thermodynamics of organic chemical partition in soils. 2. nonlinear partition of substituted...
TRANSCRIPT

Environ. Sci. Technol. 1994, 28, 996-1002
Thermodynamics of Organic Chemical Partition in Soils. 2. Nonlinear Partition of Substituted Phenylureas from Aqueous Solution
Frank C. Spurlock' and James W. Biggar
Hydrologic Sciences, Department of Land, Air, and Water Resources, University of California, Davis, Davis, California 956 16
A study was conducted to evaluate the sorption behavior of five substituted phenylureas of varying hydrophobicity over wide ranges in equilibrium solution concentration. The three sorbents were of similar texture with soil organic carbon ranging from 1 to 5%. Sorption was thermody- namically evaluated in terms of nonlinear partitioning into soil organic matter. Similar to observations of linear sorption of nonpolar compounds, sorbed-phase activity coefficients calculated at the limit of sorbate solubility (corrected for crystal energy) were well correlated with molar volume of the sorbates. In all cases, sorbate residual free energies were found to decrease dramatically as sorbed-phase concentration decreased, demonstrating the presence of strong specific sorbate-sorbent interactions at low sorbed-phase loadings. Nonlinearity was quanti- tatively related to the variation of sorbate partial molar free energy in the sorbed phase.
Introduction
Hydrophobic sorption theory has been successful in explaining observed correlations between sorbate (super- cooled) solubility and sorption to humic substances as quantified by Koc, the organic carbon-normalized linear partition coefficient (1-3). These solubility-Koc relation- ships are important because they provide a means to estimate sorbatesorbent partition coefficients when direct measurement is either inconvenient or not possible. However, the reality that many organic compounds demonstrate nonlinear sorption allows only approximate estimation of sorption from solubility (4, 5). Nonlinear sorption indicates that the fundamental process for certain compounds is more complex than hydrophobic theory suggests (5, 6). Although it has been said that nonionic organic compounds generally sorb linearly (3), it is also commonly observed that organic chemicals with polar functional groups (OCPFGs) sorb nonlinearly (2,5). There has been considerable interest in the sorption behavior of nonpolar chemicals (1-3, 7, 8). While these compounds represent important classes of pollutants, many degra- dative processes (e.g., oxidation and hydrolysis) produce products that are more polar than the parent compound. In addition, many anthropogenic OCPFGs are commonly introduced into environmental systems. Therefore, it is important to thermodynamically characterize the sorption behavior of OCPFGs. Elucidation of the sorption ther- modynamics of these more polar compounds may provide insight into certain anomalous aspects of their behavior including nonlinearity, their relatively slow sorption- desorption kinetics (9, IO), and apparent isotherm non- singularity (11).
Interactions between Substituted Phenylureas and Soil Constituents. Previous reports concerning interac- tions between the nonionic substituted phenylureas and soil minerals are mixed. Significant sorption of the nonionic ureas to "pure" clay minerals has been reported
996 Environ. Sci. Technol.. Vol. 28, No. 6, 1994
(12, 131, but the organic carbon content of the sorbents were not assessed. Other studies have found significant amounts of organic carbon (up to -0.3%) in various commercial clay minerals (14-18), which may have an influence on results for pure mineral sorption studies (14, 19). Associations between humic materials and clay surfaces appear to inhibit sorbate access to mineral surfaces (2, 12, 20, 21). In addition, data from isolated mineral sorption experiments are not generally indicative of sorption in natural soils (2, 12, 22).
Spectroscopic data indicate that, in contrast to simple substituted ureas, substituted phenylureas are excluded from the interlayer regions of Ca-, Ni-, and Al-montmo- rillonite and that interactions between the phenylureas and the clay are restricted to external surfaces (23). Surface area differences between external surfaces (edges and faces) and internal surfaces (interlayer) are significant. Total surface area of montmorillonite (an expanding clay) is about 800 m2 gm-', whereas that of kaolinite (a nonexpanding clay) is on the order of 10-20 m2 gm-l(24).
Specific interactions have been observed between substituted phenylureas and humic acids (25). In addition to hydrogen bonding, it appears that electron donor- acceptor processes involving free radicals have a prominent role in phenylurea-humic interactions. Charge-transfer interactions between the ureas (donors) and quinone moieties (acceptors) have been suggested (25).
