a novel carbon monoxide-releasing molecule fully...
TRANSCRIPT

Published Ahead of Print 12 December 2011. 10.1128/AAC.05571-11.
2012, 56(3):1281. DOI:Antimicrob. Agents Chemother. PamplonaRomão, Maria M. Mota, Gonçalo J. L. Bernardes and AnaNeres, João D. Seixas, Afonso C. Fernandes, Carlos C. Ana C. Pena, Nuno Penacho, Liliana Mancio-Silva, Rita MalariaMolecule Fully Protects Mice from Severe A Novel Carbon Monoxide-Releasing
http://aac.asm.org/content/56/3/1281Updated information and services can be found at:
These include:
SUPPLEMENTAL MATERIAL
mlhttp://aac.asm.org/content/suppl/2012/02/09/56.3.1281.DC1.ht
REFERENCEShttp://aac.asm.org/content/56/3/1281#ref-list-1at:
This article cites 46 articles, 12 of which can be accessed free
CONTENT ALERTS more»articles cite this article),
Receive: RSS Feeds, eTOCs, free email alerts (when new
http://aac.asm.org/site/misc/reprints.xhtmlInformation about commercial reprint orders: http://journals.asm.org/site/subscriptions/To subscribe to to another ASM Journal go to:
on February 15, 2012 by E
TH
-BIB
LIOT
HE
K Z
UR
ICH
http://aac.asm.org/
Dow
nloaded from

A Novel Carbon Monoxide-Releasing Molecule Fully Protects Micefrom Severe Malaria
Ana C. Pena,a Nuno Penacho,b Liliana Mancio-Silva,a,c Rita Neres,a João D. Seixas,b,d Afonso C. Fernandes,e Carlos C. Romão,b,d
Maria M. Mota,c Gonçalo J. L. Bernardes,b,f and Ana Pamplonaa
Unidade de Imunologia Molecular, Instituto de Medicina Molecular, Universidade de Lisboa, Lisbon, Portugala; Alfama Lda., Porto Salvo, Portugalb; Unidade de Malária,Instituto de Medicina Molecular, Universidade de Lisboa, Lisbon, Portugalc; Instituto de Tecnologia Química e Biológica da Universidade Nova de Lisboa, Oeiras, Portugald;Instituto de Medicina Molecular, Universidade de Lisboa, Lisbon, Portugale; and Swiss Federal Institute of Technology (ETH Zürich), Department of Chemistry and AppliedBiosciences, Zürich, Switzerlandf
Severe forms of malaria infection, such as cerebral malaria (CM) and acute lung injury (ALI), are mainly caused by the apicompl-exan parasite Plasmodium falciparum. Primary therapy with quinine or artemisinin derivatives is generally effective in control-ling P. falciparum parasitemia, but mortality from CM and other forms of severe malaria remains unacceptably high. Herein, wereport the design and synthesis of a novel carbon monoxide-releasing molecule (CO-RM; ALF492) that fully protects miceagainst experimental CM (ECM) and ALI. ALF492 enables controlled CO delivery in vivo without affecting oxygen transport byhemoglobin, the major limitation in CO inhalation therapy. The protective effect is CO dependent and induces the expression ofheme oxygenase-1, which contributes to the observed protection. Importantly, when used in combination with the antimalarialdrug artesunate, ALF492 is an effective adjunctive and adjuvant treatment for ECM, conferring protection after the onset of se-vere disease. This study paves the way for the potential use of CO-RMs, such as ALF492, as adjunctive/adjuvant treatment in se-vere forms of malaria infection.
Malaria remains a devastating global health problem, resultingin up to 800,000 annual deaths (29, 34, 39, 46). Plasmodium
falciparum causes the most severe forms of malaria infection, suchas cerebral malaria (CM) and acute lung injury (ALI) (41). Thecase-fatality rate in severe malaria treated with either artemisininor quinine derivatives remains unacceptably high; CM is amongthe deadliest syndromes, with 13 to 21% mortality, even afterantimalarial treatment (19). In an effort to reduce malaria-relatedmortality, numerous adjunctive/adjuvant therapies complement-ing treatment to an antimalarial therapy have been suggested andtested (21). However, to date, there is still no effective adjunctive/adjuvant therapy for severe malaria.
Animal models have been an indispensable tool for malariaimmunopathogenesis research, drug testing, and vaccine develop-ment. Various mouse-parasite combinations can lead to differentmalaria-associated syndromes. In particular, P. berghei ANKAstrain can induce in rodents distinct malaria pathologies, such ascerebral malaria and acute lung injury (11, 32). The relevance ofthese models to understanding of human malaria has been ques-tioned due to some differences in clinical features between humanand mouse severe malaria, particularly in cerebral malaria. Whilethese models individually may not reflect all the features of specificsyndromes, globally they have been crucial to understanding thepathogenesis of severe malaria (13, 23).
Using the P. berghei ANKA cerebral malaria model, we haveshown that heme oxygenase-1 (HO-1) is a key protective geneagainst the development of CM in mice. Moreover, inhalation ofcarbon monoxide (CO), one of the end products of HO-1 activity,fully prevented CM and malaria-associated ALI (M-AALI) inci-dence in C57BL/6 mice (11, 32). Research conducted in otherexperimental models has further shown that HO-1/CO displaycytoprotective and anti-inflammatory properties that are benefi-cial for the resolution of acute inflammation (17, 24).
CO holds great promise as a therapeutic agent (28, 36). However,
the safety and practicability of the application of CO gas in the clinicremain questionable due to its toxicity and the need for highly con-trolled medical facilities. Thus, CO-releasing molecules (CO-RMs)have been put forward as a valid alternative to CO gas (reviewed inreferences 28 and 36). Among the early-developed and still widelyused CO-RMs in experimental models are the lipid-soluble CORM-2[Ru(CO)3Cl2]2 and the water-soluble CORM-3 [Ru(CO)3Cl2(H2NCH2CO)2] models. Neither CORM-2 nor CORM-3 elevatescarboxyhemoglobin (COHb) levels in blood after in vivo administra-tion (9). Most importantly, substantial protective effects similar tothose observed for CO inhalation have been reported using theseCO-RMs in various experimental models of disease, such as bacterialinfection, vascular dysfunction, and thermal and ischemia-reperfusion injury (1, 9, 22). These CO-RMs lack some key drug-likeproperties, including adequate solubility, stability, and tissue distri-bution after administration, but serve as a proof of concept and offera safer and more efficient modality to deliver CO than CO gas admin-istration.
Building upon this knowledge, we report the design and syn-thesis of a novel CO-RM (ALF492) that displays improved watersolubility, is able to transfer CO to heme proteins, does not inducethe formation of measurable levels of COHb, and distributes invivo with a significant affinity for the liver. The bioactivity and key
Received 23 August 2011 Returned for modification 10 October 2011Accepted 2 December 2011
Published ahead of print 12 December 2011
Address correspondence to Gonçalo J. L. Bernardes, [email protected], or Ana Pamplona, [email protected].
Supplemental material for this article may be found at http://aac.asm.org/.
