thesis corrections 13 9 - lib-phds1.weizmann.ac.il of bid recombinant adenoviruses and infection of...
TRANSCRIPT

1
Thesis for the degree
Doctor of Philosophy
By
Iris Kamer
Advisor
Prof Atan Gross
August, 2008
Submitted to the Scientific Council of the
Weizmann Institute of Science
Rehovot, Israel
DNA בתגובה התאית לנזקי BIDתפקידו של החלבון
חבור לשם קבלת התואר
דוקטור לפילוסופיה
מאת
איריס קמר
ח"תשס, אב
ת שלמוגש למועצה המדעי
מכון ויצמן למדע
The role of full-length BID in the
DNA damage response
מנחה
איתן גרוס' ופפר

2
Table of contents
Table of contents ......................................................................................................................2 List of figures............................................................................................................................5 Abbreviations ...........................................................................................................................6 Abstract.....................................................................................................................................7 Scientific background ..............................................................................................................9
The BCL-2 family................................................................................................................10 BID.......................................................................................................................................12
The objective of the research ................................................................................................15 Materials and Methods..........................................................................................................16
Tissue culture.......................................................................................................................16 Mouse embryonic fibroblasts ...........................................................................................16 hTERT transformation of primary MEFs ........................................................................16 Generation of hTERT BID-/- stable clones expressing wtBID or the BID-S61A/S78A mutant ..............................................................................................................................17 Preparation of BID recombinant adenoviruses and infection of MEFs ..........................17 Human cell lines and transient transfection ....................................................................18 HeLa BID KD cells ..........................................................................................................18 ATM KD cells...................................................................................................................18 A-T Lymphoblasts ............................................................................................................18 A-T Fibroblasts ................................................................................................................18 Treatments........................................................................................................................18 Clonogenic survival assays..............................................................................................19 Immunocytochemistry (immunofluoresence) ...................................................................19
FACS analysis......................................................................................................................19 BrdU labeling and analysis .............................................................................................19 Cell viability assays .........................................................................................................20 Cell cycle assays ..............................................................................................................20 Apoptosis assays ..............................................................................................................20 Synchronization of cells in G1-S phase ...........................................................................20 Determination of mitochondrial membrane potential .....................................................21 Determination of cellular ROS levels ..............................................................................21
Proteins analysis ..................................................................................................................21 Generation of phospho-specific antibodies .....................................................................21 HA-affinity chromatography............................................................................................21 Cross-linking with BS3 .....................................................................................................21 Cross-linking with Formaldehyde....................................................................................22 Formaldehyde treatment and subcellular fractionation ..................................................22 Western blot .....................................................................................................................23 Alkaline or potato-acid phosphatase treatment ...............................................................23 Purification of the HA-BID cross-linked complex ...........................................................23 Mass spectrometry analysis .............................................................................................24

3
Chapter I - Pro-apoptotic BID is an ATM effector in the DNA damage response..........25 Introduction..........................................................................................................................25
ATM and the response of cells to DNA DSBs .................................................................25 BCL-2 family members and the response to DSBs ..........................................................26
Results..................................................................................................................................28 BID is important for DNA damage-induced apoptosis....................................................28 DSBs induce the phosphorylation of BID, and this phosphorylation is mediated by the ATM kinase ......................................................................................................................30 Mouse and human BID are phosphorylated on PIKK consensus sites............................32 Characterization of endogenous BID phosphorylation by using the phospho-specific BID antibodies .........................................................................................................................35 Phosphorylation of S61 and S78 is transient, rapid and occurs many hours before the onset of Etop- induced apoptosis .....................................................................................37 Phosphorylation of S78 does not depend on phosphorylation of S61 .............................38 The phosphorylation of BID occurs in response to extremely low, non-apoptotic levels of IR and it is dose-dependent..............................................................................................39 BID-/- MEFs expressing a non-phosphorylatable BID mutant (S61A/S78A) are more susceptible to Etop- induced apoptosis than those expressing wtBID.............................40 BID-/- MEFs fail to accumulate in the S and G2 phases of the cell cycle following Etop treatment ..........................................................................................................................41 BID-/- MEFs expressing BID-S61A/S78A do not accumulate in the S phase following Etop treatment..................................................................................................................44 Phosphorylation of BID is not cell cycle dependent ........................................................46 BID-/- MEFs show a delayed time course of cell death following low levels of DNA damage.............................................................................................................................48 Knocking down the expression of BID in HeLa cells partially impairs Etop-induced S phase arrest......................................................................................................................49 Cellular BID partially localizes to the nucleus................................................................50 BID might be involved in the immediate cellular response to DNA damage ..................52 CDC25A degradation seems to be less efficient in BID-/- and BID-S61A/S78A MEFs ...54 The phosphorylation of BID is Chk2-independent...........................................................55
Chapter II – Identification of proteins that interact with phosphorylated BID ..............56 Introduction..........................................................................................................................56 Results..................................................................................................................................57
BID is found as part of a 50KDa cross-linked complex in healthy cells and in cells treated with DNA damage................................................................................................57
Chapter III - ATM, mitochondria function and apoptosis ................................................64 Introduction..........................................................................................................................64 Results..................................................................................................................................67
Α−Τ Cells have higher basal ∆ψm (hyperpolarized) compared to cells expressing wt ATM..................................................................................................................................67 A-T cells are more resistant to Etop-induced mitochondrial depolarization and apoptosis compared to cells expressing wtATM..............................................................69 ROS levels are higher in Α−Τ cells compared to control cells .......................................72 Α−Τ cells grow slower compared to control cells...........................................................73

4
Discussion................................................................................................................................74 BID, ATM and the DNA damage response.........................................................................74 ATM and the mitochondria..................................................................................................81
References...............................................................................................................................83 Publications ............................................................................................................................90

5
List of figures Figure1 : Schematic of the extrinsic and the intrinsic apoptotic pathways. ...........................10 Figure2 : BID is important for DNA damage-induced apoptosis...........................................29 Figure3 : DNA DSBs induce the phosphorylation of BID, and this phosphorylation is
mediated by the ATM kinase. .................................................................................31 Figure4 : Mouse BID is phosphorylated on S61 and S78 whereas human BID is
phosphorylated only on S78....................................................................................34 Figure5 : Characterization of endogenous BID phosphorylation using the anti-phospho S61
and S78 Abs. ...........................................................................................................36 Figure6 : Phosphorylation of S61 and S78 is transient, rapid and occurs hours before the
onset of Etop-induced apoptosis .............................................................................37 Figure7 : Phosphorylation of S78 does not depend on the phosphorylation on S61..............38 Figure8 : Phosphorylation of BID occurs in response to non-apoptotic levels of IR and it is
dose-dependent........................................................................................................39 Figure9 : BID-S61A/S78A clones are more susceptible to Etop induced apoptosis than
wtBID clones...........................................................................................................40 Figure10 : BID-/- MEFs fail to arrest in the S phase following Etop treatment........................42 Figure11 : BID is required for S phase arrest following DNA damage. ..................................43 Figure12 : BID-/- MEFs expressing BID-S61A/S78A do not accumulate in the S phase
following Etop treatment.........................................................................................45 Figure13 : BID phosphorylation is not cell cycle dependent. ..................................................47 Figure14 : BID-/- MEFs show a delayed time course of cell death following low levels of
DNA damage...........................................................................................................48 Figure15 : Knocking down the levels of BID protein in HeLa cells partially impairs Etop-
induced S phase arrest.............................................................................................49 Figure16 : Mouse BID is partially localized to the nucleus. ....................................................51 Figure17 : BID might be involved in the immediate cellular response to DNA damage.........53 Figure18 : CDC25A degradation is more efficient in cells expressing wt BID. ......................54 Figure19 : The phosphorylation of BID is Chk2-independent .................................................55 Figure20 : Cross-linking with BS3 results in appearance of specific complexes. ....................58 Figure21 : Flag-PRX6 and BID-HA co-immunoprecipitate. ...................................................59 Figure22 : BID is found as a part of a cross-linked complex in healthy and in Etop treated
cells. ........................................................................................................................60 Figure23 : N-terminal HA tagged BID forms the 50 KDa complex. .......................................62 Figure24 : N-terminal HA tagged BID is localized to the nucleus in transfected 293 cells.....63 Figure25 : Α−Τ cells have higher ∆ψm.....................................................................................68 Figure26 : Mitochondria depolarization and cell death are lower in A-T cells post Etop
treatment..................................................................................................................70 Figure27 : IR-induced mitochondria depolarization and cell death are similar in A-T and WT
cells. ........................................................................................................................71 Figure28 : ROS levels are higher in Α−Τ cells compared to control cells. ..............................72 Figure29 : Α−Τ cells grow slower compared to control cells. .................................................73 Figure30 : Model for the dual role of BID. ..............................................................................80

6
Abbreviations Ab Antibody
ATM ataxia-telangiectasia mutated
BH BCL-2 Homology
BrdU Bromodeoxyuridine (5-bromo-2-deoxyuridine)
BS3 bis(sulfosuccinimidyl) suberate
Cyt c Cytochrome c
DDR DNA damage response
DiOC6 3,3’-dihexyloxacarbocynine iodide
DSBs Double strand breaks
Etop Etoposide
FACS Fluorescence-activated cell sorter
FL-BID Full-length BID
HA Hemagglutinin
IMM Inner mitochondrial membrane
IMS Intermembrane space
IP Immunoprecipitation
IR Ionizing radiation
KD Knock down
KO Knock out
MEFs Mouse embryonic fibroblasts
MS Mass spectrometry
MTCH2 Mitochondrial carrier homolog 2
OMM Outer mitochondrial membrane
PI Propidium iodide
ROS Reactive oxygen species
SDS-PAGE Sodium dedocyl sulphate-Polyacrylamid gel electrophoresis
tBID truncated BID
TNFα Tumor necrosis factor α
∆Ψm mitochondrial membrane potential

7
Abstract DNA damage leads to the activation of the ATM and ATR kinases, which in turn cause either
cell cycle arrest and DNA repair or apoptosis. In the first part of my thesis, we have
demonstrated that the pro-apoptotic BID protein is phosphorylated by ATM in response to
DNA damage, and that phosphorylation occurs in mouse BID on two ATM consensus sites
(Serine 61 and Serine 78). Interestingly, BID-/- cells failed to accumulate in the S phase of the
cell cycle following treatment with the topoisomerase II poison etoposide (Etop);
reintroducing wild-type BID restored accumulation. In contrast, introducing a non-
phosphorylatable BID mutant (BID-S61A/S78A) did not restore accumulation in the S phase,
and resulted in an increase in cellular sensitivity to Etop-induced apoptosis. These results
implicate BID as an ATM effector, and raise the possibility that pro-apoptotic BID may also
play a pro-survival role by inducing cell cycle arrest. Next, we explored the mechanism by
which BID regulates cell cycle arrest. For this, we took a biochemical approach to isolate
protein(s) that interact with BID following DNA damage. Using cross-linkers, we found that
BID is part of a 50 KDa complex in healthy and Etop-treated cells. Importantly, we found
that the phosphorylated form of BID is also detected in this complex. We are now at the stage
of scaling up the amount of the complex to identify its components.
In the second part of my thesis, we explored the connection between ATM, mitochondria and
apoptosis. At high levels of DNA damage, cells initiate an apoptotic process, which is closely
linked to mitochondrial function. However, the link between ATM and mitochondrial events,
is still largely unknown. We started to explore this link by measuring mitochondrial function
and apoptosis in cells originated from A-T patients expressing mutant ATM or ATM-/- mice.
We found that in these cells the mitochondria were hyperpolarized (higher levels of
mitochondria membrane potential). In addition, these cells were found to be less susceptible
to Etop-induced depolarization and cell death. We also found that these cells exhibited higher
levels of reactive oxygen species (ROS) and grew slower. Importantly, introducing wt ATM
into A-T cells restored the normal mitochondrial and apoptotic parameters. Thus, ATM
seems to be involved in regulating the function of mitochondria.

8
In summary, this study reveals 1) that the BH3-only BID protein, a molecule that was
previously considered active only as a pro-apoptotic factor, also plays a pro-survival role as
an ATM effector and 2) a novel link between ATM, a kinase considered to be active only in
the nucleus, and mitochondrial function.

9
Scientific background Apoptosis (programmed cell death - PCD) is an evolutionary conserved cell suicide
mechanism that plays a crucial role in various biological processes, including development,
maintenance of homeostasis, and removal of unwanted cells [1]. Abnormal resistance to
apoptosis can lead to disorders such as autoimmunity or cancer due to the persistence of
mutated cells [2]. In contrast, enhanced apoptosis contributes to acute diseases or chronic
pathologies such as neurodegenerative diseases [3]. Cells undergoing PCD assume
morphological features, which include membrane blabbing, chromatin condensation, and
nuclear fragmentation [4]. The genetic pathway that regulates apoptosis has been
characterized, and it appears to be conserved from the nematode C. elegans to mammals.
BCL-2 proteins are the major regulators of the apoptotic pathways [5], and caspase proteases
are the major executioners of this process [6] .
Two major apoptotic pathways have been identified in mammals: the intrinsic and extrinsic
pathways (Fig 1). In the extrinsic pathway, apoptosis is initiated through activation of certain
cell surface receptors. The best characterized are members of the TNF/Fas receptor family.
Once engaged by ligand, these receptors initiate the formation of the death inducing signaling
complex (DISC), which leads to activation of caspase-8 which then activates the downstream
caspase-3 [7]. The cell-intrinsic apoptotic pathway is triggered by death receptor-independent
stimuli such as UV-irradiation, viruses and DNA damage reagents as etoposide (Etop). This
pathway involves the activation of pro-apoptotic BCL-2 family members, which act by
inducing organelle dysfunction, of which mitochondrial dysfunction is the best characterized
[8]. At the mitochondria, pro-apoptotic BCL-2 family members induce the release of
Cytochrome c (Cyt c) and other intermembrane space (IMS) proteins to the cytosol. In the
cytosol, Cyt c together with Apaf-1 and caspase-9 form the apoptosome complex where
caspase-9 is activated. Caspase-9 activates caspase-3, which leads to apoptotic cell death [8].
Under certain circumstances, the extrinsic pathway uses the mitochondrial pathway to
amplify caspase activation. This connection is done trough BID, which is cleaved by caspase-
8 (Fig 1).

10
Figure1 : Schematic of the extrinsic and the intrinsic apoptotic pathways. In the extrinsic pathway, activation of the TNF/Fas death receptor induces DISC formation that results in direct activation of caspases. In the intrinsic pathway, the BCL-2 family members are the major players. At the mitochondria, these proteins induce the release of Cyt c, resulting in the formation of the apoptosome and caspase activation. The extrinsic and intrinsic pathways are connected by BID.
The BCL-2 family
The BCL-2 family of proteins constitutes a critical intracellular checkpoint in the intrinsic
pathway of apoptosis. The founding member, BCL-2, was discovered at the chromosomal
break-point in human B-cell lymphoma [9]. Expression of BCL-2 proved to block cell death
following several apoptotic stimuli [10, 11]. By screening for BCL-2 interacting molecules,
several members of the BCL-2 family have been identified. Currently, up to 30 members of
the family were identified [12-14]. These members can be divided into three main sub-
classes, defined in part by the homology shared within four conserved regions termed BCL-2
homology (BH) 1-4 domains. These domains roughly correspond to α helices, which dictate
structure and function. The first group includes the anti-apoptotic molecules (e.g. BCL-2,
BCL-XL), which carry all four conserved domains (BH1-4). The second group consists of the
multi-domain pro-apoptotic molecules (e.g. BAX, BAK), which carry three BH domains
(BH1-3). The third group (which is a sub-group of the pro-apoptotic members) carries only
the BH3 death domain. This group includes among others BID [13, 15].

11
Many of the BCL-2 family members are found at the mitochondria membrane either
constitutively or following a death signal. Most BCL-2 family members contain a C-terminal
hydrophobic stretch of amino acids that anchors them to membranes. Although anti-apoptotic
molecules such as BCL-2 appear to be exclusively membrane bound, particularly to
mitochondria, pro-apoptotic molecules are cytosolic and translocate to mitochondria during
apoptosis. Translocation of these proteins is triggered by specific post-translational
modifications such as dephosphorylation. For example, BAD is sequestered in the cytosol
bound to 14-3-3, and upon survival factor withdrawal it undergoes dephosphorylation and
translocates to the mitochondria [16]. It seems that the BCL-2 family members regulate
apoptosis mainly via the mitochondrial pathway. Mitochondria seem to play an important
role in apoptosis, since alteration of their membrane potential (∆Ψm) and the production of
reactive oxygen species (ROS) are early events in several apoptotic pathways. Furthermore,
during apoptosis, IMS proteins are released into the cytosol, among them Cyt c, which
initiates formation of the apoptosome leading to caspase-9 activation [17-19].
One of the major characteristics of the BCL-2 family members is their tendency to form
homo- as well as heterodimers. Mutagenesis studies have revealed that the BH domains are
important for these interactions. The BH1, BH2 and BH3 domains of the anti-apoptotic
members are required to heterodimerize with the pro-apoptotic members to repress cell death
[20]. On the other hand, only the BH3 domain of pro-apoptotic members is required to
heterodimerize with the anti-apoptotic molecules, and to promote cell death. Early findings
support the notion that the ratio of pro-apoptotic to anti-apoptotic molecules dictates the
susceptibility of cells to a death signal [21]. It seems that the multi domain pro-apoptotic
proteins possess two potentially independent mechanisms for promoting cell death. One
mechanism relies upon their ability to suppress the function of anti-apoptotic proteins through
heterodimerization. The other is a heterodimerization-independent function. For example,
BAX can induce cell death independent of its interaction with BCL-2 [22]. Several BH3
mutants of BAX that fail to bind BCL-2 are nevertheless still capable of inducing apoptosis
[23]. Furthermore, activation of BAX appears to involve its homodimerization [24]. A
proposed theory suggests that the BCL-2 family members form channels in the mitochondrial
membrane. This model originated from the structural similarity between the BCL-XL

12
structure and structure of the pore forming region of bacterial toxins [25]. Moreover, it was
demonstrated that recombinant BAX forms channels in artificial membranes allowing the
passage of large macromolecules [26-29].
BCL-2 family members have essential roles in the mouse from early embryogenesis through
to adult tissue homeostasis. The nervous system, haematopoietic tissues and spermatogenesis
are particularly dependent on BCL-2 family protein regulation [30]. Several members of the
two pro- and anti-apoptotic classes have been knocked out in mice to reveal their
physiological roles, redundancy and interactions in vivo. In many cases there are phenotypes
of abnormal cell death, like in the case of knockout of the anti-apoptotic proteins BCL-2,
BCL-XL and BCL-W, or hyperplasia and increase in cell resistance to apoptotic stimuli, like
in the case of BAX, BIM, BID and NOXA knockouts [30].
BID
The pro-apoptotic BCL-2 family proteins can be further divided into those with multiple
BCL-2 homology (BH) domains, such as BAX and BAK, and those with only one type of BH
domain, the BH3-only proteins. The BH3-only proteins are essentially the sentinels to various
apoptotic signals [31]. For example, BAD is sensitive to growth factor deprivation; PUMA
and Noxa are sensitive to DNA damage; and BIM is sensitive to DNA damage, cytokine
deprivation and glucocorticoids. When activated by these signals, the BH3-only molecules
transmit the death signals to the mitochondria to initiate the mitochondria apoptosis pathway.
BID (BH3-interacting domain death agonist) is one of the BH3-only proteins. It is a 22 KDa
protein and it was first cloned in a screen based on its interaction with BCL-2 and BAX. BID
is phylogenetically conserved [32, 33]. Human BID shows 72.3% homology to murine BID
at the amino acid level. BID is widely expressed in various tissues, with the highest level
being in the kidney [32] and in organs of the hematopoetic system (Unpublished data, Gross
lab). In general, full length BID is quite stable and is a long-lived protein, but caspase-8
cleaved truncated BID (tBID) has a half-life of less than 1.5 h [34].
BID was initially found to be cleaved and activated by caspase-8 following death receptor
activation and thus considered specific to the death receptor pathway. Furthermore, the death

13
activity of BID can be inhibited by BCL-2, suggesting that BID is acting via the
mitochondrial pathway. BID lacks a COOH-terminal membrane-anchoring segment, and
therefore is mainly found in the cytosol. Following activation of the TNF or Fas death
receptors, BID is cleaved by caspase-8 at Asp-59, to produce a p15 carboxy-terminal
fragment (truncated BID; tBID) that translocates to the mitochondria and induces Cyt c
release [13, 23, 35, 36]. Studies in recent years indicate that BID can be cleaved by other
proteases such as Granzyme B, Calpains and Cathepsins. These proteases are first activated
in response to many types of stimuli, including death receptor activation, cytotoxic T cell
attack, ischemia/reperfusion injury and lysosome damage [34]. These observations indicate
that BID is in general a sentinel to protease activation resulting from various injury stimuli.
As such, BID serves a critical role in connecting these stimuli to the mitochondria, thus
allowing the death process to be advanced and amplified.
Although most studies in the field emphasize the importance of BID cleavage in order to
activate it, there are several studies that demonstrated an apoptotic role for full-length (FL)
BID [32, 37-39]. A study that was conducted in our lab has demonstrated that a caspase-8
non-cleavable BID mutant (ncBID) is a potent inducer of apoptosis in mouse embryonic
fibroblasts (MEFs) [37]. These studies were performed with recombinant adenoviruses
carrying a tetracycline-inducible ncBID, wtBID or GFP vector and it was demonstrated that
cell death in these instances was due specifically to overexpression of nc/wtBID, since
overexpression of GFP had little effect on the viability of cells. It was also shown that both
ncBID and wtBID were much less effective than tBID in inducing Cyt c release, but only
slightly less effective in inducing apoptosis. Expression of non-apoptotic levels of both
ncBID and wtBID in BID-/- MEFs induced a similar and significant enhancement in apoptosis
in response to a variety of death signals. The most prominent cell death was with DNA-
damaging reagents such as Etop [a topoisomerase II inhibitor that forms DNA double strand
breaks (DSB)] and cisplatin [that forms covalent adducts with DNA]. In the same study from
our lab it was found that BID-/- MEFs are much less susceptible to apoptosis induced by Etop
and ionizing radiation (IR), which are two treatments known to induce DSBs in DNA, and
that ncBID was capable of restoring sensitivity to these cells. In addition, endogenous BID
was found to be rapidly phosphorylated in response to Etop and IR. Finally, BID was not

14
phosphorylated in ATM-/- MEFs, indicating that phosphorylation of BID is mediated by the
ATM kinase. Based on these studies, we hypothesized that FL-BID plays a role in the DSB
DNA damage pathway.

