a missense mutation in tbce causes progressive motor neuronopathy in mice
TRANSCRIPT

Mice that are homozygous with respect to the progressive motorneuronopathy (pmn) mutation (chromosome 13) develop a pro-gressive caudio-cranial degeneration of their motor axons fromthe age of two weeks and die four to six weeks after birth1. Themutation is fully penetrant, and expressivity does not depend onthe genetic background. Based on its pathological features, thepmn mutation has been considered an excellent model for theautosomal recessive proximal childhood form of spinal muscular
atrophy (SMA). Previously, we demonstrated that the genesresponsible for these disorders were not orthologous2,3. Here, weidentify the pmn mutation as resulting in a Trp524Gly substitu-tion at the last residue of the tubulin-specific chaperone e (Tbce)protein that leads to decreased protein stability. Electronmicroscopy of the sciatic and phrenic nerves of affected miceshowed a reduced number of microtubules, probably due todefective stabilization. Transgenic complementation with a wild-
type Tbce cDNA restored a normal phe-notype in mutant mice. Our observationsindicate that Tbce is critical for the main-tenance of microtubules in mouse motoraxons, and suggest that altered functionof tubulin cofactors might be implicatedin human motor neuron diseases.We have previously mapped the pmnlocus to a region of 0.3 cM on mousechromosome 13, established a BAC con-tig of 850 kb covering the interval andidentified four candidate genes based ontheir expression patterns4. Tbce seemedthe most probable candidate on the basisof its function. The formation andmaintenance of neurons relies on theassembly of an organized microtubulenetwork that is the predominant com-ponent of the neural cytoskeleton, andTbce is one of the cofactors involved intubulin folding, an essential step inmicrotubule assembly5–8.
Using a combination of 5′ rapidamplification of cDNA ends (RACE),searches of the expressed sequence tag(EST) database and RT–PCR analyses,we determined the nucleotide sequenceof a 1.8-kb cDNA containing the entireTbce coding sequence. To identify muta-tions in Tbce, we sequenced PCR prod-ucts derived from brain and testis cDNAof control and pmn/pmn mice. We iden-tified two nucleotide differences: aC1043T transition resulting in anAla348Val substitution and a T1570Gtransversion resulting in a Trp524Glysubstitution at the last residue of the
Fig. 1 Sequence analysis ofpmn mutation. a, Chro-matograms showing the poly-morphism observed atnucleotide 1043 (arrow) of theTbce coding sequence in dif-ferent mouse strains (indicatedon the left), which leads to aconservative amino-acidexchange (Ala348Val). b, Chro-matograms showing the muta-tion found in nucleotide 1570(arrow) of the Tbce codingsequence in the pmn allele.Only pmn/pmn mice harbor aT1570 residue at this position;all other mouse strains that wetested have a G1570 residue.The pmn mutation results in aTrp→Gly amino-acid substitu-tion at the last residue of theprotein. c, Multiple proteinalignment of partial amino-acid sequences of TBCE in the mouse, human, rat and bovine, showing thatthe carboxy terminus is highly conserved between the four species. Arrow denotes the last amino acid(position 524), mutated in pmn/pmn mice.
letter
nature genetics • volume 32 • november 2002 443
A missense mutation in Tbce causes progressive motorneuronopathy in mice
Natalia Martin1, Jean Jaubert1, Pierre Gounon2, Eduardo Salido3, Georg Haase4, Marek Szatanik1
& Jean-Louis Guénet1
1Unité de Génétique des Mammifères, Institut Pasteur, 25 Rue du Docteur Roux, F-75724 Paris Cedex 15, France. 2Station centrale de Microscopie électronique,Institut Pasteur, Paris, France. 3Unidad de Investigacion, Hospital Universitario Canarias, Tenerife, Spain. 4Institut National de la Santé et de la RechercheMédicale, Institut de Biologie du Développement de Marseille (Centre National de la Recherche Scientifique, Institut National de la Santé et de la RechercheMédicale and Université de la Méditerranée), Campus de Luminy, Marseille, France. Correspondence should be addressed to J.-L.G. (e-mail: [email protected]).
a b
c
Published online 21 October 2002; doi:10.1038/ng1016
©20
02 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reg
enet
ics
protein. The Ala348Val substitution was a mere polymorphismas it was also found in 3 of 13 mouse inbred strains studied,including NMRI/Pan, the strain in which the pmn mutation wasdiscovered (Fig. 1a). The T1570G transversion, in contrast, wasabsent in the same 13 mouse strains (Fig. 1b), and we found theTrp524 residue to be conserved in bovine, human and rat orthol-ogous sequences (Fig. 1c), suggesting that it is the pmn mutation.
