2019 11 14 snp & variation suite gwas編)
TRANSCRIPT

SNP & Variation Suite (GWAS編)
フィルジェン株式会社 バイオサイエンス部
1
2019年11月14日 臨床ゲノム情報解析ハンズオントレーニング
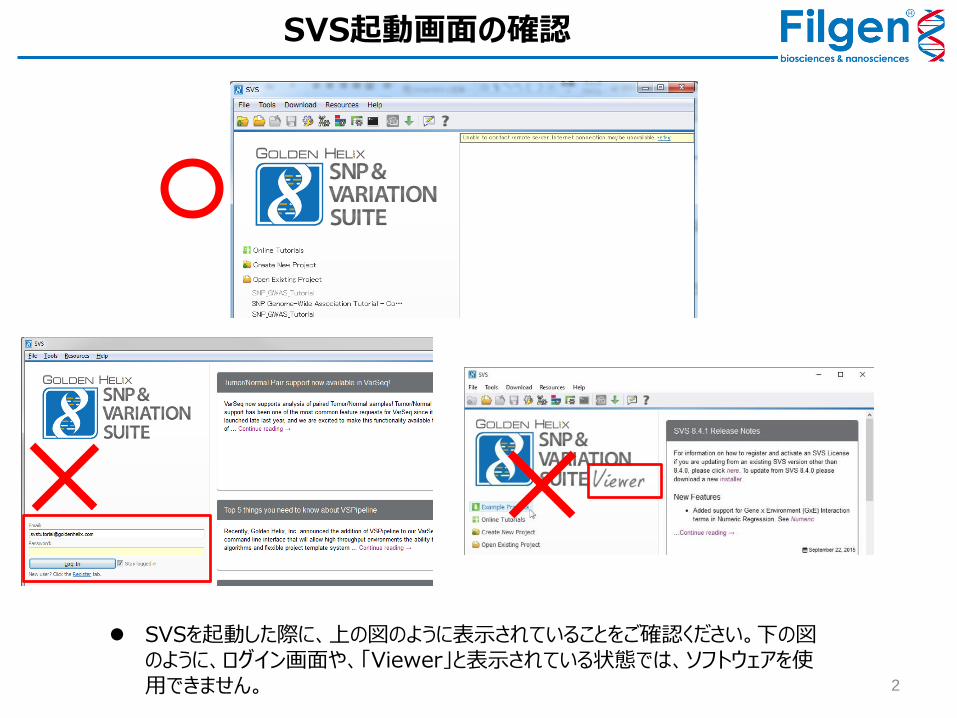
2
SVSを起動した際に、上の図のように表示されていることをご確認ください。下の図のように、ログイン画面や、「Viewer」と表示されている状態では、ソフトウェアを使用できません。
SVS起動画面の確認

3
本日使用するデータ
USBメモリで配布した「SVS」フォルダを、PC上の任意の場所(デスクトップなど)にコピーしてください。
SVSフォルダ内に、上記3つのフォルダが入っていることを確認してください。
GWAS_10samples - データインポートの練習に使用するファイルが格納されている。 実際の解析には使用しない。
Other_files - データインポートの際に必要となるライブラリーファイルなどが 収められている。
SNP_GWAS_Tutorial - 本日解析に使用する、あらかじめデータインポート済みの GWASデータが納められている。

4
SVS上より、Tools -> Open Folder -> AppData Folderをクリックし、フォルダを開きます。
「Other_files」内のMapping250K_Nsp.cdfをAffyLibraryFilesフォルダに、Affy 500K Marker Map - na32 2011_07_15.dsmをMarkerMapsフォルダにコピーしてください。
本日使用するデータ
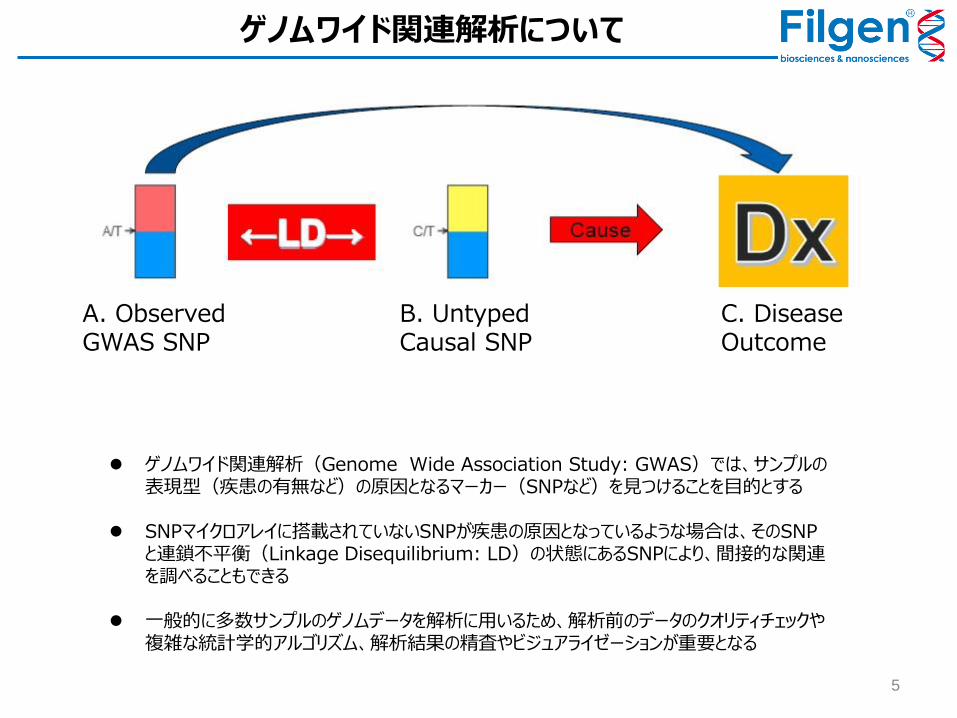
5
A. Observed GWAS SNP
B. Untyped Causal SNP
C. Disease Outcome
ゲノムワイド関連解析(Genome Wide Association Study: GWAS)では、サンプルの表現型(疾患の有無など)の原因となるマーカー(SNPなど)を見つけることを目的とする
SNPマイクロアレイに搭載されていないSNPが疾患の原因となっているような場合は、そのSNPと連鎖不平衡(Linkage Disequilibrium: LD)の状態にあるSNPにより、間接的な関連を調べることもできる
一般的に多数サンプルのゲノムデータを解析に用いるため、解析前のデータのクオリティチェックや複雑な統計学的アルゴリズム、解析結果の精査やビジュアライゼーションが重要となる
ゲノムワイド関連解析について

6
Import
• Microarray Data • Phenotype Data
• Sample QC • Marker QC
• Manhattan Plot
ゲノムワイド関連解析ワークフロー
• Genotype Association Test
QC
Test
Review