Sorption of Substituted Phenylureas in Soil. Ex- perimental observations of nonlinear sorption to humic materials have been reported in the literature for a number of compounds, including the substituted phenylureas (26- 33). The nonlinear sorption of the ureas is commonly described in terms of the Freundlich isotherm:
where c (pg mL-l) and s [pg (g of soil-l)] are solution and sorbed concentrations, kF is the Freundlich constant (pgl-N mLN gm-l), and N is the Freundlich exponent.
Phytotoxicity and sorption of the substituted phenyl- ureas are both highly correlated with soil organic matter content and uncorrelated to sorbent clay content across broad ranges of sorbent composition (22, 33-39). The independence of diuron sorption to soil particle size has been reported within a single soil by Nkedi-Kizza et al. (38). They found that nonlinear sorption was observed on a whole Webster soil and five particle size separates of the soil ( N < 0.8), and kF values varied (6 < kF < 41) among the separates. However, Freundlich constants normalized to organic carbon yielded a nearly constant value (kF/OC = 691, mn-l = 111, n = 6).
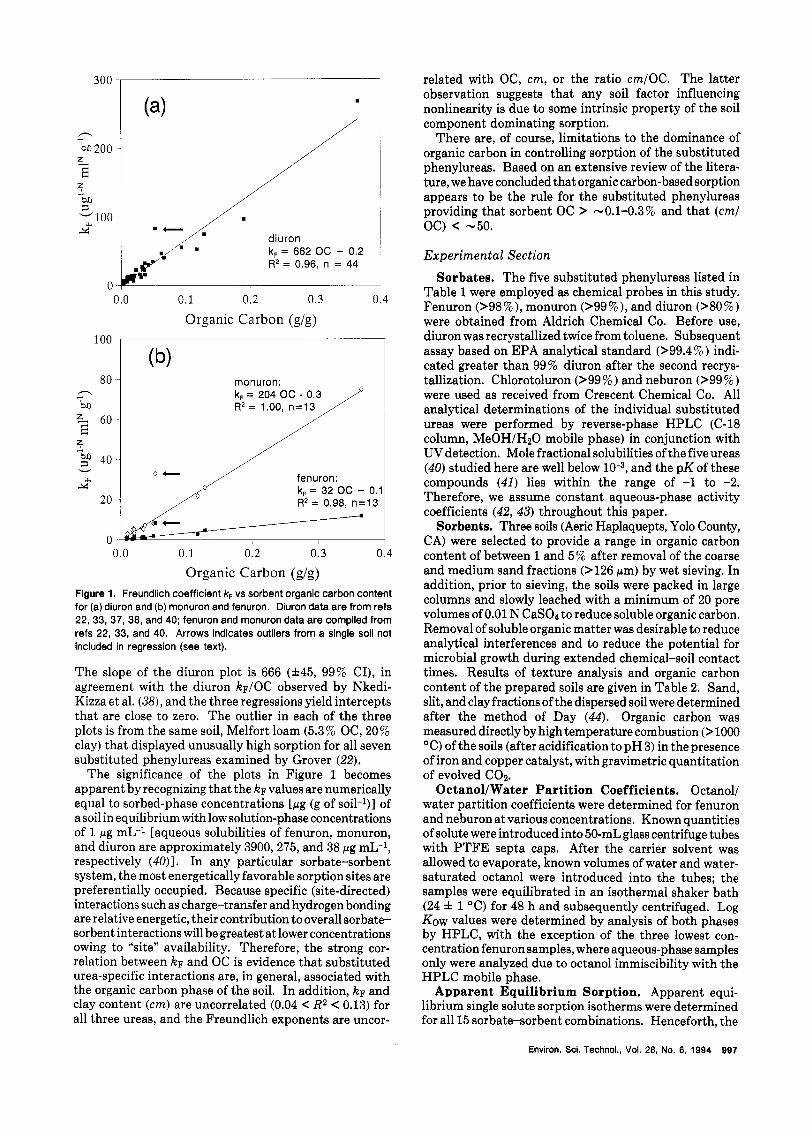
The relationship between k~ and OC appears to be quite general for the phenylureas. Figure 1 illustrates the linear relationship between kF and OC for fenuron, monuron, and diuron. The data were complied from five studies (22,33,37,38,40) and represent a diverse group of sediment and soil sorbents (0.2 < %OC < 37, 0.0 < %clay < 95).