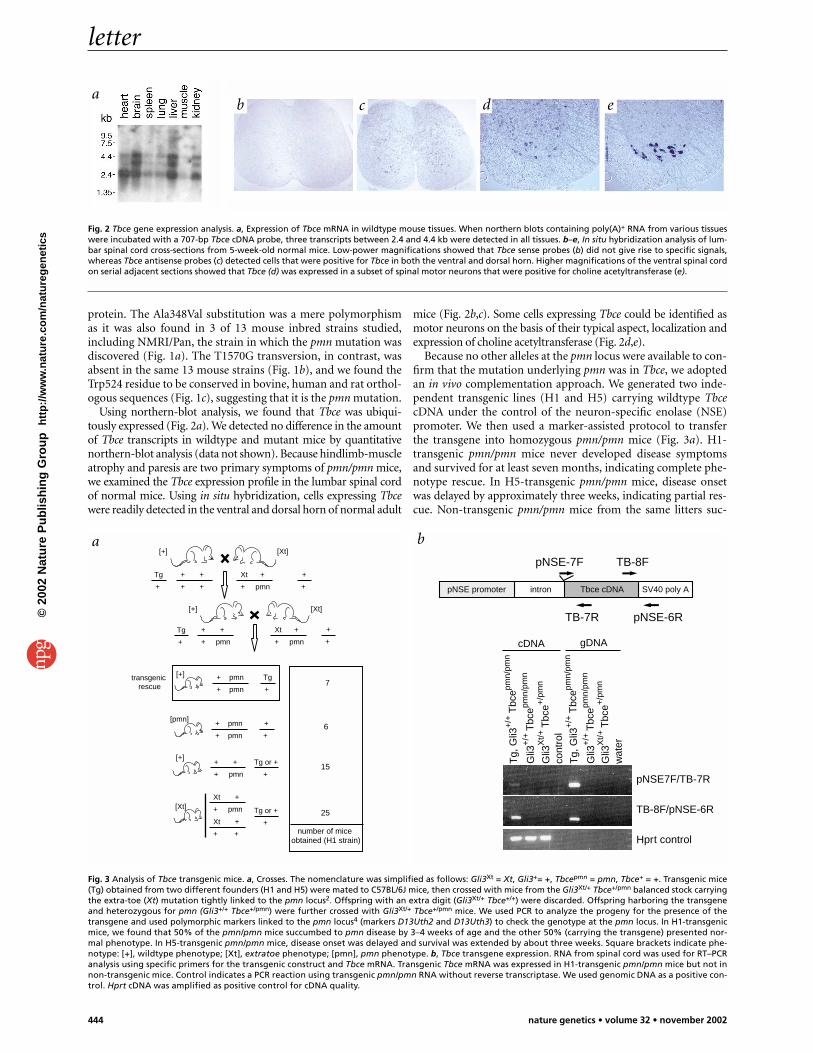
Using northern-blot analysis, we found that Tbce was ubiqui-tously expressed (Fig. 2a). We detected no difference in the amountof Tbce transcripts in wildtype and mutant mice by quantitativenorthern-blot analysis (data not shown). Because hindlimb-muscleatrophy and paresis are two primary symptoms of pmn/pmn mice,we examined the Tbce expression profile in the lumbar spinal cordof normal mice. Using in situ hybridization, cells expressing Tbcewere readily detected in the ventral and dorsal horn of normal adult
mice (Fig. 2b,c). Some cells expressing Tbce could be identified asmotor neurons on the basis of their typical aspect, localization andexpression of choline acetyltransferase (Fig. 2d,e).
Because no other alleles at the pmn locus were available to con-firm that the mutation underlying pmn was in Tbce, we adoptedan in vivo complementation approach. We generated two inde-pendent transgenic lines (H1 and H5) carrying wildtype TbcecDNA under the control of the neuron-specific enolase (NSE)promoter. We then used a marker-assisted protocol to transferthe transgene into homozygous pmn/pmn mice (Fig. 3a). H1-transgenic pmn/pmn mice never developed disease symptomsand survived for at least seven months, indicating complete phe-notype rescue. In H5-transgenic pmn/pmn mice, disease onsetwas delayed by approximately three weeks, indicating partial res-cue. Non-transgenic pmn/pmn mice from the same litters suc-
Fig. 2 Tbce gene expression analysis. a, Expression of Tbce mRNA in wildtype mouse tissues. When northern blots containing poly(A)+ RNA from various tissueswere incubated with a 707-bp Tbce cDNA probe, three transcripts between 2.4 and 4.4 kb were detected in all tissues. b–e, In situ hybridization analysis of lum-bar spinal cord cross-sections from 5-week-old normal mice. Low-power magnifications showed that Tbce sense probes (b) did not give rise to specific signals,whereas Tbce antisense probes (c) detected cells that were positive for Tbce in both the ventral and dorsal horn. Higher magnifications of the ventral spinal cordon serial adjacent sections showed that Tbce (d) was expressed in a subset of spinal motor neurons that were positive for choline acetyltransferase (e).
