211 al-0, 1 - digital library/67531/metadc332212/m2/1/high... · 211 m9/j al-0, 3/ /1 reduction...
TRANSCRIPT

211 M9/J
Al-0, 3/ /1
REDUCTION PATHWAYS IN CYCLOPENTADIENYL RHENIUM DICARBONYL
DIBROMIDE DERIVATIVES AND INDENYL RHENIUM TRICARBONYL:
SYNTHESIS, STRUCTURE, AND REACTIVITY OF ANIONIC
CYCLOPENTADIENYL RHENIUM COMPLEXES.
RING ATTACK VS. METAL-HALOGEN EXCHANGE.
DISSERTATION
Presented to the Graduate Council of the
University of North Texas in Partial
Fulfillment of the Requirements
For the Degree of
DOCTOR OF PHILOSOPHY
By
Sang Woo Lee, B.S., M.S.
Denton, Texas
December, 1989

Lee, Sang Woo, Reduction Pathways in Cvclopentadienvl
Rhenium Dicarbonvl Dibromide Derivatives and Indenvl Rhenium
Tricarbonvl; Synthesis. Structure, and Reactivity of Anionic
Cvclopentadienvl Rhenium Complexes. Ring Attack vs. Metal-
Haloaen Exchange. Doctor of Philosophy (Chemistry),
December, 1989, 178 pp., 19 tables, 30 figures, 74 titles.
The reactions of diagonal and lateral Cp'Re(CO)2Br2
(where Cp' = n5-C5H5, rj5-C5Me5) and (r/
5-CgH7)Re(CO)3 with
reducing agents have been examined. Hydride reduction at
-78 °C is observed to occur at the Cp ring in both
CpRe(CO)2Br2 isomers, affording a thermally unstable
4 -[(rj -C5Hg)Re(CO)2Br2] complex. The product of hydride ring
attack has been characterized by low-temperature IR and *H
NMR measurements in addition to 13C NOE and heteronuclear 2D
NMR measurements. Reaction of lateral CpRe(CO)2Br2 with
either MeLi or PhLi affords both Cp-ring attack and metal-
halogen exchange, [CpRe(CO)2Br]" (1) while t-BuLi reacts
exclusively via metal-halogen exchange. diag-CpRe(CO)2Br2
reacts with the above lithium reagents to yield the same
metal-halogen exchange anion. Analogous reactions using
diag- and lat-Cp*Re(CO) 2Br2 (where Cp* = r^-CgMe^) afford
only the corresponding rhenium metal-halogen exchange anion,
[Cp*Re(CO)2Br] (2). The molecular structures of

l-[Li/15-Crown-5] and 2-PPP were established by X-ray
crystallography. l-[Li/15-Crown-5] crystallizes in the
monoclinic space group P2j with a = 10.860(4) A, b -
13.116(5) A, C = 7.417(3) A, 0 = 105.26(3)°, V = 1018.7(3)
A , and Z = 2. 2-PPP crystallizes in the orthorhombic space
group Pbca with a = 20.646(5) A, b = 17.690(5) A, c -
17.553(3) A, and z = 8.
Solution FT-IR studies of 2 in THF reveal the presence
of only solvent-separated io
Li+, K+, or PPP+ from -70 °C
room temperature displays a
>n pairs when the gegencation is
to room temperature. 2-Na at
39:61 mixture of carbonyl
oxygen-sodium and solvent-separated ion pairs, respectively.
These ion pairs reveals a reversible temperature-dependent
equilibrium. The equilibrium constant has been determined
by IR band shape analysis over the temperature range -70 °C
to room temperature and values of AH and AS are reported.
The reaction of the ring-attacked complex,
diag-[(rj4-C5H6)Re(CO)2Br2]" with PPh3, P(OPh)3, or Me3CNC
leads to the formation of the CpRe(CO)2L. Treatment of
[Cp'Re(CO)2Br]" with methyltriflate, TFA, and magic ethyl
yields the corresponding diag-Cp'Re(CO)2Br(R) (R = CH3, H,
C2H5) complexes based on in situ IR analysis. All of these
functionalized complexes decomposed in solution over a
period of days to give Cp'Re(CO)3 as the only isolable

product (20-30 %).
The reaction of the [Cp,Re(C0)2Br]" with Bu3SnH at
60 °C leads to the formation of diag-Cp'Re(CO)2(SnBuj)2,
which was also synthesized independently by the
deprotonation of diag-Cp'Re(CO)2H2 with EtjN in the presence
of BugSnBr at room temperature.
The reaction of Cp'Re(CO) 2Br2 with Bu-jSnH at room
temperature was discovered to afford the dihydride in
excellent yield and, thus represents an improved synthetic
route for the synthesis of diag-Cp'Re(CO)*>H2.
The hydride reduction of (rj5-CgH7)Re(CO)3 at room
temperature leads to the immediate formation of
5
l(W -CgH7)Re(CO)2H] complex, which has been characterized
by IR analysis and *H and 13C NMR spectroscopy.

ACKNOWLEDGMENTS
The author wishes to express his sincere gratitude to
Professor Michael G. Richmond for his kind guidance,
encouragement, and assistance in directing my research, and
for serving as the chairman of my advisory committee. I
appreciate Dr. M. Schwartz and S. P. Wang for helpful
discussions and the use of their band area program, and
Messrs. Don Ellington and George Delong for NMR assistance.
Drs. K. E. Daugherty, R. D. Thomas, and D. A. Kunz are
thanked for serving on my advisory commitee.
I am especially grateful to my parents and my parents-
in-law for their dedication and support throughout the
years.
X am very grateful to my wife, Oc—Kee, sons, Leonard
and Joseph for their patience, never-ending encouragement,
and special love during the past years and throughout this
work.
Finally, I thank the University of North Texas for
support in the form of a teaching assistantship and the
Robert A. Welch Foundation of Houston, Texas for partial
financial support.
i n

TABLE OF CONTENTS
page
ACKNOWLEDGMENTS ii;L
LIST OF TABLES i x
LIST OF FIGURES # # . x i
CHAPTER
I. INTRODUCTION . # ±
II. EXPERIMENTAL
A. General Procedures 20
1. Techniques .. 20
2. Instrumentation 20
B. Materials 21
1. Solvents 21
2. Reagents .. . . 21
C. Methods 22
1. Attached Proton Test (APT) 22
2. Heteronuclear Proton-Carbon Chemical
Shift Correction (HETCOR) 23
3. Crystallographic Analysis 23
4. Band-Shape Analysis 24
D. Preparation of Compounds 25
1. Preparation of the Sealed NMR Tube Reactions 25
IV

page
2. General Procedure for the Preparations of
Aryl Rhenium Tricarbonyl Derivatives,
Cp*Re(CO)3 (Cp' « Cp, MeCp, Cp*, CgH7) .. 26
a. CpRe(CO)3 26
b. MeCpRe(CO)3 27
c. Cp*Re(CO)3 27
d. CgH7Re(CO)3 28
3. General Procedure for the Preparations
of Cyclopentadienyl Rhenium Dicarbonyl
Dibromide Derivatives, Cp'Re(CO)2Br2
(Cp» = Cp, MeCp, Cp*) 29
a. CpRe(CO) 2Br2 29
b. MeCpRe(CO)2Br2 3 0
c. Cp*Re(CO)2Br2 31
d. (d5-Cp)Re(CO)2Br2 3 2
4. diag-[(r?4-C5H6)Re(CO)2Br2][Li] 32
5. [CpRe(CO)£Br][Li] 33
6. [Cp*Re(CO)2Br][Li] 33
7. [CpRe(CO)2Br][M]
(M = Li (15-Crovm-5), PPP) 34
8. [MeCpRe(CO)2Br] [PPP] 34
9. [Cp*Re(CO)2Br] [PPP] 35
10. Reaction of Cp»Re(CO)2Br2 with One
Electron Reducing Agents (Cp1 = Cp, Cp*) .. 36

page
11. Reaction of Cp'Re(CO) 2Br2 with Grignard Reagents (Cp* = Cp, Cp*) 37
• CgHgCpRe (CO) 2 3-7
13. General Procedure for the Preparations
of Cyclopentadienyl Rhenium Dicarbonyl
Alkyl Bromide Derivatives,
CpRe(CO)2(R)Br (R « H, CH3, CgHg) 3 8
a. diag-CpRe(CO)2(H)Br 39
b. diag-CpRe (CO) 2 (CH3) Br 39
c. diag-CpRe (CO) 2 (C2H5) Br 39
d. diag-Cp*Re(CO)2(H)Br 4 0
e. diag-Cp*Re(CO)2(CH3)Br 4 0
f. diag-Cp*Re(CO)2(C2H5)Br 4 i
14. [C9H7Re(CO)2H] [Li] 4 1
15. diag-Cp'Re(CO)2H2 (Cp« = Cp, Cp*) 4 2
16. Cp'Re(CO)2(SnBu3)2 (Cp' = Cp, Cp*) 42
17. CpRe(CO)2L (L = PPh3, P(OPh)3,
(CH3)3CNC) 4 3
18. BrRe (CO) 5 4 4
19. (d5-Cp)Tl 4 4
20. (dg-Cp) Re (CO) 3 4 5
E. Thermodynamic Measurements 46
1. The Ion Pairing Eguilibrium in 2-Na, SSIP vs. CIP 4 6
VI

page
F. Kinetic Studies 47
1. Reaction of diag-CpRe(CO)2Br2 with
2. Isomerization of lat-CpRe(CO)2Br2 to diag-CpRe(CO)2Br2 in the
Presence of EtjSiH 4 7
III. RESULTS AND DISCUSSION
A. Aryl Rhenium Tricarbonyl Derivatives,
Cp'Re(CO)3 (Cp» = Cp, MeCp, Cp*, CgH7) 49
B. Cyclopentadienyl Rhenium Dicarbonyl Dibromide Derivatives, Cp'RefCOUBr? (Cp» = Cp, MeCp, Cp*) 5 5
C. Bromorheniumpentacarbonyl, BrRe(CO)g 62
D. Deuterated Cyclopentadienyl Rhenium Tricarbonyl, (d5-Cp)Re(CO)3 6 3
E. Deuterated Cyclopentadienyl Rhenium
Dicarbonyl Dibromide, (d5-Cp)Re(CO)2Br2 64
F. Hydride Reduction of diag-CpRe(CO)2Br2. Synthesis and Characterization of the Thermally Unstable Ring-Attacked Product, diag-[(n4-C5H6)Re(CO)2Br2]" 65
G. Reduction of Diagonal and Lateral CpRe(CO)2Br2: Ring Attack vs. Metal-Halogen Exchange 74
X-Ray Crystallographic Structure of [CpRe(CO)2Br][Li/l5-Crown-5] 8 3
I. Reactivity and Stability Studies of Anionic Cyclopentadienyl Complexes 94
Vll

page
J. Reduction of Diagonal and Lateral Cp*Re(CO)2Br2 with RLi, RMgX, and Trialkylborohydrides 113
K. X-Ray Crystallographic Structure of [Cp*Re(CO)2Br][PPP] 1 2 2
L. Reactivity and Stability Studies of Anionic Pentamethylcyclopentadienyl Rhenium Complex, [Cp*Re(CO)2Br][Li] 137
M. Reduction Studies Using One-Electron
Reducing Agents About Cp'Re(CO)2Br2 (Cp1 = Cp, Cp*) 138
N. Thermodynamic Study of the Ion Pairing
Equilibrium for [Cp*Re(CO)2Br][Na] 142
0. Reaction of Cp'Re(CO)2Br2 with Bu3SnH
and Et3SiH (Cp' = Cp, Cp*) 148
P. Hydride Reduction of Indenyl Rhenium Tricarbonyl: Synthesis and Character-ization of [(n -CgH7)Re(CO)2H]" 162
REFERENCES 167
Vlll

LIST OF TABLES
T a b l e Page
1. XR and Proton NMR Data for Aryl Rhenium
Tricarbonyl Derivatives, Cp»Re(CO)3 50
13 2. c NMR Chemical Shifts for Aryl Rhenium
Tricarbonyl Derivatives, Cp'Re(CO)3 51
3. IR and Proton NMR Data for Cyclopentadienyl Rhenium
Dicarbonyl Dibromide Derivatives, Cp'Re(CO)2Br2 .. 57
13
4. C NMR Chemical Shifts for Cyclopentadienyl
Rhenium Dicarbonyl Dibromide Derivatives,
Cp 'Re (CO) 2Br2 58
5. Anionic Cyclopentadienyl Rhenium Complexes with Reducing Agents: Ring Attack (R.A) vs. Metal-Halogen Exchange (M) 78
6. X-Ray Crystallographic and Data Processing
Parameters for [CpRe(CO)2Br][Li/15-Crown-5] ... 84
7. Table of Positional Parameters and Their Estimated Standard Deviations for
[CpRe(CO)2Br] | Li/15-Crown-5] 85
8. Bond Distances in Angstroms for
[CpRe(CO)2Br][Li/15-Crown-5] 8 9
9. Bond Angles in Degrees for
[CpRe(CO)2Br][Li/l5-Crown-5] 9 0
10. X—Ray Crystallographic and Data Processing
Parameters for [Li/15-Crown-5][Re04] 99
ix

13.
14.
Table Page
11. Table of Positional Parameters and Their Estimated Standard Deviations for [Li/15-Crown-5] [Re04] . 1 0 Q
12. Bond Distances in Angstroms for
[Li/15-Crown-5][Re04] 1 Q 3
Bond Angles in Degrees for [Li/15-Crown-5][Re04]. 104
X-Ray Crystallographic and Data Processing Parameters for [Cp*Re(CO)2Br] [Ph4PJ 1 2 3
15. Positional Parameters for Non-Hydrogen Atoms
for [Cp*Re(CO)2Br][Ph4P] with Estimated
Standard Deviations in Parenthesis 1 24
16. Bond Distances in Angstroms for
[Cp*Re(CO)2Br][Ph4P] 1 2 9
17. Bond Angles in Degrees for [Cp*Re(CO)2Br][Ph4P] . 131
18. Equilibrium Parameters for the Conversion of 2-Na from CIP into SSIPa
• • • 145
19. Selected Bond Dissociation Energies 160

LIST OF FIGURES
Figure P a g e
1. Methane and Carbon Dioxide Redox Coupling Scheme for the Production of Acetic Acid. Pathway (A): methane oxidative addition; pathway (B): carbon dioxide insertion; pathway (C): reductive elimination and repeat catalytic cycle 10
2. Infrared spectra of the carbonyl region for (a)
CgH/Re^OJs anc* (k) CPRe(CO)3. Both spectra
were recorded at 25 °C in cyclohexane. 53
3. *H NMR spectrum of CgHyRefCO)^ at 25 °C in
CDC13 54
4. Infrared spectra of the carbonyl region for (a)
diag-CpRe(CO)2Br2 and (b) lat-CpRe(CO)2Br2.
Both spectra were recorded at 25 °C in CH2C12. ... 61
5. Infrared Spectra of the carbonyl region
(a) [r?4-C5H6)Re(CO)2Br2] [Li] and (b)
C (-*7 -C5Hg)Re(CO)2Br2] [Li] with 5 equiv. of 15-Crown-5. Both spectra were recorded at
-70 °C in THF 66
6. NMR spectra of (a) [ (J74-C5Hg)Re(CO) 2Br2]"
and (b) [ (rj4-C5H5D)Re(CO)2Br2]~ at -70 °c
in d8-THF 6 8
7. (a) *^C{*H} NMR spectrum and (b) *3c NMR spectrum
using spin-echo J-modulation spectroscopy (APT)
of diag-[ (rj4-C5H6)Re(CO) 2Br2]". All spectra
were recorded at -70 °C in dg-THF 69
X I

Figure P a g e
8. Heteronuclear chemical shift correlation
spectrum of diag-[ (t?4-C5H6)Re(CO) gBr-]' at
-70 °C in dg-THF 7 1
9. Long-range heteronuclear chemical shift
correlation of diag-[ (r?4-C5H6)Re(CO)2Br2]" at
-70 °C in dg-THF. . 7 3
10. Infrared spectrum of the carbonyl region for
[CpRe(CO)2Br]" at -70 °C in THF 75
11. Infrared spectra of the carbonyl region for
(a) lat-[(T74-C5H6)Re(CO)2Br2]"
(b) lat-[ (n*-C5H5Me)Re(CO)2Br2]"
(c) lat-[ (r?4-C5H5Ph)Re(CO)2Br2]"
All spectra were recorded at -70 °C in THF 79
12. Infrared spectra of the carbonyl region for
(a) lat-[ (»?4-C5H6)Re(CO)2Br2] [Li]
(b) lat-[ (n4-CgHg)Re(CO) 2Br2] [Li] with 5 equiv.
of 15-Crown-5. Both spectra were recorded at •70 °C in THF. 80
13. Infrared spectra of the carbonyl region for (a)
[CpRe(C0)2Br][Li] and (b) [CpRe(CO)2Br][MgBr]
Both spectra were recorded at -70 °C in THF. ... 82
14. Perspective view (ORTEP plot) of
[CpRe(CO)2Br] [Li/15-Crown-5] showing the atom labeling (hydrogen atoms are omitted for clarity) . 8 7
15. Top view (ORTEP plot) of [CpRe(CO)2Br]"
showing the Cp ring-rhenium bond lengths 92
16. Infrared spectrum of the carbonyl region for
diag-CpRe(CO)2(SnBu3)2 at 25 °C in THF 97
Xll

Figure Page
17. Perspective view (ORTEP plot) of
[Li/l5-Crown-5][ReO^] showing the atom labeling
(hydrogen atoms are omitted for clarity). .... 102
18. Infrared spectrum of the carbonyl region for [Cp*Re(CO)2Br]- at -70 °C in THF. 1 1 5
13 1 19. C{ H) NMR spectrum of [Cp*Re(CO)2Br] [PPP]
at 25 °C in CD2C12 1 1 7
20. Infrared spectra of the carbonyl region for (a)
[Cp*Re(C0)2Br][Li] and (b) [Cp*Re(CO)2Br][MgBr].
Both spectra were recorded at -70 °c in THF. .. u s
21. Perspective view (ORTEP plot) of [Cp*Re(CO)2Br]" showing the atom labeling (hydrogen atoms are omitted for clarity) 1 2 7
22. Perspective view (ORTEP plot) of PPP+ gegenion showing the atom labeling (hydrogen atoms are omitted for clarity) 1 2 8
23. Top view of (a) lat-Cp*Re(CO)2I2 and (b)
[Cp*Re(CO)2Br]" showing the Cp* ring-rhenium bond lengths (A). Distances for the former complex are taken from ref. 32. 134
24. Infrared spectra of the carbonyl region for
[Cp*Re(CO)2Br][Na] in THF as a function of
temperature: (1) -70 °C, (2) -30 °c, and (3)
25 C. The inset displays the asymmetric
carbonyl region (1825-1725 cm"1) for the experimental spectrum ( ) of 2-Na recorded
at -70 °C, resolved bands (....) for the contact and solvent-separated ions, and the theoretical curve ( ) for both ion pairs 1 4 4
x m

Figure Page
25. Plot of In Keg + 1 vs. 1/T for the
contract solvent-separated ion pair
equilibrium for [Cp*Re(CO)2Br] [Na] in THF 146
26. Infrared spectra of the carbonyl region for
(a) diag-CpRefCOJgfSnBu^
(b) lat-Cp*Re(CO)2(SnBu3)2
Both spectra were recorded at 25 °C in
cyclohexane 152
11 0 27. The Sn NMR spectrum of diag-CpRe(CO)2(SnBuj)^
in CDClj solution (0.1 M) at 25 °C 153
28. Absorbances changes for lat-CpRe(CO)2Br2 in
toluene at 40 °C in the presence of EtgSiH. .. 156
29. Absorbances changes for diag-CpRe(CO)2Br2
with EtjSiH (10-fold excess) at 40 °C
in toluene 158
30. *H NMR spectrum of [ (r}5-CgH7)Re(CO) 2H]" at 25 °C
in dg-THF 164
xiv

CHAPTER I
INTRODUCTION
The objective of these investigations, which were
carried out in partial fulfillment for a Ph.D degree in
chemistry and are reported in this dissertation, comprises
the synthesis, structure, and reactivity of anionic
cyclopentadienylrhenium compounds and the reduction pathways
in cyclopentadienylrhenium dicarbonyl dibromides. The
chemistry of organo-transition-metal compounds pertinent to
this work and the literature on hydrocarbon C-H bond
activation and C02 insertion into the M-H bond are reviewed
in this section.
One of the most interesting goals of homogeneous
organo-transition-metal chemistry is the possibility of
carrying out selective chemical transformations on, or
functionalizing, unreactive materials such as saturated
hydrocarbons. The homogeneous activation of C-H bonds by
metal complexes, especially at saturated carbon centers, has
long been recognized as a great interest and challenging
objective.*
Initial observations of oxidative addition to arene C-H

bonds in 1965 provided the impetus to look for complexes
that would activate the weaker sp3-hybridized alkane C-H
bonds.2 However, this goal was not achieved until nearly
two decades later and was accompanied by an intermediate
period in which much confusion occurred concerning the
thermodynamic feasibility of the C-H bond activation
3
reaction. in this account, mechanistic studies with a
series of homogeneous rhodium organometallic complexes are
summarized and have provided for the first time a
comparative evaluation of the relative equilibrium constants
and rates of reaction for both alkane and arene hydrocarbon
activation (Eq. 1).
R-H m
\ Since Chatt observed the first clear example of simple
oxidative addition of the C-H bond of naphthalene to a
ruthenium metal center,2 hydrocarbon activation has become
the subject of many transition-metal studies. From the
point of view of quantity and availability, alkanes (satura-
ted hydrocarbons) would also be very attractive feedstocks
for catalytic synthesis of organic molecules since they are
major constituents of natural gas, petroleum, and coal

3
liquefaction processes. However, they have not received
much use in synthetic processes and as feedstocks in
catalytic reactions.
Saturated hydrocarbons are among the most ubiquitous
and chemically stable of all organic materials due to the
strong C-H bond (~ 99 kcal/mole) and C-C bond (~ 83 kcal/
mole) energies, but they can be activated in special cases.
As a result of their relative inertness, saturated hydro-
carbons have a long history of activation by nonmetallic
reagents and methods. Hydrocarbon thermal reactions and
combustion have been studied, and there are well-known free
radical reactions (e.g., autoxidation, photochlorination)
which serve to functionalize these materials.^ More
recently, reagents such as ozone, superacids, fluorine, and
H2O2 have been used to activate hydrocarbons.® However,
these reactions often require large amounts of energy
(either light, heat, or chemical) and are usually very
unselective. Therefore, attention has been focused on the
discovery of soluble organo-transition-metal complexes
capable of inserting a metal center into the C-H bond of
alkanes (Eq. 1). During the past 15 years many examples of
intramolecular C-H oxidative addition [that is, the metal
and reacting C-H bond are located in the same molecule
(Eq. 2)] have been discovered.^ Some specific examples of
this process are depicted in Equations 3-5.

M - CHgR — — ^ H - M - CHR (2)
/ CPPh3)3l
rC1 ( P P| l 3, 2 ( c l ) I/^__^ (3)
•I CH2CHe3
CH2CHe3
CMe4 LgPt^ L (4)
-Bu) /H ( t
HC13 + (t-Bu)2P(CH2)5P(t-Bu)2 H ' — j — ( 5 ) ^fe(t-Bu)
CM = Ir, Rh)
The largest group involves so called ortho-metallation
processes, in which insertion takes place into the C-H bond
of an aromatic ring attached to an atom directly bound to
the metal (Eq. 3). Some cases are also known of insertion
into a C-H bond of an alkyl chain located in the same
molecule as the metal (Eq. 4 and 5). Obviously, the
proximity of the reacting C-H bond to the metal center is a
critical factor favoring such cyclometallation processes.
Many examples of homogeneous intermolecular activation

of saturated hydrocarbons by metal complexes are also
reported.
In the late 1960's and early 1970's Shilov and his
group reported reactions involving both alkane H/D exchange
and conversion to chlorides and acetates catalyzed by
soluble platinum salts (Eq. 6).*e>^
DOAc RD
Ptncl„ / ,B. KB — ^ (6>
Pt'»Cln RC1
However, somewhat elevated temperatures (100-120 °C) are
required for these reactions. Little else is known about
their mechanism including whether they are most likely
colloidal and involving metal (Pt) cations similar to the
superacid reactions.
In early 1980*s Crabtree's group® at Yale, and Felkin
and coworkers at Gif-Sur-Yvette in France^ reported two very
interesting and unique iridium- and rhenium-induced
dehydrogenation processes shown in Eq. 7 and Eq. 8.
Crabtree has provided convincing evidence that these
reactions are completely homogeneous and has proposed that
they are initiated by insertion of the metal center into an
alkane C-H bond, but it has so far been difficult to

6
[lrH 2s 2L 2]+
+ t-BuCH=CH 2 [CpIrHL 2]+ + t - B u C H 2 C H 3 (7)
(S = Acetone, L = PPh 3)
L2ReH7 + t-BuCH=CH 2 CpR«L 2H 2 + t-BuCHgCHj (8)
determine this conclusively since the reactions are quite
complicated. These reactions involve multiple hydrogen loss
in the saturated hydrocarbon substrate and require an added
alkene as a hydrogen acceptor.
In solution, a few relatively electron-rich complexes
have been demonstrated to undergo insertion into C-H bonds
activated by adjacent functional groups of organic compounds
having C-H bonds with low bond energy or high acidity (Eq. 9
and 10); however, the metal centers in these molecules
.H CH 3(CO)CH 3
(dmpe)^^ ^ (dmpe)2M^ o - ArH ^ C H 2 - C - C H 3 (9)
(N = Fe, Ru)
h v J ^ C H 2 s i M ® 3 C p 2 W H 2 + M e 4 S i — y C ° (10)

7
apparently react with C-H bonds in their own ligands more
rapidly than with saturated hydrocarbons.lc>10
Recently, a few examples of the reaction shown in Eq. 1
have been demonstrated between organo-transition-metal
complexes and completely saturated hydrocarbons (alkanes) in
homogeneous solution under photolytic conditions (Eq. 11).
11-13 In 1982 Bergman and Janowicz were the first to
[CpMLH2] o r
hv RH jr [CpHL] Cp(L)Ji^
H (11)
[CpML(CO)] —
(Cp» = Cp, Cp*? M = I r , Rh? L as CO, PMe3)
synthesize an iridium complex which successfully converts
alkanes into hydrido(alkyl)metal complexes in high yield at
room temperature. Evidence has been obtained that this C-H
insertion (oxidative addition) reaction proceeds through a
simple three-center transition state and does not involve
organic free radicals as intermediates. In accordance with
this, the intermediate [Cp*IrL] reacts most rapidly with C-H
bonds such as those at primary carbon centers, in small
organic rings, and in aromatic rings. Reductive elimination
of the hydrocarbon from the hydrido(alkyl)metal complexes
can be induced photochemically or by heat, regenerating the
reactive intermediate [Cp*ML], which is then capable of
attacking the C-H bond of other hydrocarbons. Oxidative

8
addition of the corresponding rhodium complexes [Cp*RhL] to
alkane C-H bonds has also been observed, although the
products formed in this case are much less stable, and
undergo facile reductive elimination. These recent
observations provide an incentive for reexamining the
factors which have been assumed to control the rate of
reaction of transition metal complexes with C-H bonds,
notably the need for electron-rich metals and the close
proximity of reacting centers.
On the other hand, further functionalization of these
hydrocarbon activated complexes, Cp*M(CO)(R) (H), as a route
to fine organic chemicals has not been very successful to
date. These complexes can potentially react with C02 to
afford the corresponding metallocarboxylic acid,
Cp*M(CO)(R)(CO2H), via CO2 insertion into the metal-hydride
bond. This is attractive since the reductive elimination of
the methyl and metalloacid groups can be envisioned to give
the commodity chemical acetic acid and regenerate the metal
catalyst in the case of R = methyl (coordinatively unsatur-
complex, [Cp*M(CO) ]). Such an insertion reaction
(i.e., M-H + CO2 * M—CO2H) is well documented in other
metal-hydride complexes,^ but no analogous Cp*M(CO)(CHg) (H)
insertion chemistry has been studied in depth. Further
reaction of this complex with additional methane and CO2
ensures a cyclic process for this overall methane oxidation

9
and CC>2 reduction scheme as shown in Figure 1.
This directed synthesis of acetic acids is of commer-
cial importance due to its use as a major industrial
chemical which is necessary for the oxidation of para-xylene
to terephthalic acid (nylon polymer precursor), production
of polyvinyl alcohol and polyvinyl acetate polymers, pharma-
ceuticals, pesticides, dyes, and acetic anhydride synthesis
which is used to prepare cellulose acetate and aspirin.^
Collectively, these uses of acetic acid represent a multi-
million dollar industry for the whole world. Although
acetic acid has been produced in relatively large quantities
by fermentation and sundry catalytic processes, it was not
until Monsanto discovered the low pressure carbonylation of
methanol using rhodium catalysts in 197116 that an
industrial process showed such high yields (> 90%) and
reaction rates in the enzymatic range.^ However, the
Monsanto process suffers from being a petroleum-based
17 18
process. * Therfore, an alternative process based on
abundant reagents remains desirable. The use of methane and
carbon dioxide as chemical feedstocks remains highly
attractive due to their inexpensive nature and vast
abundance.
Therefore, the main goal of my research was to
synthesize the analogous rhenium-hydride complexes [i.e.,
Cp*Re(C0)2(CHg) (H) ] which are isoelectronic and isolobal^

10
coordlnately unsaturated intermediate
Cp*M(CO)2
-CO hv
[Cp*M(CO)]
1 *ch3-h
CHj-H
[Cp*M(CO)]
acetic acid
Cp*
,.M,
oc'y
ch3
Cp"
"iM\ o i \ y o c " 7 C-OH
CH,
CHr-C-OH
Figure 1. Methane and Carbon Dioxide Redox Coupling Scheme for the Production of Acetic Acid. Pathway (A): methane oxidative addition? pathway (B): carbon dioxide insertion; pathway (C): reductive elimination and repeat catalytic cycle.