Copyright © 2012, American Society for Microbiology. All Rights Reserved.
doi:10.1128/AAC.05571-11
0066-4804/12/$12.00 Antimicrobial Agents and Chemotherapy p. 1281–1290 aac.asm.org 1281
on February 15, 2012 by E
TH
-BIB
LIOT
HE
K Z
UR
ICH
http://aac.asm.org/
Dow
nloaded from

therapeutic features of ALF492 are shown in two distinct modelsof severe malaria. Importantly, our data show the use of this com-pound as a promising adjunctive/adjuvant therapy during theacute phase of cerebral malaria.
MATERIALS AND METHODSReagents. For the in vivo studies, artesunate (AS) was obtained from Sigma-Aldrich. CORM-3, fac-Ru(CO)3Cl(NH2CH2COO) or fac-Ru(CO)3Cl(glycinate), was synthesized as described previously (9), and ALF466 [tetrak-is(dimethyl sulfoxide)dichlororuthenium(II)] and tricarbonyldichloro-ruthenium(II) dimer (CORM-2) were purchased from Strem Chemicals,Inc., and Sigma-Aldrich, respectively. �-D-Thiogalactopyranoside was pur-chased from Carbosynth. Hen egg white lysozyme C (HEWL) (7) (ProteinData Bank accession number 3b6l; UniProtKB/Swiss-Prot database accessionnumber P00698; calculated average isotopic mass � 1,4305.1 [43]) was usedas a model protein for the interaction with Ru(CO)3Cl2(Gal-S-Me) (whereGal-S-Me is methylthiogalactopyranoside) and CORM-3.
Synthesis of tricarbonyldichloro(thiogalactopyranoside)rutheni-um(II) (ALF492). Tricarbonyldichlororuthenium(II) dimer (0.52 g, 1.02mmol) was dissolved in anhydrous methanol (30 ml) and transferred viacannula to a solution of �-D-thiogalactopyranoside (0.42 g, 2.04 mmol) inanhydrous methanol (30 ml). A slightly pale yellow clear solution wasformed and stirred for 24 h at room temperature under a nitrogen atmo-sphere. The solution was concentrated, and diethyl ether was added. Thewhite precipitate was filtered, washed with diethyl ether (30 ml, threetimes), and dried. The residue was partially dissolved in diethyl ether andfrozen in liquid nitrogen. The solid was crushed and stirred. A whitepowder formed and was filtered and dried to give tricarbonylchloro(thio-galactopyranoside)ruthenium(II) (348 mg, 73%); the compound wasstored under nitrogen; �max (KBr disc cm�1) 2139 (s, C'O), 2060 (s,C'O) cm�1; �H (400 MHz, D2O) 2.23 (3H, s, CH3), 3.58 to 75 (6H, m),4.38 (1H, d, J � 9.2 Hz, H-1). Found: C, 25.81%; H, 2.94%, S, 6.80%.C10H14Cl2O8RuS requires C, 25.76%; H, 3.03%; S, 6.88%.
Mice. C57BL/6 and DBA-2 wild-type mice were purchased fromCharles River Laboratories International Inc. (Barcelona, Spain) andhoused in the pathogen-free facilities of the Instituto de Medicina Molec-ular (IMM). The animal facility of IMM complies with European Guide-line 86/609/EC and Portuguese law (Portaria 1005/92) and follows theFederation of European Laboratory Animal Science Associations(FELASA) guidelines and recommendations concerning laboratory ani-mal welfare. All animals are bred and/or maintained in accordance withthe European, Portuguese, and institutional guidelines.
Parasites, infection, and disease assessment. We used infected redblood cells (RBCs) with green fluorescent protein (GFP)-transgenic P.berghei ANKA (14) and P. berghei ANKA to infect mice. Cryopreservedparasites were passed once through C57BL/6 or DBA-2 mice, before beingused to infect experimental animals. We infected 6- to 8-week-oldC57BL/6 or DBA-2 mice (sex matched in each experiment) by intraperi-toneal (i.p.) injection of 106 infected RBCs. We monitored the infectedmice daily for clinical symptoms of experimental cerebral malaria (ECM),including hemi- or paraplegia, head deviation, tendency to roll over onstimulation, ataxia and convulsions, and ALI, including dyspnea. Miceshowing severe signs of ECM at day 5, 6, or 7 postinfection (p.i.) and ALIbetween days 7 and 9 were sacrificed. Parasitemia was assessed by flowcytometry, using tail blood, for mice infected with GFP-expressing P.berghei ANKA as previously described (32). Mean parasitemia is ex-pressed as the percentage of infected red blood cells. For mice infectedwith non-GFP-expressing P. berghei, parasitemia was assessed daily bymicroscopic counting of Giemsa-stained thin blood smears. Mean para-sitemia is expressed as the percentage of infected red blood cells. Survivalis expressed as a percentage.
Experimental cerebral malaria clinical assessment. In order to eval-uate the clinical presentation of experimental cerebral malaria, we used aclassification of clinical stages from 0 to 4 (6, 15). Briefly, stage 0 indicatesno detectable clinical symptoms; stage 1, ruffled fur; stage 2, ruffled fur
and unbalancing; stage 3, limb paralysis and respiratory distress; and stage4, convulsions, coma, or death. The mice were clinically classified beforeand after the treatment to evaluate the clinical recovery.
In vivo treatments. ALF466 and ALF492 were solubilized inphosphate-buffered saline (PBS; 1�). The concentration of ALF492 usedin this study was based on previous reports of studies done with CORM-2(8, 9), optimized to our experimental models and considering the differ-ent chemical structures of these compounds. The concentration used forALF492 was 36.7 mg/kg of body weight, administered intravenously (i.v.).ALF466 was given at an equimolar concentration of ALF492. As a vehiclecontrol, we used a solution of PBS administered i.v. Artesunate is pre-sented as a powder of artesunic acid. AS solution was prepared by dissolv-ing 60 mg of anhydrous artesunic acid in 1 ml of sodium bicarbonate (5%)to form sodium artesunate, which was then diluted in 5 ml of NaCl(0.9%). We administrated the AS solution (i.p.) at 40 mg/kg/day, as de-scribed previously (6). We started AS treatment when the infected non-treated mice presented clinical stage 1 for experimental cerebral malaria.
Quantification of COHb in peripheral blood. Blood was collectedfrom the tail of the mice, placed into capillary tubes (VWR) with heparin(100 IU/ml in PBS, 1�; LEO Pharma Inc.), and transferred into AVOXi-meter 4000 cuvettes (ITC), where the levels of COHb, oxyhemoglobin(O2Hb), and methemoglobin (MetHb) were measured in a portableAVOXimeter 4000 CO oximeter (ITC). Results are shown as the meanpercentage of total hemoglobin species in circulation.
Determination of CO in tissues. We quantified CO in different tissuesas previously described (45). Briefly, CO was liberated as gas in a closedvial by adding 25 �l of water and 5 �l of sulfosalicylic acid (SSA; 30%[wt/vol]) to 30 �l of diluted sample, after being homogenized. The vialswere incubated on ice for at least 10 min before being analyzed. The gas inthe headspace of the vials was analyzed quantitatively with a gas chro-matograph (GC) equipped with a reducing-compound photometry(RCP) detector (Peak Laboratories, Mountain View, CA), which allowsthe CO in gas to be quantified to concentrations as low as 1 to 2 ppb. Theamount of CO was calculated using a calibration curve prepared from COstandards.
