a new model for utilising chemical diversity from natural sources
TRANSCRIPT

A New Model for Utilising Chemical Diversity fromNatural Sources
Antony D. Bussn and Mark S. ButlerMerLion Pharmaceuticals, Singapore Science Park II, Singapore
Strategy, Management and Health Policy
Enabling
Technology,
Genomics,
Proteomics
Preclinical
Research
Preclinical Development
Toxicology, Formulation
Drug Delivery,
Pharmacokinetics
Clinical Development
Phases I-III
Regulatory, Quality,
Manufacturing
Postmarketing
Phase IV
Abstract Although combinatorial chemistry technology has enabled large libraries of compounds to bescreened for biological activity and synthetic modifications to be effected rapidly, the reality is that qualityleads and development candidates for many drug targets still prove difficult to find. This is particularlyrelevant to antiinfective, oncology, and immunomodulation targets and also those involving protein–protein interactions. In the past, natural products have led to excellent drugs for precisely these therapeuticareas, but the difficulties of identifying and utilising such compounds compared to less complex moleculessynthesised using combinatorial chemistry techniques then led many to question their value in modern-day drug discovery. In fact, in the early 1990s, a significant number of pharmaceutical companies reducedor even terminated the natural product-based drug discovery activities that had served them so well in thepast. Nevertheless a renaissance appears to be at hand. The pressing need to fill drug developmentpipelines, combined with attractive outsourcing opportunities, have prompted many of the very samecompanies to reevaluate the utility of natural products as sources of novel drug leads. Through a judiciouscombination of new technologies and compelling business model, at least one Singapore-based companyis offering cost-effective solutions for accessing the unique chemical diversity found in Nature. Drug Dev.Res. 62:362–370, 2004. �c 2004 Wiley-Liss, Inc.
Key words: natural products; high-throughput screening; dereplication; business model; drug discovery
INTRODUCTION
To date, natural products have served thepharmaceutical industry and ultimately, mankind verywell [Sneader, 1996; Shu, 1998; Drews, 2000a; Grableyand Thiericke, 2000; Newman et al., 2000; Buss et al.,2003]. Microbial-derived compounds, for example,have been responsible for doubling our life expectancyduring the past century by providing the source of mostantibiotics [Demain, 1999]. Such compounds go farbeyond the familiar beta-lactams and include an arrayof chemistries; aminoglycosides, tetracyclines, glyco-peptides, lipopeptides, and macrolides. The macrolidefermentation product, erythromycin (1), has provedinvaluable for treating bacterial infections in patientswith beta-lactam hypersensitivity, and the semisyn-thetic derivatives, azithromycin (2) and clarithromycin
(3), represent important treatments for erythromycin-resistant strains of bacteria [Zhanel et al., 2001 (Fig. 1)].
New classes of natural products are still appear-ing on the market. The new ‘‘ketolide’’ antibiotic fromAventis, telithromycin (4), has been introduced re-cently for treating respiratory-tract infections [Zhanelet al., 2002]. The first of a new class of lipopeptideantibiotics, daptomycin (5), received approval from theUS Food and Drug Administration (FDA) in 2003 for
DDR
nCorrespondence to: Dr. Antony Buss, MerLion Pharma-ceuticals, 1 Science Park Road, The Capricorn #05-01, SingaporeScience Park II, Singapore 117528.E-mail: [email protected]
Published Online in Wiley InterScience (www.interscience.wiley.com) DOI: 10.1002/ddr.10389
DRUG DEVELOPMENT RESEARCH 62:362–370 (2004)
�c 2004 Wiley-Liss, Inc.

