a phylogenomic approach for studying plastid...
TRANSCRIPT

December 15, 2008 11:45 WSPC - Proceedings Trim Size: 9.75in x 6.5in MoustafaChan˙CameraReadyBW
A PHYLOGENOMIC APPROACH FOR STUDYING PLASTIDENDOSYMBIOSIS
AHMED MOUSTAFA1* CHEONG XIN CHAN2*[email protected] [email protected]
MEGAN DANFORTH2 DAVID ZEAR2 HIBA AHMED2
[email protected] [email protected] [email protected]
NAGNATH JADHAV2 TREVOR SAVAGE2 DEBASHISH BHATTACHARYA1,2
[email protected] [email protected] [email protected]
*These authors contributed equally to this work.1Interdisciplinary Genetics Program, University of Iowa, Iowa City, IA 52242, U.S.A.2Department of Biology and Roy J. Carter Center for Comparative Genomics, Uni-versity of Iowa, Iowa City, IA 52242, U.S.A.
Gene transfer is a major contributing factor to functional innovation in genomes. En-dosymbiotic gene transfer (EGT) is a specific instance of lateral gene transfer (LGT) in
which genetic materials are acquired by the host genome from an endosymbiont that hasbeen engulfed and retained in the cytoplasm. Here we present a comprehensive approachfor detecting gene transfer within a phylogenetic framework. We applied the approach
to examine EGT of red algal genes into Thalassiosira pseudonana, a free-living diatomfor which a complete genome sequence has recently been determined. Out of 11,390 pre-dicted protein-coding sequences from the genome of T. pseudonana, 124 (1.1%, clusteredinto 80 gene families) are inferred to be of red algal origin (bootstrap support ≥ 75%).
Of these 80 gene families, 22 (27.5%) encode novel, unknown functions. We found 21.3%of the gene families to putatively encode non-plastid-targeted proteins. Our results sug-gest that EGT of red algal genes provides a relatively minor contribution to the nucleargenome of the diatom, but the transferred genes have functions that extend beyond pho-
tosynthesis. This assertion awaits experimental validation. Whereas the current study isfocused within the context of secondary endosymbiosis, our approach can be applied tolarge-scale detection of gene transfer in any system.
Keywords: phylogenomics; endosymbiotic gene transfer; lateral gene transfer; plastid;
chromalveolates.
1. Introduction
Lateral gene transfer (LGT) is a phenomenon in which genetic materials are trans-mitted between non-lineal individuals (e.g., between two different strains or species).This phenomenon is one of the major mechanisms for functional innovation in thegenomes of prokaryotes [1, 2] and eukaryotes [3, 4], as well as for the acquisitionof new virulence genes in pathogens [5]. Therefore, the elucidation of gene transferevents will enhance our understanding of how genomes evolve. Here we present asystematic approach for detecting LGT within the context of plastid endosymbiosis.
Genome Informatics 21: 165-176 (2008)
165

December 15, 2008 11:45 WSPC - Proceedings Trim Size: 9.75in x 6.5in MoustafaChan˙CameraReadyBW
166 A. Moustafa et al.
1.1. Plastid endosymbiosis and gene transfer
The origin and establishment of the photosynthetic organelle (plastid) in algaeand plants are important for understanding biotic evolution because these taxaform the primary food source for all life on earth. The endosymbiosis hypothesispostulates that the plastid originated from the ancient engulfment and retainmentof a free-living cyanobacterium (the endosymbiont) by a heterotrophic, unicellularprotist. This ancestral photosynthetic eukaryote diversified into the red, green, andglaucophyte algae [6, 7]. Subsequent to this, a secondary endosymbiosis occurred,in which a red alga, that had gained its photosynthetic capability from primaryendosymbiosis, was itself engulfed by a non-photosynthetic protist, giving rise tothe progenitor of the eukaryote supergroup Chromalveolata [7, 8]. The process ofendosymbiosis and the origin of plastid are detailed in [9–11] and Figure 1 in [6].The phenomenon of endosymbiosis led to the transfer of genetic material from theendosymbiont to the host nuclear genome via endosymbiotic gene transfer (EGT),which is a specific case of LGT.
Chromalveolata is one of the six major “supergroups” of eukaryotes. This lineageconsists of a taxonomically diverse group of species that are of high ecological andeconomic importance, including diatoms, seaweeds, dinoflagellates, and the malariaparasite Plasmodium. Our group has previously demonstrated EGT (and LGT) inchromalveolate genomes [3, 12–14], but the extent of EGT from red algae into chro-malveolates, vis-a-vis secondary endosymbiosis, has not been studied in a rigorousmanner.
Among the chromalveolates, diatoms are unicellular eukaryotes and one of theprimary contributors to the marine food chain. The diatoms are estimated to gen-erate ≈ 40% of the organic carbon produced annually in the sea [15]. These taxaaffect the flux of atmospheric carbon dioxide into the oceans, which in turn haseffects on global climate [16]. Recently, the genome of the free-living diatom Tha-lassiosira pseudonana was sequenced to completion [17]. Using the available genomicsequences, here we present a rigorous, phylogenomic pipeline to examine the extentof EGT of red algal genes in T. pseudonana, and investigate if these transferredgenes are restricted to photosynthesis-related functions.