letter
444 nature genetics • volume 32 • november 2002
a
[Xt]
[Xt][+]
[+]
+ +
+ +
Xt +
+ pmn
Xt +
+ pmn
Tg
+
Tg
+
+
+
+
+
+ +
+ pmn
[pmn] +
+
+ pmn
+ pmn
Tg
+
+ pmn
+ pmn
transgenic rescue
[+]
Tg or +
+
[+]+ +
+ pmn
Xt +
+ pmn
Xt +
+ +
[Xt] Tg or +
+ 25
15
7
6
number of mice obtained (H1 strain)
SV40 poly ApNSE promoter intron Tbce cDNA
pNSE-7F
TB-7R
TB-8F
pNSE-6R
cont
rol
Tg,
Gli3
+/+
Tbc
epmn/
pmn
Tg,
Gli3
+/+
Tbc
epmn/
pmn
Gli3
+/+
Tbc
epmn/
pmn
Gli3
+/+
Tbc
epmn/
pmn
Gli3
Xt/+
Tbc
e+/p
mn
Gli3
Xt/+
Tbc
e+/p
mn
wat
er
pNSE7F/TB-7R
TB-8F/pNSE-6R
Hprt control
cDNA gDNA
Fig. 3 Analysis of Tbce transgenic mice. a, Crosses. The nomenclature was simplified as follows: Gli3Xt = Xt, Gli3+= +, Tbcepmn = pmn, Tbce+ = +. Transgenic mice(Tg) obtained from two different founders (H1 and H5) were mated to C57BL/6J mice, then crossed with mice from the Gli3Xt/+ Tbce+/pmn balanced stock carryingthe extra-toe (Xt) mutation tightly linked to the pmn locus2. Offspring with an extra digit (Gli3Xt/+ Tbce+/+) were discarded. Offspring harboring the transgeneand heterozygous for pmn (Gli3+/+ Tbce+/pmn) were further crossed with Gli3Xt/+ Tbce+/pmn mice. We used PCR to analyze the progeny for the presence of thetransgene and used polymorphic markers linked to the pmn locus4 (markers D13Uth2 and D13Uth3) to check the genotype at the pmn locus. In H1-transgenicmice, we found that 50% of the pmn/pmn mice succumbed to pmn disease by 3–4 weeks of age and the other 50% (carrying the transgene) presented nor-mal phenotype. In H5-transgenic pmn/pmn mice, disease onset was delayed and survival was extended by about three weeks. Square brackets indicate phe-notype: [+], wildtype phenotype; [Xt], extratoe phenotype; [pmn], pmn phenotype. b, Tbce transgene expression. RNA from spinal cord was used for RT–PCRanalysis using specific primers for the transgenic construct and Tbce mRNA. Transgenic Tbce mRNA was expressed in H1-transgenic pmn/pmn mice but not innon-transgenic mice. Control indicates a PCR reaction using transgenic pmn/pmn RNA without reverse transcriptase. We used genomic DNA as a positive con-trol. Hprt cDNA was amplified as positive control for cDNA quality.
b c d e
a b
©20
02 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reg
enet
ics

cumbed to typical progressive motor neuronopathy before fiveweeks of age. We verified expression of the transgenic mRNA inspinal cord of H1-transgenic pmn/pmn mice (Fig. 3b) and ana-lyzed nerves of the same mice by electron microscopy (see below).
