a distal enhancer controls cytokine-dependent human gene ... · downstream effects of pro- and...
TRANSCRIPT

This article is available online at http://www.jlr.org Journal of Lipid Research Volume 54, 2013 1915
Copyright © 2013 by the American Society for Biochemistry and Molecular Biology, Inc.
mice, which display age-dependent renal concentrating defects, enlarged hearts, defects in female reproduction (comparable to COX-2 � / � mice), and small intestine ulcerative lesions ( 3, 4 ). When challenged with lipopoly-saccharide , HCl ( 5 ), butylated hydroxytoluene ( 6 ), or urethane ( 7 ), these mice have attenuated lung infl am-mation and tumor formation. Most recently, in vitro stud-ies, animal models, and human brain imaging analysis have also linked cPLA 2 to the onset and maintenance of Alzheimer’s disease pathogenesis ( 8 ).
As the rate-limiting step in eicosanoid production, cPLA 2 � activity has also been coupled to events control-ling signal transduction ( 9 ), apoptosis ( 10 ), infl ammation ( 11 ), and cancer ( 12 ). The downstream eicosanoids have also been associated with a number of infl ammatory diseases, including asthma ( 13 ), atherosclerosis, sepsis, Crohn’s dis-ease ( 14 ), and rheumatoid arthritis ( 15 ), demonstrating the critical roles these bioactive lipids have in normal cel-lular function and disease pathologies.
It is well documented that the expression and enzymatic activity of cPLA 2 � are increased by numerous mediators, including interleukin (IL)-1 � ( 15, 16 ), tumor necrosis fac-tor (TNF)- � , interferon (IFN)- � ( 17 ), macrophage colony stimulating factor (M-CSF) ( 18 ), and oncogenic Ras ( 19 ), with enzymatic activity requiring calcium increases and protein phosphorylation ( 4 ). Wu et al. ( 20 ) initially char-acterized the basal expression of the human cPLA 2 � pro-moter with complementary studies on the rat promoter by Tay et al. ( 21 ). A number of regulatory factors have been implicated directly and indirectly in the regulation of
Abstract Specifi c control of group IVA cytosolic phospho-lipase A 2 (cPLA 2 � or PLA2G4A) expression modulates arachidonic acid production, thus tightly regulating the downstream effects of pro- and anti-infl ammatory eico-sanoids. The signifi cance of this pathway in human disease is apparent in a range of pathologies from infl ammation to tumorigenesis. While much of the regulation of cPLA 2 � has focused on posttranslational phosphorylation of the pro-tein, studies on transcriptional regulation of this gene have focused only on proximal promoter regions. We have identi-fi ed a DNase I hypersensitive site encompassing a 5 ′ distal enhancer element containing a highly conserved consensus AP-1 site involved in transcriptional activation of cPLA 2 � by interleukin (IL)-1 � . Chromatin immunoprecipitation (ChIP), knockdown, knockout, and overexpression analyses have shown that c-Jun acts both in a negative and positive regulatory role. Transcriptional activation of cPLA 2 � occurs through the phosphorylation of c-Jun in conjunction with increased association of C/EBP � with the distal novel en-hancer. The association of C/EBP � with the transcriptional activation complex does not require an obvious DNA bind-ing site. These data provide new and important contribu-tions to the understanding of cPLA 2 � regulation at the transcriptional level, with implications for eicosanoid me-tabolism, cellular signaling, and disease pathogenesis. —Bickford, J. S., D. E. Beachy, K. J. Newsom, S. J. Barilovits, J.-D. H. Herlihy, X. Qiu, J. N. Walters, N. Li, and H. S. Nick. A distal enhancer controls cytokine-dependent human cPLA 2 � gene expression. J. Lipid Res. 2013. 54: 1915–1926.
Supplementary key words group IVA phospholipase A2 • gene regu-lation • c-Jun • CCAAT/enhancer-binding protein �
As the apex of the eicosanoid pathway, group IVA cyto-solic phospholipase A 2 , cPLA 2 � /PLA2G4A, is responsible for liberation of arachidonic acid from the sn-2 position of membrane phospholipids, leading to prostaglandin and leukotriene biosynthesis ( 1, 2 ). The physiological rele-vance of this gene is supported by studies in cPLA 2 � � / �
This work was supported by National Institutes of Health Grants R37-HL-067456 and RO1-HL-39593 (to H.S.N.).
Manuscript received 28 February 2013 and in revised form 27 March 2013.
Published, JLR Papers in Press, April 2, 2013 DOI 10.1194/jlr.M037382
A distal enhancer controls cytokine-dependent human cPLA 2 � gene expression
Justin S. Bickford , * Dawn E. Beachy , * Kimberly J. Newsom , * Sarah J. Barilovits , * ,† John-David H. Herlihy , * Xiaolei Qiu , * ,† Jewell N. Walters , * ,† Ning Li , * ,† and Harry S. Nick 1, * ,†
Department of Neuroscience* and Department of Biochemistry and Molecular Biology, † University of Florida , Gainesville, FL
Abbreviations: C/EBP � , CCAAT/enhancer-binding protein � ; ChIP, chromatin immunoprecipitation; COX, cyclooxygenase; cPLA 2 � , cyto-solic phospholipase A2 alpha; DAPA, DNA affi nity purifi cation assay; EMSA, electrophoretic mobility shift assay; HFL-1, human fetal lung fi broblast; hGH, human growth hormone; HS, hypersensitive ; IFN, interferon; IL, interleukin; MEF, mouse embryonic fi broblast; mPGES, microsomal prostaglandin E synthase; Pol II, RNA polymerase II; siRNA, small interfering RNA; TK, thymidine kinase; TNF, tumor necrosis factor; WT, wild-type.
1 To whom correspondence should be addressed. e-mail: hnick@ufl .edu
The online version of this article (available at http://www.jlr.org) contains supplementary data in the form of three fi gures.
by guest, on July 13, 2018w
ww
.jlr.orgD
ownloaded from
.html http://www.jlr.org/content/suppl/2013/04/02/jlr.M037382.DC1Supplemental Material can be found at:

1916 Journal of Lipid Research Volume 54, 2013
Cell culture Human fetal lung fi broblast (HFL-1) cells or a human bron-
choepithelial cell line (S9) ( 34 ) were maintained in nutrient mix-ture F12 Ham Kaighn’s modifi cation (F12K) at pH 7.4 supplemented with 25 mM NaHCO3, 4 mM glutamine, antibiotic/antimycotic (ABAM) (Gibco) and 10% FBS (Atlanta Biologicals) at 37°C with 5% CO 2 . Wild-type (WT) and knockout mouse embryonic fi bro-blasts (MEF) were grown in DMEM (CellGro) with identical sup-plements. The C/EBP � � / � cell line was provided by Dr. P. Johnson, National Institutes of Health, via Dr. Michael Kilberg (University of Florida), and the c-Jun � / � and c-Jun overexpressing cell lines ( 35, 36 ) were kindly provided by Dr. David A. F. Gillespie (University of Glasgow, United Kingdom).
DNase I hypersensitive (HS) site and Southern blot analysis Ten 150 mm dishes of HFL-1 cells were grown to 75% confl u-
ency, then either left untreated or treated for 8 h with IL-1 � . Cell permeabilization, DNase I treatment, and Southern analysis were performed as previously described ( 37, 38 ). Following DNase I treatment, cell lysates were incubated overnight at 50°C, and ge-nomic DNA was isolated by organic extraction, alcohol precipita-tion, and resuspension in TE. The DNA samples were digested with BamHI, and 30 µg of each sample were fractionated on a 0.8% HGT agarose gel followed by alkaline denaturation, elec-trotransfer to a nylon membrane, and UV cross-linking. The membrane was hybridized at 61°C with an end-specifi c single copy 32 P-radiolabeled DNA probe, generated by PCR from a hu-man cPLA 2 � genomic clone that indirectly end-labeled the 3 ′ end of the 18.8 kb BamHI fragment as depicted in Fig. 1 .
RNA isolation, real-time RT-PCR, and northern analysis Total RNA was purifi ed using the Qiagen RNeasy Mini Kit with
DNase I digestion or by the Chomczynski and Sacchi method with modifi cations as previously described ( 39 ). For real-time PCR anal-ysis, cDNA was generated using a SuperScript First-Strand synthesis kit (Invitrogen) as per the manufacturer’s instructions. Two micro-liters of the resulting cDNA was used for real-time PCR analysis using SYBR Green Supermix with ROX (Bio-Rad) on a Bio-Rad iCycler and analyzed by the � � C T method normalized to cyclophi-lin A as described previously ( 40 ). Crossing threshold (CT) values from both cyclophilin A and respective target genes were used in the � � CT method to calculate relative fold inductions, which were statistically analyzed as described below ( 40 ). Real-time primers were as follows: cPLA 2 � , F: (5 ′ -CGT GAT GTG CCT GTG GTA GC -3 ′ ) R: (5 ′ -TCT GGA AAA TCA GGG TGA GAA TAC -3 ′ ); cyclo-philin A, F: (5 ′ -CAT CCT AAA GCA TAC GGG TCC-3 ′ ) R: (5 ′ -GCT GGT CTT GCC ATT CCT G-3 ′ ); human growth hormone, F: (5 ′ -GAA CCC CCA GAC CTC CCT-3 ′ ) R: (5 ′ - CAT CTT CCA GCC TCC CCA T-3 ′ ). For northern analysis, 10–20 � g of total RNA was dena-tured and fractionated on 1% agarose, 6% formaldehyde gel, then electrotransferred to a Zeta-Probe blotting membrane (Bio-Rad) and UV cross-linked. Membranes were hybridized with a gene-spe-cifi c 32 P-radiolabeled probe generated by random primer exten-sion (Invitrogen) for human growth hormone (hGH), cPLA 2 � , or the large ribosomal subunit protein, L7a. The membrane was washed at 65°C and exposed to X-ray fi lm. Densitometry was per-formed with ImageJ software (http://imagej.nih.gov/ij/). Rela-tive fold inductions were determined as untreated cells compared with IL-1 � -treated samples, normalized to the ribosomal protein L7a loading control.