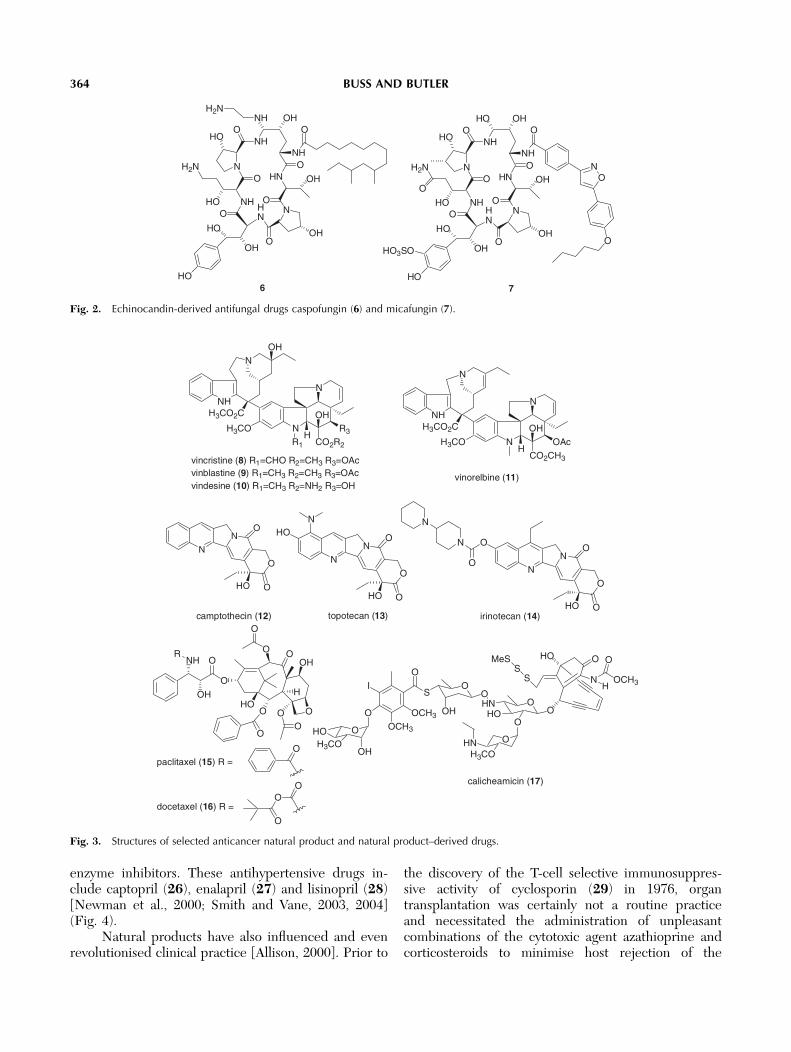
the treatment of skin and soft tissue infections [Raja etal., 2003]. The lipopeptide group of glucan synthaseinhibitors, known as the echinocandins and pneumo-candins, led chemists at Merck and Fujisawa to preparethe semisynthetic antifungal agents caspofungin (6)and micafungin (7), respectively (Fig. 2). These drugswere launched on the market within the past 3 years forthe treatment of aspergillosis and candidiasis infec-tions. The echinocandins represent the first new classof antifungal drugs to reach the market in more than 15years [Denning, 2003].
Natural products have also served the oncologyfield well. The ‘‘vinca’’ alkaloids, vincristine (8) andvinblastine (9), isolated from the rosy periwinkle plant,Catharanthus roseus, were introduced onto the marketby Lilly in the early 1960s and were followed by thesemisynthetic derivatives, vindesine (10) and vinorel-bine (11) [Sneader, 1996] (Fig. 3). Then camecamptothecin (12), a topoisomerase I inhibitor isolatedfrom the Chinese tree Camptotheca acuminata, whichled to the synthetic anticancer agents topotecan (13)and irinotecan (14). Even the structurally complex andpoorly soluble paclitaxel (15), initially isolated in tinyquantities from the Pacific yew tree (Taxus baccata),was marketed successfully by Bristol-Myers Squibb andled Aventis to prepare and market the more soluble,semisynthetic derivative, docetaxel (16) [Cragg andNewman, 2004]. The first antibody-targeted che-motherapeutic was introduced in 2000 by Wyeth forthe treatment of acute myeloid leukaemia; gemtuzu-
mab ozogamicin consists of a recombinant humanisedantibody (targets leukemic blast cells) conjugated tocalicheamicin (17), a potent cytotoxic enediyne naturalproduct [Abou-Gharbia, 2002].
Of note is the fact that between 1981 and 2002,more than 60% of all small-molecule prescription drugsintroduced onto the market could be attributed tonatural products or their derivatives [Newman et al.,2003]. Many of these compounds are neither anti-biotics nor anticancer agents. For example, mevastatin(18) and lovastatin (19) are fungal-derived metabolitesthat inhibit hepatic hydroxymethyl glutaryl coenzyme Areductase and led to the remarkable cholesterol-lowering family of drugs known as the ‘‘statins’’ (Fig.4). Lovastatin (19) was marketed first by Merck in1987, followed by simvastatin (20) (with a minorchemical modification) and pravastatin (21) (a bio-transformation product) by Sankyo/Squibb in 1989.The first completely synthetic version was fluvastatin(22) (launched by Novartis in 1994), followed byatorvastatin (23) from Pfizer in 1997. Atorvastatin (23)is now the world’s largest-selling drug (with sales ofalmost US$8 billion in 2002). The latest ‘‘statins’’ to belaunched on the market are rosuvastatin (25) (Sankyo)and pitavastatin (24) (AstraZeneca) [Tobert, 2003;Iglesias and Diez, 2003].
Staying with cardiovascular drugs, a peptidecomponent (teprotide) from the venom of the Brazilianpit viper led to another very successful series ofsynthetic compounds known as angiotensin-converting
OO
O
OH
O
OHO N
O
HO OH
O
OCH3
OH
erythromycin (1)
OO
O
OCH3
O
O
O
O
N
NN
N
OHO
N
telithromycin (4)
OO
O
OCH3
O
OHO N
O
HO OH
O
OCH3
OH
clarithromycin (3)
O
N
O
OHHO OH
O
O
O
OHO N
OCH3
OH
azithromycin (2)
daptomycin (5)
NH
NH
O
O CO2H
HN
NH
O
H2N
OHN
O
NH
O
HO2C
HN
OHO
HNO
OHN
O
O
NHO
HN CO2HOH2NOC
NHO
NHO
CO2H
H2N
O
HN
H
H
Fig. 1. Structures of selected natural product and natural product–derived antibacterial drugs.