2. A phylogenomic approach for inferring phylogenies
With the increasing amount of available genome data, phylogenomics, the intersec-tion of evolutionary and genomic approaches [18], has become a key instrument instudying genomes on a gene-by-gene basis. This is done primarily by the automatedgeneration and inspection of phylogenetic trees. In many recent studies, phyloge-nomics has been employed to answer various questions including, e.g., prediction ofbiochemical gene functions [19], evolution of gene functions [20], detection of genetransfer events [1, 3], and resolution of complex taxonomic relationships [13].
Our phylogenomic pipeline consists of four basic steps as shown in Figure 1.First, homologous genes for the target sequences are identified (step 1) using WU-

December 15, 2008 11:45 WSPC - Proceedings Trim Size: 9.75in x 6.5in MoustafaChan˙CameraReadyBW
A Phylogenomic Approach for Studying Plastid Endosymbiosis 167
(PERL)WU-
BLAST
(query)
(target)
Export
FASTA
FASTA(MySQL)
Database XMLParsing
(Java & PERL)
FASTAIdenti�cation of homologous genes
Alignment(e.g. MUSCLE)
Multiplesequence
alignment
Re!nement & conversion
(Java)PHYLIP
Phylogeny inference(e.g. RAxML)
PHYLIPPhylogeny
sorting(PhyloSort)
Phylogenyinference
Patterns ofinterest
Topological analysis of phylogeny
1
Musequli
2
Phyinfe
yy3
Topolol i
4
Fig. 1. A schematic diagram of the phylogenomic pipeline: functional components and data flow.
BLAST (http://blast.wustl.edu/) searches against a database containing sequencescollected from public resources, e.g. NCBI (http://www.ncbi.nlm.nih.gov/) andJGI (http://www.jgi.doe.gov/). We used WU-BLAST because this program showshigher time-efficiency than the original BLAST algorithm [21]. Following this, mul-tiple sequence alignment (step 2) is performed for each homologous gene family priorto phylogeny inference (step 3). We used MUSCLE [22] to align the sequences, andboth neighbor-joining (NJ) [23] and maximum likelihood (ML) [24] to reconstructthe phylogenies, because these yield high accuracy in a reasonably short periodof time [22, 24]. However, other approaches for sequence alignment and phylogenyinference can easily be incorporated into our pipeline. Finally, once the phylogenyfor each gene family is obtained, these can be searched for topological patterns ofinterest (step 4). In the current study, we used PhyloSort [25] to sort and examinemonophyletic relationships between chromalveolates and other taxa of interest.
2.1. Analysis of EGT in Thalassiosira pseudonana
We obtained all 11,390 predicted protein-coding sequences from the complete Tha-lassiosira pseudonana genome from JGI (http://www.jgi.gov/). We performed apreliminary screening using BLAST (at e-value ≤ 0.001) for sequences that arehighly similar to and thus possibly share a common ancestry (i.e., homologous) withthe genes in red algae. Using 5,014 protein sequences from the complete genomeof the red alga Cyanidioschyzon merolae [26], we found 4,894 (43.0% of 11,390)protein-coding sequences in T. pseudonana to have homologs in C. merolae.
These protein-coding sequences were used as input in our phylogenomic pipelinethat utilizes our local database, which consists of 2,555,575 sequences from 62 eu-karyote genomes, inclusive of complete and partial expressed sequence tag (EST)sequences spanning Plantae, chromalveolates, Rhizaria, excavates, animals, fungi,and Amoebozoa, and 500 complete bacterial genomes. Initially, the phylogenetic
December 15, 2008 11:45 WSPC - Proceedings Trim Size: 9.75in x 6.5in MoustafaChan˙CameraReadyBW
168 A. Moustafa et al.
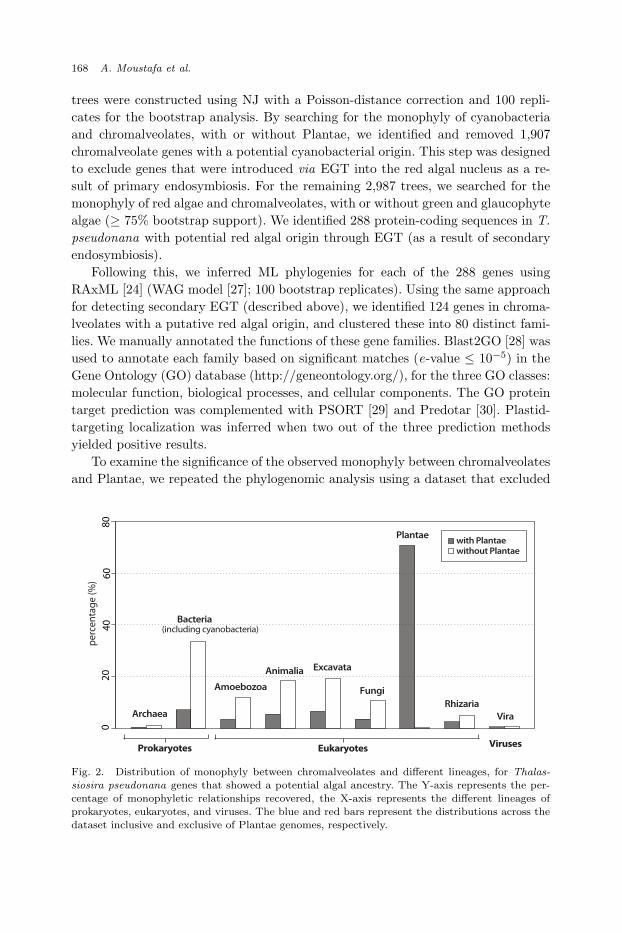
trees were constructed using NJ with a Poisson-distance correction and 100 repli-cates for the bootstrap analysis. By searching for the monophyly of cyanobacteriaand chromalveolates, with or without Plantae, we identified and removed 1,907chromalveolate genes with a potential cyanobacterial origin. This step was designedto exclude genes that were introduced via EGT into the red algal nucleus as a re-sult of primary endosymbiosis. For the remaining 2,987 trees, we searched for themonophyly of red algae and chromalveolates, with or without green and glaucophytealgae (≥ 75% bootstrap support). We identified 288 protein-coding sequences in T.pseudonana with potential red algal origin through EGT (as a result of secondaryendosymbiosis).