7
Import
データインポート
本事例ではGolden Helix社の提供するSNP GWAS Tutorial用の下記サンプルデータを使用します。 使用するデータ(GEOに登録されているAffymetrix 500K arrayデータ, 565例) ADS(自閉症スペクトラム)患者: 282例 健常者: 283例 解析対象SNP数: 499,264
上記データをそのままソフトウェアにインポートすると時間がかかるため、すでにインポート済みのSNP_GWAS_Tutorialのデータを使用します。そのためインポートについては、データ量を減らした10サンプルのみを使って説明いたします。
QC
Test
Review

8
1. メイン画面の「Create New Project」をクリック
2. 任意のプロジェクト名を入力し、またGenome Assemblyが「Homo sapiens (Human), GRCh37 (hg19)
(2 2009)」となっていることを確認したら「OK」をクリックして、プロジェクト画面を開く
プロジェクトの作成

9
1. プロジェクト画面より、Import -> Affymetrix -> CHPをクリック
2. CHPファイルのインポート画面で「Add Files」をクリックし、GWAS_10samplesフォルダ内の10個のCHPファイルを選択
3. インポート画面に10個のファイルが表示されたら、「OK」をクリック
Genotypeデータのインポート
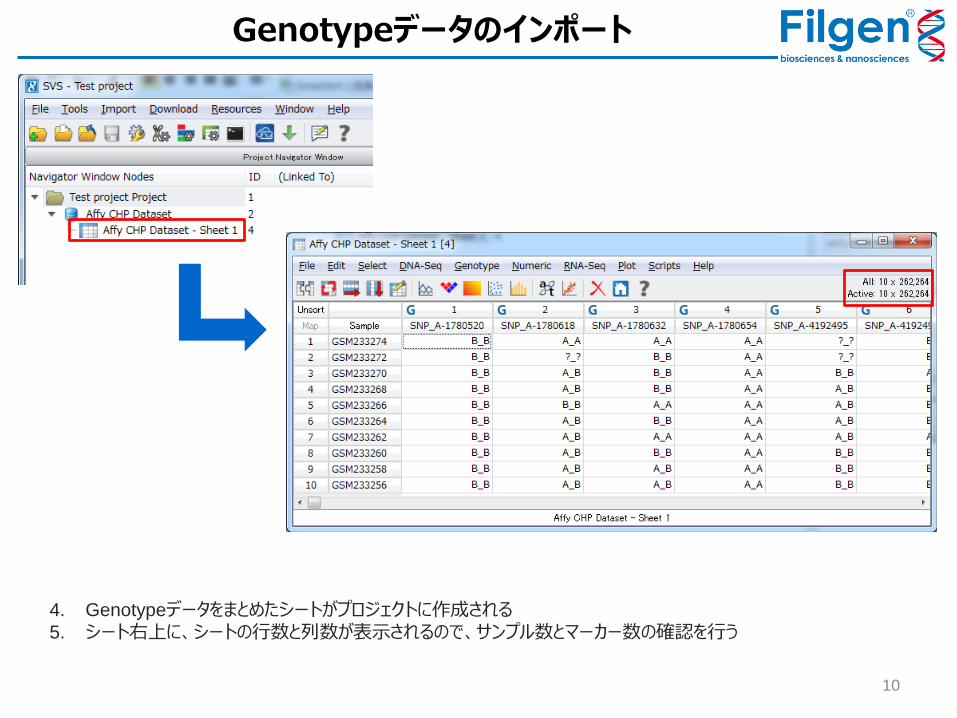
10
Genotypeデータのインポート
4. Genotypeデータをまとめたシートがプロジェクトに作成される
5. シート右上に、シートの行数と列数が表示されるので、サンプル数とマーカー数の確認を行う
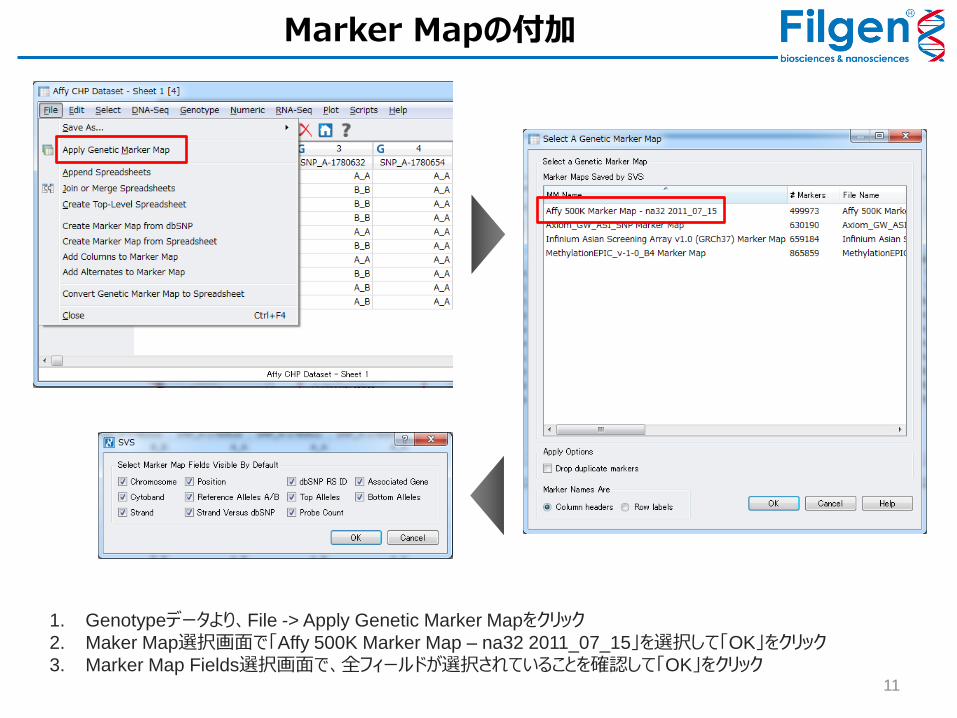
11
1. Genotypeデータより、File -> Apply Genetic Marker Mapをクリック
2. Maker Map選択画面で「Affy 500K Marker Map – na32 2011_07_15」を選択して「OK」をクリック
3. Marker Map Fields選択画面で、全フィールドが選択されていることを確認して「OK」をクリック
Marker Mapの付加

