document
TRANSCRIPT

52 NATURE BIOTECHNOLOGY VOL 18 JANUARY 2000 http://biotech.nature.com
RESEARCH ARTICLES
Stabilization of proteins encapsulated ininjectable poly (lactide-co-glycolide)
Gaozhong Zhu1,3, Susan R. Mallery2, and Steven P. Schwendeman1*
1Colleges of Pharmacy and 2of Dentistry, The Ohio State University, Columbus, OH 43210. 3Current address: Department of Pharmaceutical Science andTechnology, Biogen Inc., 14 Cambridge Center, Cambridge, MA 02142. *Corresponding author ([email protected]).
Received 14 April 1999; accepted 22 September 1999
Controlled release from biodegradable polymers is a novel approach to replace daily painful injections ofprotein drugs. A major obstacle to development of these polymers is the need to retain the structure andbiological activity of encapsulated proteins during months of incubation under physiological conditions. Weencapsulated bovine serum albumin (BSA) in injectable poly(DL-lactide-co-glycolide) (PLGA) 50/50 cylindri-cal implants and determined the mechanism of BSA instability. Simulations of the polymer microclimaterevealed that moisture and acidic pH (<3) triggered unfolding of encapsulated BSA, resulting in peptidebond hydrolysis and noncovalent aggregation. To neutralize the acids liberated by the biodegradable lac-tic/glycolic acid-based polyester, we coincorporated into the polymer an antacid, Mg(OH)2, which increasedmicroclimate pH and prevented BSA structural losses and aggregation for over one month. We successful-ly applied this stabilization approach in both cylinder- and microsphere-injectable configurations and fordelivery of angiogenic basic fibroblast growth factor and bone-regenerating bone morphogenetic protein-2.
Keywords: protein delivery, aggregation, stabilization, poly(DL-lactide-co-glycolide, bovine serum albumin.
Since protein delivery from polymers was first discovered1, researchhas focused on developing polymer formulations widely applicablefor delivery of protein drugs. Efforts to this end have intensifiedrecently as hundreds of recombinant proteins are in the pipeline forUS Food and Drug Administration (Rockville, MD) approval2, andprotein delivery generally requires daily injections3. Frequent dosingis clinically undesirable because of patient discomfort, psychologicaldistress, and poor compliance with self-injections. To reduce injec-tion frequency, peptide and protein drugs are encapsulated inbiodegradable polymers, which are processed into a form that is eas-ily administered through a syringe needle. Current preparations onthe market for the delivery of small peptides can reduce injections toonce every one to three months depending on the polymer implantsize4–6. During this incubation time, large globular proteins mustremain encapsulated at physiological temperature, which poses sig-nificant challenges regarding retention of both the structuralintegrity and biological activity of the protein7.
Two injectable polymer configurations are currently used todeliver peptides and proteins: spherical particles approximately1–100 µm in diameter, commonly referred to as “microspheres,” andsingle cylindrical implants approximately 0.8–1.5 mm in diameter,which we term “millicylinders.” Both configurations are preparedfrom the biocompatible polyesters formed from lactic and glycolicacids (PLGAs) commonly used in resorbable sutures, and each hasdistinct advantages and disadvantages8. Several potential sources ofirreversible inactivation of proteins encapsulated in PLGAs havebeen identified7. These include: (1) elevated levels of moisture, pro-viding sufficient protein mobility for reactivity; (2) an acidic micro-climate induced by acidic degradation products and carboxylic acidend groups of PLGA; and (3) adsorption of the protein to the poly-mer surface, which may catalyze protein unfolding and aggregation.
Few have been successful in stabilizing proteins encapsulated inPLGAs9–12. We report the mechanism of instability of a model pro-tein, bovine serum albumin (BSA), in PLGA millicylinders. Thismechanistic analysis suggested a general approach to stabilize pro-
teins encapsulated in PLGA, which was tested and found to work forBSA, basic fibroblast growth factor (bFGF), and bone morphogenet-ic protein-2 (BMP-2), and for both millicylinder and microspheregeometries.
Results and discussionIrreversible instability of BSA in PLGA. To investigate BSA stabilityin the polymer, we encapsulated BSA in PLGA 50/50 millicylinders(15% BSA wt/wt). Then, we monitored the release of BSA in aphosphate buffer and the simultaneous loss of soluble BSA encap-sulated in the polymer due to aggregation. Bovine serum albuminrelease was incomplete (only 20%) with a corresponding growth ofinsoluble aggregates in the polymer (Fig. 1A and B, circles). By 28days, most initially encapsulated BSA (∼ 80%) had become insolu-ble and unreleasable from the polymer. This insoluble fraction was98% soluble in 6 M urea (Table 1), indicating that the aggregateswere noncovalent instead of common disulfide-bonded aggregates,which require reducing agent to dissolve13. The soluble BSA recov-ered from the polymer (28 days of incubation) was examined bySDS–PAGE. Several peptide fragments (e.g., 55, 40, and 25 kDa)accompanied the dimeric and trimeric BSA (Fig. 1D), the precur-sors of insoluble aggregates. Therefore, the analysis revealed twosalient features of the instability of encapsulated BSA: the forma-tion of insoluble, noncovalent aggregates by hydrophobic interac-tions and peptide bond hydrolysis.
Simulating BSA instability in the polymer. Primary deleteriouscharacteristics in the polymer microclimate are expected to consistof one or more of the following: elevated moisture, acidic pH, andthe polymer surface7. To test this hypothesis, we simulated BSAinstability in the PLGA microclimate. If one or more of the chosenconditions adequately describe the microclimate, we would expectto create a denatured state of BSA similar to that observed for encap-sulated BSA with instability occurring over similar time scales.
Polymer acidity was simulated by exposing BSA, which had beenlyophilized from pH 2−5 solutions, to a humid environment (86%
© 2000 Nature America Inc. • http://biotech.nature.com©
200
0 N
atu
re A
mer
ica
Inc.