Following this, we inferred ML phylogenies for each of the 288 genes usingRAxML [24] (WAG model [27]; 100 bootstrap replicates). Using the same approachfor detecting secondary EGT (described above), we identified 124 genes in chroma-lveolates with a putative red algal origin, and clustered these into 80 distinct fami-lies. We manually annotated the functions of these gene families. Blast2GO [28] wasused to annotate each family based on significant matches (e-value ≤ 10−5) in theGene Ontology (GO) database (http://geneontology.org/), for the three GO classes:molecular function, biological processes, and cellular components. The GO proteintarget prediction was complemented with PSORT [29] and Predotar [30]. Plastid-targeting localization was inferred when two out of the three prediction methodsyielded positive results.
To examine the significance of the observed monophyly between chromalveolatesand Plantae, we repeated the phylogenomic analysis using a dataset that excluded
pe
rce
nta
ge
(%
)
with Plantaewithout Plantae
Archaea
Bacteria(including cyanobacteria)
Amoebozoa
Animalia Excavata
Fungi
Plantae
Rhizaria
Vira
Prokaryotes Eukaryotes Viruses
020
40
60
80
Fig. 2. Distribution of monophyly between chromalveolates and different lineages, for Thalas-siosira pseudonana genes that showed a potential algal ancestry. The Y-axis represents the per-centage of monophyletic relationships recovered, the X-axis represents the different lineages ofprokaryotes, eukaryotes, and viruses. The blue and red bars represent the distributions across the
dataset inclusive and exclusive of Plantae genomes, respectively.

December 15, 2008 11:45 WSPC - Proceedings Trim Size: 9.75in x 6.5in MoustafaChan˙CameraReadyBW
A Phylogenomic Approach for Studying Plastid Endosymbiosis 169
Plantae genomes (glaucophytes, red, and green algae), and compared the observedmonophyly between chromalveolates and the other lineages, with the existing results(dataset inclusive of Plantae genomes). As shown in Figure 2, the distributions ofthe observed monophyly between chromalveolates and non-Plantae are not signifi-cantly different between the two instances, i.e., when Plantae genomes are includedor not (Kolmogorov-Smirnov test [31], p-value > 0.05). This finding suggests thatthe observed monophyletic relationship between chromalveolates and Plantae isnon-random, and not biased by a secondary or tertiary association between chro-malveolates and the other lineages. The strong association between chromalveolatesand Bacteria (33.6%) in the dataset that excluded Plantae genomes can be explainedby the presence of cyanobacterial genes, which have originated via primary EGT(most of which are of plastid function). The (cyano)bacterial association with di-atom genes can therefore be explained by endosymbiosis and not by other scenariosthat involve LGT from prokaryotes.
3. EGT of red algal genes in Thalassiosira pseudonana
We observe 124 (1.1% of the total 11,390) protein-coding sequences from the genomeof T. pseudonana to have a red algal origin. The phylogenetic trees built with eachof these genes and their respective homologs show monophyly of the red algae andchromalveolates with bootstrap support ≥ 75%. The genes are clustered into 80 pu-tative families (Table 1). Among these gene families, 40 (50.0%) are well-annotatedwith gene ontologies (complete annotation for ≥ 90% of the sequences in each fam-ily), whereas 18 (22.5%) are partially annotated (complete annotation for < 90%of the sequences in each family). The remaining 22 (27.5%) are either incompletelyannotated or have no significant match in the gene ontology database. We considerthese 22 gene families to encode novel, unknown functions in the diatom.
The majority of genes from T. pseudonana in each of these families is primarilyrepresented by single-copy sequences (58, 72.5%), with some containing two (14,17.5%) or three (6, 7.5%) gene copies. There are two families in which the geneis highly duplicated within the genome of T. pseudonana. These are the ABC-1domain protein (7 copies) and light-harvesting protein (13 copies). As shown inthe last column of Table 1, 23 (28.8%) of the 80 gene families putatively code forproteins targeted to the plastid, 21 (26.3%) putatively code for proteins targetedto multiple organelles with the majority going to the plastid, 19 (23.8%) of theproteins are potentially targeted to multiple organelles with the minority being theplastid, whereas the remainder (17, 21.3%) putatively code for proteins that are nottargeted to the plastid. In parallel with gene ontology analysis, we do not observe aN-terminal extension in the bacterial homologs of these 17 eukaryotic gene families,suggesting that these genes are not targeted to membrane-bounded organelles. Thefamilies in which the gene copy is highly duplicated in T. pseudonana are foundto be targeted to multiple organelles in the cell (including the mitochondrion andnucleus) and are not restricted to the plastid.