15
The objective of the research To define the role of FL-BID and specifically of BID phosphorylation in the ATM-dependent
cellular response to DSB DNA damage.
The results of the research are presented in three chapters:
Chapter I - Characterization of BID’s phosphorylation and function in cells.
Chapter II - Identification of proteins that interact with phosphorylated BID.
Chapter III - Exploring the connection between ATM and mitochondria function.

16
Materials and Methods
Tissue culture
Mouse embryonic fibroblasts
BID-/- mice (originally kept on a mixed C57BL/6 x 129Sv background) had been bred to wild
type C57BL/6 mice twelve times in order to obtain animals that are F12 on a C57BL/6
background. BID-/- MEFs were generated from the F12 mice. BID+/+ and BID-/- primary
MEFs were prepared from 11-13 day-old embryos, and maintained in ISCOVE’s medium
containing 10% fetal bovine serum (MEF medium). Atm/Arf double knockout and
Atm+/+Arf-/- MEFs were obtained from Chuck J. Sherr (St. Jude Children's Research
Hospital).
hTERT transformation of primary MEFs
All the studies with BID+/+ and BID-/- MEFs described in this research were performed with
hTERT-immortalized MEFs. Immortalization of primary MEFs was performed by
transformation with hTERT (the catalytic subunit of human telomerase). PA317 packaging
cells stably producing pBABE-puro hTERT viral particles (a generous gift from Tej Pandita,
Washington University) were grown to 80% confluence, rinsed and the medium was then
replaced with complete MEF medium. The cells were incubated for 16 hrs and the medium
was collected and filtered through a 0.45 µm filter. The infecting media were stored at -80˚C
until use. Primary BID-/- and BID+/+ MEFs were grown for 3 passages and then infected at
~50% confluence with 3 ml infecting media mixed with 3 ml MEF media and 4 µg/ml
polybrene (Sigma). The cells were then incubated for 16 hrs, rinsed and incubated in fresh
medium for an additional 8 hrs. The cells were infected again as described above, rinsed and
incubated in fresh medium for an additional 48 hrs. The cells were then split 1:3 and grown
for 4 days in a selection medium containing 1 µg/ml puromycin. After selection, the cells
were washed once and incubated with MEF medium (without puromycin). Stable clones were
collected 14 to 18 days post-infection, and their propagation took 3-to-4 months.

17
Generation of hTERT BID-/- stable clones expressing wtBID or the BID-S61A/S78A
mutant
ψNX cells (a 293T cell line carrying an ecotropic packaging plasmid) were seeded in a 100
mm plate at 60% confluence. The next day, the medium was replaced and cells were
incubated with a transfection cocktail containing 15 µg retroviral vector (pBABE-wtBID or
pBABE-BID-S61A/S78A) prepared using a calcium phosphate kit (Promega). Cells were
incubated for 5 hrs with the cocktail and rinsed; the medium was then replaced, and the cells
reincubated in fresh medium. The following day, the conditioned medium containing the
retroviruses was collected and filtered through a 0.45 µm filter. The viruses were divided into
aliquots and frozen at -80˚C. For infection of MEFs, cells were grown in a 60mm plate and
incubated for 4 hrs at 37˚C in 3 ml of retroviral supernatant, supplemented with 16 µg/ml
polybrene. After that, 7 ml of DMEM containing 10% FCS was added and 24 hrs later, the
medium was replaced with fresh medium containing 10% FCS and 1 µg/ml puromycin.
Puromycin was replaced every day for three days. On the fourth day, cells were seeded (100
cells per 10cm dish) and grown until single clones appeared.
Preparation of BID recombinant adenoviruses and infection of MEFs
For the expression of BID in BID-/- MEFs we have produced adenovirus vectors expressing
proteins under the control of tetracycline (tet)-regulatable promoters (“tet-on”) as previously
described [37]. Briefly, in these constructs, which rely on the reverse tet transactivator
(rtTA), the E1 region of the virus was replaced with either wild-type (wt) BID or GFP.
Viruses were grown using 293T cells. Virus preparations were made from freeze/thaw lysis
of the cells, and virus titers were done on 293T cells. In experiments, cells were generally
seeded at 70-80% confluence. Cells were infected with an MOI (multiplicity of infection) of
100 with BID containing virus and the rtTA containing virus. 1 µg/ml doxycycline (a
synthetic analog of tetracycline; Sigma) was added to the medium 12-to-15 hrs post infection
to activate gene expression from the tet-inducible promoter. Efficiency of infection was
determined using the recombinant adenovirus carrying the inducible expression vector of
GFP and was in the range of 70-to-90%.

18
Human cell lines and transient transfection
293, a human embryonic kidney cell line, and HeLa, a human cervical adenocarcinoma cell
line were maintained in 10% fetal bovine serum. Transient transfections were performed by
using a calcium phosphate kit (Promega) or with lipofectamine 2000 (Gibco BRL).
HeLa BID KD cells
Human cervical adenocarcinoma cell line (HeLa) was stably transfected with BID SiRNA or
scrambled SiRNA as a control. These cells were generated in the lab of Dr. Jochen Prehn,
Dublin.
ATM KD cells
Stable ATM knocked down HeLa and 293 cells were generated by the siRNA approach using
the pRETRO-SUPER viral vector. The cells were grown under selection of 10 µg/ml
puromycin and 200 µg/ml hygromycin. These cells were generated by Prof. Yossi Shiloh,
Tel-Aviv University.
A-T Lymphoblasts
A-T lymphoblasts from an A-T patient along with lymphoblasts from a healthy individual
(obtained from Prof. Yossi Shiloh, Tel Aviv University) were cultured in RPMI medium
supplemented with 15% FCS, 100 units/ml penicillin, 100 mg/ml streptomycin and 2 mM
glutamine.
A-T Fibroblasts
SV40 transformed A-T fibroblasts stably transfected with an empty vector or an ATM vector
were obtained from Prof. Yossi Shiloh, Tel Aviv University [40]. The cells were cultured in
complete DMEM supplemented with 100 mg/ml hygromycin.
Treatments
Etop (0.1 M stock solution in dimethyl sulfoxide), Cisplatin (10 mg/ml stock), TNFα (40
ng/ml), Staurosporine ( 4 mM stock) and Thapsigargin (2 mM) were all purchased at Sigma.

19
Clonogenic survival assays
Cells were seeded at a density of 1000 cells per well (6 well plate). The next day, cells were
treated with the indicated DNA damage reagent, the medium was replaced with fresh
medium, and cells were incubated for 10 days. Once colonies were formed, cells were fixed
in 70% methanol and stained with 0.5% crystal violet. The percent of colony survival was
calculated as the ratio of the number of colonies after DNA damage to the number of colonies
in untreated cells.
Immunocytochemistry (immunofluoresence)
For immunocytochemistry, cells were grown on glass cover slips. At the designated time
points, the cells were fixed with 4% paraformaldehyde in PBS for 10 min and permeabilized
with 0.2% Triton X-100 in PBS for 5 min. For blocking, the cells were incubated in PBS
containing 0.1% Triton and 3% BSA for 1 hr at room temperature. For immunostaining, cells
were incubated overnight at 4˚C with either anti-murine BID Abs or the anti-pS61/pS78 Abs
in blocking solution. After three washes with PBS containing 0.1% Triton, the cells were
stained for 1 hr at room temperature with Alexa 488-labeled goat anti-rabbit Abs (dilution
1:120, Molecular Probes), followed by 5 min of 4’,6-diamidino-2-phenylindole
dihydrochloride (DAPI) staining (10 µg/ml). The coverslips were mounted with elvanol, and
the cells were viewed under a Nikon fluorescence microscope at a magnification of
200x/400x. Pictures were taken with a 1310 digital camera (DVC).
FACS analysis
BrdU labeling and analysis
A total of 2 x 105 cells were treated with 20 µM Etop for 2 hrs, washed twice with PBS and
incubated in fresh medium (10% FBS) for 8 hrs. The cells were then pulsed labeled with 10
µM BrdU (Sigma; added to the medium) for 30 min, washed with PBS, fixed with cold 70%
ethanol and incubated overnight at -20˚C. The next day, cells were collected and resuspended
in 2N HCl with 0.5% Triton X-100 for 30 min at room temperature followed by
neutralization with 0.1 M Na2B4O7. Cells were then collected and incubated with anti-BrdU

20
Abs (Becton-Dickinson) for 30 min in the dark at room temperature. The cells were washed
with PBS, and stained with FITC labeled goat anti-mouse Abs (Jackson) for 30 min at room
temperature in the dark. The cells were then resuspended in PBS containing PI (5 µg/ml) and
analyzed by FACScan. To evaluate cells that were in S phase, cells were gated on the BrdU+
population and DNA content was evaluated by PI.
In the BrdU pulse-chase experiments (to follow the progress of cells through S phase), cells
were treated with 20 µM Etop for 2 hrs and labeled immediately with 10 µM BrdU for 30
min. Cells were then washed, incubated in fresh medium for the indicated time points, and
fixed for BrdU analysis as described above. The percentage of BrdU positive cells in early S
phase and in late S/G2 phase was determined by PI counterstaining.
Cell viability assays
Cell viability was determined by propidium iodide (PI) dye exclusion. PI (25 µg/ml) was
added to the cells immediately prior to analysis by FACScan (Beckton Dickinson).
Cell cycle assays
3*105 cells were seeded in 6cm dish. Cells were treated with 20 µM Etop for 2 hrs, rinsed,
and then released into drug-free medium. 8 or 24 hrs after release, cells were collected for
fixation in methanol. Following fixation, cells were washed and resuspended in PBS with 25
µg/ml propidium-iodide (PI) and 50 µg/ml RNAse a half hour before FACScan analysis.
Analysis of the cell cycle results was performed using the ModFit LT program [41].
Apoptosis assays
3*105 cells were seeded in 6cm dish or in 6 well plate. Cells were treated for the indicated
times, harvested and stained as above. The percent of cells displaying sub-G1 DNA content
was determined by FACScan analysis.
Synchronization of cells in G1-S phase
4*105 cells were seeded in 10 cm dishes and grown until they reached confluency. Cells were
trypsinized and seeded into 10 µM Aphidicolin-containing medium. The cells were incubated
for 20 hrs and released into drug free medium for further treatments.

21
Determination of mitochondrial membrane potential
3*105 cells were seeded in 6cm dish. The next day cells were incubated for 15 min at 37°C
with 3,3’-dihexyloxacarbocynine iodide [DiOC6(3), 40nM, Calbiochem] followed by
FACScan analysis.
Determination of cellular ROS levels
3*105 cells were seeded in 6cm dish. The next day cells were incubated for 15 min at 37°C
with DCF-DA (2’,7’-dichlorofluorescein) or HE (Hydroethidine) (Molecular probes) to check
levels of H2O2 and superoxide, respectively. Cells were harvested and ROS levels were
determined by FACScan analysis.
Proteins analysis
Generation of phospho-specific antibodies
Anti-pS61 and anti-pS78 were generated in collaboration with Bethyl Laboratories, Inc.
(Montgomery, TX). Briefly, immunogens were phosphorylated synthetic peptides, which
represented portions of mouse BID around either serine 61 or serine 78. Antibodies that were
not phospho-specific were removed by solid phase absorption. Antibodies that were specific
for either BID pS61 or BID pS78 were affinity-purified using the phosphopeptide
immobilized on solid support.
HA-affinity chromatography
BID-/- MEFs were infected with adenoviruses containing HA-tagged BID. The next day, the
cells were either left untreated or treated with Etop, lysed (using 1% CHAPS) and incubated
for 2 hrs with beads coupled to anti-HA antibodies (Roche). The beads are then collected and
BID-HA was eluted using an HA peptide (1mg/ml; Roche). The eluted sample was
concentrated using centricon, separated by SDS-PAGE and a sample taken to Western blot
analysis. The gel was stained using gelcod (Pierce) and bands were ssent to mass
spectrometry analysis at the Smoler proteomic center of the Technion.
Cross-linking with BS3
293T cells were transfected with pcDNA3 BID-HA and treated with Etop. Cells were than

22
harvested and treated with digitonine for 10 min on ice in digitonine buffer (20 mM HEPES,
pH 7.3, 110 mM Kacetate, 5 mM NaAcetate, 2 mM MgAcetate, 1mM EGTA, 2 mM
dithiothreitol). The membrane fraction was separated from the cytosolic fraction by
centrifugations. BS3 (bis(sulfosuccinimidyl) suberate) (Pierce) was added from a 10-fold
stock solution to a final concentration of 10 mM. The cross-linker was added either to the
cytosolic/soluble fraction or to the membrane-enriched fraction. After incubation of 30 min at
room temperature, the cross-linker was quenched by addition of 1 M Tris-HCl pH 7.5 to a
final concentration of 20 mM. The cross-linked samples were lysed with RIPA (150 mM
NaCl, 1% NP-40, 0.5% DOC, 0.1% SDS, 50 mM TRIS, pH 8.0) and IP experiment with anti-
HA antibodies was conducted (as described above) and Western blot analyzed with the
indicated antibodies.
Cross-linking with Formaldehyde
293 cells were transfected with pcDNA3 BID-HA and treated with Etop. Cells were
incubated with 1% formaldehyde for 10 min in RT. The cross-linker was quenched by
addition of 125 mM Glycine for 5 min and cells were harvested and lysed with RIPA buffer
(as described above). IP experiments with anti-HA antibodies were conducted as described
above and Western blot analyzed with the indicated antibodies.
Formaldehyde treatment and subcellular fractionation
Formaldehyde was added directly to the tissue culture media to a final concentration of 1%
and the cells were incubated for 10 min at room temperature. The cross-linking reaction was
stopped by adding glycine to a final concentration of 0.125 M and incubation at room
temperature for 5 min. Cells were then subfractionated as previously described [42]. Cells
were rinsed with wash buffer (125 mM KCl, 5 mM magnesium acetate, 5 mM EGTA, 1 mM
β−mercaptoethanol, 30 mM Tris-HCl, pH 7.5) at 4˚C, scraped from the plates, washed twice
with the same buffer and allowed to swell for 10 min in 0.5 ml swelling buffer [same as wash
buffer except that the KCl concentration was 10 mM and protease (set III; Calbiochem) and
phosphatase (set I and II; Sigma) inhibitor cocktails were added]. The cells were then lysed in
a 2-ml Wheaton Dounce glass homogenizer using 30 complete up and down cycles of a glass
“B”-type pestle. The homogenate obtained was overlaid on an equal volume of swelling
buffer containing 25% glycerol and centrifuged (600 x g at 4˚C for 5 min). The

23
upper layer of the supernatant was designated the cytosolic fraction. It should be noted that
all organelle membranes (besides the nuclear membrane) and the plasma membrane are
contained in this fraction. The nuclear pellet was washed once with swelling buffer
containing 25% glycerol and 0.1% Triton X-100. Nuclei were resuspended in sonication
buffer (100 mM NaCl, 2 mM MgCl2, 5 mM EGTA, 1 mM β−mercaptoethanol, 10 mM Tris,
pH 9.0). At this stage both the cytosolic and nuclear samples were incubated at 65˚C for 4-5
hrs to reverse formaldehyde cross-links. Nuclei were then disrupted by brief sonication.
Aliquots of nuclear and cytosolic fractions were separated by 12% or 15% SDS-PAGE and
transferred to PVDF membrane (Immun-blotTM, Bio-Rad).
Western blot
Proteins were size-fractionated by SDS-PAGE and then transferred to PVDF membranes
(BioRad). Western blots were developed by use of the enhanced chemiluminescence reagent
(Amersham Bioscience, Inc).
Alkaline or potato-acid phosphatase treatment
MEFs were treated with Etop for either 30 or 60 min, lysed in phosphatase buffer (150 mM
NaCl, 1% CHAPS, 10 mM HEPES, pH 7.5) and either left untreated or incubated for 30 min
at 37˚C with either alkaline phosphatase (1U/1µg protein; Roche) or with potato-acid
phosphatase (PAP; 1.5U/30µg protein; Sigma). The reaction with PAP was performed in a
phosphatase buffer adjusted to pH 5.5. At the end of the reaction, the lysates were analyzed
by Western blot using the indicated Abs.
Purification of the HA-BID cross-linked complex
100-300x10cm plates of 293T cells were transiently transfected with pcDNA3-HA-BID. 18
hrs post transfection, cells were cross-linked with BS3 or formaldehyde (as described above)
and lysed. The diluted lysate was incubated for 16 hrs with anti-HA Ab coupled to agarose
beads (Roche), followed by extensive washing of the beads with binding buffer containing
0.05% Tween 20. The material that remained bound to the beads was eluted by incubation
with 1 ml (1 mg/ml) HA peptide (Roche) for 15 min at 37ºC. Elution was repeated twice
more, and the three eluents were pooled and concentrated using a Centricon tube with a 3
KDa cutoff (Amicon). The concentrated material was loaded onto a single lane and separated

24
by SDS-PAGE followed by staining the gel with Imperial stain (Pierce).
Mass spectrometry analysis
The stained protein bands in the gel were cut with a clean razor blade and sent in separate
tubes to analysis. Mass spectrometry analysis was performed at the Smoler Protein Center,
Technion, Haifa.

