vasopressin-induced disruption of actin cytoskeletal organization and canalicular function in...
TRANSCRIPT

Vasopressin-Induced Disruption of Actin Cytoskeletal Organizationand Canalicular Function in Isolated Rat Hepatocyte Couplets:
Possible Involvement of Protein Kinase C
MARCELO G. ROMA,1 VICKI STONE,2 ROBERT SHAW,3 AND ROGER COLEMAN3
The effect of vasopressin (VP) on canalicular functionand hepatocellular morphology, with particular regard toactin cytoskeletal organization and the concomitant plasmamembrane bleb formation, was studied in isolated rathepatocyte couplets. VP induced the concentration-depen-dent formation of multiple plasma membrane blebs as wellas simultaneous impairment in both canalicular vacuolaraccumulation (cVA) and retention (cVR) of the fluorescentbile acid, cholyl-lysyl-fluorescein (CLF), which evaluatecouplet secretory function and tight-junction integrity, re-spectively. These effects were mimicked by the proteinkinase C (PKC) activator, phorbol dibutyrate (PDB), but notby the protein kinase A (PKA) activator, dibutyryl-cAMP.VP-induced bleb formation and canalicular dysfunctionwere fully prevented by the protein kinase inhibitor, H-7,but not by the PKA inhibitor, KT5720, further suggesting aspecific role of PKC. VP-induced alterations were alsoprevented by pretreatment with the Ca21-buffering agent,BAPTA/AM, but not with the calmodulin-dependent proteinkinase II antagonist, calmidazolium. Neither the Ca21-activated neutral protease inhibitor, leupeptin, nor theantioxidants, a-tocopherol or deferoxamine, were able toprevent either VP-induced plasma membrane blebbing orcanalicular dysfunction. The Ca21-ionophore, A23187, mim-icked the VP-induced alterations, but its harmful effectswere completely prevented by H-7. Bleb formation inducedby VP and PDB was accompanied by an extensive redistribu-tion of filamentous actin from the pericanalicular area to thecell body, and this effect was fully prevented by H-7. These
results suggest that VP-induced canalicular and cytoskeletaldysfunction is mediated by PKC and that classical (Ca21-dependent) PKC appear to be involved because intracellularCa21 is required for VP to induce its harmful effects.(HEPATOLOGY 1998;28:1031-1041.)
Vasopressin (VP), like other hormonal mediators includingangiotensin II, epinephrine, and norepinephrine, exerts itsbiological actions by inducing liberation of inositol 1,4,5-trisphosphate and diacylglycerol.1 Inositol 1,4,5-trisphos-phate mediates increase in cytosolic Ca21 ([Ca21]cyt), whereasdiacylglycerol mediates activation of protein kinase C (PKC).VP induces bile flow impairment either by PKC activation orCa21 mobilization.2-4 A variety of mechanisms were proposedto account for the cholestatic effect of VP. This hormonedecreases primary secretion in isolated rat hepatocyte cou-plets via both [Ca21]cyt elevation and PKC activation,5 indicat-ing that the effect is, at least in part, hepatocellular in origin.VP was also reported to inhibit cyclic adenosine monophos-phate (cAMP)-stimulated transcytosis vesicular transport inisolated perfused rat liver,4 a mechanism necessary forcarriers to be transported and inserted into the canalicularmembrane domain.6 Finally, VP, phorbol esters, and Ca21
agonists were all shown to increase tight-junctional permeabil-ity both in isolated perfused rat liver4,7-9 and in isolated rathepatocyte couplets.10-12 An increase of ‘‘leakiness’’ of theparacellular barrier may result in dissipation of osmoticgradients, which would lead to decreased net bile secretion.13
Hepatocytes possess a fully developed cytoskeleton com-posed of filamentous (F)-actin, several actin-associated pro-teins, and myosin II. These proteins are associated with thecanalicular membrane as well as gap and tight junctions,which form a cohesive interconnected network around thebile canaliculus.14 Functional integrity of these structures isessential for hepatocytes to secrete and retain preaccumulatedmaterial into the bile canaliculus. Indeed, one of the hepato-cellular mechanisms that was postulated to be involved incholestasis is cytoskeletal dysfunction.14,15 Furthermore, peri-canalicular actin cytoskeletal alterations appear to be acommon feature in cholestasis, because a large number ofcholestatic compounds were reported to affect pericanalicularF-actin.16
Despite the aforementioned evidence that VP impairshepatocyte secretory function and that a clear link does existbetween cholestasis and cytoskeletal dysfunction, experimen-tal studies on the effect of VP on cytoskeletal integrity andtheir relationship with canalicular function have not yet beenperformed. In this study, we attempted to achieve this goal by
Abbreviations: VP, vasopressin; cytosolic Ca21, [Ca21]cyt; PKC, protein kinase C;cAMP, cyclic adenosine monophosphate; CLF, cholyl-lysyl-fluorescein; PDB, phorbol12,13-dibutyrate; dibutyryl-cAMP, N6,2’-o-dibutyryladenosine 38:58-cyclic monophos-phate; H-7, 1-(5-isoquinolinylsulfonyl)-2-methyl piperazine; FITC, fluorescein isothio-cyanate; BAPTA/AM, 1,2-bis-(o-aminophenoxy)-ethene-N,N,N8,N8-tetra-acetate tetra-(acetomethyl)ester; cVA, canalicular vacuolar accumulation; cVR, canalicular vacuolarretention; PBS, phosphate-buffered saline; PKA, protein kinase A; DMSO, dimethylsulfoxide.
From the 1Institute of Experimental Physiology (IFISE), School of Biochemical andPharmacological Science, The University of Rosario-CONICET, Rosario, Argentina;2Biological Science, Napier University, Merchiston, Edinburgh, Scotland; and 3School ofBiochemistry, The University of Birmingham, Edgbaston, Birmingham, England.
Received January 9, 1998; accepted May 27, 1998.Dr. Roma was a recipient of a postdoctoral fellowship of The British Council-
Fundacion Antorchas Awards for Advanced Studies in the United Kingdom. Some of thelaboratory expenses were supported by The Wellcome Trust.
Address reprint requests to: Prof. Roger Coleman, School of Biochemistry, TheUniversity of Birmingham, Edgbaston, Birmingham B15 2TT, UK. Fax: 0121-414-3982.
Copyright r 1998 by the American Association for the Study of Liver Diseases.0270-9139/98/2804-0018$3.00/0
1031

analyzing the effect of VP on F-actin distribution and its moreconspicuous morphological outcome, i.e., plasma membranebleb formation, in preparations of hepatocyte couplets. Suchchanges were correlated with those observed in canalicularfunction, particularly the capability of the couplets to accumu-late and retain in their canalicular lumen the fluorescent bilesalt analogue, cholyl-lysyl-fluorescein (CLF). Finally, therespective roles of protein kinases and [Ca21]cyt on the effectsobserved were analyzed.
MATERIALS AND METHODS
Materials. CLF, kindly provided by Dr. Charles O. Mills (Birming-ham, England), was synthesized and its purity confirmed accordingto the methods of Mills et al.17; the synthetic procedures gave a highyield of CLF, which appeared as a single spot after High PerformanceThin Layer Chromatography. Collagenase type A from Clostridiumhistolyticum was purchased from Gibco (Paisley, Scotland). Leibo-vitz-15 tissue culture medium, bovine serum albumin (fraction V),leupeptin, deferoxamine mesylate, a-tocopherol, VP, phorbol 12,13-dibutyrate (PDB), phorbol 12-myristate 13-acetate, trifluorperazine,N6,28-o-dibutyryladenosine 38:58-cyclic monophosphate (dibutyryl-cAMP), 1-(5-isoquinolinylsulfonyl)-2-methylpiperazine dihydrochlo-ride (H-7), and fluorescein isothiocyanate (FITC)-labeled phalloi-din were obtained from Sigma Chemical Co. (Poole, Dorset,England). KT5720, calmidazolium, and 1,2-bis-(o-aminophenoxy)-ethene-N,N,N8,N8-tetraacetate tetra-(acetomethyl)ester (BAPTA/AM) were obtained from Alexis Co. (Bingham, Nottingham, En-gland). All other chemicals were of reagent grade.
Animals. Male Wistar rats from the Biomedical Services Unit ofthe University of Birmingham (220-250 g) were used throughout.Before the experiments, the animals were maintained on a standardlaboratory diet (41B maintenance diet, Pilsbury, Birmingham, En-gland) and tap water ad libitum. Rats were anesthetized usingKetamine hydrochloride (Ketalar; 6 mg/100 g body weight), withMedetomidine (Domitor; 25 µg/100 g body weight). Surgery wasstarted between 8:00 AM and 9:00 AM to minimize circadianvariations.