December 15, 2008 11:45 WSPC - Proceedings Trim Size: 9.75in x 6.5in MoustafaChan˙CameraReadyBW
170 A. Moustafa et al.
Table 1: Gene families showing a red algal origin in T. pseudonana. The numberof genes from the species in each family is shown. Indication whether a family
encodes for a putative plastid-targeted proteins is shown in the last column,based on GO annotations of cellular components for each family: completelyplastid-targeted (+++), targeted to multiple membrane-bounded organelleswith majority to plastid (++), targeted to multiple membrane-bounded or-
ganelles with minority being plastid (+), and not targeted in plastid at all(-).
No. ID Description No. of genes in Plastid-T. pseudonana targeted (+/-)
1 49 bile acid:sodium symporter 3 +++
2 33 sodium hydrogen exchanger 3 +++3 15 ATP-dependent CLP protease proteolytic subunit 2 +++4 21 HAD-superfamily hydrolase subfamily variant 3 2 +++5 12 protease Do 2 +++
6 24 unknown protein 2 +++7 63 2-c-methyl-d-erythritol 4-phosphate 1 +++
cytidylyltransferase8 17 3-dehydroquinate synthase 1 +++
9 50 aspartate aminotransferase 1 +++10 34 aspartate kinase 1 +++11 31 carboxyl-terminal protease 1 +++
12 57 fkbp-type peptidyl-prolyl cis-transisomerase 1 +++13 67 glycosyl transferase group 1 1 +++14 54 GTP pyrophosphokinase 1 +++15 39 monogalactosyldiacylglycerol synthase 1 +++
16 52 serine acetyltransferase 1 +++17 56 small drug exporter protein 1 +++18 45 sulfolipid (UDP-sulfoquinovose) biosynthesis protein 1 +++19 41 tRNA pseudouridine synthase a 1 +++
20 44 unknown protein 1 +++21 53 unknown protein 1 +++22 78 unknown protein 1 +++23 81 unknown protein 1 +++
24 4 light-harvesting protein 13 ++25 8 ABC-1 domain protein 7 ++26 27 phosphoglycolate phosphatase precursor 2 ++27 5 trehalose-6-phosphate synthase 2 ++
28 3 ABC family transporter 1 ++29 7 ATP-dependent RNA helicase 1 ++30 32 cysteinyl-tRNA synthase 1 ++
31 61 cytochrome C peroxidase 1 ++32 48 dihydrodipicolinate reductase 1 ++33 69 methionyl aminopeptidase 1 ++34 64 peptidyl-prolyl cis-transcyclophilin type 1 ++
35 66 RNA polymerase sigma factor 1 ++36 72 thioredoxin-1 1 ++37 28 translation elongation factor g 1 ++38 14 unknown protein 1 ++
39 18 unknown protein 1 ++40 22 unknown protein 1 ++41 26 unknown protein 1 ++
continued on next page. . .

December 15, 2008 11:45 WSPC - Proceedings Trim Size: 9.75in x 6.5in MoustafaChan˙CameraReadyBW
A Phylogenomic Approach for Studying Plastid Endosymbiosis 171
Table 1 – Continued
No. ID Description No. of genes in Plastid-T. pseudonana targeted (+/-)
42 42 unknown protein 1 ++
43 76 unknown protein 1 ++44 75 valyl-tRNA synthetase 1 ++45 55 peroxisomal membrane protein 3 +
46 23 unknown protein 3 +47 16 zinc finger protein 3 +48 62 histone deacetylase family protein 2 +49 11 hypothetical protein 2 +
50 51 phosphate phosphoenolpyruvate translocator 2 +precursor
51 43 protein phosphatase 2c related protein 2 +52 9 ABC transporter related protein 1 +
53 46 cell division protein 1 +54 74 DNA topoisomerase VI subunit a 1 +55 73 elongation factor 1 alpha 1 +56 60 GTP binding protein 1 +
57 30 HAD superfamily (subfamily ig) 5-nucleotidase 1 +58 20 heat shock protein 90 1 +59 37 homogentisate solanesyltransferase 1 +60 80 NADH dehydrogenase 1 +
61 68 ribosomal protein s7 1 +62 19 unknown protein 1 +63 79 unknown protein 1 +
64 35 p-ATPase family transporter: cation 3 -65 10 anion exchange family protein 2 -66 40 prolyl-tRNA synthase 2 -67 2 unknown protein 2 -
68 6 unknown protein 2 -69 38 amine oxidase 1 -70 59 chromodomain helicase DNA binding protein 1 -71 71 DNA topoisomerase VI subunit b 1 -
72 36 glucose-6-phosphate isomerase 1 -73 70 glycerol-3-phosphate dehydrogenase (NAD+) 1 -74 65 HSP associated protein like 1 -75 47 s-adenosyl-l-homocysteine hydrolase 1 -
76 1 unknown protein 1 -77 25 unknown protein 1 -78 29 unknown protein 1 -79 58 unknown protein 1 -
80 77 unknown protein 1 -
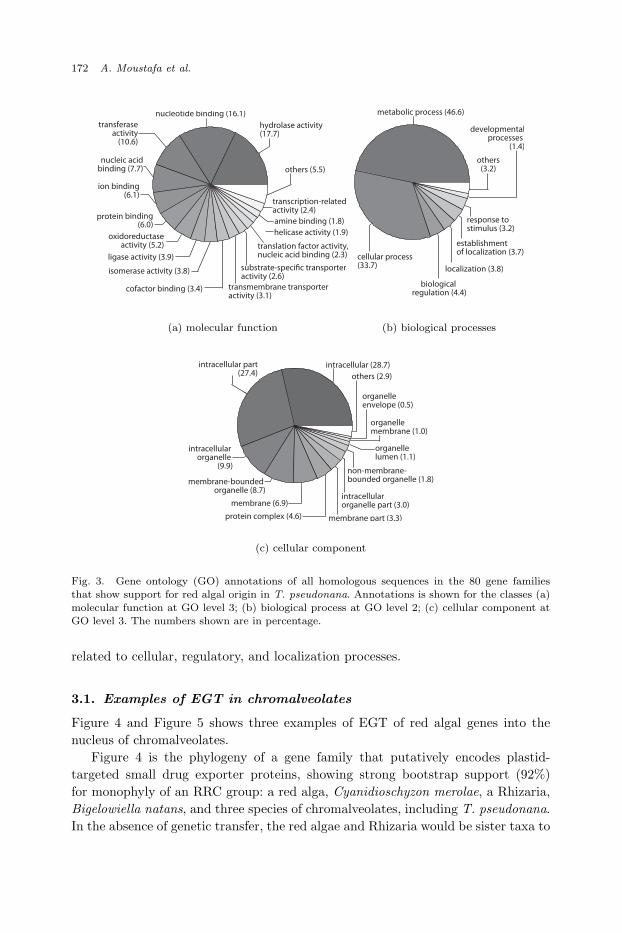