11
to the rhodium and iridium systems (Scheme 1) and to
investigate the CO^ insertion chemistry and catalytic
properties associated with these complexes. Unfortunately,
the results of the reaction of CpRe(CO)2Br2 with reducing
Scheme l
l - m -
M as R h , I r M S R e
d 8 - M L 4 ^ d 6 - M L 5
agents such as LiEt3BH and RLi (R = CH3, Ph, and t-Bu)
suggest that direct metathetical style reductions in
CpRefCOJgBrg as a route to rhenium derived hydrocarbon
activated templates are not possible unlike those observed
in the corresponding rhodium and iridium systems [e.g.,
CpMLBrg • CpMLH(R)].^ Therefore, reduction studies of
cyclopentadienylrhenium dicarbonyl dibromide derivatives
were conducted and the reactivity associated with the
resulting anionic cyclopentadienylrhenium complexes explored
and reported herein.
The cyclopentadienyl ring ligand has played an
important role in the development of organometallic

12
chemistry since the discovery of ferrocene in 1952. The
significance of the cyclopentadienyl ligand has stimulated
interest in investigating the chemistry of other closely
related ligands, such as rings in which one or more of the
hydrogen atoms are substituted by other groups.^
In 1958 the cyclopentadienylrhenium tricarbonyl, c
CpRe(CO)j (Cp = r? -C5H5), was first prepared from the
refluxing of rhenium decacarbonyl in dicyclopentadiene by
77
Green and Wilkinson. The same compound has also been
obtained from the reaction of Re(CO)gX (X = Br, CI) in THF
or benzene with CpNa or CpTl. The aryl rhenium
tricarbonyl derivatives (aryl » Cp, MeCp, Cp*, and CgHy24)
obtained from these procedures can be purified by column
chromatography over silica gel with petroleum ether or by
slow sublimation under vacuum while maintaining a small
temperature gradient. Aryl rhenium tricarbonyl derivatives
are white solids or a yellow solid in the case of the
indenyl complex (CgHy) and are air stable compounds. The structure of CpRe(CO)^ has been determined by single crystal
25
X-ray analyses. The cyclopentadienyl ring is almost
perfectly planar (largest deviation, 0.003 A). The small
variation of C-C bond lengths within the ring indicates some
tendency for delocalized bonding, which is quite expected
for molecules of this type which possess effectively

13
cylindrical symmetry.
Synthetic pathways to the deuterated cyclopentadienyl
rhenium tricarbonyl, (d5-Cp)Re(CO)3, have recently been
described in detail.26 The reaction of (d5-Cp)Tl and
BrRe(CO)g in benzene proceeds readily at 60-70 °C overnight
to give excellent yields of (d5-Cp)Re(CO)3.
Although at first glance the permethylation of
cyclopentadienyl rings might appear to be a relatively minor
alteration, it has proven to be a useful perturbation for
mechanistic: studies and has uncovered a significantly
different chemistry in many systems.27 Some of the changes
in behavior of pentamethylcyclopentadienyl complexes may be
attributed to the steric protection provided by the five
ring methyl groups. Another very important factor is the
apparent increased electron density and donor strength of
the permethy1ated ring relative to the parent Cp ring.
Therefore, methylcyclopentadienyl, pentamethylcyclopenta-
dienyl, and mdenyl coordinated rhenium carbonyl derivatives
have been prepared and the chemistry associated with these
compounds investigated and reported herein.
Cyclopentadienyl metal derivatives of the type CpMX^
are formed by many d^ transition metals of the 4d and 5d
transition series including Nb(I), Ta(I), Mo(II), W(II), and
• Four—legged piano—stool complexes of the form

14
CpMX2^2 m a¥ ®xist as two nonequivalent stereoisomers that
are commonly referred to as cis and trans isomers. We have
adopted King's nomenclature to describe the isomeric
dibromides discussed in this paper. Here the descriptors
lateral and diagonal correspond to the cis and trans
stereoisomers, respectively.
In 1969 Nesmeyanov and his coworkers first prepared
CpRe(CO>2Br2 from the reaction of bromine and CpRe(CO) in
trifluoroacetic acid,2® and subsequently the product was
shown to consist of cis and trans isomers which could be
separated.29 The diiodide complex CpRe(CO)2I2 was not
reported until 19813®'31 and may also be synthesized in cis
and trans forms from the direct reaction of CpRe(CO)3 with
I2 in dimethyl sulfoxide. The dichloride CpRe(CO)2Cl2 was
only recently reported.32 Of these dihalides, the dibromide
has received much attention in terms of chemical investiga-
33 34 ti°n ' and as a precursor in the synthesis of other four-
legged piano-stool complexes based on the CpRe fragment.35"
42
However, synthetic pathways to the corresponding
pentamethylcyclopentadienylrhenium dicarbonyl dihalides,
CP*Re(CO)2X2 (where Cp* = ^"CgMeg), have only recently been
described in detail. While the diiodide Cp*Re(CO)2I2,
initially obtained from the reaction of Cp*2Re2(CO)j with
J2' w a s t h e first compound of the Cp*Re(CO)2x2 prepared,33

15
it was not until the work of Sutton et al. that reliable and
stereoselective synthesis of these dihalide compounds were
r e p o r t e d . 3 2 , 3 3 . 4 3 , 4 4 A s such^ the reactivity of these
dihalides remains to be explored and established.
In related reactivity studies using the isomeric
dibromides, Cp'Re(CO)2Br2 (CP' ~ CP*)/ w© have observed
that a direct metathetical replacement of the bromide
ligands with RLi or RMgX reagents (where R = Me, Ph, or
t-Bu) to yield the dialkyl (aryl) complexes Cp'Re(CO)2R2
does not occur. The reaction of CpRe(CO)2Br2 with Grignard
reagents has been reported in 1974 to give compounds of the
form CpRe(CO)2(R) (X) and CpRe(CO)2R2 (where R = alkyl or
34
aryl; X = halide) . However, no mention was made of the
relationship between the product dependence and initial
dibromide stereochemistry. Furthermore, while the reported
physical data dealing with such compounds as
CpRe(CO)2Br(Me), CpRe(CO)2I(Me), and CpRe(CO)2(Me)2 are not
in question, we do not believe that these represent products
of direct alkyl/bromide exchange as we observe metal-halogen
exchange and cyclopentadienyl ring attack prior to the
formation of the above complexes (vide infra). The relative
amounts of these products (metal/halogen exchange and ring
attack) are dependent on the nature of the reducing agent
and the stereochemistry of the initial dibromide. That is,

16
diag-CpRe(CO)2Br2 and Cp*Re(CO)2Br2 (either isomer) react
exclusively with RLi and RMgX reagents to afford
[Cp'Re(CO)2Br] as a result of metal—halogen exchange while
lat-CpRe(CO)2Br2 gives both [CpRe(CO)2Br]" and 4
[ (*? -CgHgR)Re(CO)2Br2] . This latter complex derives from
RLi (R - Me or Ph) attack on the cyclopentadienyl ligand.
Although indenyl rj5-complexes of transition metals are
analogous to r?5-cyclopentadienyl species, they exhibit some
specific properties. In an indenyl ligand, a benzene ring
fused with a 5-membered ring takes part in distribution of
jr-electron density.4®
The reactivity of the rj®-indenyl rhenium tricarbonyl
has been previously studied in exchange reactions of the
coordinated CO with other p-donor ligands.46 The reactions
of (f?5-C9H7)M(CO)3 (M • Mn, Re) with strong acids (A1C13*HC1
and FSOjH) have also been studied4^ and shown to involve a
metallotropic rj5 -• rj6 rearrangement in which the metal
carbonyl group migrates from the five- to the six-membered
ring of the aromatic ligand. The reverse 17® -*• rj®
rearrangement is initiated by bases. In this study we
investigated the reduction pathways of (r?5-CgH7)Re(CO) 3 with
LiEt3BH.
Diagonal cyclopentadienylrhenium dicarbonyl dihydride,
diag-CpRe(CO)2H2 was first observed by Graham and Hoyano

17
from the photolysis of CpRe(CO)3 in the presence of H2.41a
Bergman and Yang later synthesized it in good yield by the
reduction of CpRe(CO)2Br2 with zinc and acetic acid.37b The
former method is known to give low yields while the latter
method in our hands has proven inconvenient as the yields
are dependent upon the work-up conditions. However, the
reaction of Cp'Re(CO)2Br2 (either isomer, Cp' = Cp, Cp*)
with BujSnH (2 equiv.) at room temperature was discovered to
afford the dihydride in excellent yield and, thus,
represents a new and an improved synthetic route for the
synthesis of diag-Cp'Re(CO) 2H2-
48
Graham recently reported the synthesis and properties
of a series of rhenium compounds of the type CpRe(CO)2x2
(X • germanium and tin species).31 The bis(triphenyltin)
rhenium complex, diag-CpRe(CO)2(SnPh3)2, w a s synthesized in
very poor yield (7 %) from the photolysis of CpRe(CO)3 in
the presence of HSnPhj. However, the reaction of the metal-
halogen exchange product, [CpRe(CO)2Br]", with BUjSnH at
60 °C leads to the formation of the bis(tributyltin) rhenium
complex, diag-CpRe(CO)2(SnBu3)2, which was also
independently synthesized in excellent yield by the
deprotonation of diag-CpRe(CO) 2H2 with EtjN in the presence
of Bu^SnBr at room temperature.
Recently there has been considerable interest in the

18
solution structure of the alkali metal salts of various
transition metal carbonylate anions. In particular, the
chemistry associated with cyclopentadienyl carbonylate
anions is of both mechanistic and synthetic interest. These
compounds are among the most versatile of the transition
metal anions and have been widely used to prepare many novel
and significant organometallic complexes of the transition
49
metals. Extensive studies by Edgell and coworkers on the
sodium tetracarbonyl cobaltate were the first to illustrate
the importance of ion pairing in such systems.®® It was
shown that an equilibrium existed between a tight ion pair involving a sodium-carbonyl oxygen interaction and a solvent
separated ion pair. In 1974 Pribula and Brown reported
similar ion pairs for the sodium pentacarbonylmanganate
51
system. Other workers have been concerned with the
redistribution of electronic charge in the carbonylate
itself upon changing the electrostatic potential of the
countercations,49-53 or interested in ion-pair site specifi-
city in non-symmetric carbonylates.52 The ability of v(CO)
infrared spectroscopy to detect very small structure and
electronic changes as well as the availability of a
substantial arsenal of appropriate solid—state structural
studies of metal carbonylates has promoted the development
of a new subdiscipline in ion-pairing phenomena.
Darensbourg and coworkers have extended the account of

19
ion pairing by studying the propensity of [CpM1(CO)^]" (M' =
Cr, Mo, W) to form contact ion pairs as a function of the
countercation (Li+, Na+, K+, Me^N+) and solvent (EtgO,
Et20/HMPA, THF/HMPA, and CH3CN).53 Of the three possible
proposed interactions between the metal carbonylate and the
cation shown below, and they did not observe any indication
of cation penetration of the coordination sphere yielding an
ion-paired structure (III).
° nCr V
M ^ C 0 0 C V M \ C 0 M' o c - > M ^ c o OC OC •—M OC
(i) (II) (in)
In order to establish the nature of ion pairs in
solution for [Cp*Re(C0)2Br][Na] complexes, the effect of
temperature on the equilibrium between solvent-separated ion
pairs and carbonyl oxygen contact ion pairs over the
temperature range -70 °C to room temperature was examined.
This system reveals a reversible temperature-dependent
equilibrium involving both anionic species. The equilibrium
constant for these ion pairs has been determined by IR band
shape analysis and values of AH and AS have been determined.

20
CHAPTER II
EXPERIMENTAL
A. General Procedures
1. Techniques
All manipulations were conducted under an inert
atmosphere of argon, either on a high vacuum line with
modified inert Schlenk techniques55 or in a nitrogen filled
Vacuum Atmosphere DL Series inert-atmosphere Dri-box.
2. Instrumentation
Infrared spectra were recorded on a Nicolet 20SXB FT-IR
Spectrometer in 0.1 mm NaCl cells. Low-temperature IR
spectra were recorded on the same spectrometer with a Specac
Model P/N 21.000 variable-temperature cell equipped with
inner and outer CaF£ windows. Dry ice/acetone was used as
coolant, and the reported cell temperatures, taken to be
accurate to ± 1 °c, were determined with a copper-constantan
thermocouple. *H (300 MHz) and 13C (75 MHz) NMR spectra
were obtained using a Varian VXR-300 MHz NMR Spectrometer

21
while 90 MHz *H NMR spectra were recorded on a JEOL FX90Q
NMR Spectrometer. 2D (13.1 MHz), (22.5 MHz), *19Sn (33.7
MHz) NMR spectra were recorded on the JEOL FX90Q NMR
Spectrometer. The C and H analyses were performed by
Atlantic Microlab, Atlanta, 6A.
B. Materials
1. Solvents
Tetrahydrofuran, benzene, and toluene were distilled
from purple solutions of sodium benzophenone ketyl under
argon. Aliphatic hydrocarbon solvents (CgH^, C^Hj^),
CH2CI2, Et20, and HMPA were distilled from calcium hydride
under argon.
Deuterated solvents (dg-THF and dg-benzene) were vacuum
distilled from calcium hydride while CDgC^ and CDCI3 were
distilled from P2O5 under argon and stored in Schlenk
vessels.
2. Reagents
Rhenium decacarbonyl was used as received. Deuterated
cyclopentadienylrhenium tricarbonyl was prepared from known
26 cc procedure. Pyridinium hydrobromide perbromide , sodium
naphthalide (0.5 M in THF), cobaltocene (0.25 M in THF)^,

22 CO
and t-Butyl isocyanide30 were prepared from known literature
procedures. Triphenylphosphine (Alfa) was recrystallized
from absolute ethanol. Triphenylphosphite (Aldrich) was
distilled from calcium hydride under argon. Magic ethyl was
purchased from Alfa and stored under argon in a Schlenk
vessel while methyl triflate was prepared according to the . 5 5 50
published procedure. Bu3SnH was prepared from known
literature procedures. EtjSiH was purchased from Lancaster
Synthesis Ltd. and used as received.
CH3Li (1.4 M in EtgO), PhLi (2.0 M in CgHjg/EtgO;
70/30), t-BuLi (1.7 M in pentane), sec-BuLi (1.3 M in cyclo-
hexane), MeMgBr (1.5 H in toluene/THF? 75/25), t-BuMgCl (2.0
M in THF), LiEt3BH(D) (1.0 M in THF), NaEtjBH (1.0 M in
THF), and K-Selectride (1.0 M in THF) were all purchased
from Aldrich and used as received.
C. Methods
1. Attached Proton Test (APT)
This J-modulation spin-echo experiment utilized the
standard PD-90°-D1-180°-D2-1800-D3-ACQUISITION pulse
sequence that is equipped with the Varian VXR-300
Spectrometer. The broad-band proton decoupler was gated off
during the D1 delay period and then turned back on for the

23
rest of the pulse sequence. All spectra were obtained
employing quadrature phase detection and automatic base line
correction. Alternate FIDs were corrected with 180° phase
shifts to remove DC bias between the two receiver channels.
2. Heteronuclear Proton-Carbon Chemical Shift
Correlation (HETCOR)
The heteronuclear chemical shift correlation spectrum
was acquired by using the pulse sequence of Freeman and
Morris6®, modified to provide quadrature detection in the
second frequency domain as described by Bax and Morris61.
The data was acquired as a 128 (zero filled to 512) x 2K
matrix to yield a 512 x IK F ^ data matrix that was
processed by using 2D Gaussian Apodization prior to the
second Fourier transformation.
3. Crystallographic Analysis
Data were collected on an Enraf-Nonius CAD-4 diffrac-
tometer at 24 ± 2 °C using graphite-monochromated Mo
radiation. The data were corrected for Lorentz and polari-
zation effects. The structure was solved by MULTAN6* or the
Patterson method and successive cycles of differrence
Fourier syntheses were followed by least-squares refinement.
Data with intensities less than 3a(I) were rejected, and a
non-Poisson contribution weighting scheme with an intensity

2 4
factor P set at 0.04 was used in the final stages of
refinement. P is used in the calculation of a(I) to down
weight intense reflections in the least-squares refinement.
The function minimized was S - (| FQ| -| FC|)2, where
2 - 4 ( F 0 ) 2 / [ 2 ( F 0 ) 2 ] 2 ,
S ( F Q ) 2 = [ S 2 ( C + R2B) + <P(F0)2}2]/Lp2, and S is the scan
rate, C is the total integrated peak count, R is the ratio
of scan time to background counting time, and Lp is the
Lorentz-polarization factor. Suitable crystals were grown
from a solution of THF and n-heptane (vide infra) and were
mounted in a thin-walled glass capillary and sealed under
nitrogen. All non-hydrogen atoms were refined anisotropi-
cally and scattering factors were taken from ref. 63.
4. Band-Shape Analysis
Since both the asymmetric carbonyl stretching bands of
the sodium-contact and solvent-separated ion pairs in
solution of [Cp*Re(C0)2Br][Na] complex exhibit significant
overlap, the infrared band shapes of these CO bands were
calculated using a numerical procedure in order to determine
the ratio of their areas. Absorbances were digitized from
1825 cm ^ to 1725 cm * by 2 cm~^ intervals and entered into
files on the university VAX 11 /85 computer. Following
baseline correction, the spectra were fit by a model
consisting of Lorentzian band-shapes, each characterized by

25
a peak frequency (v), maximum intensity (I), and half width
[FWHH] (A). Since the instrument resolution (2 cm'*) is far
less than the observed bandwidths (20 cm"*), it was unneces-
sary to convolute the model spectrum with a resolution
(slit) function. The parameters were varied to minimize the
squared deviation between the experimental and calculated
intensities using a standard non-linear regression
64
procedure . Given that the area of a Lorentzian peak is
proportional to the product of the bandwidth and the maximum
intensity, the area ratio of the different ion pairs is
calculated easily as A2/AJ - (I2 x A2)/(IJ x Aj) .
D. Preparation of Compounds
1. Preparation of the Sealed NMR Tube Reactions
Starting material and an appropriate deuterated solvent
(0.7 mL for 5 mm NMR tube or 3.5 mL for 10 mL NMR tube) were
transferred into the NMR tube under argon. After the tube
was shaken well, the reagent was added at -78 °C. The tube
was then freeze-pump-thaw degassed three times prior to
flame sealing.

26
2. General Procedure for the Preparations of Aryl
Rhenium Tricarbonyl Derivatives, Cp'Re(CO)3
(Cp» = Cp, MeCp, Cp*, C9H7)
A 100 mL of Fisher-Porter tube was charged with cyclo-
pentadiene dimer or freshly distilled indene, Re2(CO)10, and
a magnetic stir bar. The Fisher-Porter tube was heated up
to 210 °C over several hours employing a heating mantel as
the heat source. At this time, the pressure gauge
registered an increased pressure of 100 psi. At this time,
the heat source was turned off and the Fisher-Porter tube
was allowed to cool to room temperature. The liberated CO
and H2 gas were released from the Fisher-Porter tube. This
process was repeated until no pressure increase was
observed. The white solid material was transferred to a
fritted glass funnel and washed three times with cold
hexane. Air was pulled through the product for several
minutes to remove the hexane. The colorless powder was
analytically pure and was used directly in subsequent
reactions.
a. CpRe(CO)j
Cyclopentadiene dimer (7.4 mL, 56.0 mmole), Re2(CO)10
(10.4 g, 16.0 mmole), and a magnetic stir bar were placed
into the Fisher-Porter tube. This reaction was carried out

27
as described in general procedure. The total released
pressure of CO and H2 gas was 310 psi. The yield of
CpRe(C0)3 was 9.4 g (87 %).
IR (Cyclohexane) : 2030 (m), 1939 (s) cm"*.
^-NMR (CDCI3) : -CgHg (5.2 s, 5H) .
13C-NMR (CDC13) : -%H 5 (84.4, 5C) , -Q0 (194, 3C) .
b. MeCpRe(CO)3
Methylcyclopentadiene dimer (10.4 mL, 61.2 mmole),
Re2(CO)jQ (10.0 g, 15.3 mmole), and a magnetic stir bar were
placed into the Fisher-Porter tube. This reaction was
carried out as described in general procedure. The total
released pressure of CO and H2 gas was 300 psi. The yield
of MeCpRe(CO)3 was 9.5 g (89 %).
IR (Cyclohexane) : 2027 (m), 1935 (s) cm"*.
*H-NMR (CDCI3) : -CgU^Me (5.23 dd, J - 4.2 Hz, 4H), -CH3
(2.23 S, 3H) . 13C-NMR (CDCI3) : -£H3 (13.6), -£-Me (106.6),
-£H (83.8, 2C), -£H (83.2, 2C), -QO (194.6, 3C).
C. Cp*Re(CO)3
Freshly distilled pentametylcyclopentadiene (7.9 mL,
50.5 mmole), Re2(C0)|Q (10.0 g, 15.3 mmole), and a magnetic
stir bar were placed into the Fisher-Porter tube. This
reaction was carried out as described in general procedure.

28
The total released pressure of CO and H2 gas was 315 psi.
The yield of Cp*Re(CO)3 was 11.7 g (94 %).
IR (Cyclohexane) : 2013 (m), 1922 (s) cm"*.
*H-NMR (CDCI3) : -Qi3 (2.15 s, 15H) .
13C-NMR (CDC13) : -£H3 (10.7, 5C), -£-Me (98.4, 5C) , -Q0
(198, 3C).
d. CgH7Re(CO)3
Freshly distilled indene (6.3 mL, 53.6 xnmole),
Re2(CO)jQ (10.0 g, 15.3 mmole), and a magnetic stir bar were
placed into the Fisher-Porter tube. This reaction was
carried out as described in general procedure. The total
released pressure of CO and H2 gas was 280 psi. The yellow
crystalline material was purified by column chromatography
over silica gel with petroleum ether/C^C^ (4:1 v/v). The
yield of CgH7Re(CO)3 was 9.7 g (82 %).
IR (Cyclohexane) : 2028 (s), 1940 (s), 1933 (s) cm"*.
*H-NMR (CDC13) : -Ha (5.65 t, J - 3.2 Hz, 1H), -Hb (5.78 d,
J = 3.2 Hz, 2H), -Hc (7.10 dd, J - 3.2 Hz, 2H), -Hd (7.50
dd, J = 6.3 Hz, 2H).
13C-NMR (CDC13) : -S.0 (193, 3C) , -£j (91, 1C) , -£2 (71, 2C) ,
-C3 (108, 2C), -£4 (126, 2C), (123.5, 2C) .

29
3. General Procedure for the Preparations of
Cyclopentadienyl Rhenium Dicarbonyl Dibromide
Derivatives, Cp'Re(CO)2Br2 (Cp- - cp, MeCp, Cp*)
Cyclopentadienylrhenium tricarbonyl and pyridinium
hydrobromide perbromide were placed into a 250 mL of round
bottom flask with a magnetic stir bar, then trifluoroacetic
acid was transferred to the reaction flask. After stirring
the reaction mixture for 30 min at room temperature, the
reaction mixture was quenched by pouring it into 1000 mL of
water. The pale orange precipitate was filtered and washed
three times with water (100 mL portions). After drying
under aspirator suction, the crude product was chromato-
graphed over silica gel. Successive elution with petroleum
ether/CH2Cl2 (1:1 v/v) gave first unreacted rhenium
tricarbonyl as a colorless band, then diag-CpRe(CO)2Br2 as a
red band was separated, and finally lat-CpRe(CO)2Br2 as a
brown band was separated by elution with only CH2C12.
a. CpRe(CO)2Br2
CpRe(C0)3 (6.0 g, 18.0 mmole) and CgHgNHBr^ (6.1 g,
19.0 mmole) were placed into a 250 mL of round bottom flask,
and 55 mL of CFjCOgH was transferred to the reaction flask.
This reaction was carried out as described in general
procedure. Column chromatography gave unreacted CpRe(C0)3

30
(2.9 g, 48 % recovery), diag-CpRe(CO)2Br2 (2.8 g, 33 %
yield, 63 % conversion), and lat-CpRe(CO)2Br2 (0.9 g, 11 %
yield, 21 % conversion).
For diag-CpRe(CO)2Br2 : *H-NMR (CDClj) ; -C5H5 (5.75 s, 5H) .
13C-NMR (CDCI3) ; -£5H5 (93.9, 5C) , -£0 (182.6, 2C) .
IR (CH2C12) y 2067 (m), 2003 (s) cm"1.
For lat-CpRe(CO)2Br2 : H-NMR (CDCI3) ; -CgHg (6.15 s, 5H) .
13C-NMR (CDCI3) ; -£5H5 (94.6, 5C) , -£0 (196.4, 2C) .
IR (CH2C12) ; 2054 (s), 1983 (m) cm"1.
b. MeCpRe(CO)2Br2
MeCpRe(CO)3 (5.9 g, 16.8 mmole) and CgHgNHBr3 (5.6 g,
17.5 nunole) were placed into a 250 mL of round bottom flask,
and 50 mL of CF3C02H was transferred to the reaction flask.
This reaction was carried out as described in general
procedure. Column chromatography gave unreacted MeCpRe(CO)3
(2.6 g, 44 % recovery), diag-MeCpRe(CO)2Br2 (2.7 g, 33 %
yield, 60 % conversion), and lat-MeCpRe(CO)2Br2 (1.0 g, 12 %
yield, 22 % conversion).
For diag-MeCpRe(CO)2Br2 : -NMR (CDCI3) ; -CH3 (2.3 s, 3H),
-C5MeU4 (5.62 and 5.47 dd, each 2H). 13C-NMR (CDCI3) ;
-£H3 (13.2), -£-Me (109.3), -CH (94.5, 2C), -CH (93.4, 2C) ,
-£0 (183.6, 2C).

31
IR (CH2C12) ? 2063 (m), 2046 (m), 1998 (s) cm"1.
For lat-HeCpRe(CO)2Br2 : lH-NMR (CDClj) ; -CH3 (2.15 s, 3H),
-CgMeJfy (5.88 and 5.78 dd, each 2H). 13C-NMR (CDC13) ; -£H3
(14.1), -£-Me (111), -£H (97.9, 4C). IR (CHgClg) ; 2050
(s), 1978 (m) cm"1.
c. Cp*Re(CO)2Br2
Cp*Re(CO)3 (9.7 g, 24.0 mmole) and CgHgNHBr3 (8.0 g,
25.0 mmole) were placed into a 250 mL of round bottom flask,
and 70 mL of CF3C02H was transferred to the reaction flask.
This reaction was carried out as described in general
procedure. Column chromatography gave unreacted Cp*Re(C0)3
(5.0 g, 51 % recovery), diag-Cp*Re(CO)2Br2 (0.5 g, 4 %
yield, 8 % conversion), and lat-Cp*Re(CO)2Br2 (4.7 g, 37 %
yield, 75 % conversion).
For diag-Cp*Re(CO)2Br2 : -NMR (CDC13) ; -CE3 (1.98 s,
15H). 13C-NMR (CDC13) ; -£-Me (105, 5C), -£H3 (10.4, 5C) , -
CO (186.3, 2C) . IR (CH2C12) ; 2050 (m), 1980 (s) cm"1.
For lat-Cp*Re(CO)2Br2 : -NMR (CDC13) ? -CH3 (2.03 s, 15H).
13C-NMR (CDC13) ; -C-Me (106.8, 5C), -£H3 (10.4, 5C) , -CO
(201.5, 2C). IR (CH2C12) ; 2034 (s), 1959 (m) cm"1.

32
d. (d5-Cp)Re(CO)2Br2
(d5-Cp)Re(CO)3 (0.75 g, 2.2 mmole) and CgHgNHBrj (0.77
g, 2.4 mmole) were placed into a 100 mL of round bottom
flask, and 10 mL of CF3C02H was transferred to the reaction
flask. This reaction was carried out as described in
general procedure. Column chromatography gave unreacted
(dg-Cp)Re(CO)j (0.35 g, 47 % recovery),
diag-(d5-Cp)Re(CO)2Br2 (0.34 g, 32 % yield, 61 %
conversion), and lat-(d5-Cp)Re(CO)2Br2 (0.12 g, 11 % yield,
21 % conversion).
For diag-(d5-Cp)Re(CO)2Br2 : 13C-NMR (CDCI3) ; -£5Dg (182.5,
5C). IR (CH2C12) ; 2067 (m), 2003 (vs) cm-1.
For lat-(d5-Cp)Re(CO)2Br2 : 13C-NMR (CDCI3) ; -£5d5 (195.4,
5C). IR (CH2C12) ; 2054 (vs), 1983 (s) cm"1.
4. diag-[ (r?4-C5Hg)Re(CO)2Br2] [Li]
The thermally unstable ring-attacked anionic complex,
diag-[ (rj —<CgHg)Re(CO)2Br2] [Li], was synthesized from the
treatment of diag-CpRe(CO)2Br2 (46.7 mg, 0.1 mmole) with a
stoichiometric amount of 1.0 M LiEt3BH(D) (0.11 mL, 0.11
mmole) in 20 mL of THF at -78 ®C. The product solution
exhibited a yellow color.
1 H-NMR (dg-THF, -70 °C) : -CH2 (2.90 s, 2H), -CH (6.43 s,

33
2H), -CH (6.53 s, 2H).
13C-NMR (dg-THF, -70 °C) : -£H2 (42.6), -£H (134.3 S, 2C) ,
-£H (133.1 s, 2C), -£0 (208, 2C).
IR (THF, -70 °C) : 1884 (s), 1758 (m) cm"1.
5. [CpRe(CO)2Br][Li]
The thermally stable product of metal-halogen exchange,
[CpRe(CO)2®r][Li], was synthesized by treating
diag-CpRe(CO)2Br2 (46.7 mg, 0.1 mmole) with a stoichiometric
amount (l.o eq.) of RLi (0.11 mmole, e.g. R = Me, Ph, t-Bu)
in 20 mL of THF at -78 °C. The product solution exhibited a
yellow color.
^H-NMR (dg-THF) : -C5H5 (4.80 s, 5H).
13C-NMR (dg-THF) : -£5H5 (87.2 S, 5C) , -£0 (214.3 s, 2C) .
IR (THF) : 1882 (s), 1806 (s) cm"1.
6. [Cp*Re(C0)2Br][Li]
The thermally stable product of metal-halogen exchange,
[Cp*Re(CO)2Br][Li], was synthesized by treating
lat-Cp*Re(CO)2Br2 (53.7 mg, 0.1 mmole) with two equivalents
of LiEtjBH or one equivalent of RLi (R = Me, Ph, sec-Bu,
t-Bu) in 20 mL of THF at -78 °C. The product solution
exhibited a yellow color.