12
Marker Mapの付加
4. Marker Mapが付加されたシートが新たに作成される
5. シート左上の、緑色の「Map」をクリックすると、各マーカーの詳細情報を確認できる

13
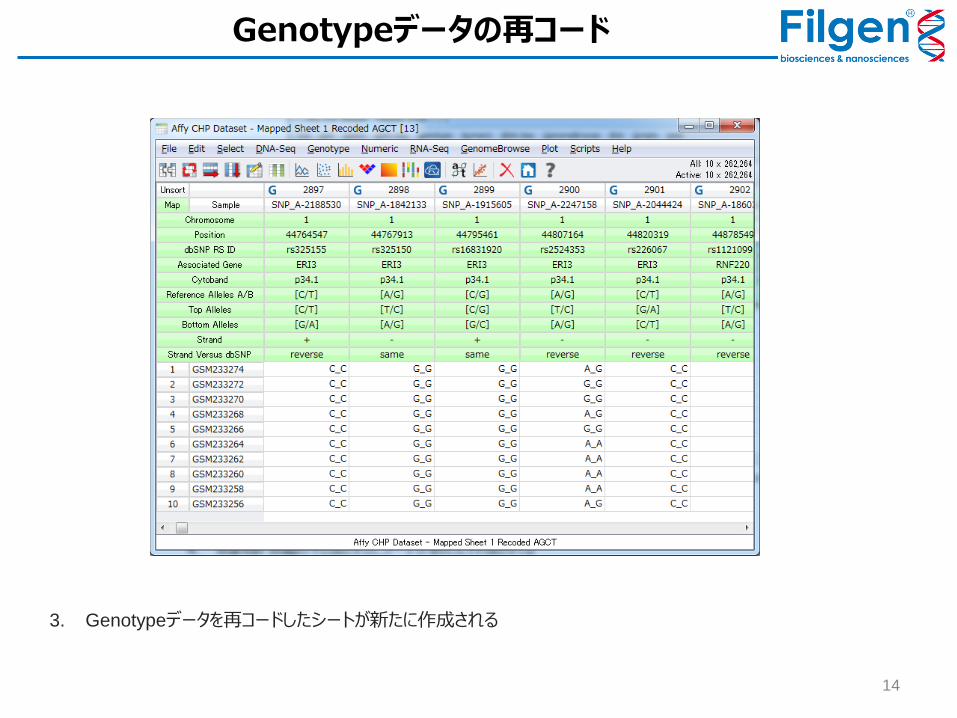
Genotypeデータの再コード
1. Marker Mapを付加したGenotypeシートを開き、Edit -> Recode -> Recode Genotypesをクリック
2. Recode Genotype画面より、Marker Mapに含まれる「Reference Alleles A/B」フィールドのデータを使用して再コードを実行するように選択し、現シートの下に新しいシートが作成されるように設定して「OK」をクリック
14
Genotypeデータの再コード
3. Genotypeデータを再コードしたシートが新たに作成される

15
Phenotypeデータのインポート
1. プロジェクト画面より、Import -> Third Partyをクリック
2. Third Partyファイルのインポート画面で「Browse」をクリックし、GWAS_10samplesフォルダ内のPhenotype_data.xlsxファイルを選択
3. 「Read Genotypic Data」のチェックを外し、「Next」をクリック
4. 続くSelect Row Labelsで、label columnとして「Sample」を選択して「OK」をクリック

16
Phenotypeデータのインポート
5. Phenotypeデータをまとめたシートがプロジェクトに作成される
17
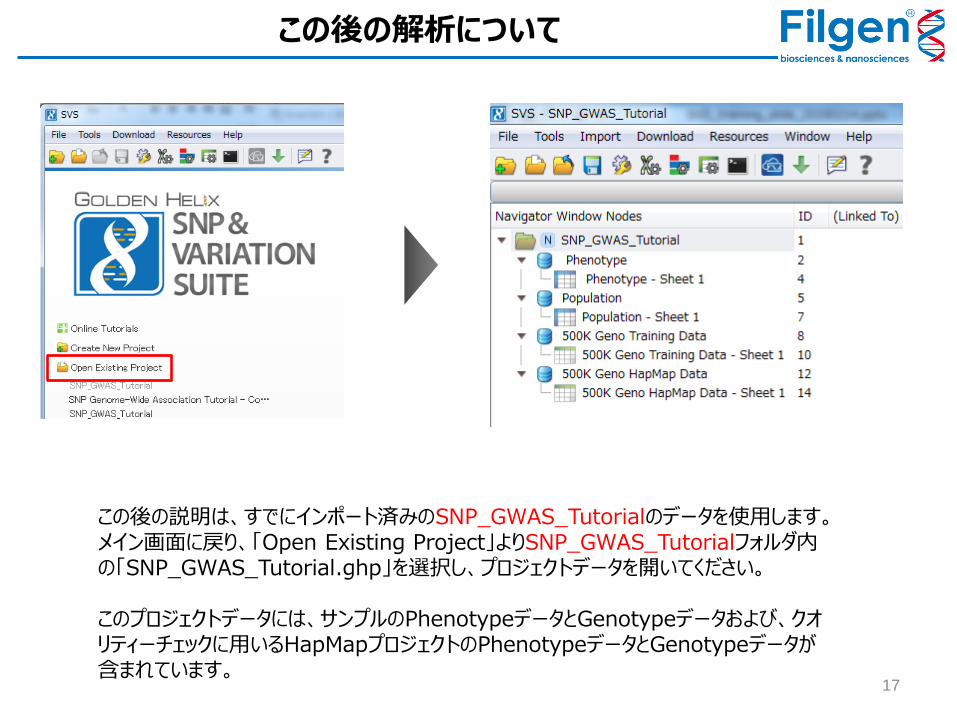
この後の解析について
この後の説明は、すでにインポート済みのSNP_GWAS_Tutorialのデータを使用します。メイン画面に戻り、「Open Existing Project」よりSNP_GWAS_Tutorialフォルダ内の「SNP_GWAS_Tutorial.ghp」を選択し、プロジェクトデータを開いてください。 このプロジェクトデータには、サンプルのPhenotypeデータとGenotypeデータおよび、クオリティーチェックに用いるHapMapプロジェクトのPhenotypeデータとGenotypeデータが含まれています。
18
Import
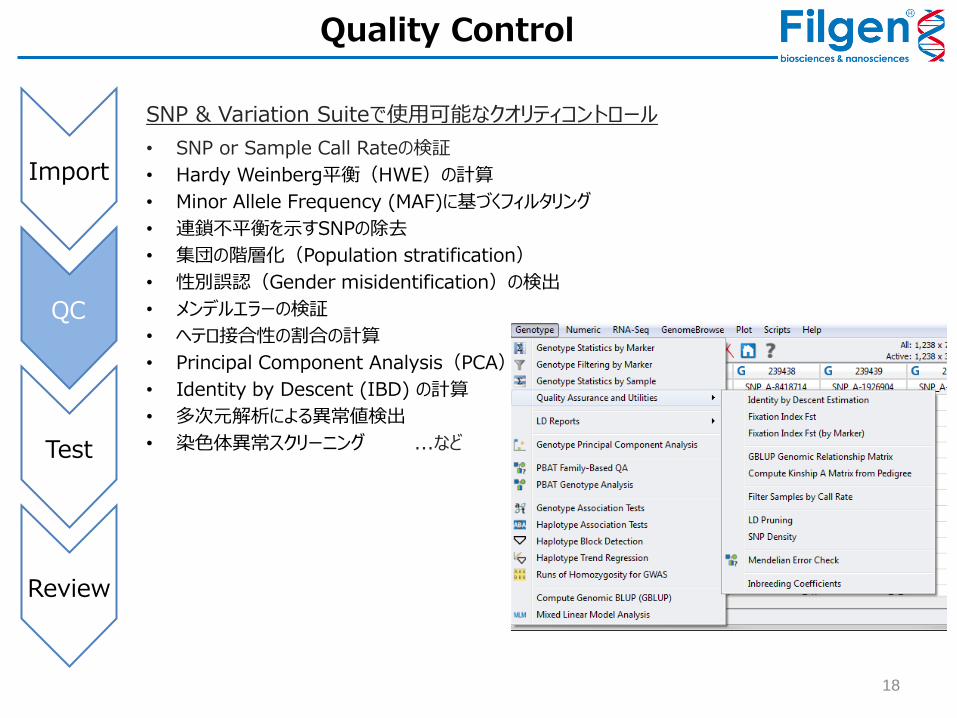
Quality Control
SNP & Variation Suiteで使用可能なクオリティコントロール
• SNP or Sample Call Rateの検証
• Hardy Weinberg平衡(HWE)の計算
• Minor Allele Frequency (MAF)に基づくフィルタリング
• 連鎖不平衡を示すSNPの除去
• 集団の階層化(Population stratification)
• 性別誤認(Gender misidentification)の検出
• メンデルエラーの検証
• ヘテロ接合性の割合の計算
• Principal Component Analysis(PCA)
• Identity by Descent (IBD) の計算
• 多次元解析による異常値検出
• 染色体異常スクリーニング ...など
QC
Test
Review
19
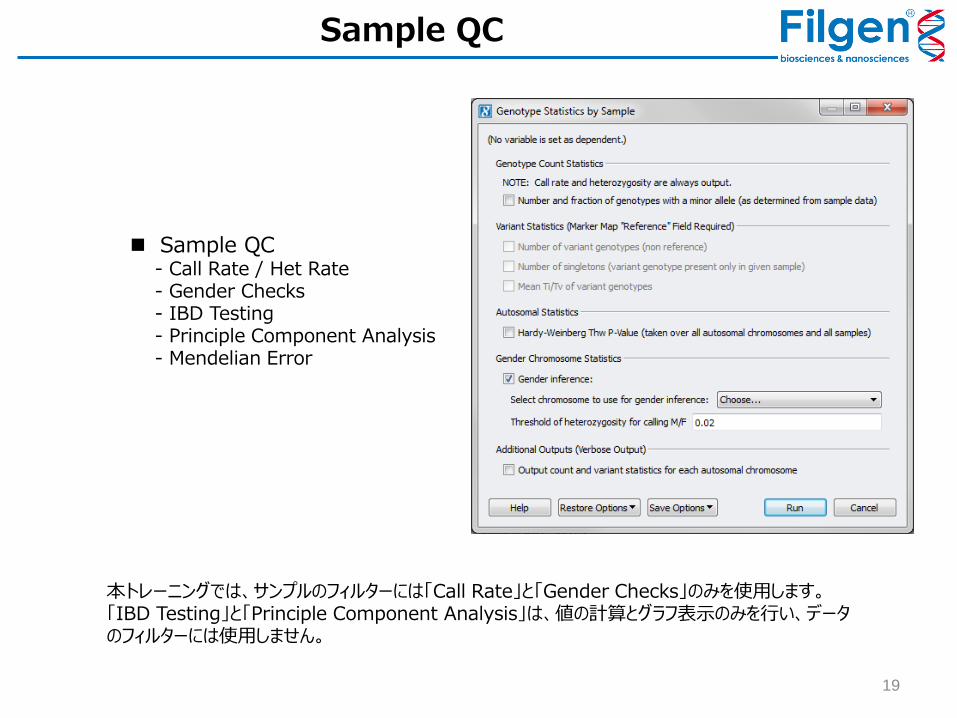
Sample QC - Call Rate / Het Rate - Gender Checks - IBD Testing - Principle Component Analysis - Mendelian Error
Sample QC
本トレーニングでは、サンプルのフィルターには「Call Rate」と「Gender Checks」のみを使用します。「IBD Testing」と「Principle Component Analysis」は、値の計算とグラフ表示のみを行い、データのフィルターには使用しません。