25
Chapter I - Pro-apoptotic BID is an ATM effector in the DNA damage
response
Introduction
ATM and the response of cells to DNA DSBs
The genome of each cell of an organism is constantly subjected to DNA damage. DNA DSBs
are a particularly deleterious form of DNA damage and if left unrepaired can result in cancer-
causing mutations or promote aging. DNA DSBs occur as a result of oxidative metabolism,
DNA replication, or V(D)J recombination during immune system maturation; they also can
arise from exogenous agents such as IR or Etop.
Following DSBs, the cell activates a survival system that allows repair and continuation of its
normal life cycle, or it may activate the apoptotic machinery in the face of extensive or
irreparable damage [43]. The mechanism of this decision is under intense investigation. One
of the major responses associated with the survival network is the temporary arrest of cell-
cycle progression, which reflects the activation of cell cycle checkpoints [44]. The best-
documented, damage-induced cell-cycle checkpoints operate in the G1/S boundary, and at the
S and G2 phases. Upon introduction of DNA DSBs, the early events involve several proteins
that are rapidly recruited to the damaged sites, where they form prominent nuclear foci.
Among them is a trimolecular complex containing the MRE11, RAD50 and NBS1 proteins
(MRN complex) that fulfills many of the criteria for a DSB sensor [45]. The concept is that
sensor molecules are the first to sense the lesion and help convey a damage signal to
transducers, which in turn deliver it to numerous downstream effectors. A prototype
transducer is ATM, which is a nuclear serine-threonine protein kinase [46].
The ATM protein was identified as the product of the gene that is mutated (lost or
inactivated) in the human genetic disorder ataxia-telangiectasia (Α−Τ) [46]. Α−Τ is
characterized by cerebellar degeneration, which leads to neuromotor dysfunction,
immunodeficiency, genomic instability, thymic and gonadal atrophy, predisposition to
lymphoreticular malignancies and sensitivity to ionizing radiation and DSB-inducing agents.

26
Cells from Α−Τ patients exhibit a variety of abnormalities, including genomic instability,
radiosensitivity, and defective activation by DSBs of cell-cycle checkpoints. ATM is a
member of a group of conserved large proteins, most of them protein kinases involved in
mediating DNA damage responses. These proteins share several motifs, among them a
domain containing a PI3-kinase signature, which gives this group the title, "PI3-kinase-
related protein kinases" (PIKKs). Additional proteins of this family are ATR (ATM and RAD
3 related), DNA-PKCs and m-TOR.
DSBs mobilize a signaling network by activating the ATM protein kinase, which, in turn,
activates this network by phosphorylating key proteins with specificity for serine/threonine
followed by glutamine [47-49]. The activation of ATM results in a rapid intermolecular
autophosphorylation of ATM on serine 1981 that causes dimer dissociation and initiation of
ATM's kinase activity [50]. It was found that nearly the entire nuclear pool of ATM
molecules was phosphorylated on Ser1981 within minutes of cellular exposure to low doses
of IR that induced only a few DSBs [50]. The rapid and strong activation of the ATM kinase
seems to be an initiating event in cellular responses to DSBs and is required for cell cycle
arrest at the G1, S and G2 phases. Several substrates of ATM that participate in these
checkpoints include p53, Mdm2 and Chk2 in the G1 phase [51], Nbs1, Brca1 and SMC1 in
the S-phase and Brca1, Chk2 and hRad17 in the G2/M phase [52]. In addition to ATM’s
versatility as a protein kinase with numerous substrates, the ATM web contains protein
kinases that are themselves capable of targeting several downstream effectors simultaneously.
Chk1 and Chk2, a checkpoint kinases, are examples of such effectors [53].
BCL-2 family members and the response to DSBs
There are several reports that connect BCL-2 family members to the non-apoptotic response
of cells to DSBs. For example, homology-directed repair of DSBs is enhanced by the anti-
apoptotic BCL-XL protein [54], while overexpression of pro-apoptotic BAX and BID, was
found to inhibit homologous-recombination DNA repair [55]. In addition, a protein essential
for DSB repair, Ku70, was demonstrated to hold BAX in an inactive state [56]. BAX and
BCL-2 were previously reported to be localized to the nucleus in certain cells [57, 58];
however their roles in this organelle remain unknown. Recently it was published that

27
exposure of cells to IR increases the expression of BCL-2 in the nucleus, which interacts and
inhibits both Ku70 and Ku86 via its BH1 and BH4 domains [59]. Removal of the BH1 or
BH4 domain abrogated its inhibitory effect, which results in the failure to block DSB repair
as well as V(D)J recombination. In addition, it was found that BCL-XL colocalizes and binds
to cdk1(cdc2) during the G2/M cell cycle checkpoint, and its overexpression stabilizes a
G2/M arrest/senescence program in surviving cells after DNA damage [60]. With respect to
BID, BID-/- mice, as they age, have been shown to spontaneously develop a clonal
malignancy closely resembling chronic myelomonocytic leukemia (CMML), which
demonstrates consistent chromosomal abnormalities [61]. These results suggested that BID
might play an unanticipated role in regulating genomic stability. Thus, both pro- and anti-
apoptotic BCL-2 family members might also play a non-apoptotic role in the response of
cells to DSBs and other forms of DNA damage.

28
Results
Most of the results presented in this chapter were published in a manuscript entitled: “Pro-
apoptotic BID is an ATM effector in the DNA damage response”. Cell 122: 593-603 (2005)
[62].
BID is important for DNA damage-induced apoptosis
To determine whether BID is required for DNA damage-induced apoptosis, we generated
hTERT-immortalized BID+/+ and BID-/- MEFs and analyzed their response to a variety of
DNA-damaging reagents: Etop, cisplatin (Cis; forms covalent adducts with the DNA), UV
(induces thymine dimers), and IR. We found that BID-/- MEFs were less susceptible than
BID+/+ MEFs to all four treatments (Fig 2A). These DNA-damaging reagents also induced
less cell death in primary BID-/- MEFs than in primary BID+/+ MEFs (data not shown),
confirming that this decreased sensitivity is not due to hTERT immortalization.
To confirm that BID-/- MEFs are indeed less sensitive than BID+/+ MEFs to DNA damage-
induced cell death, we performed clonogenic survival assays with MEFs following DNA
damage. This assay is commonly used in the field of DNA damage and can give an indication
on the cellular response to DNA damage treatment. Our studies showed that BID-/- MEFs
have increased clonogenic survival following IR (Fig 2B). To confirm that the reduced
susceptibility of BID-/- MEFs to DNA-damaging reagents was due to the absence of BID,
BID-/- MEFs were infected with recombinant adenoviruses carrying the BID vector prior to
treatment with Etop or IR. The results show that reintroduction of BID did not induce cell
death on its own but fully restored susceptibility to Etop- (and partially to IR-) induced cell
death (Fig 2C).

29
Figure2 : BID is important for DNA damage-induced apoptosis. (A) BID-/- MEFs are less susceptible than BID+/+ MEFs to apoptosis induced by DNA-damaging reagents. Dose-response/death curves of BID+/+ and BID-/- MEFs in response to treatment with the indicated doses of Etop (24 hrs), Cis (14 hrs), UV (14 hrs), and IR (24 hrs). Cell death was monitored by FACScan using propidium-iodide (PI) dye exclusion. The data represent the means ± SEM of pooled results from three independent experiments. (B) BID-/- MEFs have increased clonogenic survival compared to BID+/+ cells following DNA damage. 1000 cells from BID+/+ and BID-/- MEFs were seeded per well and irradiated with the indicated doses of IR. Cells were then incubated for 10 days and the percent of colony survival was calculated as the ratio of the number of colonies formed after IR to the number of colonies formed in untreated cells. * represent significant differences (p < 0.05) based on Student’s t test. (C) The reduced susceptibility of BID-/- MEFs to DNA-damaging reagents is due to the absence of BID. BID+/+ or BID-/- MEFs were either left untreated (N/T) or treated with either Etop (100 �M; 24 hr) (left) or ionizing radiation (IR; 100 Gy; 24 hr) (right) and cell death was monitored by FACScan using propidium-iodide (PI) dye exclusion. Alternatively, BID-/- MEFs were infected with recombinant adenoviruses carrying a tetracycline-inducible BID vector. Two hours after the addition of doxycyclin, the cultures were washed three times and treated with either Etop or IR. Cell death was monitored as described above. The data represent the means ± SEM of pooled results from three independent experiments.
A
B
**
*

30
DSBs induce the phosphorylation of BID, and this phosphorylation is mediated by the
ATM kinase
Next, we explored whether BID was modified in response to DNA damage. Western blot
analysis using anti-BID antibodies on lysates of hTERT-immortalized BID+/+ MEFs treated
with the DNA-damaging reagents (as described in Fig 2) revealed that Etop and IR, which are
known to induce DSBs in DNA, unlike Cis or UV, induced a double electrophoretic mobility
shift in BID (Fig 3A). We also treated MEFs with several other apoptotic reagents:
thapsigargin (Thaps; stress signaling from the ER, which inhibits the Ca2+ adenosine
triphosphate pump); TNFα together with actinomycin D; or with staurosporine (STS; a
kinase inhibitor), and found that none of them affected the electrophoretic mobility of BID
(Fig 3A). Similar mobility shifts have been associated with covalent modifications of
proteins, for example, as a consequence of phosphorylation.
To define whether the double electrophoretic mobility shift in BID was due to
phosphorylation, BID+/+ MEFs were treated with Etop for 30 min, lysed and either left
untreated or incubated with alkaline phosphatase for 30 min at 37˚C. Western blot analysis
using anti-BID antibodies demonstrated that treatment with alkaline phosphatase abolished
the electrophoretic mobility shifts in BID (Fig 3B), indicating that these shifts are most likely
due to phosphorylation. Taken together, these results strongly suggested that BID is rapidly
phosphorylated in response to reagents that induce DSBs.
The ATM kinase plays a pivotal role in the immediate response of cells to DSBs. To
determine whether ATM is involved in the phosphorylation of BID, we utilized MEFs
deficient in both ATM and the p19/ARF tumor suppressor gene, since loss of ARF has been
shown to reverse premature replicative arrest of Atm-null MEFs [63]. Accordingly, Atm/Arf
double knockout MEFs, as well as ATM+/+Arf-/- MEFs, were treated with Etop or IR, and
Western-blot-analyzed using anti-BID antibodies. Figure 3C shows that following Etop or IR
treatment, the slower migrating bands of BID do not appear in the ATM-deficient cells. Thus,
the presence of the ATM kinase appears to play an essential role in the process by which
Etop and IR induce phosphorylation of BID. To corroborate these findings, we took
advantage of a stable HeLa cell line in which ATM was knocked down by siRNA (In these

31
cells, the level of ATM was reduced by ~95% [64]). Both these cells and the control cells,
which carried a siRNA against LacZ, were transfected with mouse BID, exposed to Etop, and
Western-blot-analyzed using anti-BID antibodies. Exposure of control HeLa cells to Etop
induced a double electrophoretic mobility shift in BID that was absent in the ATM knocked
down cells (Fig 3D, left and middle panels). BID-/- MEFs were used as a specificity control
(Fig 3D, right panel). These results further confirm that the presence of ATM is essential for
BID phosphorylation.
Figure3 : DNA DSBs induce the phosphorylation of BID, and this phosphorylation is mediated by the
ATM kinase. (A) Etop and IR induce a double electrophoretic mobility shift in BID. BID+/+ MEFs were either left untreated (N/T), or treated with one of the indicated cell death stimuli: Etop (100 µM), IR (50 Gy), Cis (50 µM), UV (20 J/m2), Thaps (2 mM), TNFα (4ng/ml together with 2 µg/ml actinomycin D), and STS (4 µM). Cells were collected 1 hr later, lysed, and subjected to SDS-PAGE, followed by Western blot analysis using anti-BID Abs. The blot reprobed with anti-β-actin Abs to control for loading (lower panel). * marks a cross-reactive band. The question mark marks the BID double electrophoretic mobility shift. (B) Alkaline phosphatase treatment abolishes the Etop induced double electrophoretic mobility shift in BID. BID+/+ MEFs were treated with 100 µM Etop for 30 min, lysed and either left untreated (-), or treated with alkaline phosphatase (+) for 30 min at 37˚C, followed by Western blot analysis using anti-BID Abs. BID-P marks the BID double electrophoretic mobility shift. (C) The slower migrating forms of BID do not appear in ATM-deficient MEFs. Atm/Arf double knockout MEFs (ATM-/-) and ATM+/+Arf-/- MEFs (ATM+/+) were either left untreated (N/T), or treated with 100 µM Etop or 50 Gy IR, collected after 30 min, and lysed. Samples were subjected to SDS-PAGE, followed by Western blot analysis using anti-BID Abs. In the lower panel anti-β-actin Abs to control for loading. (D) Etop induced phosphorylation of exogenous BID is detected in LacZ, but not in ATM knocked down HeLa cells. Left panel: HeLa cells were transiently transfected with pcDNA3-wtBID. 18 hrs post-transfection, cells were either left untreated (-), or treated with 100 µM Etop for 30 min, collected, lysed and Western-blot-analyzed using anti-BID Abs. Middle panel: stable LacZ knocked down and stable ATM knocked down HeLa cells were transfected with pcDNA3-wtBID, treated with Etop, and analyzed as described for the left panel. Right panel: BID-/- MEFs treated with Etop for 30 min were used as a specificity control for the anti-BID Abs.

32
Mouse and human BID are phosphorylated on PIKK consensus sites
As mentioned in the Introduction, ATM is a member of the PIKK family. The common
phosphorylation sites for PIKKs are serines or threonines followed by glutamine residues
(SQ/TQ motif) [49]. Mouse BID carries two such motifs (S61Q and S78Q), whereas human
and rat BID carry only one (S78Q) (Fig 4A).
To determine whether mouse BID is phosphorylated on one or both of these sites, we mutated
each of these serines to alanines. Our initial analysis was performed in HeLa cells transfected
with either wild-type (wt) BID or with one of the BID mutants. Western blot analysis using
anti-BID antibodies indicated that treatment of the mentioned cells with Etop resulted in a
double electrophoretic mobility shift, which was abolished in the S61A mutant (Fig 4B). In
contrast, mutating the S78 site had no effect on the appearance of the two slower-migrating
bands. Thus, BID phosphorylation on S61 is likely the cause for the electrophoretic mobility
shift.
To confirm the results presented above and to establish whether S78 is also phosphorylated in
mouse BID in response to Etop, we generated phospho-specific antibodies to S61 and S78
(see Material and methods). We initially performed Western blot analysis with these
antibodies on lysates of MEFs. To define whether these antibodies recognize the
phosphorylated form of BID, BID+/+ MEFs were either not treated or treated with Etop for 30
min, lysed, and either left untreated or incubated with potato-acid phosphatase for 30 min at
37˚C. Western blot analysis demonstrated that anti-pS61 antibodies recognized a band of the
expected size of BID in Etop treated cells, and that treatment with potato-acid phosphatase
abolished this recognition (Fig 4C, left; note that these antibodies recognize an additional
~30kD protein that shares antigenicity with pS61-BID). Western blot analysis using the anti-
pS78 antibodies demonstrated that these antibodies recognized three bands (one strong band
and two very faint bands) in Etop treated cells; treatment with potato-acid phosphatase
abolished all three bands (Fig 4C, right). The bands identified with both antibodies
corresponded to BID, since they were not identified in BID-/- MEFs (Fig 4C).

33
Next, we determined whether mutation of either S61 or S78 to alanine abolished recognition
of mouse BID by the phospho-specific antibodies. These experiments were performed in
HeLa cells transfected with either wtBID, the BID-S61A mutant, or the BID-S78A mutant.
The anti-pS61 and anti-pS78 antibodies recognized BID in cells expressing wtBID and
treated with Etop, but not in cells expressing the BID-S61A or BID-S78A mutant,
respectively (Fig 4D).
As mentioned above, human BID carries only one PIKK consensus site (S78; Fig 4A). To
determine whether endogenous human BID is phosphorylated on S78, we performed Western
blot analysis with anti-human BID and anti-pS78 antibodies on lysates of 293T cells either
not treated, or treated with Etop. The anti-pS78 antibodies recognized a band of the size of
human BID only in cells treated with Etop (Fig 4E). Thus, human BID is also phosphorylated
on S78 in response to Etop.

34
Figure4 : Mouse BID is phosphorylated on S61 and S78 whereas human BID is phosphorylated only on
S78. (A) Mouse BID carries two PIKK consensus sites (S61Q and S78Q), whereas human and rat BID carry only one (S78Q). (B) Mutation of S61 to alanine abolishes the Etop induced double electrophoretic mobility shift in BID. HeLa cells were transiently transfected with pcDNA3-wtBID, pcDNA3-BID-S61A, pcDNA3-BID-S78A, or left untransfected (-). 18 hrs post-transfection, cells were either left untreated (-), or treated with 100 µM Etop for 30 min, collected, lysed and Western-blot-analyzed using anti-BID Abs. The blot was stripped and reprobed with anti-β-actin Abs to control for loading (lower panel). (C) The phospho-specific antibodies to serine 61 and serine 78 recognize endogenous BID in MEFs treated with Etop. BID+/+ or BID-/- MEFs were either left untreated (-), or treated with 100 µM Etop for 30 min (+), lysed, and Western-blot-analyzed using the phospho-specific Abs to either S61 (left) or S78 (right). Alternatively, BID+/+ MEFs treated with 100 µM Etop for 30 min were collected, treated with potato acid phosphatase (PAP; +) for 30 min at 37˚C, lysed and Western-blot-analyzed as above. The blots were stripped and reprobed with anti-β-actin Abs to control for loading (lower panels). BID-P marks the phosphorylated form of BID. * marks a cross-reactive band. (D) The anti-pS61 and anti-pS78 antibodies do not recognize BID-S61A or BID-S78A, respectively. HeLa cells were transiently transfected with pcDNA3-wtBID, pcDNA3-BID-S61A, or pcDNA3-BID-S78A. 18 hrs post-transfection, cells were either left untreated (-), or treated with 100 µM Etop for 30 min, collected, lysed and Western-blot-analyzed using either the anti-pS61 (left) or anti-pS78 (right) Abs. In the left panel, * marks a cross-reactive band, whereas in the right panel * marks the phosphorylated form of endogenous human BID. Note that the anti-pS78 Abs also recognized the lower of the three bands in HeLa cells that were not treated with Etop, indicating a basal level of phosphorylation in healthy cells. The blots were stripped and reprobed with anti-β-actin Abs to control for loading (lower panels). (E) Human BID is phosphorylated on S78 in response to Etop. 293T cells were either left untreated (-), or treated with 100 µM Etop for 1 hr (+), lysed, and equal amounts of protein were subjected to SDS-PAGE, followed by Western blot analysis using either anti-pS78 Abs (top) or anti-human BID Abs (middle). The blot was stripped and reprobed with anti-β-actin Abs to control for loading (bottom).* mark cross-reactive bands.

35
Characterization of endogenous BID phosphorylation by using the phospho-specific BID
antibodies
To determine whether endogenous mouse BID is phosphorylated on S61 and S78 in an ATM-
dependent manner, we utilized the phospho-specific antibodies for Western blot analysis of
Atm/Arf double knockout and ATM+/+Arf-/- MEFs either not treated, or treated with Etop.
This analysis demonstrated that endogenous mouse BID is phosphorylated on S61 and S78
only in ATM+/+Arf-/- MEFs treated with Etop (Fig 5A).
To show that phosphorylation of mouse BID was specific for reagents inducing DSBs, we
treated MEFs with several DNA-damaging and other apoptotic reagents (previously
described in Fig 3). Post-treatment, cells were lysed, and the phosphorylation of endogenous
mouse BID was examined by Western blot analysis using anti-pS61 antibodies. These results
demonstrated that mouse BID is phosphorylated on S61 only in response to reagents that
induce DSBs (Fig 5B).
Finally, to define whether phosphorylation of human BID was also ATM-dependent and
occurred only in response to reagents that induce DSBs, we took advantage of a stable 293T
cell line in which ATM was knocked down by siRNA (these cells were generated like the
HeLa ATM knocked down cells) [64]. These cells and the control cells, which carried a
siRNA against LacZ, were exposed to Etop, IR, UV, or STS, and Western-blot-analyzed
using anti-pS78 antibodies. Exposure of LacZ knocked down cells to Etop or IR, but not to
UV or STS, induced phosphorylation of endogenous human BID on serine 78, which did not
occur in the ATM knocked down cells (Fig 5C).