UTILISING CHEMICAL DIVERSITY FROM NATURAL SOURCES 363
enzyme inhibitors. These antihypertensive drugs in-clude captopril (26), enalapril (27) and lisinopril (28)[Newman et al., 2000; Smith and Vane, 2003, 2004](Fig. 4).
Natural products have also influenced and evenrevolutionised clinical practice [Allison, 2000]. Prior to
the discovery of the T-cell selective immunosuppres-sive activity of cyclosporin (29) in 1976, organtransplantation was certainly not a routine practiceand necessitated the administration of unpleasantcombinations of the cytotoxic agent azathioprine andcorticosteroids to minimise host rejection of the
N
HN
NH
OHNHO
N
NH
HO
H2N
H2N
HO
O
NH
O
O
OH
O
OHN
OHO
OH
HO
HO
N
HN
NH
OHHOO
N
NH
HO
H2N
HO
O
NHO
OH
O
O HN
OHO
OH
HO
O
ON
O
O
HO
HO3SO
6 7
Fig. 2. Echinocandin-derived antifungal drugs caspofungin (6) and micafungin (7).
N
N
R3
OH
CO2R2R1
H3CO
H3CO2C
N
NH
H
OH
N
N
OAc
OH
CO2CH3
H3CO
H3CO2C
N
NH
H
O
O
O
SO
OHN O
O
OHNH3CO
HOOCH3
OCH3
I
OHH3CO
HO
OH O
S
O
N
HO
OCH3
O
H
SMeS
O
O
N
N
OHO
NHO
O
O
N
N
OHO
N
O
O
O
O
HO
O
N
N
N
O
O
O
O
OH
NHR
OH
HO
O
H
O O
O
OO
O
OO
O
paclitaxel (15) R =
docetaxel (16) R =
vincristine (8) R1=CHO R2=CH3 R3=OAcvinblastine (9) R1=CH3 R2=CH3 R3=OAcvindesine (10) R1=CH3 R2=NH2 R3=OH
camptothecin (12)
calicheamicin (17)
vinorelbine (11)
irinotecan (14)topotecan (13)
Fig. 3. Structures of selected anticancer natural product and natural product–derived drugs.
364 BUSS AND BUTLER

transplanted organ (Fig. 5). Cyclosporin (29), a cyclicpolypeptide produced by fermentation of the fungusTolyplocladium inflatum, was finally approved by theFDA in 1983 and represented a major advance inimmunosuppressive therapy [Borel and Kis, 1991;Heusler and Pletscher, 2001]. Two other fermentationmetabolites have since proved invaluable additions tothe choice of immunosuppressants used to preventorgan rejection. These are the macrolides tacrolimus(30) from Fujisawa (produced by Streptomyces tsuku-baensis) [Goto et al., 1991; Dumont, 2000] andsirolimus (31) from Wyeth (produced by Streptomyceshygroscopicus) [Sehgal, 2003].
Despite its evident success in drug discovery,natural product–based drug discovery was labourintensive, costly, and slow. For example, it was notuncommon to spend many weeks or even monthspurifying an active compound, only to find that it hadbeen discovered before and reported in the literature.The discovery rate for new chemical classes of naturalproducts appeared to be diminishing and this, togetherwith problems of screening mixtures with colour-sensitive high-throughput assays and the significantchallenges of optimising relatively complex chemicalstructures, prompted many pharmaceutical companies
in the early 1990s to cut back or completely terminatetheir natural product research activities [Cordell, 2002;Rouhi, 2003a].
With sharply rising research and developmentcosts, high attrition rates, higher regulatory hurdles,tougher therapeutic challenges, and patent expiries ofmany key drugs, the pharmaceutical industry has beenstriving hard to improve productivity [Drews, 2003;PhRMA, 2003; Rawlins, 2004]. The development ofcombinatorial chemistry methodologies promised tohelp solve some of the productivity issues by allowingthe rapid synthesis of very large numbers of chemicalcompounds for drug screening purposes. In fact, manypharmaceutical companies embraced these developingtechnologies with great enthusiasm and began generat-ing huge libraries of compounds. Unfortunately, thefocus was on quantity, with insufficient attention givento quality, either in terms of compound purity, chemicaldiversity, or drugability, and it took the industry someyears to realise this mistake and begin focusing more onthe quality of their screening libraries [Balkenhohl etal., 1996; Myers, 1997; Hopkins and Groom, 2002; Leeand Breitenbucher, 2003].
With the sequencing of the human genome (aswell as those of many microbial pathogens) there has
O
O
O
OHO
H
O
O
O
OHO
H
OH
CO2H
O
O
HO
HO
H
OH
CO2HHO
NF
OH
CO2HHO
NF
HN
O
OH
CO2HHO
N N
NS
F
OO
OH
CO2HHO
N
F
O
O
O
OHO
H
N
CO2HO
HS N
CO2HO
NH
OO
N
CO2HO
NH
OHO
NH2
simvastatin (20)lovastatin (19) pravastatin (21)
fluvastatin (22) atorvastatin (23) pitavastatin (25)rosuvastatin (24)
mevastatin (18)
captopril (26) enalapril (27) Iisinopril(28)
Fig. 4. Structures of selected cardiovascular drugs derived from natural products.