Figure 3 shows the gene ontology annotations for all homologous sequences fromthe 80 gene families, for each class of (a) molecular function, (b) biological processand, (c) cellular component. As shown in the panels (a) through (c), the familiesare of diverse functions that are involved in a variety of biological processes and theencoded proteins are targeted to various compartments within the cell. The genefunctions range from biomolecule-binding, transporters, to catalytic activities. Mostof these genes are annotated to engage in metabolic processes, whereas some are
December 15, 2008 11:45 WSPC - Proceedings Trim Size: 9.75in x 6.5in MoustafaChan˙CameraReadyBW
172 A. Moustafa et al.
hydrolase activity(17.7)
nucleotide binding (16.1)
transferaseactivity
(10.6)
nucleic acid binding (7.7)
ion binding(6.1)
protein binding(6.0)
oxidoreductaseactivity (5.2)
ligase activity (3.9)
isomerase activity (3.8)
cofactor binding (3.4) transmembrane transporter activity (3.1)
substrate-speci!c transporter activity (2.6)
translation factor activity, nucleic acid binding (2.3)
helicase activity (1.9)
amine binding (1.8)
transcription-related activity (2.4)
others (5.5)
metabolic process (46.6)
cellular process(33.7) localization (3.8)
establishment of localization (3.7)
biological regulation (4.4)
response tostimulus (3.2)
developmentalprocesses
(1.4)
others(3.2)
intracellular (28.7)intracellular part(27.4)
intracellular organelle
(9.9)
membrane-bounded organelle (8.7)
membrane (6.9)
protein complex (4.6)
intracellular organelle part (3.0)
membrane part (3.3)
non-membrane-bounded organelle (1.8)
organellemembrane (1.0)
organelle envelope (0.5)
organellelumen (1.1)
others (2.9)
(a) molecular function
hydrolase activity(17.7)
nucleotide binding (16.1)
transferaseactivity
(10.6)
nucleic acid binding (7.7)
ion binding(6.1)
protein binding(6.0)
oxidoreductaseactivity (5.2)
ligase activity (3.9)
isomerase activity (3.8)
cofactor binding (3.4) transmembrane transporter activity (3.1)
substrate-speci!c transporter activity (2.6)
translation factor activity, nucleic acid binding (2.3)
helicase activity (1.9)
amine binding (1.8)
transcription-related activity (2.4)
others (5.5)
metabolic process (46.6)
cellular process(33.7) localization (3.8)
establishment of localization (3.7)
biological regulation (4.4)
response tostimulus (3.2)
developmentalprocesses
(1.4)
others(3.2)
intracellular (28.7)intracellular part(27.4)
intracellular organelle
(9.9)
membrane-bounded organelle (8.7)
membrane (6.9)
protein complex (4.6)
intracellular organelle part (3.0)
membrane part (3.3)
non-membrane-bounded organelle (1.8)
organellemembrane (1.0)
organelle envelope (0.5)
organellelumen (1.1)
others (2.9)
(b) biological processes
hydrolase activity(17.7)
nucleotide binding (16.1)
transferaseactivity
(10.6)
nucleic acid binding (7.7)
ion binding(6.1)
protein binding(6.0)
oxidoreductaseactivity (5.2)
ligase activity (3.9)
isomerase activity (3.8)
cofactor binding (3.4) transmembrane transporter activity (3.1)
substrate-speci!c transporter activity (2.6)
translation factor activity, nucleic acid binding (2.3)
helicase activity (1.9)
amine binding (1.8)
transcription-related activity (2.4)
others (5.5)
metabolic process (46.6)
cellular process(33.7) localization (3.8)
establishment of localization (3.7)
biological regulation (4.4)
response tostimulus (3.2)
developmentalprocesses
(1.4)
others(3.2)
intracellular (28.7)intracellular part(27.4)
intracellular organelle
(9.9)
membrane-bounded organelle (8.7)
membrane (6.9)
protein complex (4.6)
intracellular organelle part (3.0)
membrane part (3.3)
non-membrane-bounded organelle (1.8)
organellemembrane (1.0)
organelle envelope (0.5)
organellelumen (1.1)
others (2.9)
(c) cellular component
Fig. 3. Gene ontology (GO) annotations of all homologous sequences in the 80 gene familiesthat show support for red algal origin in T. pseudonana. Annotations is shown for the classes (a)molecular function at GO level 3; (b) biological process at GO level 2; (c) cellular component atGO level 3. The numbers shown are in percentage.
