document
TRANSCRIPT

http://immunol.nature.com • june 2001 • volume 2 no 6 • nature immunology
Stephanie A. Camacho1,William R. Heath2, Francis R. Carbone3, Nora Sarvetnick1,AgnesLeBon4, Lars Karlsson5, Per A. Peterson5 and Susan R.Webb1
We investigated how the accessory molecule interactions encountered during T cell priminginfluence T cell–mediated destruction of insulin-producing β cells and lead to type 1 diabetes.T cellreceptor (TCR)-transgenic CD4+ T cells were primed under controlled conditions in vitro beforebeing adoptively transferred into transgenic recipients expressing membrane ovalbumin under thecontrol of the rat insulin promoter (RIP-mOVA). During priming, antigen-presenting cell expressionof B7-1 without intracellular adhesion molecule 1 (ICAM-1) led to the generation of effector cellsthat migrated to the pancreata of RIP-mOVA recipients but did not cause diabetes. In contrast, whenT cells were primed with APCs expressing both B7-1 and ICAM-1, pronounced destruction of β cellsand a rapid onset of diabetes were observed. Pathogenicity was associated with T cell production ofthe macrophage-attracting chemokines CCL3 and CCL4. Thus, interactions of lymphocytefunction–associated antigen 1 with ICAM-1 during priming induce both qualitative and quantitativealterations in T effector function and induce potentially autodestructive responses.
1Department of Immunology, IMM4,The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037, USA. 2The Walter and Eliza Institute of MedicalResearch, Royal Melbourne Hospital,Victoria, Australia. 3Department of Pathology and Immunology, Monash Medical School, Prahran,Australia. 4The Edward Jenner Institute
for Vaccine Research, Compton, Newbury, Berkshire, UK. 5R.W. Johnson Pharmaceutical Research Institute, 3210 Merryfield Row, San Diego, CA 92121, USA.Correspondence should be addressed to S. R.W. ([email protected]).
A key role for ICAM-1 in generatingeffector cells mediating inflammatory
responses
Activation of naïve T cells requires not only T cell recognition of pep-tide–major histocompatibility complex (MHC) complexes but also cos-timulation provided by accessory molecule interactions. The role ofCD28 in providing costimulatory signals that promote T cell activationand survival is now well documented1–4. Accumulating evidence alsosuggests that accessory molecule interactions influence the type ofcytokines produced by activated T cells. Several accessory molecules,including CD9, lymphocyte function–associated antigen 1 (LFA-1, alsoknow as CD11a-CD18) and CD27, promote some aspects of T cell acti-vation—for example, cell division and expression of activation mark-ers—without inducing the high expression of interleukin 2 (IL-2) asso-ciated with CD28 ligation5–7. Binding of CD28 to B7 ligands inducesIL-4 as well as IL-2 production, although some evidence suggests thatbinding to B7-1 versus B7-2 may generate effector cells with distinctcytokine profiles8–15. The capacity to cause disease in murine autoim-mune models has also been associated with either B7-1 or B7-2engagement8,16. Although the mechanisms involved in the distincteffects of B7-1 versus B7-2 binding are not clear, findings are consis-tent with the theory that the type of accessory molecule engaged duringT cell responses may play a critical role in determining the type ofeffector activity displayed.
One role for LFA-1 in the regulation of effector function during pri-mary CD4+ T cell responses has been shown. Antibodies to LFA-1 lig-ands intracellular adhesion molecule 1 (ICAM-1) and/or ICAM-2 down-regulate IL-4 production by naïve CD4+ cells activated by anti-TCR and
anti-CD2817. Independent studies that used Drosophila cells expressingmurine MHC class II molecules with defined accessory molecule ligandsas antigen-presenting cells (APCs) give similar results. Naïve D011TCR–transgenic CD4+ cells stimulated with ovalbumin (OVA) peptidepresented by Drosophila APCs expressing B7 in the absence of ICAM-1show proliferative responses that are associated with high IL-4 produc-tion18. In contrast, coexpression of ICAM-1 on Drosophila APCs leads toa marked reduction in the amount of IL-4 produced after activation18.Thus, LFA-1 engagement negatively regulates expression of the geneencoding IL-4 during primary CD4 cell responses.
We addressed the question of whether LFA-1–ICAM-1 interactionsduring CD4+ cell priming have long-term inheritable functional effectsthat are apparent after secondary exposure to peptide presented by typi-cal APCs. To this end, we used a murine transgenic model for type 1 dia-betes to study the in vivo behavior of activated CD4+ cells. D011 CD4+ Tcells were primed in vitro by culture with an OVA peptide that consistedof amino acids 323–339, which was presented by Drosophila APCsexpressing H-2Ad and either B7-1 or B7-1 and ICAM-1. The activatedblast cells were adoptively transferred into mice expressing membraneovalbumin under the control of the rat insulin promoter (RIP-mOVA),which were generated as described19. Islet infiltration and diabetes inthese recipient mice were then monitored. We found that although B7-1–primed CD4+ cells migrated to the islets, they did not destroy insulin-producing β cells. In contrast, priming with APCs expressing B7-1 andICAM-1 generated effector CD4+ cells that rapidly destroyed the β cells,
ARTICLES
523
©20
01 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://im
mu
no
l.nat
ure
.co
m© 2001 Nature Publishing Group http://immunol.nature.com

nature immunology • volume 2 no 6 • june 2001 • http://immunol.nature.com
ARTICLES
leading to overt diabetes 1–2 weeks after transfer. These findings showthat the type of accessory molecules engaged during the initial primingof CD4+ T cells plays an important role in programming the subsequenteffector activity displayed by those cells in vivo. Specifically, LFA-1−ICAM-1 interactions are critical in facilitating the development ofinflammatory effector CD4+ cells.
ResultsExperimental designTo study the in vivo effector function of primed CD4+ T cells, we usedRIP-mOVA–transgenic mice as a model for type 1 diabetes. These miceserved as adoptive transfer recipients of purified OVA-specific D011CD4+ cells and diabetes (destruction of the OVA-expressing β cells)was monitored both histologically and by measuring blood glucose. Inthis system, diabetes proved to be dependent on activation of the OVA-specific CD4+ cells because transfer of naïve resting CD4+ cells did notinduce disease (Table 1). In contrast, activation of D011 CD4+ cells by3–4 days of culture with OVA peptide presented by spleen APCs repro-ducibly generated effector cells that rapidly destroyed the insulin-pro-ducing β cells in RIP-mOVA but not wild-type (WT) BALB/c recipi-ents (Table 1). The requirement for activation of the T cells beforeadoptive transfer in this system provided an opportunity to examinehow the conditions of activation influence the subsequent effectoractivity of potentially autoreactive CD4+ cells.