34
IR (THF) : 1862 (s), 1788 (m) cm'1.
7. [CpRe(CO)2BrJ[M] (M - Li(15-Crown-5), PPP)
diag-CpRe(CO)2Br2 (186.8 mg, 0.40 mmole) was dissolved
in 40 mL of THF( then 1.7 M t—BuLi (0.24 mL, 0.41 mmole) was
added to the reaction flask at -78 °c. After the reaction
mixture was allowed to warm to room temperature, a methanol
solution (1.5 mL) of PPP-C1 (165 mg, 0.44 mmole) or a 0.46 M
15-Crown-5/THF (3 mL) was added to the reaction flask,
followed by a layer of degassed n-heptane (10 mL). The
analytical sample and yellow crystals suitable for X-ray
diffraction analysis were obtained after l day at room
temperature. After the solution was decanted, the crystals
were dried under vacuum.
For [CpRe(CO)2Br][PPP], yield : 225 mg (78 %).
Anal, for CjjHgjBrOgPRe, found (calcd); C 51.00 (51.24); H
3.50 (3.44).
For [CpRe(C0)2Br][Li(15-Crown-5)], yield : 199 mg (81 %) .
8. [MeCpRe(CO)2Br][PPP]
diag-MeCpRe(CO)2Br2 (192.4 mg, 0.40 mmole) was
dissolved in 20 mL of THF, then 1.7 M t-BuLi (0.24 mL, 0.41
mmole) was added to the reaction flask at -78 °C. After the
reaction mixture was allowed to warm to room temperature, a

35
methanol solution (1.5 mL) of PPP-ci (165 mg, 0.44 mmole)
was added to the reaction flask. Degassed n-heptane (10 mL)
was gently layered on the THF reaction solution. Orange
crystals were formed immediately at the solution interface.
After solvent decantation, the crystals were collected and
dried under vacuum. Yield : 251 mg (85 %).
Anal, for C^I^BrC^PRe, found (calcd) ; c 51.27 (51.88); H
3.70 (3.65).
13C-NMR (CD2C12) : -£H3 (14.2, 1C) , -CH (79.4, 2C) , -CH
(79.0, 2C), quaternary (118.3 and 117.1 d, J_ - 90 Hz, p-c
1C), ortho and meta (130.9 and 134.7, 2C each), para (135.9,
1C), -£0 (209, 2C).
9. [Cp*Re(CO)2Br][PPP]
lat-Cp*Re(CO)2^r2 (200 mg, 0.37 mmole) was dissolved in
40 mL of THF, then 1.7 M t-BuLi (0.23 mL, 0.39 mmole) was
added to the reaction flask at -78 °C. After the reaction
mixture was allowed to warm to room temperature, a methanol
solution (2 mL) of PPP-C1 (153 mg, 0.40 mmole) was added to
the reaction flask. The solution was concentrated to 10 mL,
filtered under argon using a fine porosity frit, and then
degassed n-heptane (10 mL) was gently placed on top of the
reaction solution. The analytical sample and orange yellow
crystals suitable for X-ray diffraction analysis were

36
obtained after 2 days at room temperature. After the
solvent was decanted, the crystals were collected and dried
under vacuum. Yield : 254 mg (86 %).
Anal, for CjgHjjBrOgPRe, found (calcd); C 54.32 (54.27); H
4.43 (4.40).
IR (KBr) : 1850, 1773 cm"1, IR (CHgClg) : 1860, 1781 cm"1.
1h"NMR (CD2C12) : -CH3 (1.94 s, 15H), -CgHg (7.6-7.95 m,
20H). 13C-NMR (CD2C12) : -£H3 (11.0, 5C) , -£-CH3 (93.8,
5C), quaternary (117.3 and 118.5 d, Jp.c - 90 Hz, 1C), ortho
and meta (131.0 and 134.8, 2C each), para (136.1, 1C),
-S.0 (213.1, 2C).
10. Reaction of Cp'Re(CO)2Br2 with One Electron
Reducing Agents (Cp1 « Cp, Cp*)
In a typical experiment, a THF solution (20 mL) of
Cp'Re(CO)2Br2 (0.05 mmole, either isomer) was treated with
0.5 M sodium naphthalide (0.2 mL) or 0.25 M cobaltocene
(0.40 mL) at -78 °C. Red-brown solutions of Cp'Re(CO)2Br2
(either isomer) react instantaneously with added sodium
naphthalide to give a yellow solution containing a mixture
of carbonyl oxygen-sodium contact ions and solvent-separated
ion pairs, while cobaltocene gives only solvent-separated
ions.

37
IR (THF, -78 °C) : For [CpRe(CO)2Br][Na] (CIP) ; 1882, 1793
cm"1. [CpRe(CO)2Br]" (SSIP) ; 1882, 1808 cm'1.
For [Cp*Re(CO)2Br][Na] (CIP) ; 1863, 1776 cm"1.
[Cp*Re(CO)2Br]" (SSIP) ; 1863, 1791 cm"1.
11. Reaction of Cp'Re(CO)2Br2 with Grignard Reagents
(Cp» - Cp, Cp*)
A THF solution (20 mL) of Cp'Re(CO)2Br2 (0.05 mmole,
either isomer) was treated with 1.5 M MeMgBr (0.04 mL) or
2.0 M t-BuMgCl (0.03 mL) at -78 °C. Red-brown solutions of
Cp'Re(CO)2Br2 (either isomer) react sluggishly at -78 °C but
react instantaneously with the added Grignard reagents at
room temperature to give a yellow solution containing only
carbonyl oxygen-magnesium contact ions, [Cp'Re(CO)2Br][MgX].
IR (THF, -78 °C) : For [CpRe(CO)2Br][MgX] ; 1890, 1757 cm"1.
For [Cp*Re(CO)2Br][MgX] / 1872, 1734 cm"1.
12. C6H5CpRe(CO)3
lat-CpRe(CO) 2Br2 (467 mg, 1.0 mmole) was placed into a
Schlenk flask, then 40 mL of distilled THF was cannulated
into the Schlenk flask. This reaction mixture was next
cooled to —78 ®C. 1.0 M PhLi (1.1 mL, 1.1 mmole) was added
to the Schlenk flask using a syringe. After stirring the
reaction mixture for 20 min, the vessel was allowed to warm

38
to room temperature. The solvent was removed under
aspirator suction and the residue was dissolved in minimum
amount of CJ^C^. The crude product was column
chromatographed over silica gel. Successive elution with
petroleum ether/CHgClg (4:1 v/v) gave PhCpRe (CO) 3 (123 mg,
30 % yield) which was characterized spectroscopically.
^H-NMR (CDCI3) : -C6H5 (7.40 m, 5H) , -CH (5.43 m, 2H) , -CH
(5.79 m, 2H).
13
C-NMR (CDClj) : meta (126.6, 2C), ortho (129.0, 2C), para
(128.8, 1C), quaternary (132.0, 1C), -£H (82.1, 2C), -£H
(84.5, 2C), -£-Ph (108.8, 1C). IR (Cyclohexane) : 2028 (m), 1939 (s) cm'1.
13. General Procedure for the Preparations of Cyclo-
pentadienyl Rhenium Dicarbonyl Alkyl Bromide
Derivatives, CpRe(CO)2(R)Br (R = H, CH3, C2H5)
To a THF solution (30 mL) of the [CpRefCO^Br] [Li]
anion freshly prepared from the reaction of
diag-CpRe(CO)2Br2 (46.7 mg, 0.1 mmole) and a stoichiometric
amount (1.0 eg.) of t—BuLi was added trifluoroacetic acid,
methyl triflate (CF3SO3CH3), or magic ethyl (FSO3C2H1J) at
-78 °C, respectively. This reaction was monitored in the
carbonyl region by IR spectroscopy.

39
a. diag-CpRe(CO)2(H)Br.
The desired hydridobromide complex was immediately
formed when trifluoroacetic acid (0.10 mL, 1.25 mmole) was
added to the THF solution of [CpRe(CO)2Br][Li] anion (0.10
mmole) at -78 °c. The resulting hydridobromide complex was
shown to possess diagonal stereochemistry based on IR
spectral measurements.
IR (THF, -78 °C) : 2032 (s), 1962 (s) cm'1.
]H-NMR (dg-THF, -78 °C) : -CgHg (6.06 S, 5H) , -Re-fi (-9.03
S , 1H).
b. diag-CpRe(CO)2(CH3)Br
Methyl triflate (0.02 mL, 0.20 mmole) reacted
immediately with the [CpRe(CO)2Br][Li] anion (0.10 mmole) in
THF solution at -78 °C to afford the desired methylbromide
complex with diagonal stereochemistry.
IR (THF, -78 °C) : 2028 (m), 1954 (s) cm"1.
c. diag-CpRe(CO)2(C2H5)Br
Magic ethyl (0.02 mL, 0.20 mmole) reacted immediately
with the [CpRe(CO)2Br][Li] anion (0.10 mmole) in THF
solution at —78 to afford the desired ethylbromide
complex with diagonal stereochemistry.

40
IR (THF, -78 °C) : 2032 (m), 1959 (s) cm"1.
d. diag-Cp*Re(CO)2(H)Br
To a THF solution (30 mL) of [Cp*Re(C0)2Br][Li] anion
prepared from the reaction of lat-Cp*Re(CO)2Br2 (53.7 mg,
0.1 mmole) and a stoichiometric amount (1.0 eq.) of t-BuLi
was added trifluoroacetic acid (0.10 mL, 1.25 mmole) at
-78 °C. The desired hydridobromide complex was formed
immediately and was shown to possess diagonal stereo-
chemistry by IR measurements.
IR (THF, -78 °C) : 2018 (s), 1945 (vs) cm"1.
^H-NMR (dg-THF) : -CH3 (2.01 s, 15H), -Re-fl (-9.98 s, 1H) .
e. diag-Cp*Re(CO)2(CH3)Br
To a CH2C12 solution (10 mL) of [Cp*Re(C0)2Br][PPP] (40
mg, 0.05 mmole) was added 1.0 M methyl triflate/CH2d2 (0.06
mL, 0.06 mmole) at -78 ®C. The desired methylbromide
complex was formed immediately and was shown to possess
diagonal stereochemistry based on IR measurements.
IR (CH2C12, -78 °C) : 2023 (m), 1942 (s) cm"1.

41
f. diag-Cp*Re(CO)2(C2H5)Br
To a CH2C12 solution (10 mL) of [Cp*Re(C0)2Br][PPP] (40
mg, 0.05 mmole) was added magic ethyl (0.006 mL, 0.06 mmole)
at -78 °C. The desired ethylbromide complex was formed
immediately and was shown to possess diagonal
stereochemistry based on IR measurements.
IR (CH2C12, -78 °C) : 2020 (m), 1944 (s) cm"*.
14. [C9H7Re(CO)2H][Li]
To a THF solution (25 mL) of CgH7(CO)3 (100 mg, 0.26
mmole) was added two equivalents of LiEtjBH at room
temperature. The resulting thermally stable hydrido rhenium
anion, [CgHyRe(CO) 2H] , was characterized in situ
spectroscopically.
IR (THF) : 1992, 1906 cm"1.
JH-NMR (dg-THF) : -Re-H (-5.5 s, 1H), -Ha (6.56 t, J=3.3 Hz,
1H), -Hb (5.64 d, J—3.2 HZ, 2H), -J^ (6.42 dd, J=6.0 Hz,
2H), (7.22 dd, J= 5.9 Hz, 2H) .
13C-NMR (dg-THF) : -£0 (200, 2C), -£j (91), -£2 (71, 2C),
-£3 (HO, 2C) , -£4 (120, 2C) , (114, 2C) .

42
15. diag-Cp»Re(CO)2H2 (Cp' = Cp, Cp*)
To a benzene solution (40 mL) of Cp'Re(CO)2Br2 (2.14
mmole, either isomer) was added BU3S11H (1.20 mL, 4.40 mmole)
at room temperature. The reaction mixture was stirred
overnight at room temperature, with monitoring by IR. The
solvent was then removed under vacuum and the crude oily
product was flash column chromatographed over silica gel
under nitrogen. After the filtrate was dried under vacuum,
the pure product was crystalized in pentane at -78 °c using
a dry ice/acetone bath.
For diag-CpRe(CO)2H2 : IR (Cyclohexane) : 2022 (m), 1953 (s)
cm"1. ^-NMR (CgDg) : -Re-H (-9.68 S/ 2H) , -C^g (4.35 d,
5H) • For diag-Cp*Re(CO)2H2 : IR (Cyclohexane) : 2026 (m),
1936 (s) cm"1. !H-NMR (CgDg) : -Re-H (-9.25 s, 2H), -CH3
(1.81 s, 15H).
16. Cp'Re(CO)2(SnBU3)2 ^Cp' ™ C p'
To a benzene solution (40 mL) of Cp'Re(CO)2Br2 (2.2
mmole) was added BujSnH (1.2 mL, 4.5 mmole) at room
temperature. After the reaction mixture was stirred for
several hours with monitoring by IR, 0.7 mL of EtjN (5.0
mmole) was added to the reaction flask and stirring
continued for one additional hour. The solvent was then

43
removed under vacuum, and the crude oily product was flash
column chromatographed over silica gel with petroleum ether
under nitrogen. After the filtrate was evaporated under
vacuumm, the volatile compounds were sublimed from the
desired tin complex at 90-100 °C. The pure product was
obtained from the above sublimation residue using flash
column chromatography over silica gel with petroleum ether
under nitrogen.
For diag-CpRe(CO)2(SnBU3)2 : IR (Cyclohexane) : 1943 (m),
1890 (vs) cm"1. 119Sn-NMR (CgDg) ; -5.6 ppm.
For lat-Cp*Re(CO)2(SnBu3)2 : IR (Cyclohexane) : 1967 (vs),
1896 (s) cm"1. 119Sn-NMR (CgDg) : 8.8 ppm.
17. CpRe(CO)2L (L - PPh3, P(0Ph)3, (CH3)3CNC)
To a THF solution (30 mL) of diag-[ (rj*-C,jHg)Re(CO)2Br2]"
prepared from the reaction of diag-CpRe(CO)gBrg (93.4 mg,
0.20 mmole) and 1.0 M LiEtjBH (0.24 mL, 0.24 mmole) was
added L [0.22 mmole, i.e., L = PPh3, P(OPh)3, Me3CNC] at -78
°C. The yellow solution of diag-[ (^-CgHg)Re(CO) 2Br2]" anion
reacted instantaneously with added L to give CpRefCO^L at
-78 °C as determined by low-temperature FT-IR measurements.
IR (THF) : For CpRe(CO)2PPh3 : 1926 (s), 1844 (s) cm"1.

44
For CpRe(CO)2P(OPh)3 : 1961 (s), 1893 (s) cm"*.
For CpRe(CO)2(Me3CNC) : 1902 (s), 1850 (s) cm"1.
18. BrRe(CO)g
Re2(C0)jQ (7.0 g, 10.7 mmole) was placed into a 500 mL
of round bottom Schlenk flask equipped with a magnetic stir
bar. Freshly distilled pentane (300 mL) was transferred
into the reaction flask and bromine (0.7 mL) was then added
dropwise to the solution using a dropping funnel under a
stream of argon. A white precipitate was observed
immediately upon stirring at room temperature. The reaction
mixture was stirred at room temperature overnight and then
monitored by IR. Excess bromine was eliminated from the
reaction vessel by stirring open in the hood until all of
the orange bromine color disappeared. The solvent was
removed under vacuum, and the white powder transferred to a
sublimator and sublimed at 85-110 °C under vacuum. Yield :
8.2 g (94 %).
IR (Cyclohexane) : 2044 (vs), 1984 (m) cm"1.
19. (d5-Cp)Tl
Freshly distilled cyclopentadiene (1.2 mL, 4.9 mmole)
was added to a Schlenk flask containing a sodium deuteroxide
solution, prepared from Na metal (3.7 g, 0.16 mole) and D£0

45
(40 mL), and the reaction flask was stirred magnetically at
0 °C for 5 days in the cold room. Thallium (I) sulfate (3.0
g/ 0.01 mole) was then added to the reaction flask and
stirring was continued for 3 additional days. The yellow
precipitate was filtered, washed with 10 mL of DgO, and
dried under vacuum for 2 hr. The solid was transferred to a
water-cooled sublimation apparatus and plug of glass wool
was placed on top of the crude product. The crude (dg-Cp)Tl
was sublimed at 100-110 °C under vacuum to give a yellow
solid. The yield was 1.97 g (72 %).
20. (dg-Cp)Re(C0)3
(dg-Cp)Tl (1.74 g, 6.35 mmole) and BrRe(CO)5 (2.34 g,
5.76 mmole) were dissolved in 250 mL of Schlenk round bottom
flask containing 50 mL of benzene. The reaction mixture was
magnetically stirred at 60-70 °C for 1 day with monitoring
by IR. The benzene solution was flash column
chromatographed over silica gel to give the desired product.
The yield was 1.67 g (71 %). IR (Benzene) : 2023 (s), 1927
(vs) cm"*.

46
E. Thermodynamic Measurements
The Ion Pairing Equilibrium in 2-Na, SSIP vs. CIP
2.5 x 10"3 M THF solutions of the [Cp*Re(CO)gBr] [Na]
anion were prepared from the reaction of lat-Cp*Re(CO)2Br2
(26.85 mg, 0.05 mmole) and 0.5 M sodium naphthalide (0.24
mL, 0.12 mmole) in THF (20 mL) at -78 °C. The measurement
of the amount of contact ion pair (CIP) and solvent-
separated ion pairs (SSIP) in solution over the temperature
range of -70 °C to room temperature was assessed by FT-IR
spectroscopy. From the area associated with each asymmetric
carbonyl stretching band, the equilibrium constants were
easily determined as defined by Eq. 12.
Keq
[Cp*Re (CO) 2®r] [Na] * [Cp*Re(CO)2Br]"//Na+
CIP SSIP (12)
[8SIP] Keq s
[CIP]
The thermodynamic parameters were obtained by plotting
In Keq + 1 vs. 1/T from which the slope and intercept
afforded AH and AS, respectively. The error limits
associated with the enthalpy and entropy for ion-pairing

47
were calculated by using the available least-squares
regression program®® and should not be taken to reflect
uncertainties in sample preparation, band area intensities,
or temperature control, but rather the deviation of the data
points about the least-squares line.
F. Kinetic Studies
1. Reaction of diag-CpRe(CO)2^2 with Et3SiH
diag-CpRe(CO)2Br2 (46.7 mg, 0.1 mmole) was placed in a
30 mL Schlenk flask containing 10 mL of toluene. This
toluene solution was shaken vigorously for a few minute
prior to placement in a thermostatted bath set at 40.0 °C.
The reaction was monitored over a three day period by
following the decrease in absorbance of the reactant's
intense of asymmetric stretching CO band (2003 cm"1). The
extent of the reaction between diag-CpRe(CO)2**2 a n d Et3siH
was measured from the above data.
2. Isomerization of lat-CpRe(CO)2^*2 t o
diag-CpRefCO^B^ in the Presence of EtjSiH
lat-CpRe(CO)2Br2 (5 mg, 0.011 mmole) was placed in a 30
mL Schlenk flask containing 5 mL of toluene. This toluene
solution was shaken vigorously minute prior to placement in

48
a thermostatted bath set at 40.0 °C. A 10-fold excess of
EtgSiH (0.16 mL, 1.0 mmole) then was added to the reaction
flask at 40.0 °C. This reaction was monitored over an eight
hour period by following the decrease in absorbance of the
reactant's intense of symmetric stretching CO band (2046
cm *). The extent of the isomerization to diag-CpRe(CO)2Br2
was qualitatively measured from the above data and was shown
to be faster than the rate of bromide/hydride exchange (vide
infra).

49
CHAPTER III
RESULTS AND DISCUSSION
A. Aryl Rhenium Tricarbonyl Derivatives, Cp *Re(CO)^
(Cp' = Cp, MeCp, Cp*, cgH7)
The aryl rhenium tricarbonyl derivatives were prepared
by a modification of the procedure reported previously^"
Dirhenium decacarbonyl, Re2(CO)10, suspended in the
appropriate cyclopentadiene dimer or indene was heated up to
210 C in the Fisher—Porter tube equipped with a pressure
gauge. This method gives excellent yields of the aryl
rhenium tricarbonyl derivatives that are used for the
preparation of the necessary aryl rhenium dicarbonyl
dibromide derivatives. Equation 13 outlines the general
procedure for the synthesis of CpRe(CO)3. The aryl rhenium
Re2(CO)10 + A » 2 CpRe(CO)3 + Hg + 4 CO (13)
tricarbonyl derivatives were characterized by IR and NMR
spectroscopy. These data are summarized in Tables 1-2. The
representative IR spectra of the aryl rhenium tricarbonyl

50
- Q
i H O
• H
<#> r ^ oo
a\ 00 <n
cn oo
> i C O
fl
<d o
• H
• H G a)
« g
I a
t c ° 8
X m
O
a) c «d X a>
£ o H O &
c • H
§ 8
CO <«
CO
«d H 3
o
co CO w N—» CO i n CN H
• • CN (N
• d co - P
<w w o CO i n 00 cn CN \ o r * • • • •
i n i n i n m
CO CO CO CO 10 <w V** w * w . w cn i n CM CO o n CO CN n
o \ as as o\ H H H H H
% * * %»
*>*%
S s s CO S I M ' >—<•
o CO 00 CO CN H CN O o O o CN CN <N CN
OO
O a a) #
&
OO
O a a) «
fi-<D s
CO
O O
0) tf *
&
OO
O O
5 «
HJ c n
O
J T u Q
a
+> G 0) >
>1 a o
a (d o (d o a)
T *
• H c 0)
•8
0) 43 •M
a o
i * 0 (0
o <d C0 CQ
ro - Q

5 1
G O
•B <d o
•H u
•H c 0)
CM
CQ
a
i ft
c •H U
V 0)
0)
! 6 I X
•H Cft
C •H U
*d (I)
<1)
! s i 0) >
•H J*
(d rH P
o o
CO X o
i n O
O
CO O
CM a
u
o (x»
OH VO
OS H
VO
CO
CM •
CO CO
CO CO as as H H
o
CO
O
a
«
&
V0 •
VO O H
CO
O a
a) «
& a) S3
in
co CM H
VO CM
CO o H
CO • • • H
<?* CO CO CO CO OH
CO
O O
a) « # a -
H OH
CO
O O w 0) « t « cr»
O
C (d
(0 (d
*d a) w 3
w <d *
CO
o
4J 0
rH 04
•H
-M
> i 4-> # H A w 8 G g« <D Mi
•P _
g °
& r *
a>
g 0) o c 0)
8 8 u
H •p (0 c c a) M > a)
h - p 0 5 CO -H flj

52
derivatives are shown in Figure 2. The FT-IR spectra of
cyclopentadienylrhenium tricarbonyl derivatives exhibit two
carbonyl stretching bands (Aj + E) expected for C3y
66
symmetry. The symmetric CO stretching mode (Aj) of
cyclopentadienylrhenium derivatives appears in the range of
2030-2013 cm * while the asymmetric CO stretching mode (E)
appears in the range of 1939-1922 cm"1. Successive
substitution of methyl groups on the cyclopentadienyl ring
in the series of CpRe(CO)3, MeCpRe(C0)3, and Cp*Re(CO)3
leads to decreasingly lower CO stretching frequencies as the
cyclopentadienyl ring moiety donates more electron density
to the rhenium metal center in the order of Cp*Re(C0)3 >
MeCpRe(C0)3 > CpRe(C0)3.
On the other hand, the IR spectrum of the r?5-indenyl
derivative, CgH7Re(CO)3 (Fig. 2b) exhibits a splitting of
the E mode owing to sufficient asymmetry of the r^-indenyl-
rhenium bond as a result of deviation from idealized c3y
67 68 1
symmetry. * The H NMR spectrum of the yellow—orange
product CgHyRe(CO)3 exhibits four sets of resonances with a
relative integral ratio of 2:2:2:1 (Fig. 3). The two of
these resonances of relative intensities 2 are observed at
7.10 and 7.50 ppm with the AA'BB' spin system which are
consistent with four protons on an uncomplexed six-membered
benzenoid ring. The other two sets of resonances are

53
2000 — i 1900
v, cm i
Figure 2. Infrared spectra of the carbonyl region for
(a) CgHyRe(CO)^ and (b) CpRe (CO) . Both spectra were recorded at 25° C in cyclohexane.
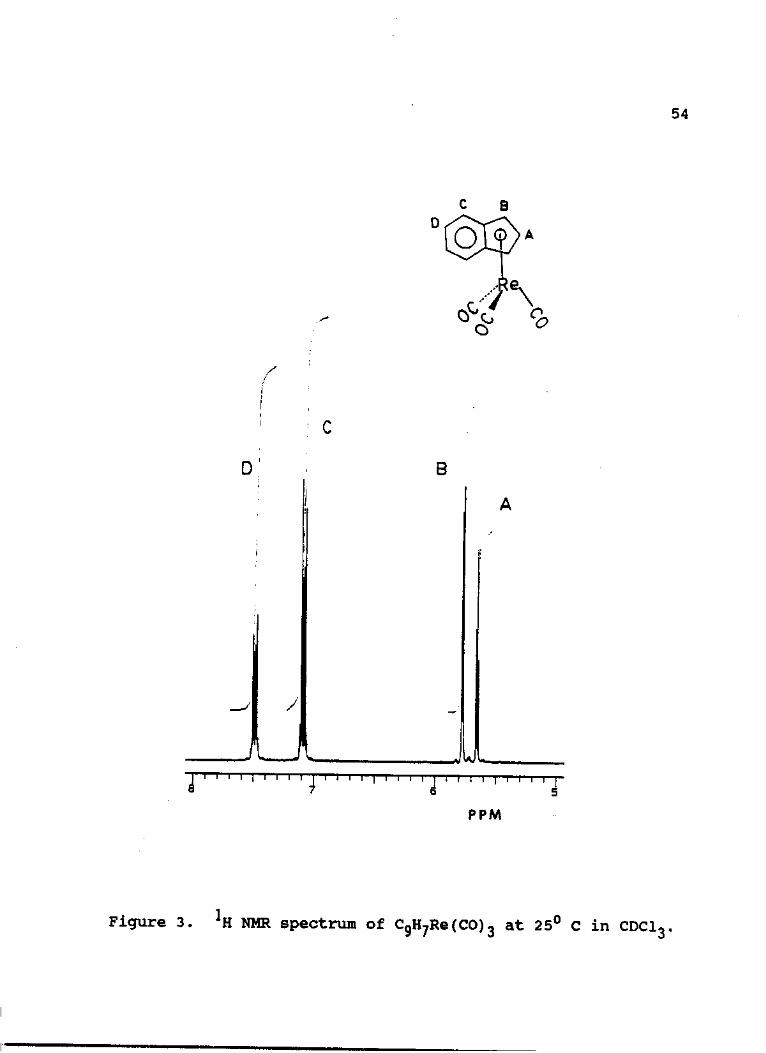
54
C B
Q > A
' T \ c K l C,
D
/
A
B
1 1 1 1 I 1 ' ' I j I ' I I | I I I I | I I I I | I I | |
PPM
| i i i i | i i i r | 6 i 6 5
Figure 3. *H NMR spectrum of CgH7Re(CO)3 at 25° C in CDC13.