Generation of promoter and enhancer constructs in human growth hormone reporter vectors
Various regions of the cPLA 2 � promoter were subcloned into the promoterless pUC12-based hGH reporter plasmid pØGH ( 41 ).
cPLA 2 � expression and activity through the proximal pro-moter, including nuclear factor (NF)- � B ( 22, 23 ), Krüppel-like factor (KLF) ( 24, 25 ), hypoxia-inducible factor (HIF-1) ( 26 ), and specifi city protein 1 (Sp1) ( 27 ). A 48 bp CA dinucleotide repeat at approximately � 200 bp has been shown to confer a negative regulatory effect on cPLA 2 � basal promoter activity ( 16, 28 ). Cowan et al. ( 29 ) have also implicated an Inr element at the transcriptional start site and a novel TBP binding site at � 30 to � 35 bp. Nem-enoff and coworkers have demonstrated that oncogenic forms of Ras ( 19 ) increase transcription of cPLA 2 � in nor-mal lung epithelial cells and non-small-cell lung carcinoma (NSCLC) cell lines through proximal promoter elements that bind lung Krüppel-like factor (LKLF) ( 24 ) and Sp1 ( 27 ) with induction, requiring the activation of the c-Jun N-terminal kinases (JNK) and extracellular signal-regulated kinases (ERK) pathways ( 30 ). Most recently, Tsou et al. ( 31 ) have implicated nucleolin through proximal c-Jun/Sp1 promoter elements in the transcriptional activation of cPLA 2 � by phorbol ester.
Chi et al. ( 32 ) have very recently argued that a puta-tive AP-1 site in the proximal promoter at approximately � 500 bp (TGATTAA), deviating from the consensus AP-1 sequence [TGA(G/C)TCA], is involved in IL-1 � -dependent induction of cPLA 2 � in rheumatoid arthritis synovial fi bro-blasts. We and others have clearly demonstrated that the human cPLA 2 � promoter (in our studies, extending to approximately � 6.8 kb) is not responsive to cytokine acti-vation ( 21–31 ). This discrepancy may result from these investigators’ use of diseased synovial fi broblasts in con-junction with IL-1 � concentrations 15 times (30 ng/ml) higher than our dosage and well beyond physiologically relevant levels. Liao et al. ( 33 ) reported a 1.4-fold response from the proximal promoter, although neither an induc-tion nor basal activity was readily detectable in any of our experiments with the promoter alone.
Given the central importance of cPLA 2 � regulation and activity in normal physiology, infl ammation, and cancer, we sought to identify a true stimulus-dependent regulatory element responsible for transcriptional regulation of the cPLA 2 � gene. In this study, we have characterized a distal DNase I hypersensitive (HS) site that harbors an IL-1 � -responsive element. Our results implicate c-Jun as a tran-scriptional repressor through direct interaction with the enhancer, while transcriptional activation is mediated through association of C/EBP � with both enhancer and promoter elements.
MATERIALS AND METHODS
Reagents FuGENE 6 transfection reagent and complete protease inhibi-
tor cocktail were purchased from Roche Applied Science, IL-1 � from R and D Systems, trichostatin A from Calbiochem, restriction enzymes from New England Biolabs, DNase I from Worthington Biochemical, and antibodies from Santa Cruz Biotechnology. The random-primers DNA labeling kit was purchased from Invitrogen, and the Qiagen RNeasy Mini Kit was purchased from Qiagen.
by guest, on July 13, 2018w
ww
.jlr.orgD
ownloaded from
.html http://www.jlr.org/content/suppl/2013/04/02/jlr.M037382.DC1Supplemental Material can be found at:
Defi ning a cytokine-dependent cPLA 2 � enhancer element 1917
BAC clones AL022147 and AL049797 (Genetics Institute, Inc., and the Sanger Center) were used as templates for PCR amplifi -cation. The � 14, � 5.5, and � 4.1 kb cPLA 2 � promoters were am-plifi ed from AL022147 using the Long Range PCR kit (Roche), with the forward primers 5 ′ -AGAGTTGGGATGGAGAAGGTTG-3 ′ , 5 ′ -TGCAAAGTGCCTGCCAGTC-3 ′ , or 5 ′ -GATG GAGA TGG CAG-TGGCAG-3 ′ , respectively, and the reverse primer 5 ′ -GCT TA-CAGTTCCCAGAGTTACC-3 ′ , and cloned into the TOPO-XL vector. The fragments were removed by BamHI using an adja-cent 5 ′ TOPO-XL vector site and a 3 ′ internal site (+36 bp) and subcloned into the BamHI site of the pØGH vector, creating the � 14, � 5.5, and � 4.1 kb hGH vectors. The � 6.8, � 4.8, and � 1.4 kb promoter constructs were generated from the � 14 kb pGH vector by restriction digest with HindIII, HincII, or XbaI, respec-tively, at an adjacent 5 ′ vector-specifi c site and an internal site, followed by religation of the resulting plasmid. The 2.3 kb fragment fl anking the DNase I HS site was amplifi ed from the � 14 kb promoter with the forward and reverse primers 5 ′ -GAGATGGTGGCAAAGGAATG-3 ′ and 5 ′ -GACG GGCG AATC AC-AAG-3 ′ , respectively, and cloned into the NdeI site upstream of either the � 1.4 kb cPLA 2 � or the minimal viral thymidine kinase (TK) promoter in an hGH reporter plasmid. The 1.6 kb enhancer fragment was derived by digestion of the 2.3 kb construct with Hin dIII followed by religation. Fragment IV was created by Quikchange mutagenesis of the 2.3 kb fragment to delete all cPLA 2 � sequences upstream of the � 9161 bp Hin dIII site. Frag-ments I, II, III, and V were created by Quikchange mutagenesis of the 1.6 kb fragment: enhancer fragment I was created by dele-tion from � 10140 to � 9341, II by deletion from � 10745 to � 10159 and from � 9632 to � 9340, III by deletion from � 10741 to � 9654, and V by deletion from � 10745 to � 10159. Site-di-rected Quikchange mutagenesis was also used to delete the puta-tive AP-1 and control sites from fragment III.
Transient transfection The cPLA 2 � promoter/enhancer fragments in the pØGH and
pTKhGH reporter plasmids were transiently transfected into HFL-1 cells at approximately 70–80% confl uency using a 1:3 ratio of FuGENE 6 (Roche) to 5 � g of reporter plasmid. To ensure equal transfection effi ciency, batch transfection was utilized, where 24 h after DNA transfection, cells were split 1:2 into 10 cm dishes. After an additional 24 h, one plate served as the control while the other plate was treated with IL-1 � (2 ng/µl) for 8 h followed by RNA isolation. Two micrograms of full-length C/EBP � /LAP* in a pcDNA3.1 expression plasmid was transfected into S9 cells. After 48 h, total RNA was collected for analysis.
Immunoblot analysis Total cellular protein (30 � g) from the indicated time points
were separated on a Tris-HCl polyacrylamide gel (Bio-Rad) and electrotransferred to a nitrocellulose membrane (Bio-Rad). The membranes were then blocked for 1 h with 8% non-fat dry milk in TBST [10 mM Tris-HCl (pH 7.5), 0.1% (v/v) Tween 20, 200 mM NaCl] at room temperature. The mem-branes were incubated overnight at 4°C with primary antibod-ies to cPLA 2 � (Santa Cruz, sc-454), c-Jun (Santa Cruz, sc-1694×), p-c-Jun (phosphorylated, Santa Cruz, sc-822), C/EBP � (Santa
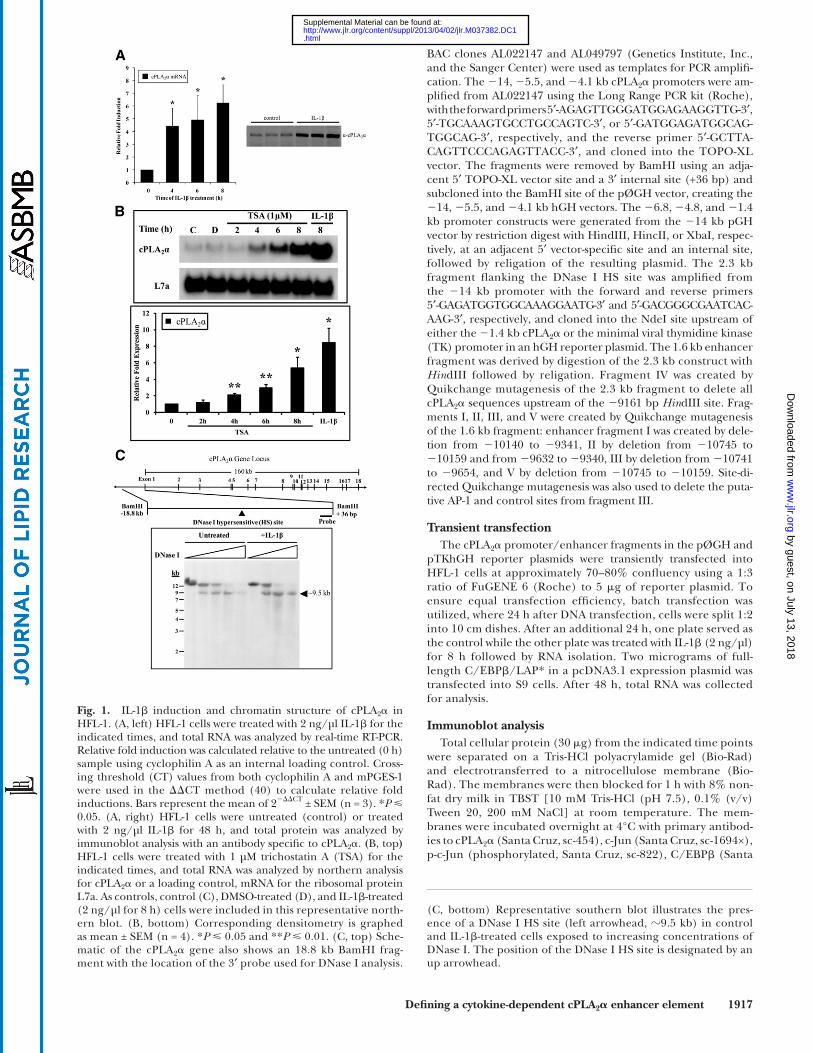
Fig. 1. IL-1 � induction and chromatin structure of cPLA 2 � in HFL-1. (A, left) HFL-1 cells were treated with 2 ng/µl IL-1 � for the indicated times, and total RNA was analyzed by real-time RT-PCR. Relative fold induction was calculated relative to the untreated (0 h) sample using cyclophilin A as an internal loading control. Cross-ing threshold (CT) values from both cyclophilin A and mPGES-1 were used in the � � CT method ( 40 ) to calculate relative fold inductions. Bars represent the mean of 2 � � � CT ± SEM (n = 3). * P 0.05. (A, right) HFL-1 cells were untreated (control) or treated with 2 ng/µl IL-1 � for 48 h, and total protein was analyzed by immunoblot analysis with an antibody specifi c to cPLA 2 � . ( B, top ) HFL-1 cells were treated with 1 µM trichostatin A (TSA) for the indicated times, and total RNA was analyzed by northern analysis for cPLA 2 � or a loading control, mRNA for the ribosomal protein L7a. As controls, control (C), DMSO-treated (D), and IL-1 � -treated (2 ng/µl for 8 h) cells were included in this representative north-ern blot. (B, bottom) Corresponding densitometry is graphed as mean ± SEM (n = 4). * P 0.05 and ** P 0.01. (C, top) Sche-matic of the cPLA 2 � gene also shows an 18.8 kb BamHI frag-ment with the location of the 3 ′ probe used for DNase I analysis.