Couplet Isolation, Enrichment, and Culture. Hepatocyte couplets wereobtained from rat liver according to the two-step collagenaseperfusion procedure described by Wilton et al.,18 adapted fromGautam et al.19 This initial preparation contained 22% 6 3% (n 518) of couplets with high viability (.89%), as assessed by theTrypan blue exclusion test. If either cell of a couplet stainedpositively with the dye, then the whole unit was considerednonviable. Couplets were further enriched from singles and mul-tiples by centrifugal elutriation as described by Wilton et al.20 Theresulting preparation, containing 69% 6 4% of couplets (n 5 18) ofhigh viability (.92%), was plated in Leibovitz-15 containing 50U/mL penicillin and 50 µg/mL streptomycin onto 35-mm plasticculture dishes (2 mL/dish) at a density of 0.5 3 105 units/mL, andincubated at 37°C for 4.5 to 5 hours. This time was described to besufficient for couplets to reach their maximal capability to transportand accumulate CLF in their canalicular vacuoles.21 Because thiscapability then remains virtually constant for at least 4 hours,18
experiments were performed within this time period.Assessment of Canalicular Function. Canalicular function was evalu-
ated quantitatively by assessing the number of couplets able toperform canalicular vacuolar accumulation (cVA) and canalicularvacuolar retention (cVR) of CLF. The former assay aims to evaluatethe capability of the couplets to secrete and accumulate thefluorescent bile salt analogue in their canalicular vacuole,18,21-23
whereas the latter one was shown to assess tight-junctional perme-ability.12
cVA of CLF was assessed by determining the percentage ofcouplets present in the field (.30), displaying sufficient fluorescentbile acid analogue to be visible in their canalicular vacuoles. For thispurpose, CLF (2 µmol/L, final concentration in the dishes) was
added to each plate and incubated at 37°C for 15 minutes. This timewas chosen because the percentage of couplets exhibiting cVA ofCLF was shown to reach an apparent steady state within this time.18
Next, CLF was removed from the media by washing twice with 2 mLof L-15 at 37°C, and cVA of CLF was subsequently assessed by usingan inverted fluorescent microscope (Olympus IMT2-RFL, OlympusOptical Ltd., London, England) equipped with a 100-W mercurylight source and an incubator to maintain the cells at 37°C duringobservation. Couplets were first focused under light microscopy toidentify the location of the couplets on the field and to simulta-neously assess blebbing of the cell membrane (see below).
To perform cVR of CLF, in a further set of dishes, using coupletsfrom the same batch, the fluorescent bile acid analogue was allowedto accumulate in the canalicular vacuole for 15 minutes at 37°C, andthen the medium was replaced by fresh medium without CLF. Then,the agent studied, or its vehicle (alone), was added and the cellswere incubated for the periods stated for each agent (see Treatmentsbelow). The percentage of couplets then capable of retainingpreaccumulated CLF following exposure to the hormonal modula-tors, etc., was then assessed by fluorescence microscopy.
Assessment of Plasma Membrane Blebbing. Plasma membrane bleb-bing was assayed immediately before cVA of CLF, i.e., following 15minutes of VP or PDB exposure. Preliminary time-course studies ofbleb formation by VP and PDB revealed that blebbing was fullydeveloped by 10 minutes of exposure to either of these agonists.Surface protrusions were identified by light microscopy using aninverted microscope (Olympus IMT2-RFL, Olympus Optical Ltd.),and expressed as the percentage of couplets (.30) exhibiting blebson their cellular surface.24-26 A couplet was designated as blebbed ifeither or both of the cells showed at least one membrane protru-sion.26
In some experiments, plasma membrane blebs were also observedby both scanning and transmission electron microscopy accordingto standard procedures involving glutaraldehyde and osmium fixa-tion and dehydratation. Cells were viewed in a Hitachi 2300scanning electron microscope or in a JEOL 1200 EX transmissionelectron microscope.
Assessment of F-Actin Distribution. Phalloidin-FITC labeling of fixed,permeabilized cells was employed to visualize cellular distributionof the F-actin cytoskeleton. Fluorescent staining of actin wasperformed using a modification of the method described by Knuttonet al.,27 as described by Wilton et al.22 Briefly, cells were fixed with3% formalin in phosphate-buffered saline (PBS) and stored at 4°Cuntil permeabilized with 0.1% Triton X-100 in PBS. F-actin waslabeled by treating the cells with phalloidin-FITC (5 µg/mL in PBS).After washing in PBS, cells were mounted in 90% glycerol-PBScontaining 2.5% (wt/vol) diazabicyclo[2,2,2]octane. The specimenswere then examined under fluorescent light (excitation wavelength,490 nm; emission wavelength, .525 nm) using a Zeiss microscope(Axiovert 350TV, Carl Zeiss Oberkochen, Ltd., Welwyn Garden City,Herts, UK), equipped with Zeiss plan-neofluar lenses. Monochromeimages taken in 1-µm steps were captured on a CCD video camera(Hamamatsu Photonic Sys. Corp., Hamamatsu City, Japan), andout-of-focus flair was removed using a deconvolution program(Micro-Tome Mac, Vaytek Ltd., Fairfield, IL). Images were analyzedto quantify the total intensity of fluorescence within the couplet and,specifically, in the pericanalicular region by using the Fluovisionprogram (Improvision, Warwick Science Park, Coventry, England).
Treatments. Changes in cVA of CLF, cVR of CLF, and blebbinginduced by VP and a number of protein kinase and Ca21 activatorsin the absence or presence of appropriate inhibitors were studied todeterminate the respective roles of the PKC- and the Ca21-dependent signal pathways in the effects induced by the hormone(Fig. 1). The compounds tested comprised (final concentrations inplate, volume of vehicle used to deliver, time of exposure): 1) VP(1028 mol/L, 1 µL in 0.05% acetic acid, 15 minutes); 2) VP 1 theprotein kinase inhibitor, H-7 (1024 mol/L, 10 µL in saline, 5minutes); 3) VP 1 the protein kinase A (PKA) inhibitor, KT5720
1032 ROMA ET AL. HEPATOLOGY October 1998

(1026 mol/L, 2 µL in dimethylsulfoxide (DMSO), 5 minutes); 4)VP 1 the [Ca21]cyt chelating agent, BAPTA/AM (2 3 1025 mol/L, 2 µL inDMSO, 30 minutes); 5) VP 1 the calmodulin-dependent proteinkinase II inhibitor, calmidazolium (5 3 1026 mol/L, 2 µL in DMSO,30 minutes); 6) PDB (1026 mol/L, 2 µL in DMSO, 15 minutes); 7)PDB 1 H-7; 8) dibutyryl-cAMP (5 3 1026 mol/L, 2 µL in DMSO, 15minutes); and 9) the Ca21-ionophore, A23187 (2.5 3 1026 mol/L, 2µL in DMSO, 30 minutes). The effect of the protein kinase or theCa21 inhibitors administered alone was also studied. For thispurpose, the cells were exposed to the inhibitors for the wholeexperimental period, i.e., the time period necessary for the inhibitorto exert its inhibitory effect plus the period of time that the activatorwould have taken to act if it had been present. Concentrations of theinhibitors were chosen by making a balance between effectivenessand specificity, according to the Ki and 50% inhibitory concentra-tions for their respective substrates, as described by the manufacturers.
Because both bleb formation and cytoskeletal disruption werereported to occur either via Ca21-mediated activation of a nonlyso-somal proteolytic system25 or by depletion of cellular thiols associ-ated with oxidative stress,24,28 the involvement of each of thesemechanisms on the effects induced by VP was also assessed. For this
purpose, the couplets were pretreated with the Ca21-dependentneutral protease inhibitor, leupeptin (100 µg/L, 50 µL in saline, 30minutes),25 or with the antioxidants, a-tocopherol (5 3 1024 mol/L,2 µL in ethanol, 1 hour) or deferoxamine (1023 mol/L, 2 µL inDMSO, 30 minutes), followed by exposure to VP (1028 mol/L) for15 minutes. The final concentrations of the vehicles (0.1% DMSOand 0.1% ethanol) had no effect on the couplets.
Stocks of all the above-mentioned compounds were prepared inthe solvents indicated above and stored in frozen aliquots until use.Because ancillary experiments showed that vehicles had no indepen-dent effect on the parameters studied, a single control groupreceiving no treatment is reported for each set of experiments.
Viability before and after each treatment was systematicallyassessed by using Trypan blue exclusion, and showed no significantchange over the whole experimental period.
Statistical Analysis. Data are expressed as means 6 SE. Means oftwo groups were compared with the Student’s t test after testing theequality of variances with an F test. Multiple means were comparedwith one-way ANOVA, followed by the Newman-Keuls multiple-range test for pairwise comparisons. Differences were consideredsignificant at P , .05.