55
observed at 5.78 and 5.65 ppm with a relative integral
ratio of 2:1 which is correspond to the three protons
attached to the five-membered ring in the rj®-indenyl ligand.
B. Cyclopentadienyl Rhenium Dicarbonyl Dibromide
Derivatives, Cp'Re(CO)2&r2 = MeCP' CP*)
Synthetic routes to the cyclopentadienylrhenium
dicarbonyl dihalide compounds, CpRe(CO)2X2 (where X = Br or
28-31
I), have been known for several years. However,
synthetic pathways to the corresponding pentamethy1eye1o-
pentadienylrhenium dicarbonyl dihalides, Cp*Re(CO)( w h e r e
Cp* » rj®-CgMeg), have only recently been described in
detail.«.«"«
The cyclopentadienylrhenium dicarbonyl dibromide
derivatives were prepared by a modification of the route
29
described by King and Reimann. The appropriate rhenium
tricarbonyl and a slight molar excess of pyridinium
hydrobromide perbromide was treated with TFA (Eq. 14). This yielded the crude isomeric dibromides which were
TFA diag-Cp'Re(CO)2Br2 Cp*Re(CO)3 + CgHeNHBr, + (14)
rt lat-Cp'Re(CO)
(Cp1 = Cp, MeCp, Cp*)

56
subsequently isolated by column chromatography over
silica gel using mixture of petroleum ether and CH^Clg.
Successive elution with petroleum ether/CHgClg (1:1 v/v)
gave first unreacted rhenium tricarbonyl as a colorless
band, then diagonal dibromides as a red band, and finally
lateral dibromides as a brown band was isolated by using
CHgClg. An increased amount of TFA led to an overall
increase in the yield of the isomeric dibromides, but use of
Br2 directly and/or longer reaction times led to an overall
decrease in the yield of the isomeric dibromides. The aryl
rhenium dicarbonyl dibromide derivatives were characterized
by IR and NMR spectroscopy. The spectral properties are
consistent with those reported by King et al.^® and Sutton
32
et al. These data are summarized in Tables 3-4. The
four-legged piano-stool complexes of the form CpMX2Y2 may
exist as two nonequivalent stereoisomers that are commonly
referred to as cis and trans isomers. We have adopted
King's nomenclature to describe the isomeric dibromides
discussed in this study. Here the descriptors lateral and
diagonal correspond to the cis and trans stereoisomers,
respectively. The diagonal designation is more acceptable
than the trans designation because no two ligands are
situated at 180° angles relative to each as would be
expected for trans isomers. The diagonal (A) and lateral

57
C o •a <d o
• H Q
Q)
co
A
a
- Q T f rH 0)
• H
i S 4
ro
CM i H U
CM
5 C
•H
o o
<*>
r o
s
£ » LO
0 1
co <
CO
<d H
I o f*4
CO H CO T • vo • as
CO H CO CM vo •
H CO CO
0 O CO CO CO CO a> 0 • • • •
CM CM H CM
•h y—S t0mSl W X cm M
w 04 0
SO <n B « • • i n i n m i n
i n i n r * H • • « X i n VO N CM
w r * CO
• • i n i n
CO n CO S «T 8* <W w w <w
r o CO 00 CO 0 ON O CO as r * CO i n O 0 \ o^ o \ o\ <}\ CM H H H H H
«—*» 8 CO S e CO e w"
w w « w s—' CO vo 0 0
vo i n vo i n i n CO 0 0 0 0 0 0 O CM CM cm CM CM CM CM
CM u co CM
O O "W Q)
a
?
t n <d
•H TJ
CM
PQ CM
O U w a) «
P4
a
<d
CM
CQ CM
O 0
0) tf & 0 £ 1 Cn <d
• H T3
CM
CQ CM
O 0
0) Pd a u Q) s 1
-M <d
rH
CM U m CM
O O
0)
•K a< a » t n (0
CM *4 PQ
CM
0 a a) «
*
8-1
4-> <a
CO rH o Q O
. p C
a) >
> . G O
• 8 <d o
• H
«P
§ fi
>1 C a)
• H * 0 <d
•P C (1) §•
rH O >1 o
a>
3 a o
* d a) CO
o <d CO a txs JQ

53
>i c o A u (d o
s
PQ
6
o o
17* C •H U
•d a) a) i a) a i <D > •H
CO B O
8
8
f o
<d H 3
0
VO VO in CO in • • • • • •
(N VO co vo VO H 00 as 00 as 00 O H H H H H ca
a* • co o\
VO
en
ca
n H
CO 0\
in
CO
0\ o H
CO on
ca
00
o o H
IT) O
00 • vo o
CM u CM
CM m u u CM PQ m CM CM o
o O o w o u s <D a» w a» w 0 a) « & as * Q* a) a< O s
1 u i
tP 1 tr> 1 cn. 1 (d •P <d -P (d •P •H <d •H ed •H (d •O iH 15 H T* rH
c (d
w <d
*d a) CO 3
W <d
oo iH U Q U
o
4J a)
-P
>1 4J "H J W § C Q) «P C •H §
Oi a
r*
-P <d
0) 43 Eh
a) o c a)
_ u CO ® H <W O o> Q M U
-P C 0) > H o CO
as
<d a u <u •p c •H

59
(B) isomers were identified by determination of the angles
69 between their two C-0 bonds through the relationship"*
<X>j
I I S / v - B ^ c C o °
B
tan20 = Ia/I- (where 20 is the angle between the two C-0 a S
bonds, I. is the area under the asymmetric v(CO) band, and
a
Is is the area under the symmetric v(CO) band with the ratio
-3 I./I. being extrapolated to infinite dilution (~ 10 M) . a S
20 m5
Symmetric Asymmetric
The above relationship is derived from the fact that an IR
band intensity is directly proportional to the dipole moment
change that occurs during the vibration. For a CO stretch-
ing vibration, a symmetric vibrational mode appears at
higher frequency than its asymmetric counterpart since more

60
energy is required to stretch two CO bands simultaneously
than to execute one CO stretch and one CO contraction.
Thus, the ratio Ia/Is in diagonal isomers is 2.55-2.73,
corresponding to an angle 29 of 116-118°. Similarly the
ratio Ia/I$ in lateral isomers is 0.71-0.80, corresponding
to an angle 29 of 80-83°. Typical IR spectra of diagonal
and lateral dibromides are shown in Figure 4. The diagonal
isomer possesses higher infrared v(C0) frequencies and a
higher field cyclopentadienyl proton NMR chemical shift than
the corresponding lateral isomers. These two isomers
exhibited differences in (A[i/(C0)]) by about 13 cm"*•for
[J/(CO)sym] and 20 cm"1 for C»/(CO)asym] in CH2C12. This can
be understood in terms of the increased competition for
rhenium d-electrons between the carbonyl groups when they
are mutually trans. They also exhibited cyclopentadienyl
proton NMR resonances differing by about 0.4 ppm. The 13c
NMR chemical shift for the CO groups is smaller by about 15
ppm in the diagonal isomers. The lateral isomers appeared
to be considerably less soluble in organic solvents than the
corresponding diagonal isomers. Both the diagonal and
lateral isomers are stable in the solid state at room
temperature with respect to isomerization. Further study of
isomerization will be discussed (vide infra).

61
OC-;>ReCBr Br CO
B
2200 2000 v, cm
1800
Figure 4. Infrared spectra of the carbonyl region for (a) diag-CpRe(CO)2Br2 and (b) lat-CpRe(CO)2Br2 Both spectra were recorded at 25° C in CHgClg.

62
C. Bromorheniumpentacarbonyl, BrRe(CO)c
The pentacarbonylrhenium halides, XRe(CO)c (X = CI Br D ' '
I), first prepared by Hieber,70'71 are precursors for the
synthesis of many novel rhenium carbonyl compounds. The
bromorheniumpentacarbonyl, BrRe(CO)5, was prepared by the
reported procedure previously.72 Dirhenium decacarbonyl was
dissolved in a 500 mL Schlenk flask containing freshly
distilled pentane (300 mL) and THF solution of bromine was
added dropwise to the reaction mixture under argon
atmosphere. A white precipitate was formed immediately upon
stirring at room temperature. After the excess of bromine
and solvent was removed, the white powder was transferred to
a sublimator and sublimed at 85-100 °C under vacuum to give
a 94 % yield of BrRe(CO)g. The compound was characterized
by IR spectroscopy. The observed medium intense symmetric
stretching band at 2044 cm"* and the very strong, intense
asymmetric stretching band at 1944 cm"* (in cyclohexane)
are in excellent agreement with the literature values.72

63
D. Deuterated Cyclopentadienyl Rhenium Tricarbonyl,
(d5-Cp)Re(CO)3
Synthetic pathways to the deuterated cyclopentadienyl
rhenium tricarbonyl, (dg-Cp)Re(CO)3, have recently been
26
described in detail. Refluxing a benzene solution of
73
(dg-Cp)Tl and BrRe(CO)j overnight gives (dg-CpJRefCOJj
according to Equation 15.
BrRe(CO)5 +
(dg-Cp)Tl
BZ
60 °C (d5-Cp)Ra(CO)3 + BrTl + 2 CO (15)
The resulting white solid was obtained in a 71 % yield. The
isotopically labelled compound was used in the preparation
of (dg-Cp)Re(CO) 2Br2* T h e isotopic purity of (dg-CpJRefCOJj
was assayed by *H NMR spectroscopy using para-methoxy-
benzene as an internal reference. The NMR spectrum,
recorded on a Varian VXR-300 MHz NMR spectrometer, revealed
a very weak, broad peak at 5.5-5.6 ppm and the integral
ratio between the residual protio cyclopentadienyl ligand
and the internal standard indicates that incorporation of
deuterium on the cyclopentadienyl ring is more than 95 %.
This compound was used directly in the preparation of
(dg-Cp)Re(CO)2Br£.

64
E. Deuterated Cyclopentadienyl Rhenium Dicarbonyl
Dibromide, (d5-Cp)Re(CO)2Br2
The deuterated cyclopentadienylrhenium dicarbonyl
dibromide, (d^-Cp)Re(CO)2Br2' w a s prepared from the reaction
(d5-Cp)Re(CO)3 and CgHgNHBr^ in TFA at room temperature.
Column chromatography gave first unreacted (d5-Cp)Re(CO)3 as
a colorless band (47%), diag-(d5-Cp)Re(CO)2Br2 as a red band
(32%), and lat-(dg-Cp)Re(CO)2Br2 as a brown band (11 %).
The isotopic purity of diag-(d5-Cp)Re(CO)2Br2 was assayed by
1h NMR spectroscopy using para-methoxybenzene as an internal
standard. The H NMR spectrum exhibited a very weak singlet
at 5.7 ppm and the integral ratio of proton chemical shifts
indicated that incorporation of deuterium on the
cyclopentadienyl ring is more than 95 %. From this
observation we can conclude that no additional hydrogen is
incorporated into the cyclopentadienyl ring during the
bromination. These deuterated CpRe(CO)2Br2 isomers
exhibited no differences in the infrared CO stretching
frequencies compared to the protio CpRe(CO)2Br2 isomers.

65
F. Hydride Reduction of diag-CpRe(CO)2Br2: Synthesis
and Characterization of the Thermally Unstable
Ring-Attacked Product, diag- [ (r^-CgHg) Re (CO) 2Br2 ]"
The reaction of diag-CpRe(CO)2Br2 with LiEtjBH shows
that cyclopentadienyl ring attack occurs according to
Equation 16 in preference to either direct metathetical
replacement of the bromide moiety or hydride attack at the
carbonyl ligand to generate the corresponding formyl
species. The low-temperature FT-IR spectrum exhibited three
carbonyl stretching bands (Fig. 5a) at 1884 (vs), 1785 (w),
and 1758 (s) cm~* Such a low energy shifting of the CO
H
rH~N LiEljBH \==*J (16)
0-.-Re-..Or THF/-78*" „ J-®r CO Vco
stretching bands of anionic species relative to neutral
compound, diag-CpRe(CO)2Br2, is directly attributed to the
increased charge density at the rhenium metal center which
manifests itself by reducing the CO stretching force
constant for each of the CO ligands.66'74 These three
carbonyl stretching bands are replaced by two new carbonyl
bands upon addition of 5 equivalents of 15-Crown-5 (1,4,7,
10,13-pentaoxacyclopentadecane) as shown in Figure 5b.

66
B
2200 2000 1800 v, cm -i
Figure 5. Infrared Spectra of the carbonyl region for (a) [ (n4-C5H6)Re(CO)2Br2][Li] and (b) [(r?4-C5H6)Re(CO)2Br2] [Li] with 5 equiv. of 15-Crown-5. Both spectra were recorded at -70 °C in THF.

67
This latter spectrum is also obtained when a THF solution of
diag-CpRe(CO)2Br2 containing 5 equivalents of 15-Crown-5 is
treated with LiEt^BH at -78 °C. These results are consis-
tent with the formation of an anionic cyclopentadienyl-
rhenium complex that displays extensive carbonyl oxygen-
lithium ion pairing in the absence of 15-Crown-5.75 > 76
The identity of this new complex was characterized by
low-temperature *H NMR spectroscopy. When a dg-THF solution
of diag-CpRe(CO) gBrg was treated with LiEt^BH at -78 °C
three new hydrogen resonances appear at 6.53, 6.43, and 2.90
ppm with an integral ratio of 2:2:2. On the other hand,
the use of LiEt^BD instead of LiEt^BH led to a decrease in
the intensity of the resonance at 2.90 ppm as shown in
Figure 6; the low-field resonances associated with the
AA'BB' spin system were unaffected yielding an observed
13 1 integral ratio of 2:2:1. A C{ H} NMR spectrum reveals two
low-field resonances at 134.3 and 133.1 ppm along with a
single resonance at 42.6 ppm which suggests the presence of
two types of olefinic carbons and a methylene moiety that
are derived from the Cp ring of diag-CpRe(CO) 2&r2 a s s^ o w n
13 in Figure 7a. A C NMR spectrum using spin-echo
77-79
J-modulation spectroscopy (APT spectrum) reveals two
downward resonances at 134.3 and 133.1 ppm and two upward
resonances at 208.6 and 42.6 ppm. Under these conditions

H
d " "
L 0 C - - R e - - B r B r ^CO
J k
D
d " H
L OC-^Rew--Br Br ^CO
^ V.
68
'' ' I . ' ' ' ' I ' ' ' ' I . ' ' ' I ' ' 1 ' I ' 1 1 1 | 1 1 < i | 1 1 i i | i B - s S.5 9.0 4.S 4.0 3 .5 3 .0 PI
F i g u r e 6 . *H NMR spec t ra o f (a) [ (r^-CgHgJRefCO) ~ and (b) [ (r?4-C5H5D)Re(CO)2Br2]" a t -70°C i n dg-THF.

69
5 mixyjjrn* 5**. p
- O ( 0
- O CD
I — 0 1
A O -o
m a>
&

70
inverted resonances would be observed for methyl and methine
carbons while normal resonances would be observed for
methylene or quaternary carbons. Among these carbon
resonances, the low-field resonance at 208.6 ppm can be
easily assigned to carbonyl carbons as shown in Figure 7b.
Finally, a chemical shift correlation spectrum
(HETCOR) was recorded to provide an accurate proton/carbon
relationship®^-®^ as shown in Figure 8. The *H NMR
resonances at 6.53/ 6.43, and 2.90 ppm were shown to 11
correlate with the 1 C NMR resonances at 133.1, 134.3, and
42.6 ppm, respectively. Based on these results we propose
that the reduction with LiEt^BH proceeds via hydride attack on the cyclopentadienyl ring of diag-CpRe(CO)£Br2 to yield
4 -diag-[ (n -CgHg)Re(CO) 28^] anion.
Additional NMR experiments were conducted in an attempt
. 4 to unequivocally demonstrate that diag-[ (r? -CgHg)Re(CO) gBrg]
anion resulted from hydride attack on the cyclopentadienyl
ring. The easiest experiment would have involved the
observation of NOE between the methylene protons and the
adjacent olefinic protons (H and H^'). Unfortunately, no
NOE was observed in the olefinic protons when the methylene
13
resonance at 2.90 ppm was irradiated. A selective C NOE
experiment was then performed using the resonance at 2.90
ppm to provide information concerning the adjacent olefinic
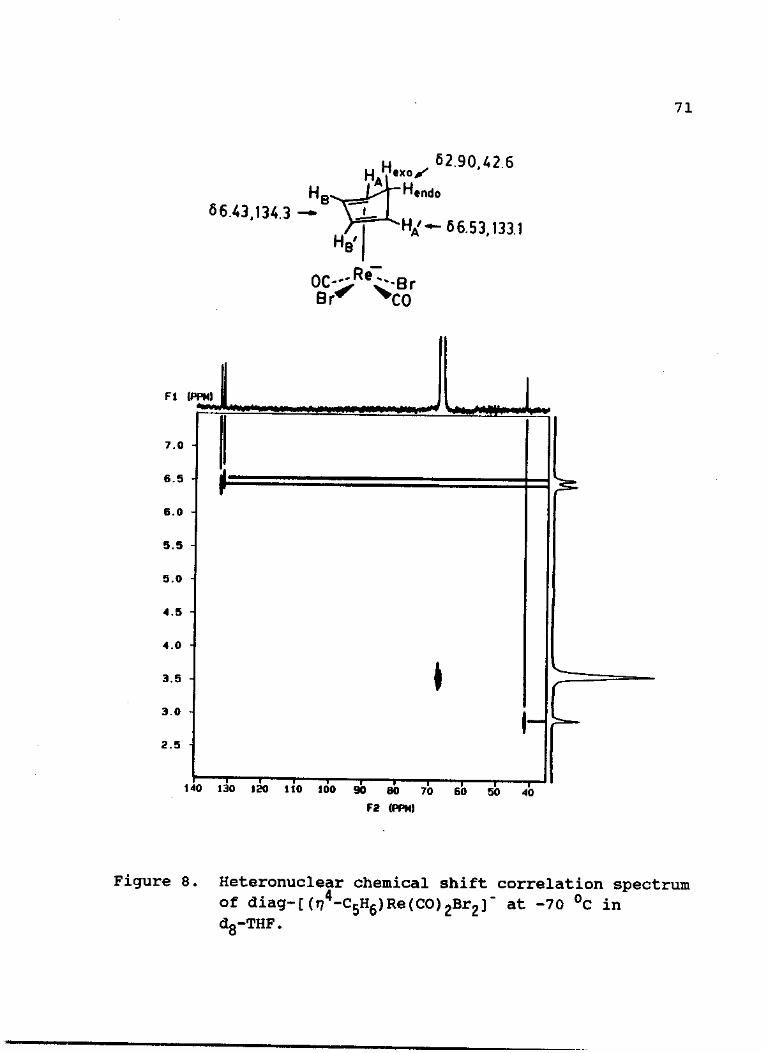
71
66.43,134.3 — \ J -
^•xoy 62.90,42.6
Hendo
Ha '— 66.53,133.1
0C--^Re^Br B r ^CO
Ft (PPM)
5.5 -
5.0 -
4.5
4.0
3.5 H
3.0
2.5 H i -
>40 "l30 120 no loo 90 BO 70 60 50~ 40~
F2 (PPM)
Figure 8. Heteronuclear chemical shift correlation spectrum of diag-[ (r74-c5H6)Re(CO)2Br2]" at -70 °C in dg-THF.

72
carbons. The 13C resonance at 133.1 ppm displayed a 15 %
enhancement in signal intensity relative to the resonance at
134.3 ppm. This indicates that the low-field resonance of
the proton AA'BB' spin system (6.53 ppm) is adjacent to the
methylene moiety. The relationship between the methylene
group and the remaining olefinic carbons (134.3 ppm) was
established by long-range *H-13C chemical shift correlation
spectroscopy using magnetization transfer delays optimized
for three-bond coupling (10 Hz).84 Figure 9 shows the long-
range coupling between the methylene protons and the
olefinic resonance at 134.3 ppm. These data collectively
support the existence of the ring-attacked anionic species,
diag-[ (r^-CgHgJRefCO^B^]". At this point we cannot rule
out polyene fluxionality (i.e., rotation about the rhenium)
in this complex, but we do believe that the initial diagonal
stereochemistry is maintained based on FT-IR spectral
comparison with the ring-attacked product from
lat-CpRe(CO).
The stereochemistry of addition of hydride or deuteride
to metal-bound rings has been established well by Bird and
Churchill in 1967.®® The examination of (r^-CgMegH) Re (CO) 3
obtained by hydride attack on (hexamethylbenzene)rhenium
tricarbonyl cation, does not display a low-frequency (2790
cm *) endo C-H stretch. Thus, the absence of a

73
^B^rsss* 66.43,134.3 —> Y »
H«xo^
OC Br / V
62.90,42.6
Hendo
HA '— 66.53,133.1
Br CO
FI 7.5
7.0
6.5 -
6.0
5.5
5.0
4.5 -
4.0
3.5
3.0 H
2.5
THF
THF
140 130 120 IfO 100 90 00 70 60 50 40
F2 (PPM)
Figure 9. Long-range heteronuclear chemical shift
correlation of diag-[ (J7*-CgHg)Re(CO) 2Br2]" at
-70 °C in dg-THF.

74
low-frequency C-H stretch in IR is taken as physical proof
of an exo hydride attack sequence. Unfortunately, we
couldn't observe this low-frequency C-H stretch by IR
spectroscopy. Alternatively, we have demonstrated the exo
addition of hydride or deuteride to the cyclopentadienyl
ring by isolation and CH NMR examination of the CpRe(CO)3
that decomposed from a reaction using LiEt^BD (vide infra).
G. Reduction of Diagonal and Lateral CpRe(CO)2Br2:
Ring Attack vs. Metal-Halogen Exchange
The reaction of diag-CpRe(CO) 2Br2 in THF at -78 °C with
various RLi reagents (1.1 eq.; R = Me, Ph, t-Bu) affords a
yellow colored solution whose infrared spectrum did not
correspond to that of CpRe(CO)2Br(R), the product of R"/Br"
34
exchange . Furthermore, the IR spectrum was inconsistent
with the presence of a formyl species and the ring-attacked
anionic species, diag-[ (rj4-C5H5R)Re(CO)2Br2]~. The IR
spectrum of the reaction exhibited two carbonyl stretching
bands at 1881 and 1806 cm"* which are consistent with the
[CpRe(CO)2Br]", the product of metal-halogen exchange.
Equation 17 gives the reaction conditions used to prepare
this compound and Figure 10 shows the resulting low-
temperature IR spectrum.

75
OC Re
Br / % CO
1900 1800 v, cm -i
Figure 10. Infrared spectrum of the carbonyl region for [CpRe(CO)2Br]" at -70 °C in THF.

76
THF + RLi . | + RBr + Li+ (17)
>Re^-Br "7» °c 0C->R« Br* ^CO Br ^CO
The identity of this new complex was also ascertained
using NMR spectroscopy. A dg-THF solution of [CpRe(CO)2Br]"
displays a single *H NMR resonance at 4.87 ppm for the
13 1
cyclopentadienyl ring. A C{ H) NMR spectrum reveals two
resonances at 87.2 and 214.3 ppm which correspond to the
cyclopentadienyl carbons and carbonyl groups, respectively.
A THF solution of [CpRe (CO) 2 Br]~ was next treated with
15-Crown-5 and methanol solution of PPP-C1 to give crystals
of [CpRe(CO)2Br][Li/15-Crown-5] and [CpRe(CO)2Br][PPP],
respectively. The latter crystals were characterized by
combustion analysis and the former crystals were employed in
the X-ray structural characterization. Complete details on
the X-ray crystal structure of [CpRefCO^Br] [Li/15-Crown- 5]
will be discussed (vide infra).
Interesting enough, the reaction of lat-CpRe(CO)£Br2 in
THF at -78 °C with various reducing agents (1.1 mole
equivalent; LiEt^BH, RLi; R = Me, Ph) gives both the product
of metal-halogen exchange, [CpRefCOJ Br]" and that of ring
attack, diag-[ (i? -CgH5R)Re(CO)2Br2]". The lateral isomer
reacts exclusively with t-BuLi to afford the [CpRe(CO)2Br]"

77
anion. The relative amounts of these anionic species are
dependent upon the nature of the reducing agent as shown in
Table 5.
The mixture of products obtained when lat-CpRe(CO)gBrg
was used was easily established by IR spectroscopy. The
low-temperature FT-IR spectrum exhibited five carbonyl
stretching bands. The two carbonyl stretching bands at 1882
and 1806 cm"* are readily assigned to the [CpRe(CO)2Br]"
anion, while the other three carbonyl stretching bands at
1938, 1906, and 1833 cm"* are similar to
lat-[ (t^-CjHgJTtefCO^Brg]" and consistent with the
lat-[ (n^-CgHgRJRefCOJgB^]" anion (Fig. 11). These three
carbonyl stretching bands are replaced by two new carbonyl
bands upon addition of 5 equivalents of 15-Crown-5 (Fig.
12). These results support the formation of an anionic
cyclopentadienylrhenium complex that displays extensive
carbonyl oxygen-lithium ion pairing in the absence of
lS-Crown-S^®'^ The identity of ring-attacked anionic
species from the lateral isomer was also characterized by
low-temperature NMR spectroscopy. When a dg-THF solution of
lat-CpRe (CO) 2^rz w a s treated with LiEtjBH at -78 °C three
new resonances appeared at 6.44, 6.34, and 2.86 ppm with an
integral ratio of 2:2:2. In comparison to
4 C (n -C5H5) Re (CO) 2 Br 2] from the diagonal isomer, the low-

78
tr 1 c S3 •H <w» 0 3 <D *d tn 0) c « <d
,a A D •p X •H w !*
w c
01 a) 0 o> X 0 0) rH iH <d ft B 6 1 0 rH u <d
• P s 9 X •H c •
0) (0 s
>
H < >t •
c a) w •H »d M (0 0 • p <d c • P 0 - P a < A H 01 a C & •H
PS 0 • •
•H co C - P 0 c •H <D C Cn < «<
•
in M 9 CO < E-»
as W 0) •H O a> ft CA o •H c o •H
c o •H •P •H
O a
CO • P §
S S •H 0 3 *0 «
CO t * G 3 O
f o
>« «
a o 00 r* i
£ A ro
«P W •H J
•H A
CO
CM M m CM
o a <w a) «
8*
<d
in B vo
a
<*> < * > <K> o o O CO in Km* <w w s £ x
•h * **
< # > <#> <#> o O O r* VO in >w *w» < «< «« • • •
Pi Pi «
i4 « I
«P
~ J: *
as (XI
CO -p W •H •J
•H CO
e
CM M PQ
CM «*"•«%
O 0 a> Pi » 1
• p
•H -H ^ d in 3 E tt u> | o i
o o
0 1
-P <d
•P •H CO G •H *0 a) u 0 co <d CD s co (d !* co 0) •H O a) a CO 0 •H c •
0 >i •H ft C 0 <d 0 <d
CO <w 0 0 k -p 'tf 0 H 0 0) ft •H CO >i
Pi Pi -P H q 1 <D D a) N P4 X5

79
H
L O C - ^ R e ^ - B r
OCT Br
Me
xd" O C - ^ R e ^ * B r
OCT Br
Ph
O C - ^ R e C * B r
O r Br
2200 2000 1800
v$ cm •i
Figure 11. Infrared spectra of the carbonyl region for (a) lat-[(f7j-C5H6)Re(CO)2Br2]-(b) lat-[(n*-C5H5Me)Re(CO)2Br2]" (c) lat-[ (r?4-C5H5Ph)Re(CO)2Br2]" All spectra were recorded at -70 °C in THF.

80
H
d " "
+ 0C--Li •—«0C
.Re-.. Br Br
H
*5" 0C->Rew*Br OCT Br
2200 2000 1800
v, cm •i
Figure 12. Infrared spectra of the carbonyl region for (a) lat-[(n4-C5H6)Re(CO)2Br2] [Li] (b) lat-[ (n4C5Hg)Re(CO)2Br2] [Li] with 5 equiv. of 15-Crown-5. Both spectra were recorded at -70 0 in THF.

81
field resonances associated with the olefinic hydrogens are
0.09 ppm upfield while the high-field resonance associated
with a methylene moiety is 0.04 ppm upfield. Reaction of
the Grignard reagent MeMgBr with the CpRefCO^B^ (either
isomer) gives [CpRe(CO)2Br]" as a result of metal-halogen
exchange. A slight excess of MeMgBr gives 50 % conversion
of [CpRefCOJgBr]" at -78 °C as determined by low-temperature
FT-IR analysis. Warming the solution to room temperature
gave the complete conversion of [CpRe(CO)2Br]" which has
been characterized in situ by IR spectroscopy.
[CpRe(CO)2Br][MgBr] exhibited carbonyl bands at 1890 and
1757 cm"*. These frequencies are different from those
observed with [CpRe(CO)2Br][Li] (vide supra). In comparison
to [CpRe(CO)2Br] [Li], the symmetric CO stretching band of
[CpRe(CO)2Br][MgBr} is shifted 9 cm"* to higher frequency
while the asymmetric CO stretching band is shifted 49 cm"*
to lower frequency (Fig. 13). This behavior is typical of a
carbonyl oxygen-MgBr+ contact ion pair where jr-electron
density is polarized towards the carbonyl oxygen involved in
ion pairing with the MgBr+ gegencation as shown in Equation
18. This interaction is expected to lower the CO stretching
frequency of the CO group involved in ion pairing with
concomitant stretching of the remaining CO group.®®
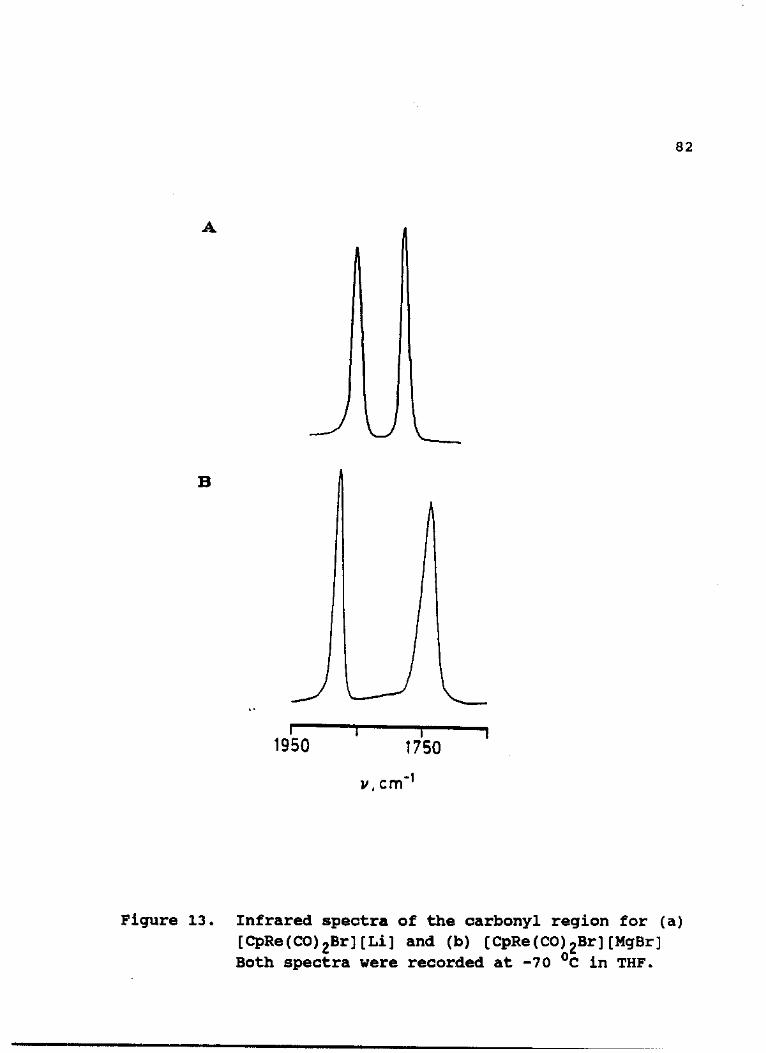
82
1950 1750
v, cm
Figure 13. Infrared spectra of the carbonyl region for (a) [CpRe(CO)2Br][Li] and (b) [CpRe(CO)2Br][MgBr] Both spectra were recorded at -70 °C in THF.