0013-936X/94/0928-0996$04.50/0 0 1994 American Chemical Society
300 1
/
- / n
'w200 ~
/ / I
0
I
I / ~
I / m I diuron k, = 662 OC + 0.2 R2 = 0.96, n = 44
0.0 0.1 0.2 0.3 0.4
Organic Carbon (g/g) I"" 1 I
monuron:
R2 = 1.00, n=13 k, = 204 OC - 0.3
3
fenuron: k, = 32 OC + 0.1
2 40 v a 2
20 R2 = 0.98, n=13 I
I
0 - .
I
0.0 0.1 0.2 0.3 0.4
Organic Carbon (g/g) Flgure 1. Freundlich coefficient kF vs sorbent organic carbon content for (a) diuron and (b) monuron and fenuron. Diuron data are from refs 22,33, 37, 38, and 40; fenuron and monuron data are compiled from refs 22, 33, and 40. Arrows indicates outliers from a single soil not included in regression (see text).
The slope of the diuron plot is 666 (zt45, 99% CI), in agreement with the diuron kF/OC observed by Nkedi- Kizza et al. (38), and the three regressions yield intercepts that are close to zero. The outlier in each of the three plots is from the same soil, Melfort loam (5.3 % OC, 20% clay) that displayed unusually high sorption for all seven substituted phenylureas examined by Grover (22).
The significance of the plots in Figure 1 becomes apparent by recognizing that the kF values are numerically equal to sorbed-phase concentrations [pg (g of soil-l)] of a soil in equilibrium with low solution-phase concentrations of 1 pg mL-l [aqueous solubilities of fenuron, monuron, and diuron are approximately 3900,275, and 38 pg mL-', respectively (4O)l. In any particular sorbate-sorbent system, the most energetically favorable sorption sites are preferentially occupied. Because specific (site-directed) interactions such as charge-transfer and hydrogen bonding are relative energetic, their contribution to overall sorbate- sorbent interactions will be greatest a t lower concentrations owing to "site" availability. Therefore, the strong cor- relation between kF and OC is evidence that substituted urea-specific interactions are, in general, associated with the organic carbon phase of the soil. In addition, kF and clay content (cm) are uncorrelated (0.04 < R2 < 0.13) for all three ureas, and the Freundlich exponents are uncor-
related with OC, cm, or the ratio crn/OC. The latter observation suggests that any soil factor influencing nonlinearity is due to some intrinsic property of the soil component dominating sorption.
There are, of course, limitations to the dominance of organic carbon in controlling sorption of the substituted phenylureas. Based on an extensive review of the litera- ture, we have concluded that organic carbon-based sorption appears to be the rule for the substituted phenylureas providing that sorbent OC > -0.1-0.3% and that (cml OC) < -50.
Experimental Section Sorbates. The five substituted phenylureas listed in
Table 1 were employed as chemical probes in this study. Fenuron (>98%), monuron (>99%), and diuron (>80%) were obtained from Aldrich Chemical Co. Before use, diuron was recrystallized twice from toluene. Subsequent assay based on EPA analytical standard (>99.4%) indi- cated greater than 99% diuron after the second recrys- tallization. Chlorotoluron (>99 %) and neburon (>99% ) were used as received from Crescent Chemical Co. All analytical determinations of the individual substituted ureas were performed by reverse-phase HPLC ((2-18 column, MeOH/H20 mobile phase) in conjunction with UV detection. Mole fractional solubilities of the five ureas (40) studied here are well below and the pK of these compounds (41) lies within the range of -1 to -2. Therefore, we assume constant aqueous-phase activity coefficients (42, 43) throughout this paper.
Sorbents. Three soils (Aeric Haplaquepts, Yo10 County, CA) were selected to provide a range in organic carbon content of between 1 and 5 % after removal of the coarse and medium sand fractions (>126 pm) by wet sieving. In addition, prior to sieving, the soils were packed in large columns and slowly leached with a minimum of 20 pore volumes of 0.01 N Cas04 to reduce soluble organic carbon. Removal of soluble organic matter was desirable to reduce analytical interferences and to reduce the potential for microbial growth during extended chemical-soil contact times. Results of texture analysis and organic carbon content of the prepared soils are given in Table 2. Sand, slit, and clay fractions of the dispersed soil were determined after the method of Day (44). Organic carbon was measured directly by high temperature combustion (>lo00 "C) of the soils (after acidification to pH 3) in the presence of iron and copper catalyst, with gravimetric quantitation of evolved COz.