20
1. 「500K Geno Training Data」を開き、Genotype -> Genotype Statistics by Sampleをクリック
2. 「Gender inference」にチェックを入れ、「Select chromosome to use for gender inference」に「X」で選択
3. 「Output count and variant statistics for each autosomal chromosome」にチェックを入れ、「Run」をクリック
Genotype Statistics by Sample

21
4. サンプルごとの各種クオリティ―データをまとめたシートと、各常染色体ごとのデータをまとめたシートがプロジェクトに作成される
Genotype Statistics by Sample
22
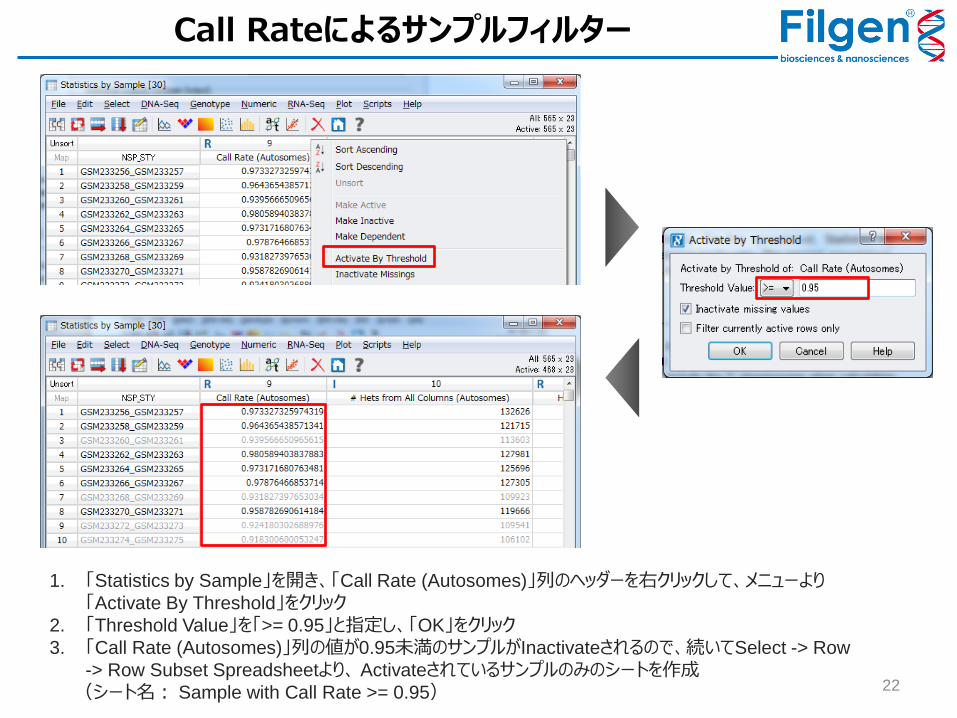
1. 「Statistics by Sample」を開き、「Call Rate (Autosomes)」列のヘッダーを右クリックして、メニューより「Activate By Threshold」をクリック
2. 「Threshold Value」を「>= 0.95」と指定し、「OK」をクリック
3. 「Call Rate (Autosomes)」列の値が0.95未満のサンプルがInactivateされるので、続いてSelect -> Row
-> Row Subset Spreadsheetより、 Activateされているサンプルのみのシートを作成
(シート名: Sample with Call Rate >= 0.95)
Call Rateによるサンプルフィルター