83
MeMgBr | or J I (18)
O C - ^ - B r OC> R*CBr ™P/-7. °C J _
6 ^ C0 OC Br ° B V Vo...i!i9Br
H. X-Ray Crystallographic Structure of
[CpRe(CO)2Br][Li/15-Crown-5]
The structure of [CpRe(CO)2Br][Li/15-Crown-5] has been
confirmed by single-crystal X-ray diffraction analysis. A
THF solution of 15-Crown-5 was added to the solution of
[CpRefCOJgBr]" from the reaction of diag-CpRe(CO)2^2 a n d
t-BuLi at room temperature. The yellow crystals suitable
for X-ray diffraction analysis were grown from the reaction
solution that had been layered with n-heptane. The X-ray
data collection and processing parameters for
[CpRe(CO)2Br][Li/15-Crown-5] are given in Table 6 and the
final fractional coordinates are listed in Table 7. In the
solid state, [CpRe(CO)£Br][Li/15-Crown-5] exists as discrete
molecules which are held together by Van der Waals forces.
The ORTEP diagram in Figure 14 shows the molecular structure
and clearly establishes the six-coordinate geometry about
the rhenium, assuming the cyclopentadienyl ring functions as
a three-coordinate ligand, and an overall geometry that is

TABLE 6. X-Ray Crystallographic and Data Processing Parameters for [CpRe(CO)2Br][Li/15-Crown-5]
84
space group
cell constants
a , A
b , A
c, A
P, A
v , A3
mol. formula
fw
formula units per cell(z)
p, g cm'1
abs. coeff (/*), cm"1
radiation (X), A
data collection method
collection range, deg
total data collected
independent data, I > 3a(I)
total variables
R
Rw
Weights W
P2 j/monoclinic
10.860(4)
13.116(5)
7.417(3)
105.26(3)
1018.7(3)
CiyHggBrLiO^Re
614.44
2
2.003
80.20
Mo Ka - 0.71073
8 - 2$
3.0° <29 < 50.0°
3782
2884
219
0.0777
0.0913
"„2
[accounting + (0.04 fft )^]

85
TABLE 7. Table of Positional Parameters and Their Estimated Standard Deviations for [CpRe(CO)2Br][Li/15-Crown-5]
Atom B(A2)
Re 0.14669(9) 0.000 0.0824(1) 2.54(1)
Br 0.1415(4) 0.1473(3) 0.3157(5) 4.97(8)
01 0.430(2) 0.000(4) 0.119(3) 6.5(5)
02 -0.292(2) -0.001(4) 0.357(3) 7.9(6)
03 0.722(3) 0.145(3) 0.083(4) 8.9(9)
04 0.537(3) 0.096(4) -0.210(4) 12(1)
05 0.479(4) 0.391(3) 0.226(4) 18(1)
06 -0.315(3) -0.134(2) 0.099(3) 5.7(60
07 0.168(2) -0.154(1) 0.325(3) 2.0(4)*
CI 0.321(20 0.011(3) 0.111(3) 3.0(5)
C2 0.100(2) 0.055(3) -0.224(3) 4.3(8)*
C3 -0.007(3) 0.085(2) -0.134(3) 2.7(6)
C4 -0.067(2) -0.009(5) -0.088(3) 4.3(6)
C5 -0.006(4) -0.099(4) -0.140(6) 8(1)
C6 0.072(3) -0.061(2) -0.217(4) 4.0(6)*
C7 0.162(4) -0.114(3) 0.279(5) 5.4(8)*
C21 0.738(6) 0.101(5) 0.402(9) 11(2)*
C22 0.771(6) 0.163(4) 0.285(5) 14(2)
C31 0.676(4) 0.221(3) -0.050(6) 10(1)
86
Table 7. continued
C32 0.608(5) 0.182(4) -0.182(8) 10(1)*
C41 0.553(4) 0.532(4) 0.361(5) 11(1)
C42 0.538(6) 0.427(5) 0.352(7) 12(2)
C51 0.419(7) 0.324(3) 0.231(5) 9(2)
C52 0.331(6) 0.290(3) 0.027(6) 7(1)
C61 -0.229(4) -0.163(3) 0.270(5) 5.6(9)
C62 -0.180(4) -0.062(4) 0.378(5) 8(1)
Li 0.600(4) 0.012(4) 0.048(5) 3.5(9)
Standard atoms were refined isotropically. Anisotropically refined atoms are given in the forms of the isotropic equivalent thermal parameter defined as: (4/3) * [a^*B(l,l) + b^*B(2,2) + c^*B(3,3) + ab(cos gamma) *B(1,2) + ac(cos beta)*B(1,3) + be(cos alpha)*B(2,3)]

87
Figure 14. Perspective view (ORTEP plot) of [CpRe(CO) 2Br] [Li/15-Crown-5] showing the atom labeling (hydrogen atoms are omitted for clarity).

88
typical of three-legged piano-stool complexes.®^
Bond distances and angles are given in Tables 8 and 9,
respectively. The 15-Crown-5 macrocycle adopts an
approximately planar arrangement of oxygen atoms in which
the lithium ion is centrally located. The five oxygen atoms
alternate above and below the plane of the best fit. The
coordination geometry about the lithium ion is that of a
slightly distorted pentagonal pyramid with the oxygen atoms
of carbonyl ligand. The two Li-0 distances in the
macrocycle complex are 2.17 and 2.19 A. The Li-0 distances
to the carbonyl ligand is shorter by average 0.12 A than
those in the macrocycle complex. The top view (Fig. 15) of
this molecule reveals an approximate mirror plane of
symmetry that is defined by the plane formed by the Re-Br
and C(3)-H bonds. This plane bisects the two carbonyl
groups and C(5)-C(6) bond of the Cp ring. The Re-Br and Re-
C(7) bond lengths are unexceptional in comparison to other
cycjLopentadienylrhenium complexes.32,44,88 T h e C(7j _0(7)
bond length is explainably short due to the poor crystal
quality and high thermal motion associated with this
molecule. The cyclopentadienyl ring is planar and regularly
delpcalized. The C(5) and C(6) carbon atoms of the Cp rings
are most nearly perpendicular to the plane defined by the
bromo-carbon bond. The C-C bond lengths of the Cp ring are
in the range of 1.26(6)-1.56(6) A with an average length

TABLE 8. Bond Distances in Angstroms for [CpRe(CO)2Br][Li/15-Crown-5]
a
89
Atoml Atom2 Distance Atoml Atom2 Distance
Re Br 2.581(4) 04 Li 2.17(6)
Re CI 1.85(2) 05 C42 1.09(6)
Re C2 2.32(3) 05 C51 1.11(7)
Re C3 2.27(3) 06 C61 1.41(4)
Re C4 2.33(2) 07 C7 0.62(4)
Re C5 2.40(4) C2 C3 1.52(4)
Re C6 2.29(3) C2 C6 1.56(5)
Re C7 2.08(4) C3 C4 1.50(6)
01 CI 1.18(3) C4 C5 1.43(7)
01 Li 2.06(5) C5 C6 1.26(6)
02 C62 1.42(5) C21 C22 1.34(9)
03 C22 1.47(5) C31 C32 1.18(6)
03 C31 1.40(5) C41 C42 1.39(8)
03 Li 2.19(7) C51 C52 1.62(6)
04 C32 1.37(7) C61 C62 1.56(6)
aNumbers in parentheses are estimated standard deviations in the least significant digits.

TABLE 9. Bond Angles in Degrees for
[CpRe(CO)gBr][Li/15-Crown-5]
90
Atoml Atom2 Atom3 Angle Atoml Atom2 Atom3 Angle
Br Re CI 93(1) C31 03 Li 114(3)
Br Re C2 112.3(9) C32 04 Li 105(3)
Br Re C3 88.5(7) C42 05 C51 123(5)
Br Re C4 103(1) Re CI 01 168(4)
Br Re C5 137(1) Re C2 C3 69(1)
Br Re C6 146.8(8) Re C2 C6 69(2)
Br Re C7 95(1) C3 C2 C6 92(2)
CI Re C2 92(1) Re C3 C2 72(2)
CI Re C3 125(1) Re C3 C4 73(1)
CI Re C4 155.0(9) C2 C3 C4 109(3)
CI Re C5 130(1) Re C4 C3 69(2)
CI Re C6 104(1) Re C4 C5 75(2)
CI Re C7 96(2) C3 C4 C5 111(3)
C2 Re C3 39(1) Re C5 C4 70(2)
C2 Re C4 64(1) Re C5 C6 70(2)
C2 Re C5 65(1) C4 C5 C6 101(4)
C2 Re C6 40(1) Re C6 C2 71(1)
C2 Re C7 151(1) Re C6 C5 79(2)
C3 Re C4 38(2) C2 C6 C5 126(4)
C3 Re C5 62(1) Re C7 07 169(5)
91
Table 9. continued
C3 Re C6 58(1) 03 C22 C21 119(5)
C3 Re C7 139(1) 03 C31 C32 107(4)
C4 Re C5 35(2) 04 C32 C31 134(5)
C4 Re C6 54(1) 05 C42 C41 120(5)
C4 Re C7 102(2) 05 C51 C52 114(4)
C5 Re C6 31(1) 06 C61 C62 107(3)
C5 Re C7 90(2) 02 C62 C61 103(3)
C6 Re C7 112(1) 01 Li 03 127(3)
CI 01 Li 159(3) 01 Li 04 101(2)
C22 03 C31 125(4) 03 Li 04 74(2)
C22 03 Li 107(3)

92
°C C2-2.32
C3-2.27
/ C4— 2.33
Figure 15. Top view (ORTEP plot) of [CpRe(CO)2Br]" showing the Cp ring-rhenium bond lengths.

93
of 1.45(4) A. The C(5)-C(6) bond length is short in
comparison to the other four C-C bond lengths of the Cp ring
and to other cyclopentadienylrhenium complexes.®®
The Re-C(ring) bond lengths range from 2.27 A to 2.40 A
which are a little longer in comparison to other penta-
methylcyclopentadienylrhenium compounds,^ suggesting
less ff-back-bonding from the rhenium metal. The disparate
Re-C(ring) bond lengths indicate that the Cp ring is tilted
away from the Li/15—Crown-5 moiety and towards the bromine
and carbonyl (C7)0. The Re-C(5) bond is the longest at
2.40(4) A and is followed by the Re-C(4) and Re-C(2) bond
lengths of 2.33(2) and 2.32(3) A, respectively. The
shortest Re-C(ring) bond lengths of 2.27(3) and 2.29(3) A
belong to the Re-C(3) and Re-C(6), respectively. These
variations in the Re-C(ring) bond lengths are not entirely
unexpected since a bulky group of Li/15-Crown-5 moiety is
bonded to 01 carbonyl oxygen atom. However, in
[Cp*Re(C0)2Br] the Re-C(ring) bond that is pseudotrans
(formally opposite) to the bromo ligand is observed to be
longer than the corresponding Re-C(ring) bonds that are
opposite to the two carbonyl ligands. This result is
readily rationalized in terms of the w-acceptor properties
of the CO ligands. Therefore, the bond elongation of
Re-C(5) indicates that this is due to steric rather than
electronic factors associated with the Cp ring and the

94
rhenium atom.
I. Reactivity and Stability Studies of Anionic
Cyclopentadienyl Complexes
The functionalization of [CpRe(CO)2Br]" anion was
examined as a potential route to complexes of the form
CpRe(CO)2Br(R). Scheme 2 shows the reactivity of
(CpRe(CO)2Br] with alkylating reagents. Treatment of
[CpRe(CO)2Br] in THF with trifluoroacetic acid afforded two
new VCO bands at 2032 (s) and 1962 (vs) cm"1 as expected for
anion protonation. From the intensity pattern of the vCO
bands it may concluded that protonation proceeds to give
CpRe(CO)2Br(H) with diagonal stereochemistry. When a dg-THF
solution of [CpRe(CO) 2Br]" was treated with CF3C02H two new
*H NMR resonances appear at 6.06 and -9.03 ppm with an
integral ratio of 5:1. Furthermore, when the solution of
CpRe(CO)2Br(H) was treated with MeLi deprotonation proceeds
to regenerate [CpRe(CO)2Br] . Our experimental observations
are consistent with the theoretical predictions of Bursten
40
et al. [CpRe(CO)2Br]" also reacts with methyl triflate
and magic ethyl to yield the corresponding
diag-CpRe(CO)2Br(R) complexes based on in situ IR analysis
[VCO for diag-CpRe (CO) 2Br (Me) in THF s 2028, 1954 cm"1 and

95
"OS
ffl
as tf">
25 r\T m o <N
# o v
I /
g o
2 .o •u
\\ 8 ®
rsi vo <y>
O CM
<n 0 erf I
z ir>
1
K z
CI
4
43 o CO
i'
% 9
<N

96
VCO for diag-CpRe(CO)2Br(C2H5) in THF : 2032, 1959 cm"1].
All of these functionalized complexes decomposed in solution
over a period of days to give CpRefCO)^ as the only isolable
product (approximately 20-30 % isolated yield).
The reaction of [CpRe(CO)2Br]" with two equivalents of
Bu^SnH at 60 °C leads to the formation of the
bis(tributyltin)rhenium complex (Eq. 19), which has also
2 BUgSnH
Re " ~ 7 T Z _ Re „ + 2 B u 3 S n B r <19> OCyr 60 °C 0C-^P*^«SnBu3 Bf ^ C 0 Bu3Sn^ V o 3
been synthesized independently by deprotonation of
diag-CpRe(CO)2H2 with Et^N in the presence of Bu^SnBr at
room temperature (vide infra). IR spectrum shows two new
VCO bands at 1936 (s) and 1880 (vs) cm"1 in THF (Fig. 16) .
THF solutions of [CpRe(CO)2Br]~ were observed to be stable
when oxygen was rigorously excluded. The presence of oxygen
has been observed to lead to complex mixtures of rhenium
oxides in addition to CpRe(CO)^. For example, the
[CpRe(CO)2Br]" anion slowly decomposed in septum-capped
vessels on the benchtop to give the oxide ReO^" as the
major products. The i/Re=0 band observed was identical to

97
£ O
vo H
0) u
•H

98
that exhibited by pure KReO^. IR analysis (KBr pellet)
revealed an intense vRe=0 band at 913 cm"* attributed to
potassium perrhenate. Prismatic crystals of
[ReO^][Li/15-Crown-5] have been isolated from decomposed
solutions of [CpRe(CO)2Br][Li/15-Crown-5], and the structure
of [ReO^][Li/15-Crown-5] has been confirmed by single-
crystal X-ray diffraction analysis. The X-ray data
collection and processing parameters are given in Table 10
and the final fractional coordinates are listed in Table 11.
The ORTEP diagram in Figure 17 shows the molecular structure
and clearly establishes the slightly distorted tetrahedral
geometry about the rhenium. Selected bond distances and
angles are given in Tables 12-13. The 15-Crown-5 macrocycle
adopts an approximately planar arrangement of oxygen atoms
in which the lithium ion is centrally located. The five
oxygen atoms alternate above and below the plane of the best
fit. The Li-0(6) bond distance is 2.00(3) A which is
shorter than the value of the K-0 bond in KReO^.90 The Re-0
bond distances and O-Re-O bond angles are unexceptional in
comparison to potassium perrhenate®^ except Re-0(9) bond
distance, 1.61(3) A, and 0(7)-Re-0(8) angle, 105.7(8)°.
Finally, the exact pathways and other products involved in
this decomposition reaction remain as unanswered questions
for further study.

99
TABLE 10. X-Ray Crystallographic and Data Processing Parameters for [Li/15-Crown-5][ReO^]
space group
a, A
b, A
c , A
crystal size, mm
V. A3
mol. formula
fw
formula units per cell(z)
p, g cm"*
abs. coeff (/*), cm"*
radiation (A), A
data collection method
collection range, deg
total data collected
independent data, I > 3a(I)
R
RW
Weights
P 2 | 2 j 2 j
9 . 2 6 9 ( 2 )
1 2 . 6 7 8 ( 3 )
1 3 . 5 7 4 ( 2 )
0 . 2 0 x 0 . 2 5 x 0 . 3 0
1 5 9 5 . 2 ( 2 )
C10H20LiO9Re
4 7 7 . 4 1
4
1 . 9 8 8
7 7 . 5
Mo Ka = 0 . 7 1 0 7 3
9-29
3 . 0 ° < 29 < 5 5 . 0 °
2108
1357
0 . 0 4 7
0 . 0 5 9
W = 4 FQ9 / 9c(Fq9)

100
TABLE 11. Table of Positional Parameters and Their Estimated Standard Deviations for [Li/15-Crown-5][ReO^]
Atoms X y z B(A2)
Re 0.15578(7) 0.14846(5) 0.11018(5) 5.03(1)
01 0.618(5) -0.063(3) 0.177(3) 28(2)*
02 0.556(3) -0.026(2) -0.006(1) 13.1(6)*
03 0.287(2) -0.046(1) -0.060(1) 11.3(5)*
04 0.207(3) -0.198(2) 0.059(2) 13.2(6)*
05 0.361(2) -0.166(1) 0.234(1) 11.0(5)*
06 0.286(1) 0.0616(9) 0.1469(8) 6.6(3)
07 0.216(2) 0.214(1) 0.010(1) 11.3(5)
08 0.136(2) 0.241(1) 0.2002(9) 9.3(4)
09 0.011(3) 0.084(2) 0.087(2) 17.1(9)*
Cll 0.667(5) -0.012(4) 0.138(3) 17(2)*
C12 0.683(4) 0.018(30 0.042(2) 14(1)*
C21 0.510(3) 0.058(2) -0.064(2) 10.8(8)*
C22 0.383(3) -0.005(2) -0.122(2) 8.8(6)*
C31 0.210(3) -0.122(2) -0.091(2) 9.0(6)*
C32 0.117(4) -0.160(2) -0.007(2) 12.5(9)*
C41 0.134(3) -0.217(2) 0.155(2) 10.5(7)*
C42 0.271(3) -0.257(2) 0.229(2) 9.8(7)*
C51 0.463(3) -0.194(2) 0.289(2) 10.5(8)*
C52 0.554(5) -0.094(3) 0.274(3) 17(1)*

101
Table 11. continued
Li 0.382(3) -0.073(2) 0.107(2) 6.1(6)*
Starred atoms were refined isotropically. Anisotropically refined atoms are given in the form of the isotropic equivalent displacement parameter defined as: (4/3) * [a2*B(l,l) + b2*B(2,2) + c**B(3,3) + ab(cos gamma) *B(1,2) + ac(cos beta)*B(1,3) + bc(cos alpha)*B(2,3)]

102
Figure 17. Perspective view (ORTEP plot) of [Li/15-Crown-5][ReO^] showing the atom labeling (hydrogen atoms are omitted for clarity).

TABLE 12. Bond Distances in Angstroms for [Li/15-Crown-5][ReO^]a
103
Atoml Atom2 distance Atoml Atom2 Distance
Re 06 1.71(1) 05 C42 1.42(3)
Re 07 1.69(2) 05 C51 1.25(3)
Re 08 1.71(1) 05 Li 2.10(4)
Re 09 1.61(3) 06 Li 2.00(3)
01 Cll 0.95(6) Cll C12 1.37(5)
01 C52 1.50(6) C21 C22 1.63(4)
02 C12 1.45(4) C31 C32 1.51(4)
02 C21 1.40(4) C41 C42 1.70(4)
02 Li 2.30(4) C51 C52 1.53(5)
03 C22 1.33(3) 01 Li 2.38(5)
03 C31 1.28(3) 03 Li 2.46(4)
04 C32 1.31(4) 04 Li 2.37(4)
04 C41 1.49(4)
aNumbers in parentheses are estimated standard deviations in the least significant digits.

104
Table 13. Bond Angles in Degrees for [Li/15-Crown-5][ReO^]
Atoml Atom2 Atom3 Angle Atoml Atom2 Atom3 Angle
06 Re 07 108.7(8) 05 C42 C41 103(2)
06 Re 08 108.2(7) 05 C51 C52 96(2)
06 Re 09 109(1) 01 Li 02 124(3)
07 Re 08 105.7(8) 01 Li 02 67(1)
07 Re 09 112(1) 01 Li 03 133(2)
08 Re 09 114(1) 01 Li 04 141(2)
Cll 01 C52 149(5) 01 Li 05 78(2)
C12 02 C21 102(2) 01 Li 06 105(2)
C22 03 C31 117(2) 02 Li 03 67(1)
C32 04 C41 112(2) 02 Li 04 118(2)
C42 05 C51 104(2) 02 Li 05 139(2)
Re 06 LI 142(1) 02 Li 06 106(1)
01 Cll C12 140(5) 03 Li 04 66(1)
02 C12 Cll 104(3) 03 Li 05 143(2)
02 C21 C22 96(2) 03 Li 06 88(1)
03 C22 C21 112(2) 04 Li 05 77(1)
03 C31 C32 108(2) 04 Li 06 110(1)
04 C32 C31 106(3) 05 Li 06 103(1)
04 C41 C42 103(2)

105
The ring-attacked product diag-[ (»7^-CgHg)Re(CO) '
(3) was next examined in the presence of donor ligands.
When THF solutions of 3, from the reaction of
diag-CpRefCO^B^ with LiEt^BH at -78 °C, were treated with
two electron donor ligands only CpRefCO^L (where L = PPhj,
P(OPh)3, Me^CNC) was observed (Eq. 20).
H
| THF
or-"R«~-Rr + L o 0C^-R
« • V B " 7 8 C csc+ Br ^C0 oc
• ^ L (20)
(L S PPh3, P(OPh)3, Me3CNC)
Reaction of 3 in THF at -78 °C with PPhj was instantaneous
and two vCO bands at 1926 (vs) and 1844 (vs) cm'* were
33 Q1 observed as expected for the known CpRe(CO)g(PPhj).
Complex 3 also react with P(OPh)3 and Me^CNC to yield the
corresponding CpRefCO^L complexes based on in situ IR
analysis [yCO for CpRe(CO)2[P(OPh)3] in THF : 1961, 1893
cm"1 and vCO for CpRe(CO)2(Me3CNC) in THF : 1902, 1850 cm"1]
Although the exact mechanism associated with this
substitution is unknown at this time, we suggest the
mechanism shown in Scheme 3.

SchttM 3
106
H(D) H
d " I -
0c--Re--Br Br ^co
m
dtH l_
oc'^R,eCBr sr I co
L 4
H(D)
0C 0C< ..-Re,
H(D)
0C->ReC:-L H ^CO
7 ox*
-Br
h(d; H
d ~ i
Oc- Re--L Br ^CO
-Br
H(D)
di* oc -Rv CO
C-H bond activation

107
The first step involves ligand attack at the rhenium center
with concomitant cyclopentadienyl ring slippage from r?4 -*• *?2
coordination. The resulting 18-electron complex 4 may then
lose bromide and reestablish the rj^-cyclopentadienyl ring.
Complex 5 is structurally similar to the aforementioned
r? -cyclopentadienyl complex 3. Further loss of bromide in 5
leads to the three-legged piano-stool complex 6 which is
coordinatively unsaturated (16-electron) and expected to be
quite reactive. We believe that an intramolecular C-H bond
activation occurs which regenerates, the ^-coordination of
cyclopentadienyl ring in complex 7. Such a C-H bond
activation scheme is supported by the fact that 6 is
isolobal to known organometallic compounds which exhibit
similar reactivity as expected for a carbene fragment (i.e.
6 19 d -MLg • q - CH2). Loss of a proton from 7 would give the
observed ligand substitution product CpRefCO^L. Supporting
evidence for this last step derives from the work of Bursten
et al. who have calculated that d* four-legged piano-stool
complexes, such as 7, should function as efficient hydronium
ion donors.*® Furthermore, Norton et al. have measured the
thermodynamic acidities of similar complexes and have shown
them to possess moderate acidities (pKa » 20-25).®*
Implicit in this activation scheme is the transfer of the
A endo C-H bond of the r? -CgHg ring to the rhenium center.

108
Assuming exo nucleophile attack on the r? -Cp ring, the exo
hydride should remain associated with resulting 77 -Cp in 7.
When Et^BD" was used to introduce an exo deuterium into 3,
followed by treatment with L, the isolated CpRe(CO)2L was
observed to contain one deuterium per Cp ring. If exo C-
H(D) activation occurs upon •+ ring conversion, then no
deuterium is expected in the Cp ring of the product.
In order to further define the reactivity of 3, 3 was
treated with t-BuLi in THF at -78 °C. Two complexes were
formed of which one was assigned to that of [CpRe(CO)2Br]".
The other complex possessed two uCO bands at 1842 (s) and
1715 (s) cm * and is tentatively assigned as the dianionic
species, [ (J7*-CgHg)Re(CO)2Br]We suggest that t-BuLi
deprotonation proceeds bimodality. Deprotonation of the
4
Tj -CjHg ring yields the dianionic species, 8. Facile loss
of bromide from dianion 8 leads to [CpRe(CO)2Br]' in 65 %
yield (by IR). Alternatively, t-BuLi reacts by metal-
halogen exchange to give [ (rj4-C5Hg)Re(CO) 2Br] "2 in 35% yield
as shown in Scheme 4.
The thermal stability of 3 was next examined. 3 was
observed to be stable for a period of six months when
maintained at -78 °C. However, decomposition of 3 is
noticeable upon warming to -30 °C, being complete by 0 °C to
afford a green solution. FT-IR analysis revealed the

109
ur> VD
O
1/ X o ®
00 I
- 8
"Vw
u
X o
I x> u. X
co X
*
m
rsT GO
* u
X <ni
O
<T
3 A CI CO
X
: ^ J
9 8
a#
°\V
g o
Of

110
22 93
presence of only CpRe(CO)^ ' which we have been able to
routinely Isolate in 25-35 % yield. While the exact
mechanism associated with this decomposition is unknown at
this time, the observation of CpRe(CO)^ (in part) provides
insight into the stereochemistry attendant upon hydride 4
attack. If we assume that the endo hydrogen of the n c5h6
ring is transferred to the rhenium metal during
94 decomposition, then the exo hydrogen must become
incorporated into the cyclopentadienyl ring of CpRe(CO)^
2 (vide supra). This was demonstrated by isolation and H NMR
examination of the CpRe(CO)^ that formed a reaction using
o
LiEtjBD. The H NMR spectrum exhibited a singlet at 5.48
ppm consistent with a dj-cyclopentadienyl moiety.
Integration against added dg-benzene or the natural
abundance solvent resonances (THF) supports the presence of
one deuterium per Cp ring and an initial exo attack of the
reducing agent. Based on these results we propose that
decomposition of 3 proceeds as in the Scheme 5.
A THF solution containing the anionic products from
lat-CpRe(CO)2Br2, lat-[ (r?4-C5H5R)Re(CO)2Br2]" and
[CpRe(CO)2Br]~, decompose similarly to afford R-CpRe(CO)^
which we have been able to routinely isolate in 25-35 %
yield as illustrated in Equation 21. It is seen that the
R-CpRe(CO)2 obtained must come from the anionic

Ill
B w
r . * ac.
a o
0) 0
1 0 01 m
92
" v g m
" 2 c o - 2
- a o
I S I U
U O
CD l
X i
9
X o
^ 3 - < " •. u GD
HI
§
4 3 O CO
© s
. *

112
ring-attacked complex lat-[ (rj*-C5H5R)Re(CO) 38^]". This
result suggests that [CpRe(CO)2Br]" may be the carbonyl
source required for R-CpRe(CO)j formation.
THF
OC Lc/ > Br Br
UEt3BH or RLi
N
d " H • L
OC- Re --Br OCT Br
decomposition
oc->R#-o<r
| RsMe.Ph.H + LiR«04
'CO
>C0
(21)

113
J. Reduction of Diagonal and Lateral, Cp*Re(CO)2Br2 with
RLi, RMgX, and Trialkylborohydrides.
The reduction behavior of diagonal and lateral
CP*Re(CO)2Br2 was investigated with RLi reagents (R = Me,
Ph, and t-Bu) in an attempt to prepare the corresponding
alkylbromide complex, Cp*Re(CO)2Br(R). Related reduction
studies are well documented in the isoelectronic Cp*Rh and
95
Cp*Ir systems. However, in all cases only the anionic
complex, [Cp*Re(C0)2Br] ,2-, was observed when one equiva-
lent of RLi was reacted with either isomer of Cp*Re(CO)2Br2
(Equation 22).
THP Cp*Re(CO)2Br2 + RLi [Cp*R«(CO)2Br] [Li] + RBr (22)
-78 C
(R •» Ni| Ph, t-Bu)
Red-brown solutions of Cp*Re(CO)2Br2 (either isomer) react
instantaneously with added RLi at -78 °C in THF to give a
yellow solution containing 2-; the essentially quantitative
conversion to 2— was easily confirmed by low—temperature
FT-IR measurements. The IR spectrum exhibited two carbonyl
stretching bands at 1863 and 1789 cm"1 which is consistent
with the ascription of 2- as an anionic three-legged piano-

114
stool complex. Such a low energy shifting of the CO
stretching bands of 2- relative to Cp*Re (CO) 2Br2 *-s directly
attributed to the increased charge density at the rhenium
center (Fig. 18). The higher frequency CO band at 1863 cm~*
is readily assigned to a symmetric CO stretching mode while
the lower frequency CO band at 1789 cm"* corresponds to the
asymmetric CO mode based on group theoretical considera-
96 tions. In such a molecule more energy is required to
stretch two CO bonds simultaneously than to execute one CO
stretch and one CO contraction. IR studies (in THF)
indicate that 2- exists as symmetrically solvated ion pairs
75 97
possessing idealized C$ symmetry. This is supported by
the spectral invariance of 2- over the temperature range
-78 °C to room temperature which serves to rule out a
dynamic equilibration between different ion pairs.®*'98
Furthermore, added Li+ (as CFjSOjLi), tetraphenylphospho-
nium chloride (PPP-C1), or 15-Crown-5 do not affect the
intensity or frequency of the initial IR spectrum of 2-,
reinforcing the existence of only solvent-separated ion
pairs in THF solution. 2- can be easily isolated in high
yield by treating [Cp*Re(CO) 2Br] [Li] with PPP-C1. The
resulting orange-yellow crystals of [Cp*Re(CO)2Br][PPP] gave
a satisfactory microanalysis and were used in the NMR
characterization of 2-. The *H-NMR spectrum of

115
oc Br
.-R
t r i 1
1900 1800 v, cm -1
Figure 18. Infrared spectrum of the carbonyl region for [Cp*Re(CO)2Br]" at -70 °C in THF.