To analyze whether the Tbce mutation resulting in theTrp524Gly substitution affected the stability of the protein, weoverexpressed wildtype or mutant Tbce tagged with FLAG at theamino terminus. Transfection of COS-7 and HeLa cells with wild-type and mutant Tbce showed that each encoded single proteins of60 kD, and that the mutant protein was less abundant than thewildtype (Fig. 4a,b). In addition, pulse–chase experiments showedthat the mutant Tbce protein was considerably less stable than thewild type. Immunoprecipitation of metabolically labeled proteinsin transfected COS-7 cells showed that a large proportion of thewildtype Tbce was still present after 24 h (data not shown),whereas most of the mutant Tbce had disappeared after 8 h (Fig.4c). Wildtype and mutant proteins synthesized in vitro also
showed subtle differences in their susceptibility to proteolysis.When 35S-labeled proteins synthesized in vitro were subjected tolimited proteolysis with trypsin, the mutant Tbce was moderatelymore susceptible to proteolysis than its wildtype counterpart overa narrow range of 3–6 µg ml–1 trypsin (Fig. 4d), consistent with aputative conformational difference between the two proteins.
To evaluate whether the observed mutation in Tbce ofpmn/pmn mice affects the expression of tubulin in peripheralnerves, we analyzed cross-sections of sciatic nerves from wild-type and pmn/pmn mice by double immunofluorescence label-ing. We used monoclonal antibodies against either the α- orβ-subunit of tubulin and polyclonal antibodies against neurofila-ment medium chain as a control. Large-caliber axons of normalmice were immunoreactive to both tubulin subunits, and weobserved a small reduction in immunoreactivity to α- and β-tubulin in the degenerating axons of pmn/pmn mice (Fig. 5a–d).
Finally, we evaluated the microtubule structure in axons byelectron microscopy. Phrenic and sciatic nerves of pmn/pmn micethat were clinically unaffected (one week old), mildly affected(two weeks old) or severely affected (three to four weeks old) wereanalyzed together with nerves of control mice and phenotypicallynormal H1-transgenic pmn/pmn mice. In phrenic nerves fromunaffected pmn/pmn mice at one week of age, all axons showednormal microtubules, but in mildly affected two-week-old mice,20% of axons showed loss of microtubules, and in severelyaffected mice, all microtubules were lost but intermediate fila-ments were still present (Fig. 6a–d). We also observed occasionalnerve-fiber collapses on 10% of the sections, probably originatingfrom degenerating motor neurons. At the same age (3–4 weeks),microtubules were still observed in 15–20% of sciatic nerve fibers
Fig. 4 Analysis of Tbce protein stabil-ity. a,b, Western-blot analysis ofCOS-7 (a) and HeLa (b) cell extractsobtained 24 h and 48 h after trans-fection with wildtype or mutatedTbce expression constructs. Theexpected fusion protein (60 kD) wasdetected in both constructs with theantibody against FLAG. Mutant Tbce(m) was less abundant in COS-7 cellsthan wildtype Tbce (wt), and in HeLacells was barely detected 24 h aftertransfection and not detected after48 h. Co-transfection with a plasmidcontaining the GFP gene was used asa transfection efficiency control, andthe band detected with GFP anti-serum is shown. Transfection with-out plasmid was used as a negativecontrol (c). c, COS-7 cells were trans-fected with either wildtype (wt) ormutated (m) Tbce cDNA, metaboli-cally labeled with TranS35-label andthen chased for 4 h and 8 h. Afterimmunoprecipitation and SDS–PAGE, we observed that the wild-type protein was intact after 8 h,whereas most of the mutant Tbcehad disappeared. d, Autoradiograph of 35S-labeled wildtype (wt) or mutant (m) Tbce proteins translated in vitro using a rabbit reticulocyte lysate andtreated with 3 µg ml–1 of trypsin for 5, 10 and 20 min.
letter
nature genetics • volume 32 • november 2002 445
Fig. 5 Immunofluorescent analysis of tubulin levels. Sciatic nerve cross-sectionsfrom 4-week-old wildtype (a,b) and pmn/pmn (c,d) littermate mice were dou-bly labeled with antibodies against α-tubulin (a,c) and neurofilament mediumchain (b,d). High α-tubulin and neurofilament levels were observed in thelarge-caliber axons from wildtype mice (asterisks). A typical aspect of axonaldegeneration, with reduced and irregular caliber fibers, was observed in thepmn/pmn mice (c,d). Large-caliber axons from pmn/pmn mice had reducedimmunoreactivity to α-tubulin (c; arrowheads), compared with neurofilamentmedium chain (d). Similar results were obtained when using antibody againstβ-tubulin (data not shown). Scale bar: 5 µm.