UTILISING CHEMICAL DIVERSITY FROM NATURAL SOURCES 365

been a significant increase in the array of potential newdrug targets available for screening [Drews, 2000b;Lindsay, 2003]. Although combinatorial chemistrytechnology has enabled very large libraries of com-pounds to be screened for biological activity andfurther synthetic modifications to be effected rapidly,the reality is that quality leads for many drug targetsstill prove difficult to find. The situation appearsparticularly critical for antiinfective, oncology, andimmunomodulation targets (and also those involvingprotein–protein interactions)Fironically, the very ther-apeutic areas for which natural products lend them-selves particularly well.
Nevertheless, a renaissance may be at hand.Through a judicious combination of new technologiesand compelling business model, a growing number ofpharmaceutical companies have again begun to utilise
the chemical diversity offered by natural products intheir drug discovery programmes, although chiralityand relative complexity remain familiar hallmarks[Strohl, 2000; Wildman, 2003; Rouhi, 2003b; Butler,2004].
DISCUSSION
Natural Product Libraries
Natural product extracts usually are generated byextracting the biomass of an organism with a singlesolvent (usually methanol) or a mixture of solvents inorder to obtain an extract with the largest number ofcompounds with varying polarity. Extracts also can begenerated by sequential extraction with nonpolar topolar solvents to produce a series of samples. Extractsgenerated using these methods are then added to
N
O
O
N
O
HN
O
N
O
N
NHO
N
O
NH
HN
O
O
N
O
N
HO
H
O
cyclosporin (29)
tacrolimus (30)
N
O
O
O
O
O
O
OH
HO
OH
O
O
O
O
sirolimus (31)
N
O
O
OH
HO
O
O
O
O
OH
O
O
O
Fig. 5. Structures of selected natural product immunosuppressive drugs.
HN
NH
HN
NH
HN
OH
O
O
O
OH O
O
OH O
3233 n = 1134 n = 13
HNNH
HN
O(CH2)n
NH
Fig. 6. Plasmepsin II inhibitors pepstatin (32) and budmunchiamines L4 (33) and L5 (34).
366 BUSS AND BUTLER

extract libraries, which in turn can be screened againstbiologically relevant targets.
Over the last few years, there has been a growingtrend to use prefractionated extracts in screeningcampaigns. The prefractionation process usually usescolumn chromatography or high performance liquidchromatography (HPLC) and produces fractions thatcan range from relatively crude fractions to mixtures ofonly a few compounds [Cardellina et al., 1993; Schmidet al., 1999; Koch et al., 2000; Eldridge et al., 2002; Jia,2003]. The advantages of using prefractionated extractsfor screening is that they allow more rapid dereplica-tion (see below), enabling compounds whose activity ismasked in a crude extract to be detected and to identifyactive components present in amounts too small todetect by extract screening. Prefractionated extractlibraries also have been generated using liquid–liquidchromatography [Alvi et al., 1995; Alvi, 2000; Ingkani-nan et al., 2000; Armbruster et al., 2001]. Theselibraries have the advantage of virtually no compoundloss due to irreversible binding to solid phases but aremore time consuming to prepare. However, there areother problems associated with prefractionated ex-tracts, such as a potential decrease in the percentage oforganism biodiversity screened, interfering/nuisancecompounds will be more frequently found, and there isa significant cost involved in the preparation ofsamples.
The ultimate prefractionated libraries are purenatural product libraries, which are usually generatedby HPLC and selected for inclusion in the library bycomparison with compound databases [Stewart et al.,2000; Bindseil et al., 2001; Abel et al., 2002]. The purenatural product library is then screened in an analogousmanner to pure compound libraries and enables activecompounds to be evaluated on an equivalent basis tosynthetic compound libraries, as well as eliminating thebioassay-guided isolation step. As with prefractiona-tion, the generation of pure compound libraries is atime-consuming and costly process, and the isolation ofselected compounds from an extract may miss minoractive components. A well-balanced natural productscreening programme should have all three librarytypesFcrude extracts, prefractionated extracts, andpure compoundsFin a ratio suitable for its needs.
Screening
Over the last 10 years, advances in high-throughput screening (HTS) have dramatically affectedthe drug discovery process [Devlin, 1997; Carrano andDonadio, 1999; Marron and Jayawickreme, 2003].Screening campaigns consisting of 100,000 to 500,000or greater individual assays can be completed routinelyin 1–2 weeks using standard 96-, 384-, or 1,536-well
microtitre plates. The total number of compounds thatcan be screened in an assay can be increasedconsiderably, and the time for the screening can bedecreased considerably by using further assay minia-turisation and robotics [Khandurina and Guttman,1998; Battersby and Trau, 2002; Garyantes, 2002;Entzeroth, 2003].