116
2-PPP in CDgClg displayed a singlet at 1.94 ppm (15H) and a
multiplet centered at 7.75 ppm (20H) consistent with the
methyl groups of the Cp* ring and the aromatic protons of
the PPP gegenion, respectively. The 13C{1H} NMR spectrum
exhibited resonances at 11.0, 93.8, and 213.1 ppm for the
methyl, carbons of Cp ring, and the carbonyl carbons of 2-
PPP as shown in Figure 19. The metal carbonyl resonance is
shifted to downfield relative to either of the lateral and
diagonal isomers of Cp*Re(C0)2Br2 as a result of increased
charge density at the rhenium center.66,74,99 Reaction of
the Grignard reagents MeMgBr or t-BuMgCl with the isomeric
dibromides of Cp*Re(C0)2Br2 gives 2-MgBr as a result of
metal-halogen exchange like that observed with CpRe(CO)2Br2.
Use of MeMgBr (slight excess) affords 2-MgBr sluggishly at
-78 °C as determined by low-temperature FT-IR analysis (10%
conversion after 30 min). Warming the solution to room
temperature led to the rapid generation of 2-MgBr (Eq. 23)
which has been characterized in situ by IR spectroscopy
(Fig. 20b). 2-MgBr exhibits CO bands at 1872 and 1734 cm"1.
These frequencies are different from those observed with
2-Li (vide supra). In comparison to 2-Li, the symmetric CO
stretching band of 2-MgBr is shifted 9 cm"1 to higher
frequency while the asymmetric CO stretching band is shifted
55 cm"1 to lower frequency.
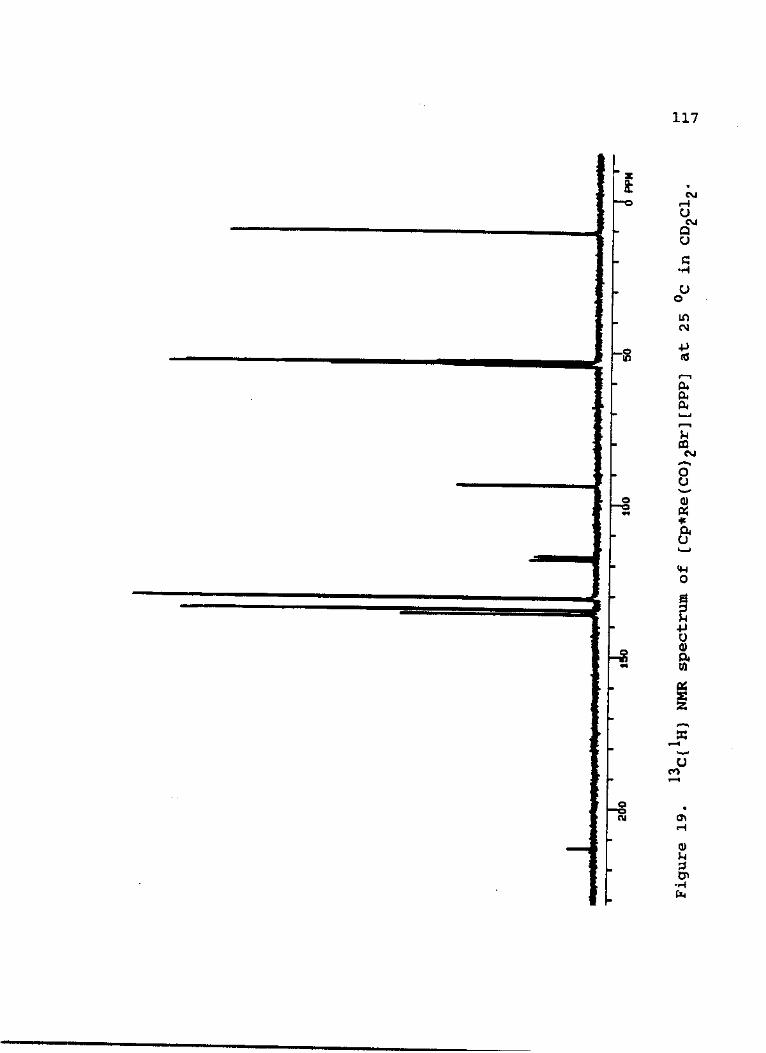
117
& CM r-4 o CM
Q a a •H
—g
a o in CM
04 04 04
U OQ CM <•—««»
O o w a) « * a a
0
1 .p o a) a w
a CO
© - © ru o\
H a) M 3 Cn
•H Cm

118
1950 1750
v, cm -i
Figure 20. Infrared spectra of the carbonyl region for (a) [Cp*Re(CO)2Br][Li] and (b) [Cp*Re(CO)2Br][MgBr], Both spectra were recorded at -70 °c in THF.

119
•RMgBr
,RBr OO^cO....;gBr (23)
(R:X s Me*Br, t-Bu:Cl)
This behavior is typical of a carbonyl oxygen-MgBr+ contact
ion pair where -electron density is polarized towards the
carbonyl oxygen involved in ion pairing with the MgBr+
gegencation as shown in Equation 23.7®>97 This interaction
is expected to lower the CO stretching frequency of the CO
group involved in ion pairing with concomitant strengthening
of the remaining CO group.®®
The reaction of lat-Cp*Re(CO)2Br2 with a stoichiomet-
ric amount (one equiv.) of t-BuMgCl is rapid at -78 °c and
gives 2- in near quantitative yield. The IR spectrum of
2 —MgCl is identical with that of 2-MgBr and suggests similar
carbonyl oxygen-MgX+ contact ion pairs. In this reaction
with t-BuMgCl the possibility exists for a bromide/chloride
exchange in the magnesium gegenion. The resulting MgBr*
cation would be expected to yield an IR spectrum of 2-MgBr
identical to that observed with MeMgBr. Unequivocal proof
for such carbonyl oxygen—MgX^ ion pairs was obtained by
examining the effect HMPA had on the solution IR spectrum of

120
2-MgX. Use of 10 mole equivalents of HMPA led to the
complete disruption of the contact ion pairs and products of
a two carbonyl band identical with 2-Li.
The reaction of lat-Cp*Re(C0)2Br2 with 1.0 mole equi-
valent of LiEtjBH or K-Selectride, [K(sec-Bu)3BH] at -78 °C
led to the immediate formation of 2- in 50 * yield as
determined by IR analysis. When the hydride concentration
was doubled, complete conversion to 2- was observed. At no
time was the corresponding bromohydride complex,
Cp*Re(CO)2Br(H), observed (vide infra). Based on these
observations we believe, in analogy to the RLi and RMgX
reactions, that the hydride reagents react with
lat-Cp*Re(CO)2Br2 to give equal molar amounts of 2- and HBr.
A subsequent, rapid reaction between the generated HBr and
excess R^BH would then account for the 1:2 rhenium/hydride
stoichiometry observed with this reaction. The hydrogen by-
product necessary for the proposed reaction was observed by
GC analysis; however, no attempt was made to quantify it.
An alternative path way involves the metathetical
replacement of a bromide ligand in lat-Cp*Re(CO)2Br2 to give
diag-Cp*Re(CO)2Br(H). Since most metal hydrides are known
to exhibit acidic character101, a fast follow-up reaction
involving metal-hydride deprotonation by additional H" would
also give 2- as outlined in Scheme 6. The IR spectra of 2-

121
m
CD
<N X
0
s A O co
m
CD •
• ** cr
O O

122
from these reactions are unexceptional when compared with
those obtained from the RLi reactions. This suggests that
the presence of R^B, an effective Lewis acid, in solution
does not perturb the local environment of the rhenium anion.
K. X-Ray Crystallographic Structure of [Cp*Re(CO)2Br][PPP]
The structure of 2-PPP has been confirmed by single-
crystal X-ray diffraction analysis. A methanol solution of
PPP-C1 was added to the solution of 2-Li from the reaction
of lat-Cp*Re(CO)2Br2 and LiEtjBH. The orange yellow
crystals suitable for X-ray analysis were grown from the
reaction solution that had been layered with n-heptane.
2-ppp exists as discrete molecules in the unit cell with no
unusually short inter- or intramolecular contacts? the PPP+
gegenion is unexception and requires no further description.
The X-ray data collection and processing parameters for
2-PPP are given in Table 14 and the final fractional coordi-
nates are listed in Table 15. The ORTEP diagrams in Figures
21-22 show the molecular structure of 2-PPP and clearly
establishes the six-coordinate geometry about the rhenium,
assuming the Cp* ring functions as a three-coordinate
ligand, and an overall geometry that is typical of three-
87
legged piano-stool complexes. Bond distances and angles
are given in Tables 16-17. The top view (Fig. 23b) of 2-

123
TABLE 14. X-Ray Crystallographic and Data Processing Parameters for [Cp*Re(CO)2Br][Ph^P].
space group
cell constants
a, A
b , A
c, A
V, A3
mol. formula
fw
formula units per cell (Z)
p, g cm"*
crystal size, mm
abs coeff (/*), cm"*
radiation (X), A
data cillection method
collection range, deg
total data collected
independent data, I > 3a(I)
total variables
R
RW
Weights W
Pbca/orthorhombic
20.646 (5)
17.690 (5)
17.555 (3)
6411.6
c36H35Br02PRe
796.76
8
1.651
0.3 x 0.2 x 0.1
51.55
Mo Ka - 0.71073
9 - 20
2.0° £ 2B < 50.0°
6233
2995
370
0.0366
0.0432
4 fj
("'counting + <0-04 fo2>2l

124
TABLE 15. Positional Parameters for Non-Hydrogen Atoms for [Cp*Re(CO)2Br][Ph4P] with Estimated Standard Deviations in Parenthesis
Atom X y z B(A2)
Re 0.64733(2) 0.10035(3) 0.61146(3) 2.904(7)
Br 0.74495(6) 0.03683(7) 0.68025(8) 4.26(3)
P 0.0909(1) 0.6040(2) 0.0984(2) 2.65(5)
01 0.5710(5) 0.1711(6) 0.7393(5) 5.7(2)
02 0.7146(5) 0.2484(6) 0.5771(6) 6.6(2)
CI 0.6035(7) 0.1453(7) 0.6965(7) 4.1(3)
C2 0.6927(6) 0.1953(7) 0.5915(7) 4.3(3)
C3 0.5981(6) 0.1021(8) 0.4961(7) 4.3(3)
C3 • 0.595(1) 0.1694(9) 0.4441(9) 7.9(4)
C4 0.5531(6) 0.0824(8) 0.5528(9) 6.1(3)
C4' 0.4887(8) 0.122(1) 0.573(1) 13.3(6)
C5 0.5714(6) 0.0079(8) 0.5848(7) 5.1(3)
C5' 0.5359(8) -0.042(1) 0.644(1) 10.7(5)
C6' 0.6648(9) -0.0888(8) 0.556(1) 7.8(5)
C6 0.6272(6) -0.0155(7) 0.5457(7) 3.6(3)
C7' 0.6994(7) 0.036(1) 0.4315(8) 6.7(4)
C7 0.6446(6) 0.0419(7) 0.4926(6) 3.8(2)
Cll 0.1250(0) 0.5125(6) 0.0820(6) 2.9(2)
C12 0.1398(6) 0.4654(7) 0.1452(7) 3.6(2)
C13 0.1675(6) 0.3928(7) 0.1327(7) 4.3(3)
T a b l e 1 5 . c o n t i n u e d
125
C14 0 . 6 7 7 0 ( 6 ) 0 . 1 3 3 0 ( 8 ) 0 . 9 4 4 7 ( 8 ) 4 . 5 ( 3
C15 0 . 1 6 0 3 ( 5 ) 0 . 4 1 6 3 ( 7 ) - 0 . 0 0 5 5 ( 7 ) 4 . 2 ( 3
C16 0 . 1 3 4 5 ( 6 ) 0 . 4 8 7 8 ( 6 ) 0 . 0 0 6 3 ( 7 ) 3 . 4 ( 2
C21 0 . 0 1 4 9 ( 5 ) 0 . 5 9 2 9 ( 6 ) 0 . 1 4 7 9 ( 6 ) 2 . 7 ( 2
C22 - 0 . 0 1 3 0 ( 6 ) 0 . 5 2 0 6 ( 7 ) 0 . 1 4 8 9 ( 7 ) 3 . 6 ( 3
C23 - 0 . 0 7 4 6 ( 6 ) 0 . 5 1 4 3 ( 7 ) 0 . 1 8 5 7 ( 7 ) 3 . 6 ( 3
C24 - 0 . 1 0 3 1 ( 6 ) 0 . 5 7 6 2 ( 8 ) 0 . 2 1 7 4 ( 7 ) 4 . 3 ( 3
C25 0 . 5 7 4 1 ( 7 ) 0 . 3 5 0 9 ( 7 ) 0 . 7 1 5 8 ( 8 ) 4 . 5 ( 3
C26 - 0 . 0 1 4 3 ( 6 ) 0 . 6 5 6 4 ( 7 ) 0 . 1 7 9 4 ( 7 ) 4 . 0 ( 3
C31 0 . 1 4 4 9 ( 5 ) 0 . 6 6 4 5 ( 6 ) 0 . 1 5 1 3 ( 6 ) 2 . 7 ( 2
C32 0 . 1 3 9 5 ( 6 ) 0 . 7 4 3 6 ( 7 ) 0 . 1 3 9 0 ( 8 ) 4 . 4 ( 3
C33 0 . 1 8 0 2 ( 7 ) 0 . 7 9 2 5 ( 8 ) 0 . 1 8 1 6 ( 8 ) 5 . 0 ( 3
C34 0 . 2 2 3 4 ( 6 ) 0 . 7 6 3 2 ( 8 ) 0 . 2 3 4 2 ( 7 ) 4 . 1 ( 3
C35 0 . 2 2 7 0 ( 6 ) 0 . 6 8 5 2 ( 8 ) 0 . 2 4 7 0 ( 7 ) 4 . 4 ( 3
C36 0 . 1 8 7 9 ( 6 ) 0 . 6 3 5 6 ( 7 ) 0 . 2 0 4 8 ( 7 ) 3 . 4 ( 3
C41 0 . 0 7 6 1 ( 5 ) 0 . 6 4 6 7 ( 6 ) 0 . 0 0 6 9 ( 6 ) 2 . 8 ( 2
C42 0 . 0 1 4 7 ( 6 ) 0 . 6 4 9 2 ( 7 ) - 0 . 0 2 2 3 ( 7 ) 3 . 6 ( 3
C43 0 . 0 0 6 1 ( 6 ) 0 . 6 7 6 1 ( 7 ) - 0 . 0 9 7 3 ( 7 ) 4 . 1 ( 3
C44 0 . 0 5 9 6 ( 7 ) 0 . 7 0 3 3 ( 8 ) - 0 . 1 3 7 3 ( 7 ) 4 . 9 ( 3
C45 0 . 6 1 9 4 ( 7 ) - 0 . 2 0 2 5 ( 9 ) 1 . 1 0 6 0 ( 8 ) 5 . 5 ( 3
C46 0 . 6 3 1 2 ( 6 ) - 0 . 1 7 2 5 ( 8 ) 1 . 0 3 1 6 ( 7 ) 4 . 6 ( 3

126
Table 15. continued
Anisotropically refined atoms are given in the form of the isotropic equivalent thermal parameter defined as: (4/3)*[a *B(1,1) + b *B(2,2) + c2*B(3,3) + ab(cos gamma) *B(1/2) + ac(cos beta)*B(l,3) + be(cos alpha)*B(2,3)J.

127
Figure 21. Perspective view (ORTEP plot) of [Cp*Re(CO)2Br] showing the atom labeling (hydrogen atoms are omitted for clarity).

128
Figure 22. Perspective view (ORTEP plot) of PPP+ gegenion showing the atom labeling (hydrogen atoms are omitted for clarity).

TABLE 16. Bond Distances in Angstroms for [Cp*Re(CO)2Br][Ph4P]
a
129
Atoml Atom2 Distance Atoml Atom2 Distance
Re Br 2.604(1) Cll C12 1.42(2)
Re CI 1.919(12) Cll C16 1.41(2)
Re C2 1.955(13) C12 C13 1.42(2)
Re C3 2.266(12) C13 C14 1.45(2)
Re C4 2.224(13) C14 C15 1.42(2)
Re C5 2.313(14) C15 C16 1.39(2)
Re C6 2.388(12) C21 C22 1.40(2)
Re C7 2.330(11) C21 C26 1.39(2)
P Cll 1.789(11) C22 C23 1.42(2)
P C21 1.803(10) C23 C24 1.36(2)
P C31 1.803(11) C24 C25 1.42(2)
P C41 1.802(11) C25 C26 1.40(2)
01 CI 1.11(2) C31 C32 1.42(2)
02 C2 1.07(2) C31 C36 1.39(2)
C3 C3 • 1.50(2) C32 C33 1.42(2)
C3 C4 1.41(2) C33 C34 1.38(2)
C3 C7 1.44(2) C34 C35 1.40(2)
C4 C4 • 1.55(2) C35 C36 1.40(2)
C4 C5 1.48(2) C41 C42 1.37(2)
C5 C5' 1.55(2) C41 C46 1.40(2)
Table 16. continued
130
C5 C6 1.40(2) C42 C43 1.41(2)
C6 * C6 1.52(2) C43 C44 1.39(2)
C6 C7 1.42(2) C44 C45 1.35(2)
C7' C7 1.56(2) C45 C46 1.43(2)
aNumbers in parentheses are estimated standard deviations in the least significant digits.

TABLE 17• Bond Angles in Degrees for [Cp*Re(CO)2Br][Ph4P]
a
131
Atoml Atom2 Atom3 Angle Atoml Atom2 Atom3 Angle
Br Re CI 100.5(4) C6 Re C7 35.1(4)
Br Re C2 94.8(4) Cll P C21 108.7(5)
Br Re C3 140.1(3) Cll P C31 112.1(5)
Br Re C4 146.1(4) Cll P C41 107.6(5)
Br Re C5 108.3(3) C21 P C31 110.8(5)
Br Re C6 89.4(3) C21 P C41 109.1(5)
Br Re C7 104.0(3) C31 P C41 108.4(5)
CI Re C2 90.5(5) Re CI 01 170(1)
CI Re C3 118.6(5) Re C2 02 175(1)
CI Re C4 90.4(5) Re C3 C3' 125(1)
CI Re C5 97.5(5) Re C3 C4 70.1(7)
CI Re C6 130.5(5) Re C3 C7 74.3(7)
CI Re C7 150.4(5) C3' C3 C4 127(1)
C2 Re C3 92.4(5) C3' C3 C7 126(1)
C2 Re C4 117.3(5) C4 C3 C7 107(1)
C2 Re C5 153.6(5) Re C4 C3 73.4(7)
C2 Re C6 137.2(4) Re C4 C4' 126(1)
C2 Re C7 103.4(5) Re C4 C5 74.2(8)
C3 Re C4 36.5(5) C3 C4 C4' 128(1)

Table 17. continued
132
C3 Re C5 61.6 5) C3 C4 C5 109(1)
C3 Re C6 60.1 4) C4 * C4 C5 123(1)
C3 Re C7 36.4 5) Re C5 C4 67.7(7)
C4 Re C5 38.1 5) Re C5 C5' 126(1)
C4 Re C6 60.1 5) Re C5 C6 75.6(7)
C4 Re C7 60.1 5) C4 C5 C5' 130(1)
C5 Re C6 34.7 4) C4 C5 C6 107(1)
C5 Re C7 59.2 4) C5 • C5 C6 123(1)
Re C6 C5 69.7 7) C12 Cll C16 122(1)
Re C6 C6' 125.7 9) Cll C12 C13 120(1)
Re C6 C7 70.2 7) C12 C13 C14 119(1)
C5 C6 C6' 128(1 C13 C14 C15 119(1)
C5 C6 C7 109(1 C14 C15 C16 123(1)
C6' C6 C7 124(1 Cll C16 C15 118(1)
Re C7 C3 69.4 7) C22 C21 C26 123(1)
Re C7 C6 74.7 7) C21 C22 C23 117(1)
Re C7 C7' 128.8 9) C22 C23 C24 120(1)
C3 C7 C6 109(1 C23 C24 C25 123(1)
C3 C7 C7' 124(1 C24 C25 C26 118(1)
C6 C7 C7 • 126(1 C21 C26 C25 119(1)
P Cll C12 119.3 8) C32 C31 C36 121(1)
P Cll C16 119.1 8) C31 C32 C33 118(1)
P C21 C22 117.9 8) C32 C33 C34 120(1)

133
Table 17. continued
p C21 C26 118.8(9) C33 C34 C35 121(1
p C31 C32 117.2(8) C34 C35 C36 120(1
p C31 C36 121.7(8) C31 C36 C35 120(1
p C41 C42 120.4(9) C42 C41 C46 124(1
p C41 C46 115.4(8) C43 C44 C45 121(1
C44 C45 C46 122(1
C41 C46 C45 115(1
aNumbers in parentheses are estimated standard deviations in the least significant digits.

134
C5 — 2.37
I C 7 - 2 . 2 6
ft C 5 - 2 . 3 1
C 6 - 2 . 3 9
C 3 - 2 . 2 7
C 7 - 2 . 3 3
C 3 - 2 . 2 0
Figure 23. Top view of (a) lat-Cp*Re(CO)2I2 and (b) [Cp*Re(C0)2Br]" showing the Cp* ring-rhenium bond lengths (A). Distances for the former complex are taken from ref. 32.

135
reveals an approximate mirror plane of symmetry that is
defined by the plane formed by the Re-Br and C(6)-C(6')
bonds. This plane bisects the two carbonyl groups and the
C(3)-C(4) bond of the Cp ring. The Re-Br, Re-carbonyl, and
C-0 bond lengths are unexceptional in comparison to other
cyclopentadienylrhenium complexes.32'44,88 The C-c bonds of
the Cp* ring range from 1.40(2) to 1.48(2) A with an average
length of 1.43(2) A in agreement with other peralkylated
cyclopentadienylrhenium complexes.32,44,103 The disparate
Re-C(ring) bond lengths indicate that the Cp* ring is tilted
away from the bromide atom and towards the two carbonyl
groups. The Re-C(6) bond is the longest at 2.388(12) A and
is followed by the Re-C(7) and Re-C(5) lengths of 2.330(11)
and 2.313(14) A, respectively. The shortest Re-C(ring)
distances of 2.266(12) and 2.224(13) A belong to the Re-C(3)
and Re-C(4), respectively. These variations in the Re-C
(ring) bond lengths are not entirely unexpected when the
crystallographic results of lat-Cp*Re(CO)2I2 and the
theoretical studies of related CpML^ complexes are
considered.104 For example, in lat-Cp*Re(CO)2I2 the
Re-C(ring) bonds that are opposite the iodide ligands are
observed to be longer than the corresponding pseudotrans
Re-C(ring) bonds as shown in Figure 23a. The bond length
asymmetry in 2- (as in lat-Cp*Re(CO)2I2) is readily
rationalized in terms of the ir-acceptor properties of the CO

136
ligands. The CO ligands accept electron density (via the
w* manifold) from the Cp* ring at the expense of the
opposite Re-C(5,6,7) bonds; this results in an elongation
and a tilting of the Cp* ring towards the carbonyl ligands.
Alternatively, Cp* ring tilting may be viewed as arising
from the interaction between metal d-orbital (d ) and v* xy
orbital (e2) of Cp* ring.104 The Cp* ring is expected to
exhibit maximum tilting in d5-ML4 fragments that possess an
eclipsed conformation. Moreover, small deviations from an
eclipsed conformation are not expected to negate the
predicted trends in Cp* ring tilting.32 Based on this, the
isolobal relationship predicts that 2- should display a
similar tilting pattern as in lat-Cp*Re(CO)2I232 and
104 CpMo(CO)3Me since the [Re(CO)2Br]" fragment is formally a
d^-MLj species and is equivalent to its isolobal d^-ML^
19
counterparts. Here, the metal fragments are derived by
treating the Cp ring as a neutral five-electron donor. An
alternative procedure would involve treating the Cp ring as
an anionic six-electron donor with concomitant charge
adjustment for the metal fragment, i.e., Re(CO)2Br and
Re(CO)2I2+. Regardless of the exact factor(s) responsible
for Cp* ring tilting, the isolobal nature of these metal
fragments underscores the predicted and experimentally
observed similarity between 2-, lat-Cp*Re(CO)2I2, and

137
CpMo(CO)jMe.
L. Reactivity and Stability Studies of Anionic
Pentamethylcyclopentadienyl Rhenium Complex,
[Cp*Re(CO)2Br][Li]
The functionalization of [CpftRefCO^Br]', 2-, was
examined as a potential route to complexes of the form
Cp*Re(C0)2Br(R). Treatment of 2- in THF with trifluoro-
acetic acid afforded two new PCO bands at 2018 and 1945 cm"*
as expected for anion protonation. From the intensity
pattern of the vCO bands it was concluded that protonation
proceeds to give Cp*Re(C0)2Br(H) with diagonal stereo-
chemistry. The *H NMR spectrum of treatment of 2- with
CF3CO2H in dg-THF displayed a singlet at 2.01 ppm (15H) and
a singlet at -9.98 ppm (1H) consistent with the methyl
groups of the Cp* ring and Re-H bond, respectively. 2- also
reacts with methyl triflate and magic ethyl in CH2CI2 to
yield the corresponding diag-Cp*Re(CO)2Br(R) complexes based
on in situ IR analysis. The low-temperature FT-IR spectrum
exhibits two carbonyl bands at 2016 (s) and 1935 (s), and
2020 (s) and 1944 (vs) cm"* in CH2CI2 consistent with the
diag-Cp*Re(CO)2Br(Me) and diag-CpARefCO^Br^Hg),
respectively. All of these functionalized complexes

138
decomposed in solution over a period of days to give
Cp*Re(CO)j as the only isolable product (about 20-30 %
isolated yield).
Finally, THF solution of 2- were observed to be stable
for a period of weeks when oxygen was rigorously excluded.
The presence of oxygen leads to complex mixtures of rhenium
oxides in addition to Cp*Re(C0)3. For example, 2-K (vide
infra) slowly decomposed in septum-capped vessels on the
benchtop to give the known oxides Cp*ReOg^ and ReO^" as the
major products. The yRe=0 band of ReO^" observed was
identical to that exihibited by pure KReO^. IR analysis
(KBr pellet) revealed intense pRe*0 bands at 909 and 877
cm"1 assignable to Cp*Re03 and an intense band at 913 cm"1
attributed to potassium perrhenate. The exact pathways and
other products involved in this decomposition reaction
remain as unanswered questions for future study.
M. Reduction Studies Using One-Electron Reducing Agents
About Cp'Re(CO)2Br2 (Cp* = Cp, Cp*)
the reduction of lateral and diagonal Cp'Re(CO)2&r2' 9'
was examined using known one-electron reducing agents in
order to probe for the intermediacy of the corresponding
19e" complex [Cp'RefCO^Brg]*, 10• Treatment of 9 (either