a b
c d
a b
c d
©20
02 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reg
enet
ics
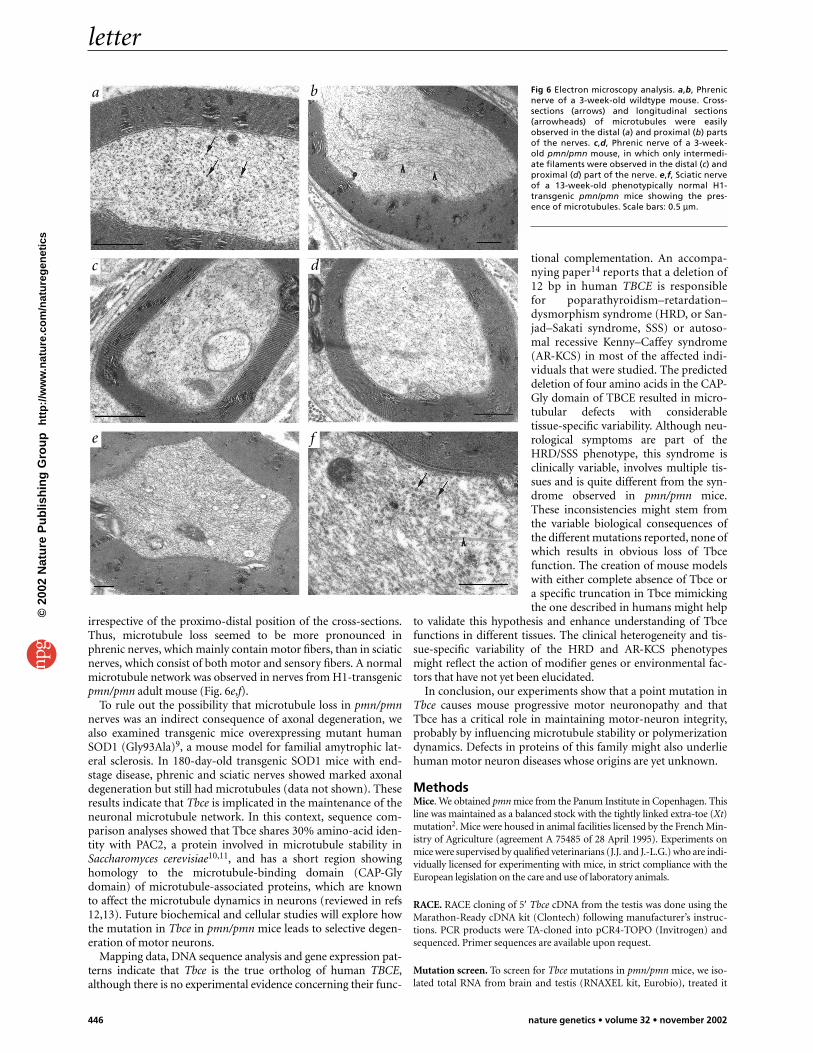
irrespective of the proximo-distal position of the cross-sections.Thus, microtubule loss seemed to be more pronounced inphrenic nerves, which mainly contain motor fibers, than in sciaticnerves, which consist of both motor and sensory fibers. A normalmicrotubule network was observed in nerves from H1-transgenicpmn/pmn adult mouse (Fig. 6e,f).
To rule out the possibility that microtubule loss in pmn/pmnnerves was an indirect consequence of axonal degeneration, wealso examined transgenic mice overexpressing mutant humanSOD1 (Gly93Ala)9, a mouse model for familial amytrophic lat-eral sclerosis. In 180-day-old transgenic SOD1 mice with end-stage disease, phrenic and sciatic nerves showed marked axonaldegeneration but still had microtubules (data not shown). Theseresults indicate that Tbce is implicated in the maintenance of theneuronal microtubule network. In this context, sequence com-parison analyses showed that Tbce shares 30% amino-acid iden-tity with PAC2, a protein involved in microtubule stability inSaccharomyces cerevisiae10,11, and has a short region showinghomology to the microtubule-binding domain (CAP-Glydomain) of microtubule-associated proteins, which are knownto affect the microtubule dynamics in neurons (reviewed in refs12,13). Future biochemical and cellular studies will explore howthe mutation in Tbce in pmn/pmn mice leads to selective degen-eration of motor neurons.