Quantification of Ru in tissues. After they were weighed, the tissuesamples were dried at 80°C overnight and then for more than 2 h at 120°C.The dried tissues were then digested by the addition of 2 ml of tetraethyl-ammonium hydroxide solution (20% [wt/wt] in water; Sigma-Aldrich, St.Louis, MO) for 24 h. After complete tissue digestion, 1 ml of water wasadded. The Ru content was analyzed by an inductively coupled plasma-atomic emission spectrometer (ICP-AES; model Ultima; Horiba JobinYvon, Longjumeau, France) using an external Ru standard method.
BBB permeability. We assessed the blood-brain barrier (BBB) perme-ability using Evan’s blue (EB) dye as described before (40). Briefly, micewere injected i.v. with 0.2 ml of PBS (1�)–2% EB (Sigma) when clinicalsymptoms of ECM were observed in mice treated with dimethyl sulfoxidevehicle solution. Mice were killed 1 h after EB injection and perfused with20 ml of PBS (1�), and the brains were weighed and placed in 2 mlformamide (Merck) for 24 h at 37°C to extract EB dye from the tissue.Absorbance was measured at a � of 620 nm (EB absorbance) in a DU 70spectrophotometer (Beckman). The concentration was calculated using astandard curve of EB dye. The data are expressed as mg of Evans blue perg of brain tissue.
Histology. For evaluation of histological features, when infected con-trol mice presented signs of ECM or ALI, the mice were deeply anesthe-tized until cessation of breathing. Livers, lungs, and brains were removedand fixed in 10% buffered formalin for 24 to 72 h. Four-micrometersections were cut from paraffin-embedded tissues and stained with hema-toxylin and eosin according to standard procedures. Histological analyseswere performed on a Leica DM 2500 microscope (Leica Microsystems).
VEGF protein level determination. Mouse vascular endothelialgrowth factor (VEGF) in plasma samples was determined using a com-mercial enzyme-linked immunosorbent assay kit (R&D Systems) follow-
Pena et al.
1282 aac.asm.org Antimicrobial Agents and Chemotherapy
on February 15, 2012 by E
TH
-BIB
LIOT
HE
K Z
UR
ICH
http://aac.asm.org/
Dow
nloaded from

ing the manufacturer’s instructions. The P. berghei ANKA-infectedDBA-2 mice were clinically classified after determining the cause of death.
Quantitative real-time RT-PCR. Mice were sacrificed, when infectedcontrol mice presented signs of ECM, and perfused intracardially withPBS to remove circulating RBCs and leukocytes from the organs. RNA wasisolated from brains, livers, and lungs using the TRIzol reagent (Invitro-gen, Life Technologies), according to the manufacturer’s recommenda-tion. The synthesis of the first-strand cDNA from the RNA templates wascarried out using a Transcriptor first-strand cDNA synthesis kit (Roche).Reverse transcription PCRs (RT-PCRs) were performed in the presence ofSYBR green (SYBR green PCR master mix; Applied Biosystems) on an ABIPrism 7500Fast apparatus (Applied Biosystems). The indicated oligonu-cleotides were used for the specific amplification of the genes for hypo-xanthine phosphoribosyltransferase (HPRT; 5=-GTTGGATACAGGCCAGACTTTGTTG-3= [forward] and 5=-GATTCAACCTTGCGCTCATCTTAGGC-3= [reverse]), HO-1 (5=-TCTCAGGGGGTCAGGTC-3= [forward]and 5=-GGAGCGGTGTCTGGGATG-3= [reverse]), P. berghei 18S rRNA(5=-AAGCATTAAATAAAGCGAATACATCCTTAC-3= [forward] and5=-GGAGATTGGTTTTGACGTTTATGTG-3= [reverse]), CD8 � chain(5=-TGCTCGAGATGTGATGAAGG-3= [forward] and 5=-TCCCCTGTTGACTGGTCATT-3= [reverse]), gamma interferon (IFN-�; 5=-CACACTGCATCTTGGCTTTG-3= [forward] and 5=-TCTGGCTCTGCAGGATTTTC-3= [reverse]); and intercellular adhesion molecule 1 (ICAM-1; 5=-CGAAGGTGGTTCTTCTGAGC-3= [forward] and 5=-GTCTGCTGAGACCCCTCTTG-3= [reverse]). The relative changes in gene expressionbetween experimental and control groups were calculated by the Pfafflmethod using the gene for HPRT as an internal control.
Assessment of antimalarial activity in vitro. Quantitative assessmentof the in vitro antimalarial activity of ALF466 and ALF492 was performedon P. berghei ANKA expressing GFP and P. falciparum 3D7 parasites, aspreviously described (38). Briefly, the drugs were dispensed in duplicateinto a 96-well plate with a series of 5-fold dilutions, with the highestconcentration being the one used in vivo (1.25 mM). Chloroquine (CQ)and PBS were used as background and baseline controls. The parasitesuspension was added to the plates and then incubated at 37°C for 24 h or48 h. P. berghei ANKA parasites expressing GFP were collected from in-fected mice (1 to 3% parasitemia). The infected blood was washed inculture medium (RPMI 1640 medium containing L-glutamine supple-mented with 25% [vol/vol] fetal bovine serum, 25 mM HEPES, 0.05mg/ml gentamicin) and added to the plates at a 1% final hematocrit. Theplates were incubated for 24 h at 37°C and examined by flow cytometry(laser, 488 nm; FACSCalibur flow cytometer) and microscopy afterGiemsa staining. Cultures of P. falciparum 3D7 parasites (in RPMI 1640medium containing L-glutamine supplemented with 0.5% [wt/vol] Albu-MAX II lipid-rich bovine serum albumin, 25 mM HEPES, 0.05 mg/mlgentamicin) were added to the plates at 0.5% final parasitemia and 4%final hematocrit. The plates were incubated for 48 h at 37°C and stored at�20°C. After thawing at room temperature, the detection reagent, con-sisting of SYBR green in lysis buffer (Tris HCl, EDTA, saponin, and Tri-ton), was dispensed into the assay plates, and the plates were incubated for1 h at 37°C and examined at 485 nm of excitation and 530 nm of emissionwith a gain setting equal to 100 using a fluorescence plate reader (Tecan).
Statistical analysis. For samples in which n was �5, statistical analyseswere performed using the unpaired Student’s t test, and when n was �5,statistical analyses were performed using the Mann-Whitney U test. Sur-vival curves were compared using the log-rank test and the Gehan-Breslow-Wilcoxon test. A P value of �0.05 was considered significant.
RESULTSA novel CO-releasing molecule (ALF492) protects mice fromECM without COHb formation. We designed and synthesized anovel water-soluble CO-RM, tricarbonyldichloro(methylthio-galactopyranoside)Ru(II) [Ru(CO)3Cl2(Gal-S-Me); ALF492],through the reaction of CORM-2 with Gal-S-Me (see the supple-mental material) (Fig. 1a). This Ru tricarbonyl complex features a
galactose (Gal)-derived ligand coordinated to the Ru center via athioether linkage to enhance water solubility and biocompatibility(5, 16, 25, 47). The presence of the Gal ligand may also confer acertain degree of specificity by targeting asialoglycoprotein recep-tors in the liver (5, 23, 45).