139
isomer) in THF at -78 °c with one equivalent of cobaltocene
or potassium naphthalide immediately gave 50 % conversion of
[Cp'RefCO^Br]" (11) which was assayed by low-temperature
FT-IR analysis. Use of two equivalents of reducing agent
afforded 11 in quantitative yield as expected for a two-
electron reduction process. The resulting two vCO bands of
the rhenium anion are identical in intensity and frequency
to 1-Li or 2-Li and suggest that negligible ion pairing is
present between 11 and gegencation (vide supra). The fact
that the 19e" radical anion is not observed indicates that
the lifetime of 10 is very short and is followed by faster
subsequent reactions to ultimately give 11. Loss of bromide
from 11 is anticipated to give the 17e" complex
[Cp'Re(CO)£Br], 12, which is then expected to accept a
second electron in a fast electron transfer step involving 106
the reducing agent. ° Examples of facile ligand loss upon
one-electron coupled with a second reduction step occurring
at less negative potential are quite common in
organometal1ic reduction reactions.106 Electron transfer
from 10 to 12 is also expected to yield 11 and regenerates
9. The importance of this pathway will depend on the
relative magnitude of the rates associated with bromide loss
and electron transfer from the 19e" complex 10. Facile loss
of bromide, compared to CO loss, is also predicted when one

140
considers the acidity of these two ligands. Here the weaker
jr-acceptor ligand preferentially dissociates so as to
minimize unfavorable metal-ligand orbital interactions in
the 19e" radical anion.^ Alternatively, # could undergo a
second electron accession to give a 20e" complex (assuming
no ring hapticity changes), followed by bromide loss to give
11 as shown in Scheme 7. We favor the former pathway based
on reports of dissociative halide loss in organic halides
and metal dimer fragmentation reaction upon one-electron
reduction*®® and note that these two schemes have precedence
in electrochemistry as they formally conform to an ECE and
109 EEC process, respectively.
Reaction of 9 in THF at -78 °C with sodium naphthalide
(2 mole equivalents) was observed to occur rapidly to give
9-Na. However, low-temperature FT-IR analysis revealed that
9- existed as a mixture of carbonyl oxygen-Na and solvent-
separated ion pairs. The existence of the former ion pairs
was easily confirmed through the addition of either
15-Crown-5 or HMPA (10 mole equivalents based on Re) which
immediately afforded an IR spectrum identical to
[Cp'RefCO^Br] [Li]. The vCO of 9-Na are readily assigned
based on their frequency and response to additives as was
done for 2-MgX. The two low frequency i/CO bands of 1-Na at
1808 and 1793 cm"* and 2-Na at 1791 and 1776 cm"* represent
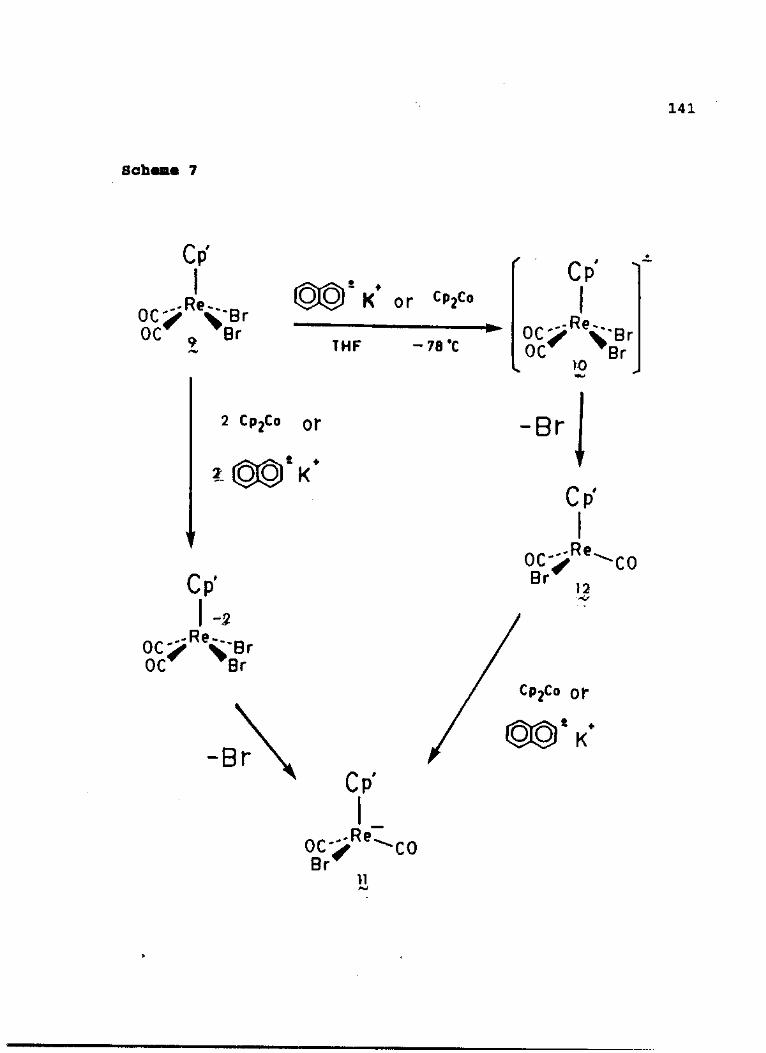
Sch«n« 7
141
CP
OC>Rev'Br Or 9 Br
K or CP2C°
THF — 78 *C
2 Cp2Co Qf
s • K
C P '
-a Dp
OCy \-Br OC Br
- B r
OCv Br
Cp
,,-Re CO
CP
OC^ReC*Br OC Br
-to
Br
C p '
0C'yRe^C0 * 1 2
cp2co or
VI

142
the asymmetric carbonyl stretching mode associated with the
solvent-separated and carbonyl oxygen-sodium contact ion
pairs, respectively. In principle, two additional vCO bands
corresponding to a symmetric carbonyl stretching for each
type of ion pairs should be observed; however, only one band
is observed at 1882 cm ^ for 1-Na and 1863 cm~^ for 2-Na,
respectively. An analogous situation involving
[CpFe(CO)2] [Na]^®® and [CpMo(CO)^] [Na]^ has been observed
and presumably is the result of coupling and/or inadequate
band resolution.
N. Thermodynamic Study of the Ion Pairing Equilibrium for
[Cp*Re(CO)2Br][Na]
Solution FT-IR studies of 2-Na (in THF) at room
temperature display a 39:61 mixture of carbonyl oxygen-
sodium and solvent-separated ion pairs, respectively.
However, low-temperature FT-IR analysis at -70 °C reveals a
73:27 mixture of ion pairs, respectively. This behavior
indicates that these ion pairs in THF solution are involved
in a reversible temperature-dependent equilibrium. The
equilibrium constant for these ion pairs has been determined
by IR band shape analysis over the temperature range -70 °C
to room temperature and values of AH and As are obtained.
.3 2.5 x 10 M THF solutions of 2-Na were used to examine the

143
effect of temperature on the equilibrium between these ion
pairs in order to quantify the thermodynamics associated
with this system. Figure 24 shows selected IR spectra for
2-Na as a function of temperature. As the temperature is
raised from -70 °C to room temperature, the equilibrium
constant is gradually shifted to the solvent-separated ion
pairs in accordance with the equilibrium shown in Eq. 12.
The ion pairing exhibited by 2-Na represents another example
of the subtle balance between the electrostatic anion/cation
interaction and the solvation of the cation by THF in
determining the extent of ion pairing in organometallic
complexes. Lithium and potassium ion pairing in 2 is
negligible based on the formation of stable Li+»nTHF
solvates and an insufficient K+ electrostatic potential
which minimizes anion/cation interactions.^5>97 Extensive
sodium ion pairing results from Na+,s intermediate
oxophilicity and electrostatic properties which favor
contact ion pairs.75>97 Band area measurements on the two,
overlapping asymmetric carbonyl stretching bands were
performed with the assumption that the area under each
absorption may be taken to be proportional to the relative
amount of each ion pair. Table 18 gives the Keq values for
the equilibrium defined in Eq. 12 from which a Van't Hoff
plot showing the variation of In Keq as a function of
temperature is readily constructed as shown in Figure 25.

e ^
144
O O
o o CO
£ o
O o CT>
© «) 0) <a M 2 > tt
CM
<# u sj o>

145
TABLE 18. Equilibrium Parameters for the Conversion of 2-Na from CIP into SSIPa.
1000/T in Keqb
3.36 0.405
3.36 0.456c
3.53 0.306
3.60 0.261
3.66 0.148
3.80 0.040
3.80 -0.012°
3.95 -0.136
4.20 -0.439
4.39 -0.571
4.59 -0.717
4.69 -0.719
4.93 -0.974
AH » 1.8 ± 0.06 kcal/mole^ AS = 7.0 ± 0.2 eu^
From 2.5 x 10-3 M [Cp*Re(CO)2Br] [Na] in THF by following the changes in the area of the 1790 and 1776 cm"1 carbonyl bands. Defined as the [area of solvent-separated ions]/ [area of carbonyl oxygen-sodium contact ions]. Multiple determination from a second separate experiment. Error limits at 95 % confidence limits.
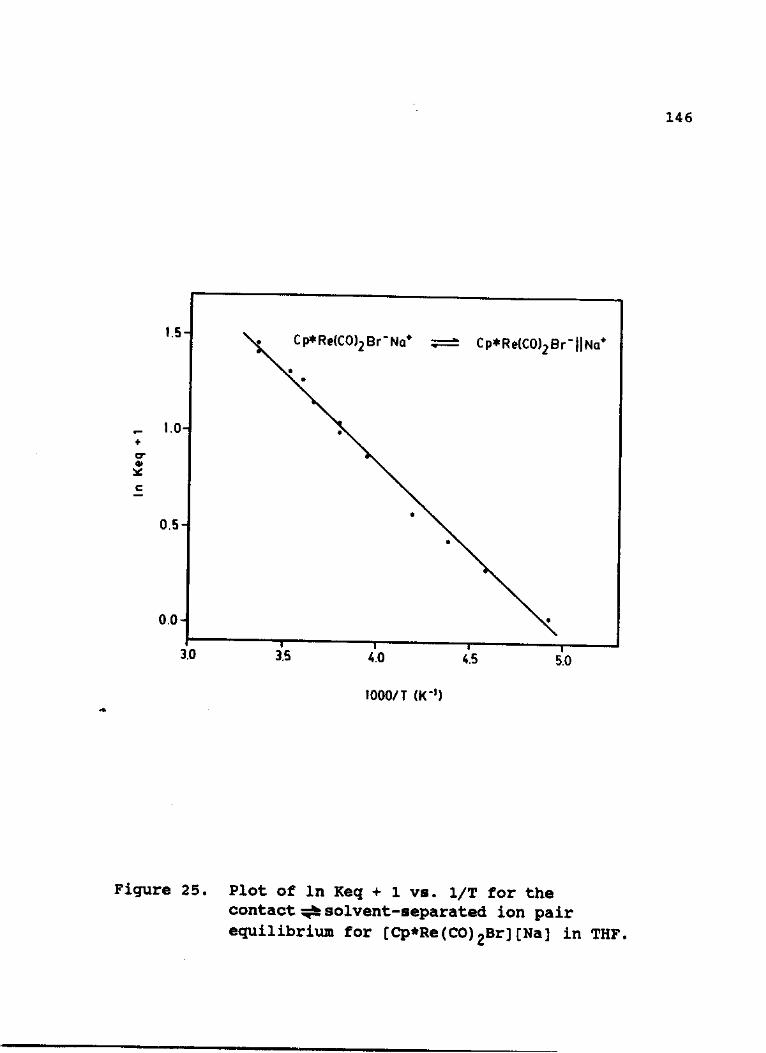
146
1.5-
- 1.0-• er XL c
0.5
0.0-
3.0
Cp*Re(CO)2Br_Na*
~r-3.5
Cp*Re(CO)2Br-||Na*
r~ 4.0
1000/T 0C')
Figure 25. Plot of In Keq + 1 vs. 1/T for the contact solvent-separated ion pair equilibrium for [Cp*Re(CO)2Br][Na] in THF,

147
Values of 1.8 ± 0.06 (kcal/mole) for AH and 7.0 ± 0.2 (eu)
for AS are computed from the slope of the straight line and
the intercept, respectively, it is immediately apparent
that the enthalpic and entropic contribution to the ion pair
equilibrium oppose each other. Low temperatures (< 263 K)
favor contact ion pairs as a result of an exothermic
electrostatic interaction between 2- and Na+ while higher
temperatures lead to solvent-separated ion pairs as the TAS
term dominates the free energy change associated with the
equilibrium. A similar temperature-dependent process has
also been reported for [Co(C0)4][Na] in THF by Edgell and
51a
Lyford. In that study the AH and AS values were observed
to be 3.7 (kcal/mole) and 4 (eu), respectively, for the
equilibrium defined analogously to our Eq. 12. Our
thermodynamic values compare well with those of Edgell and
Lyford, which indicate that the carbonyl oxygen-sodium
interaction in [Co(CO)4][Na] is stronger than in
[CpRe(CO)2Br][Na]. Finally, we note that the conversion of
contact ion pairs of 2-Na into solvent-separated ion pairs
displays a temperature response that is different from
several aromatic carbanions and complexes of macrocyclic
polyethers. For example, Jackman et al.^® have
demonstrated that sodium fluorenide exists primarily as
contact ions at room temperature (~ 95%) while potassium
fluorenide/18-Crown-6 solutions are predicted to exhibit

148
increased ion pairing as the temperature is raised above
room temperature. While an explanation concerning the
difference in AH for the conversion contact ions into
solvent-separated ions has been proposed®**, no current
theory exists that adequately explains such an inverse
temperature behavior between organic and organometallic
ions.
0. Reaction of Cp'Re(CO)2Br2 with BU3S11H and Et3SiH
(Cp' = Cp, Cp*)
The reaction of Cp'Re(CO)2Br2 with BU3S11H at room
temperature was discovered to afford the corresponding
dihydride in excellent yield and, thus, represents an
improved synthetic route for the synthesis of
Cp'Re(CO)2H2.49 Treatment of CpRe(CO)2Br2 (either isomer) in
benzene at room temperature with one equivalent of Bu^SnH
afforded four new i/CO bands at 2038 and 1970 cm"1; 2014 and
1939 cm"1 which are consistent with the diag-CpRe(CO)2Br(H)
(13) and diag-CpRe(CO)2H2 (14), respectively. Use of two
equivalents of Bu^SnH afforded complete conversion to 14
based on in situ IR analysis. These results indicate that
H"/Br"exchange occurs stepwise to produce the intermediate
complex 13, followed by a second H~/Br" exchange to give 14

149
complex (Eq. 24). The identity of 14 was also determined by
oc...r#...b I °bV \?O °£*>rV
Bu3SnH ^ 2 2 ? BujSnH J4 C°
VI I -v
oc->R#^Bf
(XT Br
OC->R*wH +" <24)
Br ^CO 2 BujSnBr
13
*H NMR spectroscopy. When a dg-benzene solution of
CpRe(CO)gBr£ (either isomer) was treated with two
equivalents of BujSnH at room temperature two new resonances
appeared at 4.35 and -9.68 ppm with an integral ratio of
5:2, respectively. Interesting enough, diag-CpRe(CO)2Br2
was observed to react faster with Bu3SnH than the
corresponding lateral isomer based on qualitative rate
measurements made using IR spectroscopy. Reaction of BugSnH
with lat-Cp*Re(CO)2Br2 was examined next. Use of one
equivalent of Bu^SnH led to the formation of two new vco
bands at 1999 and 1923 cm"1 which are readily to the
diagonal dihydride complex diag-Cp*Re(CO) 2H2 (15). The
yield of the dihydride was approximately 50 % with unreacted
dibromide accounting for the remaining material. The
intermediate complex diag-Cp*Re(CO)2Br(H) (16) was not

1 5 0
observed which is in direct contrast to the Cp analog where
the stepwise H /Br exchange was observed* This suggests
that the lifetime of the intermediate monohydride (16) is
very short and is followed by a faster subsequent H~/Br~
exchange reaction to ultimately give 15. The identity of 15
was also ascertained using NMR spectroscopy. Treatment
of lat-2 with two equivalents of Bu3SnH is dg-benzene gave
two resonances at 1.81 and -9.25 ppm with an integral ratio
of 15:2, respectively, consistent with the proposed
compound.
The dihydrides 14 and 15 could be converted directly to
the corresponding bis(tributyltin) complexes in a one-pot
synthesis using EtjN. Such a sequence undoubtedly involves
the deprotonation of the Re-H moiety to generate the
conjugate base, followed by anionic attack on the in situ
BujSnBr by-product present in solution. An analogous
reaction scheme using alkyl iodides has been examined by
37a
Bergman and Yang. The deprotonation/alkylation scheme is
shown in Equation 25.
Cp*R«(CO)2H2 +
2 Bu^SnBr
Cp* Re(CO)9(SnBu*)9 2 Bt*N . 4 C
3 , + (25) 2 Et3MHBr

151
The bis(tributyltin) derivatives were isolated by
chromatography, followed by sublimation in good to moderate
yield. IR analysis has been used to assign the stereo-
chemistry in both bis(tributyltin) compounds. Based on the
observed intensity pattern of the two vCO bands [1943 (m)
and 1890 (s) cm"1, ZOC-Re-CO - 118.4°] in the cyclopenta-
dienyl complex, diagonal stereochemistry is suggested for
CpRe(CO)2(SnBu3)2, 17 (Fig. 26a). This agrees with the
results of Graham et al. who have shown that reaction of
Me3SnCl with diag-CpRe(CO)2H2 gives diag-CpRe(CO)2(SnMe3)2.
41a
The same reaction using 15 leads to inversion of
stereochemistry and productive of the lat-Cp*Re(CO)2(SnBu3)2
(18). IR analysis reveals a reversed i/CO intensity pattern
[1967 (vs) and 1896 (s) cm"1, ZOC-Re-CO - 83°] indicative of lateral stereochemistry (Fig. 26b).
119 1
The Sn{ H) NMR spectra of both bis (tributyltin)
complexes were recorded in order to further characterize
these new complexes. The 119Sn chemical shifts of 17 and 18
were observed at -5.6 and 8.8 ppm, respectively, relative to
external Me4Sn (S - 0.0). Figure 27 displays the
representative 119Sn NMR spectrum.
Use of Et3SiH as a potential H donor was next examined
because silanes have been reported to function as reducing
agents.111 However, no reaction was observed between

152
1950 1850
v, cm -1
Figure 26. Infrared spectra of the carbonyl region for (a) diag-CpRe(CO)2(SnBu3)2 (b) lat-Cp*Re(CO)2(SnBu3)2 Both spectra were recorded at 25 °C in cyclohexane.

153
i i — i — i — i
100 0 -100
PPM
Figure 27. The Sn nmr spectrum of diag-CpRe (CO) 2 (SnBu3), at 25 °c in CDClj solution (0.1 M).

154
CpRe(CO)2Br2 (either isomer) and benzene at room
temperature. This observation stands in marked contrast to
the BUjSnH reactions which proceed readily at room
temperature. When the reaction between diag-CpRe(CO)2Br2
and EtjSiH (5 equiv.) was conducted at 60 °C the initial
products were diag-CpRe(CO)2Br(H) and CpRe(CO)3. Longer
reaction times led to an increase in the yield of the latter
compound at the expense of the former monohydride and
dibromide starting material. From IR analysis the solution
yield was 30-40 % and its formation is believed to arise
from the thermal decomposition of the monohydride (vide
supra). Equation 26 shows the proposed course of the
reaction.
CpRe(CO)2Br2 + EtgSiH diag-CpRe(CO)?Br(H) 60 °C
decomposition CpRe(CO)3
(26)
The reaction between lat-CpRe(CO)2Br2 and Et3SiH in
toluene was next examined at 40 °C where an initial fast
isomerization of the starting material to diag-CpRe(CO)2Br2
was observed, followed by a slower reaction to give

155
diag-CpRe (CO) 2Br(H) and CpRe(CO)g (vide supra). Evidence to
support an isomerization/H" exchange scheme was next probed
by examining this reaction using IR spectroscopy, if the
isomerization step does indeed precede (i.e., is faster) the
H exchange step, the rate of consumption of
lat-CpRe (CO) 2Br2 ®kould be faster than that observed with
diag-CpRe(CO)2Br2.
The isomerization reaction of lat-CpRe(CO)2Br2 was
studied in toluene solution both with and without added
silane by following the decrease in the absorbance of the
highest frequency vco band (symmetric vCO stretch) of the
starting material as a function of time. Changes in the
carbonyl absorbance (2046 cm"*) were negligible at 40 ° c in
the absence of added silane. On the other hand,
isomerization of lat-CpRe (CO) 2Br2 to diag-CpRe (CO) 2Br2 in
the presence of a measured excess of Et^SiH (10 equiv.)
proceeded rapidly at 40 °C and without any noticeable
Br /H" exchange. The isomerization reaction was complete
after eight hours at 40 °C (Fig. 2 8 ) .
Confirmatory proof for a slower H" exchange reaction
was obtained by qualitatively following the isomerization
reaction between diag-CpRe (CO) 2Br2 and Et3SiH. If the above
observations are correct, then the observed IR absorbance
changes for the consumption of diag-CpRe(CO)2Br2 should be

156
- o
o
00 E
- vo
- *4-
- CM
•sqv (00)A m
o o o
•P <d 0) c 0) 3 H 0 •p c •H
C\J CQ cm o o w 0) « a a •p •
(d E H •H
to M CO 0 4J W
m <W 0) 0 tn c a* (0 0 JS G 0
(0 a> 0 0 u c ai (0 JQ a)
£ 0 4J (0 3
g •H
oo CM 0) u §> •H

157
less than that observed with isomerization. The reaction
between diag-CpRe(CO)2Br2 and EtjSiH (10 equiv.) was studied
in toluene at 40 °C by monitoring the change in the
absorbance of the highest frequency vCO band (2000 cm"*).
Figure 29 shows these results. It is readily seen that the
consumption of diag-CpRe(CO)2Br2 is rather slow, requiring
three days for complete consumption. The consumption of
diag-CpRe(CO)2Br2 is clearly slower than the isomerization
reaction.
While these reactions with Bu^SnH and EtjSiH have not
been studied kinetically it is of interest to consider these
reactions on thermodynamic grounds in order to better
understand why the observed dihydride products formed. This
is readily done when the bond dissociation energies of the
participating reactants are examined. Equations 27 and 28
show the overall reactions under consideration.
I 2 Bu3snH + 2 Bu3snBr
0C> R'vJ3r 0 C > R , C - H U 7 )
Br ^ C 0 H ^ C 0
AH • 2 (BDE Re-H + BDE Sn-Br) - 2 (BDE Re-Br + BDE Sn-H)
AH » - 26 kcal/mole

158
-t 00
k.
CSI -<=
£ o ^
00 -4-
<£> CO
-<r cn
— CM
A •P • •H <D * C
0) eg 5 & H CQ o CNJ.p
1-5
« o° & 4.3 <G jj •h (d v bi w" 0 (Q <H 0) 3 -<D 0) tJ> c T3 2 H Xi o o (H Q> O H i <0 •9 w M -H S a„ 5 w
<n oa Q)
•sqv (oo) /i a i
•H

159
Table 19 lists the bond dissociation energies (BDE) needed
to evaluate the reactions shown in Equations 27 and 28.
While accurate measurements for the Sn-H (74 kcal/mole),
and Si-Br (96 kcal/mole) bond exist, no actual
2 Et38iH C C l T
+ 2 Et3SiBr
°bc;>%BO V%O <28>
- AH = 2 (BDE Re-H + BDE Si-Br) - 2 (BDE Re-Br + BDE Si-H)
AH - - 30 kcal/mole
measurements are available for the strengths of the Re-H and
Re-Br bonds. However, we may readily estimate the values
associated with these bonds by using similar model
compounds. For example, Cp*Ir(PMe3)(Br)-Br114 and
115
Cp2W(Br)-Br have bond dissociation energies of 76 and 72
kcal/mole, respectively. Based on these values for the M-Br
bond, a BDE value of 70 kcal/mole has been assumed for the
Re-Br bond in diag-CpRe(CO)2Br2. A similar situation exists
with respect to the BDE of the Re-H bond in
diag-CpRe(CO)2H2. Based on the reported M-H BDE values
given in Table 19 the Re-H BDE in diag-CpRe(CO) 2H2 may be

160
0) 0 G 0) k 0) «
ra H H
n in vo
w <D •H Cn o c w c o •H -P (d •H O 0 W CO •H Q
TJ C o A •g 0)
O <D H 0) CO
OH H
3 cq $
& a) c w c o •H <d •H o o (0 m
*0 c <§
0) r-4
S
(0 o M
in oo CO
vo Ol r*
vo r*
n CM oo oo
U CQ
S l l ^ ^ u « CQ
TJ a 3 o & o a
CO 0) CO
a)
M
u U £ s i w CQ « cq P* p« S. M 1 1 1 i W s CQ a c •H •H u «W <w CO CO CO CO H H s £ CO CO CO CO * * -K * a) se S 1 S3 & a o 04 o
U CQ

161
confidently estimated at -70 kcal/mole. This Re-H BDE value
is acceptable since it is known that M-H BDE's are not
particularly dependent upon the metal or the nature of the
ancillary ligands.3*'*^
The reaction in Equation 27 is calculated to proceed to
dihydride and BujSnBr based on the favorable enthalpy value
(AH » -26 kcal/mole). The driving forces in this transfor-
mation are the formation of two strong Re-H and Sn-Br bonds.
Reaction with silane (Equation 28) is also predicted to
proceed to diag-CpRe(CO) 2H2 and Et3SiBr based on an
exothermic enthalpy value of -30 kcal/mole. However, no
dihydride is observed presumably due to the fast and
irreversible decomposition reaction associated the
intermediate monohydride complex diag-CpRe(CO)2(Br)(H).
The thermodynamics associated with the bis(tributyltin)
complex diag-CpRe (CO)2(SnBuj) 2 are of interest because the
bis(tributyltin) complex is predicted to be unstable
according to Equation 29. The BDE values used for the Re-H
^7
+ 2 HBr (29) 2 Bu^SnBr |
0C-> R*CH 0C->R,^SnBu3 H ^ C 0 BuaSrr CO
AH - 2 (BDE Re-Sn + BDE H-Br) - 2 (BDE Re-H + BDE Sn-Br)
AH « 54 kcal/mole

162
and Sn-Br bonds have already been described while the BDE
value for HBr is given in Table 19. The only BDE value that
is unknown belongs to the Re-Sn bond in the product. Based
on the fact that metal-alkyl bond strengths are weaker than
the corresponding metal-hydride bonds,3a coupled with the
fact that many metal-alkyl BDE's are on the order of 20-40
kcal/mole, we have assured that the BDE of the Re-Sn bond in
diag—CpRe(CO)2(SnBUj)2 is ~40 kcal/mole. Using these values
the reaction as written in Equation 29 is observed to be
endothermic by 54 kcal/mole consistent with the observation
that no reaction occurs without the addition of EtjN (vide
supra). The base serves to drive the reaction to be right
by removing the generated HBr as [EtjNH][Br].
P. Hydride Reduction of Indeny Rhenium Tricarbonyl:
Synthesis and Characterization of [ (r?5-CgH7)Re(CO) 2H]"
The reaction of (f?5-CgH7)Re(CO)3, 19, with 1.0 mole
equiv. of LiEt^BH at room temperature led to the immediate
formation of [ (n5-C9H7)Re(CO)2H]", 20, in 50 % yield as
determined by IR analysis. When the hydride concentration
was doubled, complete conversion to 20 was observed. 20
exhibits two vCO bands at 1991 and 1906 cm"1. At no time
were any possible intermediates obtained in this reaction.

163
The identity of this new complex was characterized by NMR
spectroscopy. *H NMR spectrum of the yellow solution
obtained from the treatment of 19 with LiEtjBH in dg-THF is
shown in Figure 30. Two sets of resonances associated with
the AA'BB' spin system are observed at 7.24 and 6.43 ppm
with a relative integral ratio of 2:2 which are consistent
with four protons on an uncomplexed six-membered benzenoid
ring. The other two sets of resonances appearing at 6.55
and 5.64 ppm with a relative integral ratio of 1:2
correspond to the three protons attached to the five-
membered ring in the fj -indenyl ligand. The terminal metal
hydride resonance appeared at -5.5 ppm as expected for a
singlet with a relative intensity of one.
The reduction of (rj -CgHjJRefCOJj requires two equiva-
lents of LiEtjBH (vide supra). It is believed that one
equivalent of the hydride reagent reacts with
(T75-C9H7)Re(CO)3 to give [ (n5-CgH7)Re(CO)2H]" and Et^B as
shown in Scheme 8. A subsequent, rapid reaction between the
generated EtjB and excess R3BH" would then yield the dimer
[EtjB-H-BEtj]" and account for the 1:2 rhenium/hydride
stoichiometry observed with this reaction.107b»118 An
5 1
alternative pathway involves r) -* r? ring slippage of
indenyl ring in 19 upon hydride attack, followed by a fast
loss of CO to give 20. Since the presence of the fused

164
ift IO I
1
= 1
in
© - k
B I
*0° c •H o o in ca •P <d
K C\l
O u w 0) «
a as 0 1
IO c
o cn a) u & •H

165
Schema 8
o c / \
OCT CO
+ 2Et3BH
19
(Et3BJ2H"
R® H ocy \ oc f CO
21
benzene ring in t -CgHy complexes results in more a facile
5 3 .
r\ -*• r\ ring slippage for indenyl ligands than for
cyclopentadienyl ligands, coupled with the observation of
other slipped indenyl rings,47b we cannot rule out this

166
pathway based on the existing experimental data.
Finally, THF solutions of [ (rj5-CgH7)Re(CO) 2H]" were
observed to be stable when oxygen was rigorously excluded.
The functionalization of [ (»?5-C9H7)Re(CO)2H]" remains to be
explored.