(C, bottom) Representative southern blot illustrates the pres-ence of a DNase I HS site (left arrowhead, � 9.5 kb ) in control and IL-1 � -treated cells exposed to increasing concentrations of DNase I. The position of the DNase I HS site is designated by an up arrowhead.
by guest, on July 13, 2018w
ww
.jlr.orgD
ownloaded from
.html http://www.jlr.org/content/suppl/2013/04/02/jlr.M037382.DC1Supplemental Material can be found at:

1918 Journal of Lipid Research Volume 54, 2013
were purchased from Santa Cruz. Purifi ed DNA was used for real-time PCR analysis. We analyzed our data relative to the fraction of total isolated chromosomal DNA prior to immunoprecipita-tion (input) rather than to IgG. The data were analyzed both ways, and we strongly feel that presenting the data relative to in-put is more refl ective of any actual changes. The following prim-ers were used: enhancer region, F: (5 ′ -AGG GCA GAT GTT TCT CAG CG-3 ′ ), R: (5 ′ -GCC ATT TTA TTT TAT GTG TAT GTT CTT-3 ′ ); promoter region, F: (5 ′ -CAA ACT CCT GGT TCT AAT AAC TAA GCA-3 ′ ), R: (5 ′ -TTG CTT ACA GTT CCC AGA GTT ACC-3 ′ ).
Immunoprecipitation Due to the low sensitivity of the available serine 63-phosphory-
lated c-Jun (Santa Cruz, sc-822), c-Jun was immunoprecipitated prior to immunoblot analysis. HFL cells were treated with IL-1 � for 4 or 8 h. Cells were collected in PBS, lysed with RIPA lysis buf-fer [150 nm NaCl, 1% NP40, 1.5% DOC, 0.1% SDS, 50 mM Tri-HCl (pH 7.5)] and incubated at 4°C overnight with an antibody against c-Jun (Santa Cruz, sc-1694×). Complex capture was com-pleted by incubating with Protein A Sepharose beads at 4°C for 2 h. Complexes were washed four times with RIPA buffer, fol-lowed by immunoblot analysis with an antibody against serine 63-phosphorylated c-Jun (Santa Cruz, sc-822).
siRNA knockdown HFL-1 cells at 40% confl uency were transfected with a fi nal
concentration of 100 nM Dharmacon SMARTpool siRNA for c-Jun or C/EBP � using DharmaFECT 1 siRNA transfection re-agent. After 72 h, cells were collected and immunoblot analysis was used to assess protein knockdown, followed by real-time RT-PCR for cPLA 2 � gene expression using primers described above.
Statistical analysis Data points are the means from at least three independent
experiments, and error bars represent the standard error of the means (SEM). Statistical analyses were determined by paired or unpaired Student t -test. P 0.05 was considered statistically signifi cant.
RESULTS
Induction of cPLA 2 � and detection of DNase I hypersensitive sites in the cPLA 2 � gene locus
To establish the induction of cPLA 2 � in our cells, HFL-1 cells were treated with IL-1 � for both RNA and protein analyses. Following 4, 6, or 8 h of IL-1 � treatment, cPLA 2 � mRNA was signifi cantly induced in these cells as deter-mined by real-time RT-PCR. cPLA 2 � protein levels were elevated by 48 h of stimulation as shown by immunoblot analysis ( Fig. 1A ). It has been previously shown that induc-tion of cPLA 2 � gene expression by the proinfl ammatory cytokines IL-1 � , TNF- � and IFN- � requires de novo transcrip-tion ( 16, 44 ). To date, limited information exists regard-ing regulatory elements that mediate cytokine-dependent induction of cPLA 2 � , with most data focusing on only basal expression from the proximal promoter ( 16, 24, 27, 32 ).
The state of chromatin acetylation is known to contrib-ute to the chromatin state and the regulation of gene ex-pression. Therefore, we treated HFL-1 cells with a histone deacetylase inhibitor, trichostatin A (TSA), to determine
Cruz, sc-150), or � -actin (Sigma), washed three times with TBST, incubated with a peroxidase-conjugated secondary anti-body for 1 h, washed again three times, and subjected to ECL (Amersham).
Electrophoretic mobility shift assay Nuclear extracts were obtained by rinsing cells with ice-cold
PBS and harvesting in ice-cold PBS with protease inhibitors (Roche). Cells were pelleted by centrifugation at 300 g for 10 min at 4°C, then resuspended in lysis buffer [20 mM HEPES (pH 7.8), 10 mM KCl, 1.5 mM MgCl 2 , 0.2 M EDTA, 1 mM DTT, 1% Triton X-100, protease inhibitors]. Samples were incubated on ice for 15 min, then centrifuged at 1,000 g for 10 min at 4°C to pellet nuclei. Nuclei were resuspended in nuclear extraction buffer [20 mM HEPES (pH 7.8), 500 mM KCl, 1.5 mM MgCl 2 , 0.2 mM EDTA, 1 mM DTT, protease inhibitors]. Samples were rotated at 4°C for 30 min, then centrifuged at 16,000 g for 10 min at 4°C. The supernatant containing nuclear extract was collected, and protein concentration was determined using a BCA assay (Pierce).
Single-stranded oligonucleotides were designed to include the AP-1/CRE-like and consensus AP-1 site in the cPLA2 � enhancer region: sense, 5 ′ -GAATGCCTTGATGACCACTCTCATGA GT-CATT ATGTTTTCAT-3 ′ and antisense, 5 ′ -ATGAAAACATAA-TGACTCATGAGAGTGGTCATCAAGGCATTC-3 ′ . The sense strand was left unlabeled (for cold competitor) or biotinylated (for hot-labeled probes). Probes were annealed by mixing complementary oligonucleotides at a 1:1 ratio in annealing buffer [10 mM Tris-HCl, 1 mM EDTA, 50 mM NaCl (pH 8.0)] through a temperature gradient. For electrophoretic mobility shift assay (EMSA) reactions, 10 µg of nuclear extract was added to a mixture of binding buffer (9 mM Tris, 45 mM KCl, 100 µM DTT), 2 µg herring sperm DNA, 0.0125% glycerol, and for cold competitor reactions, 0.1 pmol unlabeled double-stranded DNA probe, then incubated at room temperature for 5 min. Biotinylated double-stranded DNA probes (0.5 fmol ) were added, followed by incubation at room temperature for 20 min. Samples were run on a 5% Tris-Glycine gel (Bio-Rad) at 200 V, then transferred to a nitrocellulose membrane. Mem-branes were probed for biotin-labeled DNA-protein complexes using a LightShift Chemiluminescent EMSA Kit (Thermo Scientifi c).
DNA affi nity purifi cation assay For DNA affi nity purifi cation assay (DAPA) ( 42 ), 5 � g of bi-
otinylated DNA double-stranded probe and 500 � g nuclear ex-tract (described above) from HFL-1 cells, S9 cells, or mouse fi broblasts was incubated with 4% streptavidin-agarose beads (Sigma). The samples were rotated at room temperature for 1 h, and then centrifuged at 1,000 g for 1 min. The supernatant was removed, and the pellet was washed four times with 1 ml ice-cold PBS, then resuspended in 100 � l SDS loading buffer. Samples were boiled for 5 min and run on a 10% Tris-HCl gel (Bio-Rad). Immunoblot analysis for c-Jun was performed as described.
Chromatin immunoprecipitation analysis Chromatin immunoprecipitation (ChIP) analysis was per-
formed as previously described with an average chromatin soni-cation size of � 500 bp ( 43 ). Briefl y, HFL-1 cells were treated with IL-1 � for 8 h followed by protein-DNA crosslinking with 1% formaldehyde for 10 min. Cell extracts were incubated overnight at 4°C with 2 � g of control rabbit IgG (sc-2027) or the following rabbit antibodies: c-Jun (sc-1694×); C/EBP � (sc-150); RNA Pol II (sc-899), p300 (sc-584×), or Sp1 (sc-59). All antibodies for ChIP
by guest, on July 13, 2018w
ww
.jlr.orgD
ownloaded from
.html http://www.jlr.org/content/suppl/2013/04/02/jlr.M037382.DC1Supplemental Material can be found at:
Defi ning a cytokine-dependent cPLA 2 � enhancer element 1919
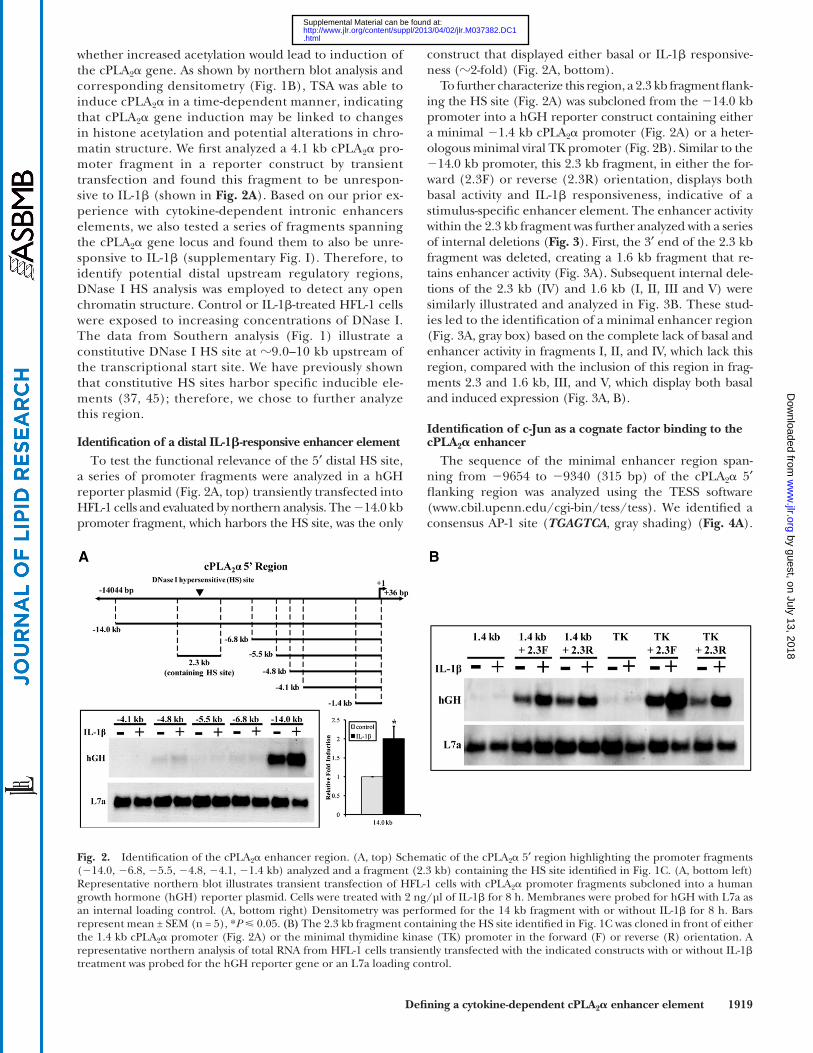
construct that displayed either basal or IL-1 � responsive-ness ( � 2-fold) ( Fig. 2A , bottom).