The biodistribution of ALF492 in tissues was assessed by quan-tifying the levels of Ru and CO (45) in the host. Liver, kidney,spleen, lung, and brain tissues of noninfected (NI) mice treated for2 days with ALF492 and ALF466 twice daily were analyzed 1 h afterthe last administration (Fig. 1b and c). In ALF492-treated mice,Ru could be detected in all organs analyzed and had a markedaffinity for the liver (Fig. 1b). In fact, the concentration of Rumeasured in the liver of ALF492-treated mice was approximately7.0 � 0.3 times higher than that measured in the liver of ALF466-treated mice (P � 0.05) (Fig. 1b). The brain was the organ wherethe levels of Ru were lower (401.8 � 17.3 times less than in theliver), suggesting a low potential for neurotoxicity (Fig. 1b). Thelevels of Ru in ALF492-treated mice were significantly higher thanthose in ALF466-treated mice for all the organs analyzed (P �0.05) (Fig. 1b), implying a lower rate of excretion of ALF492 or itsRu-containing metabolites. In addition, the measured levels ofCO for ALF466- and ALF492-treated mice were found to be sig-nificantly different in the liver and spleen, which showed the high-est levels of CO (P � 0.05) (Fig. 1c).
To assess the reactivity of ALF492 in the presence of proteins,we used the hen egg white lysozyme (HEWL) as a model protein(35). We foresaw that ALF492 could react with serum proteins ina similar manner as previously observed for CORM-3 (36) andform adducts from which CO may then be released and exert itsprotective effects. Indeed, ALF492 in the presence of HEWL formsprotein-Ru(II)(CO)2 adducts but reacts slower than CORM-3(36) (see the supplemental material). We then evaluated the COdonation capacity of ALF492 by a deoxy-myoglobin (Mb) car-bonylation assay (9). We observed that ALF492 transfers approx-imately 1 equivalent of CO to Mb after 15 min of incubation withdeoxy-Mb, as seen for Ru tricarbonyl CORM-3 (see the supple-mental material). Altogether, these data demonstrate that ALF492is capable of transferring CO to the heme of Mb, reacts with pro-teins to form protein-Ru(II)(CO)2 adducts, and preferably dis-tributes to the liver.
We next evaluated the potential therapeutic effect of ALF492 inECM. To this end, we treated P. berghei ANKA-infected C57BL/6mice twice daily on days 2 and 3 after infection with ALF492.ALF492 treatment protected 100% of P. berghei ANKA-infectedC57BL/6 mice from developing ECM, in contrast to infected con-trol and ALF466-treated mice, which died with ECM symptomsbetween days 6 and 8 after infection (P � 0.0001) (Fig. 1d). Asmall but significant arrest in parasitemia in ALF492-treated micewas also observed between days 5 and 7 after infection (P � 0.01)(Fig. 1e). This led us to investigate whether ALF492 could have adirect antiparasitic effect on P. berghei ANKA and P. falciparumparasites. To this end, we monitored the effect of ALF492 and theantimalarial CQ on the in vitro replication of the P. falciparum3D7 isolate and P. berghei ANKA for 48 h and 24 h. ALF492showed 50% inhibitory concentrations (IC50s) remarkably higherthan those of CQ: approximately 5,600- and 350-fold higher in P.falciparum 3D7 and P. berghei ANKA parasites, respectively (seeFig. S1A and B in the supplemental material). The IC50 for ALF466was not determined due to the absence of an inhibitory effect overthe range of concentrations tested. Altogether, these results show
CO-Releasing Molecule Protects from Severe Malaria
March 2012 Volume 56 Number 3 aac.asm.org 1283
on February 15, 2012 by E
TH
-BIB
LIOT
HE
K Z
UR
ICH
http://aac.asm.org/
Dow
nloaded from
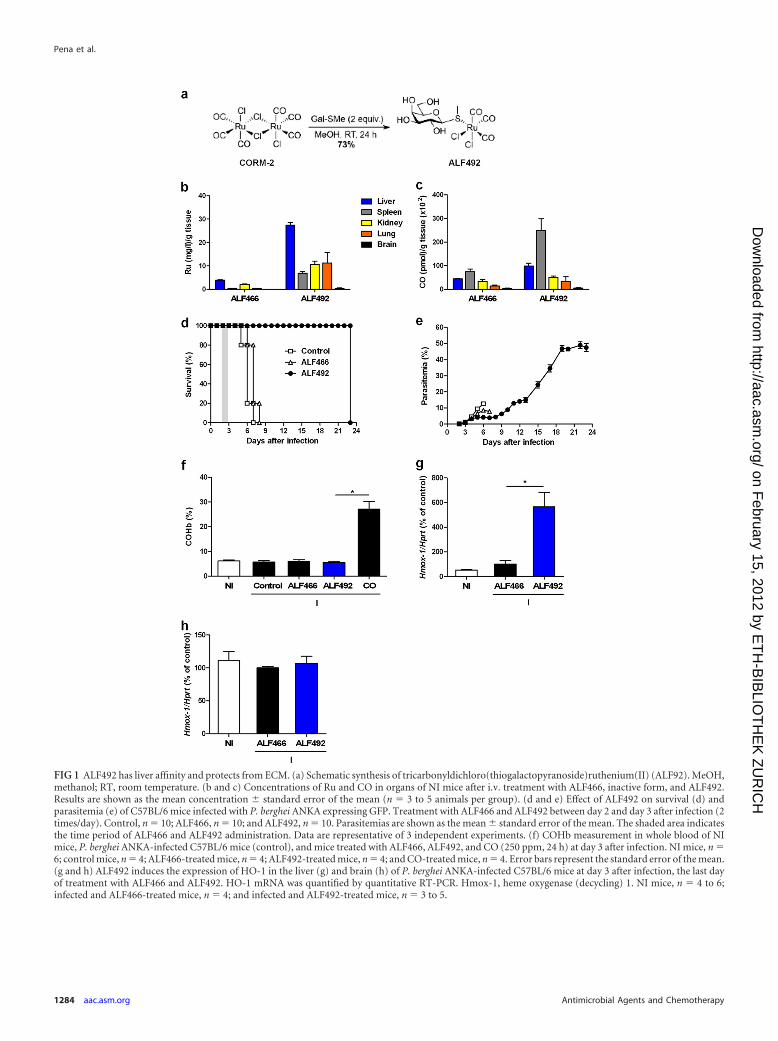
FIG 1 ALF492 has liver affinity and protects from ECM. (a) Schematic synthesis of tricarbonyldichloro(thiogalactopyranoside)ruthenium(II) (ALF92). MeOH,methanol; RT, room temperature. (b and c) Concentrations of Ru and CO in organs of NI mice after i.v. treatment with ALF466, inactive form, and ALF492.Results are shown as the mean concentration � standard error of the mean (n � 3 to 5 animals per group). (d and e) Effect of ALF492 on survival (d) andparasitemia (e) of C57BL/6 mice infected with P. berghei ANKA expressing GFP. Treatment with ALF466 and ALF492 between day 2 and day 3 after infection (2times/day). Control, n � 10; ALF466, n � 10; and ALF492, n � 10. Parasitemias are shown as the mean � standard error of the mean. The shaded area indicatesthe time period of ALF466 and ALF492 administration. Data are representative of 3 independent experiments. (f) COHb measurement in whole blood of NImice, P. berghei ANKA-infected C57BL/6 mice (control), and mice treated with ALF466, ALF492, and CO (250 ppm, 24 h) at day 3 after infection. NI mice, n �6; control mice, n � 4; ALF466-treated mice, n � 4; ALF492-treated mice, n � 4; and CO-treated mice, n � 4. Error bars represent the standard error of the mean.(g and h) ALF492 induces the expression of HO-1 in the liver (g) and brain (h) of P. berghei ANKA-infected C57BL/6 mice at day 3 after infection, the last dayof treatment with ALF466 and ALF492. HO-1 mRNA was quantified by quantitative RT-PCR. Hmox-1, heme oxygenase (decycling) 1. NI mice, n � 4 to 6;infected and ALF466-treated mice, n � 4; and infected and ALF492-treated mice, n � 3 to 5.