23
シートの結合
1. 「Sample with Call Rate >= 0.95」を開き、File -> Join or Merge Spreadsheetをクリック
2. 「Navigator Window Chooser 」で「Phenotype - Sheet 1」を選択し、「OK」をクリック
3. 「Join or Merge Spreadsheets」において、「Spread as Child of」の「Current spreadsheet」が選択されていることを確認した後、 「OK」をクリック

24
ヒストグラム表示
1. 「Samples with Call Rate >= 0.95 + Phenotype - Sheet 1」を開き、 「Het Rate from All columns (Chr. X)」列のヘッダーを右クリックして、メニューより「Plot Histogram」をクリック
2. 「Het Rate from All columns (Chr. X)」列のデータのヒストグラムが表示される

25
3. データのグループ分けのサイズや、Phenotypeデータに含まれるカテゴリーの色分け表示などの設定を行う
+
ヒストグラム表示

26
性別誤認サンプルのフィルター
1. 「Samples with Call Rate >= 0.95 + Phenotype - Sheet 1」を開き、 Select -> Compare and Activate
by Column Agreementをクリック
2. 「Compare Columns」にて「Add Columns」をクリックし、「Inferred Gender」と「Gender」を選択し、「OK」をクリック
3. 「Compare Columns」にて「Row with matching data values」と「Row with differing data values」にチェックを入れ、「OK」をクリック

27
4. 性別データが一致するサンプルと、一致しないサンプルのそれぞれのリストが出力される
性別誤認サンプルのフィルター

28
5. 「Rows with matching values in columns Inferred Gender and Gender」を開き、 Select -> Apply
Current Selection to Second Spreadsheetをクリック
6. 「Apply Filter to Spreadsheet」にて、「Apply filtered」を「rows」と選択し、「Select Sheet」をクリックして「500K Geno Training Data - Sheet 1」を指定し、「OK」をクリック
7. 新たに作成された「500K Geno Training Data - Sheet 2」を開き、 Select -> Row -> Row Subset
Spreadsheetをクリックして、Activateされているサンプルのみのシートを作成
(シート名: Subset - Samples with Call Rate >= 0.95 and Matched Gender)
性別誤認サンプルのフィルター

29
Cryptic Relatedness
今回使用しているサンプルデータは、すべて血縁関係のないものを用いていますが、隠れた血縁関係を検出するために、アレルを同じ祖先から受け継いでいることを示す「Identity by Descent (IBD)」の計算を行います。 IBDの計算を行うと、サンプル間の血縁関係や、サンプルの重複またはコンタミネーションなどを検出することができます。 IBDの計算は、一般的にはゲノム上の全SNPを使用するのではなく、連鎖不平衡(Linkage Disequilibrium: LD)の状態にあるSNPを除外し、SNP数を削減してから行います。

30
1. 「Subset - Samples with Call Rate >= 0.95 and Matched Gender」を開き、 Genotype -> Quality
Assurance and Utilities -> LD Pruningをクリック
2. 「LD Pruning」にて、「Window Size」を「100」と指定し、「OK」をクリック
3. 計算が終了したら、Select -> Column -> Column Subset Spreadsheetより、 ActivateされているSNPのみのシートを作成
4. ActivateされているSNPのみのシートより、さらにSelect -> Activate by Chromosomesをクリックし、「X」のみ選択を外して「OK」をクリック
(シート名: Pruned SNP Subset)
LD Pruning

31
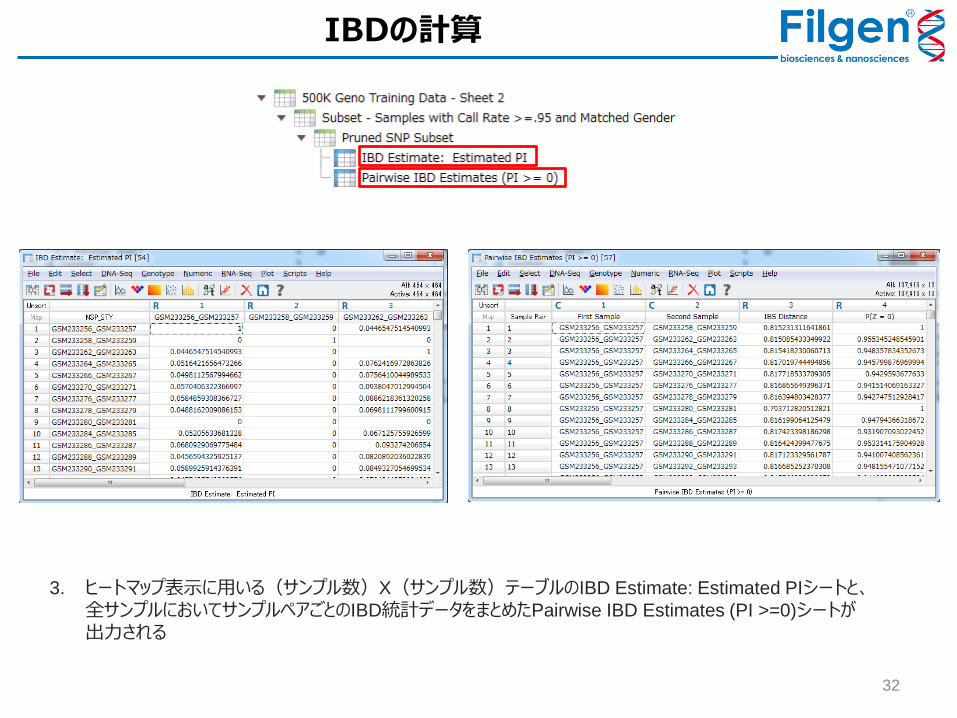
1. 「Pruned SNP Subset」を開き、 Genotype -> Quality Assurance and Utilities -> Identity by Descent
Estimationをクリック
2. 「Estimate Identity by Descent」にて、「Output IBS distances ((IBS 2 + 0.5*IBS 1)/# non-missing
markers)」と「Output untransformed estimates of P(Z=0), P(Z=1), and P(Z=2)」のチェックを外し、「Output PI = P(Z=1)/2 + P(Z=2)」と「Output all pairs where PI >=」にチェックを入れ、値を「0」に指定してから「Run」をクリック
IBDの計算
32
3. ヒートマップ表示に用いる(サンプル数)X(サンプル数)テーブルのIBD Estimate: Estimated PIシートと、全サンプルにおいてサンプルペアごとのIBD統計データをまとめたPairwise IBD Estimates (PI >=0)シートが出力される
IBDの計算