To further characterize this region, a 2.3 kb fragment fl ank-ing the HS site ( Fig. 2A ) was subcloned from the � 14.0 kb promoter into a hGH reporter construct containing either a minimal � 1.4 kb cPLA 2 � promoter ( Fig. 2A ) or a heter-ologous minimal viral TK promoter ( Fig. 2B ). Similar to the � 14.0 kb promoter, this 2.3 kb fragment, in either the for-ward (2.3F) or reverse (2.3R) orientation, displays both basal activity and IL-1 � responsiveness, indicative of a stimulus-specifi c enhancer element. The enhancer activity within the 2.3 kb fragment was further analyzed with a series of internal deletions ( Fig. 3 ). First, the 3 ′ end of the 2.3 kb fragment was deleted, creating a 1.6 kb fragment that re-tains enhancer activity ( Fig. 3A ). Subsequent internal dele-tions of the 2.3 kb (IV) and 1.6 kb (I, II, III and V) were similarly illustrated and analyzed in Fig. 3B . These stud-ies led to the identifi cation of a minimal enhancer region ( Fig. 3A , gray box) based on the complete lack of basal and enhancer activity in fragments I, II, and IV, which lack this region, compared with the inclusion of this region in frag-ments 2.3 and 1.6 kb, III, and V, which display both basal and induced expression ( Fig. 3A, B ).
Identifi cation of c-Jun as a cognate factor binding to the cPLA 2 � enhancer
The sequence of the minimal enhancer region span-ning from � 9654 to � 9340 (315 bp) of the cPLA 2 � 5 ′ fl anking region was analyzed using the TESS software (www.cbil.upenn.edu/cgi-bin/tess/tess). We identifi ed a consensus AP-1 site ( TGAGTCA , gray shading) ( Fig. 4A ).
whether increased acetylation would lead to induction of the cPLA 2 � gene. As shown by northern blot analysis and corresponding densitometry ( Fig. 1B ), TSA was able to induce cPLA 2 � in a time-dependent manner, indicating that cPLA 2 � gene induction may be linked to changes in histone acetylation and potential alterations in chro-matin structure. We fi rst analyzed a 4.1 kb cPLA 2 � pro-moter fragment in a reporter construct by transient transfection and found this fragment to be unrespon-sive to IL-1 � (shown in Fig. 2A ). Based on our prior ex-perience with cytokine-dependent intronic enhancers elements, we also tested a series of fragments spanning the cPLA 2 � gene locus and found them to also be unre-sponsive to IL-1 � (supplementary Fig. I). Therefore, to identify potential distal upstream regulatory regions, DNase I HS analysis was employed to detect any open chromatin structure. Control or IL-1 � -treated HFL-1 cells were exposed to increasing concentrations of DNase I. The data from Southern analysis ( Fig. 1 ) illustrate a constitutive DNase I HS site at � 9.0–10 kb upstream of the transcriptional start site. We have previously shown that constitutive HS sites harbor specifi c inducible ele-ments ( 37, 45 ); therefore, we chose to further analyze this region.
Identifi cation of a distal IL-1 � -responsive enhancer element To test the functional relevance of the 5 ′ distal HS site,
a series of promoter fragments were analyzed in a hGH reporter plasmid ( Fig. 2A , top) transiently transfected into HFL-1 cells and evaluated by northern analysis. The � 14.0 kb promoter fragment, which harbors the HS site, was the only
Fig. 2. Identifi cation of the cPLA 2 � enhancer region. (A, top) Schematic of the cPLA 2 � 5 ′ region highlighting the promoter fragments ( � 14.0, � 6.8, � 5.5, � 4.8, � 4.1, � 1.4 kb) analyzed and a fragment (2.3 kb) containing the HS site identifi ed in Fig. 1C . (A, bottom left) Representative northern blot illustrates transient transfection of HFL-1 cells with cPLA 2 � promoter fragments subcloned into a human growth hormone (hGH) reporter plasmid. Cells were treated with 2 ng/µl of IL-1 � for 8 h. Membranes were probed for hGH with L7a as an internal loading control. (A, bottom right) Densitometry was performed for the 14 kb fragment with or without IL-1 � for 8 h. Bars represent mean ± SEM (n = 5), * P 0.05. ( B ) The 2.3 kb fragment containing the HS site identifi ed in Fig. 1C was cloned in front of either the 1.4 kb cPLA 2 � promoter ( Fig. 2A ) or the minimal thymidine kinase (TK) promoter in the forward (F) or reverse (R) orientation. A representative northern analysis of total RNA from HFL-1 cells transiently transfected with the indicated constructs with or without IL-1 � treatment was probed for the hGH reporter gene or an L7a loading control.
by guest, on July 13, 2018w
ww
.jlr.orgD
ownloaded from
.html http://www.jlr.org/content/suppl/2013/04/02/jlr.M037382.DC1Supplemental Material can be found at:

1920 Journal of Lipid Research Volume 54, 2013
complex from HFL-1 nuclear extracts that was inhibited by unlabeled cold competitor. To demonstrate that this complex contained c-Jun, we utilized DAPA ( 42 ) to ana-lyze complexes using the same oligonucleotide binding site with nuclear extracts from both HFL-1 cells and a hu-man bronchopulmonary epithelial cell line, S9, specifi -cally demonstrating reactivity with a c-Jun antibody by immunoblot analysis ( Fig. 4C , bottom).
To determine the in vivo response of this element to c-Jun, the aforementioned constructs were cotransfected along with the prominent AP-1 family member c-Jun. Over-expression of human c-Jun induces reporter gene expres-sion when the 2.3 kb cPLA 2 � enhancer fragment is coupled to either the cPLA 2 � (1.4 kb) or the minimal TK promoter ( Fig. 4D ). Furthermore, we also observed an increase in reporter activity from fragment III ( Fig. 3B ) cloned in front of the TK promoter in response to c-Jun overexpression ( Fig. 4D ). These data demonstrate that c-Jun can func-tion to induce cPLA 2 � gene expression through the identi-fi ed enhancer region, which includes the highly conserved AP-1 site (supplementary Fig. II).
Having linked c-Jun with a specifi c binding site within the enhancer, we then sought to determine the effect of IL-1 � on c-Jun abundance and phosphorylation. IL-1 � treatment did not affect the levels of c-Jun protein in HFL-1 cells based on immunoblot analysis ( Fig. 4E , top). It is well documented, however, that phosphorylation of c-Jun at serine 63 is required for both gene derepression ( 46–49 ) and activation ( 50, 51 ); therefore, we evaluated the effect of IL-1 � on c-Jun phosphorylation at S63. Due to the low affi nity of the available phospho-serine 63 anti-body, total c-Jun was fi rst immunoprecipitated to obtain suffi cient proteins levels for detection by immunoblot analysis with this antibody. We observed a time-dependent increase in c-Jun phosphorylation at serine 63 following IL-1 � treatment, consistent with the role of c-Jun in dere-pression and activation ( Fig. 4E , bottom).
Analysis of the enhancer and promoter regions by ChIP Having implied that c-Jun is capable of interacting with
the putative AP-1 site in vitro by EMSA and DAPA analyses ( Fig. 4C ), we next addressed the association of c-Jun with the cPLA 2 � enhancer element in situ by ChIP analysis. ChIP analysis of c-Jun showed a constitutive association with the enhancer ( Fig. 5A ), whereas we were unable to identify c-Jun at the promoter. Although IL-1 � does not elicit a change in the cellular levels ( Fig. 4E , top) or abun-dance of c-Jun at the enhancer ( Fig. 5A ), IL-1 � does cause a time-dependent increase in c-Jun phosphorylation at S63, linking this posttranslation modifi cation with the role of this transcription factor in cPLA 2 � gene expression. Due to the low affi nity of the S63 phospho-c-Jun antibody, some of our ChIP results for S63 phospho-c-Jun demon-strated occupancy at the enhancer, whereas others did not (data not shown). Therefore, in conjunction with increased S63 phosphorylation in response to IL-1 � ( Fig. 4E , bottom), we believe that it is likely that a fraction of c-Jun at the en-hancer is similarly phosphorylated, consistent with its role in derepression and activation.
The selective deletion of either a control upstream (con-trol A) or downstream (control B) site relative to the AP-1 sequence had no effect on expression levels ( Fig. 4A, B ). On the other hand, the selective deletion of the AP-1 consensus site ( Fig. 4A , bold letters) caused a striking loss of both basal and IL-1 � -dependent enhancer activity, as shown in a representative northern analysis ( Fig. 4B , right). The importance of this AP-1 sequence is further demonstrated by a strikingly high level of cross-species conservation illustrated by the alignment provided in sup-plementary Fig. II. This alignment also provides a strong argument for the critical importance of this AP-1 site and the homology found in the surrounding sequences.