Pena et al.
1284 aac.asm.org Antimicrobial Agents and Chemotherapy
on February 15, 2012 by E
TH
-BIB
LIOT
HE
K Z
UR
ICH
http://aac.asm.org/
Dow
nloaded from

that the therapeutic administration of ALF492, while not havingan antiparasitic effect, has a significant impact on the overall out-come of the infection (Fig. 1d). CO inhalation at a dose necessaryto obtain similar protective effects for ECM (250 ppm for 24 h,starting at day 2 after infection) induced 30.3% � 2% COHbformation (P � 0.05), which is an unacceptably high level forhumans. Remarkably, ALF492 fully protected mice against ECMwithout causing a measurable increase in COHb levels in theblood. The COHb levels in ALF492-treated mice were similar tothose observed for NI, infected control, and ALF466-treated mice(Fig. 1f). Moreover, treatment with ALF492 per se does not in-crease hepatotoxicity or oxidative stress, as levels of alanine ami-notransferase and glutathione were comparable to those of theinfected controls (data not shown).
Taken together, these data demonstrate that ALF492 fully pro-tects mice from ECM onset without affecting oxygen transport byhemoglobin, thereby overcoming the main adverse effect of COgas therapy.
ALF492 induces expression of HO-1. In a previous report, wehave shown that HO-1 induction reduced CM incidence in P.berghei ANKA-infected C57BL/6 mice (32). ALF492 distributespreferentially to the liver, which is considered a mediator of sys-temic and local innate immunity and has been implicated in theregulation of genes that contribute to the control of inflammation,such as HO-1 (31). We asked whether ALF492 could modulate theexpression of HO-1 and thus contribute to the observed protec-tion against ECM. Expression of HO-1 mRNA was significantlyupregulated in the livers of P. berghei ANKA-infected ALF492-treated C57BL/6 mice compared to noninfected and infectedALF466-treated mice (11.1 � 2.3-fold and 5.7 � 1.2-fold, respec-tively; P � 0.05) (Fig. 1g). Moreover, the expression of HO-1mRNA in the brains of infected ALF492-treated mice at day 3 afterinfection was not significantly different from that in the brains ofnoninfected and infected ALF466-treated mice (Fig. 1h). Theseresults show that treatment with ALF492 induced the upregula-tion of HO-1 expression in the livers of infected mice, thus con-tributing to the control of the systemic inflammatory response ofthe host to P. berghei ANKA infection.
ALF492 prevents BBB disruption and neuroinflammation.BBB disruption is a hallmark of ECM (40) and has been reportedin human CM (26, 40). P. berghei ANKA-infected nontreated andALF466-treated C57BL/6 mice showed BBB disruption (Fig. 2a;see Fig. S2 in the supplemental material), as measured by Evansblue accumulation in brain parenchyma, i.e., 4.3- and 6.7-foldincreases, respectively, compared to NI mice (P � 0.01). ALF492-treated mice did not show any evidence of BBB disruption, as thelevels of Evans blue accumulation were similar to those in NI mice(Fig. 2a; see Fig. S2 in the supplemental material). Inhibition ofBBB disruption is known to contribute to the suppression of ECMdevelopment (12, 40). Furthermore, several reports have un-equivocally demonstrated that the development of ECM in P. ber-ghei ANKA-infected mice is dependent on the recruitment of Tcells, mainly CD8� T cells (3, 4). More recently, it was shown thataccumulation of CD8� T cells in the brain is not sufficient for thedevelopment of ECM in C57BL/6 mice, but the concomitant pres-ence of parasitized red blood cells (pRBCs) is necessary for thepathology onset (2). Both pRBC accumulation and CD8-�-chainmRNA expression in the brain were significantly lower inALF492-treated mice than ALF466-treated mice, which showedclear signs of CM (P � 0.01) (Fig. 2b and c). During ECM, proin-
flammatory cytokines, such as IFN-�, and adhesion molecules,such as ICAM-1, are upregulated and play a decisive role in thepathogenesis of ECM (10, 12, 33). Importantly, treatment withALF492 reduced IFN-� mRNA expression compared to that ininfected ALF466-treated mice (P � 0.01) (Fig. 2d) and decreasedICAM-1 expression 2-fold (P � 0.01, ALF492-treated versusALF466-treated mice), when assessed at day 6 after infection(Fig. 2e).
ALF492 treatment also prevented the neuropathologic featuresassociated with ECM (2, 3, 30). Brains from infected and ALF466-treated P. berghei ANKA-infected mice showed evidence of micro-vascular congestion with pRBCs and leukocytes and hemorrhagicfoci. In contrast, ALF492-treated infected mice showed less hem-orrhage, and the vessels had lower levels of accumulation ofpRBCs and leukocytes (Fig. 2f to i). Overall, these results show thatALF492 treatment prevents BBB permeability and decreases con-gestion, hemorrhage, and neuroinflammation in the brains of in-fected mice.
ALF492 protects mice from developing malaria-associatedALI. We evaluated the protective effect of ALF492 in a model ofmalaria-associated acute lung injury (M-AALI) (11). The M-AALImodel, based on the infection of DBA-2 mice with P. bergheiANKA, is characterized by dyspnea, airway obstruction, hypox-emia, pulmonary exudate, and elevated VEGF levels in plasma,followed by death between days 7 and 12 after infection (11).None of P. berghei ANKA-infected DBA-2 mice treated twice dailywith ALF492 between days 2 and 4 after infection developedM-AALI. In the control groups of nontreated and ALF466-treatedinfected mice, 83% and 67% of the mice, respectively, died, dis-playing M-AALI symptoms such as dyspnea, respiratory insuffi-ciency (as first symptoms) (Fig. 3a), and pulmonary exudate whenthey were analyzed postmortem. Moreover, VEGF levels were sig-nificantly lower in infected mice treated with ALF492 (P � 0.001;Fig. 3b) (11). Histological examination of lung tissue from in-fected and untreated mice and infected and ALF466- and ALF492-treated mice showed significant differences in vascular congestionwith pRBCs (Fig. 3c to e). In sum, our data show that treatmentwith ALF492 significantly improves the infection outcome in theM-AALI model.