33
1. 「IBD Estimate: Estimated PI」を開き、Plot -> Heat Map (Uniform)をクリック
2. ヒートマップが表示される
IBDプロットの表示

34
3. 色彩の表示などの設定を行う
IBDプロットの表示

35
Population Stratification
GWASでは、サンプルの人種・民族などの違いなどを評価するために、主成分分析(Principal Component Analysis: PCA)が用いられます。 本トレーニングでは、より人種・民族の違いを分かりやすくするために、解析サンプルのデータに加え、HapMapプロジェクトのヨーロッパ、アジア、アフリカ人のサンプルデータとともに主成分分析を行います。
36
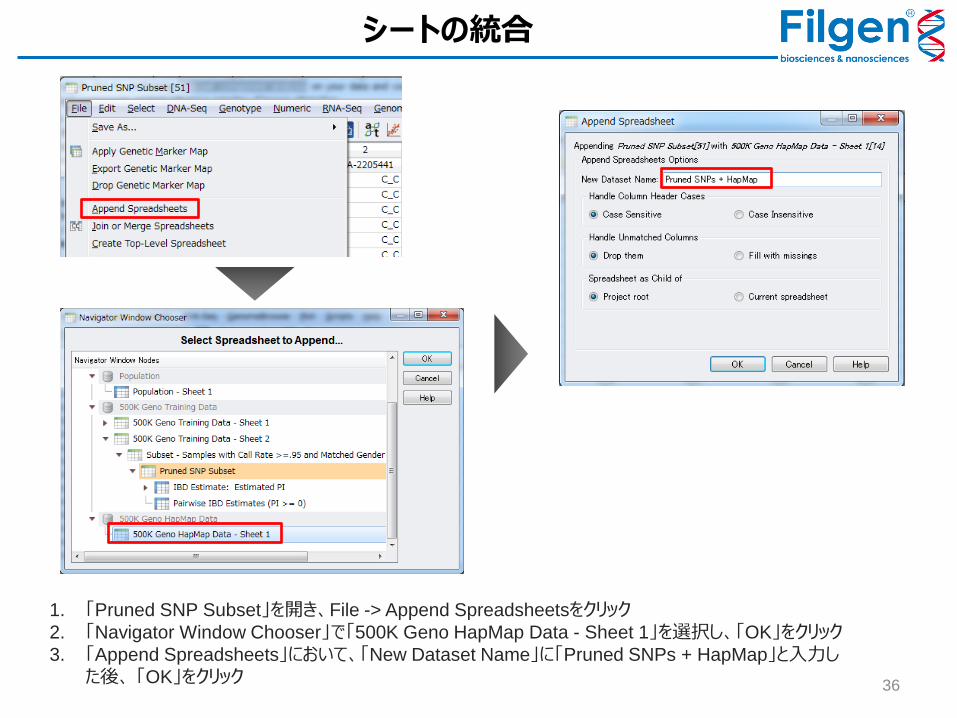
1. 「Pruned SNP Subset」を開き、File -> Append Spreadsheetsをクリック
2. 「Navigator Window Chooser」で「500K Geno HapMap Data - Sheet 1」を選択し、「OK」をクリック
3. 「Append Spreadsheets」において、「New Dataset Name」に「Pruned SNPs + HapMap」と入力した後、 「OK」をクリック
シートの統合
37
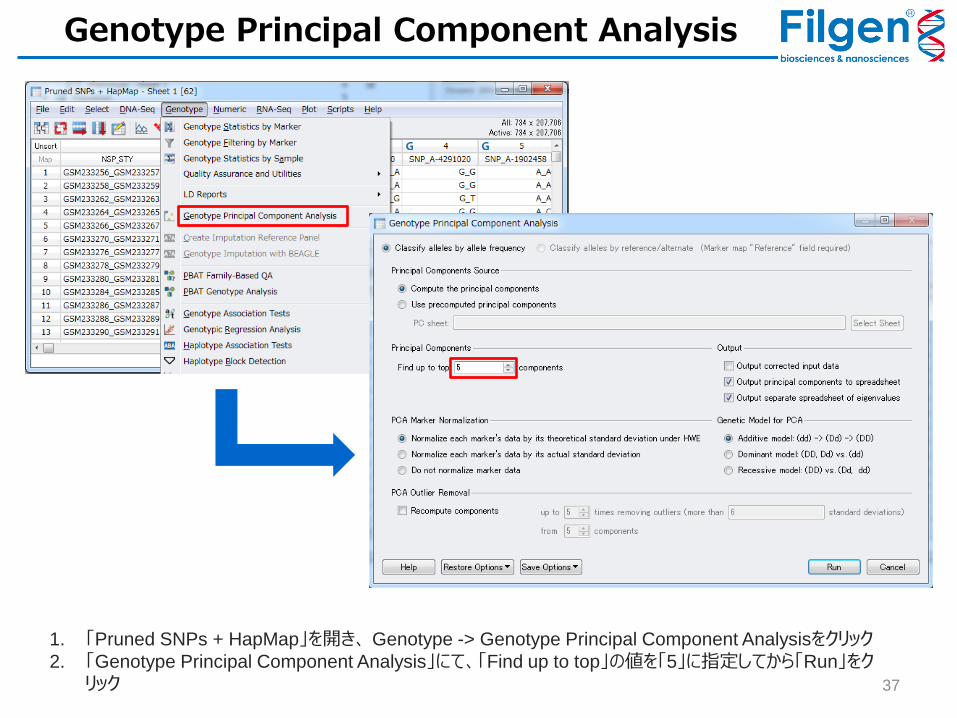
1. 「Pruned SNPs + HapMap」を開き、 Genotype -> Genotype Principal Component Analysisをクリック
2. 「Genotype Principal Component Analysis」にて、「Find up to top」の値を「5」に指定してから「Run」をクリック
Genotype Principal Component Analysis

38
3. 主成分プロット表示に用いる、サンプルごとの主成分データをまとめたPrincipal Components (Additive
Model)シートと、各主成分の固有値をまとめたPC Eigenvalues (Additive Model)シートが出力される
Genotype Principal Component Analysis

39
1. 「Population - Sheet 1」を開き、File -> Join or Merge Spreadsheetsをクリック
2. 「Navigator Window Chooser」で「Principal Components (Additive Model)」を選択し、「OK」をクリック
3. 「Join or Merge Spreadsheets」において、「Spreadsheet as Child of」に「Current spreadsheet」を選択した後、 「OK」をクリック
シートの結合

40
1. 「Population + Principal Components (Additive Model) 」を開き、Plot -> XY Scatter Plotsをクリック
2. 「XY Scatter Parameters」で、左側のX軸に「EV = 35.8391」、右側のY軸に「EV = 15.5366」を選択して「Plot」をクリック
3. スキャッタープロットが表示される
PCAプロットの表示