To control the conditions of priming, we used as APCs DrosophilaSchneider SC2 cells transfected with murine MHC class II H-2Ad mol-ecules and either B7-1 alone (SC2.Ad.B7-1) or B7-1 and ICAM-1(SC2.Ad.B7-1.ICAM-1)18. Purified D011 CD4+ cells cultured withSC2.Ad.B7-1 cells stimulated strong peptide-dependent proliferativeresponses that were similar in magnitude to those induced by culturewith T-depleted spleen APCs. However, the concentration of peptiderequired for this response was generally higher than that required forresponses to T-depleted spleen APCs (Fig. 1). Culture with peptide pre-sented by SC2.Ad.B7-1.ICAM-1 cells also elicited strong proliferativeresponses that peaked at lower concentrations of peptide, which con-firmed previous findings that ICAM-1 lowers the threshold for CD4+ Tcell activation20–24. Because CD4+ cells proliferated well in response to0.1–2 µM of OVA peptide presented by either SC2.Ad.B7-1 or SC2.Ad.B7-1.ICAM-1 cells, these concentrations of peptide were used to acti-vate D011 cells before adoptive transfer into RIP-mOVA mice. Theresults were similar with all concentrations of peptide tested.
Generation of diabetogenic effector cellsWe studied the capacity of D011 CD4+ cells, activated by culture with0.1–2 µM of OVA peptide and either SC2.Ad.B7-1 or SC2.Ad.B7-1.ICAM-1 cells, to induce disease in RIP-mOVA recipients. Viable blastcells recovered after 3–4 days of culture were depleted of residual trans-fected SC2 cells by passage over Percoll density gradients and injectedinto lightly irradiated (500 cGy) RIP-mOVA mice. Recipients weremonitored for disease on a weekly basis by measuring blood glucose.CD4+ cells stimulated with peptide presented by SC2.Ad.B7-1.ICAM-1cells consistently induced disease in RIP-mOVA recipients within 1–2weeks after transfer (Table 2). However, adoptive transfer of CD4+ cellsprimed with SC2.Ad.B7-1 cells did not induce detectable diabetes,regardless of the OVA peptide concentration, even after up to 12 weeksafter transfer. Diabetes was not observed in WT nontransgenic recipientsof either SC2.Ad.B7-1– or SC2.Ad.B7-1.ICAM-1–primed cells.
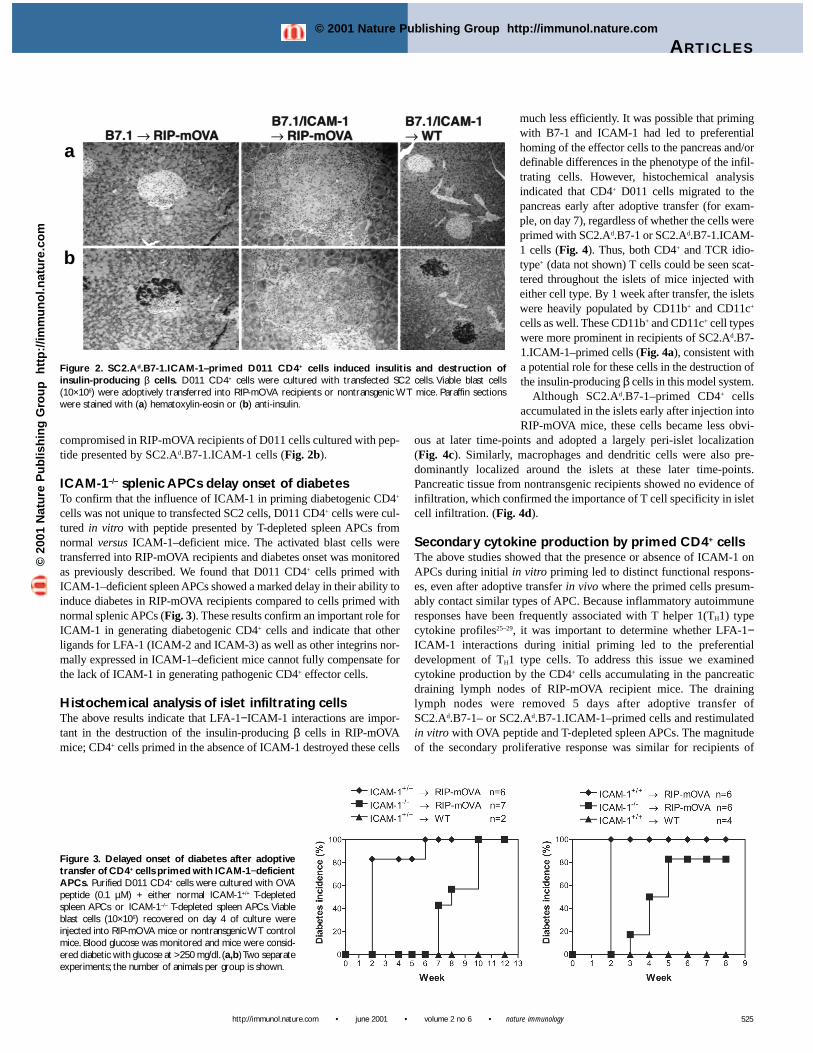
Islet infiltration was assessed by examining hematoxylin-eosin–stained pancreatic sections. Infiltration appeared more marked inrecipients of SC2.Ad.B7-1.ICAM-1–primed cells compared to thosereceiving SC2.Ad.B7-1–primed cells (Fig. 2a). The insulin-producing βcells in these latter mice were intact, as determined by staining withanti-insulin (Fig. 2b). This data supported the previous results, whichshowed no evidence of diabetes in mice receiving D011 cells primedwith SC2.Ad.B7-1 cells. In contrast, insulin production was severely
524
Table 1. Activated DO11 CD4+ T cells induce diabetes inRIP-mOVA mice
CD4+ T cells Recipient Incidence
Naïve RIP-mOVA 0/3Naïve WT 0/3Activated RIP-mOVA 49/50Activated WT 0/24— RIP-mOVA 0/28
Ten million naïve DO11 CD4+ T cells, or T cells cultured for 4 days in the presenceof 0.1 µM OVA peptide and splenic APCs, were transferred into irradiated (500cGy) RIP-mOVA mice. Blood glucose was monitored on a weekly basis and animalswere considered hyperglycemic after two consecutive nonfasting blood glucosereadings higher than 250 mg/dl. Diabetes onset generally occurred within 1–2weeks after transfer of activated cells; healthy mice were monitored on a weeklybasis for 8–16 weeks. Data are from ten experiments.
Table 2. B7-1–primed cells do not induce diabetes in RIP-mOVA recipients; coexpression of ICAM-1 reconstitutespathogenicity
APCs Recipient Incidence %
SC2.Ad.B7-1.ICAM-1 RIP-mOVA 31/32 97SC2.Ad.B7-1 RIP-mOVA 0/26 0SC2.Ad.B7-1.ICAM-1 WT 0/10 0SC2.Ad.B7-1 WT 0/6 0
D011 CD4+ T cells (10×106) cultured for 4 days with OVA peptide and transfectedSC2 cells were adoptively transferred into irradiated (500 cGy) RIP-mOVA mice orWT BALB/cJ mice. Blood glucose was monitored on a weekly basis and animalswere considered diabetic after two consecutive blood glucose readings above 250mg/dl. Diabetes onset generally occurred 1–3 weeks after transfer of SC2.Ad.B7-1.ICAM-1–primed cells. Healthy mice were monitored on a weekly basis for 8–16weeks.The data are pooled from seven experiments.