FIG. 1. Schematic representation of the activation of the PKC- and the Ca21-dependent signal-transducing pathways by VP. The activators (white arrows)and inhibitors (crosses) used to either stimulate or block these two signal pathways are also depicted. Once bound to the receptor, VP activates a G protein,which, in turn, activates a phosphoinositide-specific phospholipase C. This enzyme hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2), and, by doingso, two messengers are formed, namely inositol 1,4,5, trisphosphate (IP3) and diacylglycerol. Other sources of diacylglycerol formation like G protein–mediatedactivation of phospholipase D are also possible, but are not included in the scheme for the sake of clarity. IP3 induces release, from intracellular stores into thecytosol, of Ca21, which then binds to the intracellular protein, calmodulin. The complex Ca21-calmodulin activates, among others, a Ca21-calmodulin–dependent protein kinase (Ca21-calmodulin–dependent PK), which initiates a cascade of protein phosphorylation that triggers a number of intracellularprocesses. Diacylglycerol, in turn, activates PKC by increasing the affinity of the enzyme for Ca21, an ion that is essential for classical (Ca21-dependent) PKCactivation. PKC, like Ca21-calmodulin–dependent PK, triggers a phosphorylation cascade that controls a number of intracellular events. We mimicked theintracellular Ca21 increase induced by IP3 by using the Ca21-ionophore, A23187, which introduces extracellular Ca21 into the cells. On the other hand, thediacylglycerol effect was mimicked by using the phorbol ester phorbol, PDB, a specific PKC activator. The Ca21-dependent signal pathway was blocked at twolevels, namely preventing Ca21-calmodulin formation (by adding BAPTA/AM, a specific intracellular Ca21 chelator) or inhibiting Ca21-calmodulin–dependent PK (with calmidazolium). On the other hand, the PKC-dependent pathway was inhibited by using the protein kinase inhibitor, H-7.
HEPATOLOGY Vol. 28, No. 4, 1998 ROMA ET AL. 1033
RESULTS
Effect of VP and PDB on Canalicular Function and PlasmaMembrane Blebbing. Under control conditions, 74.1% 6 1.3% ofthe couplets exhibited cVA of CLF following 15 minutes ofexposure to the fluorescent bile acid analog (Table 1). Onceremoved from the incubation medium, 70.3% 6 1.9% of thecouplets retained the fluorescent bile acid analog in their canalicu-lar vacuoles (cVR of CLF assay) for at least 30 minutes. Untreatedcells showed a low background blebbing (,15%), which con-sisted usually in a single small bleb per couplet (type 1 bleb). Thisbasal blebbing was comparable with, or lower than, previouslyreported values,25,26,29 and was most likely a result of membranedamage or metabolic alterations suffered during the isolationprocedure (e.g., high local collagenase digestion or transitoryshortage in oxygen supply).
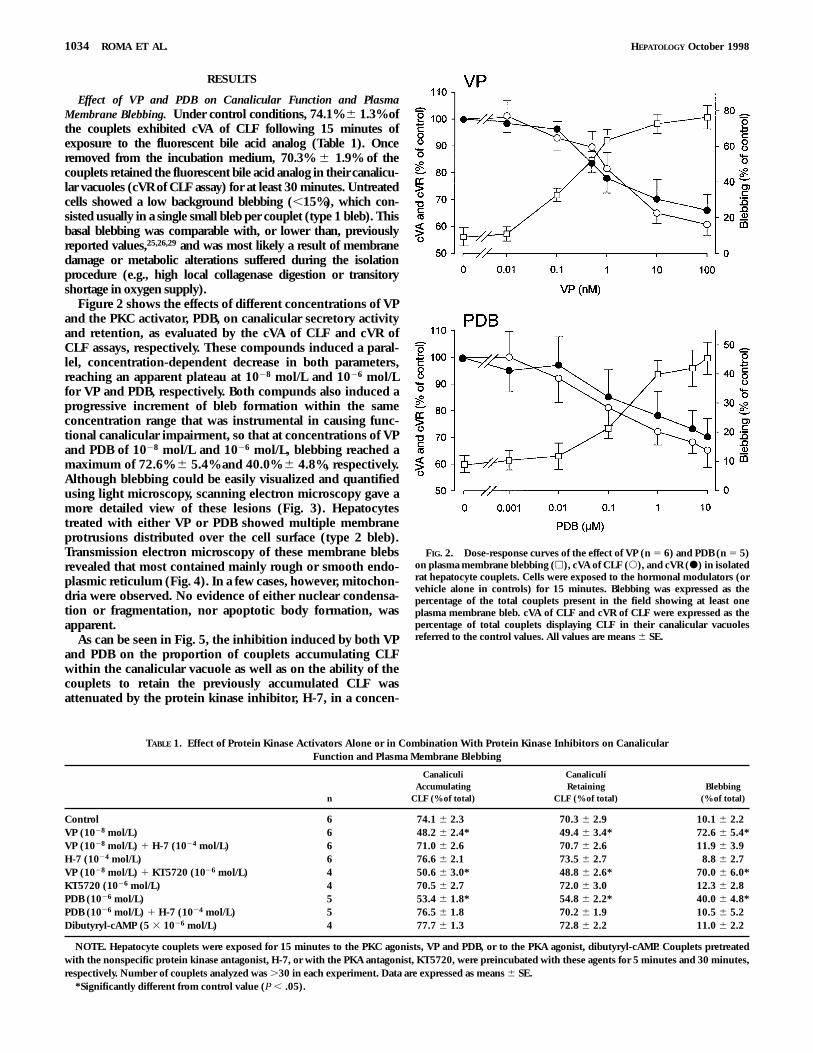
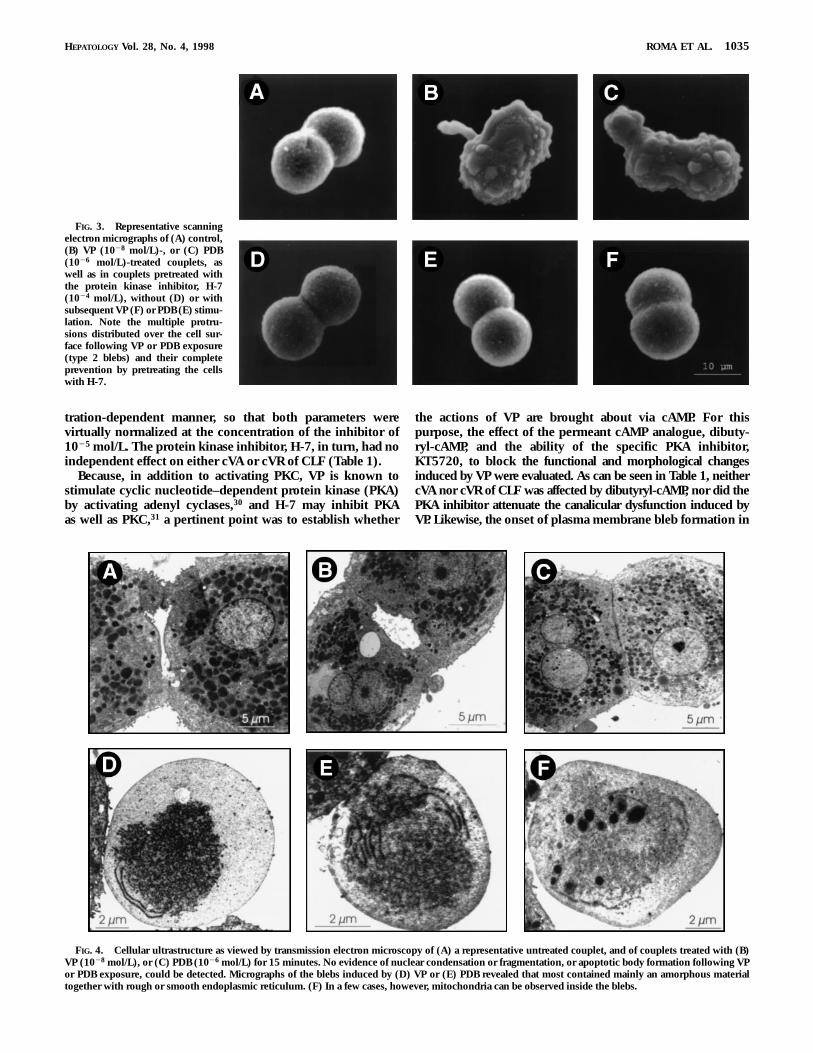
Figure 2 shows the effects of different concentrations of VPand the PKC activator, PDB, on canalicular secretory activityand retention, as evaluated by the cVA of CLF and cVR ofCLF assays, respectively. These compounds induced a paral-lel, concentration-dependent decrease in both parameters,reaching an apparent plateau at 1028 mol/L and 1026 mol/Lfor VP and PDB, respectively. Both compunds also induced aprogressive increment of bleb formation within the sameconcentration range that was instrumental in causing func-tional canalicular impairment, so that at concentrations of VPand PDB of 1028 mol/L and 1026 mol/L, blebbing reached amaximum of 72.6% 6 5.4% and 40.0% 6 4.8%, respectively.Although blebbing could be easily visualized and quantifiedusing light microscopy, scanning electron microscopy gave amore detailed view of these lesions (Fig. 3). Hepatocytestreated with either VP or PDB showed multiple membraneprotrusions distributed over the cell surface (type 2 bleb).Transmission electron microscopy of these membrane blebsrevealed that most contained mainly rough or smooth endo-plasmic reticulum (Fig. 4). In a few cases, however, mitochon-dria were observed. No evidence of either nuclear condensa-tion or fragmentation, nor apoptotic body formation, wasapparent.