• h
ttp
://b
iote
ch.n
atu
re.c
om

NATURE BIOTECHNOLOGY VOL 18 JANUARY 2000 http://biotech.nature.com 53
RESEARCH ARTICLES
relative humidity [RH] and 37°C) for one week. Of the conditionstested, only the highly acidic simulation (pH 2) induced aggregation(43 ± 6% insoluble BSA, n = 3). For pH ≥ 3, no aggregation wasobserved (data not shown). To probe for possible similarities withaggregation in the polymer, the kinetics and mechanism of BSAaggregation under the pH 2 condition were examined. Acid-inducedaggregates of BSA formed over 10 days (Fig. 1C) and were soluble indenaturing solvent (Table 1). Moreover, the denatured BSA pro-duced in the simulation and in the polymer microclimate exhibitednearly identical peptide bond fragmentation, as shown bySDS–PAGE (Fig. 1D). Hence, incubation of moist protein(lyophilized at pH 2) produced a denatured state in BSA that wasidentical to that when encapsulated (Table 1). The aggregate type(noncovalent), peptide bond fragments (e.g., 25, 40, 55 kDa), andaggregation time scale (e.g., ∼ 10 days) were equivalent.
Moisture affected the aggregation rate but not the mechanism.For example, if little water was added to the acidic BSA powder, vari-able aggregation levels were observed. For water:protein weightratios of 0−0.2, no aggregates were observed after one week at 37°C;for a 0.8−1.0 ratio, aggregates were produced (about 70% insolubleBSA), which were >94% soluble in 6 M urea (data not shown). Tosimulate BSA adsorption, BSA solutions (pH 2−7) were incubated at37°C for one week with blank PLGA microspheres or fine polymerpowder (as obtained from the manufacturer) to provide a polymersurface. Negligible adsorption (<2%) was recorded in any of thesesimulations (data not shown).
Developing a hypothesis for the mechanism of instability ofencapsulated BSA. Acidity commonly develops in PLGAs14−19
because of accumulation of acidic degradation products upon poly-ester hydrolysis. A highly acidic pH (<3) was also required for non-covalent aggregate formation in our polymer microclimate simula-tions. Thus, the added 15% BSA did not significantly alter the acidicpH (e.g., by creating water channels in the polymer to allow bufferions to diffuse in or by buffering pH itself). Instead, the encapsulatedBSA was rehydrated at a pH that caused the protein to unfold (BSAundergoes a conformational transition from the F to E isoform at pH2.7 [ref. 20]), providing the driving force for noncovalent aggrega-tion via hydrophobic interactions. Peptide bond hydrolysis is alsoparticularly rapid at acidic pH or when labile peptide bonds (e.g.,Asp-Xaa linkage21) become exposed during unfolding.
Developing a rational stabilization approach using Mg(OH)2.Our mechanistic observations suggested a rational approach to sta-bilize encapsulated BSA. As BSA aggregation did not occur in micro-climate simulations at pH ≥ 3, and peptide bond hydrolysis would beinhibited at higher pH, we tried an established method of increasingthe microclimate pH in poly(ortho esters), a polymer cousin ofPLGA. To stabilize the ortho ester bond and to extend the releasetime of drugs, Mg(OH)2 is routinely incorporated into poly(orthoesters)22. Consequently, to test whether Mg(OH)2 could inhibit BSAinstability, we coencapsulated the salt with BSA at 0.5% and 3% byweight. Then, we compared release and aggregation kinetics of thesesamples with those observed for the 0% base control. As predicted,as more Mg(OH)2 was added, more BSA was released and less encap-sulated BSA became insoluble (Fig. 1A and B). For the 3% Mg(OH)2
preparation, the aggregation was virtually eliminated.To examine hydrolysis extent and any other potential structural
alterations of BSA, SDS–PAGE, isoelectric focusing (IEF), circulardichroism (CD), and fluorescence spectroscopy were employed tocharacterize the encapsulated and released BSA from theMg(OH)2/PLGA (Fig. 2). The large degree of fragmentation thatoccurred in the absence of Mg(OH)2 had largely disappeared (Fig.2A). Some faint low-molecular-weight bands were noticeable, par-ticularly in the residual encapsulated BSA after 28 days, but thesewere small relative to the monomeric band.
Table 2. Neutralization effect of Mg(OH)2 on the erosion behaviorof 15% BSA/PLGA millicylinders.
No salt 3% Mg(OH)2
Noncovalent aggregates (%)a 65 ± 8 2.0 ± 0.4Water uptake (%)b 48 ± 2 106 ± 4PLGA degradation t1/2 (days)c 16.0 25.1pH of mediumd 3.5 7.0
aExtracted from the devices after incubation in PBST at 37°C for two weeks(mean ± s.e.m., n = 3).bDetermined by weighing the wet and dry devices after incubation in PBST at37°C for one week (mean ± s.e.m., n = 3).ct1/2 is the time when the PLGA Mr (determined by GPC) was reduced to half ofthe original Mr during incubation in PBST at 37°C.dPBST medium containing 5 mg polymer device after incubation at 37°C forfour weeks. No medium pH change was detected in either sample for the firstthree weeks.
Table 1. Comparison of BSA instability under simulated andencapsulated conditions.
Encapsulateda Simulatedb
Time to 50% 12 days 7 daysaggregation
Aggregates soluble in >98% >94%denaturing solventc
Peptide fragmentationd 25, 40, and 55 kDa 25, 40, and 55 kDa
a15% BSA in PLGA millicylinders incubated in PBST at 37°C.bLyophilized BSA at pH 2 incubated under 86% RH at 37°C.cPBST containing 6 M urea and 1 mM EDTA.dFrom SDS–PAGE of BSA samples treated with SDS and β-mercap-toethanol.