41
4. Phenotypeデータに含まれるカテゴリーの色分け表示などの設定を行う
PCAプロットの表示

42
Marker QC
Marker QC /Filtering - Call Rate / HWE - Minor Allele Frequency - LD Pruning - Genomic Annotations
ここからは、Sample QCを行った「500K Geno Training Data - Sheet 2」シートを使用し、引き続きMarker QCを実施します。

43
1. 「Phenotype - Sheet 1」を開き、File -> Join or Merge Spreadsheetsをクリック
2. 「Navigator Window Chooser」で「500K Geno Training Data - Sheet 2」を選択し、「OK」をクリック
3. 「Join or Merge Spreadsheets」において、デフォルト設定のまま「OK」をクリック
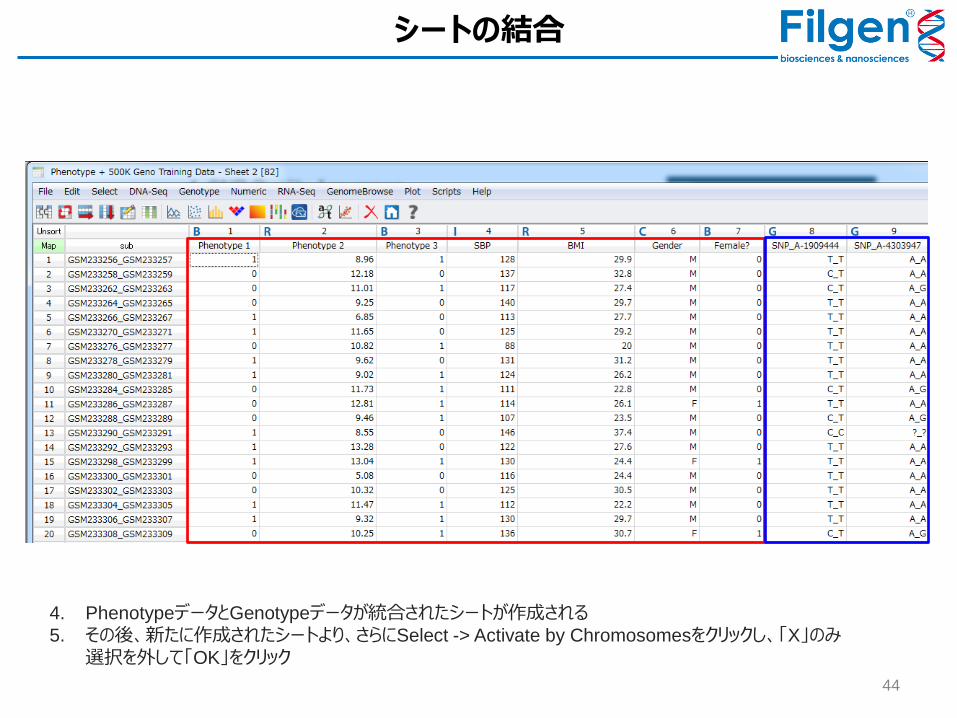
シートの結合
44
4. PhenotypeデータとGenotypeデータが統合されたシートが作成される
5. その後、新たに作成されたシートより、さらにSelect -> Activate by Chromosomesをクリックし、「X」のみ選択を外して「OK」をクリック
シートの結合

45
1. 「Phenotype + 500K Geno Training Data - Sheet 1」を開き、「Phenotype」列のヘッダーをクリック
2. 「Phenotype」列のデータが赤紫色に着色される
独立変数の選択

46
1. 「Phenotype - 500K Geno Training Data - Sheet 1」を開き、Genotype -> Genotype Filtering by
Markerをクリック
2. 「Genotype Filtering by Marker」にて、「Drop if call rate < 0.9」「Drop if Minor Allele Frequency
(MAF) < 0.01」「Perform HWE filtering based on: Controls」「Drop if Fisher’s exact test for
HWE P-Value < 0.0001」と指定し、「Run」をクリック
3. 「Phenotype - 500K Geno Training Data - Sheet 1」を開き、Select -> Subset Active Dataをクリック
(シート名:Filtered Data for Association Testing)
Genotype Filtering by Marker
47
Import
QC
Test
Review
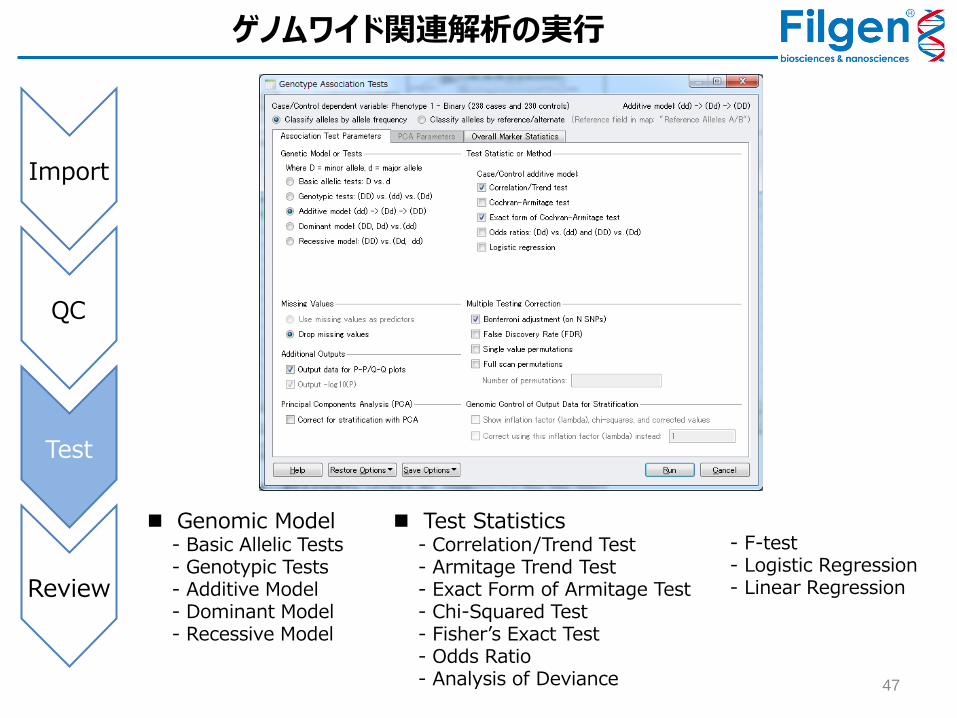
Genomic Model - Basic Allelic Tests - Genotypic Tests - Additive Model - Dominant Model - Recessive Model
Test Statistics - Correlation/Trend Test - Armitage Trend Test - Exact Form of Armitage Test - Chi-Squared Test - Fisher’s Exact Test - Odds Ratio - Analysis of Deviance
- F-test - Logistic Regression - Linear Regression
ゲノムワイド関連解析の実行

48
DD Dd dd
Case a b c
Control d e f
DD Dd dd
Case a b c
Control d e f
DD + Dd dd
Case a + b c
Control d + e f
DD Dd + dd
Case a b + c
Control d e + f
(D:マイナーアリル、d: メジャーアリル、a~f: 遺伝型数) 頻度の低いアリル(マイナーアリル)数の、疾患への影響に合わせて、遺伝学モデルを選択する
Full Data
Additive Model (dd -> Dd -> DD)
Dominant Model ((DD + Dd) vs (dd))
Recessive Model ((DD) vs (Dd + dd))
Genetic Model