To analyze the relevance of this particular site in vitro, we utilized a biotinylated oligonucleotide fl anking the AP-1 site in an EMSA ( Fig. 4C , top); we identifi ed a shifted
Fig. 3. Deletion analysis of the 2.3 kb enhancer fragment. (A, top) A schematic of the 2.3 kb fragment harboring the distal en-hancer as well as a Hin dIII-digested 1.6 kb subfragment. The lines indicate the regions present in each construct while the deleted regions are depicted as a box with an open triangle. (A, bottom) The 2.3 kb and 1.6 kb fragments were analyzed in an hGH reporter plasmid containing either the TK or 1.4 kb cPLA 2 � promoter by transient transfection in HFL-1 cells treated with 2 ng/µl of IL-1 � for 8 h. A representative northern blot illustrates the activity of these fragments. ( B, top) A schematic of deletions made within the 2.3 kb (IV) or 1.6 kb (I, II, III, V) enhancer fragments. (B, bottom) Representative northern analysis of the indicated enhancer sub-fragments driving expression of the TK promoter (top) or cPLA 2 � 1.4 kb promoter (bottom) with or without IL-1 � treatment. Mem-branes were probed for the hGH reporter gene and the L7a load-ing control.
by guest, on July 13, 2018w
ww
.jlr.orgD
ownloaded from
.html http://www.jlr.org/content/suppl/2013/04/02/jlr.M037382.DC1Supplemental Material can be found at:

Defi ning a cytokine-dependent cPLA 2 � enhancer element 1921
Fig. 4. Characterization of the cPLA 2 � enhancer. (A) Sequence of fragment III ( Fig. 2C ) containing the enhancer region from 9654 to 9340 bp upstream of the transcription start site. The consensus AP-1 ( TGAGTCA , gray box) is shown as underlined italicized text. The black bold text indicates the nucleotides at the AP-1 site and the control regions that were deleted for analysis in (B). ( B ) Representative northern blots illustrate the constructs containing the TK promoter with intact region III ( Fig. 3B ) or the site-specifi c deletions (A) with or without IL-1 � . Resulting membranes were probed for hGH and L7a. ( C, top) A representative autoradiogram of an EMSA using a fragment contain-ing the indicated AP-1 site depicts a shifted complex in the presence of nuclear extract (NE) isolated from HFL-1 cells. A cold unlabeled competitor was used to demonstrate specifi c interaction. (C, bottom) A representative autoradiogram of a DAPA shows the specifi c interac-tion of c-Jun with the fragment used above with extracts from HFL-1 cells, human bronchoepithelial cells (S9) with or without c-Jun over-expression, and a negative mouse fi broblast cell line. The resulting membrane was probed with an antibody specifi c to c-Jun. ( D ) An enhancer fragment, 2.3 kb or fragment III, was cloned in front of either the endogenous 1.4 kb cPLA 2 � promoter or the TK promoter with or without overexpression of human c-Jun. Total RNA was analyzed by real-time RT-PCR. Bars represent the mean of 2 � � � CT ± SEM (4 n 6). * P 0.05. ( E, top) Representative immunoblot of HFL-1 cells treated with IL-1 � for the indicated times and probed with antibodies specifi c to c-Jun or a � -actin. (E, bottom) HFL-1 extracts from control and IL-1 � -treated cells were immunoprecipitated with an antibody to c-Jun, and resulting protein was subjected to immunoblot analysis with an antibody specifi c to c-Jun phosphorylated at serine-63.
by guest, on July 13, 2018w
ww
.jlr.orgD
ownloaded from
.html http://www.jlr.org/content/suppl/2013/04/02/jlr.M037382.DC1Supplemental Material can be found at:

1922 Journal of Lipid Research Volume 54, 2013
knockdown of C/EBP � and c-Jun by their respective siRNAs ( Fig. 6A , left). IL-1 � -dependent induction of cPLA 2 � mRNA levels at 8 h was inhibited more than 50% by C/EBP � siRNA, whereas c-Jun knockdown showed no effect ( Fig. 6A , right) despite substantial knockdown of c-Jun protein levels. A po-tential explanation for the ineffectiveness of c-Jun siRNA on cPLA 2 � induction may be due to redundancy among the large AP-1 family (containing members from the Jun, Fos, and ATF subfamilies). We therefore evaluated the effects of overexpression of various AP-1 family members (JunB, JunD, FosB, c-Fos, and ATF2) on enhancer fragment III coupled to either the TK or endogenous promoter (supple-mentary Fig. III). All of these AP-1 family members were able to induce either promoter/enhancer construct to vary-ing degrees, demonstrating redundancy and thus explain-ing the inability of c-Jun knockdown to affect cPLA 2 � induction. In addition, it would appear that any member of the AP-1 family can replace c-Jun at this site, thus providing no obvious route for analysis of occupancy by any specifi c member of this large family.
To further establish the functional importance of C/EBP � and c-Jun with respect to IL-1 � induction of cPLA 2 � , we evaluated the cytokine response in two sepa-rate WT MEF cell lines and MEFs derived from C/EBP � � / � ( 53 ) and c-Jun � / � mice ( 35, 36 ). While siRNA provided a signifi cant decrease in protein levels for each factor ( Fig. 6A , left), the use of knockout MEFs allowed for com-plete gene ablation. Real-time RT-PCR of total RNA in control or IL-1 � -treated cells for 8 h demonstrated an identical and statistically signifi cant increase in cPLA 2 � mRNA levels in both WT MEF cell lines ( Fig. 6B ). Consis-tent with the siRNA results ( Fig. 6A , right), we observed no increase in cPLA 2 � mRNA expression in response to IL-1 � exposure in the C/EBP � � / � cells compared with the in-duction in WT MEFs ( Fig. 6B ). The combination of ChIP,
We also evaluated C/EBP � by ChIP analysis because of its involvement in transcriptional activation of other mem-bers of the eicosanoid pathway, specifi cally cyclooxyge-nase (COX)-2 and microsomal prostaglandin E synthase (mPGES), based on our studies and others ( 38, 52 ). We found that both c-Jun and C/EBP � were constitutively as-sociated with the enhancer in control cells and that the association of C/EBP � increased more than 2-fold follow-ing 8 h of IL-1 � treatment ( Fig. 5A ). Similarly, we observed a � 2-fold IL-1 � -dependent increase in RNA polymerase II (Pol II) association with the enhancer along with a low level of interaction with p300, while IgG and Sp1 served as negative controls. We next evaluated the occupancy of these factors at the promoter region and found that the association of Pol II and C/EBP � with this region was in-creased after 8 h of cytokine treatment ( Fig. 5B ), with a small but signifi cant association of p300 and Sp1 com-pared with IgG, as previously documented ( 23, 31 ). Pol II was also found inducibly associated with the promoter at 4 h of cytokine exposure ( Fig. 5B , inset), as would be ex-pected given the induction of cPLA 2 � mRNA by IL-1 � at 4 h ( Fig. 1A ). Of note, our enhancer fragment, which con-tained all the necessary sequences for IL-1 � -dependent induction ( Fig. 4 ), lacked any identifi able C/EBP � con-sensus-binding sequence. Similarly, we also searched within � 500 bp of the promoter region sampled by our ChIP analysis and found no C/EBP � binding sites. Together these results indicate that C/EBP � associated with these regions independent of direct DNA binding.
Functional role of C/EBP � and c-Jun on cPLA 2 � gene expression
To address the functional roles of C/EBP � and c-Jun, HFL-1 cells were transfected with specifi c siRNA pools for each gene. Immunoblot analysis demonstrated equivalent
Fig. 5. Identifi cation of transcription factors associated with the cPLA 2 � enhancer fragment. HFL cells were treated with IL-1 � for the indicated times, followed by ChIP analysis as described in Materials and Methods. (A) ChIP analysis was performed at 8 h using primers specifi c to the cPLA 2 � enhancer region with the indicated antibody. Bars represent the signal relative to the total input ± SEM as deter-mined by real-time PCR (n 3). ( B) ChIP analysis was performed at 8 h or 4 h (inset) using primers specifi c to the cPLA 2 � promoter region with the indicated antibody. Bars represent the signal relative to the total input ± SEM as determined by real-time PCR (n 3). * P 0.05 and † P 0.05 compared with control (gray bar) or IgG, respectively.
by guest, on July 13, 2018w
ww
.jlr.orgD
ownloaded from
.html http://www.jlr.org/content/suppl/2013/04/02/jlr.M037382.DC1Supplemental Material can be found at:

Defi ning a cytokine-dependent cPLA 2 � enhancer element 1923
tance of this AP-1 site. We demonstrated that c-Jun binds to the AP-1 site based on both in vitro EMSA and DAPA analyses ( Fig. 4C ). ChIP analysis further demonstrated the constitutive association of c-Jun in vivo with the enhancer in both basal and induced cells ( Fig. 5A ). A transiently transfected reporter construct containing the enhancer was transcriptionally activated in response to c-Jun overex-pression ( Fig. 4D ). IL-1 � did not affect the occupancy of c-Jun at the enhancer nor did it alter cellular levels of c-Jun. However, we did observe a time-dependent increase of c-Jun phosphorylation at serine 63 of the transactivation domain in response to IL-1 � ( Fig. 5C ). The phosphoryla-tion of this serine residue has been strongly associated with c-Jun activation ( 56 ). We would argue that phospho-rylation of c-Jun and its cognate AP-1 site are clearly criti-cal to cPLA 2 � induction by cytokines.
Regarding c-Jun occupancy, as shown at the enhancer re-gion by ChIP analysis in Fig. 5A , c-Jun was always present at the enhancer in control and IL-1 � -treated cells, and we saw no c-Jun binding at the promoter. We believe, based on the knockout data in Fig. 6B showing that cPLA 2 � expression was signifi cantly increased in c-Jun � / � cells, that c-Jun is part of a repressive complex in the basal state on the endogenous gene. Our data support a mechanism where IL-1 � -dependent phosphorylation of endogenous c-Jun at serine 63 ( Fig. 4E ) causes the dissociation of a repressive complex allowing phos-pho-c-Jun to participate in gene activation. c-Jun’s role as an activator is supported by the ability of c-Jun overexpression to cause transcriptional activation through the enhancer cou-pled to either the TK or 1.4 kb cPLA 2 � promoter ( Fig. 4D ). In addition, we have shown that numerous members of the AP-1 family when overexpressed can induce reporter gene expression through the enhancer coupled to either of these promoters (supplementary Fig. III). Therefore, c-Jun can function as a repressor in the basal state and participate in the cytokine-dependent induction, while the other AP-1 fam-ily members can function as activators alone or redundantly in the absence of c-Jun ( Fig. 6B ).
c-Jun’s role in a repressive complex is substantiated by similar results in numerous other genes ( 46–49 ). In fact, recent studies have demonstrated that the repressive com-plex interacts with c-Jun to repress gene expression ( 50, 51 ). Ogawa et al. ( 51 ) have shown that repressor com-plexes on AP-1 target genes specifi cally require the pres-ence of c-Jun, which is consistent with our ChIP data demonstrating constitutive occupancy of c-Jun at the en-hancer. In addition, these investigators have demonstrated that the role of the corepressor complex is to block the exchange of c-Jun for active c-Jun/AP-1 heterodimers, which is also consistent with our data on the ability of other AP-1 family members to induce cPLA 2 � (supplementary Fig. III). Furthermore, these studies have shown that the release of the repressor complex occurs specifi cally in re-sponse to c-Jun phosphorylation at serine 63, consistent with our data demonstrating IL-1 � -dependent phosphory-lation of c-Jun at serine 63 ( Fig. 4E ). Therefore, we have been able to demonstrate that c-Jun can function both to maintain cPLA 2 � in the off state and to serve as an activa-tor upon cytokine stimulation. The function of c-Jun as a
knockdown, and knockout studies demonstrate the criti-cal involvement of C/EBP � in the IL-1 � -dependent induc-tion of cPLA 2 � , through a mechanism independent of DNA binding.