The data presented above show that treatment with ALF492protects P. berghei ANKA-infected mice from death caused byECM and M-AALI when administered before symptoms of dis-ease were observed. However, to be clinically relevant for humans,ALF492 should show therapeutic activity after the onset of disease,either alone or in combination with antimalarial drugs. Thus, wedecided to test ALF492 as an adjunctive and adjuvant therapyduring the acute phase of ECM. Artesunate (AS) is the primarytreatment in severe malaria, is generally effective in controlling P.falciparum parasitemia, and has previously been used to treat P.berghei ANKA-infected mice (6, 37, 44). Therefore, we assessedthe effect of the combination of ALF492 and AS on parasite clear-ance and clinical recovery from ECM. We tested two AS andALF492 combinations after the onset of CM on day 5 after infec-tion: (i) adjunctive therapy, in which AS and ALF492 were admin-istered at the same time for 2 days (day 5 [d5] and d6 after infec-tion), followed by a 3-day treatment (d7 to d9 after infection) withjust ALF492, or (ii) adjuvant therapy, in which AS was adminis-tered alone on the first 2 days (d5 and d6 after infection), followedby ALF492 administration for 3 more days (d7 to d9 after infec-tion). We started the AS treatment when the infected mice (con-
CO-Releasing Molecule Protects from Severe Malaria
March 2012 Volume 56 Number 3 aac.asm.org 1285
on February 15, 2012 by E
TH
-BIB
LIOT
HE
K Z
UR
ICH
http://aac.asm.org/
Dow
nloaded from

trol) showed the initial stage of ECM (ECM score, 1) (Fig. 4c). Allinfected nontreated mice died of ECM by day 6 after infection(Fig. 4a and c). The effect of antimalarial treatment with AS alonewas shown by the decrease of parasitemia from 6.8% � 0.3% to0.59% � 0.06% (at days 5 and 9 postinfection, respectively). AS
treatment alone delayed, but in most cases did not prevent, deathby CM (Fig. 4b and c). Nine out of 11 (82%) AS-treated mice diedwith ECM between days 12 and 13 after infection (Fig. 4a). Micetreated simultaneously with AS and ALF492 under the adjunctiveprotocol showed a significant increase in survival (83%) com-
FIG 2 ALF492 reduces parasite accumulation in the brain and neuroinflammation. BBB permeability (a), parasite 18S rRNA (b), CD8-� (c), IFN-� (d), andICAM-1 (e) mRNA expression were quantified by qRT-PCR. NI mice, n � 4; infected and ALF466-treated mice, n � 4; and infected and ALF492-treated mice,n � 5. Evans blue quantification is shown as the mean �g of EB per g of brain tissue � standard error of the mean. NI mice, n � 4; ALF466-treated mice, n � 5;and ALF492-treated mice, n � 5. NI mice, infected and ALF466-treated mice, and ALF492-treated mice were sacrificed when the control group, ALF466-treatedmice, showed signs of ECM, and brains were harvested after intracardiac perfusion. (f to i) Semiquantification of histological findings in hematoxylin andeosin-stained brain sections analyzed at the same time that the data for panels a to e were analyzed, using a blinded score system. Dot plots compare the numberof animals assigned severity scores from 1 (less severe) to 3 (most severe) in infected, infected and ALF466-treated, and ALF92-treated mice. Images arerepresentative of 3 to 8 mice. Bar, 100 �m.
Pena et al.
1286 aac.asm.org Antimicrobial Agents and Chemotherapy
on February 15, 2012 by E
TH
-BIB
LIOT
HE
K Z
UR
ICH
http://aac.asm.org/
Dow
nloaded from

pared with the AS-treated group (18%) (P � 0.01) (Fig. 4a). Theinfected group treated with the AS and ALF492 combination un-der the adjuvant protocol showed an improved survival fromECM of 67% (P � 0.01 versus AS-treated mice) (Fig. 4a). Since noantimalarial agents were administered after day 7, mice that didnot have ECM still developed hyperparasitemia and anemia(�30% parasitemia) and were sacrificed 3 weeks after infection(Fig. 4a). During the administration of AS, between days 5 and 6,ALF492 did not interfere with the action of AS in vivo (Fig. 4b).These results show that this combination therapy (AS-ALF492)used as an adjunctive/adjuvant therapy after the onset of ECM cansignificantly improve survival compared to that for AS-treatedmice.
DISCUSSION
We have previously shown that administration by inhalation of thepotent anti-inflammatory molecule CO suppresses the pathogenesisof CM and M-AALI in mice (11, 30, 32). In this study, we report thedevelopment of a novel, water-soluble, drug-like CO-RM, tricarbon-yldichloro(thiogalactopyranoside)Ru(II) (ALF492), its protective ef-fect on CM and M-AALI, and its effect as an adjunctive/adjuvantagent when used in combination with the antimalarial AS for thetreatment of ECM.
By changing the nature of the ligand in the Ru core scaffold andmetal-ligand linkage, we succeeded in synthesizing an alternativeCO-RM, ALF492, which is water soluble and capable of transferring
FIG 3 ALF492 protects mice from M-AALI. (a) Survival of P. berghei ANKA-infected DBA-2 mice receiving no treatment or treated i.v. with ALF466 and ALF492between day 2 and day 3 after infection (2 times/day). Infected mice, n � 5; infected and ALF466-treated mice, n � 9; infected and ALF492-treated mice, n � 7.Parasitemias are shown as the mean � standard error of the mean. (b) Levels of VEGF protein in the plasma of P. berghei ANKA-infected DBA-2 mice with ALIsymptoms, ALF466-treated mice, and ALF492-treated mice compared to NI mice. NI mice, n � 3, infected mice with ALI, n � 3; infected and ALF466-treatedmice, n � 5; and infected and ALF492-treated mice, n � 5. Results are shown as the mean concentration � standard error of the mean. (c to e) Semiquantificationof the histological findings in hematoxylin and eosin-stained lung sections, analyzed at the same time that the data for panel b were analyzed, using a blinded scoresystem. Dot plots show the number of infected (ALI), infected and ALF466-treated (ALI), and ALF492-treated mice assigned severity scores from 1 (less severe)to 3 (most severe). Images are representative of 4 to 6 mice. Bar, 100 �m.
CO-Releasing Molecule Protects from Severe Malaria
March 2012 Volume 56 Number 3 aac.asm.org 1287
on February 15, 2012 by E
TH
-BIB
LIOT
HE
K Z
UR
ICH
http://aac.asm.org/
Dow
nloaded from

CO to heme proteins and protects mice from death caused by severemalaria. In contrast, the known water-soluble CORM-3 was less ac-tive and could protect only 50% of mice from ECM (data not shown).CO inhalation suppressed the pathogenesis of CM and M-AALI inmice but produced unacceptable levels of COHb (11, 32). COHb isroutinely used to assess CO toxicity in humans, and transient levels of10% or less are considered safe (28). In contrast, we showed thatALF492, at therapeutic concentrations, did not induce the formationof measurable levels of COHb while preserving the protective effectsseen with inhaled CO (32).