Figure 1. SC2.Ad.B7-1 and SC2.Ad.B7-1.ICAM-1 cells elicit strong prolifer-ative responses that are equivalent in magnitude to T-depleted spleencells. Purified OVA-specific D011 CD4+ T cells (4×104) were cultured with variousconcentrations of OVA peptide + either 2×105 SC2.Ad.B7-1 (B7.1) or SC2.Ad.B7-1.ICAM-1 (B7.1 +ICAM-1) cells or 5×105 T-depleted spleen (T-S) APCs. Proliferationwas determined by [3H]thymidine uptake on days 3–5. The peak response, shownhere, occurred on day 4. Data are means of triplicate cultures.
©20
01 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://im
mu
no
l.nat
ure
.co
m© 2001 Nature Publishing Group http://immunol.nature.com
ARTICLES
http://immunol.nature.com • june 2001 • volume 2 no 6 • nature immunology
compromised in RIP-mOVA recipients of D011 cells cultured with pep-tide presented by SC2.Ad.B7-1.ICAM-1 cells (Fig. 2b).
ICAM-1–/– splenic APCs delay onset of diabetesTo confirm that the influence of ICAM-1 in priming diabetogenic CD4+
cells was not unique to transfected SC2 cells, D011 CD4+ cells were cul-tured in vitro with peptide presented by T-depleted spleen APCs fromnormal versus ICAM-1–deficient mice. The activated blast cells weretransferred into RIP-mOVA recipients and diabetes onset was monitoredas previously described. We found that D011 CD4+ cells primed withICAM-1–deficient spleen APCs showed a marked delay in their ability toinduce diabetes in RIP-mOVA recipients compared to cells primed withnormal splenic APCs (Fig. 3). These results confirm an important role forICAM-1 in generating diabetogenic CD4+ cells and indicate that otherligands for LFA-1 (ICAM-2 and ICAM-3) as well as other integrins nor-mally expressed in ICAM-1–deficient mice cannot fully compensate forthe lack of ICAM-1 in generating pathogenic CD4+ effector cells.
Histochemical analysis of islet infiltrating cellsThe above results indicate that LFA-1−ICAM-1 interactions are impor-tant in the destruction of the insulin-producing β cells in RIP-mOVAmice; CD4+ cells primed in the absence of ICAM-1 destroyed these cells
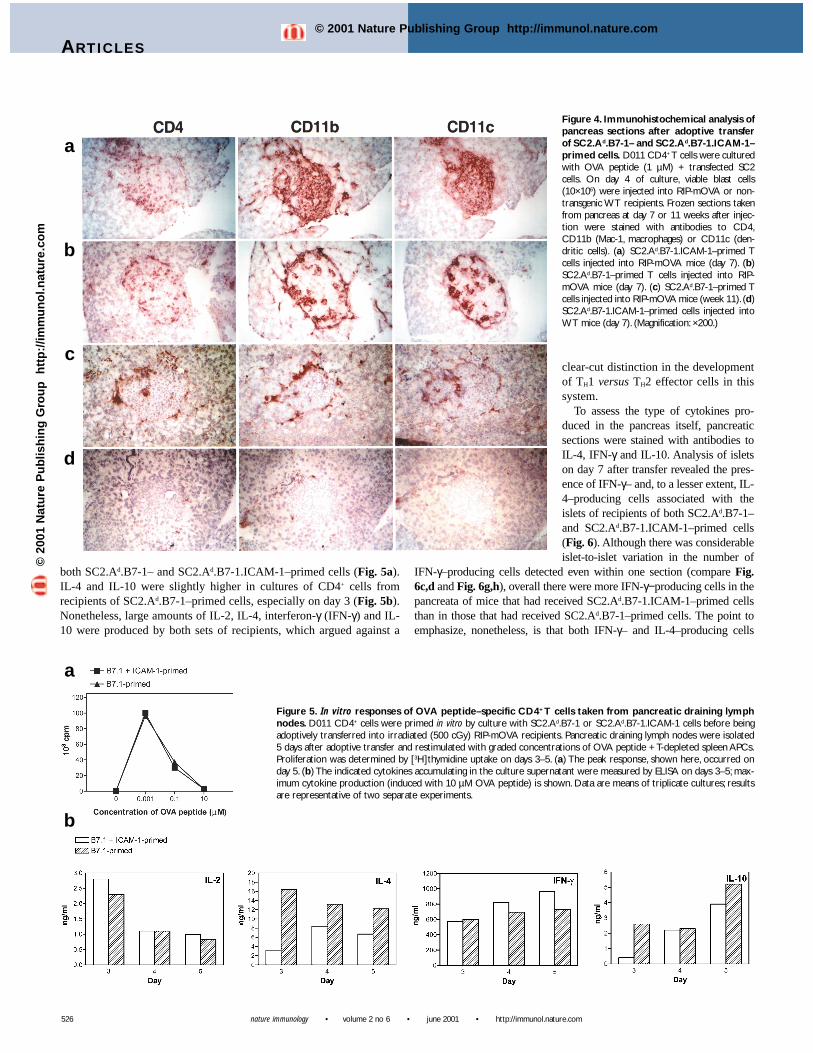
much less efficiently. It was possible that primingwith B7-1 and ICAM-1 had led to preferentialhoming of the effector cells to the pancreas and/ordefinable differences in the phenotype of the infil-trating cells. However, histochemical analysisindicated that CD4+ D011 cells migrated to thepancreas early after adoptive transfer (for exam-ple, on day 7), regardless of whether the cells wereprimed with SC2.Ad.B7-1 or SC2.Ad.B7-1.ICAM-1 cells (Fig. 4). Thus, both CD4+ and TCR idio-type+ (data not shown) T cells could be seen scat-tered throughout the islets of mice injected witheither cell type. By 1 week after transfer, the isletswere heavily populated by CD11b+ and CD11c+
cells as well. These CD11b+ and CD11c+ cell typeswere more prominent in recipients of SC2.Ad.B7-1.ICAM-1–primed cells (Fig. 4a), consistent witha potential role for these cells in the destruction ofthe insulin-producing β cells in this model system.
Although SC2.Ad.B7-1–primed CD4+ cellsaccumulated in the islets early after injection intoRIP-mOVA mice, these cells became less obvi-
ous at later time-points and adopted a largely peri-islet localization(Fig. 4c). Similarly, macrophages and dendritic cells were also pre-dominantly localized around the islets at these later time-points.Pancreatic tissue from nontransgenic recipients showed no evidence ofinfiltration, which confirmed the importance of T cell specificity in isletcell infiltration. (Fig. 4d).