Mapping data, DNA sequence analysis and gene expression pat-terns indicate that Tbce is the true ortholog of human TBCE,although there is no experimental evidence concerning their func-
tional complementation. An accompa-nying paper14 reports that a deletion of12 bp in human TBCE is responsiblefor poparathyroidism–retardation–dysmorphism syndrome (HRD, or San-jad–Sakati syndrome, SSS) or autoso-mal recessive Kenny–Caffey syndrome(AR-KCS) in most of the affected indi-viduals that were studied. The predicteddeletion of four amino acids in the CAP-Gly domain of TBCE resulted in micro-tubular defects with considerabletissue-specific variability. Although neu-rological symptoms are part of theHRD/SSS phenotype, this syndrome isclinically variable, involves multiple tis-sues and is quite different from the syn-drome observed in pmn/pmn mice.These inconsistencies might stem fromthe variable biological consequences ofthe different mutations reported, none ofwhich results in obvious loss of Tbcefunction. The creation of mouse modelswith either complete absence of Tbce ora specific truncation in Tbce mimickingthe one described in humans might help
to validate this hypothesis and enhance understanding of Tbcefunctions in different tissues. The clinical heterogeneity and tis-sue-specific variability of the HRD and AR-KCS phenotypesmight reflect the action of modifier genes or environmental fac-tors that have not yet been elucidated.
In conclusion, our experiments show that a point mutation inTbce causes mouse progressive motor neuronopathy and thatTbce has a critical role in maintaining motor-neuron integrity,probably by influencing microtubule stability or polymerizationdynamics. Defects in proteins of this family might also underliehuman motor neuron diseases whose origins are yet unknown.
MethodsMice. We obtained pmn mice from the Panum Institute in Copenhagen. Thisline was maintained as a balanced stock with the tightly linked extra-toe (Xt)mutation2. Mice were housed in animal facilities licensed by the French Min-istry of Agriculture (agreement A 75485 of 28 April 1995). Experiments onmice were supervised by qualified veterinarians (J.J. and J.-L.G.) who are indi-vidually licensed for experimenting with mice, in strict compliance with theEuropean legislation on the care and use of laboratory animals.
RACE. RACE cloning of 5′ Tbce cDNA from the testis was done using theMarathon-Ready cDNA kit (Clontech) following manufacturer’s instruc-tions. PCR products were TA-cloned into pCR4-TOPO (Invitrogen) andsequenced. Primer sequences are available upon request.
Mutation screen. To screen for Tbce mutations in pmn/pmn mice, we iso-lated total RNA from brain and testis (RNAXEL kit, Eurobio), treated it
Fig 6 Electron microscopy analysis. a,b, Phrenicnerve of a 3-week-old wildtype mouse. Cross-sections (arrows) and longitudinal sections(arrowheads) of microtubules were easilyobserved in the distal (a) and proximal (b) partsof the nerves. c,d, Phrenic nerve of a 3-week-old pmn/pmn mouse, in which only intermedi-ate filaments were observed in the distal (c) andproximal (d) part of the nerve. e,f, Sciatic nerveof a 13-week-old phenotypically normal H1-transgenic pmn/pmn mice showing the pres-ence of microtubules. Scale bars: 0.5 µm.
letter
446 nature genetics • volume 32 • november 2002
a b
c d
e f
©20
02 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reg
enet
ics

letter
nature genetics • volume 32 • november 2002 447
with DNase (Invitrogen) and reverse transcribed it (Superscript II, Gibco).We used single-stranded cDNA with gene-specific primers to amplify over-lapping products of 200–700 bp by PCR (Advantage-HF 2 PCR Kit, Clon-tech). We gel-purified the PCR products, subcloned them in pCR-TOPOvectors (Invitrogen) and sequenced the clones. For each allele, we clonedand sequenced at least three independently generated PCR products.Primer sequences are available upon request.
Transgenic mice. We constructed the pNSE-Ex4·mTBCE plasmid by insert-ing a full-length Tbce cDNA obtained by PCR (HF2-Kit, Clontech) into theHindIII site of the pNSE-Ex4 vector15. To prepare DNA for oocyte microin-jection, the vector was digested with SalI, and the fragment of roughly 6.7 kbwas isolated in an agarose gel (QiaexII kit, Qiagen). Microinjections ofC57BL/6J × DBA/2J F2 hybrid oocytes were done by the Pasteur TransgenicCore Facility. We analyzed tail-extracted DNA of the progeny by PCR.
Northern-blot and RT–PCR analyses. We probed multiple-tissue north-ern blots (Clontech) with a Tbce cDNA probe of 707 bp (generated withprimers Tbce-1F and -2R) following the manufacturer’s recommenda-tions. The probe was labeled by random priming and hybridized for 1 h at68 °C in ExpressHyb (Clontech). After washing, we detected and quanti-fied signals with a Storm 840 phosphorimager. To assess transgene expres-sion, we isolated total RNA from spinal cord (RNAXEL kit, Eurobio), treat-ed it with DNase (Invitrogen) and reverse transcribed it (Superscript II,Gibco). Single-stranded cDNA was used in PCR. Primer sequences areavailable upon request.