This work also provides evidence that ALF492 treatment in theECM model induces the expression of HO-1. We have previouslyshown that the induction of HO-1 protected mice from develop-ing ECM (32). Additionally, in a model of chronic intestinal in-flammation, it was shown that CO could ameliorate chronic mu-rine colitis through an HO-1-dependent pathway (18). Our datastrongly suggest that HO-1 mediates a significant component ofthe anti-inflammatory action of ALF492 in ECM. ECM is charac-terized not only by an exacerbated parasite-mediated inflamma-
tory response but also by pRBCs, unparasitized RBCs sequesteredin the microvasculature of the brain, and coagulation and micro-circulation dysfunction (42).
Despite the introduction of new antimalarial agents, such asartemisinin derivatives (e.g., artesunate), these drugs take at least12 to 18 h to kill parasites (27). Still, deaths from severe malariamay occur within the first 24 h after hospital admission (19). Thus,administration of adjunctive therapies in the first 24 h is critical toreduce mortality. Children who survive the acute episode of CMoften have long-term cognitive (�25%) and neurological (1.1 to4.4%) deficits (19, 41). The use of adjunctive therapies that wouldreduce neurological injury may prove essential to reduce this bur-den. A series of adjunctive therapies such as anti-tumor necrosisfactor alpha agents, iron chelators (such as desferrioxamine), anddichloroacetate (which stimulates pyruvate dehydrogenase and soreduces lactate) has been assessed in randomized clinical trials(reviewed in reference 21).
ALF492 is an effective adjunctive and adjuvant agent whenused in combination with the antimalarial AS for the treatment of
FIG 4 ALF492 is a potential adjunctive/adjuvant therapy for ECM. Survival (a) and parasitemia (b) of C57BL/6 mice infected with P. berghei ANKA expressingGFP, treated with AS (d5 to d6) (AS), AS (d5 to d6) and ALF492 (d5 to d9) (AS � ALF492), or AS (d5 to d6) and ALF492 (d8 to d9) (AS ¡ ALF492) are shown.Survival was monitored over a 24-day period. Data are representative of 2 independent experiments. The treatment with AS started when the infected mice(control) showed a score of 1 (ruffled fur), the initial stage of ECM. Overall survival was significantly improved by ALF492 treatment (P � 0.01). Data representthe mean � standard error of the mean. (c) Each ECM clinical stage (no detectable symptoms, ruffled fur, ruffled fur and motor impairment, respiratory distress,and convulsions and/or coma) was given a score (0, 1, 2, 3, and 4). Mice were graphically ranked on the basis of symptoms presented after day 5 of infection.
Pena et al.
1288 aac.asm.org Antimicrobial Agents and Chemotherapy
on February 15, 2012 by E
TH
-BIB
LIOT
HE
K Z
UR
ICH
http://aac.asm.org/
Dow
nloaded from

ECM. Furthermore, treatment with ALF492 protected mice fromM-AALI, decreasing significantly the levels of VEGF in the circu-lation. Therefore, we should not exclude the possibility thatALF492 may also be used as an adjunctive/adjuvant therapy in thispathology, since the only treatments shown to improve survivaland reduce mortality for patients with ALI have been supportivecare strategies (20).
In summary, the present study discloses the anti-inflammatoryand cytoprotective effects of a novel Ru tricarbonyl CO-RM inECM and M-AALI models. This study highlights the therapeuticpotential of ALF492 in targeting pharmacologically the expressionof HO-1, which has been shown to play crucial cytoprotective,immunomodulatory, and anti-inflammatory roles. Our workclearly demonstrates that CO delivered from a Ru tricarbonylwater-soluble molecule can induce protection similar to that seenwith CO gas therapy, but without its toxic effects (elevated COHblevels). Altogether, this work represents an important preclinicalproof of principle for CO-RMs as a new class of drugs to treatsevere forms of malaria. Thus, we should stress that transfer ofknowledge on the cytoprotective role of ALF492 or other CO-RMsto a potential therapeutic application in human severe malariashould be made with the adequate cautions.
ACKNOWLEDGMENTS
We thank Bruno Silva-Santos (Molecular Immunology Unit [IMM]) forlaboratory infrastructure, scientific advice, and critical review of the man-uscript; Dario Neri (ETH Zürich) for collaborative support (mass spec-trometry facilities); Walter Blätter (Alfama Inc.) and Filipa da Cruz(IMM) for critical review of the manuscript; Andreia Nascimento Pinto(Instituto de Histologia e Biologia do Desenvolvimento, Faculdade deMedicina de Lisboa) for help in histopathology studies; Carla Rodrigues(REQUIMTE) for the ICP-AES Ru quantification; and the IMM animalfacility for animal care.
This work was supported by project grant PTDC/SAU-MII/64125/2006 (FCT) to A.P. and Alfama Inc. A.P. holds a Ciência 2008 position ofthe Portuguese Ministry of Science and Technology. We thank FCT(IMM/BI/47-2008 to A.C.P.; SFRH/BI/51054/2010 to R.N.) and EMBO(ALTF 960-2009 to L.M.-S.) for the award of fellowships. The NMR spec-trometers are part of the National NMR Network and were purchased inthe framework of the National Program for Scientific Re-equipment, con-tract REDE/1517/RMN/2005, with funds from POCI 2010 (FEDER) andthe Fundação para a Ciência e a Tecnologia (FCT).
The work from this paper has been filed by Alfama Inc. as a patentapplication for the use of ruthenium tricarbonyl carbon monoxide-releasing molecules (CO-RMs) to treat severe forms of malaria.
REFERENCES1. Alcaraz MJ, Guillen MI, Ferrandiz ML, Megias J, Motterlini R. 2008.
Carbon monoxide-releasing molecules: a pharmacological expedient tocounteract inflammation. Curr. Pharm. Des. 14:465– 472.
2. Baptista FG, et al. 2010. Accumulation of Plasmodium berghei-infectedred blood cells in the brain is crucial for the development of cerebralmalaria in mice. Infect. Immun. 78:4033– 4039.
3. Belnoue E, et al. 2002. On the pathogenic role of brain-sequesteredalphabeta CD8� T cells in experimental cerebral malaria. J. Immunol.169:6369 – 6375.
4. Berendt AR, Tumer GD, Newbold CI. 1994. Cerebral malaria: the se-questration hypothesis. Parasitol. Today 10:412– 414.
5. Bernardes GJL, et al. 2010. Design, synthesis and biological evaluation ofcarbohydrate-functionalized cyclodextrins and liposomes for hepatocyte-specific targeting. Org. Biomol. Chem. 8:4987– 4996.
6. Bienvenu AL, Ferrandiz J, Kaiser K, Latour C, Picot S. 2008. Artesunate-erythropoietin combination for murine cerebral malaria treatment. ActaTrop. 106:104 –108.
7. Canfield RE. 1963. The amino acid sequence of egg white lysozyme. J.Biol. Chem. 238:2698 –2707.
8. Chen B, et al. 2009. Carbon monoxide rescues heme oxygenase-1-deficient mice from arterial thrombosis in allogeneic aortic transplanta-tion. Am. J. Pathol. 175:422– 429.
9. Clark JE, et al. 2003. Cardioprotective actions by a water-soluble carbonmonoxide-releasing molecule. Circ. Res. 93:e2– e8.
10. de Kossodo S, Grau GE. 1993. Profiles of cytokine production in relationwith susceptibility to cerebral malaria. J. Immunol. 151:4811– 4820.