Secondary cytokine production by primed CD4+ cellsThe above studies showed that the presence or absence of ICAM-1 onAPCs during initial in vitro priming led to distinct functional respons-es, even after adoptive transfer in vivo where the primed cells presum-ably contact similar types of APC. Because inflammatory autoimmuneresponses have been frequently associated with T helper 1(TH1) typecytokine profiles25–29, it was important to determine whether LFA-1−ICAM-1 interactions during initial priming led to the preferentialdevelopment of TH1 type cells. To address this issue we examinedcytokine production by the CD4+ cells accumulating in the pancreaticdraining lymph nodes of RIP-mOVA recipient mice. The draininglymph nodes were removed 5 days after adoptive transfer ofSC2.Ad.B7-1– or SC2.Ad.B7-1.ICAM-1–primed cells and restimulatedin vitro with OVA peptide and T-depleted spleen APCs. The magnitudeof the secondary proliferative response was similar for recipients of
525
Figure 2. SC2.Ad.B7-1.ICAM-1–primed D011 CD4+ cells induced insulitis and destruction ofinsulin-producing β cells. D011 CD4+ cells were cultured with transfected SC2 cells. Viable blast cells(10×106) were adoptively transferred into RIP-mOVA recipients or nontransgenic WT mice. Paraffin sectionswere stained with (a) hematoxylin-eosin or (b) anti-insulin.
a
b
Figure 3. Delayed onset of diabetes after adoptivetransfer of CD4+ cells primed with ICAM-1−deficientAPCs. Purified D011 CD4+ cells were cultured with OVApeptide (0.1 µM) + either normal ICAM-1+/+ T-depletedspleen APCs or ICAM-1–/– T-depleted spleen APCs. Viableblast cells (10×106) recovered on day 4 of culture wereinjected into RIP-mOVA mice or nontransgenic WT controlmice. Blood glucose was monitored and mice were consid-ered diabetic with glucose at >250 mg/dl. (a,b) Two separateexperiments; the number of animals per group is shown.
©20
01 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://im
mu
no
l.nat
ure
.co
m© 2001 Nature Publishing Group http://immunol.nature.com
nature immunology • volume 2 no 6 • june 2001 • http://immunol.nature.com
ARTICLES
both SC2.Ad.B7-1– and SC2.Ad.B7-1.ICAM-1–primed cells (Fig. 5a).IL-4 and IL-10 were slightly higher in cultures of CD4+ cells fromrecipients of SC2.Ad.B7-1–primed cells, especially on day 3 (Fig. 5b).Nonetheless, large amounts of IL-2, IL-4, interferon-γ (IFN-γ) and IL-10 were produced by both sets of recipients, which argued against a
clear-cut distinction in the developmentof TH1 versus TH2 effector cells in thissystem.
To assess the type of cytokines pro-duced in the pancreas itself, pancreaticsections were stained with antibodies toIL-4, IFN-γ and IL-10. Analysis of isletson day 7 after transfer revealed the pres-ence of IFN-γ– and, to a lesser extent, IL-4–producing cells associated with theislets of recipients of both SC2.Ad.B7-1–and SC2.Ad.B7-1.ICAM-1–primed cells(Fig. 6). Although there was considerableislet-to-islet variation in the number of
IFN-γ–producing cells detected even within one section (compare Fig.6c,d and Fig. 6g,h), overall there were more IFN-γ−producing cells in thepancreata of mice that had received SC2.Ad.B7-1.ICAM-1–primed cellsthan in those that had received SC2.Ad.B7-1–primed cells. The point toemphasize, nonetheless, is that both IFN-γ– and IL-4–producing cells
526
a
b
Figure 4. Immunohistochemical analysis ofpancreas sections after adoptive transferof SC2.Ad.B7-1– and SC2.Ad.B7-1.ICAM-1–primed cells. D011 CD4+ T cells were culturedwith OVA peptide (1 µM) + transfected SC2cells. On day 4 of culture, viable blast cells(10×106) were injected into RIP-mOVA or non-transgenic WT recipients. Frozen sections takenfrom pancreas at day 7 or 11 weeks after injec-tion were stained with antibodies to CD4,CD11b (Mac-1, macrophages) or CD11c (den-dritic cells). (a) SC2.Ad.B7-1.ICAM-1–primed Tcells injected into RIP-mOVA mice (day 7). (b)SC2.Ad.B7-1–primed T cells injected into RIP-mOVA mice (day 7). (c) SC2.Ad.B7-1–primed Tcells injected into RIP-mOVA mice (week 11). (d)SC2.Ad.B7-1.ICAM-1–primed cells injected intoWT mice (day 7). (Magnification: ×200.)
a
b
c
d
Figure 5. In vitro responses of OVA peptide–specific CD4+ T cells taken from pancreatic draining lymphnodes. D011 CD4+ cells were primed in vitro by culture with SC2.Ad.B7-1 or SC2.Ad.B7-1.ICAM-1 cells before beingadoptively transferred into irradiated (500 cGy) RIP-mOVA recipients. Pancreatic draining lymph nodes were isolated5 days after adoptive transfer and restimulated with graded concentrations of OVA peptide + T-depleted spleen APCs.Proliferation was determined by [3H]thymidine uptake on days 3–5. (a) The peak response, shown here, occurred onday 5. (b) The indicated cytokines accumulating in the culture supernatant were measured by ELISA on days 3–5; max-imum cytokine production (induced with 10 µM OVA peptide) is shown. Data are means of triplicate cultures; resultsare representative of two separate experiments.
©20
01 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://im
mu
no
l.nat
ure
.co
m© 2001 Nature Publishing Group http://immunol.nature.com
DiscussionOur data argue that accessory molecules play a key role in determiningthe long-term effector function of activated CD4+ T cells. Priming naïveCD4+ T cells with APCs expressing B7-1 alone generated benign effec-tor cells that did not significantly damage insulin-producing β cells inRIP-mOVA recipients. Coexpression of ICAM-1 and B7-1 on APCs,however, promoted the development of highly pathogenic effectorCD4+ cells that rapidly destroyed β cells. The marked differences in thebehavior of these two primed CD4+ cell populations were notable giventhat the transfected SC2 cells, which are maintained at room tempera-ture, survive poorly at 37 °C, rarely remaining viable for more than 24h. Thus, the accessory molecule interactions that determine the func-tional differences in vivo must take place very early during T cell acti-vation. Although SC2.Ad.B7-1–primed CD4+ cells did not cause dia-betes in this system, migration to the islets was extensive, especiallyearly after adoptive transfer into RIP-mOVA mice.
It is important to determine whether the priming commitmentobserved in these studies is immutable. Although the influence of the invitro priming conditions was not reversed upon encountering APCs inthe adoptive transfer hosts, it is possible that secondary stimulation invitro with distinct accessory molecule interactions could convert thefunctional phenotype of the resulting effector cells. The results of a pre-liminary experiment suggest that reversibility may be limited becauseSC2.Ad.B7-1–primed CD4+ cells that were subsequently restimulatedwith SC2.Ad.B7-1.ICAM-1 APCs did not induce diabetes upon adop-tive transfer. However, more investigative work needs to be done.