Once the initial screen, usually referred to as theprimary screen, has been completed, primary positivesnumbering anywhere from just a few, to hundreds ofpositive samples are generated. Efficient prioritisationof these samples in an industrial setting is imperative toeliminate any unnecessary replication of work. One wayof achieving this before any chemical analysis is to useorthogonal assays to help eliminate assay interference/false positives and secondary assays to make sure theprimary positive extracts have the desired biologicalprofile [Walters and Namchuk, 2003].
An example of an efficient HTS system from ourlaboratory using this approach was recently describedfor the antimalarial target plasmepsin II by Flotow etal. [2002]. The plasmepsins are aspartyl proteases,which cleave haemoglobin and are essential for parasitesurvival [Coombs et al., 2001; Boss et al., 2003].Plasmepsin II is an enzyme that has been wellcharacterised and was available in sufficient quantitiesfor development of a high-throughput screen. Twocomplementary assays in different screening formats,FRET [Allen et al., 2000] and FP [Owicki, 2000], weredeveloped. The FRET assay format measures theexcitation of a fluorophore and its proximity to aquencher. In the plasmepsin II FRET assay, cleavage ofa peptide with a fluorophore and quencher on each endby plasmepsin II will lead to increase in emission.Therefore, inhibition of plasmepsin II would result inno peptide cleavage and no extra fluorescence. Becauseof the nature of the readout of the FRET screen,coloured compounds can give a false-positive response,whereas autofluorescent compounds can give negativeresults. The FP assay format measures the ratiometricdifference of the fluorescent polarisation betweenbound and unbound ligands. Fluorescently taggedligands bound to a protein will rotate more slowlycompared to the free ligand and will therefore showmore polarisation. In the plasmepsin II FP assay, apeptide tagged with fluorescein was bound to theprotein avidin. Therefore, cleavage of the peptide withplasmepsin II will lead to an increase of theconcentration of cleaved peptide and, as a conse-quence, a decrease in fluorescent polarisation. FPassays are relatively independent of concentration andhave less interference by colour and background. TheFRET assay was chosen as the primary assay because ofits fast, robust, and cost-effective properties.
UTILISING CHEMICAL DIVERSITY FROM NATURAL SOURCES 367

The primary positive extracts from the FRETassay were then cross screened using the FP assay toeliminate false-positive colour compounds and againstthe aspartyl proteases cathepsin D and pepsin forselectively profiling. The use of these two orthogonalscreens allowed the generation of a high-quality set ofextracts devoid of coloured interference. These FRETand FP assays were also used in tandem during thebioassay-guided fractionation to guard against fluores-cent compounds that could mask compounds ofinterest by showing a false-negative effect in FRET.The effectiveness of the orthogonal screening strategywas demonstrated by the identification of the knownaspartyl protease inhibitor pepstatin (32) [Umezawa etal., 1970; Morishima et al., 1970], also used as thestandard inhibitor in this assay, from a majority of theactive Actinomycetes extracts (Fig. 6).
During this HTS campaign to look for inhibitorsof plasmepsin II, the methanol extracts from both thestem bark and leaves of Albizia adinocephala (Legu-minosae) were found to be active in both the FRETand FP assays. Bioassay-guided fractionation of thestem bark extract led to the isolation of two newmacrocyclic spermine alkaloids, budmunchiamines L4(33) and L5 (34), details of which can found in thepaper by Ovenden et al. [2002].
Dereplication, Isolation, and Structure Elucidation
Dereplication, which is the process of recognisingand eliminating previously isolated substances presentin an extract, has been a concern in natural productchemistry since the beginning of antibiotic research[Cordell et al., 1997; VanMiddlesworth and Cannell,1998]. The general procedure most often used involvesthe separation of extracts using reversed-phase HPLCand postcolumn splitting of the eluent into variousdetectors and a fraction collector [Constant andBeecher, 1995; Cordell and Shin, 1999; Lee and Kerns,1999; Bobzin et al., 2000; Wolfender et al., 2001]. Thedata obtained from the postcolumn detectors for theactive fractions are then analysed for common inter-fering compounds and known inhibitors using com-mercial and in-house databases. This process helps tominimise ‘‘reinventing the wheel’’ and as such, savesinvaluable research time and money. The dereplicationprocess is also important in helping to prioritiseextracts for chemical isolation and to group samplesthat contain unknown active components with similardereplication profiles. For example, the assignment ofsimilar extracts to one or a few chemists reduces thepossibility of different chemists independently isolatingand identifying the same active component. Once theactive components of the extract have been identified,
MS/MS can be used to establish their presence in theother similar dereplicated extracts.
Instrumentation that facilitates the isolation andstructure elucidation of natural products has improvedsignificantly over the last 20 years, and these processesthat were once considered bottlenecks have becomemuch faster and more manageable. For example,bioassay-guided fractionation using HTS techniquescombined with HPLC automation and improvedcolumn technology has allowed our laboratory toisolate most pure bioactive compounds from crudeextracts after dereplication using on average two tothree bioassay-guided iterations (typically 2 weeks).Difficult isolation projects raise the average isolationtime to 3 weeks, although this includes the extra timeneeded for further microbial refermentation. Advancesin nuclear magnetic resonance (NMR) automation,magnetic field strengths, probe technology, and pulsesequences allow quick acquisition of high-quality NMRdata, which has enabled the structure elucidation ofnatural products to become routine for experiencedchemists.