36
Figure5 : Characterization of endogenous BID phosphorylation using the anti-phospho S61 and S78
Abs. (A) Endogenous mouse BID is phosphorylated on S61 and on S78 in an ATM-dependent manner. Atm/Arf double knockout MEFs (ATM-/-) and ATM+/+ARF-/- MEFs (ATM+/+) were either left untreated (-), or treated with 100 µM Etop for 30 min, collected, lysed, and Western-blot-analyzed using either anti-pS61 (left) or anti-pS78 (right) Abs. The blots were stripped and reprobed with anti-β-actin Abs to control for loading (lower panels). * marks a cross-reactive band. (B) Mouse BID is phosphorylated on S61 only in response to reagents that induce double-strand breaks in DNA. BID+/+ MEFs were either left untreated (N/T), or treated with the death stimuli indicated in Fig 2A. Cells were collected 1 hr later, lysed, and subjected to SDS-PAGE followed by Western blot analysis using anti-pS61 Abs (top). The blot was stripped and reprobed with anti-β-actin Abs to control for loading (bottom). * marks a cross-reactive band. (C) Phosphorylation of S78 in endogenous human BID is ATM-dependent, and occurs only in response to reagents that induce DSBs in DNA. Stable LacZ knocked down and stable ATM knocked down 293T cells were either left untreated (N/T), or treated with Etop (100 µM), IR (50 Gy), UV (20 J/m2), or STS (4 µM). Cells were collected after 1 hr, lysed, and subjected to SDS-PAGE followed by Western blot analysis using anti-pS78 Abs (top). The blots were stripped and reprobed with anti-β-actin Abs to control for loading (bottom).

37
Phosphorylation of S61 and S78 is transient, rapid and occurs many hours before the onset
of Etop- induced apoptosis
To determine the time course of endogenous mouse BID phosphorylation, BID+/+ MEFs were
treated with Etop and Western-blot-analyzed with the anti-BID, the anti-pS61, or the anti-
pS78 antibodies. Phosphorylation of S61 was detected by 15 min (the first time point
analyzed), reached a peak at 1 hr, and was reduced by 3 hrs post Etop treatment (Fig 6A).
Phosphorylation of S78 was also transient (peak at 2-3 hrs), though was somewhat delayed,
compared to phosphorylation of the S61 site. Next, we determined when apoptosis began in
MEFs treated with Etop, and found that the onset of apoptosis occurred between 8 and 12 hrs
following Etop treatment (Fig 6B). These cells were also Western-blot-analyzed with anti-
cleaved caspase-3 Abs, and as expected, the appearance of cleaved caspase-3 was detected 8
hrs post Etop treatment (Fig 6B, lower panel). Thus, BID phosphorylation occurs many hours
prior to the onset of apoptosis.
Figure6 : Phosphorylation of S61 and S78 is transient, rapid and occurs hours before the onset of Etop-
induced apoptosis (A) Time course of Etop-induced phosphorylation of endogenous mouse BID on S61 and S78. BID+/+ MEFs were either left untreated (N/T), or treated with 100 µM Etop, collected at the indicated time points, lysed, and equal amounts of protein were subjected to SDS-PAGE followed by Western blot analysis using either anti-BID (top), anti-pS61 (middle top), or anti-pS78 (middle bottom) Abs. The blot was stripped and reprobed with anti-β-actin Abs to control for loading (bottom). * marks a cross-reactive band. (B) Time course of Etop induced apoptosis of MEFs. BID+/+ MEFs were treated with 100 µM Etop, collected at the indicated time points, and cell death was monitored by PI dye exclusion assay in FACS. These samples were also Western-blot-analyzed using anti-cleaved caspase 3 Abs (lower panel).

38
Phosphorylation of S78 does not depend on phosphorylation of S61
Since the time course of S78 phosphorylation is slower then S61 phosphorylation (Fig 6A),
we decided to check whether the phosphorylation on S78 requires initial phosphorylation of
S61. BID-/- MEFs were infected with wtBID or BID-S61A. 18hrs post infection, cells were
either left untreated or exposed to Etop for different time points. Cells were lysed, collected
and Western-blot-analysed using phospho-specific Abs to S78 (Fig 7). The results clearly
show that phosphorylation of S78 occurs in cells transfected with BID-S61A (lower panel).
However, as expected, there is no electrophoretic mobility shift when using this mutant since
phosphorylation on S61 is the cause for this shift (see Fig 4B). Moreover, the time course of
phosphorylation of S78 in these cells was similar to the time course of wtBID
phosphorylation. Thus, phosphorylation of S78 does not require the initial phosphorylation of
S61.
Figure7 : Phosphorylation of S78 does not depend on the phosphorylation on S61. BID-/- MEFs were infected with WT BID or BID-S61A. Cells were treated with 100 µM Etop for the indicated time points, and lysed; equal amounts of protein were subjected to SDS-PAGE followed by Western blot analysis using anti-pS78 Abs.

39
The phosphorylation of BID occurs in response to extremely low, non-apoptotic levels of
IR and it is dose-dependent
We previously determined that phosphorylation of BID on both S61 and S78 occurs several
hours prior to the onset of apoptosis (Fig 6). We therefore speculated that phosphorylation
might also occur in response to extremely low levels of IR, which do not result in apoptosis.
Indeed, we found that a 25-fold lower dose of IR (0.2 Gy) was sufficient to induce
phosphorylation of BID (Fig 8A). The level of BID phosphorylation increased with IR levels.
Since the phosphorylation on S78 is slower, we treated cells with IR (from 1 Gy up to 20 Gy)
and checked phosphorylation on S78 after 1 hr. The results show that also the
phosphorylation on S78 is detectable at low levels of IR and is increased with the increase of
IR levels (Fig 8B). The same results were obtained with increasing levels of Etop (data not
shown). Thus, these results suggested that phosphorylated BID might play a non-
apoptotic/pro-survival role in the DNA damage response.
Figure8 : Phosphorylation of BID occurs in response to non-apoptotic levels of IR and it is dose-
dependent. (A) BID+/+ or BID-/- MEFs were either left untreated (N/T), or treated with the indicated doses of IR, collected 30 min later, lysed, and analyzed with anti-pS61 Abs. (B). BID+/+ or BID-/- MEFs were either left untreated (N/T), or treated with the indicated doses of IR, collected 1 hour later, lysed, and analyzed with anti-pS78 Abs.

40
BID-/- MEFs expressing a non-phosphorylatable BID mutant (S61A/S78A) are more
susceptible to Etop- induced apoptosis than those expressing wtBID
To explore the role of BID phosphorylation in cells, we generated wtBID and BID-
S61A/S78A stable clones by retroviral infection of BID-/- MEFs. First, we assessed the levels
of apoptosis in both type of clones either not treated, or treated with Etop. Our results
indicated that Etop induced a significantly higher rate of apoptosis in the BID-S61A/S78A
clones than in wtBID clones (Fig 9A). Of note, UV and TNFα, that do not induce
phosphorylation of BID, did not induce increased apoptosis in the mutant BID clones (Fig
9A). Western blot analysis using anti-BID antibodies indicated that the increase in apoptosis
seen in the mutant BID clones in response to Etop was not due to either higher levels of
expression of mutant BID, or to its enhanced cleavage to tBID (Fig 9B). Thus, the mutant
BID clones were found to be more susceptible to apoptosis induced solely by a reagent that
leads to DNA DSBs. This result can imply that the phosphorylation of BID either suppresses
a pro-apoptotic function or induces a pro-survival function of BID.
Figure9 : BID-S61A/S78A clones are more susceptible to Etop induced apoptosis than wtBID clones. (A) wtBID or BID-S61A/S78A clones were either left untreated (N/T), or treated with Etop (50 µM; 18 hrs), UV (20 J/m2; 24 hrs), or TNFα (4 ng/ml together 2 µg/ml with actinomycin D; 4.5 hrs). Cell death was monitored by FACS using PI. The data represent the means ± SEM of pooled results from three independent experiments. (B) The enhanced death obtained with the BID-S61A/S78A clones is not due to higher expression of mutant BID or to more cleavage to tBID. The wtBID and mutant BID clones were treated either with 50 µM Etop for 18 hrs, or with TNFα/ActD for 4.5 hrs, lysed, and subjected to SDS-PAGE, followed by Western blot analysis using anti-BID Abs. For the TNFα treatment, only clones #1 are shown. The blots were stripped and reprobed with anti-β-actin Abs to control for loading (lower panels).
A B

41
BID-/- MEFs fail to accumulate in the S and G2 phases of the cell cycle following Etop
treatment
The functional consequences of certain ATM phosphorylation events include activation of
cell cycle checkpoints, which result in temporary arrest of cell cycle progression to enable
DNA repair [44]. Since we concluded that phosphorylation of BID is not related directly to
the activation of apoptosis but might have a pro-survival role, we decided to examine the
possible involvement of BID in cell cycle arrest following DNA DSBs. First, we performed
cell cycle analyses on BID+/+ and BID-/- MEFs either not treated, or treated with a long
exposure to Etop. Surprisingly, exposure of MEFs to 20 µM Etop for 16 hrs resulted in
massive accumulation of BID+/+ MEFs in the S phase of the cell cycle, whereas such an
accumulation was not observed in the BID-/- MEFs (Fig 10A). These results suggest that BID
is important for the accumulation of cells in the S phase of the cell cycle following DSB
DNA damage. To examine the response of cells to more moderate DNA damage, we exposed
BID+/+ and BID-/- MEFs to 20 µM Etop for only 2 hrs, and released the cells into drug-free
medium after 8 and 24 hrs. We found that Etop induced accumulation of BID+/+ MEFs in the
S and G2 phases of the cell cycle after 8 hrs, whereas such an accumulation was not observed
in the BID-/- MEFs (Fig 10B). After 24 hrs, BID-/- MEFs did show a slight accumulation in S
and G2 phases.
To determine whether BID is required for S phase arrest following DNA damage, we
performed double labeling experiments with BrdU and PI to determine the level of DNA
synthesis and to follow the progress of cells through S phase after Etop treatment. The results
demonstrated that BID+/+ MEFs show a decrease in DNA synthesis (less BrdU positive cells),
whereas BID-/- MEFs fail to show a decrease in DNA synthesis (Fig 11A). Moreover, BID-/-
MEFs are not delayed in their progression form S to G2/M (Fig 11B). Taken together, these
experiments indicate that BID is required for S phase arrest following Etop treatment.

42
Figure10 : BID-/- MEFs fail to arrest in the S phase following Etop treatment (A) BID-/- MEFs do not accumulate in the S phase following long exposure to Etop. BID+/+ or BID-/- MEFs were either left untreated (NT), or treated with 20 µM Etop for 16 hrs. The DNA content was analyzed by FACS as described in the material and methods. The actual raw data from a representative experiment together with multi-line plots generated by the ModFit LT computer software program appear in the left panels. The red histograms represent the percent of cells in the G1 and G2/M phases, and the hatched histograms represent cells in S phase. The exact percentage of cells S phase is shown in the right panel. The data represent the means ± SEM os pooled results from three independent experiments. (B) BID+/+ or BID-/- MEFs were left untreated (NT), or treated with 20 µM Etop for 2 hrs, rinsed, and then released into drug-free medium. At the indicated time points, the DNA content was analyzed by FACS. The actual raw data from a representative experiment together with multi-line plots generated by the ModFit LT computer software program appear in the left panels, as describe above. The exact percentage of cells in each phase of the cell cycle is shown in the right three panels. The data represent the means ± SEM of pooled results from three independent experiments.

43
Figure11 : BID is required for S phase arrest following DNA damage. (A) BID-/- MEFs fail to decrease DNA synthesis following Etop treatment. BID+/+ or BID-/- MEFs were left untreated (NT), or treated with 20 µM Etop for 2 hrs, rinsed, and then released into drug-free medium (upper schema). 8 or 24 hrs after release, cells were pulse-labeled with BrdU for 30 min to determine DNA synthesis. The percentage of BrdU positive cells was determined by FACS. The data represent the means ± SEM os pooled results from three independent experiments. (B) BID-/- MEFs are not delayed in their progression from S to G2/M following Etop treatment. BID+/+ (left) or BID-/- MEFs (right) were either left untreated (NT), or treated with 20 µM Etop for 2 hrs, and labeled with BrdU for 30 min. Cells were then rinsed, incubated in fresh medium for the indicated time periods, and fixed. The percentage of BrdU positive cells in early S phase and in late S/G2 phase was determined by FACS. The data represent the means ± SEM of pooled results from three independent experiments.

44
BID-/- MEFs expressing BID-S61A/S78A do not accumulate in the S phase following Etop
treatment
To assess whether ATM is regulating BID’s ability to induce cell cycle arrest, we used the
wtBID and a BID-S61A/S78A stable clones described above. We initially confirmed that
BID (in the wtBID clones) was phosphorylated on S61 and S78 in response to Etop, and that
BID-S61A/S78A (in the mutant BID clones) was not (Fig 12A).
Next, we performed cell cycle analysis on two of the wtBID and two of the BID-S61A/S78A
stable clones. As shown in Figure 12B, Etop induced accumulation of the wtBID clones in
the S and G2 phases of the cell cycle (as measured 8 and 24 hrs after release into drug-free
medium), whereas the mutant BID clones bypassed accumulation in the S phase and rapidly
accumulated in the G2 phase. Thus, the mutant BID cells were found to be impaired in their
ability to temporarily arrest in S phase following DSB DNA damage.

45
Figure12 : BID-/- MEFs expressing BID-S61A/S78A do not accumulate in the S phase following Etop
treatment (A) BID-S61A/S78A expressed in BID-/- MEFs is not recognized by the phospho-specific Abs. BID-/- MEFs stably expressing either wtBID or the BID-S61A/S78A mutant were treated with 20 µM Etop for 1 hr, lysed, and subjected to SDS-PAGE, followed by Western blot analysis using either anti-BID Abs (left panel) or the phospho-specific Abs (middle and right panels). The blots were reprobed with anti-β-actin Abs to control for loading (lower panels). * mark cross-reactive bands. (B) BID-/- MEFs stably expressing BID-S61A/S78A do not accumulate in the S phase following Etop treatment. BID-/- MEFs stably expressing either wtBID or BID-S61A/S78A (two clones from each) were treated with 20 µM Etop for 2 hrs, rinsed and then released into drug-free medium. At the times indicated, the DNA content was analyzed by FACS. The actual raw data from a representative experiment together with multi-line plots generated by the ModFit LT computer software program appear in the top panel. The red histograms represent the percent of cells in the G1 and G2/M phases, and the hatched histograms represent the percent of cells in S phase. The exact percentage of cells in each phase of the cell cycle (in each of the four clones) is shown in the bottom three panels. The data represent the means ± SEM of pooled results from three independent experiments.

46
Phosphorylation of BID is not cell cycle dependent
We demonstrated that BID phosphorylation is important for S phase arrest in cells treated
with Etop (Fig 12). There are known proteins that are involved in cell cycle progression and
therefore their activity is controlled throughout the different phases of the cell cycle (e.g.,
Chk1) [65]. To assess whether phosphorylation of BID is regulated in the different stages of
the cell cycle and specifically in S phase, we synchronized cells in the G1/S boundary using
aphidicolin (a DNA polymerase α inhibitor). BID+/+ MEFs incubated with aphidicolin for 16
hrs, washed and released into drug-free medium. At the indicated time points, cell cycle
analysis was measured by FACS. As shown in Figure 13A, almost all cells were
synchronized in the G1/S boundary (time point 0) and 4 hours after release into drug-free
medium all the cells were already in S phase. To determine the effect of cell cycle
progression on BID phosphorylation, synchronized cells were treated with Etop for 1 hr, and
the level of BID phosphorylation in the different phases was determined. As shown in Figure
13B, the phosphorylation of BID on S61 and S78 did not change in the different phases.
Interestingly, synchronized cells that were not treated with Etop also showed phosphorylation
on BID, especially on S78 (Figure 13B). This phosphorylation is due to replicative stress that
is induced by aphidicolin, as previously described by Zinkel et al. [66].

47
Figure13 : BID phosphorylation is not cell cycle dependent. (A) Synchronization of cells using aphidicolin. BID+/+ MEFs were incubated with aphidicolin for 16 hrs, washed and released into drug-free medium. At the indicated time points, cell cycle analysis was done by FACS. In the upper panel histogram of DNA content as was measured with CellQuest software. The exact percentage of cells in each phase of the cell cycle is shown in the lower panel. (B) The level of BID phosphorylation on S61 and S78 does not increase during S phase. BID+/+ MEFs were incubated with aphidicolin for 16 hrs, washed and released into drug-free medium. At the indicated time points, cells were treated for 1 hr with 50 µM Etop. Cells were collected, lysed and equal amounts of protein were subjected to SDS-PAGE, followed by Western blot analysis using the phospho-specific Abs. * mark cross-reactive bands.

48
BID-/- MEFs show a delayed time course of cell death following low levels of DNA damage
Our results above demonstrate that BID is important for DNA damage-induced apoptosis on
one hand and for cell cycle arrest and inhibition of apoptosis on the other hand. Temporary
arrest of the cell cycle enables the cell to estimate and repair the damage. We assumed that
cells that do not arrest after exposure to low levels of Etop would die due to unrepaired DNA
damage. Thus, we monitored the levels of cell death in BID+/+and BID-/- MEFs several days
after exposure to lower levels of Etop and shorter duration. BID+/+ and BID-/- MEFs were
exposed to 20 µM Etop for 2 hrs, washed, and cell death was determined 2, 3, 4 and 5 days
post treatment. As expected, BID+/+ MEFs were more susceptible to Etop compared to BID-/-
MEFs (Fig 14). At first, it seemed that BID-/- MEFs are totally resistant to the treatment, but
after 5 days there was a sharp increase in cell death in the BID-/- MEFs. The levels of cell
death are relatively low, since this is the same mild treatment that we used to assess
differences in cell cycle arrest (Fig 10B). These results suggest that although BID is
important for Etop-induced apoptosis, it is also important for the arrest of cells in the S phase,
and therfore BID-/- cells eventually die. The lack of BID protein in these cells can explain the
slower kinetics in cell death compared to wt cells.
Figure14 : BID-/- MEFs show a delayed time course of cell death following low levels of DNA damage BID+/+ and BID-/- MEFs were treated with 20 µM Etop for 2 hrs, rinsed and then released into drug-free medium. Cell death was monitored by FACS using propidium-iodide (PI) dye exclusion at the indicated time points.