Octanol/Water Partition Coefficients. Octanol/ water partition coefficients were determined for fenuron and neburon a t various concentrations. Known quantities of solute were introduced into 50-mL glass centrifuge tubes with PTFE septa caps. After the carrier solvent was allowed to evaporate, known volumes of water and water- saturated octanol were introduced into the tubes; the samples were equilibrated in an isothermal shaker bath (24 f 1 "C) for 48 h and subsequently centrifuged. Log Kow values were determined by analysis of both phases by HPLC, with the exception of the three lowest con- centration fenuron samples, where aqueous-phase samples only were analyzed due to octanol immiscibility with the HPLC mobile phase.
Apparent Equilibrium Sorption. Apparent equi- librium single solute sorption isotherms were determined for all 15 sorbate-sorbent combinations. Henceforth, the
Environ. Scl. Technol., Vol. 28, No. 8, 1994 997

-.
Table 1. Sorbate Propertiese
compound solubility loglo In KO,, Vl (pgimL) KO, In yw mole fraction (cm3irnol)
fenuron l,l-dimethyl-3-phenylurea 3900 1.00 5.318 4.22 159 monuron l,l-dimethyl-3-(p-chlorophenyl)urea 275 2.12 7.273 6.77 173 chlorotoluron l,l-dimethyl-3-(3-chloro-p-tolyl)urea 90 2.54 8.999 7.74 192
neburon 3-(3,4-dichlorophenyl)-l-methyl-l-n-butylurea 5.2 4.22 12.285 11.62 236 diuron 1,1-dimethyl-3-(3,4-dichlorophenyl)urea 38 2.81 9.697 8.36 iaa
[I From ref 40, molar volume from modified Lebas method (45).
Table 2. Sorbents and Selected Properties8
soil series use history clay (<2 pm) silt (2-50 pm) sand (50-126 pm) % oc (hn-l)
a Sycamore row crop, >20 years 28 48 24 1.03 (zkO.11, n = 5) 3.42 (zk0.07, n = 4) b Sycamore grassland, >20 years 32 56 13
C Sycamore oak woodland 30 47 24 4.92 (*0.07, n = 4)
[I From ref 40.
qualifying term “apparent” will be omitted while recog- nizing that sorption processes have sometimes been observed to display a significant kinetic component. The assumption of equilibrium here is taken from the stand- point of practical laboratory-scale investigation. Lacking definitive criteria, equilibration times were chosen to meet or exceed our estimated times to 95% desorptiue equi- librium based on an ongoing kinetic study, falling with the range of 80 h (fenuron) to 25 days (neburon). When possible, the soiliwater ratios were chosen such that -40- 60% of the compound would be sorbed at equilibrium. For the sorption experiments, precise quantities of sorbate dissolved in methanol were introduced into 40-mL boro- silicate glass vials with Teflon-lined septa caps using a Hamilton syringe dispenser equipped with gas-tight syringes. After the solvent was allowed to evaporate, appropriate quantities of soil and 0.01 N CaCl2 solution containing 400 mg L-l NaN3 bacteriostat were added, with each spike level performed in duplicate. The samples were then vigorously agitated in an isothermal shaker bath (24 f 1 “C) during the equilibration period and subsequently centrifuged to separate phases. Sorbed-phase concentra- tions were calculated by difference, with total extractions performed on selected samples to verify mass balance. Independent experiments showed no measurable sorption to vial walls or septa for fenuron or neburon. Sorption to vial components were assumed negligible for the remaining compounds. No coeluting peaks were found in aqueous or ethyl acetate soil extracts, and mass balance recoveries indicated that any losses were negligible (fenuron recov- ered, X = 97.1 %, u,-~ = L O % , n = 4; monuron recovered, X = 103.092, cn-l = 3.092, n = 8; chlorotoluron recovered, X = 96.476, = 1.392, n = 6; neburon recovered, X = 101.0 76 , un-l = 0.893, n = 6). Mass balance was assumed for the diuron experiments (no diuron mass balance extractions were performed).