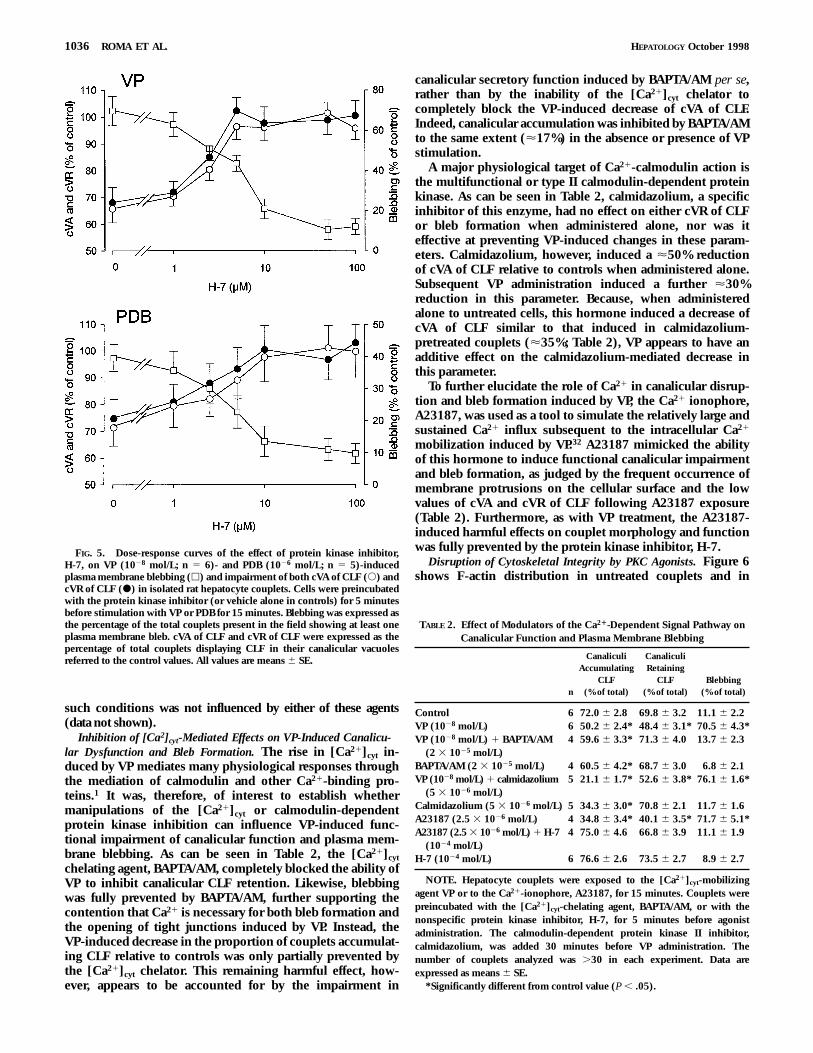
As can be seen in Fig. 5, the inhibition induced by both VPand PDB on the proportion of couplets accumulating CLFwithin the canalicular vacuole as well as on the ability of thecouplets to retain the previously accumulated CLF wasattenuated by the protein kinase inhibitor, H-7, in a concen-
FIG. 2. Dose-response curves of the effect of VP (n 5 6) and PDB (n 5 5)on plasma membrane blebbing (h), cVA of CLF (s), and cVR (d) in isolatedrat hepatocyte couplets. Cells were exposed to the hormonal modulators (orvehicle alone in controls) for 15 minutes. Blebbing was expressed as thepercentage of the total couplets present in the field showing at least oneplasma membrane bleb. cVA of CLF and cVR of CLF were expressed as thepercentage of total couplets displaying CLF in their canalicular vacuolesreferred to the control values. All values are means 6 SE.
TABLE 1. Effect of Protein Kinase Activators Alone or in Combination With Protein Kinase Inhibitors on CanalicularFunction and Plasma Membrane Blebbing
n
CanaliculiAccumulating
CLF (% of total)
CanaliculiRetaining
CLF (% of total)Blebbing
(% of total)
Control 6 74.1 6 2.3 70.3 6 2.9 10.1 6 2.2VP (1028 mol/L) 6 48.2 6 2.4* 49.4 6 3.4* 72.6 6 5.4*VP (1028 mol/L) 1 H-7 (1024 mol/L) 6 71.0 6 2.6 70.7 6 2.6 11.9 6 3.9H-7 (1024 mol/L) 6 76.6 6 2.1 73.5 6 2.7 8.8 6 2.7VP (1028 mol/L) 1 KT5720 (1026 mol/L) 4 50.6 6 3.0* 48.8 6 2.6* 70.0 6 6.0*KT5720 (1026 mol/L) 4 70.5 6 2.7 72.0 6 3.0 12.3 6 2.8PDB (1026 mol/L) 5 53.4 6 1.8* 54.8 6 2.2* 40.0 6 4.8*PDB (1026 mol/L) 1 H-7 (1024 mol/L) 5 76.5 6 1.8 70.2 6 1.9 10.5 6 5.2Dibutyryl-cAMP (5 3 1026 mol/L) 4 77.7 6 1.3 72.8 6 2.2 11.0 6 2.2
NOTE. Hepatocyte couplets were exposed for 15 minutes to the PKC agonists, VP and PDB, or to the PKA agonist, dibutyryl-cAMP. Couplets pretreatedwith the nonspecific protein kinase antagonist, H-7, or with the PKA antagonist, KT5720, were preincubated with these agents for 5 minutes and 30 minutes,respectively. Number of couplets analyzed was .30 in each experiment. Data are expressed as means 6 SE.
*Significantly different from control value (P , .05).
1034 ROMA ET AL. HEPATOLOGY October 1998
tration-dependent manner, so that both parameters werevirtually normalized at the concentration of the inhibitor of1025 mol/L. The protein kinase inhibitor, H-7, in turn, had noindependent effect on either cVA or cVR of CLF (Table 1).
Because, in addition to activating PKC, VP is known tostimulate cyclic nucleotide–dependent protein kinase (PKA)by activating adenyl cyclases,30 and H-7 may inhibit PKAas well as PKC,31 a pertinent point was to establish whether
the actions of VP are brought about via cAMP. For thispurpose, the effect of the permeant cAMP analogue, dibuty-ryl-cAMP, and the ability of the specific PKA inhibitor,KT5720, to block the functional and morphological changesinduced by VP were evaluated. As can be seen in Table 1, neithercVA nor cVR of CLF was affected by dibutyryl-cAMP, nor did thePKA inhibitor attenuate the canalicular dysfunction induced byVP. Likewise, the onset of plasma membrane bleb formation in
FIG. 4. Cellular ultrastructure as viewed by transmission electron microscopy of (A) a representative untreated couplet, and of couplets treated with (B)VP (1028 mol/L), or (C) PDB (1026 mol/L) for 15 minutes. No evidence of nuclear condensation or fragmentation, or apoptotic body formation following VPor PDB exposure, could be detected. Micrographs of the blebs induced by (D) VP or (E) PDB revealed that most contained mainly an amorphous materialtogether with rough or smooth endoplasmic reticulum. (F) In a few cases, however, mitochondria can be observed inside the blebs.
FIG. 3. Representative scanningelectron micrographs of (A) control,(B) VP (1028 mol/L)-, or (C) PDB(1026 mol/L)-treated couplets, aswell as in couplets pretreated withthe protein kinase inhibitor, H-7(1024 mol/L), without (D) or withsubsequent VP (F) or PDB (E) stimu-lation. Note the multiple protru-sions distributed over the cell sur-face following VP or PDB exposure(type 2 blebs) and their completeprevention by pretreating the cellswith H-7.
HEPATOLOGY Vol. 28, No. 4, 1998 ROMA ET AL. 1035
such conditions was not influenced by either of these agents(data not shown).
Inhibition of [Ca2]cyt-Mediated Effects on VP-Induced Canalicu-lar Dysfunction and Bleb Formation. The rise in [Ca21]cyt in-duced by VP mediates many physiological responses throughthe mediation of calmodulin and other Ca21-binding pro-teins.1 It was, therefore, of interest to establish whethermanipulations of the [Ca21]cyt or calmodulin-dependentprotein kinase inhibition can influence VP-induced func-tional impairment of canalicular function and plasma mem-brane blebbing. As can be seen in Table 2, the [Ca21]cyt
chelating agent, BAPTA/AM, completely blocked the ability ofVP to inhibit canalicular CLF retention. Likewise, blebbingwas fully prevented by BAPTA/AM, further supporting thecontention that Ca21 is necessary for both bleb formation andthe opening of tight junctions induced by VP. Instead, theVP-induced decrease in the proportion of couplets accumulat-ing CLF relative to controls was only partially prevented bythe [Ca21]cyt chelator. This remaining harmful effect, how-ever, appears to be accounted for by the impairment in
canalicular secretory function induced by BAPTA/AM per se,rather than by the inability of the [Ca21]cyt chelator tocompletely block the VP-induced decrease of cVA of CLF.Indeed, canalicular accumulation was inhibited by BAPTA/AMto the same extent (<17%) in the absence or presence of VPstimulation.