Figure 1. Effect of Mg(OH)2 content on BSA release kinetics (A) andencapsulated BSA aggregation kinetics (B) during incubation of thePLGA implants at 37°C in PBST. Millicylinders were loaded with 15%BSA and 0% (P), 0.5% (L), and 3.0% (H) Mg(OH)2. (mean ± s.e.m., n =3). (C) Aggregation kinetics of BSA in a simulated polymermicroclimate. BSA was lyophilized from pH 2 solution and exposed to37°C and 86% RH (mean ± s.e.m., n = 2). (D) SDS–PAGE of denaturedBSA from 15% BSA/PLGA (lanes 2, 5, and 7) and from the simulation(lanes 3, 6, and 8). Lane 1: high-molecular-weight markers (reduced);lane 2: insoluble BSA from the polymer (reduced); lane 3: insolubleBSA from the simulation (reduced); lane 4: nonencapsulatedstandard BSA (nonreduced); lane 5: insoluble BSA from the polymer(nonreduced); lane 6: insoluble BSA from the simulation(nonreduced); lane 7: soluble BSA from the polymer (nonreduced);lane 8: soluble BSA from the simulation (nonreduced).
A B
C D
© 2000 Nature America Inc. • http://biotech.nature.com©
200
0 N
atu
re A
mer
ica
Inc.
• h
ttp
://b
iote
ch.n
atu
re.c
om
54 NATURE BIOTECHNOLOGY VOL 18 JANUARY 2000 http://biotech.nature.com
RESEARCH ARTICLES
To test for the most common route of protein degrada-tion deamidation the released and encapsulated BSA was exam-ined by IEF. No alteration of the protein’s pI (4.7) was detected (Fig.2B). Likewise, no alterations in secondary or tertiary structure ofBSA were noticeable in the CD (Fig. 2C) and fluorescence spectra(Fig. 2D). Hence, the structure of BSA was retained (>90%) fromthe Mg(OH)2/PLGA preparation for a period of one month.
To confirm that the Mg(OH)2 actually neutralized the acidicmicroclimate in the polymer, we examined changes in polymer ero-sion of BSA-loaded millicylinders on addition of Mg(OH)2. The fol-lowing alterations in polymer erosion would indicate that the saltinduced a microclimate pH increase: (1) a polymer water contentincrease due to increased osmotic pressure generated by magnesiumcarboxylate salts and ionization of polymer end groups (2) a poly-mer degradation half-time increase resulting from inhibition ofacid-catalyzed polyester hydrolysis23, and (3) an inhibited release ofsequestered acidic moieties (high-molecular-weight PLGA releasesdegradation products into the erosion medium only after the molec-ular weight declines to a critical value24). As seen in Table 2, eachtrend was observed. When the base was added to 15% BSA/PLGAmillicylinders, polymer water content doubled, degradation half-time was extended from 16 days to 25 days, and fewer acidic specieswere released (phosphate-buffered saline, 0.02% Tween 80 [PBST]pH 7.4 dropped to pH 7.0 instead of 3.5 after four weeks).
The basicity of the encapsulated salt also affected microclimatepH. For example, when a stronger base, Ca(OH)2, was substituted
for Mg(OH)2, after two weeks incubation disulfide-bonded aggre-gates of BSA began to form (mean ± s.e.m.: 11 ± 1% disulfide bond-ed; 3.9 ± 0.1% noncovalent bonded for 3% Ca[OH]2; n = 3). Theformation of disulfide bonds requires a free thiolate, revealing a neu-tral to basic microclimate pH. When a weaker base, ZnCO3, wasused, the noncovalent aggregation increased, suggesting a lowermicroclimate pH (after two weeks, 10 ± 1% noncovalent aggregatesfor 3% ZnCO3; 2.0 ± 0.4% for 3% Mg[OH]2) with negligible disul-fide-bonded aggregation. (A weight basis was used instead of amolar basis for the salt comparison, because pores in the polymerare saturated with salt and the weight basis controls percolation ofthe salt throughout the polymer.) Finally, coencapsulation of a neu-tral salt (e.g., NaCl) was also unsuccessful in preventing BSA aggre-gation (after two weeks, 24 ± 1% noncovalent aggregates for 3%NaCl), and any partial stabilization was consistent with polymerwater content increases induced by the salt.
Generality of the use of basic salts to improve stability of pro-teins encapsulated in PLGA. To test the generality of our stabiliza-tion approach, we encapsulated two growth factors, recombinanthuman bFGF and BMP-2, in PLGA millicylinders, and formulatedBSA in PLGA microspheres. Basic fibroblast growth factor is cur-rently being tested for wound healing, osteogenesis, and diabeticulcers25. Like most therapeutic proteins, bFGF’s in vivo serum half-life is very short (<3 min)26, making it a good candidate for con-trolled release27–30. Another important application is to inject BMP-2with a carrier matrix to induce bone regeneration, which is a
Figure 2. Structural analyses of BSA from incubated PLGA millicylinders by SDS–PAGE (A), IEF (B), CD spectra (C), and fluorescence emissionspectra (D). In SDS–PAGE and IEF analyses, lanes 1–5 are standard BSA, released BSA on day 1, days 5–7, and days 21–28, and residual BSA onday 28, respectively. In CD and fluorescence emission spectra, samples include: standard BSA ( ), BSA heat denatured in PBST (90ºC for 30min) (---), released BSA samples from day 1 (——), days 5–7 (– – –), days 16–20 (---), days 21–28 (⋅⋅⋅⋅⋅⋅), and residual BSA (−⋅−⋅). The IEF of heatdenatured BSA was distinguishable from standard BSA (data not shown). Samples for fluorescence were 50 µg/ml.