Soil extractions for mass balance determinations re- quired development of a novel extraction method taking advantage of the polyelectrolytic nature of soil organic matter. Preliminary extraction recovery experiments investigated several solvents (methanol, acetone, dichlo- romethane, and others) and extraction procedures (shak- ing, sonication, reflux, soxhlet) and generally demonstrated low and variable recoveries for extended chemical-soil exposure times (>1-30 days). The following procedure was developed for soil extraction. After phase separation
of the water-soil batch samples by centrifugation and sampling of supernatant solution (volume removed de- termined gravimetrically), the wet soil ( - 1.5 g depending on the experiment) was quantitatively transferred to a 500-mL amber jar and then mixed with 100 mL of aqueous dispersant solution containing 4.2 g L-’ sodium meta- phosphate, 0.5 g L-l sodium bicarbonate, and 0.3 g L-l sodium carbonate. After a 20-min sonication, 50 mL of ethyl acetate was added, and the mixture was vigorously shaken on an orbital shaker for 48 h. A total of 30 g of sodium chloride was then added, followed by 30 min further agitation. The resulting emulsion was centrifuged (30 min X 6000g) to separate phases; an aliquot of the clear upper ethyl acetate layer was taken and dried over anhydrous sodium sulfate. Prior to analysis, the solvent was evapo- rated under dry N2, and the sample was reconstituted in HPLC mobile phase. In calculating the final recovery, a correction is made for the dissolution of ethyl acetate in the aqueous sodium chloride phase (measured here as 1.4 mL of ethyl acetate/100 mL of aqueous sodium chloride solution).
Resul ts and Discussion
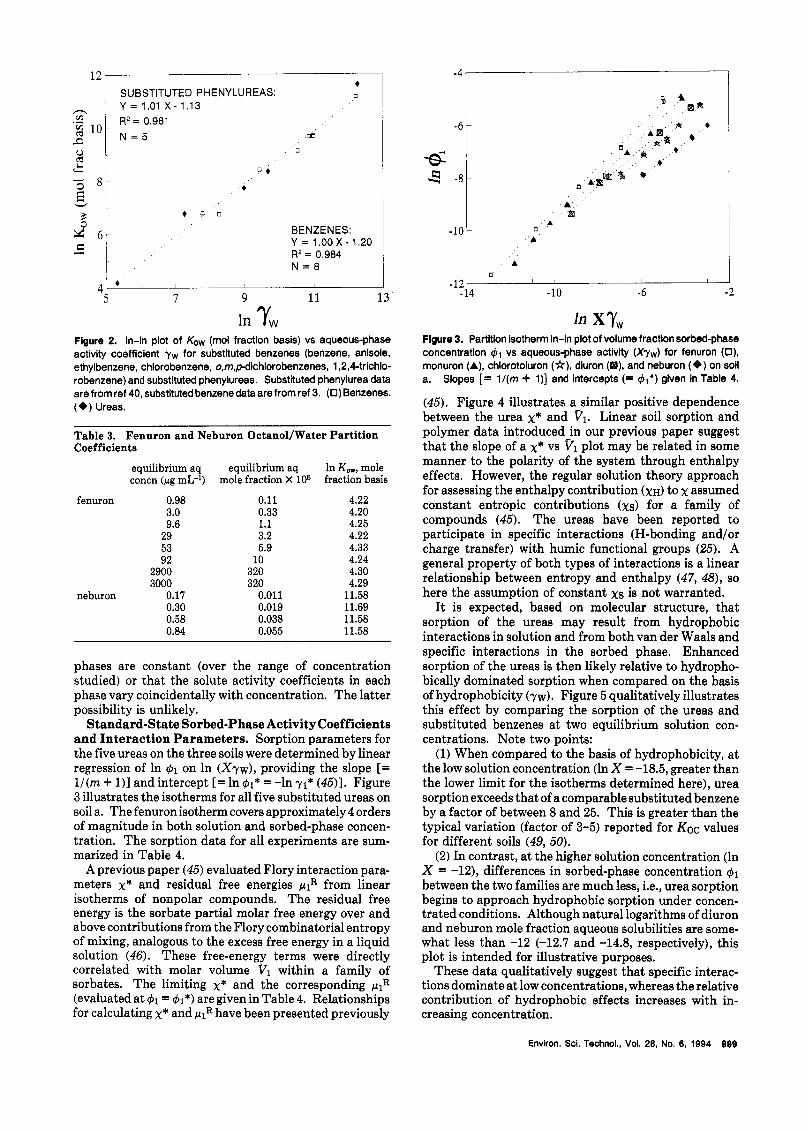
Before evaluating the sorption of the substituted phenylureas, it will be instructive to first compare their 1-octanol/water partitioning with that of nonpolar com- pounds without polar functional groups. Substituted benzenes are an appropriate group of compounds for comparison because these five ureas vary primarily by degree of substitution on the aromatic ring. Figure 2 is a In-ln plot of Kow (octanol/water partition coefficient, mole fraction basis) vs aqueous-phase activity coefficient yw (a measure of hydrophobicity) for alkyl- and chloro- substituted benzenes and the ureas. It is evident that the ureas and substituted benzenes display similar octanoli water partitioning when compared on the basis of hydro- phobicity. The observed slope of both plots is =1, showing that, in the water/octanol system, partitioning of these compounds is hydrophobically driven.