A Compelling Business Model
With quality leads for many drug targets remain-ing elusive, pharmaceutical and biotechnology compa-nies have begun to seriously reconsider accessingchemical diversity from natural sources. However,having terminated in-house natural product–baseddrug discovery activities, the reentry barrier, even forthe larger pharmaceutical companies, appears high andunattractive. The rapid pace of relevant technologicaldevelopments (as described in the preceding sectionsof this paper) and associated costs at this high-riskstage of the discovery process only serve to reinforcethis barrier. Nevertheless, the situation representsa substantial opportunity for smaller, nimble,natural product–focused enterprises to offer cost-effective solutions for accessing novel natural productchemistry through collaborative drug discoverypartnerships.
One example of a company offering suchcollaborative partnerships or outsourcing opportunitiesis MerLion Pharmaceuticals, a Singapore-based com-pany with a very large, diverse collection of naturalproduct samples, together with advanced HTS andchemistry capabilities (www.merlionpharma.com). Thecompany transfers selected bioassays from partners onan exclusive basis to its laboratories in Singapore. Theassays are used to screen its natural product collection,and pure, active compounds are supplied to thecorresponding partner for further evaluation and leadidentification. The additional, key ingredient for thecompany is a business model that provides low initial
368 BUSS AND BUTLER

costs (and low risk) for its collaboration partners andfuture revenue streams secured through preagreedmilestone and royalty payments only when leadcompounds are identified and progressed through thedrug development process.
It is through this kind of collaborative partner-ship, involving minimal upfront costs, that companieswith good, validated therapeutic targets can readilyaccess and utilise the rich chemical diversity fromnatural sources to enhance their drug discoverycapabilities.
REFERENCES
Abel U, Koch C, Speitling M, Hansske FG. 2002. Modern methodsto produce natural-product libraries. Curr Opin Chem Biol6:453–458.
Abou-Gharbia M. 2002. Optimization of natural product leads:discovery of MylotargTM, CCI-779 and GAR-936. In: Sener B,editor. Biodiversity: biomolecular aspects of biodiversity andinnovation utilization. New York: Kluwer Academic. p 63–70.
Allen M, Reeves J, Mellor G. 2000. High throughput fluorescencepolarization: a homogeneous alternative to radioligand binding forcell surface receptors. J Biomol Screen 5:63–69.
Allison AC. 2000. Immuno suppressive drugs: the first 50 years anda glance forward. Immunopharmacology 47:63–83.
Alvi KA. 2000. A strategy for rapid identification of noveltherapeutic leads from natural products. In: Cutler SJ, CutlerHG, editors. Biologically active natural products: pharmaceuti-cals. New York: CRC Press. p 185–195.
Alvi KA, Peterson J, Hofmann B. 1995. Rapid identification ofelaiophylin and geldanamycin in Streptomyces fermentationbroths using CPC coupled with a photodiode array detector andLC-MS methodologies. J Ind Microbiol 15:80–84.
Armbruster JA, Borris RP, Jiminez Q, Zamora N, Tamayo-Castillo G,Harris GH. 2001. Separation of crude plant extracts with highspeed CCC for primary screening in drug discovery. J LiqChromatogr Rel Technol 24:1827–1840.
Balkenhohl F, von dem Bussche-Hunnefeld C, Lansky A, Zechel C.1996. Combinatorial synthesis of small organic molecules. AngewChem Int Ed 35:2289–2337.
Battersby BJ, Trau M. 2002. Novel miniaturized systems in high-throughput screening. Trends Biotechnol 20:167–173.
Bindseil KU, Jakupovic J, Wolf D, Lavayre J, Leboul J, van der PylD. 2001. Pure compound libraries; a new perspective for naturalproduct based drug discovery. Drug Discov Today 6:840–847.
Bobzin SC, Yang S, Kasten TP. 2000. LC-NMR: a new tool toexpedite the dereplication and identification of natural products. JInd Microbiol Biotechnol 25:342–345.
Borel JF, Kis ZL. 1991. The discovery and development ofcyclosporine (Sandimmune). Transplant Proc 23:1867–1874.
Boss C, Richard-Bildstein S, Weller T, Fischli W, Meyer S, BinkertC. 2003. Inhibitors of the Plasmodium falciparum parasiteaspartic protease plasmepsin II as potential antimalarial agents.Curr Med Chem 10:883–907.
Buss AD, Cox B, Waigh RD. 2003. Natural products as leads fornew pharmaceuticals. In: Abraham DJ, editor. Burger’s medicinalchemistry and drug discovery. Sixth edition, Vol. 1: Drugdiscovery. Hoboken, NJ: John Wiley & Sons. p 847–900.
Butler MS. 2004. The role of natural product chemistry in drugdiscovery. J Nat Prod 67; In press.