In situ hybridization and immunofluorescent analyses. Mice were deeplyanesthetized with xylazine and ketamine, and perfused intracardially with4% (w/v) paraformaldehyde in phosphate-buffered saline (PBS), pH 7.4.Peripheral nerves and spinal cord were dissected out, postfixed, cryoprotect-ed for 48 h with 30% (w/v) sucrose in PBS, frozen in Tissue-Tek andprocessed for in situ hybridization or immunofluorescence. Briefly, lumbarspinal cord cross-sections (20 µm) were hybridized overnight at 70 °C withdigoxigenin-labeled RNA probes. We synthesized antisense or sense probesfor Tbce (length 707 bp) or for choline acetyltransferase by in vitro transcrip-tion. Sections were reacted with alkaline phosphatase–coupled antibodiesagainst digoxigenin (Boehringer), the antibodies were revealed withNBT/BCIP, and the sections were dehydrated and analyzed on a Zeiss Axio-phot microscope. We incubated cross-sections of sciatic nerves (7 µm) withpolyclonal antibodies against neurofilament medium chain (Chemicon,Ab1987) and monoclonal antibodies against either α-tubulin (Sigma, cloneT5168) or βIII-tubulin (Babco, clone TUJ1). After incubation with Alexa-488 and Texas-Red–coupled secondary antibodies (Jackson Research Labo-ratories), we mounted sections with Vectashield/DAPI (Vector). We exam-ined nerves from mutant and control mice (n = 3 each) in parallel on a ZeissLSM 410 confocal microscope using identical parameters.
Expression vectors and protein analysis. We amplified pmn/pmn andwildtype Tbce cDNA by PCR (HF-2-Clontech kit) and cloned both TbcecDNAs into the pCMV-Tag2 vector (Stratagene). For western-blot analysis,we transiently transfected (Lipofectamine plus, Gibco) COS-7 and HeLacells with 2 µg of the pmn/pmn or wildtype plasmid plus 1 µg of pHook3-GFP plasmid as control (gift from D. Feigelstock). Three independentexperiments were carried out (2 dishes each time). Cells were harvestedafter 24 h and 48 h and lysed in RIPA buffer (300 mM NaCl, 10mM Tris-HCl, pH 7.4, 5 mM EDTA, 1% (v/v) Nonidet P-40) supplemented withprotease inhibitors (Complete cocktail, Roche). We subjected 10 µg of cel-lular extracts to SDS–PAGE (4–12% NuPAGE Bis-Tris gels, Invitrogen)and transferred them to nitrocellulose membranes (Invitrogen). Proteinswere detected with monoclonal antibody against FLAG M2 (Stratagene,1:1,000) or GFP antiserum (Invitrogen, 1:5,000) using chemiluminescence(ECL; Amersham).
For pulse–chase experiments, we transfected (Lipofectamine plus,Gibco) COS-7 cells (0.8 million per well) with 2 µg of the pmn/pmn orwildtype plasmid plus 1 µg pcDNA-lacZ control plasmid. Eight indepen-dent experiments were done. Thirty-six hours after transfection, cells werestarved in Met– Cys– serum-free medium for 30 min, labeled with 40 µCiTranS35-label (ICN) for 20 min and cultured in complete DMEM mediumfor different chase periods. We sonicated cell pellets in 200 µl buffer (50mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100) with
protease inhibitors (Complete, Roche) and immunoprecipitated 3 millioncpm of each sample with agarose slurry containing antibody against FLAG(Sigma) following manufacturer’s directions.
For limited proteolysis analysis, we synthesized 35S-labeled Tbce pro-teins in vitro using the rabbit reticulocyte lysate transcription–transla-tion system (TnT, Promega). We incubated 10 µl TnT products withtrypsin at 30 °C for 5–20 min and analyzed products by 9% SDS–PAGEand fluorography. We did ten independent experiments in which trypsinconcentrations ranged from 0.6–12 µg ml–1 to find the optimal concen-tration of trypsin in which pmn/pmn and wildtype Tbce behaved differ-ently (3 or 6 µg ml–1), and then repeated the experiment five times usingthese conditions.