Conversely, we observed a higher level of induction of cPLA 2 � mRNA levels following IL-1 � treatment in the c-Jun � / � MEFs compared with both WT MEF cell lines. We believe that the higher level of induction in the c-Jun � / � MEFs resulted from both the lack of repressive effects of unphosphorylated c-Jun and the redundancy afforded by other AP-1 family members (supplementary Fig. III). Consistent with this observation, stable high-level over-expression of c-Jun in the c-Jun � / � cells prevented the IL-1 � -dependent induction compared with the WT MEFs or the c-Jun � / � cells ( Fig. 6B ). This may result from the in-ability of endogenous kinase to phosphorylate overex-pressed levels of c-Jun, with S63 phospho-c-Jun being a prerequisite for derepression. The c-Jun � / � cell lines were kindly provided by the laboratory of Dr. David Gillespie, who also verifi ed the levels of c-Jun in these cell lines by immunoblot analysis ( 35, 36 ).
As C/EBP � -overexpressing MEFs were unavailable, we transiently overexpressed the full-length isoform of C/EBP � in S9 cells to evaluate endogenous cPLA 2 � gene expression. We utilized the human bronchoepithelial cell line S9, as we have found that this cell line can be more effi ciently trans-fected by plasmid, which is the only manner by which we can observe effects on endogenous gene expression. As shown ( Fig. 6C ), C/EBP � expression is capable of causing a greater than 2.5-fold increase in endogenous cPLA 2 � levels.
DISCUSSION
Many studies have established a direct link between cPLA 2 � activity and a wide variety of physiological and path-ological events with critical roles in apoptosis, infl amma-tion, and cancer ( 54, 55 ). The regulation of cPLA 2 � has previously focused on posttranslational, site-specifi c phos-phorylation events; however, numerous studies have docu-mented events associated with transcriptional regulation ( 4 ). Until now, all studies have focused solely on the proxi-mal promoter region where, in our hands, sequences ex-tending as far as 6.8 kb upstream of the transcriptional initiation site show no response to cytokines in lung cells. In this study, DNase I HS site analysis ( Fig. 1 ) has led to the discovery of a 5 ′ distal IL-1 � -responsive cPLA 2 � enhancer element at approximately � 9.5 kb ( Figs. 2–4 ). Our data demonstrate that the enhancer is required for and contrib-utes to both basal and IL-1 � -dependent gene expression. We believe that our results provide functionally and physi-ologically relevant data identifying a cytokine-responsive enhancer element for the human cPLA 2 � gene.
A perfect consensus AP-1 site within the enhancer is re-quired for both basal and induced levels of reporter gene expression based on deletion analysis ( Fig. 4A, B ). This AP-1 site and the surrounding DNA sequences are strik-ingly conserved across a highly diverse range of mammals (supplementary Fig. II), further substantiating the impor-
by guest, on July 13, 2018w
ww
.jlr.orgD
ownloaded from
.html http://www.jlr.org/content/suppl/2013/04/02/jlr.M037382.DC1Supplemental Material can be found at:

1924 Journal of Lipid Research Volume 54, 2013
Fig. 6. Effects of knockdown, knockout, and overexpression of c-Jun and C/EBP � on endogenous cPLA 2 � expression. (A) HFL-1 cells were transfected with the indicated siRNA. (A, left) Isolated protein was subjected to immunoblot analysis and probed with anti-bodies specifi c for c-Jun or C/EBP � . (A, right) Isolated RNA from siRNA and IL-1 � -treated HFL-1 cells was used for real-time RT-PCR for cPLA 2 � expression. Bars represent mean 2 � � � CT ± SEM (n = 3). ( B ) Total RNA from two wild-type MEF cell lines (WT I and WT II), C/EBP � � / � MEF, c-Jun � / � MEF, and c-Jun � / � MEFs overexpressing c-Jun (c-Jun+) with or without IL-1 � treatment was analyzed by real-time RT-PCR for cPLA 2 � expression. Bars represent mean 2 � � � CT ± SEM (n 3). * P 0.05 compared with control cells (gray bar); ,† P 0.05 compared with IL-1 � -treated WT cells (black bar); ,♦ P 0.05 compared with IL-1 � treated c-Jun � / � cells. ( C ) S9 human bron-choepithelial cells were transfected with a pcDNA3.1 expression plasmid containing the full-length isoform of human C/EBP � . Iso-lated RNA was used for real-time RT-PCR for cPLA 2 � expression. Bars represent mean 2 - � � CT ± SEM (n = 3); * P 0.05.
repressor in the basal state is consistent with the extremely low levels of basal expression of cPLA 2 � that we observed, which is reasonable considering the potentially detrimental
impact of high levels of phospholipase expression on nor-mal membrane integrity.
Our ChIP data demonstrated association of C/EBP � with both the enhancer and promoter regions ( Fig. 5A, B ). Most importantly, its levels increased at both sites following cytokine stimulation. Overexpression of C/EBP � caused a greater than 2.5-fold increase in endogenous cPLA 2 � mRNA levels ( Fig. 6C ). Furthermore, functional studies using either siRNA knockdown or knockout cell lines dem-onstrated that C/EBP � is a transcriptional activator neces-sary for IL-1 � -dependent induction ( Fig. 6A, B ). These data strongly establish the importance of this transcription factor to cPLA 2 � gene expression. The literature clearly demonstrates that C/EBP � /p300 complexes are recruited to regulatory elements, thus facilitating assembly of the preinitiation complex (PIC) ( 57, 58 ). This is consistent with our data, which show an inducible association of Pol II with the distal enhancer and proximal promoter re-gions as well as an association of p300 with the enhancer. We searched the region surrounding the enhancer that was tested by our ChIP analysis (within approximately 500–1000 bp based on chromatin sonication and primer location), but we did not identify an acceptable binding site. Thus, we believe that C/EBP � is not directly binding DNA because no consensus sequence for C/EBP � exists within the fragments that have enhancer activity. In addi-tion, our deletion of the AP-1 site completely abolished cytokine induction; therefore, we believe that C/EBP � is an activator of cPLA 2 � and that its activity is mediated through interaction with the transcription complex (pos-sibly p300, which it is known to interact with) independent of DNA binding. This is consistent with several studies that demonstrate that C/EBP � and other C/EBPs can exert transcriptional effects independent of direct DNA binding ( 59–62 ). This is not an uncommon transcriptional regula-tory theme in that steroid receptors ( 63–65 ), the aryl hy-drocarbon receptor (AhR) ( 66 ), FOXA1 ( 67 ), and the thyroid hormone receptor ( 68 ) can all function to regu-late gene expression through protein-protein interac-tions independent of direct DNA binding. Analogous to our results on C/EBP � , ChIP analysis has shown that the PHOX2A transcription factor can also associate with and regulate the human � 3 nicotinic receptor subunit promoter through a DNA-independent mechanism ( 69 ). Furthermore, genomewide ChIP-chip arrays and ChIP-seq data for AhR indicates that � 50% of the AhR-enriched regions lack any dioxin response element (DRE) ( 70 ). We therefore believe that C/EBP � functions as a tran-scriptional activator through interaction with a large transcription complex, including p300, independent of DNA binding.
In summary, in the basal state c-Jun is bound to the enhancer as a repressor while C/EBP � is associated with both the promoter and enhancer regions. Upon cytokine stimulation, c-Jun becomes phosphorylated, which allows it to function as an activator, and additional C/EBP � is recruited to both regulatory regions. Our data identify the fi rst documented cytokine-dependent enhancer element for the human cPLA 2 � gene and the critical role of c-Jun
by guest, on July 13, 2018w
ww
.jlr.orgD
ownloaded from
.html http://www.jlr.org/content/suppl/2013/04/02/jlr.M037382.DC1Supplemental Material can be found at:

Defi ning a cytokine-dependent cPLA 2 � enhancer element 1925
fi broblasts. Evidence for their roles in the production of prosta-glandin E2. Arthritis Rheum. 37 : 653 – 661 .
16 . Dolan-O’Keefe , M. , V. Chow , J. Monnier , G. A. Visner , and H. S. Nick . 2000 . Transcriptional regulation and structural organization of the human cytosolic phospholipase A(2) gene. Am. J. Physiol. Lung Cell. Mol. Physiol. 278 : L649 – L657 .
17 . Wu , T. , S. J. Levine , M. G. Lawrence , C. Logun , C. W. Angus , and J. H. Shelhamer . 1994 . Interferon-gamma induces the synthesis and activation of cytosolic phospholipase A2. J. Clin. Invest. 93 : 571 – 577 .
18 . Nakamura , T. , L. L. Lin , S. Kharbanda , J. Knopf , and D. Kufe . 1992 . Macrophage colony stimulating factor activates phosphatidyl-choline hydrolysis by cytoplasmic phospholipase A2. EMBO J. 11 : 4917 – 4922 .
19 . Heasley , L. E. , S. Thaler , M. Nicks , B. Price , K. Skorecki , and R. A. Nemenoff . 1997 . Induction of cytosolic phospholipase A2 by onco-genic Ras in human non-small cell lung cancer. J. Biol. Chem. 272 : 14501 – 14504 .
20 . Wu , T. , T. Ikezono , C. W. Angus , and J. H. Shelhamer . 1994 . Characterization of the promoter for the human 85 kDa cytosolic phospholipase A2 gene. Nucleic Acids Res. 22 : 5093 – 5098 .
21 . Tay , A. , P. Maxwell , Z. Li , H. Goldberg , and K. Skorecki . 1994 . Isolation of promoter for cytosolic phospholipase A2 (cPLA2). Biochim. Biophys. Acta . 1217 : 345 – 347 .