49
Knocking down the expression of BID in HeLa cells partially impairs Etop-induced S
phase arrest
Up until now, all our cell cycle experiments were done on hTERT-immortalized MEFs. To
assess the relevance of our results in another cell type, we used the human HeLa cell line.
These cells were stably transfected with either an SiRNA to BID or a scrambled SiRNA as a
control (Fig 15A; these cells were generated by Dr. Jochen Prehn, Dublin). We performed
cell cycle analysis on these cells that were either left untreated or treated with Etop. Etop
induced accumulation of 74% of HeLa control cells in S phase whereas only 57% of the BID
knock-down cells accumulated in the S phase (Fig 15B). There were also differences in the
G2/M phases (5% in control cells versus 15% in BID knockdown cells). These results show
that knocking down the expression of BID influences the response of HeLa cells to Etop and
that fewer cells accumulate in the S phase. The results we obtained earlier in MEFs were
more significant, probably because of the complete absence of BID.
Figure15 : Knocking down the levels of BID protein in HeLa cells partially impairs Etop-induced S phase
arrest (A) Western blot analysis showing the expression level of human BID in stable HeLa control RNAi and BID RNAi cells. (B) HeLa BID knockdown and control cells were either left untreated (NT) or treated with 100 µM Etop. 16 hrs post treatment cells were harvested for cell cycle analysis using FACS and the percentage of cells in each phase is shown. * represent significant differences (p<0.05) based on Student’s t test.
*

50
Cellular BID partially localizes to the nucleus
Next, we analyzed the cellular location of BID and its phosphorylated form. To determine the
location of BID in healthy cells, we performed immunofluoresence studies with BID+/+ and
BID-/- MEFs using anti-BID antibodies. Surprisingly, in these studies we obtained positive
staining of BID also in the nucleus (Fig 16A, left top panel). These antibodies specifically
recognized BID, since we obtained background staining in BID-/- MEFs (Fig 16A, left lower
panel). To determine whether Etop treatment leads to a change in the staining pattern of BID,
BID+/+ and BID-/- MEFs were treated with Etop for 3 hrs prior to fixation. These studies
indicated that Etop did not change the staining pattern of BID (Fig 16A, middle panel).
To assess whether BID indeed localizes to the nucleus in healthy MEFs, we performed
subcellular fractionations followed by Western blot analysis using anti-BID antibodies. In
these experiments, cellular BID was detected only in the soluble/cytoplasmic fraction (Fig
16B, left top panel). MEK and BAX (cytosolic proteins) and lamin B (a nuclear protein) were
used as markers to confirm the purity of the nuclear/cytoplasmic fractions (Fig 16B, left
bottom panel). These results together with the immunofluoresence results suggested that BID
might be loosely associated with the nuclear fraction, and that cellular disruption leads to its
dissociation from this fraction. To examine this possibility, cells were treated with
formaldehyde as a cross-linker prior to cellular disruption. These experiments demonstrated
that a small fraction of cellular BID was localized to the nuclear fraction (Fig 16B, top right
panel). These results suggest that BID is loosely associated with the nuclear fraction by
interaction with another protein(s) or with DNA, and that cross-linking is required to preserve
this interaction. To determine whether Etop treatment leads to a change in the levels of
nuclear BID, cells were treated with Etop for 1 and 3 hrs, and then treated with formaldehyde
followed by subcellular fractionation. We found that Etop did not change the levels of BID
associated with the nuclear fraction (data not shown). To determine the cellular location of
the phosphorylated forms of BID, we performed immunofluoresence studies with the
phospho-specific antibodies using BID+/+ and BID-/- MEFs either not treated or treated with
Etop for 30 min. These antibodies detected an increase in nuclear fluorescence in BID+/+
MEFs treated with Etop; however, a similar increase was detected in BID-/- MEFs (data not

51
shown). Thus, these antibodies cross-react with other phospho-S/T-Q proteins. Nonetheless,
the fact that BID partially localizes to the nucleus, and that all currently identified substrates
of ATM are nuclear proteins, suggested that BID is phosphorylated in the nucleus.
Figure16 : Mouse BID is partially localized to the nucleus. (A) Positive staining of BID in the nucleus of healthy MEFs. BID+/+ MEFs (left, top and middle panels) or BID-
/- MEFs (left, bottom) grown on glass cover slips, were either left untreated, or treated with 100 µM Etop for 3 hrs, fixed and immunostained with anti-BID Abs (green), as described in the Material and methods. The nuclei were visualized by DAPI staining (blue; right panels). (B) A small fraction of cellular BID is detected in the nuclear fraction. BID+/+ MEFs were either left untreated (-), or treated with formaldehyde (+), and subfractionated as described in the Material and methods. Aliquots of the cytosolic (C) and nuclear (N) fractions were subjected to SDS-PAGE followed by Western blot analysis using anti-BID (top), anti-MEK (middle top), anti-BAX (middle bottom), and anti-lamin B (bottom) Abs. * marks a cross-reactive band that might represent a modified form of BID.

52
BID might be involved in the immediate cellular response to DNA damage
To assess whether BID is involved in the immediate cellular response to DNA damage, we
next determined the earliest point at which BID in MEFs was phosphorylated, following
exposure to IR. Phosphorylation of S61 was detectable immediately after exposure of cells to
50 Gy IR (Fig 17A). To check other cellular markers that are known to be involved in the
immediate response to DNA damage we monitored the phosphorylation of H2AX. H2AX is
rapidly phosphorylated by ATM in the chromatin micro-environment surrounding a DNA
DSB [67, 68]. Phosphorylated H2AX, also named γH2AX, forms discrete foci at the sites of
DSBs, facilitates the recruitment of damage-responsive proteins and chromatin remodeling
complexes to the sites of DNA damage, and influences both the efficiency and fidelity of
DSB repair. We exposed BID+/+ and BID-/- MEFs to 20 µM Etop for 2 hrs, washed the cells,
and measured the levels of γH2AX 0.5, 3, 6 and 24 hrs post treatment. We found that Etop
induced the phosphorylation of H2AX in all time points that were checked and that the levels
of phosphorylated H2AX appeared to be higher in BID+/+ MEFs than in BID-/- MEFs (Fig
17B). Moreover, 24 hrs post Etop treatment there was a decrease in γH2AX levels detected in
BID-/- but not in BID+/+ MEFs. Similar differences appeared in MEFs treated with IR: 2 hrs
post irradiation, γH2AX levels appeared to be higher in BID+/+ MEFs than in BID-/- MEFs,
whereas 20 hrs post Etop wash γH2AX was still detected in BID+/+ but non in BID-/- MEFs
(Fig 17C). Since γH2AX is present at DNA break foci, we performed immunofluoresence
study with α−γH2AX Abs. BID+/+ and BID-/- MEFs were either not treated or treated with 20
µM Etop for 2 hrs, washed and analyzed for γH2AX foci formation 0.5 and 2 hrs post
treatment. As expected from the Western blot analysis results, foci formation in BID+/+ MEFs
appeared earlier and stronger than in BID-/- MEFs (Fig 17D). The most prominent difference
was 30 min post Etop wash (Fig 17D, middle panel). These results suggest that BID is also
participating in the very early stages of the DSB DNA damage response. No differences in
γH2AX foci formation were detected between BID-S61/S78A and wtBID clones (data not
shown), arguing that phosphorylation of BID is not required for this process.

53
Figure17 : BID might be involved in the immediate cellular response to DNA damage (A) Phosphorylation of BID occurs very rapidly following IR. BID+/+ MEFs were either left untreated (N/T), or treated with 50 Gy IR, collected 5 min later, and lysed. Equal amounts of protein were subjected to SDS-PAGE followed by Western blot analysis using either anti-BID Abs (top) or anti-pS61 Abs (middle). The blot was stripped and reprobed with anti-α-actin Abs to control for loading (bottom). (B) The level of γH2AX is higher in BID+/+ MEFs treated with Etop compare to BID-/- MEFs. BID+/+ MEFs (right) and BID-/- MEFs (left) were either left untreated or treated with 20 µM Etop for 2 hrs. Cells were rinsed and then released into drug-free medium for the indicated time points. Level of γH2AX was determined by western blot analysis using α γH2AX Abs. (C) The level of γH2AX is higher in BID+/+ MEFs treated with IR compare to BID-/- MEFs. BID+/+ MEFs (right) and BID-/- MEFs (left) were exposed to 50 Gy. 2 and 20 hrs post irradiation cells were lysed and the level of γH2AX was determined by western blot analysis using α γH2AX Abs. (D) γH2AX focus formation in BID+/+ MEFs is more pronounced than in BID-/- MEFs. γH2AX focus formation in BID+/+ MEFs (right) and BID-/- MEFs (left) grown on glass cover slips. Cells were either left untreated or treated with 20 µM Etop for 2 hrs. Cells were rinsed and then released into drug-free medium for 0.5 hr and 2 hrs, before fixation, permeabilization, and staining with anti-γH2AX antibody.

54
CDC25A degradation seems to be less efficient in BID-/- and BID-S61A/S78A MEFs
It is well established that to enable S phase entry CDC25A phosphatase activates the cyclin-
dependent kinase 2 (Cdk2) needed for DNA synthesis. CDC25A is degraded in response to
DNA damage or stalled replication, and loss of CDC25A prevents dephosphorylation of
Cdk2 and leads to a transient blockade of DNA replication (S phase arrest) [69]. Since we
found that BID-/- MEFs fail to accumulate in the S and G2 phases of the cell cycle following
Etop treatment, we checked whether BID is involved in regulating the levels of CDC25A.
BID+/+ and BID-/- MEFs were either left untreated or treated with 20µM Etop for 1 hr, and
CDC25A levels were monitored 0.5, 1.5 and 4 hrs post Etop wash. In BID+/+ cells, CDC25A
degradation was detected immediately post Etop exposure (Fig 18A). In BID-/- MEFs
CDC25A was degraded immediately after Etop exposure but it didn’t seem to be degraded
further as in BID+/+ cells. We also checked the degradation of CDC25A in the wtBID and
BID-S61A/S78A clones and found less degradation in the BID mutant clones (Fig 18B).
Thus, BID might be acting upstream of CDC25A and affecting its degradation. This effect
can consequently explain, at least in part, the impaired S phase arrest of the BID-/- and the
BID-S61A/S78A cells.
Figure18 : CDC25A degradation is more efficient in cells expressing wt BID. (A) Kinetics of Etop induced degradation of CDC25A in BID+/+ and BID-/- MEFs. Cells were either left untreated or treated with 20 µM Etop for 1 hr. Cells were rinsed and then released into drug-free medium for the indicated time points. Level of CDC25A was determined by western blot analysis using α CDC25A Abs. (B) BID-/- MEFs stably expressing either wtBID or BID-S61A/S78A were treated and analyzed as above.

55
The phosphorylation of BID is Chk2-independent
CDC25A degradation requires both ATM and the Chk2-mediated phosphorylation of
CDC25A on serine 123. This phosphorylation is an inhibitory phosphorylation and is
important for the arrest of cells in the S phase. To check whether the phosphorylation of BID
is also Chk2 dependent and is part of the ATM-Chk2- CDC25A -Cdk2 pathway. Chk2+/+ and
Chk2-/- MEFs were either left untreated or treated with 20 µM Etop for 5 min and up to 2 hrs.
BID phosphorylation was determined by Western blot analysis using the BID-phospho-
specific Abs. As shown in Figure 19, BID phosphorylation is not Chk2-dependent and occurs
in the same kinetics in Chk2+/+ and Chk2-/- MEFs.
Figure19 : The phosphorylation of BID is Chk2-independent Phosphorylation of BID is Chk2-independent. Chk2+/+ and Chk2-/- MEFs were either left untreated or treated with 50 µM Etop for the indicated time points. Cells were lysed and analyzed for BID phosphorylation levels by Western blot analysis using the anti pS78 Abs (upper panel) and anti-pS61 Abs (lower panel).

56
Chapter II – Identification of proteins that interact with phosphorylated BID
Introduction
It is well established that BH3-only proteins are sentinels of intracellular damage [70]. These
proteins are held at cellular locations in which they can sense a specific kind of cellular
damage, and once activated, translocate to the mitochondria to communicate the damage
signal. As presented above, a portion of BID localizes to the nucleus. In addition, all
currently identified substrates of ATM are nuclear proteins. Thus, it is most likely that BID is
phosphorylated in the nucleus. We proposed that BID functions as a nuclear effector of ATM,
and that this function(s) requires interaction with another protein(s) in the nucleus or at
another cellular location.
There are a number of reports describing proteins that interact with FL-BID. Two of them
reported that BID interacts and is phosphorylated by casein kinase I and II (CKI/II), and that
this phosphorylation decreases BID’s sensitivity to caspase-8 cleavage and protects cells
from Fas-induced apoptosis [71, 72]. Another report found that PACS-2, which is a sorting
protein that controls the ER–mitochondria axis, binds FL-BID and shuttles it to the
mitochondria [73]. In addition, this PACS-2-dependent trafficking of full-length BID to the
mitochondria seems to promote cell death by inducing the formation of tBID. Recently,
another report showed that FL-BID directly regulates the activation of BAX during TNFα-
induced apoptosis in ASTC-a-1 cells [74]. All the interactions described above relate to the
function of BID as a pro-apoptotic protein and none of them relate to its cell cycle arrest
function in the DNA damage response.
To understand the mechanism by which phosphorylated BID regulates cell cycle arrest and
its connection to apoptosis after DNA damage, we took a biochemical approach to purify
proteins that associate with BID. We used two different cellular systems in order to identify
possible interactors of BID: 1) Infection of BID-/- MEFs with adeno-BID-HA, Etop treatment
followed by IP with anti-HA antibodies; 2) 293T cells transfected with pcDNA3-BID-HA,
treated with Etop, followed by crosslinking and then IP with anti-HA antibodies.
Since the experiments using infection of BID-/- MEFs with adeno-BID-HA did not result in

57
meaningful results and the background of non-specific proteins was very high (data not
shown), we decided to use cross-linkers. The use of cross-linkers enables the capture of close
interactions in intact cells.
Results
BID is found as part of a 50KDa cross-linked complex in healthy cells and in cells treated
with DNA damage
We used the primary amine-reactive non-reversable cross linker BS3 and formaldehyde to
identify proteins that associate with BID. These cross-linkers induce nucleophilic attack of
the amino group of lysine and subsequent covalent bonding via the cross-linker. This
biochemical process allows the capture of interactors that otherwise could not be detected
under harsh lysis conditions.
1) Cross-linking using BS3- 293 cells were transfected with pcDNA3-BID-HA and treated
with Etop. Cells were than harvested and treated with digitonine for 10 min on ice. The
membrane fraction was separated from the cytosolic fraction by centrifugation and each were
incubated with BS3 for 30 min. Western blot analysis showed that most of the BID monomer
and complexes appeared in the cytosolic enriched fraction (data not shown). The anti-HA
antibodies detected a ~50 KDa band that may represent a complex of BID-HA with another
protein(s) (Fig 20A). An IP experiment in large scale with anti-HA antibodies was conducted
on the cytosolic fraction and the ~50 KDa band with several additional bands were sent to
MS analysis (Fig 20B).

58
Figure20 : Cross-linking with BS3 results in appearance of specific complexes. (A) 293 cells transfected with pcDNA3-BID-HA. 16 hrs post transfection cells were treated with 50µM Etop for 90 min. Cells were permeabilized with digitonine. BS3cross linker was added to the cytosolic enriched-fraction and IP was conducted using agarose beads coupled to α HA Abs. The beads were then collected and BID-HA and cross-linked complexes composed of BID-HA are eluted using an HA peptide. Western blot analysis of control sample (no cross-linker; -) and cross-linked sample (+) using α HA Abs. (B) 200X10cm dishes of 293 cells were transfected with pcDNA3-BID-HA. 16 hrs post transfection cells were treated with 50 µM Etop for 90 min, and permeabilized with digitonine. BS3cross linker was added to the cytosolic enriched-fraction and IP was conducted using agarose beads coupled to α HA Abs. The beads were then collected and BID-HA and cross-linked complexes composed of BID-HA were eluted using an HA peptide. The eluted sample was concentrated using centricon, and separated by SDS-PAGE (4-20%, Pierce). The gel was stained using Imperial stain (Pierce) and bands that may represent BID complexes were sent to MS.
The use of cross-linkers in a non-reverse manner, provided us with a useful tool to estimate
the size of the interacting protein(s). The MS results showed that BID-HA (25 KDa protein)
was present in the ~50 KDa band. Another protein that was identified in this band in two
separate experiments was peroxiredoxin 6 (PRX6). PRX6 is a 25 KDa protein, that reduces
H2O2 and phospholipid hydroperoxides by redox-active cysteines and is part of the
peroxiredoxin family [75]. Proteins from this family are conserved from bacteria to
mammals, and there are six subgroups of PRX enzymes (PRX1–6) in mammals. Two other
members of this family – PRX1 and PRX3, were identified also in the first experiments done
without the cross linkers, but their interaction with BID was not confirmed (data not shown).
PRX6 is mainly a cytosolic protein but is also found in the nucleus and mitochondria [76]. To
verify the interaction between BID and PRX6, we transfected 293 cells with Flag-PRX6 with
or without BID-HA and checked their interaction by cross-linking followed by co-

59
IP. Co-transfection of Flag-PRX6 and BID-HA, followed by cross-linking, IP with α HA Abs
and Western blot with anti-Flag Abs resulted in detection of Flag-PRX6 monomer (30 KDa)
and a cross-linkable higher complex (55 KDa) (Fig 21, lanes 5 and 6). As expected,
expression of each of these proteins alone gave no bands (Fig 21, lanes 1-4). These results
suggest that BID-HA, which is a 25 KDa protein and Flag-PRX6, which is a 30 KDa protein,
seem to interact and form a 55 KDa cross-linkable complex. This experiment will be repeated
with all the necessary controls to confirm this interaction.
Figure21 : Flag-PRX6 and BID-HA co-immunoprecipitate. Treatment of lysates of 293 cells expressing BID-HA or Flag-PRX6 with BS3 results in appearance of complexes. These complexes are recognized by αFlag Abs. 293 cells were transfected with BID-HA and/or Flag-PRX6. 16 hrs post transfection cells were permeabilized with digitonine. BS3cross linker was added to the cytosolic enriched-fraction and IP was conducted using α HA Abs. BID-HA and cross-linked complexes are eluted using an HA peptide. Western blot analysis using α Flag Abs was conducted.
2) Cross linking using formaldehyde – formaldehyde has been widely used to study binding
of specific proteins to DNA elements in intact cells. Formaldehyde as a cross-linker, has an
advantage because it can penetrate the membrane of living cells and it can therefore capture
protein interactions without cell disruption. In addition, it has a very short spacer (2Å) and
therefore it can cross-link proteins in close proximity.
293 cells transfected with pcDNA3-BID-HA and either treated or not with Etop, were treated
with 1% formaldehyde for 10 min at RT. The reaction was stopped using glycine for 5 min
and cells were harvested and lysed with RIPA buffer. A pull-down assay was conducted
using beads coupled to anti-HA antibodies. The bound proteins were eluted with an HA

60
peptide and separated by SDS-PAGE. Western blot analysis using α HA antibodies showed
again the appearance of a ~50 KDa band (Fig 22). This band was also detected by the anti-
phospho BID antibodies (α pS61, α pS78; Fig 22; middle and right panels) suggesting the
presence of phosphorylated BID in the complex. Of note, the relativelly high basal
phosphorylation we detected in cells that were not treated with Etop is probably due to the
transfection of cells. In addition, the ~50 KDa band appeared also in non-treated cells,
suggesting that DNA damage is not required to form this complex.
Figure22 : BID is found as a part of a cross-linked complex in healthy and in Etop treated cells. 293 cells were transfected with pcDNA3-BID-HA. Cells were left untreated or treated with 50 µM Etop. Cells were exposed to 1% formaldehyde and lysed in RIPA. A pull-down assay was conducted using beads coupled to α HA Abs. The BID-HA eluted with HA peptide and the sample was separated by a 10% SDS-PAGE. Western blot analysis using α HA, α pS61 and α pS78 Abs revealed bands that may represent complexes composed of BID and phosphorylated BID with other protein(s).
Next, we scaled up this experiment and detected the ~50 KDa band in a gel stained with
Imperial stain (data not shown). The ~50 KDa band was sent to MS analysis and the proteins
listed in the table below are the major ones detected (as determined by the % covergae of the
protein by peptides) with suitable low Mw.