A B C D
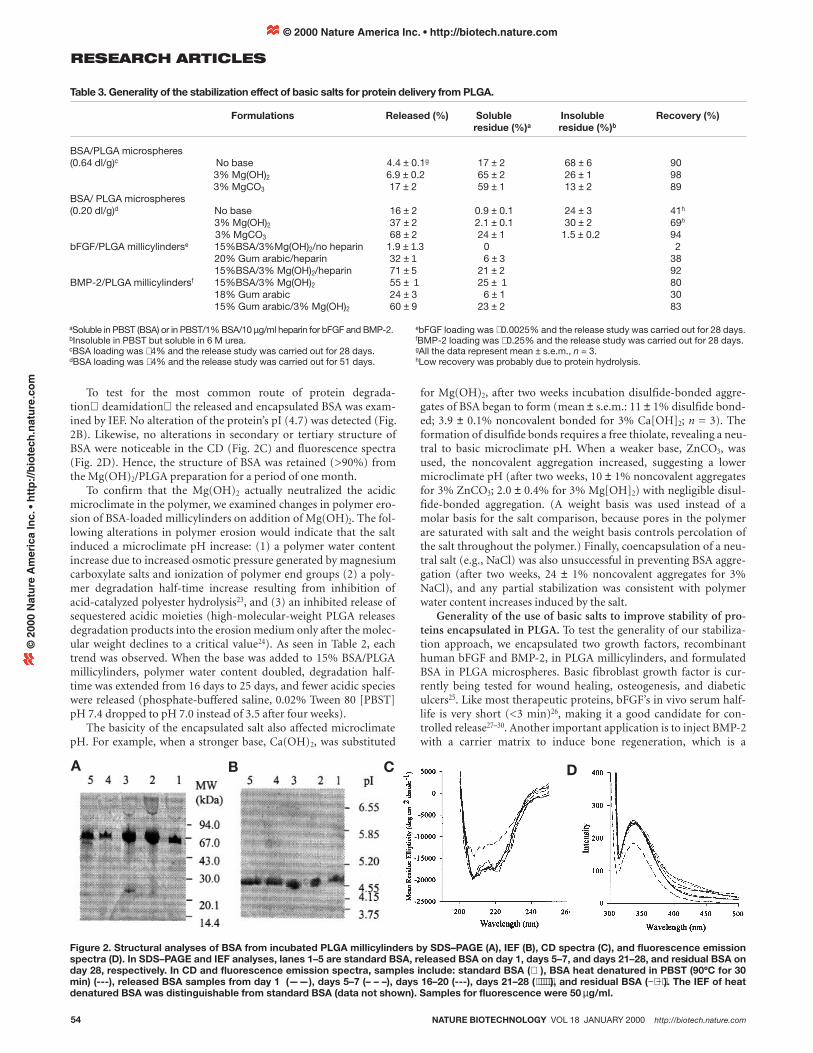
Table 3. Generality of the stabilization effect of basic salts for protein delivery from PLGA.
Formulations Released (%) Soluble Insoluble Recovery (%)residue (%)a residue (%)b
BSA/PLGA microspheres(0.64 dl/g)c No base 4.4 ± 0.1g 17 ± 2 68 ± 6 90
3% Mg(OH)2 6.9 ± 0.2 65 ± 2 26 ± 1 983% MgCO3 17 ± 2 59 ± 1 13 ± 2 89
BSA/ PLGA microspheres(0.20 dl/g)d No base 16 ± 2 0.9 ± 0.1 24 ± 3 41h
3% Mg(OH)2 37 ± 2 2.1 ± 0.1 30 ± 2 69h
3% MgCO3 68 ± 2 24 ± 1 1.5 ± 0.2 94bFGF/PLGA millicylinderse 15%BSA/3%Mg(OH)2/no heparin 1.9 ± 1.3 0 2
20% Gum arabic/heparin 32 ± 1 6 ± 3 3815%BSA/3% Mg(OH)2/heparin 71 ± 5 21 ± 2 92
BMP-2/PLGA millicylindersf 15%BSA/3% Mg(OH)2 55 ± 1 25 ± 1 8018% Gum arabic 24 ± 3 6 ± 1 3015% Gum arabic/3% Mg(OH)2 60 ± 9 23 ± 2 83
aSoluble in PBST (BSA) or in PBST/1% BSA/10 µg/ml heparin for bFGF and BMP-2.bInsoluble in PBST but soluble in 6 M urea.cBSA loading was ∼ 4% and the release study was carried out for 28 days.dBSA loading was ∼ 4% and the release study was carried out for 51 days.
ebFGF loading was ∼ 0.0025% and the release study was carried out for 28 days.fBMP-2 loading was ∼ 0.25% and the release study was carried out for 28 days.gAll the data represent mean ± s.e.m., n = 3.hLow recovery was probably due to protein hydrolysis.
© 2000 Nature America Inc. • http://biotech.nature.com©
200
0 N
atu
re A
mer
ica
Inc.
• h
ttp
://b
iote
ch.n
atu
re.c
om

NATURE BIOTECHNOLOGY VOL 18 JANUARY 2000 http://biotech.nature.com 55
RESEARCH ARTICLES
promising alternative to bone grafting31,32. PLGA is now considered agood candidate to overcome the difficulties with conventional BMP-2 carriers, including inactivated collagenous bone matrix33,34.Moreover, in contrast to millicylinders, microspheres have severaladvantages, particularly to reduce pain of injections and to simplifyadministration8. PLGA 50/50 microspheres also frequently developan acidic microclimate, according to recent findings15−18.
Before encapsulating bFGF, we considered several instabilitypathways characteristic of this protein. Basic fibroblast growth factorbinds heparin, and when unbound, loses activity rapidly35,36. Itadheres avidly to glass and plastic surfaces29 and disulfide exchangeof bFGF is catalyzed by trace metals (e.g., those remaining from thepolymerization of polymer7). Therefore, we selected five additivesfor encapsulation of bFGF. The 3% Mg(OH)2/15% BSA combina-tion was suitable for neutralizing the acidic microclimate. High BSAconcentrations may also inhibit adsorption of bFGF to PLGA29. Weadded heparin at a weight ratio of 1:1 (heparin to bFGF) to enhancebFGF stability36, and EDTA to chelate trace heavy metals. Sucrosewas used to retain bFGF structure in the solid state25.
As predicted, when bFGF was encapsulated (∼ 0.0025%) in theMg(OH)2/BSA/PLGA millicylinders, the growth factor was released(Fig. 3A, filled circles) in a fashion similar to that observed for BSA(Fig. 1A). Over 28 days, 71% of immunoreactive bFGF was detectedin the release medium and 21% remained in the polymer (Table 3),accounting for approximately 92% of initially encapsulated bFGF. Ifmillicylinders did not contain both heparin and Mg(OH)2/BSA,bFGF lost immunoreactivity. For example, when heparin wasremoved from the stabilized formulation, only 2% bFGF wasreleased over one month with no immunoreactive bFGF in the resid-ual fraction (Table 3). Similarly, when 20% arabic gum was substi-tuted for 3% Mg(OH)2/15% BSA, no bFGF was observed in therelease medium after four days and only 38% was accounted for inboth the release and residual fraction. (A 0% Mg[OH]2 and 15%BSA control was not performed because of BSA aggregation.)