In light of the likely ability of LFA-1–ICAM-1 to strengthen TCR-peptide-MHC interactions24,42–45, the functional differences betweenSC2.Ad.B7-1– and SC2.Ad.B7-1.ICAM-1–primed CD4+ cells could be areflection of differences in the strength of TCR-peptide activation.According to this view, strong activation of T cells would lead to dia-betes, whereas weak activation would generate effectors that are poorlydestructive. A number of observations make this possibility unlikely.First, priming with a range of OVA peptide concentrations from 0.1–2µM did not overcome the functional differences in the primed CD4+ cells.This was the case despite the fact that 0.1 µM was the optimal concen-tration for stimulating proliferative responses of SC2.Ad.B7-1.ICAM-1–primed cells and 1 µM was optimal for proliferation of SC2.Ad.B7-1–primed cells. In other studies, we found that high concentrations ofpeptide during priming (for example, 10 µM) actually decreased thepathogenicity of OVA peptide–reactive CD4+ cells, presumably reflecting
ARTICLES
http://immunol.nature.com • june 2001 • volume 2 no 6 • nature immunology
were seen in recipients of SC2.Ad.B7-1– and SC2.Ad.B7-1.ICAM-1–primed cells. In contrast, IL-10–producing cells were not detected inany of these sections (data not shown).
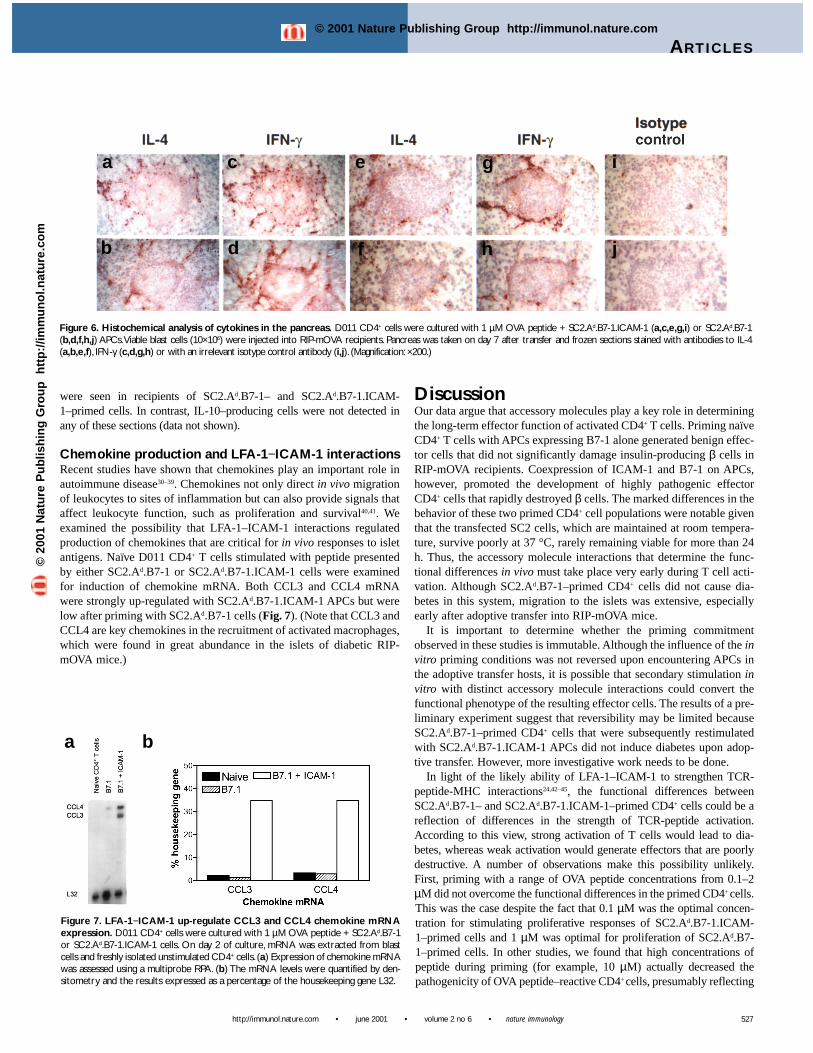
Chemokine production and LFA-1−ICAM-1 interactionsRecent studies have shown that chemokines play an important role inautoimmune disease30–39. Chemokines not only direct in vivo migrationof leukocytes to sites of inflammation but can also provide signals thataffect leukocyte function, such as proliferation and survival40,41. Weexamined the possibility that LFA-1–ICAM-1 interactions regulatedproduction of chemokines that are critical for in vivo responses to isletantigens. Naïve D011 CD4+ T cells stimulated with peptide presentedby either SC2.Ad.B7-1 or SC2.Ad.B7-1.ICAM-1 cells were examinedfor induction of chemokine mRNA. Both CCL3 and CCL4 mRNAwere strongly up-regulated with SC2.Ad.B7-1.ICAM-1 APCs but werelow after priming with SC2.Ad.B7-1 cells (Fig. 7). (Note that CCL3 andCCL4 are key chemokines in the recruitment of activated macrophages,which were found in great abundance in the islets of diabetic RIP-mOVA mice.)
527
a b
Figure 7. LFA-1−ICAM-1 up-regulate CCL3 and CCL4 chemokine mRNAexpression. D011 CD4+ cells were cultured with 1 µM OVA peptide + SC2.Ad.B7-1or SC2.Ad.B7-1.ICAM-1 cells. On day 2 of culture, mRNA was extracted from blastcells and freshly isolated unstimulated CD4+ cells. (a) Expression of chemokine mRNAwas assessed using a multiprobe RPA. (b) The mRNA levels were quantified by den-sitometry and the results expressed as a percentage of the housekeeping gene L32.
Figure 6. Histochemical analysis of cytokines in the pancreas. D011 CD4+ cells were cultured with 1 µM OVA peptide + SC2.Ad.B7-1.ICAM-1 (a,c,e,g,i) or SC2.Ad.B7-1(b,d,f,h,j) APCs.Viable blast cells (10×106) were injected into RIP-mOVA recipients. Pancreas was taken on day 7 after transfer and frozen sections stained with antibodies to IL-4(a,b,e,f), IFN-γ (c,d,g,h) or with an irrelevant isotype control antibody (i,j). (Magnification: ×200.)
a
b
c
d
e
f
g
h
i
j
©20
01 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://im
mu
no
l.nat
ure
.co
m© 2001 Nature Publishing Group http://immunol.nature.com

nature immunology • volume 2 no 6 • june 2001 • http://immunol.nature.com
ARTICLES
high zone tolerance (S. R. Webb et al., unpublished findings). Theseresults are inconsistent with a strictly adhesion-strengthening role forLFA-1. It is also important to note that both SC2.Ad.B7-1– andSC2.Ad.B7-1.ICAM-1–primed effector cells were similarly activated inthat they expressed comparable amounts of CD25, CD44 and L-selectin(data not shown). In addition, adoptive transfer of carboxyfluoresceindiacetate succinimidyl diester–labeled cells did not reveal any markeddifferences in the in vivo proliferative responses of SC2.Ad.B7-1– versusSC2.Ad.B7-1.ICAM-1–primed cells (data not shown), which suggeststhat both cell types are represented in roughly equivalent numbers invivo.