61
As in previous experiments, proteins that belong to the Peroxiredoxin family were identified.
This time it was PRX1 and PRX2, compared to PRX6 that was identified in the experiments
with BS3 (Fig 21).
Another protein that drew our attention was Mitochondrial Carrier Homolog 2 (MTCH2).
MTCH2 was identified in our lab as part of a cross-linked complex with tBID in cells
signaled to die by TNFα [77, 78]. MTCH2 is a novel and previously uncharacterized protein,
which is related to a family of mitochondrial carrier proteins that catalyze the transport of
metabolites across the inner mitochondrial membrane. The function(s) of MTCH2 and its
relatives are still largely unknown.
To confirm that MTCH2 indeed interacts with FL-BID (and not with tBID, that is formed in
these cells), we constructed a new plasmid in which the HA tag is located in the N-terminus
of BID. Use of this plasmid eliminates the possibility that tBID, which is a C-terminal
fragment of BID, will be “pulled down” by the α HA Abs. Western blot analysis of cells
transfected with the C-terminal tagged BID and the N-terminal tagged BID showed that the
N-terminal tagged BID also forms the ~50 KDa complex (Fig 23). However, the pattern of
bands in the two samples was slightly different and in the C-terminal tagged BID lane two
separated bands around 50 KDa appeared (Fig 23, right panel). Taken together, it is possible
that FL-BID interacts with one or more of the proteins listed in the table and large scale
experiments using N-terminus HA-BID followed by MS will clarify this issue.
Identified proteins in cross linked band
(~50 KDa)
% Coverage of the protein KDa
BID 60% 25
MTCH2 49% 30
PRX1 33% 22
PRX2 23% 22
VDAC2 11% 33
BAX 19% 22

62
Figure23 : N-terminal HA tagged BID forms the 50 KDa complex. 293 cells were transfected with BID tagged with HA in its N-terminal (N) and BID tagged in its C-terminal (C). Cells were exposed to 1% formaldehyde and lysed in RIPA. A pull-down assay was conducted using beads coupled to α HA Abs. The BID-HA eluted with HA peptide and the sample was separated by a 10% SDS-PAGE. Western blot analysis of the samples using α BID and α HA Abs revealed that in the C-terminal tagged BID there is an appearance of two separated bands around 50 KDa (two arrows), whereas in the N-terminal-tagged BID there is an appearance of only the upper band (one arrow). As expected, tBID was present only in the C-terminal tagged BID sample.
We have previously showed in MEFs that BID is partially localized to the nucleus and that
cross-linking with formaldehyde was required to detect BID in the nuclear fraction upon sub-
cellular fractionation (Fig 16). Based on these findings we suggested that BID is loosely
associated with the nuclear fraction by interaction with another protein(s) or with DNA. To
check whether BID was also localized to the nucleus in 293 cells transfected with BID-HA,
we performed immunofluoresence experiments with α HA Abs. As shown in Figure 24A,
BID-HA is also detected in the nucleus. To be sure that we are not missing proteins that
interact with BID in the nucleus (and that remained in the insoluble fraction) we sonicated the
cell lysates prior to IP, and indeed, there was an increase in yield and the appearance of
additional bands (Fig 24B). Interestingly, in cells treated with Etop there was an additional
band that appeared above the ~50KDa band. In the near future, we will add a sub-cellular
fractionation step (to analyze the nuclear fraction) and upscale the experiment for MS
analysis.

63
Figure24 : N-terminal HA tagged BID is localized to the nucleus in transfected 293 cells. (A) 293 cells transfected with BID HA were grown on glass cover slips, fixed and immunostained with anti-HA Abs as described in the Material and methods. (B) 293 cells were transfected with BID tagged with HA in its N-terminus. Cells were treated with Etop for 1 hr and exposed to 1% formaldehyde and lysed in RIPA. Cell lysates were left untreated or sonicated for 10 sec on ice. A pull-down assay was conducted using beads coupled to α HA Abs. The HA-BID eluted with HA peptide and the sample was separated by a 10% SDS-PAGE. Western blot analysis of the samples using α HA Abs revealed a higher protein yield and additional bands in the sonicated sample (arrow).

64
Chapter III - ATM, mitochondria function and apoptosis
Introduction
DNA damage activates cell cycle checkpoints that enable either cell cycle arrest followed by
DNA repair, or apoptosis (in the case of high levels of damage). The ATM kinase seems to
be a major player in this “life versus death” decision. Activation of ATM results in the
activation of Chk2 [79]. Chk2 can then phosphorylates and inhibit Cdc25C, contributing to
maintenance of G2 for cells in S or G2, and phosphorylate p53. Additional phosphorylation
of p53 by ATM, is required for activation of p53 as a transcription factor [80]. Activated p53
then induces transcription of BAX, PUMA and NOXA to initiate apoptosis in certain cell
types and induces p21 to cause inhibition of Cdk’s and G1 cell cycle arrest [80].
Cells lacking active ATM suffer abnormalities in cell cycle checkpoints after IR [81, 82]. In
addition, sensitivity to IR or radio-mimetic chemicals, and radioresistant DNA synthesis
(RDS) are hallmarks of the Α−Τ cellular phenotype [40, 83, 84]. On the contrary, in spite of
their increased radiosensitivity, it was also found that irradiated Α−Τ cells are unable to
undergo mitochondrial collapse and caspase-3 activation. These data suggest that ATM is
necessary for the initiation of molecular pathway(s) leading to IR-induced apoptosis [85].
Apoptosis is a wide signaling response that includes many cellular events. Some central
events occur at the mitochondria, including the release of caspase activators (such as
Cytochrome c), changes in electron transport, loss of mitochondrial transmembrane potential,
altered levels of reactive oxygen species (ROS) and activation of pro- and anti-apoptotic
proteins [8, 86-89]. Apart of the “apoptotic link” between ATM and the mitochondria, there
is strong evidence that mitochondria dysfunction occurs early and acts causally in
neurodegenerative diseases [90]. In addition, mitochondrial-DNA mutations and oxidative
stress contribute to aging, the greatest risk factor for neurodegenerative diseases.
In this part of my studies, we further explored the link between ATM and mitochondria by
examining several mitochondrial parameters in cells lacking ATM. The following
mitochondrial parameters were analyzed:

65
1) Mitochondrial transmembrane potential (∆ψm) - The mitochondrial respiratory chain
produces energy that is stored as an electrochemical gradient which consists of a
transmembrane electrical potential (∆ψm), negative inside of about 180-200 mV. This energy
is then able to drive the synthesis of ATP. Partial inner mitochondrial membranes
permeabilization disrupts mitochondrial ion and volume homeostasis and dissipates ∆ψm
causing depolarization of the mitochondrial membrane (reviewed in [89]). The process of
membrane permeabilization is considered as the “point-of-no-return” in numerous models of
programmed cell death. It affects the inner and outer mitochondrial membranes (IM and OM,
respectively) to a variable degree. OM permeabilization effect the release of proteins that are
normally confined to the mitochondrial intermembrane space (IMS), including caspase
activators and caspase-independent death effectors. The assessment of early mitochondrial
alterations allows for the identification of cells that are committed to die but have not
displayed yet the apoptotic phenotype [91].
2) ROS levels – The majority of ROS within eukaryotic cells is thought to be derived from
the mitochondria as by-products during the generation of ATP, through the process of
oxidative phosphorylation [89]. To generate electrochemical gradient, oxidative
phosphorylation complexes pump protons from the matrix to the intermembrane space, thus
leading to the formation of ∆ψm as well as to the generation of ROS. In healthy cells, ROS
are kept at harmless levels by the activity of both non-enzymatic and enzymatic antioxidant
systems. If not properly controlled, ROS can damage mitochondria leading to the production
of more ROS and more defective mitochondria via a vicious cycle. ROS can cause severe
damage to cellular molecules, especially DNA [92]. Since ATM is a central protein involved
in the DNA damage response, it might be involved in oxidative defense through the induction
of anti-oxidative systems mediated by catalase, peroxidases, SOD and reductases [92].
It has been found that Α−Τ patients have significantly reduced levels of total antioxidant
capacity [93] and that ATM-deficient cells are more sensitive to oxidative stress than normal
cells. It is also assumed that an increase in ROS may contribute to neurodegenration and
premature aging observed in Α−Τ patients [92]. Increase in ROS levels in Α−Τ cells puts the
cells under constant oxidative stress. ATM may have an additional role in sensing/modulating

66
redox homeostasis. The basis for the observed tissue specificity of the oxidative damage in
A-T is not clear.

67
Results
In this chapter, I will present preliminary results regarding the possible connection between
ATM and mitochondrial function.
Α−Τ Cells have higher basal ∆ψm (hyperpolarized) compared to cells expressing wt ATM
We initially assessed ∆ψm in three cell lines with impaired ATM activity using the
fluorescence dye DiOC6 (3,3-dihexyloxacarbocyanine): lymphoblasts from an Α−Τ patient
compared to healthy lymphoblasts, ATM-/- MEFs compared to ATM+/+ MEFs and Α−Τ human
fibroblasts compared to Α−Τ human fibroblasts expressing wtATM. We found that all three
lines with impaired ATM activity had higher basal ∆ψm (hyperpolarized) compared to three
controls (Fig 25A-C). This phenomenon was described earlier in the literature in several
human cancer cell lines that exhibited higher ∆ψm compared to normal cells [94]. In that
study, it was demonstrated that the hyperpolarized state of mitochondria contributed to
apoptosis resistance.

68
Figure25 : Α−Τ cells have higher ∆ψm Cells were incubated with DIOC6 for 15 min to determine the mitochondrial membrane potential. (A) On the left, overlay of FACS histogram showing the fluorescence of DIOC6 (FL-1) measured in ATM+/+ MEFs (purple) compared to ATM-/- MEFs (green). On the right, charts that represents the mean DIOC6 fluorescence that was measured in 3 repeats. (B) WT versus Α−Τ lymphoblasts. (C) Α−Τ fibroblasts expressing ATM compared to Α−Τ fibroblasts. * represent significant differences (p<0.05) based on Student’s t test.
*
*
*

69
A-T cells are more resistant to Etop-induced mitochondrial depolarization and apoptosis
compared to cells expressing wtATM
Next, we checked whether hyperpolarization is compatible with apoptosis resistance. One of
the earliest markers of apoptosis in cells is the loss of ∆ψm (depolarization), and thus we
checked whether there are any differences in the loss of ∆ψm after exposure of cells to
apoptotic stimuli. Cells were treated with 5µM Etop for 24h and incubated with DiOC6.
FACS analysis revealed that mitochondria from WT cells showed significantly stronger
depolarization (low ∆ψm) compared to Α−Τ cells (Fig 26A, top panels). The same differences
were detected in Α−Τ cells compared to Α−Τ+ATM cells (Fig 26A, bottom panels). To
detremine whether the differences in depolarization correlated with cell death post Etop
treatment, we measured PI dye-exclusion by FACS analysis. Indeed, WT cells and
Α−Τ+ATM cells showed higher levels of cell death compared to Α−Τ cells (Fig 26B). The
relative resistance of Α−Τ cells to Etop treatment was surprising since one of the hallmarks of
Α−Τ cells is increase sensitivity to DNA damage caused by IR. To explore whether Etop and
IR affected the cells in similar manner, we checked ∆ψm and cell death in response to IR.
Cells were irradiated with 10 Gy, incubated for 18 and 40 hrs, and low ∆ψm. and cell death
were determined by FACS analysis as described above. As opposed to Etop treatment, there
were no significant differences in mitochondrial depolarization (Fig 27A) and in cell death
(Fig 27B) between the lines. Thus, in these cells ATM seems to be important for Etop-
induced mitochondrial depolarization and cell death, but not for IR.

70
Figure26 : Mitochondria depolarization and cell death are lower in A-T cells post Etop treatment Cells were treated with 5µM Etop for 24 hrs. Mitochondrial membrane potential was measured by DIOC6. (A) Overlay of FACS histograms of WT lymphoblasts and Α−Τ+ATM fibroblasts (left panels, purple line) and Α−Τ lymphoblasts and A-T fibroblasts (green line) showing the distribution of DIOC6 fluorescence. On the right charts represent mitochondrial depolarization as was determined by the percentage of cells exhibiting low DIOC6 fluorescence (gated as M1 in the left histogram). * represent significant differences (p<0.05) based on Student’s t test. (C) Measurement of cell death as was determined by PI exclusion assay using FACS of the above Etop treated cells. * represent significant differences (p<0.05) based on Student’s t test.
M1
* *
*
*
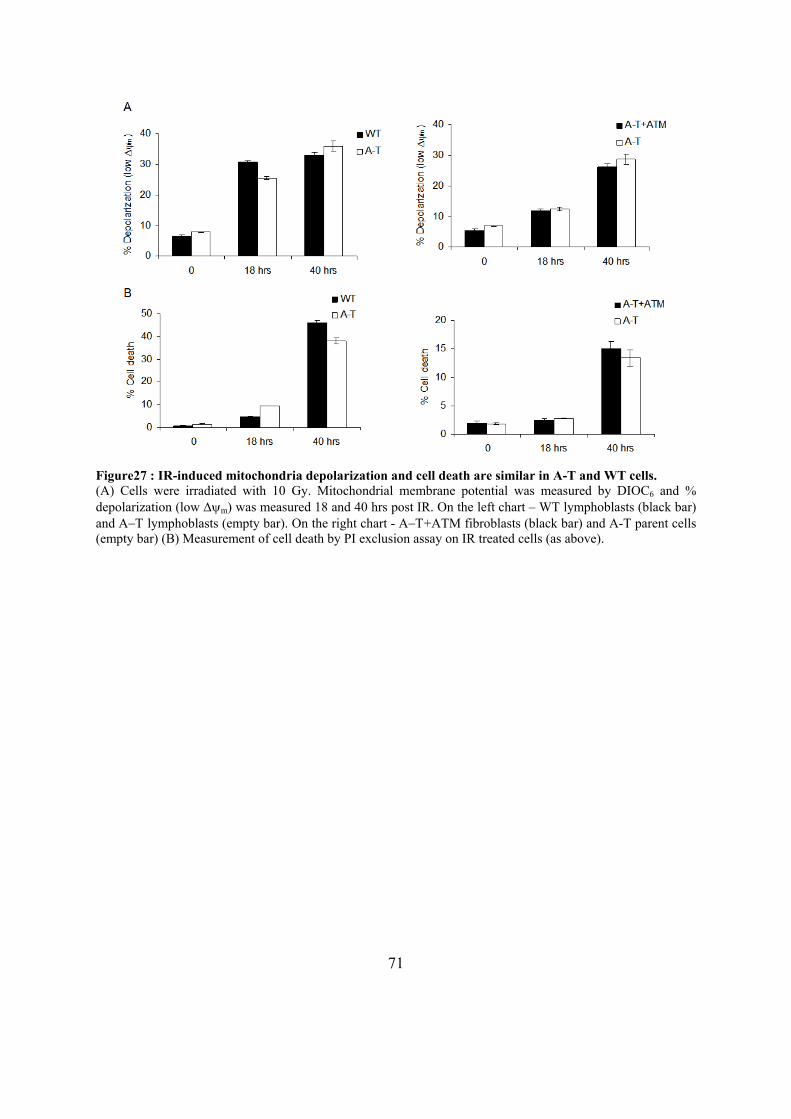
71
Figure27 : IR-induced mitochondria depolarization and cell death are similar in A-T and WT cells. (A) Cells were irradiated with 10 Gy. Mitochondrial membrane potential was measured by DIOC6 and % depolarization (low ∆ψm) was measured 18 and 40 hrs post IR. On the left chart – WT lymphoblasts (black bar) and Α−Τ lymphoblasts (empty bar). On the right chart - Α−Τ+ATM fibroblasts (black bar) and A-T parent cells (empty bar) (B) Measurement of cell death by PI exclusion assay on IR treated cells (as above).

72
ROS levels are higher in Α−Τ cells compared to control cells
As mentioned previously, Α−Τ cells and tissues have increased levels of ROS. Next, we
checked whether we could detect higher ROS levels in the A-T lines. To asses the levels of
superoxide and H2O2, cells in growing phase were incubated with HE (Hydroethidine) and
DCF-DA (2’,7’-dichlorofluorescein) respectively, and taken for FACS analysis. Indeed, we
found that these lines had increased H2O2 and superoxide (Fig 28) compared to the control
lines. These results suggest that ATM plays a role in controlling cellular ROS levels or that
mitochondria of Α−Τ cells display abnormalities that can influence ROS levels in these cells.
Figure28 : ROS levels are higher in Α−Τ cells compared to control cells. Cells in growing phase were incubated with DCF-DA (upper charts) and HE (bottom charts) and analyzed by FACS to assess levels of H2O2 and superoxide, respectively. WT lymphoblasts (black bars) compared to Α−Τ lymphoblasts (empty bar). Α−Τ fibroblasts expressing WT ATM (black bars) compared to the parent Α−Τ fibroblasts (empty bars). * represent significant differences (p<0.05) based on Student’s t test.
* *
*

73
Α−Τ cells grow slower compared to control cells
Mitochondria play a central role in energy metabolism and therefore can effect cellular
growth. In addition, it was found that ATM is also required for efficient G1- to S-phase
transition during cell-cycle progression and that ATM-/- cells prematurely senesce and that
their p21 protein levels are chronically high [95]. Thus we next checked the growth rate of
the different cell lines, and found that the A-Τ lines grew slower compared to the controls
(Fig 29).
Figure29 : Α−Τ cells grow slower compared to control cells. Cell growth in WT and Α−Τ lymphoblasts (left chart) and A-T and Α−Τ+ATM fibroblasts (right chart). The data represent the means ± SEM of pooled results from three independent experiments.

