficient for in vitro cellulose synthesis. Proc Natl Acad Sci USA110(44):17856–17861.
15. Cifuentes C, Bulone V, Emons AMC (2010) Biosynthesis of callose and cellulose bydetergent extracts of tobacco cell membranes and quantification of the polymerssynthesized in vitro. J Integr Plant Biol 52(2):221–233.
16. Lai-Kee-Him J, et al. (2002) In vitro versus in vivo cellulose microfibrils from plantprimary wall synthases: Structural differences. J Biol Chem 277(40):36931–36939.
17. Cho SH, et al. (2015) In vitro synthesis of cellulose microfibrils by a membrane proteinfrom protoplasts of the non-vascular plant Physcomitrella patens. Biochem J 470(2):195–205.
18. Gray MC, Converse AO, Wyman CE (2003) Sugar monomer and oligomer solubility:Data and predictions for application to biomass hydrolysis. Appl Biochem Biotechnol105 -108:179–193.
19. Yang H, Zimmer J, Yingling YG, Kubicki JD (2015) How cellulose elongates–a QM/MMstudy of the molecular mechanism of cellulose polymerization in bacterial CESA.J Phys Chem B 119(22):6525–6535.
20. Brown C, Leijon F, Bulone V (2012) Radiometric and spectrophotometric in vitro assaysof glycosyltransferases involved in plant cell wall carbohydrate biosynthesis. NatProtoc 7(9):1634–1650.
21. Tlapak-Simmons VL, Baron CA, Weigel PH (2004) Characterization of the purifiedhyaluronan synthase from Streptococcus equisimilis. Biochemistry 43(28):9234–9242.
22. Kang MS, et al. (1984) Isolation of chitin synthetase from Saccharomyces cerevisiae.Purification of an enzyme by entrapment in the reaction product. J Biol Chem259(23):14966–14972.
23. Liew CW, Sun H, Hunter T, Day CL (2010) RING domain dimerization is essential forRNF4 function. Biochem J 431(1):23–29.
24. Kurek I, Kawagoe Y, Jacob-Wilk D, Doblin M, Delmer D (2002) Dimerization of cottonfiber cellulose synthase catalytic subunits occurs via oxidation of the zinc-bindingdomains. Proc Natl Acad Sci USA 99(17):11109–11114.
25. Maji S, Mehrotra R, Mehrotra S (2013) Extraction of high quality cellulose from thestem of Calotropis procera. South Asian J Exp Biol 3(3):113–118.
26. Updegraff DM (1969) Semimicro determination of cellulose in biological materials.Anal Biochem 32(3):420–424.
27. Thomas LH, et al. (2013) Structure of cellulose microfibrils in primary cell walls fromcollenchyma. Plant Physiol 161(1):465–476.
28. Fernandes AN, et al. (2011) Nanostructure of cellulose microfibrils in spruce wood.Proc Natl Acad Sci USA 108(47):E1195–E1203.
29. Herth W (1983) Arrays of plasma-membrane “rosettes” involved in cellulose micro-fibril formation of Spirogyra. Planta 159(4):347–356.
30. Roberts AW, Roberts EM, Haigler CH (2012) Moss cell walls: Structure and bio-synthesis. Front Plant Sci 3:166.
31. Vandavasi VG, et al. (2016) A structural study of CESA1 catalytic domain of Arabidopsiscellulose synthesis complex: Evidence for CESA trimers. Plant Physiol 170(1):123–135.
32. Nixon BT, et al. (2016) Comparative structural and computational analysis supportseighteen cellulose synthases in the plant cellulose synthesis complex. Sci Rep 6:28696.
33. Vain T, et al. (2014) The cellulase KORRIGAN is part of the cellulose synthase complex.Plant Physiol 165(4):1521–1532.
34. Matthysse AG, Thomas DL, White AR (1995) Mechanism of cellulose synthesis inAgrobacterium tumefaciens. J Bacteriol 177(4):1076–1081.
35. Peng L, Kawagoe Y, Hogan P, Delmer D (2002) Sitosterol-beta-glucoside as primer forcellulose synthesis in plants. Science 295(5552):147–150.
36. Grillitsch K, et al. (2014) Isolation and characterization of the plasma membrane fromthe yeast Pichia pastoris. Biochim Biophys Acta 1838(7):1889–1897.
37. Morgan JLW, McNamara JT, Zimmer J (2014) Mechanism of activation of bacterialcellulose synthase by cyclic di-GMP. Nat Struct Mol Biol 21(5):489–496.
38. Vergara CE, Carpita NC (2001) Beta-D-glycan synthases and the CesA gene family:Lessons to be learned from the mixed-linkage (1–>3),(1–>4)beta-D-glucan synthase.Plant Mol Biol 47(1-2):145–160.
39. Hill JL, Jr, Hammudi MB, Tien M (2014) The Arabidopsis cellulose synthase complex: Aproposed hexamer of CESA trimers in an equimolar stoichiometry. Plant Cell 26(12):4834–4842.
40. Gonneau M, Desprez T, Guillot A, Vernhettes S, Höfte H (2014) Catalytic subunitstoichiometry within the cellulose synthase complex. Plant Physiol 166(4):1709–1712.
41. Bernsel A, Viklund H, Hennerdal A, Elofsson A (2009) TOPCONS: Consensus predictionof membrane protein topology. Nucleic Acids Res 37(Web Server issue):W465–468.
42. Djerbi SAH, et al. (2004) Identification and expression analysis of genes encodingputative cellulose synthases (CesA) in the hybrid aspen, Populus tremula (L.) xP-tremuloides (Michx.). Cellulose 11:301–312.
Purushotham et al. PNAS | October 4, 2016 | vol. 113 | no. 40 | 11365
PLANTBIOLO
GY