The imprinting of CD4+ cell effector activity appears to occur largelyindependently of the differentiation and commitment of effector cells tothe TH1 versus TH2 subsets, which has been defined on the basis of qual-itative differences in cytokine production. Although IL-4 was producedin larger amounts by SC2.Ad.B7-1–primed T cells and IFN-γ–producingcells were more prevalent in the pancreata of mice whose T cells havebeen primed with SC2.Ad.B7-1.ICAM-1 cells, there was no qualitative-ly distinct pattern of cytokine production by the two populations.Notwithstanding the potential importance of these slight quantitativedifferences in cytokine profiles, the results suggest that additional fac-tors contribute to the pathogenicity of CD4+ cells. This view is consis-tent with increasing evidence that raises questions about the extent towhich cytokine imbalance alone can account for autoimmunity25,28,46.
In accounting for how LFA-1–ICAM-1 interactions control the path-ogenicity of CD4+ effector cells, it is notable that SC2.Ad.B7-1.ICAM-1–primed CD4+ cells produced much higher amounts of CCL3 andCCL4 chemokines than SC2.Ad.B7-1–primed cells. This findingimplies that LFA-1–ICAM-1 interactions during priming have qualita-tive effects on CD4+ cell gene transcription as well as the quantitativeeffects mentioned above for cytokines. These qualitative effects areespecially pronounced at the level of chemokine genes. It is well estab-lished that CCL3 and CCL4 are macrophage attractants30,35 and thatmacrophages are implicated in the destruction of islet β cells25,47,48. Inaddition, CD4+ cell priming with ICAM-1+ APCs augmentedmacrophage accumulation in islets. Thus, the pathogenicity of ICAM-1+–primed CD4+ cells may be largely attributable to increased localsynthesis of critical chemokines in the vicinity of the islets, althoughthis does not exclude that LFA-1 may also regulate the induction ofadditional inflammatory gene products. Further work must be done toassess these possibilities. In summary, the data in this paper show thatthe cell-cell interactions encountered during initial T cell activation canhave radical effects on subsequent expression of effector function. Inparticular, effector cell activity leading to destruction of pancreatic βcells appears to be highly dependent on LFA-1-ICAM-1 interactionsduring T cell priming.
MethodsMice. DO11 TCR transgenic mice (provided by K. Murphy and D. Loh), ICAM-1–deficientmice (Jackson Laboratories, Bar Harbor, ME) and RIP-mOVA mice were bred and main-tained at The Scripps Research Institute. DO11 mice were backcrossed to BALB/cJ micefor more than 6 years and then maintained on a BALB/cJ background. RIP-mOVA miceexpress a transgene that encodes a membrane form of OVA that is under the control of therat insulin promoter19 and have been backcrossed for ten generations to BALB/cJ mice. Allexperiments were done in compliance with institutional guidelines as approved by theInstitutional Animal Care and Use Committee of The Scripps Research Institute.
Drosophila cell lines. SC2.Ad.B7-1.ICAM-1 and SC2.Ad.B7-1 cells were generated asdescribed18,20. Briefly, cDNA clones for B7-1, ICAM-1 and H-2Ad α and β chains wereexpressed in Drosophila Schneider SC2 cells under the control of the metallothionein pro-moter. Cell lines expressing the desired proteins were selected by growing in Schneider’sDrosophila medium (Gibco-BRL, Gaithersburg, MD) containing 5% fetal calf serum(FCS), antibiotics and geneticin (500 µg/ml), followed by several rounds of cell sorting.
Drosophila cells were grown at room temperature and expression of the transfected proteinswas induced by the addition of CuSO4 (1 mM).
Cell purification. CD4+ cells were purified from DO11 lymph nodes by passage over nylonwool columns followed by antibody- and complement-mediated killing of cells bearingCD8 (with mAb 3.168), H-2Ad (with mAb MK.D6), H-2E (with mAb 14-4-4) and heat-sta-ble antigen (HSA) (with mAb J11d), as described18. The purity of CD4+ T cells was moni-tored by [3H]thymidine uptake during culture with OVA peptide in the absence of addedAPCs. T-depleted spleen APCs were prepared by treatment of spleen cell suspensions withantibodies to T cells (anti-CD8, mAb 3.168; anti-CD4, mAb RL172; anti-Thy1.2, mAb J1j)and complement to deplete T cells.
Proliferation assays. Purified CD4+ cells were cultured in microtiter wells at a concentra-tion of 4×104 cells/well + 2×105 transfected SC2 cells or 5×105 mitomycin C–treated T-depleted spleen APCs and OVA peptide. RPMI 1640 culture medium was supplementedwith 10% FCS, 5% NCTC 109, 5×10-5 M 2-mercaptoethanol, glutamine, Hepes buffer andantibiotics. Culture wells were pulsed with [3H]thymidine (1 µCi) 12–18 h before collect-ing on glass fiber filters and counting in an EG&G Wallac Microbeta TriLux scintillationcounter (San Fransisco, CA). All cultures were set up in triplicate. As Drosophila SC2 cellsdie within 24 h of culture at 37 °C, any contribution of SC2 cell division to the measuredproliferative responses was excluded.
For secondary responses, 1×106 DO11 CD4+ cells were cultured for 10–12 days in culturemedium (5 ml) with OVA peptide (1 µM) and 4×106 transfected SC2 cells. Activated cellswere collected and purified by passage over density gradients before culture in microtiterwells (4×104 cells/well) with 5×105 mitomycin C–treated T-depleted spleen APCs and peptide.
Adoptive transfer studies. Activated CD4+ T cells were cultured as described for sec-ondary responses and collected after 4 days of stimulation. After removal of transfected SC2cells by passage over density gradients, 10×106 viable blast cells were injected intra-venously into lightly irradiated (500 cGy) RIP-mOVA mice19. Blood glucose was monitoredon a weekly basis with an Accu-Chek Advantage blood glucose monitor (BoehringerMannheim, Indianapolis, IN). Mice were considered diabetic after two consecutive non-fasting blood glucose readings of >250 mg/dl.
Immunohistochemistry. Mice were killed by CO2 asphyxiation. The pancreata were halvedand fixed either in 10% neutral-buffered formalin and embedded in paraffin or snap-frozenin OCT compound (Tissue Tek, Torrance, CA) in liquid nitrogen for cryosectioning.Formalin-fixed sections were deparaffinized with xylene and alcohol and either stained withhematoxylin and eosin or used for detecting insulin production. Insulin labeling was donewith anti–porcine insulin (Dako, Carpinteria, CA) and biotinylated anti–guinea pigimmunoglobulin (Vector Laboratories, Burlingame, CA) followed by treatment with theVectastain ABC peroxidase kit (Vector Laboratories). Labeling was developed with the sub-strate 3′,3′diaminobenzidine (Sigma, St. Louis, MI).
For CD4 (with GK1.5), CD11b (with M1/70, BD PharMingen, San Diego, CA) andCD11c (PharMingen) labeling, 7 µm acetone-fixed cryosections were incubated with theprimary antibody and biotinylated anti-rat (Jackson Laboratories) or anti-hamster (forCD11c, Jackson Laboratories) and ExtrAvidin -alkaline phosphatase (Sigma) and visual-ized with Fast Violet (Sigma).
For cytokine detection, acetone-fixed frozen sections were incubated overnight withanti–IFN-γ (XMG1.2, PharMingen), anti–IL-4 (BVB6/24G2, PharMingen) or isotype-matched control antibody (R3-34, PharMingen) in PBS with 1% bovine serum albumin con-taining 0.05% saponin (Sigma)49,50. The specificity of labeling was confirmed with the useof nontransgenic normal pancreas, which was not labeled by any of these antibodies, andwith the use of isotype-matched control antibodies. Sections were washed and treated asdescribed above for lineage-specific antibodies. All sections were lightly counterstainedwith hematoxylin.
In vitro cytokine production. Samples of culture supernatant of the secondary cultureswere removed before pulsing with [3H]thymidine. The accumulation of cytokines in the cul-ture supernatant was measured by a sandwich ELISA assay, as described18. Capture anti-bodies and biotinylated detection antibodies were both from PharMingen. Recombinantcytokines (Genzyme, Cambridge, MA or PharMingen) were used for standard curves.
Analysis of chemokine mRNA by RNase protection assay (RPA). After purified DO11CD4+ cells were cultured with transfected SC2 cells, total RNA was extracted from collect-ed cells with the use of Stat60 (TEL-Test “B”)18. The RiboQuant Multi-Probe RPA System(PharMingen) was used to detect chemokine mRNA according to the manufacturer’sinstructions and as described18,51. RNA was hybridized to labeled probes for 13 h at 56 °Cand unhybridized probes and RNA were digested by incubating with 100 µl of RNase A(192 ng/ml) and RNase T1 (600 U/ml) for 45 min at 37 °C. Phenol + chloroform–extractedprobe-target duplexes were separated by PAGE and visualized by autoradiography. Theintensity of the radioactive bands was measured with a PhosphorImager SI (MolecularDynamics, Eugene, OR). The net cpm per uridine triphosphate residue was calculated, asdescribed18,51, with the formula:
(cpm of cytokine band) – (cpm background around the band).number of U residues in the specific riboprobe
528
©20
01 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://im
mu
no
l.nat
ure
.co
m© 2001 Nature Publishing Group http://immunol.nature.com

ARTICLES
http://immunol.nature.com • june 2001 • volume 2 no 6 • nature immunology
The results are expressed as a percentage of the L32 housekeeping gene.
Acknowledgements
We thank B. Marchand for typing the manuscript and D. Redondo for technical assis-tance. Supported by grants CA41993, CA25803 and AI39664 from the United StatesPublic Health Service, a grant from the Juvenile Diabetes Foundation and a grant from R.W. Johnson Pharmaceutical Research Institute.
Received 18 December 2000; accepted 19 March 2001.
1. Lenschow, D. J.,Walunas,T. L. & Bluestone, J.A. CD28/B7 system of T cell co-stimulation. Annu. Rev.Immunol. 14, 233–258 (1996).
2. Linsley, P. S. & Ledbetter, J.A.The role of the CD28 receptor during T cell responses to antigen.Annu. Rev. Immunol. 11, 191–212 (1993).
3. Allison, J. P. & Krummel, M. F.The Yin and Yang of T cell costimulation. Science 270, 932–933 (1995).4. Schwartz, R. H. Costimulation of T lymphocytes:The role of CD28, CTLA-4, and B7/BB1 in
Interleukin-2 Production and Immunotherapy. Cell 71, 1065–1068 (1992).5. Hintzen, R. Q. et al. Engagement of CD27 with its ligand CD70 provides a second signal for T cell
activation. J. Immunol. 154, 2612–2623 (1995).6. Yashiro,Y. et al. A fundamental difference in the capacity to induce proliferation of naive T cells
between CD28 and other co-stimulatory molecules. Eur. J. Immunol. 28, 926–935 (1998).7. Zuckerman, L.A., Pullen, L. & Miller, J. Functional consequences of costimulation by ICAM-1 on IL-2
gene expression and T cell activation. J. Immunol. 160, 3259–3268 (1998).8. Kuchroo,V. K. et al. B7-1 and B7-2 costimulatory molecules activate differentially the Th1/Th2 devel-
opmental pathways:Application to autoimmune disease therapy. Cell 80, 707–718 (1995).9. Freeman, G. J. et al. B7-1 and B7-2 do not deliver identical costimulatory signals, since B7-2 but not
B7-1 preferentially costimulates the initial production of IL-4. Immunity 2, 523–532 (1995).10. Gause,W., Urban, J., Linsley, P. & Lu, P. Role of B7 signaling in the differentiation of naive CD4+ T cells
to effector interleukin-4-producing T helper cells. Immunol. Res. 14, 176–178 (1995).11. King, C. L., Stupi, R. J., Craighead, N., June, C. H. & Thyponitis, G. CD28 activation promotes Th2 sub-
set differentiation by human CD4+ cells. Eur. J. Immunol. 25, 587–595 (1995).12. Lenschow, D. J. et al. CD28/B7 regulation of Th1 and Th2 subsets in the development of autoim-
mune diabetes. Immunity 5, 285–293 (1996).13. Ranger,A. M., Das, M. P., Kuchroo,V. K. & Glimcher, L. H. B7-2 (CD86) is essential for the develop-
ment of IL-4-producing T cells. Int. Immunol. 8, 1549–1560 (1996).14. Rulifson, I. C., Sperling,A. I., Fields, P. E., Fitch, F.W. & Bluestone, J.A. CD28 costimulation promotes
the production of Th2 cytokines. J. Immunol. 158, 658–665 (1997).15. Rodriguez-Palmero, M., Hara,T.,Thumbs,A. & Hunig,T.Triggering of T cell proliferation through
CD28 induces GATA-3 and promotes T helper type 2 differentiation in vitro and in vivo. Eur. J.Immunol. 29, 3914–3924 (1999).
16. Lenschow, D. J. et al. Differential effects of anti-B7-1 and anti-B7.2 monoclonal antibody treat-ment on the development of diabetes in the nonobese diabetic mouse. J. Exp. Med. 181,1145–1155 (1995).
17. Salomon, B. & Bluestone, J.A. LFA-1 interaction with ICAM-1 and ICAM-2 regulates Th2 cytokineproduction. J. Immunol. 161, 5138–5142 (1998).
18. Luksch, C. R. et al. Intercellular adhesion molecule-1 inhibits interleukin-4 production by naïve Tcells. Proc. Natl Acad. Sci. USA 96, 3023–3028 (1999).
19. Kurts, C. et al. Constitutive class I-restricted exogenous presentation of self antigens in vivo. J. Exp.Med. 184, 923–930 (1996).
20. Cai, Z. et al. Transfected Drosophila cells as a probe for defining the minimal requirements for stimu-lating unprimed CD8+ T cells. Proc. Natl Acad. Sci. USA 93, 14736–14741 (1996).
21. van Seventer, G.A., Shimizu,Y., Horgan, K. J. & Shaw, S.The LFA-1 ligand ICAM-1 provides an impor-tant costimulatory signal for T cell receptor-mediated activation of resting T cells. J. Immunol. 144,4579–4586 (1990).
22. Dubey, C., Croft, M. & Swain, S. Costimulatory requirements of naive CD4+ T cells. ICAM-1 or B7-1can costimulate naive CD4 T cell activation but both are required for optimum response. J. Immunol.155, 45–57 (1995).
23. Damle, N. K., Klussman, K., Linsley, P. S. & Aruffo,A. Differential costimulatory effects of adhesionmolecules B7, ICAM-1, LFA-3, and VCAM-1 on resting and antigen-primed CD4+ T lymphocytes. J.Immunol. 148, 1985–1992 (1992).
24. Bachmann, M. F. et al. Distinct roles for LFA-1 and CD28 during activation of naive T cells: adhesionversus costimulation. Immunity 7, 549–557 (1997).
25. Rabinovitch,A.An update on cytokines in the pathogenesis of insulin-dependent diabetes mellitus.
Diabetes Metab. Rev. 14, 129–151 (1998).26. Delovitch,T. L. & Singh, B.The nonobese diabetic mouse as a model of autoimmune diabetes: immune
dysregulation gets the NOD. Immunity 7, 727–738 (1997). [Erratum in Immunity 8, 531 (1998).]27. Falcone, M. & Sarvetnick, N.The effect of local production of cytokines in the pathogenesis of
insulin-dependent diabetes mellitus. Clin. Immunol. 90, 2–9 (1999).28. Lafaille, J. J.The role of helper T cell subsets in autoimmune diseases. Cytokine Growth Factor Rev. 9,
139–151 (1998).29. Liblau, R. S., Singer, S. M. & McDevitt, H. O.Th1 and Th2 CD4+ T cells in the pathogenesis of organ-
specific autoimmune diseases. Immunol.Today 16, 34–38 (1995).30. DeVries, M. E., Ran, L. & Kelvin, D. J. On the edge: the physiological and pathophysiological role of
chemokines during inflammatory and immunological responses. Semin. Immunol. 11, 95–104 (1999).31. Schimmer, R. C. et al. Streptococcal cell wall-induced arthritis. Requirements for neutrophils, P-
selectin, intercellular adhesion molecule-1, and macrophage-inflammatory protein-2. J. Immunol. 159,4103–4108 (1997).
32. Schrier, D. J., Schimmer, R. C., Flory, C. M.,Tung, D. K. & Ward, P.A. Role of chemokines and cytokinesin a reactivation model of arthritis in rats induced by injection with streptococcal cell walls. J.Leukoc. Biol. 63, 359–363 (1998).
33. Plater-Zyberk, C., Hoogewerf,A. J., Proudfoot,A. E., Power, C.A. & Wells,T. N. Effect of a CCchemokine receptor antagonist on collagen induced arthritis in DBA/1 mice. Immunol. Lett. 57,117–120 (1997).
34. Gong, J. H., Ratkay, L. G.,Waterfield, J. D. & Clark-Lewis, I.An antagonist of monocyte chemoattractantprotein 1 (MCP-1) inhibits arthritis in the MRL-lpr mouse model. J. Exp. Med. 186, 131–137 (1997).
35. Grewal, I. S. et al. Transgenic monocyte chemoattractant protein-1 (MCP-1) in pancreatic islets pro-duces monocyte-rich insulitis without diabetes: abrogation by a second transgene expressing sys-temic MCP-1. J. Immunol. 159, 401–408 (1997).
36. Karpus,W. J. et al. An important role for the chemokine macrophage inflammatory protein-1 α inthe pathogenesis of the T cell-mediated autoimmune disease, experimental autoimmuneencephalomyelitis. J. Immunol. 155, 5003–5010 (1995).
37. Godiska, R., Chantry, D., Dietsch, G. N. & Gray, P.W. Chemokine expression in murine experimentalallergic encephalomyelitis. J. Neuroimmunol. 58, 167–176 (1995).
38. Glabinski,A. R.,Tuohy,V. K. & Ransohoff, R. M. Expression of chemokines RANTES, MIP-1α and GRO-α correlates with inflammation in acute experimental autoimmune encephalomyelitis.Neuroimmunomodulation 5, 166–171 (1998).
39. Bradley, L. M. et al. Islet-specific Th1, but not Th2, cells secrete multiple chemokines and promoterapid induction of autoimmune diabetes. J. Immunol. 162, 2511–2520 (1999).
40. Nelson, P. J. & Krensky,A. M. Chemokines, lymphocytes and viruses: what goes around, comesaround. Curr. Opin. Immunol. 10, 265–270 (1998).
41. Zlotnik,A., Morales, J. & Hedrick, J.A. Recent advances in chemokines and chemokine receptors. Crit.Rev. Immunol. 19, 1–47 (1999).
42. Kuhlman, P., Moy,V.T., Lollo, B.A. & Brian,A.A.The accessory function of murine intercellular adhe-sion molecule-1 in T lymphocyte activation. Contributions of adhesion and co-activation. J. Immunol.146, 1773–1782 (1991).
43. Brunmark,A. & O’Rourke,A. M.Augmentation of mature CD4+ T cell responses to isolated antigenicclass II proteins by fibronectin and intercellular adhesion molecule-1. J. Immunol. 159, 1676–1685 (1997).
44. van Seventer, G.A., Semnani, R.T., Palmer, E. M., McRae, B. L. & van Seventer, J. M. Integrins and Thelper cell activation. Transplant Proc. 30, 4270–4274 (1998).
45. Dustin, M. L. et al. TCR-mediated adhesion of T cell hybridomas to planar bilayers containing purifiedMHC class II/peptide complexes and receptor shedding during detachment. J. Immunol. 157,2014–2021 (1996).
46. Suri,A. & Katz, J. D. Dissecting the role of CD4+ T cells in autoimmune diabetes through the use ofTCR transgenic mice. Immunol. Rev. 169, 55–65 (1999).
47. Jun, H. S.,Yoon, C. S., Zbytnuik, L., van Rooijen, N. & Yoon, J.W.The role of macrophages in T cell-mediated autoimmune diabetes in nonobese diabetic mice. J. Exp. Med. 189, 347–358 (1999).
48. Benoist, C. & Mathis, D. Cell death mediators in autoimmune diabetes—no shortage of suspects. Cell89, 1–3 (1997).
49. Vyth-Dreese, F.A. et al. Localization in situ of costimulatory molecules and cytokines in B-cell non-Hodgkin’s lymphoma. Immunology 94, 580–586 (1998).
50. Rothe, H., Hibino,T., Itoh,Y., Kolb, H. & Martin, S. Systemic production of interferon-γ inducing factor(IGIF) versus local IFN-γ expression involved in the development of Th1 insulitis in NOD mice. J.Autoimmun. 10, 251–256 (1997).
51. Sabzevari, H., Propp, S., Kono, D. H. & Theofilopoulos,A. N. G1 arrest and high expression of cyclinkinase and apoptosis inhibitors in accumulated activated/memory phenotype CD4+ cells of olderlupus mice. Eur. J. Immunol. 27, 1901–1910 (1997)
529
©20
01 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://im
mu
no
l.nat
ure
.co
m© 2001 Nature Publishing Group http://immunol.nature.com