74
Discussion
BID, ATM and the DNA damage response
In the present study, we demonstrated that DSB DNA-damaging reagents induced an
immediate and transient phosphorylation of BID. In addition, we showed that this
phosphorylation was mediated by the ATM kinase. Most importantly, we demonstrated that
BID-/- MEFs failed to delay the cell cycle in response to Etop, and that introduction of a non-
phosphorylatable BID mutant restored delay in the G2 but not in the S phase. Moreover, in
response to Etop, the mutant BID cells entered apoptosis more readily than the wild-type BID
cells [62].
Our current and previous findings demonstrate that in MEFs BID is important for apoptosis
induced by a variety of DNA-damaging reagents (Fig 2 and [37]). Phosphorylation of BID
seems to effect its apoptotic function since BID-/- MEFs expressing the BID-S61A/S78A
mutant are more susceptible than BID-/- MEFs expressing wild-type BID to Etop-induced
apoptosis (Fig 9). Thus, ATM-mediated BID phosphorylation might serve as a mechanism to
inhibit BID’s apoptotic activity or alternatively as a mechanism to activate a pro-survival
activity of BID.
If phosphorylation was inhibiting the apoptotic activity of BID, then what would likely be the
molecular basis of such an inhibition? The only known apoptotic function of BID lies in its
ability to induce the release of pro-apoptotic factors from the mitochondria (e.g., Cytochrome
c). tBID is much more efficient than FL-BID in inducing Cytochrome c release; it would
therefore be expected that ATM-mediated BID phosphorylation inhibits the cleavage of BID,
since casein kinase 1 and/or 2-mediated phosphorylation of BID was demonstrated to inhibit
its cleavage [71]. However, our findings suggest that ATM-mediated phosphorylation of BID
is not related to BID cleavage, for the following reasons: Phosphorylation occurs many hours
before the activation of caspases and the onset of apoptosis (Fig 6). A relatively small amount
of BID is phosphorylated (Fig 3), and phosphorylation occurs also in response to extremely
low, non-apoptotic levels of DNA damage (Fig 8). Moreover, the BID-S61A/S78A mutant
was not found to be more susceptible to cleavage than wtBID (Fig 9). Lastly, though TNFα

75
relies on the generation of tBID to induce/enhance apoptosis [96], the BID-S61A/S78A
mutant clones are not more susceptible to TNFα-induced apoptosis than the wtBID clones
(Fig 9). Thus, BID phosphorylation does not seem to serve as a mechanism to inhibit BID’s
apoptotic activity.
The results presented in Figures 10 and 11 suggest that ATM-mediated phosphorylation of
BID regulates a novel, pro-survival function of BID related to cell cycle arrest following
DNA DSBs. We first found that Etop induced accumulation of BID+/+ MEFs in the S and G2
phases of the cell cycle, whereas such an accumulation was not observed in the BID-/- MEFs
(Fig 10). Moreover, using BrdU labeling we have demonstrated that BID is specifically
required for S phase arrest (Fig 11). We then reintroduced wild-type BID into BID-/- cells,
and found that this addition restored the ability to accumulate in the S and G2 phases of the
cell cycle. Introducing the non-phosphorylatable BID mutant into BID-/- cells, however,
restored accumulation only in the G2 phase (Fig 12). These findings imply that BID
phosphorylation plays an important role in S phase arrest.
To demonstrate the BID-dependent S phase checkpoint we have used relatively high levels of
Etop, suggesting that this function of BID is associated with high levels of genotoxic stress in
fibroblasts. On the other hand, Zinkel at al. have demonstrated that in myeloid and in
activated T cells this function of BID is associated with lower levels of genotoxic stress [66].
Hematopoetic cells are primed to undergo cell cycle arrest and apoptosis following treatment
with DNA-damaging reagents, whereas fibroblasts are relatively resistant to DNA-damaging
reagents and prevent proliferation of mutations by entering into long-term G1 or G2 arrest
[51]. Thus, the activation threshold and the biological impact of the BID-dependent S phase
checkpoint function may vary largely among different cell types.
How might the involvement of BID in S phase arrest be related to apoptosis? Following DNA
DSBs, the cell may decide to activate a survival system (mainly through the ATM kinase,
which induces cell cycle arrest and DNA repair), or in the face of extensive or irreparable
damage, the cell may activate the apoptotic machinery. The results presented in Fig 9
demonstrate that BID-/- MEFs expressing the BID-S61A/S78A mutant are more susceptible

76
than BID-/- MEFs expressing wild-type BID to Etop-induced apoptosis, but not to UV- or to
TNFα-, induced apoptosis. Thus, it appears that the non-phosphorylatable BID mutant
sensitizes BID-/- MEFs to apoptosis induced only by reagents that induce DSBs in DNA.
These results, together with the cell cycle results (Figs 10 to 12) suggest that the impaired
ability of the mutant BID cells to induce cell cycle arrest results in increased sensitivity to
DSB DNA damage.
If BID is indeed capable of playing a pro-survival role in the response of cells to DNA DSBs,
then why are the BID-/- MEFs less sensitive than BID+/+ MEFs to apoptosis induced by Etop
and IR (Fig 2)? BID may play both a pro-survival and a pro-apoptotic role in this pathway.
Based on our finding that the phosphorylation of BID occurs in response to extremely low
levels of IR, and increases in an IR dose-dependent manner (Fig 8), we propose that BID acts
as a sentinel of DNA DSBs. BID might translate the damage into either cell cycle arrest/DNA
repair processes (at low levels of damage) or apoptosis (at high levels of damage). In
addition, we found that BID-/- cells that were exposed to relatively low Etop levels, eventually
die after several days (Fig 14). These results demonstrate that the lack of cell cycle arrest and
probably accumulation of unrepaired DNA damage eventually leads to death. In a parallel
study that was done by Zinkel et al., it was found that myeloid progenitor cells (MPCs)
treated with either hydroxyurea or mitomycin c, agents that induce DNA damage through
replicative stress, fail to accumulate in S phase and displayed increased genomic instability
and increased cell death [66]. These results demonstrate that the action of BID as a pro-
apoptotic or as a pro-survival protein is influenced from the cell type, the kind of treatment
and the levels of a given treatment.
To examine the role of BID phosphorylation in another cell type, we used the human HeLa
cell line. We found that knocking-down the expression of BID in HeLa cells partially impairs
Etop-induced S phase arrest (Fig 15). These results indicated that a partial reduction in the
levels of BID is sufficient to impair DNA-damage-induced S phase arrest.
Up until now, BID has been considered a cytosolic protein. Since signaling proteins involved
in cell cycle checkpoints often function in the nucleus and so are proteins that are

77
phosphorylated by ATM, we examined the subcellular location of BID, and found that it is
partially localized to the nucleus. Immunofluoresence studies using anti-BID antibodies and
subcellular fractionations of MEFs after treatment with cross-linker showed that a fraction of
BID is localized to the nucleus (Fig 16). Since cellular disruption avoided the detection of
BID in the nucleus, we suggested that BID is loosely associated with the nuclear fraction by
interaction with another protein(s) or with DNA, and that cross-linking is required to preserve
this interaction. Moreover, in a published study that was conducted by Galia Oberkovitz
(another Ph.D. student in the lab), it was found that Etop and IR induce the nuclear export of
BID and that this nucleocytoplasmic shuttling of BID is involved in regulating its activity
following DNA damage. Fusing BID to a nuclear localization signal (NLS) inhibited its
nuclear export, and impaired is ability to induce S phase arrest and cell death [97]. Zinkel et
al. also found that BID in MPCs is localized to the nuclear fraction and more specifically to
the chromatin fraction [66]. Thus, BID is most likely phosphorylated by ATM in the nucleus,
and the phosphorylated form might need to exit the nucleus to execute its function in the
DNA damage response.
The very rapid kinetics of BID phosphorylation positions it as part of a wide web of proteins
that are involved in the immediate response to DSB DNA damage (Fig 17). Another
important protein in the early response to DNA damage is H2AX. This protein is rapidly
phosphorylated and recruited to the site of DSB and forms foci. We found that H2AX
phosphorylation and foci formation in BID+/+ MEFs appeared earlier and stronger than in
BID-/- MEFs. These results suggest that BID participates in a much earlier stage of the DNA
damage response in addition to its involvement in the late response related to cell cycle
regulation.
Protein phosphorylation is widely recognized as the major mechanism that controls cell cycle
progression. In addition, the activity of proteins that regulate cell cycle progression is
controlled throughout the different phases of cell cycle. Although BID was found to regulate
the arrest of cells in S phase, the phosphorylation of BID was found to be cell cycle-
independent when using synchronized cells (Fig 13). Examination of cell cycle-dependent
downstream event such as degradation of CDC25A, which is directly effecting S phase

78
progression, revealed less degradation in BID-/- MEFs compared to BID+/+ cells (Fig 18).
Similar results were seen in BID-/- MEFs expressing the non-phosphorylatable BID mutant.
These results connect the phosphorylation of BID to one of the most well characterized
pathways in the response of cells to DNA damage: the ATM-Chk2-CDC25A pathway. Of
note, the Chk2 kinase phosphorylates many down stream substrates in this pathway, such as
p53 and CDC25A, but it is not necessary for the phosphorylation of BID (Fig 19). Therefore,
we still do not know where to position BID in this pathway. In addition, there are new ATM
substrates identified all the time that expand the web and branches of the DDR [98].
One way to define BID’s position in this expanding pathway is to identify proteins that
interact with BID upon DNA damage. We found that BID is part of a ~50 KDa complex in
both healthy cells and in cells treated with Etop. The detection of this complex was possible
due to the use of cross-linkers that stabilized this interaction prior to cell disruption (Figs 20
and 22). Importantly phosphorylated BID was detected in this complex prior and following
Etop treatment (Fig 22). MS analysis of the ~50 KDa band revealed a list of mitochondrial
proteins that included MTCH2, which was previously identified in our lab as a tBID
interactor [77]. These results suggest that either FL-BID indeed interacts with MTCH2 at the
mitochondria or that pulling down MTCH2 was a result of tBID generation in the cells. To
eliminate the involvement of tBID in the pull-down experiments, HA tag was fused to the N-
terminus of BID, and the ~50 KDa cross-linked band appeared again (Fig 23). These results
indicate that FL-BID forms a complex with another protein or that the appeared band can also
represent a BID homodimer. Future studies will determine the identity of this band. Since the
results regarding BID’s localization imply that BID may execute its function in the nucleus
possibly by interaction with a nuclear protein, we are in the process of an experiment using
formaldehyde to identify a nuclear BID complex.
Another protein that was identified by MS results as part of the ~50 KDa complex is PRX6
(Fig 21). This protein is an anti-oxidant, and our initial studies suggest that BID interacts
with this protein. It is now to be discovered whether this interaction is physiologicaly relevant
and whether it is connected to the DNA damage response and to cell cycle regulation by BID.
It is possible that we succeeded in “fishing” another interactor of BID that participates in the

79
response of cells to the elevated levels of ROS following Etop treatment.
In summary, following DNA damage, the cell activates a survival system that allows cell
cycle arrest/DNA repair and continuation of its normal life cycle, or it may activate the
apoptotic machinery in the face of extensive or irreparable damage. Our study reveals that the
BH3-only BID protein, a molecule that was previously considered to be active only as a pro-
apoptotic factor, also plays a pro-survival role as an ATM effector (see model in Fig 30). If
BID is indeed playing both a pro-apoptotic and a pro-survival function in the DNA damage
pathway, then it is an excellent candidate to link DNA repair processes and apoptosis. Indeed,
Zinkel et al.previously showed that BID-/- primary activated T cells and myeloid progenitor
cells demonstrate increased chromosomal damage in response to mitomycin c [66]. Galia
Oberkovitz, another Ph.D student in the lab, has comfirmed these finding, and further showed
that ATM-mediated BID phosphorylation is important for preserving genomic stability.
Recently, two studies regarding proteins that are known as apoptotic regulators – Apaf-1 and
Aven, showed that these proteins have a new role in cell cycle regulation. The apoptotic
protein Apaf-1 was demonstrated to regulate S phase arrest in response to DNA damage by
acting downstream of ATM/ATR and upstream of Chk1 [99] It was found that its new role
can be separated from its apoptotic activity since Apaf-1 lacking the proapoptotic CARD
domain (unable to activate caspase-9) induces S phase arrest and Chk1 phosphorylation.
Similarly, in the case of BID, ATM-mediated BID phosphorylation occurs at extremely low
levels of DNA damage and within minutes following the damage. In addition, Zinkel et al.
showed that BID lacking its BH3 death domain is capable of inducing S phase arrest [66]. In
contrast to the pro-apoptotic activities of Apaf-1 and BID, Aven is known as an apoptotic
inhibitor, and it was found to function as an ATM activator to inhibit G2/M progression
[100]. Aven was found to bind ATM and overexpression of Aven in cycling Xenopus egg
extracts prevented mitotic entry and induced phosphorylation of ATM and its substrates.
There are also similarities regarding protein localization: Aven and BID were found to
partially localized to the nucleus in healthy cells, whereas Apaf-1 was found to translocate to
the nucleus upon DNA damage. Thus BID, along with Apaf-1 and Aven demonstrate the
ability of proteins to act as double agents – in regulating apoptosis on one hand, and cell

80
cycle progression on the other hand. It is yet to be determined how these proteins function in
this “life versus death” decision.
To define the importance of ATM-mediated BID phosphorylation in-vivo, Hagit Niv (a
former post doc in the lab) generated a BID knock-in mouse, in which the endogenous BID
gene has been replaced with a gene that drives the expression of a non-phosphorylatable BID
protein (BIDS61A/S78A). Using cells from these mice it was found that BIDS61A/S78A primary T
and B cells demonstrate a defect in the intra S-phase DNA damage checkpoint, increased
chromosomal damage, and increased apoptosis in response to the DNA interstrand cross-
linking agent mitomycin c. Thus, BID's S phase arrest function seems to be critical for T and
B cells to preserve genomic stability and to survive following replication stress.
These results further strengthen the link between BID and the DNA damage response, and
future studies will determine the exact role and importance of BID phosphorylation in
mammalian organisms.
Figure30 : Model for the dual role of BID. Following Etop treatment BID is being phosphorylates in the nucleus on two SQ sites in an ATM-dependent manner. This phosphorylation is required to abrogate apoptosis and for the arrest of cells in S phase. BID was found to be loosely associated with the nuclear fraction probably by interaction with another protein(s). Indeed, we found BID as a part of a 50 KDa complex in healthy and treated cells. The interacting protein is still not known. In addition, it was found that at the mitochondria ∆ψm, ROS levels and apoptosis are regulated in an ATM-dependent manner. Whether it is regulating mitochondrial function directly or indirectly is yet to be discovered.

81
ATM and the mitochondria
Cell cycle checkpoints, which lead to survival and apoptosis, are different processes that must
be coordinated. In the case of high levels of damage, cells initiate an apoptotic pathway
which both in its physiological and pathological occurrence, is closely linked to
mitochondrial structure and function. The link between ATM, which stands at the top of the
signaling pathway, and mitochondrial events, which are downstream in the apoptotic
signaling pathway is still largely unknown. In a separate project we measured mitochondrial
function and apoptosis in cells lacking a functional ATM protein. We found that cells
expressing mutant ATM or lacking the ATM protein have hyperpolarized mitochondria (Fig
25). Hyperpolarization of mitochondria was described earlier in the literature in several
human cancer cell lines that exhibited higher ∆ψm compared to normal cells [94]. It is
assumed that hyperpolarization of mitochondria contribute to apoptosis resistance in these
cells. Indeed, we found that Α−Τ cells are less susceptible to Etop-induced depolarization of
mitochondria and this correlated with resistance to Etop-induced cell death (Fig 26).
Interestingly, the response of the cells to IR did not result in any significant differences
between Α−Τ and WT cells (Fig 27). These results suggest that the response of cells to IR is
different in these cells. Other features of these cells were higher ROS levels and slower
growth rate, which both can be the outcome of mitochondrial dysfunction. Introduction of
wtATM into Α−Τ cells restored the normal ∆ψm and the sensitivity to Etop (Fig 26). In a
recently published paper it was found that mitochondria in Α−Τ cells showed un-uniformed
dispersion in the cell and they tended to polarize at one end of the cell, a phenomena usually
exhibited by lymphoblastoid cells derived from patients suffering from known mitochondrial
diseases [101]. Another study demonstrated that Etop increases mitochondrial biogenesis in
an ATM-dependent manner [102]. In contrast to our results, these two published papers
showed that Α−Τ lymphoblastoid cells exhibit a lower ∆ψm [101, 102]. They suggested that
in the absence of ATM, DSBs induced by Etop triggers a cell death pathway involving
depolarization of mitochondria. To explore the controversies between our studies and to
further establish our findings, there is a need to investigate the differences in mitochondria
function in Α−Τ cells compared to WT cells. To do so we will compare the rate of respiration
and ATP levels in these cells. It will also be interesting to explore the possible connection

82
between high ROS levels, high ∆ψm and apoptosis in Α−Τ cells (for example by using
antioxidants). Another important aspect of this research will be to understand how ATM,
which is a nuclear protein, regulates mitochondria function (see model in Fig 30). Recently it
has been reported that ATM localizes to the microsomal fraction (containing mitochondria)
[101]. Thus, ATM might affect mitochondria parameters by being localized to mitochondria.

83
References
1. Jacobson, M.D., Weil, M., and Raff, M.C. (1997). Programmed cell death in animal development. Cell 88, 347-354.
2. Green, D.R., and Evan, G.I. (2002). A matter of life and death. Cancer Cell 1, 19-30. 3. Thompson, C.B. (1995). Apoptosis in the pathogenesis and treatment of disease.
Science 267, 1456-1462. 4. Lockshin, R.A., and Zakeri, Z. (1994). Programmed cell death: early changes in
metamorphosing cells. Biochem Cell Biol 72, 589-596. 5. Puthalakath, H., Huang, D.C., O'Reilly, L.A., King, S.M., and Strasser, A. (1999).
The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol Cell 3, 287-296.
6. Thornberry, N.A., and Lazebnik, Y. (1998). Caspases: enemies within. Science 281, 1312-1316.
7. Medema, J.P., Scaffidi, C., Kischkel, F.C., Shevchenko, A., Mann, M., Krammer, P.H., and Peter, M.E. (1997). FLICE is activated by association with the CD95 death-inducing signaling complex (DISC). Embo J 16, 2794-2804.
8. Wang, X. (2001). The expanding role of mitochondria in apoptosis. Genes Dev 15, 2922-2933.
9. Bakhshi, A., Jensen, J.P., Goldman, P., Wright, J.J., McBride, O.W., Epstein, A.L., and Korsmeyer, S.J. (1985). Cloning the chromosomal breakpoint of t(14;18) human lymphomas: clustering around JH on chromosome 14 and near a transcriptional unit on 18. Cell 41, 899-906.
10. McDonnell, T.J., Deane, N., Platt, F.M., Nunez, G., Jaeger, U., McKearn, J.P., and Korsmeyer, S.J. (1989). bcl-2-immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell 57, 79-88.
11. Vaux, D.L., Cory, S., and Adams, J.M. (1988). Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 335, 440-442.
12. Adams, J.M., and Cory, S. (2001). Life-or-death decisions by the Bcl-2 protein family. Trends Biochem Sci 26, 61-66.
13. Gross, A., McDonnell, J.M., and Korsmeyer, S.J. (1999). BCL-2 family members and the mitochondria in apoptosis. Genes Dev 13, 1899-1911.
14. Puthalakath, H., and Strasser, A. (2002). Keeping killers on a tight leash: transcriptional and post-translational control of the pro-apoptotic activity of BH3-only proteins. Cell Death Differ 9, 505-512.
15. Yin, X.M. (2000). Signal transduction mediated by Bid, a pro-death Bcl-2 family proteins, connects the death receptor and mitochondria apoptosis pathways. Cell Res 10, 161-167.
16. Zha, J., Harada, H., Yang, E., Jockel, J., and Korsmeyer, S.J. (1996). Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell 87, 619-628.
17. Nicholson, D.W., and Thornberry, N.A. (2003). Apoptosis. Life and death decisions. Science 299, 214-215.

84
18. Li, P., Nijhawan, D., Budihardjo, I., Srinivasula, S.M., Ahmad, M., Alnemri, E.S., and Wang, X. (1997). Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91, 479-489.
19. Rodriguez, J., and Lazebnik, Y. (1999). Caspase-9 and APAF-1 form an active holoenzyme. Genes Dev 13, 3179-3184.
20. Yin, X.M., Oltvai, Z.N., and Korsmeyer, S.J. (1994). BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimerization with Bax. Nature 369, 321-323.
21. Oltvai, Z.N., Milliman, C.L., and Korsmeyer, S.J. (1993). Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 74, 609-619.
22. Knudson, C.M., and Korsmeyer, S.J. (1997). Bcl-2 and Bax function independently to regulate cell death. Nat Genet 16, 358-363.
23. Luo, X., Budihardjo, I., Zou, H., Slaughter, C., and Wang, X. (1998). Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94, 481-490.
24. Gross, A., Jockel, J., Wei, M.C., and Korsmeyer, S.J. (1998). Enforced dimerization of BAX results in its translocation, mitochondrial dysfunction and apoptosis. Embo J 17, 3878-3885.
25. Muchmore, S.W., Sattler, M., Liang, H., Meadows, R.P., Harlan, J.E., Yoon, H.S., Nettesheim, D., Chang, B.S., Thompson, C.B., Wong, S.L., Ng, S.L., and Fesik, S.W. (1996). X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature 381, 335-341.
26. Schendel, S.L., Montal, M., and Reed, J.C. (1998). Bcl-2 family proteins as ion-channels. Cell Death Differ 5, 372-380.
27. Saito, M., Korsmeyer, S.J., and Schlesinger, P.H. (2000). BAX-dependent transport of cytochrome c reconstituted in pure liposomes. Nat Cell Biol 2, 553-555.
28. Kuwana, T., Mackey, M.R., Perkins, G., Ellisman, M.H., Latterich, M., Schneiter, R., Green, D.R., and Newmeyer, D.D. (2002). Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 111, 331-342.
29. Schlesinger, P.H., Gross, A., Yin, X.M., Yamamoto, K., Saito, M., Waksman, G., and Korsmeyer, S.J. (1997). Comparison of the ion channel characteristics of proapoptotic BAX and antiapoptotic BCL-2. Proc Natl Acad Sci U S A 94, 11357-11362.
30. Youle, R.J., and Strasser, A. (2008). The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9, 47-59.
31. Strasser, A. (2005). The role of BH3-only proteins in the immune system. Nat Rev Immunol 5, 189-200.
32. Wang, K., Yin, X.M., Chao, D.T., Milliman, C.L., and Korsmeyer, S.J. (1996). BID: a novel BH3 domain-only death agonist. Genes Dev 10, 2859-2869.
33. Degli Esposti, M. (2002). Sequence and functional similarities between pro-apoptotic Bid and plant lipid transfer proteins. Biochim Biophys Acta 1553, 331-340.
34. Yin, X.M. (2006). Bid, a BH3-only multi-functional molecule, is at the cross road of life and death. Gene 369, 7-19.
35. Gross, A., Yin, X.M., Wang, K., Wei, M.C., Jockel, J., Milliman, C., Erdjument-Bromage, H., Tempst, P., and Korsmeyer, S.J. (1999). Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor- R1/Fas death. J Biol Chem 274, 1156-1163.