Cardellina JH II, Munro MH, Fuller RW, Manfredi KP, McKee TC,Tischler M, Bokesch HR, Gustafson KR, Beutler JA, Boyd MR.1993. A chemical screening strategy for the dereplication andprioritization of HIV-inhibitory aqueous natural products extracts.J Nat Prod 56:1123–1129.
Carrano L, Donadio S. 1999. Key ingredients for efficient high-throughput screening. In: Miertus S, Fassina G, editors.Combinatorial chemistry and technology: principles, methods,and applications. New York: Marcel Dekker. p 233–250.
Constant HL, Beecher CWW. 1995. A method for the dereplicationof natural products extracts using electrospray HPLC/MS. NatProd Lett 6:193–195.
Coombs GH, Goldberg DE, Klemba M, Berry C, Kay J, MottramJC. 2001. Aspartic proteases of Plasmodium falciparum and otherparasitic protozoa as drug targets. Trends Parasitol 17:532–537.
Cordell GA. 2002. Natural products in drug discoveryFcreating anew vision. Phytochem Rev 1:261–273.
Cordell GA, Shin YG. 1999. Finding a needle in the haystack. Thedereplication of natural product extracts. Pure Appl Chem71:1089–1094.
Cordell GA, Beecher CWW, Seo EX, Long L, Kinghorn AD,Pezzuto JM, Constant HL, Chai HB, Fang L, Cui B, Slowing-Barillas K. 1997. The dereplication of plant-derived naturalproducts. In: Atta-ur-Rahman, editor. Studies in natural productschemistry, Vol. 19: structure and chemistry (Part E) (studiesin natural product chemistry series). Amsterdam: Elsevier.p 747–749.
Cragg GM, Newman DJ. 2004. A tale of two tumor targets:topoisomerase I and tubulin. The Wall and Wani contribution tocancer chemotherapy. J Nat Prod 67:232–244.
Demain AL. 1999. Pharmaceutically active secondary metabolites ofmicroorganisms. Appl Microbiol Biotechnol 52:455–463.
Denning DW. 2003. Echinocandin antifungal drugs. Lancet362:1142–1151.
Devlin JP, editor. 1997. High throughput screening: the discovery ofbioactive substances. New York: Marcel Dekker.
Drews J. 2000a. Drug discovery: a historical perspective. Science287:1960–1964.
Drews J. 2000b. Drug discovery todayFand tomorrow. DrugDiscov Today 5:2–4.
Drews J. 2003. Strategic trends in the drug industry. Drug DiscovToday 8:411–420.
Dumont FJ. 2000. FK506, an immunosuppressant targetingcalcineurin function. Curr Med Chem 7:731–748.
Eldridge GR, Vervoort HC, Lee CM, Cremin PA, Williams CT,Hart SM, Goering MG, O’Neil-Johnson M, Zeng L. 2002.High-throughput method for the production and analysis oflarge natural product libraries for drug discovery. Anal Chem74:3963–3971.
Entzeroth M. 2003. Emerging trends in high-throughput screening.Curr Opin Pharmacol 3:522–529.
Flotow H, Leong Cladd, Buss AD. 2002. Development of aplasmepsin II fluorescence polarization assay suitable for highthroughput antimalarial drug discovery. J Biomol Screen 7:367–371.
Garyantes TK. 2002. 1536-well assay plates: when do they makesense? Drug Discov Today 7:489–490.
UTILISING CHEMICAL DIVERSITY FROM NATURAL SOURCES 369

Goto T, Kino T, Hatanaka H, Okuhara M, Kohsaka M, Aoki H,Imanaka H. 1991. FK 506: historical perspectives. TransplantProc 23:2713–2717.
Grabley S, Thiericke R. 2000. The impact of natural products ondrug discovery. In: Grabley S, Thiericke R, editors. Drugdiscovery from nature. Berlin: Springer. p 3–37.
Heusler K, Pletscher A. 2001. The controversial early history ofcyclosporin. Swiss Med Wkly 131:299–302.
Hopkins AL, Groom CR. 2002. The druggable genome. Nat RevDrug Discov 1:727–730.
Iglesias P, Diez JJ. 2003. New drugs for the treatment ofhypercholesterolaemia. Expert Opin Investig Drugs 12:1777–1789.
Ingkaninan K, Hazekamp A, Hoek AC, Balconi S, Verpoorte R.2000. Application of centrifugal partition chromatography in ageneral separation and dereplication procedure for plant extracts.J Liq Chromatogr Rel Technol 23:2195–2208.
Jia Q. 2003. Generating and screening a natural product library forcyclooxygenase and lipoxygenase dual inhibitors. In: Atta-ur-Rahman, editor. Studies in natural products chemistry: bioactivenatural products (Part J) (studies in natural product chemistryseries). Amsterdam: Elsevier. p 643–718.
Khandurina J, Guttman A. 1998. High-throughput screening.Advances in robotics and miniaturization continue to acceleratedrug lead identification. Curr Opin Chem Biol 6:359–366.