To increase the capacity of the polymer to deliver bFGF, weincreased bFGF loading to 0.01% and sucrose loading to 2.3%. ThebFGF release initially was much slower and later exhibited a linearrelease profile up to four weeks (Fig. 3A, filled squares). The releaseof BSA from the same preparation in PBST was retarded similarly(data not shown). Thus, sucrose can be used to slow down release ofboth BSA and bFGF from the polymer, probably by increasing vis-cosity of aqueous pores in the polymer.
Although >90% recovery by ELISA during release indicatesretention of bFGF immunoreactivity (Table 3), this fraction is notnecessarily bioactive. To test this, we examined whether encapsulat-
ed and released growth factor induced cell proliferation(as indicated by [3H]thymidine incorporation37). Theencapsulation procedure did not affect the biologicalactivity of bFGF (time 0, Fig. 3B). Some small inactivationapparently occurred during release, but 65–85% bioactivi-ty of bFGF was retained over the entire one-month releaseexperiment.
To similarly stabilize encapsulated BMP-2, we againused 3% Mg(OH)2 and 15% BSA to neutralize microcli-mate acidity in PLGA millicylinders containing BMP-2(0.25% loading). No other additives were used. We alsoincluded a second positive control using the protein sub-stitute, gum arabic (i.e., 3% Mg[OH]2, 15% gum arabic,0.25% BMP-2). In both cases, controlled release wasobserved over 28 days (data not shown) resulting in arecovery (released + soluble residue fraction) of >80%(Table 3). In contrast, when base was removed from theformulation (18% gum arabic), only 30% protein wasrecovered by immunoassay.
To test our approach in microspheres, we examinedwhether encapsulated BSA undergoes the acid-induced mechanismof instability and if so, whether basic additives inhibit the mechanism.As expected, BSA also forms noncovalent aggregates (∼ 25–70%)when encapsulated in PLGA microspheres (Table 3), confirming thatan acidic microclimate also commonly develops in PLGA 50/50microspheres. It has been suggested that BSA becomes unstable inPLGA microspheres primarily by protein adsorption to the poly-mer38. This conclusion was strongly weighed on the SDS-induced lib-eration of previously unreleasable BSA from the polymer. We foundthat the SDS buffer used in the SDS–PAGE (Fig. 1D) dissolves encap-sulated noncovalent aggregates, which may explain the reportedrelease of sequestered BSA from the polymer. Therefore, we concludethat protein adsorption, consistent with our simulations, is not thepredominant source of BSA instability in PLGA microspheres.
Whereas the BSA instability mechanism in microspheres wassimilar to that observed in millicylinders, coencapsulation ofMg(OH)2 was only moderately successful to inhibit BSA aggregationin microspheres. For example, the soluble fraction of BSA in PLGAmicrospheres (0.64 dl/g) decreased from 68% without base to 26%with Mg(OH)2 (Table 3). This modest BSA stability increase afford-ed by Mg(OH)2, and our previous studies illustrating a heteroge-neous pH distribution in Mg(OH)2/PLGA microspheres (no pro-tein)15, suggest that the basic additive could not diffuse to all acidicprotein pores in the polymer. To overcome this problem, we turnedto another base, MgCO3, which has basicity equivalent to that ofMg(OH)2, but which is about 10-fold more water soluble, to facili-tate base diffusion. The more soluble salt inhibited BSA aggregationat a level similar to the inhibition attained in millicylinders withMg(OH)2. For the medium-molecular-weight PLGA (0.64 dl/g),aggregation was held to just 13% over 28 days with 89% recovery(Table 3). Remarkably, coencapsulation of MgCO3 in low-molecu-lar-weight PLGA (0.20 dl/g) resulted in a reduction of BSA aggrega-tion to just 1.5% over 51 days with 94% recovery (Table 3). This lat-ter preparation controlled release of BSA slowly and continuouslyover the entire experiment after a 32% burst (data not shown).
We conclude that the acidic microclimate in PLGA delivery sys-tems (including microspheres) is a common source of instability ofencapsulated proteins. Poorly water-soluble basic salts can be usedto neutralize the polymer microclimate pH to levels necessary toretain the structure and biological activity of encapsulated acid-labile proteins.
Experimental protocolReagents. Poly(DL-lactide-co-glycolide) 50/50 (inherent viscosity of 0.20,0.63, and 0.64 dl/g in hexafluoroisopropanol solution) was from
Figure 3. (A) Controlled release of bFGF. PLGA millicylinders were loaded with 3%Mg(OH)2/0.0025% bFGF/0.0025% heparin/0.01% EDTA/0.6% sucrose/14.4% BSA(P) and 3% Mg(OH)2/0.01% bFGF/0.01% heparin/0.01% EDTA/2.3% sucrose/12.7%BSA (slow-releasing, L) (± s.e.m., n = 3). (B) Evaluation of biological activity of bFGFsamples from the slow-releasing millicylinders. Bioactivity (%) = concentrationdetermined by bioassay/concentration determined by ELISA × 100%.
A B
© 2000 Nature America Inc. • http://biotech.nature.com©
200
0 N
atu
re A
mer
ica
Inc.
• h
ttp
://b
iote
ch.n
atu
re.c
om

56 NATURE BIOTECHNOLOGY VOL 18 JANUARY 2000 http://biotech.nature.com
RESEARCH ARTICLES
Birmingham Polymers (Birmingham, AL). Recombinant human bFGF andBMP-2 were supplied by Scios (Sunnyvale, CA) and Orthogene (Fremont,CA), respectively. Bovine serum albumin (A-3059), heparin (H-3393), andgum arabic were from Sigma (St. Louis, MO). Fine Mg(OH)2, MgCO3, andCa(OH)2 (<5 µm) powders were from Aldrich (Milwaukee, WI) and ZnCO3
(<5 µm) was from ICN Biomedical (Aurora, OH). Cell culture reagents werefrom GIBCO Life Technologies (Rockville, MD). All other chemicals were ofanalytical grade or purer and purchased from commercial suppliers.