85
36. Li, H., Zhu, H., Xu, C.J., and Yuan, J. (1998). Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94, 491-501.
37. Sarig, R., Zaltsman, Y., Marcellus, R.C., Flavell, R., Mak, T.W., and Gross, A. (2003). BID-D59A is a potent inducer of apoptosis in primary embryonic fibroblasts. J Biol Chem 278, 10707-10715.
38. Valentijn, A.J., and Gilmore, A.P. (2004). Translocation of full-length Bid to mitochondria during anoikis. J Biol Chem 279, 32848-32857.
39. Konig, H.G., Rehm, M., Gudorf, D., Krajewski, S., Gross, A., Ward, M.W., and Prehn, J.H. (2007). Full length Bid is sufficient to induce apoptosis of cultured rat hippocampal neurons. BMC Cell Biol 8, 7.
40. Ziv, Y., Bar-Shira, A., Pecker, I., Russell, P., Jorgensen, T.J., Tsarfati, I., and Shiloh, Y. (1997). Recombinant ATM protein complements the cellular A-T phenotype. Oncogene 15, 159-167.
41. Tripathi, A.K., Chaturvedi, R., Ahmad, R., Asim, M., Sawlani, K.K., Singh, M.K., Tripathi, P., and Tekwani, B.L. (2003). Flow cytometric analysis of aneuploidy and S-phase fraction in chronic myeloid leukemia patients: role in early detection of accelerated phase. Leuk Res 27, 899-902.
42. Eldar, H., Ben-Chaim, J., and Livneh, E. (1992). Deletions in the regulatory or kinase domains of protein kinase C-alpha cause association with the cell nucleus. Exp Cell Res 202, 259-266.
43. Kaina, B. (2003). DNA damage-triggered apoptosis: critical role of DNA repair, double-strand breaks, cell proliferation and signaling. Biochem Pharmacol 66, 1547-1554.
44. Iliakis, G., Wang, Y., Guan, J., and Wang, H. (2003). DNA damage checkpoint control in cells exposed to ionizing radiation. Oncogene 22, 5834-5847.
45. D'Amours, D., and Jackson, S.P. (2002). The Mre11 complex: at the crossroads of dna repair and checkpoint signalling. Nat Rev Mol Cell Biol 3, 317-327.
46. Shiloh, Y. (2003). ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 3, 155-168.
47. Lees-Miller, S.P., Sakaguchi, K., Ullrich, S.J., Appella, E., and Anderson, C.W. (1992). Human DNA-activated protein kinase phosphorylates serines 15 and 37 in the amino-terminal transactivation domain of human p53. Mol Cell Biol 12, 5041-5049.
48. O'Neill, T., Dwyer, A.J., Ziv, Y., Chan, D.W., Lees-Miller, S.P., Abraham, R.H., Lai, J.H., Hill, D., Shiloh, Y., Cantley, L.C., and Rathbun, G.A. (2000). Utilization of oriented peptide libraries to identify substrate motifs selected by ATM. J Biol Chem 275, 22719-22727.
49. Kim, S.T., Lim, D.S., Canman, C.E., and Kastan, M.B. (1999). Substrate specificities and identification of putative substrates of ATM kinase family members. J Biol Chem 274, 37538-37543.
50. Bakkenist, C.J., and Kastan, M.B. (2003). DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421, 499-506.
51. Di Leonardo, A., Linke, S.P., Clarkin, K., and Wahl, G.M. (1994). DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes Dev 8, 2540-2551.
52. Kastan, M.B., and Bartek, J. (2004). Cell-cycle checkpoints and cancer. Nature 432, 316-323.
53. Bartek, J., and Lukas, J. (2003). Chk1 and Chk2 kinases in checkpoint control and

86
cancer. Cancer Cell 3, 421-429. 54. Wiese, C., Pierce, A.J., Gauny, S.S., Jasin, M., and Kronenberg, A. (2002). Gene
conversion is strongly induced in human cells by double-strand breaks and is modulated by the expression of BCL-x(L). Cancer Res 62, 1279-1283.
55. Dumay, A., Laulier, C., Bertrand, P., Saintigny, Y., Lebrun, F., Vayssiere, J.L., and Lopez, B.S. (2006). Bax and Bid, two proapoptotic Bcl-2 family members, inhibit homologous recombination, independently of apoptosis regulation. Oncogene 25, 3196-3205.
56. Sawada, M., Sun, W., Hayes, P., Leskov, K., Boothman, D.A., and Matsuyama, S. (2003). Ku70 suppresses the apoptotic translocation of Bax to mitochondria. Nat Cell Biol 5, 320-329.
57. Krajewski, S., Tanaka, S., Takayama, S., Schibler, M.J., Fenton, W., and Reed, J.C. (1993). Investigation of the subcellular distribution of the bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res 53, 4701-4714.
58. Hoetelmans, R., van Slooten, H.J., Keijzer, R., Erkeland, S., van de Velde, C.J., and Dierendonck, J.H. (2000). Bcl-2 and Bax proteins are present in interphase nuclei of mammalian cells. Cell Death Differ 7, 384-392.
59. Wang, Q., Gao, F., May, W.S., Zhang, Y., Flagg, T., and Deng, X. (2008). Bcl2 negatively regulates DNA double-strand-break repair through a nonhomologous end-joining pathway. Mol Cell 29, 488-498.
60. Schmitt, E., Beauchemin, M., and Bertrand, R. (2007). Nuclear colocalization and interaction between bcl-xL and cdk1(cdc2) during G2/M cell-cycle checkpoint. Oncogene 26, 5851-5865.
61. Zinkel, S.S., Ong, C.C., Ferguson, D.O., Iwasaki, H., Akashi, K., Bronson, R.T., Kutok, J.L., Alt, F.W., and Korsmeyer, S.J. (2003). Proapoptotic BID is required for myeloid homeostasis and tumor suppression. Genes Dev 17, 229-239.
62. Kamer, I., Sarig, R., Zaltsman, Y., Niv, H., Oberkovitz, G., Regev, L., Haimovich, G., Lerenthal, Y., Marcellus, R.C., and Gross, A. (2005). Proapoptotic BID is an ATM effector in the DNA-damage response. Cell 122, 593-603.
63. Kamijo, T., van de Kamp, E., Chong, M.J., Zindy, F., Diehl, J.A., Sherr, C.J., and McKinnon, P.J. (1999). Loss of the ARF tumor suppressor reverses premature replicative arrest but not radiation hypersensitivity arising from disabled atm function. Cancer Res 59, 2464-2469.
64. Elkon, R., Rashi-Elkeles, S., Lerenthal, Y., Linhart, C., Tenne, T., Amariglio, N., Rechavi, G., Shamir, R., and Shiloh, Y. (2005). Dissection of a DNA-damage-induced transcriptional network using a combination of microarrays, RNA interference and computational promoter analysis. Genome Biol 6, R43.
65. Kaneko, Y.S., Watanabe, N., Morisaki, H., Akita, H., Fujimoto, A., Tominaga, K., Terasawa, M., Tachibana, A., Ikeda, K., and Nakanishi, M. (1999). Cell-cycle-dependent and ATM-independent expression of human Chk1 kinase. Oncogene 18, 3673-3681.
66. Zinkel, S.S., Hurov, K.E., Ong, C., Abtahi, F.M., Gross, A., and Korsmeyer, S.J. (2005). A role for proapoptotic BID in the DNA-damage response. Cell 122, 579-591.
67. Burma, S., Chen, B.P., Murphy, M., Kurimasa, A., and Chen, D.J. (2001). ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem 276, 42462-42467.

87
68. Thiriet, C., and Hayes, J.J. (2005). Chromatin in need of a fix: phosphorylation of H2AX connects chromatin to DNA repair. Mol Cell 18, 617-622.
69. Falck, J., Mailand, N., Syljuasen, R.G., Bartek, J., and Lukas, J. (2001). The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 410, 842-847.
70. Danial, N.N., and Korsmeyer, S.J. (2004). Cell death: critical control points. Cell 116, 205-219.
71. Desagher, S., Osen-Sand, A., Montessuit, S., Magnenat, E., Vilbois, F., Hochmann, A., Journot, L., Antonsson, B., and Martinou, J.C. (2001). Phosphorylation of bid by casein kinases I and II regulates its cleavage by caspase 8. Mol Cell 8, 601-611.
72. Olsen, B.B., Petersen, J., and Issinger, O.G. (2006). BID, an interaction partner of protein kinase CK2alpha. Biol Chem 387, 441-449.
73. Simmen, T., Aslan, J.E., Blagoveshchenskaya, A.D., Thomas, L., Wan, L., Xiang, Y., Feliciangeli, S.F., Hung, C.H., Crump, C.M., and Thomas, G. (2005). PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. Embo J 24, 717-729.
74. Pei, Y., Xing, D., Gao, X., Liu, L., and Chen, T. (2007). Real-time monitoring full length bid interacting with Bax during TNF-alpha-induced apoptosis. Apoptosis 12, 1681-1690.
75. Manevich, Y., and Fisher, A.B. (2005). Peroxiredoxin 6, a 1-Cys peroxiredoxin, functions in antioxidant defense and lung phospholipid metabolism. Free Radic Biol Med 38, 1422-1432.
76. Kumin, A., Huber, C., Rulicke, T., Wolf, E., and Werner, S. (2006). Peroxiredoxin 6 is a potent cytoprotective enzyme in the epidermis. Am J Pathol 169, 1194-1205.
77. Grinberg, M., Schwarz, M., Zaltsman, Y., Eini, T., Niv, H., Pietrokovski, S., and Gross, A. (2005). Mitochondrial carrier homolog 2 is a target of tBID in cells signaled to die by tumor necrosis factor alpha. Mol Cell Biol 25, 4579-4590.
78. Gross, A. (2005). Mitochondrial carrier homolog 2: a clue to cracking the BCL-2 family riddle? J Bioenerg Biomembr 37, 113-119.
79. Matsuoka, S., Huang, M., and Elledge, S.J. (1998). Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 282, 1893-1897.
80. Hirao, A., Kong, Y.Y., Matsuoka, S., Wakeham, A., Ruland, J., Yoshida, H., Liu, D., Elledge, S.J., and Mak, T.W. (2000). DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science 287, 1824-1827.
81. Beamish, H., Khanna, K.K., and Lavin, M.F. (1994). Ionizing radiation and cell cycle progression in ataxia telangiectasia. Radiat Res 138, S130-133.
82. Beamish, H., and Lavin, M.F. (1994). Radiosensitivity in ataxia-telangiectasia: anomalies in radiation-induced cell cycle delay. Int J Radiat Biol 65, 175-184.
83. Meyn, M.S. (1995). Ataxia-telangiectasia and cellular responses to DNA damage. Cancer Res 55, 5991-6001.
84. Thacker, J. (1994). Cellular radiosensitivity in ataxia-telangiectasia. Int J Radiat Biol 66, S87-96.
85. Vit, J.P., Moustacchi, E., and Rosselli, F. (2000). ATM protein is required for radiation-induced apoptosis and acts before mitochondrial collapse. Int J Radiat Biol 76, 841-851.
86. Green, D.R., and Reed, J.C. (1998). Mitochondria and apoptosis. Science 281, 1309-1312.

88
87. Giorgio, M., Migliaccio, E., Orsini, F., Paolucci, D., Moroni, M., Contursi, C., Pelliccia, G., Luzi, L., Minucci, S., Marcaccio, M., Pinton, P., Rizzuto, R., Bernardi, P., Paolucci, F., and Pelicci, P.G. (2005). Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 122, 221-233.
88. Gottlieb, E., Armour, S.M., Harris, M.H., and Thompson, C.B. (2003). Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ 10, 709-717.
89. Kroemer, G., Galluzzi, L., and Brenner, C. (2007). Mitochondrial membrane permeabilization in cell death. Physiol Rev 87, 99-163.
90. Lin, M.T., and Beal, M.F. (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787-795.
91. Galluzzi, L., Zamzami, N., de La Motte Rouge, T., Lemaire, C., Brenner, C., and Kroemer, G. (2007). Methods for the assessment of mitochondrial membrane permeabilization in apoptosis. Apoptosis 12, 803-813.
92. Barzilai, A., Rotman, G., and Shiloh, Y. (2002). ATM deficiency and oxidative stress: a new dimension of defective response to DNA damage. DNA Repair (Amst) 1, 3-25.
93. Reichenbach, J., Schubert, R., Schwan, C., Muller, K., Bohles, H.J., and Zielen, S. (1999). Anti-oxidative capacity in patients with ataxia telangiectasia. Clin Exp Immunol 117, 535-539.
94. Bonnet, S., Archer, S.L., Allalunis-Turner, J., Haromy, A., Beaulieu, C., Thompson, R., Lee, C.T., Lopaschuk, G.D., Puttagunta, L., Bonnet, S., Harry, G., Hashimoto, K., Porter, C.J., Andrade, M.A., Thebaud, B., and Michelakis, E.D. (2007). A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11, 37-51.
95. Xu, Y., and Baltimore, D. (1996). Dual roles of ATM in the cellular response to radiation and in cell growth control. Genes Dev 10, 2401-2410.
96. Yin, X.M., Wang, K., Gross, A., Zhao, Y., Zinkel, S., Klocke, B., Roth, K.A., and Korsmeyer, S.J. (1999). Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature 400, 886-891.
97. Oberkovitz, G., Regev, L., and Gross, A. (2007). Nucleocytoplasmic shuttling of BID is involved in regulating its activities in the DNA-damage response. Cell Death Differ 14, 1628-1634.
98. Matsuoka, S., Ballif, B.A., Smogorzewska, A., McDonald, E.R., 3rd, Hurov, K.E., Luo, J., Bakalarski, C.E., Zhao, Z., Solimini, N., Lerenthal, Y., Shiloh, Y., Gygi, S.P., and Elledge, S.J. (2007). ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160-1166.
99. Zermati, Y., Mouhamad, S., Stergiou, L., Besse, B., Galluzzi, L., Boehrer, S., Pauleau, A.L., Rosselli, F., D'Amelio, M., Amendola, R., Castedo, M., Hengartner, M., Soria, J.C., Cecconi, F., and Kroemer, G. (2007). Nonapoptotic role for Apaf-1 in the DNA damage checkpoint. Mol Cell 28, 624-637.
100. Guo, J.Y., Yamada, A., Kajino, T., Wu, J.Q., Tang, W., Freel, C.D., Feng, J., Chau, B.N., Wang, M.Z., Margolis, S.S., Yoo, H.Y., Wang, X.F., Dunphy, W.G., Irusta, P.M., Hardwick, J.M., and Kornbluth, S. (2008). Aven-Dependent Activation of ATM Following DNA Damage. Curr Biol 18, 933-942.
101. Ambrose, M., Goldstine, J.V., and Gatti, R.A. (2007). Intrinsic mitochondrial

89
dysfunction in ATM-deficient lymphoblastoid cells. Hum Mol Genet 16, 2154-2164. 102. Fu, X., Wan, S., Lyu, Y.L., Liu, L.F., and Qi, H. (2008). Etoposide induces ATM-
dependent mitochondrial biogenesis through AMPK activation. PLoS ONE 3, e2009.

90
Publications
Kamer, I., Sarig, R., Zaltsman, Y., Niv, H., Oberkovitz, G., Regev, L., Haimovich, G.,
Lerenthal, Y., Marcellus, R.C., and Gross, A. (2005). Proapoptotic BID is an ATM effector in
the DNA-damage response. Cell 122, 593-603.

91
תקציר
) אפפטוזיס( שיכולים לעורר בתא תגובה של מוות תאי ATR- וATM גורם לשפעול של חלבוני הקינאז DNAנזק ל
עובר BCL-2- ה ממשפחתBIDהראנו שהחלבון האפפטוטי , בחלק הראשון של התיזה שלי. או לעצירת מחזור התא
לא עצרו -/-BIDמצאנו שתאי . 78S-ו S61 על שני אתרי זירחוןDNA- בתגובה לנזק לATMי "פוספורילציה ע
. DNAגדיליים ב - שגורם לשברים דוEtop שלהם בתגובה לטיפול בחומרDNA-את מחזור התא ואת שיכפול ה
בעוד שהחזרה ) S-שלב ה (DNA-ב סינטזת ה לתאים אלו הביאה לעצירת מחזור התא בשלBIDהחזרה של החלבון
. לא הביאה לעצירת מחזור התא אלא למוות אפפטוטי של התאים, ATMי "שאינו עובר זירחון ע, מוטנטBIDשל
הינו בעל תפקיד חיוני בשרידות של תאים ובבקרה , שידוע כחלבון אפפטוטיBID-התוצאות הללו מצביעות על כך ש
. מבקר את התקדמות מחזור התאBIDבחנו את המכניזם בו , בשלב הבא. DNA-זק לשל עצירת מחזור התא אחרי נ
ומאפשרים את תפקודו BIDלשם כך נקטנו בגישה ביוכימית על מנת לבודד חלבונים שעוברים אינטרקציה עם
50 - מהווה חלק מקומפלקס בגודל שלBID-מצאנו ש, cross-linkers -תוך שימוש ב. DNA-בתגובה לנזק ל
KDaבתאים לא מטופלים ובתאים שטופלו ב -Etop . כמו כן מצאנו שהחלבון המזורחן מהווה גם חלק מהקומפלקס
על מנת לזהות את החלבונים המצויים בקומפלקס בכוונתנו לערוך ניסוי בקנה מידה גדול ולשלוח את הקומפלקס . ל"הנ
. י ספקטרומטריה"לזיהוי ע
הקשר בין . פעילות מיטוכונדריאלית ואפפטוזיס, ATMהאפשרי בין בחלק השני של העבודה חקרנו את הקשר
אינו אקטיבי או בתאים מעכברים בהם אין כלל ATMבהם , T-Aהגורמים השונים נבדק בתאים שמקורם בחולי
ATM . מצאנו שממברנת המיטוכונדריה בתאים אלו הינה בעלת פוטנציאל חשמלי גבוה יותר מתאי הביקורת המכילים
תאים אלו נמצאו כרגישים יותר לנפילה של פוטנציאל ממברנת המיטוכונדריה אחרי , בנוסף. אקטיביATMחלבון
מצאנו שתאים אלו היו פחות רגישים למוות מושרה , בהתאמה. תהליך המתרחש בזמן אפפטוזיס, Etop-טיפול ב
Etop .ראדיקלים חופשיים(מצאנו שבתאים אלו קיימת רמה גבוהה יותר של מולקולות חמצן ריאקטיביות , בנוסף( ,
. תאים אלו הראו גם קצב גדילה איטי יותר. שמקורן העיקרי בתא הוא בפעילות הנשימה המתבצעת במיטוכונדריה
רמטרים המיטוכנדריאלים אקטיבי הראה שהפATM המקודד לחלבון DNA אליהם הוחדר T-Aשימוש בתאי
שידוע כחלבון גרעיני משפיע על תפקוד ATM-לכן נראה ש. חזרו לרמות נורמליותEtop -ורגישות התאים ל
. המיטוכונדריה בתא
ישנה פעילות בה הוא , חלבון שהיה ידוע כבעל פעילות אפפטוטית בלבד, BID -ל מצא של"המחקר הנ, לסיכום
בפעילות זו . DNA-גדיליים ב-יוני להישרדות תאים אחרי נזק הגורם לשברים דומשמש בתא בתפקיד של חלבון הח
BIDחשוב לעצירת מחזור התא בשלב ה -S ולעיכוב Apoptosis.