167
REFERENCES
1. (a) Halpern, J. Adv. Chem. Ser, 1966, 7Q., 1. (b) Halpern, J. Discuss. Faradav Sac. 1968, 7, 46. (c) Parshall, G. W. Acc. Chem. Rea. 1975, 5, 114. (d) Parshall, G. W. Catalysis (Chemical Society Specialist Periodical Report) 1977, 1, 325. (e) Shilov, A. E.; Steinman, A. A. Coord. Chem. Rev. 1977, Zk, 97. (f) Webster, D. E. Adv. Oraanomet. che^T 1977, 147.
2. Chatt, J.; Davidson, J. M. J. Chem. Soc. 1965, 843.
3. (a) Halpern, J. Inorcr. Chim. Acta. 1985, 100. 41. (b) Crabtree, R. H. Chem. Rev. 1985, ££, 245. (c) Shilov, A. E. "Activation of Saturated Hydrocarbons by Transition Metal Complexes", D. Reidel Publishing Company: Boston, 1984.
4. (a) Walling, C. "Free Radicals in Solution", Wiley: New ¥ork, 1957. (b) Huyser, E. S. "Free Radical Chain Reactions", Wiley: New York, 1970. (c) Pryor, W. A. "Free Radicals". McGraw-Hill: New York, 1966.
5. (a) Cohen, Z.; Keinan, E.? Mazur, Y.? Haim, V. T.
Qrqt Chefflt 1975, 2141. (b) Deno, N. C.J Jedziniak, E. J.? Messer, L. A.; Meyer, M. D.; Stroud, S. G.; Tomezsko, E. S. Tetrahedron 1977, 12, 2503. (c) Olah, G. A.? Parker, D. G.; Yoneda, N. Ancrew. Chem. Int. Ed. Engl. 1978, H , 909. (d) Rozen, S.;
Gal, C.; Faust, Y. J. Am. Chem. Soc. 1980, 102. 6861.
6. (a) Bruce, M. I. Anqew. Chem. Int. Ed. Rnci. 1977, l£,
73. (b) Foley, P.; Discosimo, R.; Whitesides, G. M. Am. Chem. Soc. 1980, 102. 6713. (c) Whitesides, G.
M- Pyre AFPlt Chemt 19«1, 52, 287. (d) Reamey, R. H.; Whitesides, G. M. J. Am. Chem. Soc. 1984, lag, 81. (e) Calabrese, J. c . ; Colton, M. C.; Herskovitz, T.; Klabunde, U.; Parshall, G. W.; Thorn, D. L.; Tulip, T. H- Annt y, Y, A<?<Pdt ggit 1983, U S , 302. (f) Parshall, G- W. CHEM. TECH. 1984, 628. (g) Anderson, R. A.;

168
Jones, R. A.; Wilkinson, G. J. Chem. Soc.. Dalton Transt 1978, 446. (h) Werner, H.; Werner, R. J. Oraanomet. Chem. 1981, 209. C60.
7. Shilov, A . E. Pure APPI. Chem. 1 9 7 8 , 725.
8. (a) Crabtree, R. H.? Mellea, M. F.? Mihelcic, J. M.; Quirk, J. M. J. Am. Chem. Soc. 1982, 1 M , 107. (b) Crabtree, R. H. CHEM. TECH. 1982, 506. (c) Crabtree, R. H.; Demou, P. C.; Eden, D.; Mihelcic, J. M.; Parnell, C. A.; Quirk, J. M.? Morris, G. E. J. Am. Chem. Soc. 1982, 1M/ 6994. (d) Crabtree, R. H.; Parnell, C. P.; Uriarte, R. J. Oraanometallics 1984, 2, 816.
9. (a) Baudry, D.; Ephritikhine, M.; Felkin, H. J. chem. Soc.. Chem. Commun. 1980, 1243; 1982, 606; 1982, 1235. (b) Baudry, D.; Ephritikhine, M.; Holmes-Smith, R. J. Chem. Soc.. Chem. Commun. 1983, 788.
10. (a) Berry, M.; Elmitt, K.; Green, M. L. H. J. Chem. Soc.. Dalton Trans. 1979, 1950. (b) Bradly, M. G.; Roberts, D. A.; Geoffroy, G. L. J. Am. Chem. Soc. 1981, 103. 379. (c) Ittel, S. D.; Tolman, C. T.; English, A. 0.; Jesson, J. P. J. Am. Chem. Soc. 1978, 100. 7577.
11. Hoyano, J. K.; Graham, W. A. G. J. Am. Chem. Soc. 1982, IQi, 3723; 1983, lfi£, 7190.
12. (a) Janowicz, A. H.; Bergman, R. G. J. Am. Chem. Soc. 1983, 105. 3020. (b) Wax, M. J.; Stryker, J. M.; Buchanan, J. M.; Kovac, C. A.; Bergman, R. G. J. Am. Chem. Soc. 1984, 1££, 1121.
13. (a) Jones, W. D.; Feher, F. J. J. Am. Chem. Soc. 1984, JJ2£, 1650. (b) Jones, W. D.; Feher, F. J. Oraanometallics 1983, 2, 562.
14. For reviews on C02 activation by organometallic complexes and the manifestations of the "Greenhouse Effect", see: (a) Behr, A. in "Catalysis in Cj Chemistry", Keim, W.; Ed.; D. Reidel Publishing Co.: Boston, 1983; CH. 6. (b) Volpin, M. E.; Kolomnikov, I. S. Pure APPI. Chem. 1 9 7 3 , 22, 567. (c) Woodwell, G. M.

169
Sclent. Amer. 1978, 233., 34. (d) Volpin, M. E. ? Kolomnikov, I. s. in "Organometallic Reactions", Becker, E. S.; Tsutsui, M., Ed.? Wiley: New York,
Vplt—1/ 313—386. (e) Sneeden, R. P. A. in "Comprehensive Organometallic Chemistry", Wilkinson, G.; Stone, F. G.; Abel, E. W., Ed., Pergamon Press: London, 1982; CH 50. (f) Evans, G. 0.; Walter, W. F.; Mills, D. R.; Streit, C. A. J. Orcranomet. Cham. 1978, Ml/ C34. (g) Forshner, T.; Menard, K.; Culter, A. J. Chem. Soc.. Chem. Commun. 1984, 121. (h) Felkin, H.; Knowles, P. J. J. Oraanomet. Chem. 1972, 21, C14. (i) Felkin, H.; Knowles, P. J.; Meunier, B. J, QrqanPPett 1978, 146, 151. (j) Davies, S. G. ; Green, M. L. H. J. Chem. Soc.. Dalton Trans. 1978, 1510.
15. Lowry, R. P.; Auguilo, A. Hydrocarbon Proc. 1974, 103.
16. Roth, J. F.; Craddock, J. H.? Hershman, A.; Paulik, F. E- CHEM. TECH. 1971, 1, 600.
17. Foster, D. Adv. Oraanomet. Chem. 1979, 12, 255.
18. Broich, F.; Hofermann, H.; Hunsmann, W.; Simmrock, H. Erdol Kohle-Erdaas Petrochemie. 1963, 1&, 272.
19. (a) Hoffmann, R. Science (Washington. D. c.\ 1981, 211, 995. (b) Hoffmann, R. Anaew. Chem. Int. Ed. Enqlt 1982, 21, 711.
20. For other analogous processes employing iridium complexes and reviews on C-H bond activation, see: (a) Newman, L. J.; Bergman, R. G. J. Am. Chem. Soc. 1985, 5314. (b) Faller, J. W.; Felkin, H. Oraanometa11ics 1985, ±, 1488. (c) Burk, M. J.; Crabtree, R. H.; Parnell, C. P.; Uriarte, R. J. Qrqan<?ffletain<?? 1984, 2, 8I6. (d) Bergman, R. G. ggj^n<r? 1984, 221, 902. (e) Ephritikhine, M.
NQUVt Chlfflt 1986, lfl, 9. (f) Janowicz, A. H.; Periana, R. A.; Buchanan, J. M.? Kovac, C. A.; Stryker, J. M.; Wax, M. J.? Bergamn, R. G. Pure APPI. Chem. 1984, 108. 1537.

170
21. Fagan, P. J.; Manriquez, M. J.; Maries, T. J. in "0rganometalli.es of Elements"; D. Reidal: Pordrecht, 1979, p. 113.
22. Green, H. L. H.; Wilkinson, G. J. chem. Soc. 1958, 4314-17.
23. Nesmeyanov, A. V.; Anisimov, K. N.? Koloboba, N. E.; Baryshikov, L. I. Izv. Akad. Nauk SSSP. ntd. Khim. Nauk 1963, 193.
24. Zdanovich, V. I.? Lobanova, I. v.; Kolobova, N. E. IZYf Akad. Nauk SSSR. Ser. KMm. 1984, H i , 2149.
25. Fitzpatrick, P. J.; Le Page, Y.; Sedman, J.; Butler, I. s* Inora. Chem. 1981, 2852.
26. Heah, P. C.; Poatton, A. T.; Gladysz, J. A. J. Am. Chem. Soc. 1986, 108. 1185.
27. (a) Bercaw, J. E.; Marvich, R. H.; Bell, L. G.; Brintzinger, H. H. J. Am. Chem. Soc. 1972, M , 1219. (b) Bercaw, J. E. J. Am. Chem. Soc. 1974, 9£, 5087. (c) Thomas, J. L.; Brintzinger, H. H. J. Am. chem.
1972, 91, 1386. (d) Thomas, J. L. J. Am. Chem. Soc. 1973, 25, 1838.
28. Nesmeyanov, A. N.? Kolobova, N. E.; Makarov, Y. V.; Anisimov, K. N. Izv. Akad. Nauk sssr. ser. Khim. 1969, 1826.
29. (a) King, R. B.; Reimann, R. H.; Darensbourg, D. J.
1975, 21, C23. (b) King, R. B.; Reimann, R. H. Inora. chem. 1976, 15, 179.
30. Kolobova, N. E.; Valueva, Z. P.; Kazimirchuk, E. I. IZVf Akad. Nauk SSSR. Ser. Khim. 1981, 408.
31. Dong, D. F.; Hoyno, J. K.? Grasham, W. A. G.; Can. J. Chem. 1981, ££, 1455.
32. Einstein, F. W. B.; Klahn-Oliva, A. H.; Sutton, D.; Tyers, K. G. Oraanometallica 1986, 5, 53.

171
33. Lokshin, B. V.; Rusach, E. B.; Valueva, Z. P.; Ginzburg, A. G.; Kolobova, N. E. J. Oraanomet. Chem. 1975, U22, 535.
34. Nesmeyanov, A. N.? Kolobova, N. E.; Makarov, Y. V. ; Anisimov, K. N. Zh. Obshch. Khim. 1974, M , 2222.
35. Aleksandrov, G. G.; Struchkov, Y. T.; Makarou, Y. V. Zht KhiWt 1973, n , 86.
36. Nesmeyanov, A. N.; Kolovoba, N. E.; Makarov, Y. V.; Kazimirchuk, E. I.; Anisimov, K. N. Izv. Akad. Nauk SSSR. Ser. Khim. 1976, 159.
37. (a) Yang, G. K.; Bergman, R. G. J. Am. Chem. Soc. 1983, 1££, 6500. (b) Yang, G. K.; Bergman, R. G. Oraanometallics 1985, ±, 129.
38. Casey, C. P.; Rutter, E. W., Jr.? Haller, K. J. J. Am. Chem. Soc. 1987, 1££, 6886.
39. Kristjansdottir, S. S.? Moody, A. E.; Weberg, R. T.; Norton, J. R. Oraanometallics 1988, 7, 1983.
40. Bursten, B. E.? Gatter, M. G. Oraanometallics 1984, 3, 941-943.
41. For examples of other four-legged piano-stool complexes based on the CpRe fragment, see: (a) Hoyano, J. K.; Graham, W. A. G. Oraanometallics 1982, 1, 783. (b) Fischer, E. O. ? Frank, A. Chem. Ber. 1978, 3740. (c) Bergman, R. G.? Seidler, P. F.? Wenzel, T. T. J. Am. Chem. Soc. 1985, 1£I,4359. (d) Wenzel, T. T.; Bergman, R. G. J. Am. Chem. Soc. 1986, I M , 4856. (e) Graham, W. A. G. J. Oraanomet. Chem. 1986, 300. 81. (f) Pasman, P.? Snel, J. J. M. J. Oraanomet. Chem. 1986, 2Q1, 329. (g) Jones, W. D.? Maguire, J. A. Qrqangffigtalligg 1986, 5, 590. (h) Baudry, D. H.; Ephretikhine, M. J. Chem. Soc.. Chem. Commun. 1980, 249. (i) Goldberg, K. I.; Bergman, R. G. Oraanometallics 1987, £, 430. (j) ibid, J. Am. Chem.
1989, H I , 1285.
42. Hoyano, J. K.? Graham, W. A. G. J. Chem. Soc.. Chem. Commun. 1982, 27.

172
43. (a) Klahn-Oliva, A. H.? Singer, R. D.; Sutton, D. J. Afflt Cheffit g9<rt 1986, 1M, 3107. (b) For related chemistry, see: Klahn, A. H.; Sutton, D. Oraanometallics 1989, &, 198.
44. Barrientos-Penna, C. F.? Klahn-Oliva, A. H.; Sutton, D. Oraanometallics 1985, 1, 367.
45. Einstein, F. W. B.; Jones, R. H.; Klahn-Oliva, A. H.; Sutton, D. Oraanometallics 1986, 5, 2476.
46. King, R. B.; Efraty, A. J. Oraanomet. Chem. 1970, 22, 527.
47. (a) Casey, C. P.; O'Connor, J. M. Oraanometallics 1985, 1, 384. (b) O'Connor, J. M.; Casey, C. P. Chem. Rev. 1987, £7, 307.
48. Yezernitzkaya, M. G.? Lokshin, B. V.; Zdanovich, V.I.; Lobanova, I.A. J. Oraanomet. Chem. 1985, 222,363.
49. Lee, S. W.; Richmond, M. 6. Unpublished results.
50. King, R. B. Adv. Oraanomet. Chem. 1964, 2, 157.
51. (a) Edgell, W. F.; Lyford, J., IV. J. Am. Chem. Soc. 1971, 21, 6407. (b) Edgell, W. F.; Hegde, S.; Barbetta, A. J. Am. Chem. Soc. 1978, lfifl, 1406. (c) Edgell, W. F.? Chanjamsri, S. J. Am. Chem. Soc. 1980, 102. 147.
52. Pribula, C. D.? Brown, T. L. J. Oraanomet. Chem. 1974, 21/ 415.
53. (a) Darensbourg, M. Y•j Darensbourg, D. J.y Burns, D* r Drew, D. A. J. Am. Chem. Soc. 1976, M , 3127. (b) Darensbourg, M. Y.; Darensbourg, D. J.; Barros, H. L. C. Inoro. Chem. 1978, 12, 297. (c) Darensbourg, M. Y.; Hanckel, J. M. J. Oraanomet. £h£BL. 1981, 212, C9; Qrqanpffigtalligg 1982, 1, 82.
54. Darensbourg, H. Y.; Jimenez, P.? Sackett, J. R.? Hanckel, J. M.; Kump, R. L. J. Am. Chem. Soc. 1982, 1M» 1521.

173
55. Schriver, D. F. "The Manipulation of Air-sensitive Compounds", McGraw-Hill: Yew York, 1969.
56. Fieser, L. F.; Fieser, M. "Reagnets for Organic Synthesis", Wiley: New York, 1967: Vol.1, p. 967.
57. Eisch, J. J.; King, R. B. "Organometallic Syntheses", Academic Press: New York, 1965: pp. 70-71.
58. Weber, W. P.; Gokel, G. W. ? Ugi, I. K. Ancrew. Chem. I n t * E a , E n g l , 1 9 7 2 , £ .
59. Kuivila, H. G. and Beumel, O. F. Jr., J. Am. Chem., Soc. 1 9 6 1 , SI, 1246.
60. Freeman, R.? Morris, G. A. J. Chem. Soc.. Chem. CQPPVmt 1 9 7 8 , 6 8 4 .
61. Bax, A.; Morris, G. A. J. Maan. Reson. 1981, 42, 501.
62. Germain, G.: Main, P.; Wolfson, M. N. Acta Crvstlloqr.. Sect. A: Crvst. Phvs. Diffr. Theor. Gen. Crystalloar. 1971, hZL, 368.
63. International Tables for X-Ray Crystallography; Kynoch: Birmingham, England, 1974, Vol. IV.
64. Program STEPT, Chander, J. P., Oklahoma State Univ. Quantum Chemistry Exchange Program, #307.
65. Gordon, A. J.; Ford, R. A. "The Chemist's Companion. A Handbook of Practical Data, Techniques, and References", Wiley: New York, 1976.
66. Cotton, F. A.? Kraihanzel, C. S.; J. Am. Chem. Soc.. 1962, M # 4432.
67. King, R. B. ? Efraty A. J. Oraanomet. Chem. 1969, 2SL, 264.
68. Fayer, M. D.; Harris, C. B. Inora. Chem. 1969, fi, 2792.
69. (a) Brown, T. L.? Darensbourg, D. J. Inora. Chem. 1967, £, 971. (b) Beck, W.; Melnikoff, A.; Stahl, R. Anaew. Chem. 1965, 21, 719. (c) Mannig, A. R. J. Chem. Soc. 1967, &, 1984.

174
70. Hieber, W.; Schulten, H. Z. Anoq. Alia. Chen. 1939, Ml, 164.
71. Hieber, W.? Schuh, R.; Fuchs, H. Z. Anora. Alia. Chem. 1 9 4 1 , 21£, 243.
72. Schmit, S. P.; Troger, W. C.; Basolo, F. Inora. Syn. 1 9 8 5 , Vol. 21, 41.
73. Anderson, G. K.? Cross, R. J.; Phillips, I. G. J. Chem. Soc.. Chem. Commun. 1978, 709.
74. Cotton, F. A.; Wilkinson, G. "Advanced Inorganic Chemistry", 3rd Ed.; Wiley: New York, 1972; Chapter 22.
75. Darensbourg, M. Y. Proa. Inora. Chem. 1985, 21, 221.
76. Pannel, K. H.; Raghu Veer, K. S.; DelBene, J. E.; Nathan, F. J. Am. Chem. Soc. 1987, 109. 4890.
77. Brown, D. W.; Nakashima, T. T.; Robenstein, D. L., J. Maan. Reson. 1981, 41, 302.
78. Jakobsen, H. J.; Sorensen, 0. W.; Brey, W. S.; Kanyha, P. J. Maan. Reson. 1982, Ifi, 328.
79. Robenstein, D. L.; Nakashima, T. T. Anal. Chem. 1979, 11, 1465A.
80. Maudsley, A. A.; Huller, L.; Ernst, R. R. J. Maan. Reson. 1977, 21, 463.
81. Bodenhausen, G.; Freeman, R. J. Maan. Reson. 1977, 21, 471.
82. Ibid. J. Am. Chem. Soc. 1978, 100. 320.
83. Benn, R.; Gunther, H. Anaew. Chem. Int. Ed. Enal. 1 9 8 3 , 21, 350.
84. Martin, G. E.; Zekter, A. S. Maan. Reson. Chem. 1988, 21, 631.
85. Bird, P. H.; Churchill, M. R. Chem. Commun. 1967, 777.
86. Cotton, F. A.; Wing, R. M. Inora. Chem. 1965, 4, 1328.

175
87. (a) Elian, M.; Hoffmann, R. Inora. Chen. 1975, 1±, 1058. (b) Elian, M; Chem, M. M. L.; Hingos, D. M. P.; Hoffmann, R. InS£SU_eh61IU. 1976, 15, 1148. (c) Schilling, B. E. R.; Hoffmann, R.; Lichtenberger, D. L. J. Am. Chem. Soe. 1979, 101. 585.
88. (a) Smith, R. A.; Bennett, M. J. Acta Crvstallor.
B/ 1977, fill, 1113. (b) Harrison, W.; Trotter, J« Jt Chem. SQC.. Dalton Trans. 1972, 678. (c) Kreissel, F. R.; Friedrich, P. Anaew. Chem. Int. Erf. Engl* 1977, l£, 543. (d) Einstein, F. W. B.; Tyers, K. G.; Sutton, D. Oraanometa11 i as 1985, 489. (e) Barrientos-penna, C. F.; Einstein, F. W. B.; Jones, T.; Sutton, D. Inora. Chem. 1982, 21, 2578.
89. Fitzpatrick, P. J.? Le Page, Y.; Butler, I. S. Acta ClYSt- 1981, fi21, 1052.
90. (a) Krebs, B.; Hasse, K. D. Acta Crvst. 1976, B32. 1334. (b) Lock, C. J. L.; Turner, G. Acta Crvst. 1975, fill/ 1764.
91. (a) Lokshin, B. v.; Rusach, E. B.; Kolobova, N. E.; Makarov, Y. v.? Ustynuk, N. A.; Zdanovich, V. I.; Zhakaeva, A. Z.; Setkina, V. N. J. Qraanomet. chem. 1976, JLQSL# 353. (b) Lokshin, B. V.; Pasinsky, A. A.; Kolobova, N. E.; Anisimov, K. N.; Makarov, Y. V. J. Qraanomet. Chem. ia73r 315.
92. Kristjansdottir, S. S.; Moody, A. E.; Weberg, R. T.y Norton, J. R. Orqanometallles. 1988, 7, 1783.
93. (a) Nesmeyanov, A. N.? Anisimov, K. N.? Kolobova, N. E.; Varyshikov, L. I. Izv. Akad. Nauk SSSR. Ser. Khim. 1963, 193. (b) Tam, W.; Lin, G. Y.; Wong, W. K.; Kiel, W. A.; Wong, W. K.; Gladysz, J. A. J. Am. chem. §<?g. 1982, 141.
94. (a) Reger, D. L.? Belmore, K. A.; Atwood, J. L.; Hunter, W. E. J. Am. Chem. Soc. 1983, 105. 5710. (b) Bullock, R. M.; Headford, C. E. L.? Kegley, S. E.; Norton, J. R. J. Am. Chem. Soc. 1985, 107. 727. (c) McNally, J. p.; Gluek, D.? Cooper, N. J. J. Am. chem.
1988, Hfl, 4838. (d) Jones, W. D.; Maguire, J. A. Oraanometa11les 1987, 1301.

176
95. (a) Jones, W. D.? Feher, F. I. J. Am. Chem. Soc. 1982, 101, 4240? 1985, lfll, 620? 1986, 10&, 4814. (b) Janowicz, A. H. ? Bergman, R. G. J. Am. Chem. soc. 1983, 105, 3929. (c) Periana, R. A.? Bergman, R. G. J. Am. Chem. Soc. 1986, 1£&, 7332? 7346. (d) Jones, W. D.? Feher, F. J. Inora. Chem. 1984, 21, 2376. (e) Periana, R. A.? Bergman, R. G. Oraanometalllcs 1984, 2, 508.
96. Lukehart, C. M. "Fundamental Transition Metal Organometal1ic Chemistry1", Brooks/Cole Publishing Co.: Monterey, CA. 1985.
97. Marcus, Y. "Ion Solvation", Wily: New York, 1985.
98. (a) Pannell, K. H.? Jackson, D. J. Am. Chem. Soc. 1976, 4443. (b) Hogen-Esch, T. E. ? Smid, J. J. Am. Chem. Soc. 1966, M , 307? 318. (c) Takaki, U. ? Hogen-Esch, T. E.? Smid, J. J. Am. Chem. Soc. 1971, 22, 6760. (d) Chan, L. L.? Wong, K. H.? Smid, J. J. Am. Chem. Soc. 1970, 22, 1955.
99. Mann, B. E. ? Taylor, B. F. "13C NMR Data for Organometallic Compounds", Academic Press: New York, 1981.
100. (a) Coates, G. E.? Wade, K. "Organometallic Compounds ; The Main Group Elements", 3rd Ed.? Methuen & Co.: London, 1967, Vol. 1. (b) Ashby, E. C. Quart. Rev. 1967, 21, 259. (c) Ashby, E. C. Pure APPI. Chem. 1980 , 52., 545.
101. (a) Schunn, R. A. in "Transition Metal Hydrides". Ed. by Muetterties, E. L.? Marcel Dekker: New York, 1971? CH. 5. (b) Edidin, R. T.? Sullivan, J. M.? Norton, J.
J. Am. Chem. Soc. 1987, 102, 3945. (c) Jordan, R. F.? Norton, J. R. ACS Svmp. Ser. 1982, No. 198, 403, and references therein, (d) Moore, E. J.? Sullivan, J. M. ? Norton, J. R. J. Am. Chem. Soc. 1986, IM, 2257. (e) Walker, H. W.? Kresge, C. T.? Pearson, R. G.? Ford, P* C. J. Am. Chem. Soc. 1979, 101, 7428. (f) Walker, H. W.? Pearson, R. G.? Ford, P. C. J. Am. chem. Soc. 1983, 10£, 1179.

177
102. Muller, A.; Krickemeyer, E.; Boggle, H. A. Anora. Allgt Ch lRt 1987. $§4, 61.
103. (a) Okuda, J.; Herdtweck, E.? Herrmann, W. A. Inora. Chem. 1988, 21, 1254. (b) Herrmann, W. A.; Floel, M.; Herdtweck, E. J. Oraanomet. Chem. 1988, 2 M , 321. (c) Hermann, W. A.; Fischer, R. A.; Herdtweck, E. Anaew. Chem. Int. Ed. Enal. 1988, 21, 1509? 1987, 23., 1263.
104. Kubacek, P.; Hoffamnn, R.? Havlas, Z. Oraanometallics 1982, 1, 180.
105. (a) Hermann, W. A.; Serrano, R.; Bock, H. Anaew. Chem. Int. Ed. Enal. 1984, 21, 383. (b) Hermann, W. A.; Voss, E.; Floel, M. J. Oraanomet. Chem. 1985, 231, C5.
106. (a) Amatore, C.; Verpeaux, J, N.; Krusic, P. J. Oraanometallics 1988, 2, 2426. (b) Kelly, R. S.; Geiger, W. E. Oraanometallics 1987, £, 1432. (c) Reike, R. D.? Henry, W. P.? Arney, J. S. Inora. Chem. 1987, 23., 420. (d) Cyr, J. C.; DeGray, J.A.; Gosser, D. K.? Lee, E. S.; Reiger, P. H. Oraanometallics 1985.
950. (e) Leong, V. S.; Cooper, N. J. Oraanometallics 1987, 3, 2000; 1988, I, 2080. (f) Maher, J. M.; Beatty, R. P.; Cooper, N. J. PrqanQmetalligg 1985, i, 1354.
107. (a) Albright, T. A.; Burdett, J. K.; Whango, M. H. "Orbital Interactions in Chemistry," Wiley: New York, 1985; pp. 282-288. (b) Purcell, K. F.; Kotz, J. C. "Inorganic Chemistry", W. B. Saunders Co.: Philadelphia, 1977; CH. 9.
108. (a) Saveant, J. M. &<?<?, Cheit 1980, 12, 323. (b) Andrieux, C. P.; Merz, A.; Seveant, J. M. J. Am. Chem. Soc. 1985, 107. 6097. (c) Andriex, C. P.; Gallardo, I.; Seveant, J. M.; Su, K. B. J. Am. Chem. Soc. 1986, 108. 638. (d) Kadish, K. M.; Lacombe, D. A.; Anderson, J. E. Inora. Chem. 1986, 25. 2074; 2246. (e) Moulton, R.; Weidman, T. W.? Vollhardt, K. P. C.; Bard, A. J. In<?rq. chemt i98«, 25., 1846. (f) Dessy, r. e.; Weissman, P. H.; Pohl, R. L. J. Am. Chem. Soc. 1966, 88. 5117. (g) Connelly, N. G.; Geiger, W. E. Adv. Qrqan<?roett chgmt 1984, 21, i*

178
109. (a) Bard, A. J.? Faulkner, L. R. "Electrochemical Methods'*, Wiley: New York, 1980; CH. 11. (b) Nicholson, R. S.; Shain, I. Anal. Cham. 1964, 2£, 706; 1965, 12, 178.
110. (a) Grutzner, J. B.; Lawlor, J. M.; Jackman, L. M. J, Afflt Chgffit ?<?<?, 1972, 21, 2306. (b) Ashby, E. C. ; Dobbs, F. R.; Hopkins, H. P., Jr. J. Am. Chem. Soc. 1973, 25, 2823.
111. (a) Hudlicky, M. "Reductions in Organic Chemistry", Ellis Horwood: Chichester, 1984. (b) Kogure, T.; Ojima, I. J. Orqanomet. Chem. 1982, 221, 249. (c) Ojima, I.; Kogure, T. "Rev. Silicon, Germanium, Tin, and Lead Compounds", 1981, £, 6. (d) Ojima, I.; Yamamoto, K.; Kumada, M. "Aspects of Homogeneous Catalysis Vol. 3", D. Reidel Pub. Co.: Holland, 1977; CH. 3.
112. Jackson, R. A. J. Orqanomet. Chem. 1979, 1££, 17.
113. Colvin, E. W. Butterworths Monographs in Chemistry and Chemical Engineering "Silicon in Organic Synthesis", 1981, p 4.
114. Nolan, S. P.; Hoff, C. D.; Stoutland, P. O.; Newman, L. J.; Buchanan, J. M.; Bergman, R. G.; Peters, K. S. Jt Afflt Chem, §Q9t 1987, 1 £ 2 , 3143.
115. Calado, J. C. G.; Dias, A. R.; Salem, M. S.; Marthinho Simoes, J. A. Jt Chgffit , PaltOfl Iran? 1981, 1174.
116. Morrison, R. T.; Boyd, R. N. "Organic Chemistry", 3rd Ed.; Allyn and Bacon: Boston, 1974; CH. 1.
117. Connor, J . A. "Topics in Current Chemistry", 71. Springer-Verlag: New York, 1977; pp71-110.
118. (a) James, B. D.; Wallbridge, M. G. H. Proa. Inora. Chem. 1970, H , 99. (b) Brown, H. C.; Khuri, A.; Krishnamurthy, S. J. Am. Chem. Soc. 1977, 92, 6237.