The assumption of a constant aqueous-phase activity coefficient is supported by the Kow data in Table 3. The octanol/water partition coefficient (mole fraction basis) is equal to the ratio of solute activity coefficients in the aqueous and octanol phases (40). Therefore, the constant Kow indicates that solute activity coefficients in both
998 Environ. Sci. Technol., Vol. 28, No. 6, 1994
12 I I -- r 1 SUBSTITUTED PHENYLUREAS: Y = l . O l X - 1 . 1 3
.$ lol r150.981
e 0 . (d
o *
* * n o
* I 0
m
0 1
BENZENES: Y = 1 .oox-1 .20 R2 = 0.984 N = 8 I
I
I 9 11 13 4 *
5
Flgure 2. In-In plot of Kow (mol fraction basis) vs aqueous-phase activity coefficient yw for Substituted benzenes (benzene, anisole, ethylbenzene, chlorobenzene, o,m,pdtchiorobenzenes, 1,2,44richlo- robenzene) and substituted phenylureas. Substituted phenylurea data are from ref 40, substituted benzene data are from ref 3. (0) Benzenes. (e) Ureas.
Table 3. Fenuron and Neburon Octanol/Water Partition Coefficients
equilibrium aq equilibrium aq In K,, mole concn (pg mL-1) mole fraction x 106 fraction basis
fenuron 0.98 0.11 4.22 3.0 0.33 4.20 9.6 1.1 4.25
29 3.2 4.22 53 5.9 4.33 92 10 4.24
2900 320 4.30 3000 320 4.29
neburon 0.17 0.011 11.58 0.30 0.019 11.69 0.58 0.038 11.58 0.84 0.055 11.58
phases are constant (over the range of concentration studied) or that the solute activity coefficients in each phase vary coincidentally with concentration. The latter possibility is unlikely.
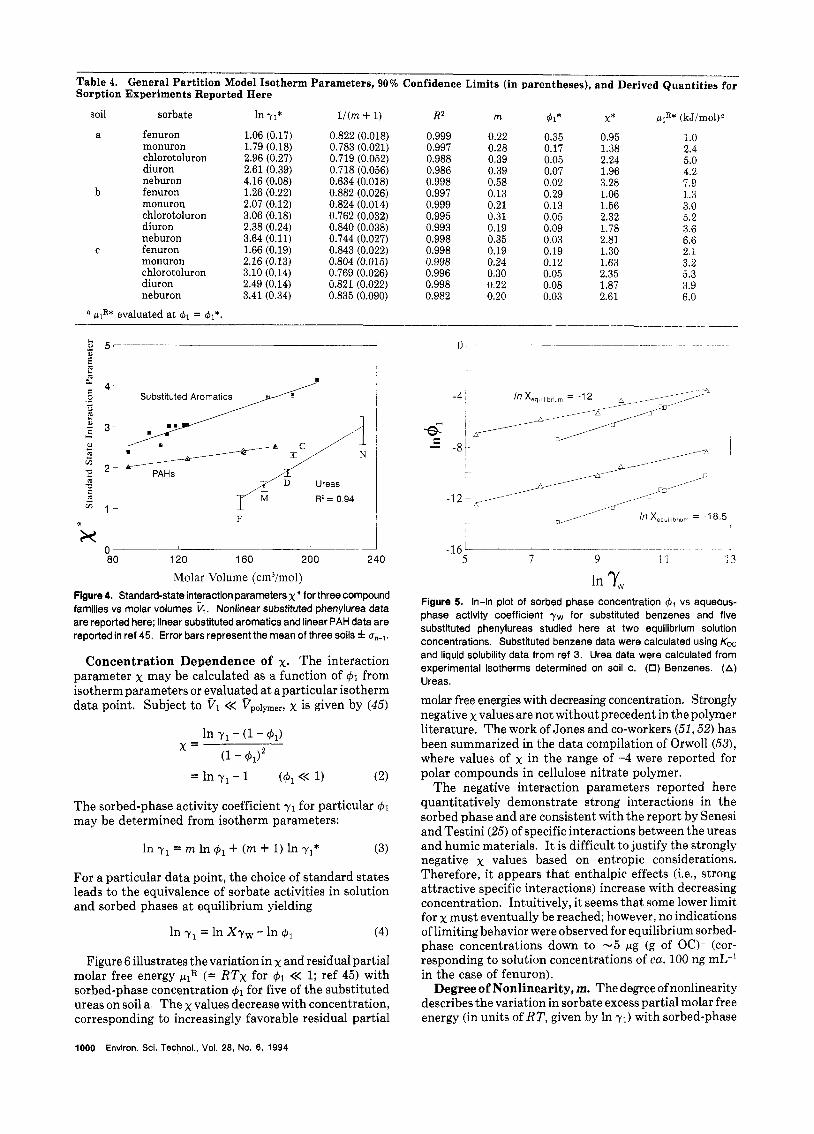
Standard-State Sorbed-Phase Activity Coefficients and Interaction Parameters. Sorption parameters for the five ureas on the three soils were determined by linear regression of In 41 on In (Xyw), providing the slope [= l / (m + 1)l and intercept [= In 41* = -In y1* (4511. Figure 3 illustrates the isotherms for all five substituted ureas on soil a. The fenuron isotherm covers approximately4 orders of magnitude in both solution and sorbed-phase concen- tration. The sorption data for all experiments are sum- marized in Table 4.
A previous paper (45) evaluated Flory interaction para- meters x* and residual free energies plR from linear isotherms of nonpolar compounds. The residual free energy is the sorbate partial molar free energy over and above contributions from the Flory combinatorial entropy of mixing, analogous to the excess free energy in a liquid solution (46). These free-energy terms were directly correlated with molar volume VI within a family of sorbates. The limiting x* and the corresponding plR (evaluated a t 41 = 41*) are given in Table 4. Relationships for calculating x* and plR have been presented previously