Morphological analysis of microtubules in axons. Mutant pmn/pmnmice, normal control mice, H1-transgenic rescued pmn/pmn mice ortransgenic mice overexpressing human SOD1 with the Gly93Ala mutation(line G1H) were killed with an overdose of Avertin. We fixed phrenicnerves in situ for 1 h with 2.5% (w/v) glutaraldehyde (EM grade, Sigma) in0.1 M cacodylate buffer, pH 7.5, containing 0.8% (w/v) tannic acid, andthen divided the nerves into distal and proximal parts. We briefly fixed sci-atic nerves in situ, dissected them out, stretched them for 1 h in fixative andthen divided them into distal, median and proximal parts. Samples werethen postfixed with 1% (w/v) osmium tetroxide in the same buffer, dehy-drated with an acetone series and embedded in epoxy resin. Thin sectionswere conventionally prepared and observed with a Philips CM12 electronmicroscope. To evaluate the extent of the syndrome, we checked 30–65cross-sections of nerves for the presence of microtubules.
GenBank accession numbers. Tbce mRNA sequence: AY082332.
AcknowledgmentsWe thank C. Babinet, M. Cohen-Tannoudji and C. Henderson for helpfuladvice, J. Segre for critical reading of our manuscript, U. Maskos forproviding the pNSE plasmid, L. Salas-Cortes for assistance in cell culture andV. Guyot for generation of transgenic mice. This work was supported bygrants from the Association Française contre les Myopathies. N.M. benefitedfrom a fellowship of the Association Française contre les Myopathies.
Competing interests statementThe authors declare that they have no competing financial interests.
Received 29 July; accepted 12 September 2002.
1. Schmalbruch, H.M.D., Jensen, H., Bjearg, M., Kamienniecka, Z. & Kurland, L.B.S. Anew mouse mutant with progressive motor neuronopathy. J. Neuropathol. Exp.Neurol. 50, 192–204 (1991).
2. Bueno Brunialti, A.L., Poirier, C., Schmalbruch, H. & Guénet, J.-L. The mousemutation progressive motor neuronopathy (pmn) maps to chromosome 13.Genomics 29, 131–135 (1995).
3. Viollet, L. et al. cDNA isolation, expression, and chromosomal localization of themouse survival motor neuron gene (Smn). Genomics 40, 185–188 (1997).
4. Martin, N., Jaubert, J., Glaser, P., Szatanik, M. & Guénet, J.-L. Genetic and physicaldelineation of the region overlapping the progressive motor neuronopathy(pmn) locus on mouse chromosome 13. Genomics 75, 9–16 (2001).
5. Radcliffe, P.A., Hirata, D., Vardy, L. & Toda, T. Functional dissection and hierarchyof tubulin-folding cofactor homologues in fission yeast. Mol. Biol. Cell 10,2987–3001 (1999).
6. Lewis, S.A., Tian, G., Vainberg, I.E. & Cowan, N.J. Chaperonin-mediated folding ofactin and tubulin. J. Cell Biol. 132, 1–4 (1996).
7. Tian, G. et al. Pathway leading to correctly folded β-tubulin. Cell 86, 287–296(1996).
8. Grishchuk, E.L. & McIntosh, J.R. Sto1p, a fission yeast protein similar to tubulinfolding Cofactor E, plays an essential role in mitotic microtubule assembly. J. CellScience 112, 1979–1988 (1999).
9. Gurney, M.E. et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264, 1772–1775 (1994).
10. Hoyt, M.A., Macke, J.P., Roberts, B.T. & Geiser, J.R. Saccharomyces cerevisiae PAC2functions with CIN1, 2 and 4 in a pathway leading to normal microtubulestability. Genetics 146, 849–857 (1997).
11. Fleming, J.A., Vega, L.R. & Solomon, F. Function of tubulin-binding proteins invivo. Genetics 156, 69–80 (2000).
12. Hirokawa, N. Microtubule organization and dynamics dependent onmicrotubule-associated proteins. Current Opinion in Cell Biology 6, 74–81 (1994).
13. Hunter, A.W. & Wordeman, L. How motor proteins influence microtubulepolymerisation dynamics. J. Cell Science 113, 4379–4389 (2000).
14. Parvari, R. et al. Mutation of TBCE causes hypoparathyroidism–retardation–dysmorphism syndrome and autosomal recessive Kenny–Caffey Syndrome.Nature Genet. 32, 448–442 (2002).
15. Mucke, L. et al. Synaptotrophic effects of human amyloid beta protein precursorsin the cortex of transgenic mice. Brain Res. 666, 151–167 (1994).
©20
02 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reg
enet
ics