A major physiological target of Ca21-calmodulin action isthe multifunctional or type II calmodulin-dependent proteinkinase. As can be seen in Table 2, calmidazolium, a specificinhibitor of this enzyme, had no effect on either cVR of CLFor bleb formation when administered alone, nor was iteffective at preventing VP-induced changes in these param-eters. Calmidazolium, however, induced a <50% reductionof cVA of CLF relative to controls when administered alone.Subsequent VP administration induced a further <30%reduction in this parameter. Because, when administeredalone to untreated cells, this hormone induced a decrease ofcVA of CLF similar to that induced in calmidazolium-pretreated couplets (<35%; Table 2), VP appears to have anadditive effect on the calmidazolium-mediated decrease inthis parameter.
To further elucidate the role of Ca21 in canalicular disrup-tion and bleb formation induced by VP, the Ca21 ionophore,A23187, was used as a tool to simulate the relatively large andsustained Ca21 influx subsequent to the intracellular Ca21
mobilization induced by VP.32 A23187 mimicked the abilityof this hormone to induce functional canalicular impairmentand bleb formation, as judged by the frequent occurrence ofmembrane protrusions on the cellular surface and the lowvalues of cVA and cVR of CLF following A23187 exposure(Table 2). Furthermore, as with VP treatment, the A23187-induced harmful effects on couplet morphology and functionwas fully prevented by the protein kinase inhibitor, H-7.
Disruption of Cytoskeletal Integrity by PKC Agonists. Figure 6shows F-actin distribution in untreated couplets and in
FIG. 5. Dose-response curves of the effect of protein kinase inhibitor,H-7, on VP (1028 mol/L; n 5 6)- and PDB (1026 mol/L; n 5 5)-inducedplasma membrane blebbing (h) and impairment of both cVA of CLF (s) andcVR of CLF (d) in isolated rat hepatocyte couplets. Cells were preincubatedwith the protein kinase inhibitor (or vehicle alone in controls) for 5 minutesbefore stimulation with VP or PDB for 15 minutes. Blebbing was expressed asthe percentage of the total couplets present in the field showing at least oneplasma membrane bleb. cVA of CLF and cVR of CLF were expressed as thepercentage of total couplets displaying CLF in their canalicular vacuolesreferred to the control values. All values are means 6 SE.
TABLE 2. Effect of Modulators of the Ca21-Dependent Signal Pathway onCanalicular Function and Plasma Membrane Blebbing
n
CanaliculiAccumulating
CLF(% of total)
CanaliculiRetaining
CLF(% of total)
Blebbing(% of total)
Control 6 72.0 6 2.8 69.8 6 3.2 11.1 6 2.2VP (1028 mol/L) 6 50.2 6 2.4* 48.4 6 3.1* 70.5 6 4.3*VP (1028 mol/L) 1 BAPTA/AM
(2 3 1025 mol/L)4 59.6 6 3.3* 71.3 6 4.0 13.7 6 2.3
BAPTA/AM (2 3 1025 mol/L) 4 60.5 6 4.2* 68.7 6 3.0 6.8 6 2.1VP (1028 mol/L) 1 calmidazolium
(5 3 1026 mol/L)5 21.1 6 1.7* 52.6 6 3.8* 76.1 6 1.6*
Calmidazolium (5 3 1026 mol/L) 5 34.3 6 3.0* 70.8 6 2.1 11.7 6 1.6A23187 (2.5 3 1026 mol/L) 4 34.8 6 3.4* 40.1 6 3.5* 71.7 6 5.1*A23187 (2.5 3 1026 mol/L) 1 H-7
(1024 mol/L)4 75.0 6 4.6 66.8 6 3.9 11.1 6 1.9
H-7 (1024 mol/L) 6 76.6 6 2.6 73.5 6 2.7 8.9 6 2.7
NOTE. Hepatocyte couplets were exposed to the [Ca21]cyt-mobilizingagent VP or to the Ca21-ionophore, A23187, for 15 minutes. Couplets werepreincubated with the [Ca21]cyt-chelating agent, BAPTA/AM, or with thenonspecific protein kinase inhibitor, H-7, for 5 minutes before agonistadministration. The calmodulin-dependent protein kinase II inhibitor,calmidazolium, was added 30 minutes before VP administration. Thenumber of couplets analyzed was .30 in each experiment. Data areexpressed as means 6 SE.
*Significantly different from control value (P , .05).
1036 ROMA ET AL. HEPATOLOGY October 1998
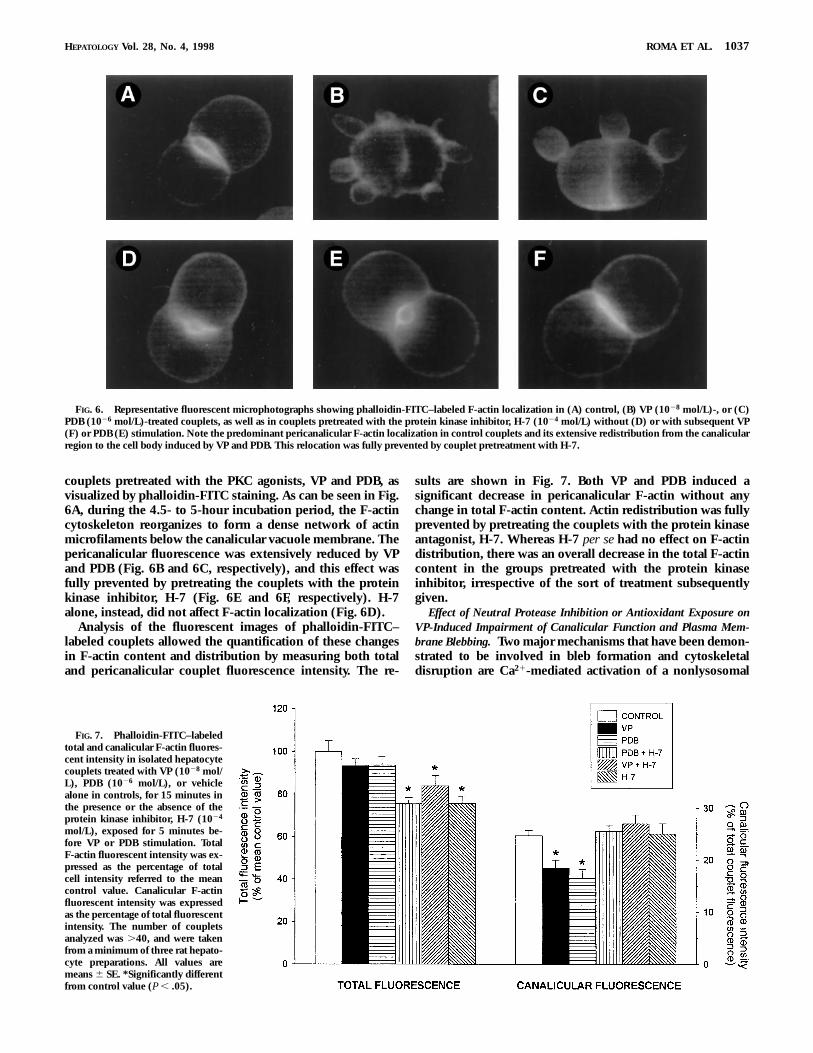
couplets pretreated with the PKC agonists, VP and PDB, asvisualized by phalloidin-FITC staining. As can be seen in Fig.6A, during the 4.5- to 5-hour incubation period, the F-actincytoskeleton reorganizes to form a dense network of actinmicrofilaments below the canalicular vacuole membrane. Thepericanalicular fluorescence was extensively reduced by VPand PDB (Fig. 6B and 6C, respectively), and this effect wasfully prevented by pretreating the couplets with the proteinkinase inhibitor, H-7 (Fig. 6E and 6F, respectively). H-7alone, instead, did not affect F-actin localization (Fig. 6D).
Analysis of the fluorescent images of phalloidin-FITC–labeled couplets allowed the quantification of these changesin F-actin content and distribution by measuring both totaland pericanalicular couplet fluorescence intensity. The re-
sults are shown in Fig. 7. Both VP and PDB induced asignificant decrease in pericanalicular F-actin without anychange in total F-actin content. Actin redistribution was fullyprevented by pretreating the couplets with the protein kinaseantagonist, H-7. Whereas H-7 per se had no effect on F-actindistribution, there was an overall decrease in the total F-actincontent in the groups pretreated with the protein kinaseinhibitor, irrespective of the sort of treatment subsequentlygiven.