related to cellular, regulatory, and localization processes.
3.1. Examples of EGT in chromalveolates
Figure 4 and Figure 5 shows three examples of EGT of red algal genes into thenucleus of chromalveolates.
Figure 4 is the phylogeny of a gene family that putatively encodes plastid-targeted small drug exporter proteins, showing strong bootstrap support (92%)for monophyly of an RRC group: a red alga, Cyanidioschyzon merolae, a Rhizaria,Bigelowiella natans, and three species of chromalveolates, including T. pseudonana.In the absence of genetic transfer, the red algae and Rhizaria would be sister taxa to

December 15, 2008 11:45 WSPC - Proceedings Trim Size: 9.75in x 6.5in MoustafaChan˙CameraReadyBW
A Phylogenomic Approach for Studying Plastid Endosymbiosis 173
98
32
48
92
100
51
28 74
100
100
Arabidopsis thaliana
Cyanidioschyzon merolae
Bigelowiella natans
Thalassiosira pseudonana
Phaeodactylum tricornutum
Aureococcus anophage!erens
Dehalococcoides sp.
Synechococcus elongatus
Thermus thermophilus
Bacteroides capillosus
Physcomitrella patens
Oryza sativa
0.8
Chromalveolates
Red alga
Rhizaria
Bacteria
Chloro�exi
Cyanobacteria
Deinococci
Bacteroidetes
Green alga
Plants
Firmicutes
Firmicutes
Fig. 4. A maximum likelihood phylogeny showing an example of EGT of an annotated plastid-targeted protein from red algae to T. pseudonana (monophyly support for chromalveolates andred algae). Numbers shown are bootstrap support values for each node. The scale bar is shown in
unit of substitution per site.
the green algae. This phylogeny implies EGT between the ancestral lineage of thered algae to the ancestral lineage of chromalveolates. In addition, the RRC groupingalso forms a monophyletic relationship with all gene copies present in bacteria (boot-strap support 100%), suggesting that the transferred gene is of an ancient bacterialorigin. The observation supports the notion of plastid endosymbiosis that plastidsin chromalveolates originated from red algae, which in turn are of a cyanobacterialorigin.
In contrast, Figure 5 shows the phylogenies of (a) a plastid-targeted gene familyand (b) a non-plastid-targeted gene famaily of unknown (and likely novel) functions.In the gene phylogeny shown in Figure 5(a), three species of red algae form the sistertaxa with three species of chromalveolates rather than with the green algae. Themonophyly of red algae and chromalveolates is strongly supported at bootstrap sup-port 100%. Although the gene function is unknown, this family putatively encodesproteins targeted only to plastids and might therefore play roles in the process ofphotosynthesis. For the gene phylogeny shown in 5(b), homologous sequences areabsent in a large number of lineages. A non-EGT explanation would involve manygene loss events along a large number of lineages. The most parsimonious explana-tion for such a gene phylogeny is an EGT event from the ancestral lineage of thered alga Cyanidioschyzon merolae to the ancestral lineage of the chromalveolates.