49
Case/Control, Additive model
Case/Control, Dominant model
Quantitative, Additive model
本トレーニングでは、Case/Control表現型データを用いた「Additive model」で計算を実行します
Test Statistics

50
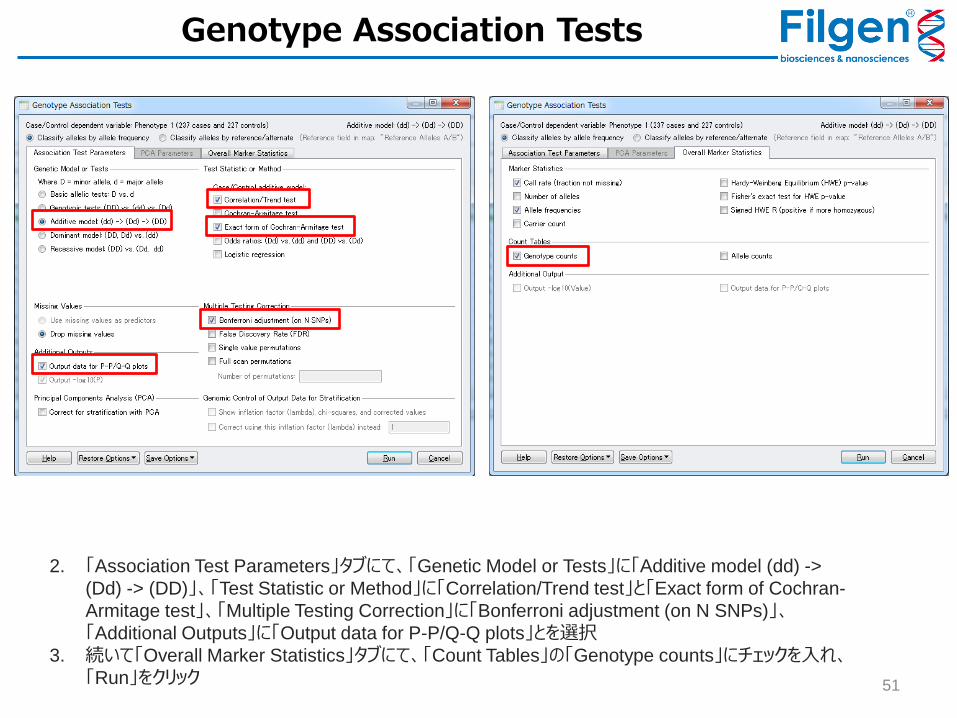
1. 「Filtered Data for Association Testing」を開き、Genotype -> Genotype Association Testsをクリック
Genotype Association Tests
51
2. 「Association Test Parameters」タブにて、「Genetic Model or Tests」に「Additive model (dd) ->
(Dd) -> (DD)」、「Test Statistic or Method」に「Correlation/Trend test」と「Exact form of Cochran-
Armitage test」、「Multiple Testing Correction」に「Bonferroni adjustment (on N SNPs)」、「Additional Outputs」に「Output data for P-P/Q-Q plots」とを選択
3. 続いて「Overall Marker Statistics」タブにて、「Count Tables」の「Genotype counts」にチェックを入れ、
「Run」をクリック
Genotype Association Tests
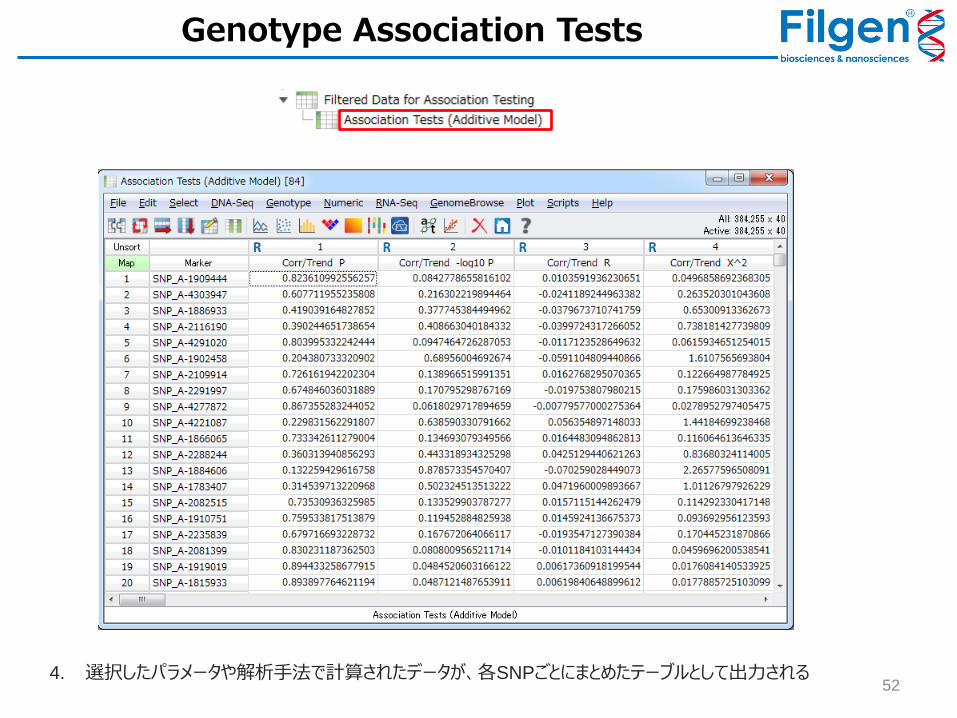
52 4. 選択したパラメータや解析手法で計算されたデータが、各SNPごとにまとめたテーブルとして出力される
Genotype Association Tests

53
1. 「Association Tests (Additive Model)」を開き、Plot -> XY Scatter Plotsをクリック
2. 「XY Scatter Parameters」で、左側のX軸に「Corr/Trend expected X^2」、右側のY軸に「Corr/Trend X^2」を選択して「Plot」をクリック
3. スキャッタープロットが表示される
Q-Qプロットの表示

54
4. グラフ上の直線の表示などの設定を行う
Q-Qプロットの表示

55
Import
QC
Test
Review
解析データのグラフ表示
本トレーニングでは、Genotype Association Testsで計算したSNPごとの有意確率を、ゲノム上にプロットしたマンハッタンプロットを作成します

56
1. 「Association Tests (Additive Model)」を開き、 「Corr/Trend –log10 P」列のヘッダーを右クリックして、メニューより「Plot Variable in Genome Browse」をクリック
2. 「Corr/Trend –log10 P」列のデータをゲノム上にプロットしたグラフが表示される
マンハッタンプロットの表示

57
3. 染色体ごとの色分け表示などの設定を行う
マンハッタンプロットの表示