Effect of Neutral Protease Inhibition or Antioxidant Exposure onVP-Induced Impairment of Canalicular Function and Plasma Mem-brane Blebbing. Two major mechanisms that have been demon-strated to be involved in bleb formation and cytoskeletaldisruption are Ca21-mediated activation of a nonlysosomal
FIG. 6. Representative fluorescent microphotographs showing phalloidin-FITC–labeled F-actin localization in (A) control, (B) VP (1028 mol/L)-, or (C)PDB (1026 mol/L)-treated couplets, as well as in couplets pretreated with the protein kinase inhibitor, H-7 (1024 mol/L) without (D) or with subsequent VP(F) or PDB (E) stimulation. Note the predominant pericanalicular F-actin localization in control couplets and its extensive redistribution from the canalicularregion to the cell body induced by VP and PDB. This relocation was fully prevented by couplet pretreatment with H-7.
FIG. 7. Phalloidin-FITC–labeledtotal and canalicular F-actin fluores-cent intensity in isolated hepatocytecouplets treated with VP (1028 mol/L), PDB (1026 mol/L), or vehiclealone in controls, for 15 minutes inthe presence or the absence of theprotein kinase inhibitor, H-7 (1024
mol/L), exposed for 5 minutes be-fore VP or PDB stimulation. TotalF-actin fluorescent intensity was ex-pressed as the percentage of totalcell intensity referred to the meancontrol value. Canalicular F-actinfluorescent intensity was expressedas the percentage of total fluorescentintensity. The number of coupletsanalyzed was .40, and were takenfrom a minimum of three rat hepato-cyte preparations. All values aremeans 6 SE. *Significantly differentfrom control value (P , .05).
HEPATOLOGY Vol. 28, No. 4, 1998 ROMA ET AL. 1037

proteolytic system,25 and the depletion of cellular thiolsassociated with oxidative stress.24,28 It was of interest, there-fore, to determine whether either of these mechanisms wasinvolved in VP-induced blebbing. As can be seen in Table 3,neither the Ca21-activated neutral protease inhibitor, leupep-tin, nor the antioxidants, a-tocopherol (a free radical scaven-ger) or deferoxamine (an Fe21-chelator), was able to preventeither VP-induced plasma membrane blebbing or canaliculardysfunction.
DISCUSSION
Effect of VP on Plasma Membrane Bleb Formation and Canalicu-lar Function. In the present study, we have shown that VP wasinstrumental in inducing actin cytoskeletal disruption, F-actin being redistributed from the pericanalicular area to thecell body. This rearrangement was associated with extensivebleb formation and the simultaneous impairment of thecouplets ability to accumulate and retain preaccumulatedmaterial into the canalicular lumen, as evaluated by the cVAof CLF and cVR of CLF assays, respectively. These functionaland morphological changes appeared to parallel each other,because both effects were visualized within the same range ofdoses (see Fig. 2).
Bleb formation is a common morphological feature of bothhepatocellular injury and apoptosis.28 Transmission electronmicroscopy studies have shown that blebs produced bynecrotic cells contain amorphous material, together withrough and smooth endoplasmic reticulum, whereas the blebsproduced by apoptotic cells also contain larger organelles,such as mitochondria, lysosomes, and peroxisomes.28,33 Fur-thermore, in contrast to necrotic cells, apoptotic cells exhibitnuclear condensation and fragmentation, organelle relocationand compaction, and ultimately, cell separation into mem-brane-bound fragments referred to as ‘‘apoptotic bodies.’’33
Although the morphological pattern observed following VPor PDB administration was rather compatible with thatobserved in necrosis (see Fig. 4), the presence of mitochon-dria in some blebs suggests that apoptosis can be involved. It
must be kept in mind, however, that necrosis and apoptosiscannot be distinguished from each other only on the basis ofhistological criteria. At certain stages, both modes of celldeath share some morphological characteristics.33 Further-more, both necrosis and apoptosis may occur in response tothe same injurious agents or circumstances depending on theseverity and time scale of the injury.33 A definitive conclusionabout the nature of this process will therefore require furtherstudies employing more specific tests of apoptosis at variabletime exposure, like fluorescent in situ nick end labeling ofcleaved DNA debris strands or enzyme-linked immunosor-bent assay quantification of DNA fragmentation.
Different molecular mechanisms of bleb formation havebeen proposed, including alterations in thiol and Ca21
homeostasis, adenosine triphosphate depletion, and alter-ations in the content and composition of membrane lipids.28
However, the demonstration that toxins that alter microfila-ments directly also induce extensive plasma membraneblebbing,34 led to the assumption that alterations of thestructure and function of the cytoskeleton could represent acommon mechanism of bleb formation.28 This contentionwas further supported by the finding that many of thecompounds known to induce blebbing simultaneously dis-rupted the cytoskeletal integrity.24,26,35 Cytoskeletal dysfunc-tion can, in turn, lead to cholestasis.14 Different mechanismshave been suggested to account for this cholestatic effect,including impaired canalicular contractions, which are respon-sible for peristaltic flow of bile within the biliary tree,15 arrestof the vesicular transport,15 and finally, increased tight-junctional permeability.36 Bearing all of the aforementionedin mind, it is possible to speculate that a link exists betweenVP-induced hepatocyte surface alterations, cytoskeletal dis-ruption, and functional alterations in the hepatocyte capabil-ity to secrete and/or retain preaccumulated material in thebile canaliculus.
Secretory dysfunction was evaluated as a whole in hepato-cyte couplets by assessing cVA of CLF. This parameter is theend result of a number of processes, namely basolateraluptake of the bile acid analogue, its intracellular transport, itssecretion across the canalicular membrane, and, finally, itsretention in the canalicular vacuole. The last of theseprocesses requires integrity of the structures that seal thecanaliculus, and may be specifically evaluated through thecVR of CLF assay.12 Our results that cVA of CLF fell in parallelwith cVR of CLF following VP administration suggest thatthe impairment of tight-junctional permeability is a major, ifnot the only, mechanism by which this hormone inducesimpairment of CLF accumulation. This is in line withprevious findings showing that VP increases tight-junctionalpermeability in both isolated perfused rat liver4,7,9 and inisolated rat hepatocyte couplets.10-12
Role of PKC in VP-Induced Impairment of Canalicular Functionand Plasma Membrane Bleb Formation. VP is known to activatePKC by stimulating the formation of diacylglycerol, a specificPKC activator.1 This process involves interaction of VP withtwo kinds of receptors, namely V1a and V2.37 In addition toactivating PKC, V2 also activates PKA by stimulating adenylcyclases.30
Our results suggest that PKC activation plays an importantrole in VP-induced impairment of canalicular function andplasma membrane bleb formation. In fact, the PKC agonist,PDB, induced a pattern of canalicular dysfunction andcytoskeletal disruption similar to that induced by VP, CLF
TABLE 3. Effect of Leupeptin and the Antioxidants, a-Tocopherol andDeferoxamine, on VP-Induced Impairment of Canalicular Function and
Plasma Membrane Blebbing
n
CanaliculiAccumulating
CLF(% of total)
CanaliculiRetaining
CLF(% of total)
Blebbing(% of total)
Control 6 69.8 6 2.6 71.9 6 1.8 12.0 6 1.4VP (1028 mol/L) 6 47.7 6 3.0* 51.1 6 2.8* 70.3 6 4.5*VP (1028 mol/L) 1 leupeptin
(100 mg/L)4 50.3 6 2.1* 49.9 6 3.0* 57.0 6 5.2*
Leupeptin (100 mg/L) 4 66.9 6 3.3 70.5 6 2.7 13.5 6 1.4VP (1028 mol/L) 1 a-tocopherol 4 47.5 6 3.2* 52.1 6 3.3* 71.0 6 4.1*a-Tocopherol (5 3 1024 mol/L) 4 74.0 6 2.5 73.2 6 3.5 10.1 6 0.8VP (1028 mol/L) 1 deferoxamine
(1023 mol/L)4 48.7 6 2.4* 49.3 6 3.2* 72.2 6 1.0*
Deferoxamine (1023 mol/L) 4 73.6 6 2.7 73.8 6 2.6 11.8 6 3.3
NOTE. Hepatocyte couplets were preincubated with the Ca21-activatedneutral protease inhibitor, leupeptin, for 30 minutes or with the antioxi-dants, deferoxamine and a-tocopherol, for 1 hour and 30 minutes, respec-tively. Cells were subsequently exposed to VP for 15 minutes. The number ofcouplets analyzed was .30 in each experiment. Data are expressed asmeans 6 SE.
*Significantly different from control value (P , .05).