Koch C, Neumann T, Thiericke R, Grabley S. 2000. A centralnatural product poolFnew approach in drug discovery strategies.In: Grabley S, Thiericke R, editors. Drug discovery from nature.Berlin: Springer. p 51–55.
Lee A, Breitenbucher JG. 2003. The impact of combinatorialchemistry on drug discovery. Curr Opin Drug Discov Devel 6:494–508.
Lee MS, Kerns EH. 1999. LC/MS applications in drug develop-ment. Mass Spectrom Rev 18:187–279.
Lindsay MA. 2003. Target discovery. Nat Rev Drug Discov 2:831–838.
Marron BE, Jayawickreme CK. 2003. Going to the well no more:lawn format assays for ultra-high-throughput screening. CurrOpin Chem Biol 7:395–401.
Morishima H, Takita T, Aoyagi T, Takeuchi T, Umezawa H. 1970.The structure of pepstatin. J Antibiot 23:263–265.
Myers PL. 1997. Will combinatorial chemistry deliver realmedicines? Curr Opin Biotechnol 8:701–707.
Newman DJ, Cragg GM, Snader KM. 2000. The influence of naturalproducts upon drug discovery. Nat Prod Rep 17:215–234.
Newman DJ, Cragg GM, Snader KM. 2003. Natural products assources of new drugs over the period 1981-2002. J Nat Prod66:1022–1037.
Ovenden SPB, Cao S, Leong C, Flotow H, Gupta MP, Buss AD,Butler MS. 2002. Spermine alkaloids from Albizia adinocephalawith activity against Plasmodium falciparum plasmepsin II.Phytochemistry 60:175–177.
Owicki JC. 2000. Fluorescence polarization and anisotropy in highthroughput screening: perspectives and primer. J Biomol Screen5:297–306.
PhRMA. 2003. Pharmaceutical industry profile 2003. Washington,DC: Pharmaceutical Research and Manufacturers of America(PhRMA) (available on-line at http://www.phrma.org/publications/publications/profile02/index.cfm).
Raja A, LaBonte J, Lebbos J, Kirkpatrick P. 2003. Daptomycin. NatRev Drug Discov 2:943–944.
Rawlins MD. 2004. Cutting the cost of drug development? Nat RevDrug Discov 3:360–364.
Rouhi AM. 2003a. Rediscovering natural products. Chem Eng News81:77–91.
Rouhi AM. 2003b. Betting on natural products for cures. Chem EngNews 81:93–103.
Schmid I, Sattler I, Grabley S, Thiericke R. 1999. Natural productsin high throughput screening: automated high-quality samplepreparation. J Biomol Screen 4:15–25.
Sehgal SN. 2003. Sirolimus: its discovery, biological properties, andmechanism of action. Transplant Proc 35:7S–14S.
Shu YZ. 1998. Recent natural products based drug development: apharmaceutical industry perspective. J Nat Prod 61:1053–1071.
Smith CG, Vane JR. 2003. The discovery of captopril. FASEB J17:788–789.
Smith CG, Vane JR. 2004. The discovery of captopril: a reply.FASEB J 18:935.
Sneader W. 1996. Drug prototypes and their exploitation. Chiche-ster, UK: Wiley.
Stewart M, Nash RJ, Chicarelli-Robinson MI. 2000. Production of adiverse library of plant natural products for bioassays. In: OleszekW, Marston A, editors. Saponins in food, feedstuffs and medicinalplants. Boston: Kluwer Academic Publishers. p 73–77.
Strohl WR. 2000. The role of natural products in a modern drugdiscovery program. Drug Discov Today 5:39–41.
Tobert JA. 2003. Lovastatin and beyond: the history of the HMG-CoA reductase inhibitors. Nat Rev Drug Discov 2:517–526.
Umezawa H, Aoyagi T, Morishima H, Matsuzaki M, Hamada M.1970. Pepstatin, a new pepsin inhibitor produced by Actinomy-cetes. J Antibiot 23:259–262.
VanMiddlesworth N, Cannell RJP. 1998. Dereplication and partialpurification of natural products. In: Cannell RJP, editor. Naturalproduct isolation; methods in biotechnology, Vol. 4. Totowa:Humana Press. p 279–327.
Walters WP, Namchuk M. 2003. Designing screens: how to makeyour hits a hit. Nat Rev Drug Discov 2:259–266.
Wildman HG. 2003. The fall and rise of natural products screeningfor drug discovery. Fungal Diversity 13:221–231.
Wolfender JL, Ndjoko K, Hostettmann K. 2001. The potentialof LC-NMR in phytochemical analysis. Phytochem Anal12:2–22.
Zhanel GG, Dueck M, Hoban DJ, Vercaigne LM, Embil JM, GinAS, Karlowsky JA. 2001. Review of macrolides and ketolides:focus on respiratory tract infections. Drugs 61:443–498.
Zhanel GG, Walters M, Noreddin A, Vercaigne LM, Wierzbowski A,Embil JM, Gin AS, Douthwaite S, Hoban DJ. 2002. The ketolides:a critical review. Drugs 62:1771–1804.
370 BUSS AND BUTLER