22 . Lee , C. W. , C. C. Lin , I. T. Lee , H. C. Lee , and C. M. Yang . 2011 . Activation and induction of cytosolic phospholipase A2 by TNF-alpha mediated through Nox2, MAPKs, NF-kappaB, and p300 in human tracheal smooth muscle cells. J. Cell. Physiol. 226 : 2103 – 2114 .
23 . Chi , P. L. , S. F. Luo , H. L. Hsieh , I. T. Lee , L. D. Hsiao , Y. L. Chen , and C. M. Yang . 2011 . Cytosolic phospholipase A2 induction and prostaglandin E2 release by interleukin-1beta via the myeloid dif-ferentiation factor 88-dependent pathway and cooperation of p300, Akt, and NF-kappaB activity in human rheumatoid arthritis synovial fi broblasts. Arthritis Rheum. 63 : 2905 – 2917 .
24 . Wick , M. J. , S. Blaine , V. Van Putten , M. Saavedra , and R. A. Nemenoff . 2005 . Lung Kruppel-like factor (LKLF) is a transcrip-tional activator of the cytosolic phospholipase A2 alpha promoter. Biochem. J. 387 : 239 – 246 .
25 . Buttar , N. S. , C. J. DeMars , G. Lomberk , S. Rizvi , J. Bonilla-Velez , S. Achra , S. Rashtak , K. K. Wang , M. E. Fernandez-Zapico , and R. Urrutia . 2010 . Distinct role of Kruppel-like factor 11 in the regulation of prostaglandin E2 biosynthesis. J. Biol. Chem. 285 : 11433 – 11444 .
26 . Alexandrov , P. N. , J. G. Cui , and W. J. Lukiw . 2006 . Hypoxia-sensitive domain in the human cytosolic phospholipase A2 pro-moter. Neuroreport . 17 : 303 – 307 .
27 . Blaine , S. A. , M. Wick , C. Dessev , and R. A. Nemenoff . 2001 . Induction of cPLA2 in lung epithelial cells and non-small cell lung cancer is mediated by Sp1 and c-Jun. J. Biol. Chem. 276 : 42737 – 42743 .
28 . Sokolowska , M. , M. Borowiec , A. Ptasinska , M. Cieslak , J. H. Shelhamer , M. L. Kowalski , and R. Pawliczak . 2007 . 85-kDa cytoso-lic phospholipase A2 group IValpha gene promoter polymorphisms in patients with severe asthma: a gene expression and case-control study. Clin. Exp. Immunol. 150 : 124 – 131 .
29 . Cowan , M. J. , X. L. Yao , R. Pawliczak , X. Huang , C. Logun , P. Madara , S. Alsaaty , T. Wu , and J. H. Shelhamer . 2004 . The role of TFIID, the initiator element and a novel 5 ′ TFIID binding site in the transcriptional control of the TATA-less human cytosolic phospholipase A2-alpha promoter. Biochim. Biophys. Acta . 1680 : 145 – 157 .
30 . Van Putten , V. , Z. Refaat , C. Dessev , S. Blaine , M. Wick , L. Butterfi eld , S. Y. Han , L. E. Heasley , and R. A. Nemenoff . 2001 . Induction of cytosolic phospholipase A2 by oncogenic Ras is me-diated through the JNK and ERK pathways in rat epithelial cells. J. Biol. Chem. 276 : 1226 – 1232 .
31 . Tsou , J. H. , K. Y. Chang , W. C. Wang , J. T. Tseng , W. C. Su , L. Y. Hung , W. C. Chang , and B. K. Chen . 2008 . Nucleolin regulates c-Jun/Sp1-dependent transcriptional activation of cPLA2alpha in phorbol ester-treated non-small cell lung cancer A549 cells. Nucleic Acids Res. 36 : 217 – 227 .
32 . Chi , P. L. , Y. W. Chen , L. D. Hsiao , Y. L. Chen , and C. M. Yang . 2012 . Heme oxygenase 1 attenuates interleukin-1beta-induced cytosolic phospholipase A2 expression via a decrease in NADPH oxidase/re-active oxygen species/activator protein 1 activation in rheumatoid arthritis synovial fi broblasts. Arthritis Rheum. 64 : 2114 – 2125 .
and C/EBP � . It is interesting that both c-Jun and C/EBP � have been found to regulate COX-2, the subsequent enzyme in the eicosanoid pathway ( 71, 72 ). Analogously, our pre-vious studies established C/EBP � in the cytokine-depen-dent regulation of mPGES-1 ( 38 ), a terminal synthase in the prostaglandin arm of the eicosanoid pathway. As these are the fi rst results to address the cytokine-dependent reg-ulation of cPLA 2 � , the role of these cognate transcription factors may have important implications in the overall reg-ulation of eicosanoid biosynthesis.
The authors thank the members of the Nick and Kilberg laboratories for helpful discussions. The authors also thank Dr. Shermi Liang for assistance with fi gures.
REFERENCES
1 . Burke , J. E. , and E. A. Dennis . 2009 . Phospholipase A2 biochemis-try. Cardiovasc. Drugs Ther. 23 : 49 – 59 .
2 . Murakami , M. , Y. Taketomi , Y. Miki , H. Sato , T. Hirabayashi , and K. Yamamoto . 2011 . Recent progress in phospholipase A(2) research: from cells to animals to humans. Prog. Lipid Res. 50 : 152 – 192 .
3 . Bonventre , J. V. , Z. Huang , M. R. Taheri , E. O’Leary , E. Li , M. A. Moskowitz , and A. Sapirstein . 1997 . Reduced fertility and pos-tischaemic brain injury in mice defi cient in cytosolic phospholipase A2. Nature . 390 : 622 – 625 .
4 . Ghosh , M. , D. E. Tucker , S. A. Burchett , and C. C. Leslie . 2006 . Properties of the Group IV phospholipase A2 family. Prog. Lipid Res. 45 : 487 – 510 .
5 . Nagase , T. , N. Uozumi , S. Ishii , K. Kume , T. Izumi , Y. Ouchi , and T. Shimizu . 2000 . Acute lung injury by sepsis and acid aspira-tion: a key role for cytosolic phospholipase A2. Nat. Immunol. 1 : 42 – 46 .
6 . Meyer , A. M. , L. D. Dwyer-Nield , G. Hurteau , R. L. Keith , Y. Ouyang , B. M. Freed , L. R. Kisley , M. W. Geraci , J. V. Bonventre , R. A. Nemenoff , et al . 2006 . Attenuation of the pulmonary infl am-matory response following butylated hydroxytoluene treatment of cytosolic phospholipase A2 null mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 290 : L1260 – L1266 .
7 . Meyer , A. M. , L. D. Dwyer-Nield , G. J. Hurteau , R. L. Keith , E. O’Leary , M. You , J. V. Bonventre , R. A. Nemenoff , and A. M. Malkinson . 2004 . Decreased lung tumorigenesis in mice genetically defi cient in cyto-solic phospholipase A2. Carcinogenesis . 25 : 1517 – 1524 .
8 . Gentile , M. T. , M. G. Reccia , P. P. Sorrentino , E. Vitale , G. Sorrentino , A. A. Puca , and L. Colucci-D’Amato . 2012 . Role of cyto-solic calcium-dependent phospholipase A2 in Alzheimer’s disease pathogenesis. Mol. Neurobiol. 45 : 596 – 604 .
9 . Burke , J. E. , and E. A. Dennis . 2009 . Phospholipase A2 structure/function, mechanism, and signaling. J. Lipid Res. 50 ( Suppl. ): S237 – S242 .
10 . Li , G. , W. Chen , C. Han , and T. Wu . 2011 . Cytosolic phospholipase A(2)alpha protects against Fas- but not LPS-induced liver injury. J. Hepatol. 55 : 1281 – 1290 .
11 . Linkous , A. , and E. Yazlovitskaya . 2010 . Cytosolic phospholi-pase A2 as a mediator of disease pathogenesis. Cell. Microbiol. 12 : 1369 – 1377 .
12 . Tosato , G. , M. Segarra , and O. Salvucci . 2010 . Cytosolic phospho-lipase A2{alpha} and cancer: a role in tumor angiogenesis. J. Natl. Cancer Inst. 102 : 1377 – 1379 .
13 . Fahy , J. V. , D. B. Corry , and H. A. Boushey . 2000 . Airway infl amma-tion and remodeling in asthma. Curr. Opin. Pulm. Med. 6 : 15 – 20 .
14 . Lilja , I. , K. Smedh , G. Olaison , R. Sjodahl , C. Tagesson , and C. Gustafson-Svard . 1995 . Phospholipase A2 gene expression and ac-tivity in histologically normal ileal mucosa and in Crohn’s ileitis. Gut . 37 : 380 – 385 .
15 . Hulkower , K. I. , S. J. Wertheimer , W. Levin , J. W. Coffey , C. M. Anderson , T. Chen , D. L. DeWitt , R. M. Crowl , W. C. Hope , and D. W. Morgan . 1994 . Interleukin-1 beta induces cytosolic phospho-lipase A2 and prostaglandin H synthase in rheumatoid synovial
by guest, on July 13, 2018w
ww
.jlr.orgD
ownloaded from
.html http://www.jlr.org/content/suppl/2013/04/02/jlr.M037382.DC1Supplemental Material can be found at:

1926 Journal of Lipid Research Volume 54, 2013
52 . Wu , K. K. , J. Y. Liou , and K. Cieslik . 2005 . Transcriptional con-trol of COX-2 via C/EBPbeta. Arterioscler. Thromb. Vasc. Biol. 25 : 679 – 685 .
53 . Sterneck , E. , L. Tessarollo , and P. F. Johnson . 1997 . An essen-tial role for C/EBPbeta in female reproduction. Genes Dev. 11 : 2153 – 2162 .
54 . Duan , L. , H. Gan , J. Arm , and H. G. Remold . 2001 . Cytosolic phospholipase A2 participates with TNF-alpha in the induction of apoptosis of human macrophages infected with Mycobacterium tu-berculosis H37Ra. J. Immunol. 166 : 7469 – 7476 .
55 . Panini , S. R. , L. Yang , A. E. Rusinol , M. S. Sinensky , J. V. Bonventre , and C. C. Leslie . 2001 . Arachidonate metabolism and the signaling pathway of induction of apoptosis by oxidized LDL/oxysterol. J. Lipid Res. 42 : 1678 – 1686 .
56 . Davis , R. J. 2000 . Signal transduction by the JNK group of MAP kinases. Cell . 103 : 239 – 252 .