1038 ROMA ET AL. HEPATOLOGY October 1998

retention being affected roughly to the same extent as CLFaccumulation. Essentially, the same results were obtainedwhen phorbol 12-myristate 13-acetate, another PKC activa-tor, was administered (data not shown), further eliminating anonspecific effect of PDB on the parameters affected. Further-more, the protein kinase antagonist, H-7, prevented both VP-and PDB-induced morphological and functional alterationsin the concentration range that was instrumental in inhibit-ing protein kinases.31 Taken together, these results addsupport to the contention that tight-junctional permeabilityis the preferential target of VP action via PKC activation, andthat cytoskeletal disruption would play a crucial role in thisphenomenon. PKA activation, instead, appears not to beinvolved, because the permeant cAMP-analogue, dibutyryl-cAMP, did not induce any change in canalicular function andbleb formation, nor did the PKA inhibitor, KT5720, block theVP-induced alterations (see Table 1). These results are in linewith the view that hepatocytes only express V receptors butnot V receptors for VP, and only the latter activates adenylcyclase.37
Interestingly, hyperphosphorylation of proteins caused byokadaic acid–induced inhibition of phosphatases 1 and 2A,two of the major phosphatases responsible for the serine/threonine dephosphorylation in mammalian cells, was shownto disrupt the hepatocyte cytoskeleton, as suggested by theformation of a homogenate rather than of structurally intactghosts after electrodisruption.38 Likewise, microcystin-LR,another protein phosphatase inhibitor, was reported to dis-turb the ultrastructural organization of both actin and inter-mediate filaments in hepatocytes.39 For inhibition of theseprotein phosphatases to induce protein phosphorylation,however, there must be an active involvement of proteinkinases to carry out the attachment of the phosphate groupsin the first place. One likely candidate is PKC. Indeed, PKCactivation has been shown to induce extensive actin filamentreorganization in a variety of cell lines,40-42 presumably by thephosphorylation of actin43,44 as well as other actin-associatedproteins such as vinculin, filamin, a-actinin, and calspec-tin.41,44-46 Furthermore, in addition to the effect on actinconformation, PKC activation was reported to induce changesin other major cytoskeletal elements such as intermediatefilament proteins47-49 and tubulin.42 Finally, PKC was re-ported to have a role in hepatocyte apoptosis induced by anumber of compounds such as the bile salt, glycochenodeoxy-cholate,50 or the microtubule antagonists, vinblastine andcolchicine.51
Role of [Ca21]cyt in VP-Induced Impairment of Canalicular Func-tion and Plasma Membrane Bleb Formation. [Ca21]cyt plays amajor role in bile formation. VP, as well as [Ca21]cyt elevationindependently of VP action, induces a decrease in bile flow inthe isolated perfused rat liver.2-4 Dissipation of osmoticgradients induced by an increase of ‘‘leakiness’’ of theparacellular barrier appears to play a key role in thisphenomenon. Indeed, VP as well as a number of agentsaffecting Ca21 mobilization such as the Ca21-ionophore,A23187, and the inhibitor of microsomal Ca21 sequestration,t-butyl-benzohydroquinone, were all shown to increase tight-junctional permeability in both isolated perfused rat liver4,7-9
and in isolated rat hepatocyte couplets.10-12 Finally, thefinding that similar manipulations leading to [Ca21]cyt eleva-tion induce bleb formation (see Table 2 and Nicotera,25
Stone,26,29 and Orrenius52) leads to the proposal that Ca21
may be involved in the cytoskeletal alterations associated
with bleb formation. Until now, two different Ca21-depen-dent mechanisms have been proposed to account for thesealterations. Increased [Ca21]cyt may cause actin microfila-ments to dissociate from a-actinin, which serves as anintermediate in the association of microfilaments with actin-binding proteins in the plasma membrane.53 Alternatively,Ca21 can also activate nonlysosomal proteases, which cancleave actin-binding proteins and lead to dissociation of thecytoskeleton from the plasma membrane.25 This latter alterna-tive seems not to be the case with VP, however, because theCa21-activated neutral protease inhibitor, leupeptin, wasineffective at preventing the alterations induced by thishormone (see Table 3).
In line with the view that [Ca21]cyt elevation plays a crucialrole in VP-induced functional and morphological impair-ment, the buffering of the rise of [Ca21]cyt induced by VPusing BAPTA/AM as a Ca21 chelator afforded completeprotection on the harmful effects induced by the hormone(see Table 2). However, our finding that both VP- andA23187-induced bleb formation and canalicular dysfunctionwere fully prevented by the protein kinase inhibitor, H-7,suggests that [Ca21]cyt elevation acts indirectly through PKC.This is conceivable, because classical PKC requires Ca21 forits activation and the Ca21- and PKC-mediated signallingpathways often act synergistically with each other to elicitmaximal cellular responses.32 Other putative effectors of theCa21-activated signalling cascade downstream of the Ca21-calmodulin–dependent protein kinase II appear not to beinvolved because calmidazolium, a specific inhibitor of thisenzyme, was not able to prevent these effects. Instead, thiskinase activity appears to be required for CLF to be trans-ported to the bile canaliculus, because cVA of this bile acidanalogue was impaired following either [Ca21]cyt chelation orcalmidazolium administration. Requirement of Ca21 for thenormal translocation and/or canalicular secretion, but not forthe basolateral uptake, of bile salts had been previouslysuggested,54 although the precise site on the Ca21-activatedsignalling pathway at which this cation is required has notbeen assessed. Our results point to a preferential role ofCa21-calmodulin–dependent protein kinase II in hepatobili-ary bile acid transport.
In conclusion, our results show that VP induces a numberof morphological alterations including cytoskeletal disrup-tion and the concomitant formation of plasma membraneblebs. These events were associated with functional impair-ment of the canalicular ability to accumulate bile salts, whichwas caused, at least in part, by an impaired capability of thetight junctions to retain the bile salts in the bile canaliculus.Because all the evidence points to a preferential role for thePKC signalling pathway in such events, our study wouldprovide a new intracellular mechanism for cytoskeleton-associated functional and morphological alterations in hepa-tocyte injury, i.e., PKC activation. We hope that furtherstudies on the involvement of this protein in other forms oftoxic injury will further expand our current understanding ofthe mechanisms and regulatory aspects of hepatotoxicity andits prevention by hormonal modulation.
Acknowledgment: The authors thank Lesley Tomkins andPeter Whittle for assistance with the electron microscopy, andJose Manuel Pellegrino for his assistance in graphic design.
HEPATOLOGY Vol. 28, No. 4, 1998 ROMA ET AL. 1039

REFERENCES
1. Exton JH. Role of inositol trisphosphate and diacylglycerol in theregulation of liver function. In: Arias IM, Jacoby WB, Popper H, eds. TheLiver: Biology and Pathobiology. New York: Raven, 1988:785-791.
2. Corasanti JG, Smith ND, Gordon ER, Boyer JL. Protein kinase C agonistsinhibit bile secretion independently of effects on the microcirculation inthe isolated perfused rat liver. HEPATOLOGY 1989;10:8-13.
3. Nathanson MH, Gautam A, Bruck R, Isales C, Boyer JL. Effect of Ca21
agonists on cytosolic Ca21 and on bile secretion in the isolated perfusedrat liver. HEPATOLOGY 1992;15:107-116.
4. Bruck R, Nathanson MH, Roelofsen H, Boyer JL. Effects of protein kinaseC and cytosolic Ca21 on exocytosis in the isolated perfused rat liver.HEPATOLOGY 1994;20:1032-1040.
5. Nathanson MH, Gautam A, Boyer JL. Effect of vasopressin and phorboldibutyrate on bile secretion in treated rat hepatocyte couplets [Abstract].Gastroenterology 1990;98:A615.
6. Boyer JL, Soroka CJ. Vesicle targeting to the apical domain regulated bileexcretory function in isolated rat hepatocyte couplets. Gastroenterology1995;109:1600-1611.
7. Lowe PJ, Miyai K, Steinbach JH, Hardison WGM. Hormonal regulationof hepatocyte tight junctional permeability. Am J Physiol 1988;255:G454-G461.
8. Kan KS, Coleman R. The calcium ionophore A23187 increases thetight-junctional permeability in rat liver. Biochem J 1988;256:1039-1041.
9. Llopis J, Kass GEN, Duddy SK, Farrel GC, Gahm A, Orrenius S.Mobilization of the hormone-sensitive calcium pool increases tightjunctional permeability in the perfused rat liver. FEBS Lett 1991;280:84-86.
10. Nathanson MH, Gautam A, Ng OC, Boyer JL. Hormonal regulation ofparacellular permeability in isolated rat hepatocyte couplets. Am JPhysiol 1992;262:G1079-G1086.
11. Burgstahler AD, Nathanson MH. NO modulates the apicolateral cytoskel-eton of isolated hepatocytes by a PKC-dependent, cGMP-independentmechanism. Am J Physiol 1995;269:G789-G799.
12. Roma MG, Orsler DJ, Coleman R. Canalicular retention as a marker oftight junctional permeability in isolated hepatocyte couplets: effects ofprotein kinase modulation and cholestatic agents. Fund Appl Toxicol1997;37:71-81.
13. Boyer JL. Tight junctions in normal and cholestatic liver: does theparacellular pathway have functional significance? HEPATOLOGY 1983;3:614-617.
14. Phillips MJ, Satir P. The cytoskeleton of the hepatocyte: organization,relationship, and pathology. In: Arias IM, Jacoby WB, Popper H, eds. TheLiver: Biology and Pathobiology. New York: Raven, 1988:11-27.
15. Phillips MJ, Oshio C, Miyairi M, Watanabe S, Smith CR. What is actindoing in the liver cell? HEPATOLOGY 1983;3:433-436.
16. Thibault N, Claude JR, Ballet F. Actin filament alteration as a potentialmarker for cholestasis: a study in isolated rat hepatocyte couplets.Toxicology 1992;73:269-279.
17. Mills CO, Rahman K, Coleman R, Elias E. Cholyllysyl fluoresceinsynthesis, biliary excretion in vivo and during single pass perfusion inisolated rat liver. Biochim Biophys Acta 1991;1115:151-156.
18. Wilton JC, Coleman R, Lankester DJ, Chipman JK. Stability andoptimization of canalicular function in hepatocyte couplets. Cell Bio-chem Funct 1993;11:179-185.
19. Gautam A, Ng O-C, Strazzabosco M, Boyer JL. Quantitative assessmentof canalicular bile formation in isolated hepatocyte couplets usingmicroscopic optical planimetry. J Clin Invest 1989;83:565-573.
20. Wilton JC, Williams DE, Strain AJ, Parslow RA, Chipman JK, ColemanR. Purification of hepatocyte couplets by centrifugal elutriation. HEPATOL-OGY 1991;14:180-183.
21. Wilton JC, Chipman JK, Lawson CJ, Strain AJ, Coleman R. Periportal-and perivenous-enriched hepatocyte couplets: differences in canalicularactivity and in response to oxidative stress. Biochem J 1993;292:773-779.
22. Wilton JC, Matthews G N, Burgoyne RD, Mills CO, Chipman JK,Coleman R. Fluorescent choleretic and cholestatic bile salts takedifferent paths across the hepatocyte: transcytosis of glycolithocholateleads to an extensive redistribution of annexin II. J Cell Biol 1994;127:401-410.
23. Roman ID, Johnson GD, Coleman R. S-Adenosyl-L-methionine preventsdisruption of canalicular function and pericanalicular cytoskeletonintegrity caused by cyclosporin A in isolated rat hepatocyte couplets.HEPATOLOGY 1996;24:134-140.
24. Jewell SA, Bellomo G, Thor H, Orrenius S, Smith MT. Bleb formation inhepatocytes during drug metabolism is caused by disturbances in thioland calcium ion homeostasis. Science 1982;217:1257-1258.
25. Nicotera P, Hartzell P, Davis G, Orrenius S. The formation of plasmamembrane blebs in hepatocytes exposed to agents that increase cytosolicCa21 is mediated by the activation of a non-lysosomal proteolyticsystem. FEBS Lett 1986;209:139-144.
26. Stone V, Johnson GD, Wilton JC, Coleman R, Chipman JK. Effect ofoxidative stress and disruption of Ca21 homeostasis on hepatocytecanalicular function in vitro. Biochem Pharmacol 1994;47:625-632.
27. Knutton SK, Baldwin T, Williams PH, McNeish SA. Actin accumulationat sites of bacterial adhesion to tissue culture cells: basis of a newdiagnostic test for enteropathogenic and enterohemorrhagic Escherichiacoli. Infect Immun 1989;57:1290-1298.
28. Gores GJ, Hermans B, Lemasters JJ. Plasma membrane bleb formationand rupture: a common feature of hepatocellular injury. HEPATOLOGY
1990;11:690-698.29. Stone V, Lankester DJ, Wilton JC, Coleman R, Chipman JK. Toxicologi-
cal insult of canalicular function in rat hepatocyte couplets: the role ofcalcium. Toxicol In Vitro 1994;8:539-541.
30. Manning M, Sawyer WH. Discovery, development, and some uses ofvasopressin and oxytocin antagonists. J Lab Clin Med 1989;114:617-632.
31. Hidaka H, Inagaki M, Kawamoto S, Sasaki Y. Isoquinolinesulfonamides,novel and potent inhibitors of cyclic nucleotide dependent proteinkinase and protein kinase C. Biochemistry 1984;23:5036-5041.
32. Nishizuka Y. Studies and perspectives of protein kinase C. Nature1986;233:305-312.
33. Patel T, Gores GJ. Apoptosis and hepatobiliary disease. HEPATOLOGY
1995;21:1725-1741.34. Prentki M, Chaponnier C, Jeanrenaud B, Gabbiani G. Actin microfila-
ments, cell shape, and secretory processes in isolated rat hepatocytes.J Cell Biol 1979;81:592-607.
35. Nicotera P, McConkey D, Svensson S-Å, Bellomo G, Orrenius S.Correlation between cytosolic Ca21 and cytotoxicity in hepatocytesexposed to oxidative stress. Toxicology 1988;52:55-63.
36. Elias E, Hubran Z, Wade JB, Boyer JL. Phalloidin-induced cholestasis: amicrofilament-mediated change in junctional complex permeability.Proc Natl Acad Sci U S A 1980;77:2229-2233.
37. Ostrowski NL, Lolait SJ, Bradley DJ, O’Carrol A-M, Brownstein MJ,Young III WS. Distribution of V1a and V2 vasopressin receptor messen-ger ribonucleic acid in rat liver, kidney, pituitary and brain. Endocrinol-ogy 1992;131:533-535.
38. Holen I, Gordon PB, Seglen PO. Protein-kinase–dependent effects ofokadaic acid on hepatocytic autophagy and cytoskeleton integrity.Biochem J 1992;284:633-636.
39. Falconer IR, Yeung DKS. Cytoskeletal changes in hepatocytes inducedby Microcystis toxins and their relation to hyperphosphorylation of cellproteins. Chem Biol Interact 1992;81:181-196.
40. Sastrodihardjo S, Sasaki Y, Shiba Y, Kanno Y. Possible involvement ofreorganization of actin filaments induced by tumor-promoting phorbolesters, in changes in colony shape and enhancement of proliferation ofcultured epithelial cells. J Cell Physiol 1987;132:49-56.
41. Sobue K, Fujio Y, Kanda K. Tumor promoter induces reorganization ofactin filaments and calspectin (fodrin or nonerythroid spectrin) in 3T3cells. Proc Natl Acad Sci U S A 1988;85:482-486.
42. Lyass LA, Bershadsky AD, Vasilliev JM, Gelfand IM. Microtubule-dependent effect of phorbol esters on the contractility of cytoskeleton ofcultured fibroblasts. Proc Natl Acad Sci U S A 1988;85:9538-9541.
43. Phatak PD, Packman CH, Lichtman MA. Protein kinase C modulatesactin conformation in human T lymphocytes. J Immunol 1988;141:2929-2943.
44. Keller HU, Nggli V, Zimmermann A. Diacylglycerol and PMA induceactin polymerization and distinct shape changes in lymphocytes:relation to fluid pinocytosis and locomotion. J Cell Sci 1989;93:457-465.
45. Meigs JB, Wang Y-L. Reorganization of a-actinin and vinculin inducedby phorbol esters in living cells. J Cell Biol 1986;102:1430-1438.
46. Kawamoto S, Hidaka H. Ca21-activated, phospholipid-dependent pro-tein kinase catalyses the phosphorylation of actin-binding proteins.Biochem Biophys Res Commun 1984;118:736-742.
47. Giese G, Kubbies M, Traub P. Alterations in cell cycle kinetics andvimentin expression in TPA-treated, asynchronous MPC-11 mouseplasmacytoma cells. Exp Cell Res 1990;177:47-59.
1040 ROMA ET AL. HEPATOLOGY October 1998

48. Chou C-F, Omary MB. Phorbol acetate enhances the phosphorylation ofcytokeratins 8 and 18 in human colonic epithelial cells. FEBS Lett1991;282:200-204.
49. Cadrin M, McFarlane-Anderson N, Aasheim LH, Kawahara H, FranksDJ, Marceau N, French SW. Differential phosphorylation of CK 8 and CK18 by 12-O-tetradecanoyl-phorbol-13-acetate in primary cultures ofmouse hepatocytes. Cell Signal 1992;4:715-722.
50. Jones BA, Rao Y-P, Stravitz R.T, Gores GJ. Bile salt–induced apoptosis ofhepatocytes involves activation of protein kinase C. Am J Physiol1997;272:G1109-G1115.
51. Tsukidate K, Yamamoto K, Snyder JW, Farber JL. Microtubule antago-nists activate programmed cell death (apoptosis) in culture rat hepato-cytes. Am J Physiol 1993;143:918-925.
52. Orrenius S, McCabe Jr MJ, Nicotera P. Ca21-dependent mechanisms ofcytotoxicity and programmed cell death. Toxicol Lett 1992;64/65:357-364.
53. Weeds A. Actin-binding proteins. Regulators of the cell architecture andmotility. Nature 1982;296:811-816.
54. Reichen J, Berr F, Le M, Warren GH. Characterization of calciumdeprivation–induced cholestasis in the perfused rat liver. Am J Physiol1985;249:G48-G57.
HEPATOLOGY Vol. 28, No. 4, 1998 ROMA ET AL. 1041