4. Performance and limitations
We have demonstrated the use of a rigorous, computational phylogenomic approachto infer the events of gene transfer within the context of plastid endosymbiosis. Our

December 15, 2008 11:45 WSPC - Proceedings Trim Size: 9.75in x 6.5in MoustafaChan˙CameraReadyBW
174 A. Moustafa et al.
Oryza sativa
Arabidopsis thaliana
Physcomitrella patens
Chondrus crispusPorphyra yezoensis
Cyanidioschyzon merolae
Aureococcus anophage�erens
Phaeodactylum tricornutum
Thalassiosira pseudonanaChlamydomonas reinhardtii
Volvox carteri
Ostreococcus lucimarinus
Ostreococcus tauri
Micromonas RCC299
Micromonas CCMP1545
94
98
72
100
100
65
100 100
72
100
100
93
Plants
Green alga
Red algae
Chromalveolates
0.5
Green algae
Cyanophora paradoxa Glaucophyte
0.8
Cyanidioschyzon merolae Red alga
Aureococcus anophage�erens
Isochrysis galbana
Thalassiosira pseudonana
Phaeodactylum tricornutum Chromalveolates78
76
98
(a) Gene family ID 81, plastid-targeted
Oryza sativa
Arabidopsis thaliana
Physcomitrella patens
Chondrus crispusPorphyra yezoensis
Cyanidioschyzon merolae
Aureococcus anophage�erens
Phaeodactylum tricornutum
Thalassiosira pseudonanaChlamydomonas reinhardtii
Volvox carteri
Ostreococcus lucimarinus
Ostreococcus tauri
Micromonas RCC299
Micromonas CCMP1545
94
98
72
100
100
65
100 100
72
100
100
93
Plants
Green alga
Red algae
Chromalveolates
0.5
Green algae
Cyanophora paradoxa Glaucophyte
0.8
Cyanidioschyzon merolae Red alga
Aureococcus anophage�erens
Isochrysis galbana
Thalassiosira pseudonana
Phaeodactylum tricornutum Chromalveolates78
76
98
(b) Gene family ID 58, non-plastid-targeted
Fig. 5. Two maximum likelihood phylogenies showing EGT of red algal genes in T. pseudonana(monophyly support for chromalveolates and red algae). The genes are of unknown function for(a) a plastid-targeted gene family and (b) a non-plastid-targeted gene family. Numbers shown arebootstrap support values for each node. The scale bars are shown in unit of substitution per site.
approach is based on the implicit assumption that genes are transferred as a whole.The transfer of genes in smaller fragments, which introduces within-gene discrep-ancies of phylogenetic signal, might not be fully recovered using this approach. Inaddition, the efficiency of detecting phylogenetic signal can also be compromisedby sequence divergence, presence or absence of informative and/or invariant sites.Therefore, the extent of genetic transfer inferred in this study is a conservativeestimate.
In the current study, our approach shows a low false positive discovery rateof 1.23% (e.g., trees that return the incorrect monophyly of chromalveolates and

December 15, 2008 11:45 WSPC - Proceedings Trim Size: 9.75in x 6.5in MoustafaChan˙CameraReadyBW
A Phylogenomic Approach for Studying Plastid Endosymbiosis 175
animals). In a preliminary study, we generated simulated eight-taxon protein sets(sample size = 100, sequence length = 1000 amino acids) that are evolved homo-geneously at various degrees of sequence conservation. Our phylogenomic approachyielded 0% false positive in recovering the target monophyletic relationships (datanot shown), with 0.17% false negative rate in cases where sequences are highlydivergent (average substitution per site = 2). Under a more-realistic evolutionaryregime, e.g., heterogeneous evolution with varied substitution rates along the sameor different lineages, the false positive and negative rates are expected to be higher.
Based on bioinformatic predictions and analysis at a high statistical (bootstrap)confidence, our findings suggest that genes that show a history of EGT from redalgae into T. pseudonana extend beyond plastid-related (e.g., photosynthetic) func-tions, and thus these transferred genes might make a much greater impact in genomeinnovation of T. pseudonana than previously thought. Nevertheless, the extent ofsuch an impact in plastid endosymbiosis remains to be verified by experimentalapproaches. The current approach is suitable for an high-throughput detection ofwhole-gene transfer within broader biological contexts at a multi-genome scale.
5. Authors’ contributions
AM designed and implemented the phylogenomic pipeline, conducted the phyloge-nomic analysis and contributed to the preparation of the manuscript draft. CXCconducted downstream functional analysis of the gene families, wrote and preparedthe table, figures, and the manuscript draft. Both AM and CXC contributed tothe analysis of the results. MD, DZ, HA, NJ and TS conducted gene-by-gene phy-logenetic analysis to validate the results from the pipeline. DB conceived of andsupervised this study. AM, CXC and DB conceived, edited and approved the finalmanuscript.
6. Acknowledgments
This work was supported by a grant from the National Institutes of Health(R01ES013679) awarded to DB. We acknowledge the intellectual input of AdrianReyes-Prieto and Valerie Reeb (University of Iowa) in this project.
References
[1] R. G. Beiko, T. J. Harlow and M. A. Ragan, Proc. Natl. Acad. Sci. U.S.A. 102, 14332(2005).
[2] E. Lerat, V. Daubin, H. Ochman and N. A. Moran, PLoS Biology 3, Art. e130 (2005).[3] T. Nosenko and D. Bhattacharya, BMC Evol. Biol. 7, Art. 173 (2007).[4] D. Bhattacharya and T. Nosenko, J. Phycol. 44, 7 (2008).[5] V. M. D’Costa, K. M. McGrann, D. W. Hughes and G. D. Wright, Science 311, 374
(2006).[6] D. Bhattacharya, H. S. Yoon and J. D. Hackett, Bioessays 26, 50 (2004).[7] G. I. McFadden, J. Phycol. 37, 951 (2001).

December 15, 2008 11:45 WSPC - Proceedings Trim Size: 9.75in x 6.5in MoustafaChan˙CameraReadyBW
176 A. Moustafa et al.
[8] T. Cavalier-Smith, J. Eukaryot. Microbiol. 46, 347 (1999).[9] A. Reyes-Prieto, A. P. M. Weber and D. Bhattacharya, Ann. Rev. Genet. 41, 147
(2007).[10] D. Bhattacharya, J. M. Archibald, A. P. M. Weber and A. Reyes-Prieto, Bioessays
29, 1239 (2007).[11] S. B. Gould, R. F. Waller and G. I. McFadden, Annu Rev Plant Biol 59, 491 (2008).[12] J. D. Hackett, H. S. Yoon, M. B. Soares, M. F. Bonaldo, T. L. Casavant, T. E. Scheetz,
T. Nosenko and D. Bhattacharya, Curr. Biol. 14, 213 (2004).[13] J. D. Hackett, H. S. Yoon, S. Li, A. Reyes-Prieto, S. E. Rummele and D. Bhattacharya,
Mol. Biol. Evol. 24, 1702 (2007).[14] A. Reyes-Prieto, A. Moustafa and D. Bhattacharya, Curr Biol 18, 956 (2008).[15] D. M. Nelson, P. Treguer, M. A. Brzezinski, A. Leynaert and B. Queguiner, Global
Biogeochem. Cycl. 9, 359 (1995).[16] M. A. Brzezinski, C. J. Pride, V. M. Franck, D. M. Sigman, J. L. Sarmiento, K. Mat-
sumoto, N. Gruber, G. H. Rau and K. H. Coale, Geophys. Res. Lett. 29, 1564 (2002).[17] E. V. Armbrust, J. A. Berges, C. Bowler, B. R. Green, D. Martinez, N. H. Putnam,
S. G. Zhou, A. E. Allen, K. E. Apt, M. Bechner, M. A. Brzezinski, B. K. Chaal,A. Chiovitti, A. K. Davis, M. S. Demarest, J. C. Detter, T. Glavina, D. Goodstein,M. Z. Hadi, U. Hellsten, M. Hildebrand, B. D. Jenkins, J. Jurka, V. V. Kapitonov,N. Kroger, W. W. Y. Lau, T. W. Lane, F. W. Larimer, J. C. Lippmeier, S. Lu-cas, M. Medina, A. Montsant, M. Obornik, M. S. Parker, B. Palenik, G. J. Pazour,P. M. Richardson, T. A. Rynearson, M. A. Saito, D. C. Schwartz, K. Thamatrakoln,K. Valentin, A. Vardi, F. P. Wilkerson and D. S. Rokhsar, Science 306, 79 (2004).
[18] J. A. Eisen and C. M. Fraser, Science 300, 1706 (2003).[19] J. Huang, G. S. V. Aller, A. N. Taylor, J. J. Kerrigan, W. S. Liu, J. M. Trulli, Z. Lai,
D. Holmes, K. M. Aubart, J. R. Brown and M. Zalacain, J. Bacteriol. 188, 5249(2006).
[20] U. John, B. Beszteri, E. Derelle, Y. V. de Peer, B. Read, H. Moreau and A. Cembella,Protist 159, 21 (2008).
[21] S. F. Altschul, W. Gish, W. Miller, E. W. Myers and D. J. Lipman, J Mol Biol 215,403 (1990).
[22] R. C. Edgar, Nucl. Acids Res. 32, 1792 (2004).[23] N. Saitou and M. Nei, Mol. Biol. Evol. 4, 406 (1987).[24] A. Stamatakis, Bioinformatics 22, 2688 (2006).[25] A. Moustafa and D. Bhattacharya, BMC Evol. Biol. 8, Art. 6 (2008).[26] M. Matsuzaki, O. Misumi, I. T. Shin, S. Maruyama, M. Takahara, S. Y. Miyag-
ishima, T. Mori, K. Nishida, F. Yagisawa, Y. Yoshida, Y. Nishimura, S. Nakao,T. Kobayashi, Y. Momoyama, T. Higashiyama, A. Minoda, M. Sano, H. Nomoto,K. Oishi, H. Hayashi, F. Ohta, S. Nishizaka, S. Haga, S. Miura, T. Morishita,Y. Kabeya, K. Terasawa, Y. Suzuki, Y. Ishii, S. Asakawa, H. Takano, N. Ohta,H. Kuroiwa, K. Tanaka, N. Shimizu, S. Sugano, N. Sato, H. Nozaki, N. Ogasawara,Y. Kohara and T. Kuroiwa, Nature 428, 653 (2004).
[27] S. Whelan and N. Goldman, Mol. Biol. Evol. 18, 691 (2001).[28] A. Conesa, S. Gotz, J. M. Garcıa-Gomez, J. Terol, M. Talon and M. Robles, Bioin-
formatics 21, 3674 (2005).[29] P. Horton, K. J. Park, T. Obayashi, N. Fujita, H. Harada, C. Adams-Collier and
K. Nakai, Nucl. Acids Res. 35, W585 (2007).[30] I. Small, N. Peeters, F. Legeai and C. Lurin, Proteomics 4, 1581 (2004).[31] F. J. Massey, J. Am. Stat. Assoc. 46, 68 (1951).