57 . Mo , X. , E. Kowenz-Leutz , H. Xu , and A. Leutz . 2004 . Ras in-duces mediator complex exchange on C/EBP beta. Mol. Cell . 13 : 241 – 250 .
58 . Schwartz , C. , K. Beck , S. Mink , M. Schmolke , B. Budde , D. Wenning , and K. H. Klempnauer . 2003 . Recruitment of p300 by C/EBPbeta triggers phosphorylation of p300 and modulates coactivator activ-ity. EMBO J. 22 : 882 – 892 .
59 . Timchenko , N. A. , M. Wilde , and G. J. Darlington . 1999 . C/EBPalpha regulates formation of S-phase-specifi c E2F-p107 complexes in livers of newborn mice. Mol. Cell. Biol. 19 : 2936 – 2945 .
60 . Harris , T. E. , J. H. Albrecht , M. Nakanishi , and G. J. Darlington . 2001 . CCAAT/enhancer-binding protein-alpha cooperates with p21 to inhibit cyclin-dependent kinase-2 activity and induces growth arrest independent of DNA binding. J. Biol. Chem. 276 : 29200 – 29209 .
61 . Muller , C. , M. Alunni-Fabbroni , E. Kowenz-Leutz , X. Mo , M. Tommasino , and A. Leutz . 1999 . Separation of C/EBPalpha-mediated proliferation arrest and differentiation pathways. Proc. Natl. Acad. Sci. USA . 96 : 7276 – 7281 .
62 . Grant , C. , M. Nonnemacher , P. Jain , D. Pandya , B. Irish , S. C. Williams , and B. Wigdahl . 2006 . CCAAT/enhancer-binding pro-teins modulate human T cell leukemia virus type 1 long terminal repeat activation. Virology . 348 : 354 – 369 .
63 . McEwan , I. J. , A. P. Wright , and J. A. Gustafsson . 1997 . Mechanism of gene expression by the glucocorticoid receptor: role of protein-protein interactions. Bioessays . 19 : 153 – 160 .
64 . Tuckermann , J. P. , H. M. Reichardt , R. Arribas , K. H. Richter , G. Schutz , and P. Angel . 1999 . The DNA binding-independent func-tion of the glucocorticoid receptor mediates repression of AP-1-dependent genes in skin. J. Cell Biol. 147 : 1365 – 1370 .
65 . Reichardt , H. M. , K. H. Kaestner , J. Tuckermann , O. Kretz , O. Wessely , R. Bock , P. Gass , W. Schmid , P. Herrlich , P. Angel , et al . 1998 . DNA binding of the glucocorticoid receptor is not essential for survival. Cell . 93 : 531 – 541 .
66 . Patel , R. D. , I. A. Murray , C. A. Flaveny , A. Kusnadi , and G. H. Perdew . 2009 . Ah receptor represses acute-phase response gene expression without binding to its cognate response element. Lab. Invest. 89 : 695 – 707 .
67 . Minoo , P. , L. Hu , Y. Xing , N. L. Zhu , H. Chen , M. Li , Z. Borok , and C. Li . 2007 . Physical and functional interactions between ho-meodomain NKX2.1 and winged helix/forkhead FOXA1 in lung epithelial cells. Mol. Cell. Biol. 27 : 2155 – 2165 .
68 . Wulf , A. , M. G. Wetzel , M. Kebenko , M. Kroger , A. Harneit , J. Merz , and J. M. Weitzel . 2008 . The role of thyroid hormone receptor DNA binding in negative thyroid hormone-mediated gene transcription. J. Mol. Endocrinol. 41 : 25 – 34 .
69 . Benfante , R. , A. Flora , S. Di Lascio , F. Cargnin , R. Longhi , S. Colombo , F. Clementi , and D. Fornasari . 2007 . Transcription fac-tor PHOX2A regulates the human alpha3 nicotinic receptor sub-unit gene promoter. J. Biol. Chem. 282 : 13290 – 13302 .
70 . Dere , E. , R. Lo , T. Celius , J. Matthews , and T. R. Zacharewski . 2011 . Integration of genome-wide computation DRE search, AhR ChIP-chip and gene expression analyses of TCDD-elicited responses in the mouse liver. BMC Genomics . 12 : 365 .
71 . Subbaramaiah , K. , P. A. Cole , and A. J. Dannenberg . 2002 . Retinoids and carnosol suppress cyclooxygenase-2 transcription by CREB-binding protein/p300-dependent and -independent mecha-nisms. Cancer Res. 62 : 2522 – 2530 .
72 . Wu , K. K. 2006 . Transcription-based COX-2 inhibition: a therapeu-tic strategy. Thromb. Haemost. 96 : 417 – 422 .
33 . Liao , W. L. , W. C. Wang , W. C. Chang , and J. T. Tseng . 2011 . The RNA-binding protein HuR stabilizes cytosolic phospholipase A2alpha mRNA under interleukin-1beta treatment in non-small cell lung cancer A549 Cells. J. Biol. Chem. 286 : 35499 – 35508 .
34 . Egan , M. , T. Flotte , S. Afi one , R. Solow , P. L. Zeitlin , B. J. Carter , and W. B. Guggino . 1992 . Defective regulation of outwardly rec-tifying Cl- channels by protein kinase A corrected by insertion of CFTR. Nature . 358 : 581 – 584 .
35 . MacLaren , A. , E. J. Black , W. Clark , and D. A. Gillespie . 2004 . c-Jun-defi cient cells undergo premature senescence as a result of spontaneous DNA damage accumulation. Mol. Cell. Biol. 24 : 9006 – 9018 .
36 . MacLaren , A. , W. Clark , and D. A. Gillespie . 2000 . v-Jun sensitizes cells to apoptosis by a mechanism involving mitochondrial cyto-chrome C release. Oncogene . 19 : 5906 – 5918 .
37 . Mellott , J. K. , H. S. Nick , M. F. Waters , T. R. Billiar , D. A. Geller , and S. E. Chesrown . 2001 . Cytokine-induced changes in chromatin structure and in vivo footprints in the inducible NOS promoter. Am. J. Physiol. Lung Cell. Mol. Physiol. 280 : L390 – L399 .
38 . Walters , J. N. , J. S. Bickford , K. J. Newsom , D. E. Beachy , S. J. Barilovits , J. D. Herlihy , and H. S. Nick . 2012 . Regulation of human microsomal prostaglandin E synthase-1 by IL-1beta requires a distal enhancer element with a unique role for C/EBPbeta. Biochem. J. 443 : 561 – 571 .
39 . Visner , G. A. , W. C. Dougall , J. M. Wilson , I. A. Burr , and H. S. Nick . 1990 . Regulation of manganese superoxide dismutase by lipopoly-saccharide, interleukin-1, and tumor necrosis factor. Role in the acute infl ammatory response. J. Biol. Chem. 265 : 2856 – 2864 .
40 . Livak , K. J. , and T. D. Schmittgen . 2001 . Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods . 25 : 402 – 408 .
41 . Selden , R. F. , K. B. Howie , M. E. Rowe , H. M. Goodman , and D. D. Moore . 1986 . Human growth hormone as a reporter gene in regu-lation studies employing transient gene expression. Mol. Cell. Biol. 6 : 3173 – 3179 .
42 . Thiaville , M. M. , E. E. Dudenhausen , C. Zhong , Y. X. Pan , and M. S. Kilberg . 2008 . Deprivation of protein or amino acid induces C/EBPbeta synthesis and binding to amino acid response elements, but its action is not an absolute requirement for enhanced tran-scription. Biochem. J. 410 : 473 – 484 .
43 . Qiu , X. , K. J. Aiken , A. L. Chokas , D. E. Beachy , and H. S. Nick . 2008 . Distinct functions of C/EBP isoforms in the regulation of manganese superoxide dismutase during IL-1beta stimulation. J. Biol. Chem. 283: 25774–25785.
44 . Dolan-O’keefe , M. , and H. S. Nick . 1999 . Inhibition of cytoplasmic phospholipase A2 expression by glucocorticoids in rat intestinal epithelial cells. Gastroenterology . 116 : 855 – 864 .
45 . Kuo , S. , S. E. Chesrown , J. K. Mellott , R. J. Rogers , J. L. Hsu , and H. S. Nick . 1999 . In vivo architecture of the manganese superoxide dismutase promoter. J. Biol. Chem. 274 : 3345 – 3354 .
46 . Sprowles , A. , D. Robinson , Y. M. Wu , H. J. Kung , and R. Wisdom . 2005 . c-Jun controls the effi ciency of MAP kinase signaling by tran-scriptional repression of MAP kinase phosphatases. Exp. Cell Res. 308 : 459 – 468 .
47 . Pessah , M. , C. Prunier , J. Marais , N. Ferrand , A. Mazars , F. Lallemand , J. M. Gauthier , and A. Atfi . 2001 . c-Jun interacts with the corepressor TG-interacting factor (TGIF) to suppress Smad2 transcriptional activity. Proc. Natl. Acad. Sci. USA . 98 : 6198 – 6203 .
48 . Freire , G. , C. Ocampo , N. Ilbawi , A. J. Griffi n , and M. Gupta . 2007 . Overt expression of AP-1 reduces alpha myosin heavy chain expres-sion and contributes to heart failure from chronic volume over-load. J. Mol. Cell. Cardiol. 43 : 465 – 478 .
49 . Bein , K. , H. Leight , and G. D. Leikauf . 2011 . JUN-CCAAT/enhancer-binding protein complexes inhibit surfactant-associ-ated protein B promoter activity. Am. J. Respir. Cell Mol. Biol. 45 : 436 – 444 .
50 . Huang , W. , S. Ghisletti , V. Perissi , M. G. Rosenfeld , and C. K. Glass . 2009 . Transcriptional integration of TLR2 and TLR4 signaling at the NCoR derepression checkpoint. Mol. Cell . 35 : 48 – 57 .
51 . Ogawa , S. , J. Lozach , K. Jepsen , D. Sawka-Verhelle , V. Perissi , R. Sasik , D. W. Rose , R. S. Johnson , M. G. Rosenfeld , and C. K. Glass . 2004 . A nuclear receptor corepressor transcriptional check-point controlling activator protein 1-dependent gene networks re-quired for macrophage activation. Proc. Natl. Acad. Sci. USA . 101 : 14461 – 14466 .
by guest, on July 13, 2018w
ww
.jlr.orgD
ownloaded from
.html http://www.jlr.org/content/suppl/2013/04/02/jlr.M037382.DC1Supplemental Material can be found at: