tritium deposition in pine trees and soil from atmospheric releases of molecular tritium
TRANSCRIPT

Environ. Sci. Technol. 1904, 18, 358-361
annual discharge of 300 km3 (21). Some of this water intrudes into the Great Barrier Reef region through the Bligh entrance (143’30’ E, 9’20’ S) only 40 km south of the Fly River mouth. Most of the sediment load is de- posited in the ocean outside the reef.
Because of the low population density and low degree of industrialization, the potential input of PAH into the Queensland rivers is small. The water from Papua-New Guinea originates in a region free from industry. The small initial input combined with probable removal by associ- ating with depositing sediment is consistent with the very low levels observed on the Great Barrier Reef.
PAH in atmospheric emissions may also be transported in association with airborne particles. The prevailing wind direction over the Great Barrier Reef is from the ocean to the shore for most of the year. This minimizes transport of aerosols from the coast to the reef and provides a high degree of protection from any PAH emitted to the atmo- sphere along the Queensland coast.
At present the Great Barrier Reef appears to be almost pristine and free of significant PAH contamination from coastal sources. The most likely cause of significant pollution in the future would be any increase of human activity directly on or near the reef.
Acknowledgments
We thank D. W. Connell and G. Denton for collabora- tion in sampling and J. Y. Hauser for assistance with sampling and analysis.
Registry No. Anth, 120-12-7; Pyr, 129-00-0; Chry, 218-01-9; B[k]F, 207-08-9; B[a]P, 50-32-8; B[ghi]P, 191-24-2; Fn, 206-44-0; Per, 198-55-0.
Literature Cited (1) Neff, J. M.; Cox, B. A.; Dixit, D.; Anderson, J. W. Mar. Biol.
(2) Wakeham, S. G.; Schaffner, C.; Giger, W. Geochim. Cos-
(3) Rosewater, J. Indo-Pac. Mollusca 1965, I, 347-396.
(Berlin) 1976, 38, 279-290.
mochim. Acta 1980,44, 403-413.
(4) Clementson, L. A. M. Sc. Thesis, University of Melbourne,
(5) Cheder, S. N.; Gump, B. H.; Hertz, H. S.; May, W. E.; Wise,
(6) Dunn, B. P. Environ. Sci. Technol. 1976, 10, 1018-1021. (7) Hanus, J.; Guerroro, H.; Biehl, E. R.; Kenner, C. T. J. Assoc.
O f f . Anal. Chem. 1979,62, 29-35. (8 ) Mix, M. C.; Riley, R. T.; King, K. J.; Trenholm, S. R.;
Schaffer, R. L. In “Fate and Effect of Petroleum Hydro- carbons in Marine Organisms and Ecosystems”; Wolfe, D. A., Ed.; Pergammon Press: New York, 1977; pp 421-431.
(9) Pancirov, R. J.; Searl, T. D.; Brown, R. A. Adv. Chem. Ser.
(10) Pancirov, R. J.; Brown, R. A. Environ. Sci. Technol. 1977,
(11) Guerroro, H.; Biehl, E. R.; Kenner, C. T. J. Assoc. Off . Anal.
(12) Perdriau, J. Cah. Oceanogr. 1964, 16, 205-229. (13) Neff, J. M. ”Polycylic Aromatic Hydrocarbons in the
Aquatic Environment”; Applied Science Publishers: Lon- don, 1979.
(14) Fazio, T. “Proceedings of the 7th National Shellfish San- itation Workshop”; Food and Drug Administration, Division of Shellfish Sanitation: Washington, DC, 1971; pp 283-243.
(15) Cahnmann, H. J.; Karatsune, M. Anal. Chem. 1957, 29,
(16) Obana, H.; Hari, S.; Kashimoto, K. Bull. Environ. Contam. Toricol. 1981, 26, 613-620.
(17) Fassato, V. U.; Nasci, C.; Dolci, F. Mar. Environ. Res. 1979, 2, 47-53.
(18) Mackie, P. R.; Hardy, R.; Whittle, K. J.; Bruce, C.; McGill, A. S. In “Polynuclear Hydrocarbons: Chemistry and Bio- logical Effects”; Bjoresth, A.; Dennis, A. J., Eds.; Battelle Press: Columbus, OH, 1980; pp 379-394.
(19) Wolanski, E.; van Seden, D. Aust. J. Mar. Freshwater Res. 1983, 34, 49-63.
(20) Belperio, A. P. Ph.D. Thesis, James Cook University, Queensland, Australia, 1978.
(21) Wolanski, E. Conference on Environmental Engineering, Institution of Engineers, Townsville, Australia, July 1981.
Australia, 1982.
S. A. Anal. Chem. 1978,50,305-310.
1980, NO. 185, 123-142.
11, 989-992.
Chem. 1976,59,989-992.
1312-1317.
Received for review June 13,1983. Accepted November 28,1983. This research was supported by the Australian Marine Sciences and Technology Committee (AMSTAC).
Tritium Deposition in Pine Trees and Soil from Atmospheric Releases of Molecular Tritium
Clyde W. Sweet” and Charles E. Murphy, Jr.
E. I . du Pont de Nemours & Co., Savannah River Laboratory, Aiken, South Carolina 29808
w Much of the tritium found in soil and leaf litter near a chemical separations facility is incorporated into soil organic matter in a stable, nonexchangeable form. For- mation of this “bound” tritium seems to result from the uptake of molecular tritium (HT) by living pine needles. A deposition velocity ( v d ) for HT of 1.2 X cm/s was measured in pine needles. About 1% of the tritium de- posited in this way is converted into organic matter. Soil and litter microbes convert HT to HTO more rapidly ( v d = 0.01 cm/s), but no measurable organic tritium is formed.
Introduct ion Starting with the first test of a thermonuclear weapon
in 1952, man’s activities have introduced tritium into the
* Address correspondence to this author at the Department of Chemistry, Alma College, Alma, MI 48801.
global environment. About 99% of this tritium is in the form of tritiated water (HTO), most of which is now in the oceans. Much of the remaining tritium is in the molecular form (HT). This form of tritium makes up over 50% of the tritium in the troposphere (1). The global inventory of molecular tritium has been relatively constant in recent years (2). However, tritium levels may increase in the future because of increases in nuclear fuel reprocessing activities and increased tritium use in consumer products. In addition, very large amounts of molecular tritium could be used in fusion power plants so that developement of this technology would also increase atmospheric HT levels.
At the present time, little is known about the deposition and environnmental cycling of molecular tritium. Due to its low reactivity and low solubility in water, HT is con- sidered to be a less serious radiological hazard than HTO, which is readily absorbed by living organisms. However, experiments with soil (3, 4 ) have indicated that HT is
358 Environ. Sci. Technol., Vol. 18, No. 5, 1984 0013-936X/84/0918-0358$01.50/0 0 1984 American Chemical Society

readily oxidized to HTO by soil microorganisms under environmental conditions. There is some evidence that pine trees (5 ,6) can also convert HT to HTO, but the rate is much less than that in soil. Relatively high levels of tritium have been found in soil and plants after large ac- cidental releases of H T to the atmosphere (7).
In this paper, the pattern of tritium deposition in soil, litter, and vegetation in forested areas surrounding a nu- clear separations facility is examined. The significance of this pattern in relation to H T metabolism by pines and soil microbes is discussed.
Forms of Tritium Tritium can be present in vegetation and soil as a con-
stituent of water, organic matter, or soil minerals. The tritium in vegetation and soil samples that is easily re- moved by evaporation a t low temperature or by freeze- drying is in the form of HTO. Tritium that is associated with organic matter or soil minerals is more difficult to remove from samples and is often designated “bound” tritium. Nearly all of the bound tritium can be removed by heating and combustion in a furnace.
The bound tritium fraction of soil or vegetation can be further divided into an exchangeable bound fraction and a nonexchangeable bound fraction. The exchangeable fraction will rapidly come into equilibrium with the specific activity of the tritiated water in its environment, while the nonexchangeable bound fraction will retain the specific activity of the materials from which it was synthesized. Exchangeable tritium can be removed from a sample by repeated washing with tritium-free water.
In the experiments described below, HTO is defined as tritium separated from the sample as water by freeze- drying. Tritium collected as water from samples com- busted in a high-temperature oven (700 “C) immediately after freeze-drying is designated “total bound” tritium. (The tritium removed from the sample by equilibration with low tritium water is designated “exchangeable bound tritium.) Tritium collected as tritiated water from leached samples combusted in a high-temperature oven is desig- nated “nonexchangeable bound” tritium.
Methods Ten sampling locations were chosen between 0.5 and 12
km from the H-area chemical separation facility a t the Savannah River Plant (SRP) near Aiken, SC. This facility is a source of both HT and HTO. Soil cores and leaf litter samples were taken between Sept 1979 and Aug 1980. Soil cores were sectioned into 5-cm segments and stored frozen in glass jars before processing. Soil, pine litter (consisting of partially decomposed needles), and fresh pine needle samples were freeze-dried overnight. Water removed by freeze-drying was collected in a cold trap and analyzed for tritium by liquid scintillation counting (LSC). The dried samples were then placed in a combustion furnace. Water was collected in a cold trap and analyzed for tritium by LSC.
Tritium concentrations in atmospheric moisture are routinely determined by the Health Protection Depart- ment a t a number of stations. At each station, air is continuously pulled through a silica gel column. Biweekly, composite samples are analyzed for tritium by LSC. Av- erages of atmospheric moisture for 1979 and early 1980 were compared with the tritium concentrations in the soil cores and litter samples.
Pine needles were exposed to tritium under controlled conditions as follows. Freshly excised loblolly pine branches were immediately placed in a container of water and the needles sealed in a glass flask with a split rubber
Table I. Tritium in Soil near the H-Area soil tritium, nCi/m2
2 kma 8 kma soil depth,
cm HTO boundT HTO boundT litter 250 t 170 1100 t 550 60 f 20 200 i 115
0-10 4002 600 1 5 0 k 250 50 t 40 7 0 i 40 10-10 150 i 125 2 0 0 i 120 30 i 30 60 k 35 40-50 3 0 0 i 425 1 O O t 50 3 0 i 10 5 0 i 35 90-100 400 i 575 50 i 25 30 i 1 5 6 0 t 32 a Distance from H-area; five locations were sampled at
each distance.
stopper and rubber cement around the branch below the needles. For 10-15 min, a flow of moist air containing HT was directed over the needles, passed through a cold trap, and vented to the atmosphere. Photosynthesis occurs normally under these conditions (8). In most experiments, the needles were exposed to direct sunlight. Water in both the cold trap and the pine needles was analyzed for tritium.
The nonexchangeable bound tritum content of the needles was determined as follows. Two grams of dried needles was ground and equilibrated with a Wcm3 portion of tritium-free water, followed by freeze-drying. This procedure was repeated until the tritium in the water was reduced to background levels. Samples were then com- busted to recover any nonexchangeable bound tritium. The results are expressed as picocuries per milliliter of the resulting water.
The contribution of synthetic processes involving HTO to the nonexchangeable organic pool was estimated by exposing branches to HTO vapor. Small amounts of nonexchangeable bound tritium were found to be formed during control exposures to HTO vapor. This tritium was subtracted from the nonexchangeable organic tritium be- fore calculating the deposition rate of HT to the nonex- changeable tritium pool.
Results The HTO and total bound tritium concentrations of
pine litter were determined at a number of locations at varying distances from the separations facility. The results (20 data points) were analyzed by linear regression. The following are the resulting equations: HTO
In T = 4.27 - 1.10 In D r2 = 0.75 total bound tritium
In T = 5.91 - 0.97 In D r2 = 0.83 where T = tritium concentration in picocuries per milliliter and D = distance from the H-area in kilometers. These equations are plotted in Figure 1. The atmospheric HTO values from monitoring data are also shown for compari- son. Near the H-area, HTO levels in the litter may be slightly elevated relative to atmospheric moisture. How- ever, the variability in the values measured (Table I) makes it difficult to draw firm conclusions with this number of samples. Total bound tritium levels, in contrast, are much higher than either litter HTO levels or atmospheric HTO levels.
The results of analyses of soil cores (Table I) indicate that the distribution of HTO is relatively uniform in the upper meter of soil. In contrast, there are higher levels of total bound tritium in the leaf litter than in the soil.
In an attempt to determine if the total bound tritium was associated with any specific class of needle organic compounds, fresh pine needles and pine litter were treated
359 Environ. Sci. Technol., Vol. 18, No. 5, 1984
L I
HTC
1 10 Distance, krn
100
Flgure 1. Tritium in pine litter near the H-area.
Table 11. Tritium Levels in Fractions Extracted from Pine Needles (pCi/mL Water)
present
needle needle bud litter year past year
water 7 9 i 6 8 0 i 2 156 i 34 benzene- 421 i 19 456 i- 105 1334b 666 i 54
residue 204 i 10 243 i 37 423b 324 i 68 a Extracts were burned after evaporation of the solvent. One sample.
ethanola
with a 2:l mixture of benzene and ethanol. Benzene- ethanol extractives are a heterogeneous group of com- pounds including fats, oils, resins, and phenolics. The results of combustion and tritium analysis of the resulting water are given in Table 11. Total bound tritium is con- centrated in the extractive fraction. The residue was lower in tritium but still above average levels of ambient HTO (Figure 1).
Several controlled exposures were carried out by using soil, pine litter, and excised living pine branches to de- termine whether synthesis of tritium into the nonex- changeable bound tritium fraction of organic compounds contributes to the total bound tritium. Deposition ve- locities (relative deposition rates) were calculated by using the following formula:
rn
where v d = deposition velocity (cm/s), T = total tritium reacted (pCi), C = air tritium concentration (pCi/cm3), A
360 Environ. Sci. Technol., Vol. 18, No. 5, 1984
Table 111. Molecular Tritium Deposition Velocities free tritium,b bound tritium,c
cm/s cm/s soil (1.0 f 0.2) x 10-2 0 leaf litter (1.1 I 0.3) x 0 pine needlesu (1 .2 i. 0.2) X (1.0 i 0.7) x pine needles (2.6 i 0.3) X 10.’
( dark)a a Light = 100 p E m.-* s-l and dark = 0.3 p E m-2 s-’ at
Tritium in freeze-dried water plus ex- Nonexchangeable
(0.2 i 0.2) X
400-700 nm. changeable organic tritium, n = 5. organic tritium, n = 5 .
2 10-5 I I I I I I
E I i 5 : ~ ~ ~ : ~ ~ \BOUN: TRITIU;
I I 4 0 10 20 30 40 50 60
0
Air Concentration (pCi/cc x
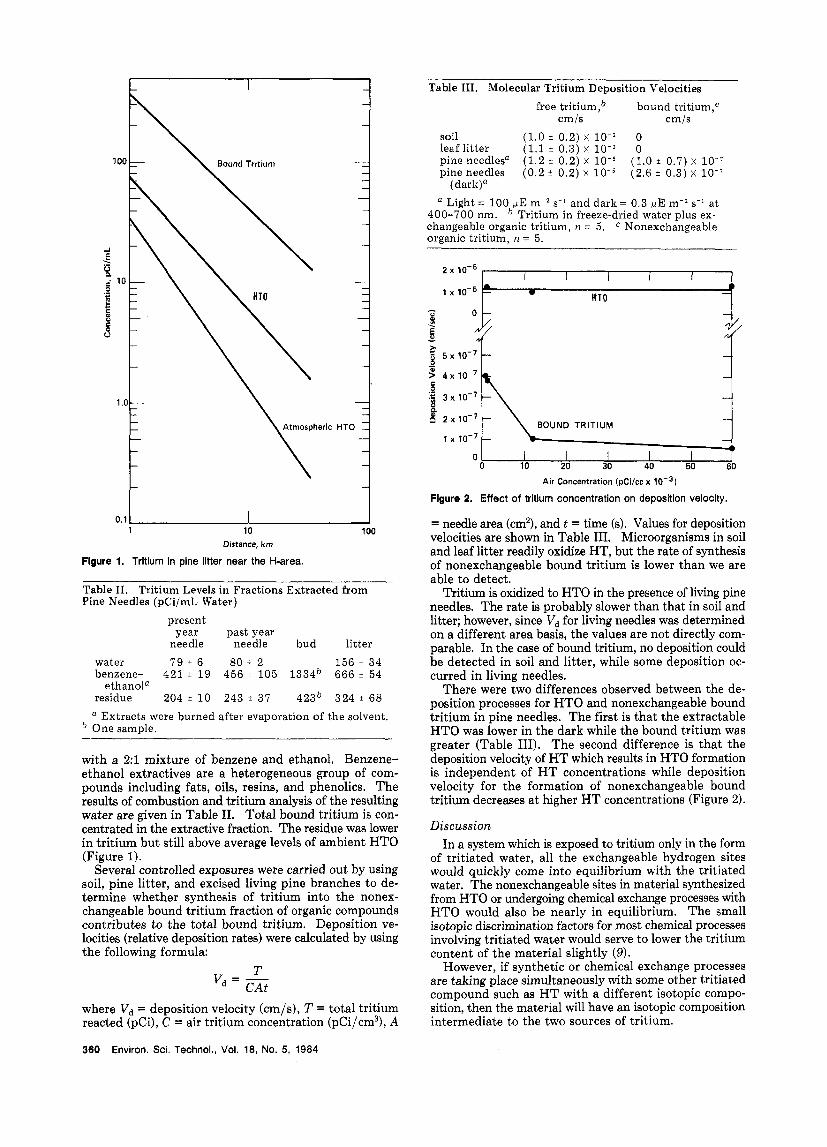
Flgure 2. Effect of tritium concentration on deposition velocity.
= needle area (cm2), and t = time (s). Values for deposition velocities are shown in Table 111. Microorganisms in soil and leaf litter readily oxidize HT, but the rate of synthesis of nonexchangeable bound tritium is lower than we are able to detect.
Tritium is oxidized to HTO in the presence of living pine needles. The rate is probably slower than that in soil and litter; however, since V, for living needles was determined on a different area basis, the values are not directly com- parable. In the case of bound tritium, no deposition could be detected in soil and litter, while some deposition oc- curred in living needles.
There were two differences observed between the de- position processes for HTO and nonexchangeable bound tritium in pine needles. The first is that the extractable HTO was lower in the dark while the bound tritium was greater (Table 111). The second difference is that the deposition velocity of H T which results in HTO formation is independent of H T concentrations while deposition velocity for the formation of nonexchangeable bound tritium decreases at higher HT concentrations (Figure 2).
Discussion In a system which is exposed to tritium only in the form
of tritiated water, all the exchangeable hydrogen sites would quickly come into equilibrium with the tritiated water. The nonexchangeable sites in material synthesized from HTO or undergoing chemical exchange processes with HTO would also be nearly in equilibrium. The small isotopic discrimination factors for most chemical processes involving tritiated water would serve to lower the tritium content of the material slightly (9).
However, if synthetic or chemical exchange processes are taking place simultaneously with some other tritiated compound such as HT with a different isotopic compo- sition, then the material will have an isotopic composition intermediate to the two sources of tritium.

It is well-known that a variety of synthetic and exchange processes in living plants and microorganisms involve water. This paper has demonstrated that H T is also in- volved in some process leading to synthesis of organic matter. The relative amount of tritium in atmospheric hydrogen is higher than that in atmospheric water despite the smaller absolute amount of tritiated hydrogen because of the much smaller amount of natural hydrogen in the atmosphere. Therfore, it is reasonable to expect the con- centration of tritium in organic matter synthesized from hydrogen to be greater than that of the water in the or- ganism which is freely exchanged with the tritiated water in the environment.
Bogen and Welford (10) found relatively high levels of tritium in the water from combustion of dried surface soils. They determined that there is a small discrimination against deuterium in the bound water fraction of soil. They inferred from this that the source of tritium in this fraction must be different than that for deuterium. We suggest that the anomalously high levels of bound tritium found in soil at SRP and elsewhere may be related to HT metabolism by plants and soil microbes.
Microorganisms in soil and surface litter can readily oxidize HT to HTO ( 3 , 4 ) . Soil uptake of H T may make a contribution to soil HTO levels adjacent to the H-area separations facility at SRP. Average levels there seem to be somewhat higher than would be expected from equil- ibration with atmospheric HTO alone (Figure 11, but the large variations in litter HTO make this conclusion un- certain. Bound tritium, however, is consistently much higher than either soil HTO or atmospheric HTO. Since there is no measurable formation of bound tritium during H T oxidation by soil and litter microbes, another process must be involved in its production.
Living pine needles are able to convert HT into both HTO and nonexchangeable organic tritium. H T uptake in pines seems to take place via two different processes. The formation of HTO is inhibited in the dark and is diffusion limited; Le., the tritium flux depends on the concentration gradient between the needle and the at- mosphere, and Vd is constant. The formation of bound tritium, on the other hand, is accelerated in the dark and is sink limited; i.e., the tritium flux is constant, and Vd varies with the concentration gradient (Figure 2). Bound tritium formation is not responsive to decreases in either internal tritium concentrations caused by stomatal closure or external tritium concentrations. This response suggests a process that is limited by the number of active sites such as an enzyme-mediated reaction.
Alternately, organically bound tritium with a high spe- cific activity could be synthesized from HTO if the site of tritium oxidation is near the site of synthesis and synthesis
is faster than HTO equilibration with the rest of the cellular water. Decreased water flux in the dark and the consequent slower HTO dilution may then explain the higher production of tritiated organics.
Although the conversion rate of HT to HTO seems to be slower in pine needles than in soil, this process may still be important for a forested area because the needle surface area of a pine stand can be 10-20 times that of the soil. In addition, wind speeds are higher in the tree canopy than at the soil surface, and higher wind speeds are known to increase HT deposition velocity (4 ) . A small fraction of the HT oxidized in pines is converted to an organically bound form. This tritium is concentrated in the extractive fraction of the needles, and needle litter is very slow to decompose (preliminary results indicate no measurable loss of bound tritium after 1 year).
Although the mechanism is unknown, oxidation of at- mospheric HT in pine needles thus results in an organically bound form of tritium which remains in the soil for many years. Samples of oak, elm, cherry, and soybean taken near a source of HT also show higher tritium activity in the bound fraction than in free water. Therefore, it appears HT may be the source of elevated bound tritium levels found in surface soils throughout North America (IO).
Registry No. Tritium, 10028-17-8.
Literature Cited (1) Burger, L. L. In “Behavior of Tritium in the Environment”;
International Atomic Energy Association: Vienna, Austria, 1979; pp 47-64.
(2) Mason, A. S.; Ostlund, H. G. In “Behavior of Tritium in the Environment”; International Atomic Energy Associa- tion: Vienna, Austria, 1979; pp 3-16.
(3) McFarlane, J. C.; Rodgers, D.; Bradley, D. V. Enuiron. Sci. Technol. 1978, 12, 590-592.
(4) Sweet, C. W.; Murphy, C. E., Jr. Environ. Sci. Technol.
(5) Murphy, C. E., Jr.; Pendergast, M. M. In “Behavior of Tritium in the Environment”; International Atomic Energy Association: Vienna, Austria, 1979; pp 361-372.
(6) Garland, S. A.; Cox, L. C. Water, Air, Soil Pollut. 1980,14,
(7) Murphy, C. E., Jr.; Watts, J. R.; Corey, J. C. Health Phys.
(8) Anclair, D. For. Sci. 1979, 25, 72-79. (9) Weston, R. E. In “Tritium”; Moghissi, A. A.; Carter, M. W.,
Eds.; Messenger Press: Las Vegas, NV, 1973; pp 289-303. (10) Bogen, D. C.; Welford, G. A. Health Phys. 1976,30,203-208.
1981,15, 1485-1487.
102-114.
1977, 33, 325-331.
Received for review June 30,1983. Accepted November 7, 1983. This paper was prepared in connection with work done under Contract DE-ACOS-76SR00001 with the U.S. Department of Energy.
Environ. Sci. Technol., Vol. 18, No. 5, 1984 361