PLGA millicylinder preparation. A suspension of sieved BSA (<90 µm)with or without basic salt in acetone-PLGA (0.63 dl/g) solution (50% wt/wt)was loaded in a syringe and extruded into silicone rubber tubing (0.8 mmi.d.) at approximately 0.1 ml/min (ref. 39). The solvent-extruded suspensionwas dried at room temperature (24 h) and then under vacuum at 45°C (24 h).For encapsulation of bFGF and BMP-2, each growth factor was combinedwith additives in 10 mM sodium phosphate buffer (pH 7.4), lyophilized to afine powder (∼ 4% moisture determined by Fisher titration), and sievedbefore extrusion and drying. All preparations had a loading efficiency invari-ably between 85% and 95%.
PLGA microsphere preparation. According to a solvent evaporationmethod40, 100 µl of 150 mg/ml BSA in 10 mM phosphate buffer (pH 7.4)were added to 1 ml of PLGA/CH2Cl2 solution (30% for 0.64 dl/g; 70% for 0.2dl/g) with or without basic salt. The mixture was homogenized at 10,000r.p.m. (1 min at 4°C), and transferred to a 2% polyvinyl alcohol (PVA) aque-ous solution. The water-in-oil-in-water emulsion was formed by vortexingthe mixture for 20 s, and the particles were hardened for 3 h in 100 ml of0.5% PVA at room temperature. Microspheres were collected by centrifuga-tion, washed with water, and lyophilized. Microspheres from both prepara-tions were spherical with a mean diameter between 60 and 70 µm. The BSAloading was approximately 4% with an encapsulation efficiency between70% and 80%.
Controlled release of proteins. Bovine serum albumin release was moni-tored in PBST. Millicylinders (∼ 10 mg) or microspheres (∼ 20 mg) wereplaced in release medium (0.5 ml) and incubated under mild agitation(37°C). To sample, the buffer was removed (by centrifugation for micros-pheres) and replaced with new medium. Protein content in release sampleswas determined by using a modified Bradford assay (Coomassie brilliant blueplus protein assay, Pierce, Rockford, IL), which is also compatible with dena-turing/reducing agents used below. The release of bFGF and BMP-2 milli-cylinders was examined similarly except that 1% BSA, 10 µg/ml heparin, and1 mM EDTA were added to the release medium to prevent protein damageonce released from the polymer.
Evaluation of BSA aggregation. Incubated polymers were removed fromrelease medium, dried, and dissolved in acetone. After centrifugation andremoval of the polymer solution, the remaining BSA pellet was reconstitutedin PBST and incubated (37°C) overnight before determining the protein con-tent; this gave a measure of the water-soluble protein encapsulated (also usedfor protein loading measurement). Any aggregate was collected by centrifu-gation and incubated (37°C for 30 min) in denaturing solvent (PBST, 6 Murea, 1 mM EDTA); analysis of protein concentration gave the amount ofnoncovalently bonded BSA aggregates41. The same procedure was repeatedwith reducing solvent (10 mM dithiothreitol in denaturing solvent) to deter-mine the amount of disulfide-bonded aggregates.
Simulations of BSA instability in the polymer microclimate. For simulat-ing pH, BSA (4 mg/ml) in a universal buffer (H3PO4, HAc, and H3BO3; 40mM each, titrated with NaOH) was lyophilized from pH 2−5 and incubated(37°C and 86% RH)41. To sample, protein was reconstituted in PBST andexamined for the aggregate type. For simulating water content, water wasadded directly to lyophilized BSA (from pH 2), sealed, and incubated (37°Cfor one week)42. To test protein adsorption, BSA (1 mg/ml) in universalbuffer (pH 2–7) was incubated for one week (37°C) with 20 mg PLGA pow-der (0.20 dl/g, <100 µm) or PLGA (0.63 dl/g) microspheres prepared by sol-vent evaporation43. Losses of BSA from solution were used to determineadsorption extent.
Structural analysis of encapsulated BSA. Fluorescence emission spectra ofBSA (300–500 nm; 240 nm/min) were obtained with a Perkin-Elmer(Norwalk, CT) LS50B luminescence spectrometer. Far ultraviolet CD spectra(200–250 nm) were recorded with a J-500A Jasco (Tokyo, Japan) spectropo-larimeter at room temperature. The integrity of protein samples was deter-mined by both SDS–PAGE and IEF gel electrophoresis, which were per-formed on a PhastSystem (Amersham Pharmacia Biotech AB, Uppsala,Sweden). PhastGel gradient 10–15 gels and IEF 3–9 gels (AmershamPharmacia Biotech AB) were used for SDS–PAGE and IEF analyses, respec-
tively. Coomassie brilliant blue staining was performed after separation.ELISA for bFGF. A 96-well plate was coated with monoclonal anti-bFGF
(Upstate Biotechnology, Waltham, MA) overnight at 4°C (refs 44 and 45).Samples or standards of bFGF in release media were added to each well andincubated at 4°C for 24 h. After washing, polyclonal rabbit anti-bFGF(Sigma) was added and left at room temperature for 2 h, followed by 2 h withanti-rabbit IgG–horseradish peroxidase (1:10,000, Sigma). The substrate o-phenylenediamine with H2O2 (Sigma Fast OPD tablet sets) was added (30min at room temperature) and the reaction was stopped with 3 M H2SO4.The product was detected (490 nm) on a plate reader (DynatechLaboratories, Chantilly, VA).
BIAcore immunoassay for BMP-2. BMP-2 was quantified with a BIAcore2000 biosensor (Biacore AB, Uppsala, Sweden). A monoclonal antibody ofBMP-2 (Orthogene) was immobilized onto a CM-5 sensor chip using theamine coupling kit (Biacore AB). Bone morphogenetic protein-2 in releasemedium was assayed over the immobilized antibody surface. Sample volumewas 30 µl and HEPES-buffered saline was the mobile phase (10 µl/min). Thesurface was regenerated (10 µl 10 mM HCl) after each sample. The antibodywas stable on the chip surface for more than one month (standard curverange, 50−1,600 ng/ml BMP-2).
Cell proliferation assay for bFGF. BALB/c 3T3 fibroblasts (25,000/well,CCL-163, American Type Culture Collection) in Dulbecco’s modifiedEagle’s medium containing 10% bovine calf serum, 50 U/ml streptomycin,and 50 µg/ml penicillin were seeded (200 µl/well) on 96-well plates37 andgrown to confluence (one week) without changing the medium. Then, bFGFsamples or standards (10 µl) in release medium were added (20 h) followedby 1 µCi of [3H]thymidine (6.7 Ci/mmol, DuPont/NEN Research Products,Boston, MA) per well (6–8 h). Cells were collected on filter paper by using aPHD cell harvester (Cambridge Technology, Cambridge, MA), resuspendedin 3 ml scintillation cocktail 3a70B (Research Products InternationalCorporation, Mount Prospect, IL), and counted (Beckman scintillationcounter, Fullerton, CA).
AcknowledgmentsWe thank Dr. John Wang of Synzyme Technologies (Irvine, CA) for his helpfulsuggestions. We are very grateful to Prof. Mark Coggeshall, from the Departmentof Microbiology of the Ohio State University for his help in the bFGF bioassayexperiment. We thank Dr. Hanne Bentz from Orthogene and Prof. JeffreyHubbell from the Swiss Federal Technology Institute for their aid in the BMP-2studies. We also thank Professor Gary Means from the Department ofBiochemistry, and Dr. Jichao Kang of the College of Pharmacy at the Ohio StateUniversity for their helpful discussions and technical assistance, respectively.This work was supported by NIH DE 12183 and a PhRMA grant to S.P.S.
1. Langer, R. & Folkman, J. Polymers for the sustained release of proteins andother macromolecules. Nature 263, 797–800 (1976).
2. Struck, M.M. Biopharmaceutical R&D success rates and development times.A new analysis provides benchmarks for the future. Bio/Technology 12,674–677 (1994).
3. Talmadge, J.E. The pharmaceutics and delivery of therapeutic polypeptidesand proteins. Adv. Drug Delivery. Rev. 10, 247–299 (1993).
4. Ogawa, Y., Okada, H., Heya, T. & Shimamota T. Controlled release of LHRHagonist, leuprolide acetate, from microcapsules: serum drug level profiles andpharmacological effects in animals. J. Pharm. Pharmcol. 41, 439–444 (1989).
5. Okada, H., Doken, Y., Ogawa, Y. & Toguchi, H. Preparation of three-monthdepot injectable microspheres of leuprorelin acetate using biodegradablepolymers. Pharm. Res. 11, 1143 (1994).
6. Dutta, A.S., Furr, B.J.A. & Hutchinson, F.G. Discovery and development ofgoserelin (Zoladex). Pharm. Med. 7, 9–28 (1993).
7. Schwendeman, S.P., Cardamone, M., Klibanov., A. & Langer, R. inMicroparticulate systems for the delivery of proteins and vaccines (eds Cohen,S. & Bernstein, H.), 1–49 (Marcel Dekker, New York; 1996).
8. Schwendeman, S.P., Costantino, H.R., Gupta, R.K. & Langer, R. in Controlleddrug delivery. (ed. Park, K.), 229–267 (American Chemical Society,Washington, D.C; 1997).
9. Johnson, O.L. et al. A month-long effect from a single injection of microencap-sulated human growth hormone. Nat. Med. 2, 795–799 (1996).
10. Cleland, J.L. & Jones, A.J.S. Stable formulations of recombinant humangrowth hormone and interferon-gamma for microencapsulation in biodegrad-able microspheres. Pharm. Res. 13, 1464–1475 (1996).
11. Schwendeman, S.P., Tobio, M., Joworowicz, M., Alonso, M.J. & Langer, R.New strategies for the microencapsulation of tetanus vaccine. J.Microencapsulation 15, 299–318 (1998).
12. Putney, S.D. & Burke, P.A. Improving protein therapeutics with sustained-release formulations. Nat. Biotechnol. 16, 153–157 (1998).
13. Costantino, H.R., Langer, R. & Klibanov, A.M. Solid-phase aggregation of pro-teins under pharmaceutically relevant conditions. J. Pharm. Sci. 83,
© 2000 Nature America Inc. • http://biotech.nature.com©
200
0 N
atu
re A
mer
ica
Inc.
• h
ttp
://b
iote
ch.n
atu
re.c
om

NATURE BIOTECHNOLOGY VOL 18 JANUARY 2000 http://biotech.nature.com 57
RESEARCH ARTICLES
1662–1669 (1994).14. Herrlinger, M. Ph.D. In vitro polymerabbau und wirksttofffreigabe von poly-DL-
laktidformlingen Thesis, Univ. Heidelberg, Germany (1994).15. Shenderova, A., Burke, T. & Schwendeman, S.P. The acidic microclimate in
poly(lactide-co-glycolide) microspheres stabilizes camptothecins. Pharm.Res. 16, 241–248 (1999).
16. Brunner, A., Mäder, K. & Göpferich, A. pH and osmotic pressure insidebiodegradable microspheres during erosion. Pharm. Res. 16, 847–853 (1999).
17. Mader, K., Bittner, B., Li, Y., Wohlauf, W. & Kissel, T. Monitoring microviscosityand microacidity of the albumin microenvironment inside degrading micropar-ticles from poly(lactide-co-glycolide) (PLG) or ABA-triblock polymers contain-ing hydrophobic poly(lactide-co-glycolide) A blocks and hydrophilic poly(eth-yleneoxide) B blocks. Pharm. Res. 15, 787–793 (1998).
18. Fu, K., Pack, D.W., Laverdiere, A., Son, S. & Langer, R. Visualization of pH indegrading polymer microspheres. Proceedings 25th International Symposiumon Controlled Resealse of Bioactive Materials 25, 150–151 (1998).
19. Mader, K., Gallez, B., Liu, K.J. & Swartz, H.M. Non-invasive in vivo characteri-zation of release processes in biodegradable polymers by low-frequency elec-tron paramagnetic resonance spectroscopy. Biomaterials 17, 457–461 (1996).
20. Peters, T. Jr. Serum albumin. Adv. Protein Chem. 37, 161–245 (1985).21. Manning, M.C., Patella, L. & Borchardt, R.T. Stability of protein pharmaceuti-
cals. Pharm. Res. 6, 903–917 (1989).22. Heller, J. Development of poly(ortho esters): a historical overview. Biomaterials
11, 659–665 (1990).23. Li, S.M., Garreau, H. & Vert, M. Structure-property relationships in the case of
the degradation of massive poly(a-hydroxy acids) in aqueous media. Part 3.Influence of the morphology of poly(L-lactic acid). J. Mater. Sci. 1, 123–130(1990).
24. Hutchinson, F.G. & Furr, B.J.A. Biodegradable polymer systems for the sus-tained release of polypeptides. J. Controlled Release13, 279–294 (1990).
25. Wang, Y.J. et al. in Formulation, characterization, and stability of protein drugs(eds Pearlman, R. & Wang, Y.J.) 141–180 (Plenum Press, New York; 1996).
26. Edelman, E.R., Nugent, M.A. & Karnovsky, M.J. Perivascular and intravenousadministration of basic fibroblast growth factor: vascular and solid organdeposition. Proc. Natl. Acad. Sci. USA 90, 1513–1517 (1993).
27. Berscht, P.C., Nies, B., Liebendorfer, A. & Kreuter, J. Incorporation of basicfibroblast growth factor into methylpyrrolidinone chitosan fleeces and deter-mination of the in vitro release characteristics. Biomaterials 15, 593–600(1994).
28. Fukunaga, K. et al. Aluminium beta-cyclodextrin sulphate a stabilizer and sus-tained-release carrier for basic fibroblast growth factor. J. Pharm. Pharmacol.46, 168–171 (1994).
29. Edelman, E.R., Mathiowitz, E., Langer, R. & Klagsbrun, M. Controlled andmodulated release of basic fibroblast growth factor. Biomaterials 12, 619–626
(1991).30. Davies, M.J. et al. In vitro assessment of the biological activity of basic fibrob-
last growth factor released from various polymers and biomatrices. Journal ofBiomaterial Applications 12, 31–56 (1997).
31. Wang, E.A. et al. Recombinant human bone morphogenetic protein inducesbone formation. Proc. Natl. Acad. Sci. USA 87, 2220–2224 (1990).
32. Welch, R.D. et al. Effect of recombinant human bone morphogenetic protein-2on fracture healing in a goat tibial fracture model. J. Bone Miner. Res. 13,1483–1490 (1998).
33. Mayer, M., Hollinger, J., Ron, E. & Wozney, J. Maxillary alveolar cleft repair indogs using recombinant human bone morphogenetic protein-2 and a polymercarrier. Plast. Reconstr. Surg. 98, 247–259 (1996).
34. Kenley, R.A. et al. Biotechnology and bone graft substitutes. Pharm. Res. 10,1393–1401 (1993).
35. Gospodarowicz, D. & Cheng, J. Heparin protects basic and acidic FGF frominactivation. J. Cell. Physiol. 128, 475–484 (1986).
36. Sommer, A. & Rifkin, D.B. Interaction of heparin with human basic fibroblastgrowth factor: protection of the angiogenic protein from proteolytic degrada-tion by a glycosaminoglycan. J. Cell. Physiol.138, 215–220 (1989).
37. Sullivan, R. & Klagsbrun, M. Purification and assay of intact human basicfibroblast growth factor using heparin-sepharose chromatography. J. Tiss.Culture Methods 10, 125–132 (1986).
38. Crotts, G., Sah, H. & Park, T.G. Adsorption determines in-vitro protein releaserate from biodegradable microspheres: quantitative analysis of surface areaduring degradation. J. Controlled Release. 47, 101–111 (1997).
39. Zhang, X., Wyss, U.P, Amsden, B. & Goosen, M.F.A. Controlled release ofalbumin from biodegradable poly(DL-lactide) cylinders. J. Controlled Release25, 61–69 (1993).
40. Cohen, S., Yoshioka, T., Lucarelli, M., Hwang, L.H. & Langer, R. Controlleddelivery systems for proteins based on poly(lactic/glycolic acid) microspheres.Pharm. Res. 8, 713–720 (1991).
41. Schwendeman, S.P. et al. Stabilization of tetanus and diphtheria toxoidsagainst moisture-induced aggregation. Proc. Natl. Acad. Sci. USA 92,11234–11238 (1995).
42. Liu, W.R., Langer, R. & Klibanov, A.M. Moisture-induced aggregation oflyophilized proteins in the solid state. Biotechnol. Bioeng. 37, 177–184 (1991).
43. Shenderova, A., Burke, T.G. & Schwendeman, S.P. Stabilization of 10-hydrox-ycamptothecin in poly(lactide-co-glycolide) microsphere delivery vehicles.Pharm. Res. 14, 1406–1413 (1997).
44. Watanabe, H. et al. A sensitive enzyme immunoassay for human basic fibrob-last growth factor. Biochem. Biophys. Res. Commun. 175, 229–235 (1991).
45. Gabra, N., Khiat, A. & Calabresi, P. Detection of elevated basic fibroblastgrowth factor during early hours of in vitro angiogenesis using a fast ELISAimmunoassay. Biochem. Biophys. Res. Commun. 205, 1423–1430 (1994).
© 2000 Nature America Inc. • http://biotech.nature.com©
200
0 N
atu
re A
mer
ica
Inc.
• h
ttp
://b
iote
ch.n
atu
re.c
om