11. Epiphanio S, et al. 2010. VEGF promotes malaria-associated acute lunginjury in mice. PLoS Pathog. 6:e1000916.
12. Favre N, et al. 1999. Role of ICAM-1 (CD54) in the development ofmurine cerebral malaria. Microbes Infect. 1:961–968.
13. Franke-Fayard B, Fonager J, Braks A, Khan SM, Janse CJ. 2010. Seques-tration and tissue accumulation of human malaria parasites: can we learnanything from rodent models of malaria? PLoS Pathog. 6:e1001032.
14. Franke-Fayard B, et al. 2004. A Plasmodium berghei reference line thatconstitutively expresses GFP at a high level throughout the complete lifecycle. Mol. Biochem. Parasitol. 137:23–33.
15. Franklin BS, et al. 2011. Therapeutical targeting of nucleic acid-sensingToll-like receptors prevents experimental cerebral malaria. Proc. Natl.Acad. Sci. U. S. A. 108:3689 –3694.
16. Gottschaldt M, Schubert US. 2009. Prospects of metal complexes periph-erally substituted with sugars in biomedicinal applications. Chem. Eur. J.15:1548 –1557.
17. Hayashi S, et al. 1999. Induction of heme oxygenase-1 suppresses venularleukocyte adhesion elicited by oxidative stress: role of bilirubin generatedby the enzyme. Circ. Res. 85:663– 671.
18. Hegazi RA, et al. 2005. Carbon monoxide ameliorates chronic murinecolitis through a heme oxygenase 1-dependent pathway. J. Exp. Med. 202:1703–1713.
19. Idro R, Jenkins NE, Newton CR. 2005. Pathogenesis, clinical features,and neurological outcome of cerebral malaria. Lancet Neurol. 4:827– 840.
20. Jain R, DalNogare A. 2006. Pharmacological therapy for acute respiratorydistress syndrome. Mayo Clin. Proc. 81:205–212.
21. John CC, Kutamba E, Mugarura K, Opoka RO. 2010. Adjunctive ther-apy for cerebral malaria and other severe forms of Plasmodium falciparummalaria. Expert Rev. Anti Infect. Ther. 8:997–1008.
22. Kim HP, Ryter SW, Choi AM. 2006. CO as a cellular signaling molecule.Annu. Rev. Pharmacol. Toxicol. 46:411– 449.
23. Langhorne J, et al. 2011. The relevance of non-human primate androdent malaria models for humans. Malar. J. 10:23.
24. Lee TS, Chau LY. 2002. Heme oxygenase-1 mediates the anti-inflam-matory effect of interleukin-10 in mice. Nat. Med. 8:240 –246.
25. Lunney J, Ashwell G. 1976. A hepatic receptor of avian origin capable ofbinding specifically modified glycoproteins. Proc. Natl. Acad. Sci. U. S. A.73:341–343.
26. Medana IM, Turner GD. 2006. Human cerebral malaria and the blood-brain barrier. Int. J. Parasitol. 36:555–568.
27. Mishra SK, Newton CR. 2009. Diagnosis and management of the neuro-logical complications of falciparum malaria. Nat. Rev. Neurol. 5:189 –198.
28. Motterlini R, Otterbein LE. 2010. The therapeutic potential of carbonmonoxide. Nat. Rev. Drug Discov. 9:728 –743.
29. Mwangi TW, Ross A, Snow RW, Marsh K. 2005. Case definitions ofclinical malaria under different transmission conditions in Kilifi District,Kenya. J. Infect. Dis. 191:1932–1939.
30. Neill AL, Hunt NH. 1992. Pathology of fatal and resolving Plasmodiumberghei cerebral malaria in mice. Parasitology 105(Pt 2):165–175.
31. Nemeth E, Baird AW, O’Farrelly C. 2009. Microanatomy of the liverimmune system. Semin. Immunopathol. 31:333–343.
32. Pamplona A, et al. 2007. Heme oxygenase-1 and carbon monoxide sup-press the pathogenesis of experimental cerebral malaria. Nat. Med. 13:703–710.
33. Rudin W, Favre N, Bordmann G, Ryffel B. 1997. Interferon-gamma isessential for the development of cerebral malaria. Eur. J. Immunol. 27:810 – 815.
34. Sachs JD. 2002. A new global effort to control malaria. Science 298:122–124.
35. Santos-Silva T, et al. 2011. CORM-3 reactivity toward proteins: the crys-tal structure of a Ru(II) dicarbonyl-lysozyme complex. J. Am. Chem. Soc.133:1192–1195.
36. Santos-Silva T, et al. 2011. Towards improved therapeutic CORMs: un-
CO-Releasing Molecule Protects from Severe Malaria
March 2012 Volume 56 Number 3 aac.asm.org 1289
on February 15, 2012 by E
TH
-BIB
LIOT
HE
K Z
UR
ICH
http://aac.asm.org/
Dow
nloaded from

derstanding the reactivity of CORM-3 with proteins. Curr. Med. Chem.18:3361–3366.
37. Sinclair D, Donegan S, Lalloo DG. 2011. Artesunate versus quinine fortreating severe malaria. Cochrane Database Syst. Rev. 3:CD005967.
38. Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M. 2004.Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob. Agents Chemother.48:1803–1806.
39. Snow RW, Guerra CA, Noor AM, Myint HY, Hay SI. 2005. The globaldistribution of clinical episodes of Plasmodium falciparum malaria. Na-ture 434:214 –217.
40. Thumwood CM, Hunt NH, Clark IA, Cowden WB. 1988. Breakdown ofthe blood-brain barrier in murine cerebral malaria. Parasitology 96(Pt3):579 –589.
41. Trampuz A, Jereb M, Muzlovic I, Prabhu RM. 2003. Clinical review:severe malaria. Crit. Care 7:315–323.
42. van der Heyde HC, Nolan J, Combes V, Gramaglia I, Grau GE. 2006. Aunified hypothesis for the genesis of cerebral malaria: sequestration, in-flammation and hemostasis leading to microcirculatory dysfunction.Trends Parasitol. 22:503–508.
43. Veros CT, Oldham NJ. 2007. Quantitative determination of lysozyme-ligand binding in the solution and gas phases by electrospray ionisationmass spectrometry. Rapid Commun. Mass Spectrom. 21:3505–3510.
44. Vivas L, et al. 2008. Anti-malarial efficacy of pyronaridine and artesunatein combination in vitro and in vivo. Acta Trop. 105:222–228.
45. Vreman HJ, Wong RJ, Kadotani T, Stevenson DK. 2005. Determinationof carbon monoxide (CO) in rodent tissue: effect of heme administrationand environmental CO exposure. Anal. Biochem. 341:280 –289.
46. WHO. 2008. World malaria report 2008. WHO, Geneva, Switzerland.47. Wu J, Nantz MH, Zern MA. 2002. Targeting hepatocytes for drug and
gene delivery: emerging novel approaches and applications. Front. Biosci.7:d717– d725.
Pena et al.
1290 aac.asm.org Antimicrobial Agents and Chemotherapy
on February 15, 2012 by E
TH
-BIB
LIOT
HE
K Z
UR
ICH
http://aac.asm.org/
Dow
nloaded from