A e
-101 A 0 A
A
-14 -10 -6 -2
In XY, Flgure 3. Partition isotherm In-in plot of volume fraction sorbed-phase concentration 4, vs aqueous-phase activity (Xyw) for fenuron (0), monuron (A), chiorotoiuron (*), diuron (B), and neburon (e) on soil a. Slopes [= l l(m + l)] and intercepts (= &*) glven in Table 4.
(45). Figure 4 illustrates a similar positive dependence between the urea x* and VI. Linear soil sorption and polymer data introduced in our previous paper suggest that the slope of a x* vs VI plot may be related in some manner to the polarity of the system through enthalpy effects. However, the regular solution theory approach for assessing the enthalpy contribution (XH) to x assumed constant entropic contributions (XS) for a family of compounds (45). The ureas have been reported to participate in specific interactions (H-bonding and/or charge transfer) with humic functional groups (25). A general property of both types of interactions is a linear relationship between entropy and enthalpy (47, 48), so here the assumption of constant xs is not warranted.
I t is expected, based on molecular structure, that sorption of the ureas may result from hydrophobic interactions in solution and from both van der Waals and specific interactions in the sorbed phase. Enhanced sorption of the ureas is then likely relative to hydropho- bically dominated sorption when compared on the basis of hydrophobicity (yw). Figure 5 qualitatively illustrates this effect by comparing the sorption of the ureas and substituted benzenes a t two equilibrium solution con- centrations. Note two points:
(1) When compared to the basis of hydrophobicity, a t the low solution concentration (In X = -18.5, greater than the lower limit for the isotherms determined here), urea sorption exceeds that of a comparable substituted benzene by a factor of between 8 and 25. This is greater than the typical variation (factor of 3-5) reported for KOC values for different soils (49, 50).
(2) In contrast, a t the higher solution concentration (In X = -121, differences in sorbed-phase concentration $1 between the two families are much less, i.e., urea sorption begins to approach hydrophobic sorption under concen- trated conditions. Although natural logarithms of diuron and neburon mole fraction aqueous solubilities are some- what less than -12 (-12.7 and -14.8, respectively), this plot is intended for illustrative purposes.
These data qualitatively suggest that specific interac- tions dominate a t low concentrations, whereas the relative contribution of hydrophobic effects increases with in- creasing concentration.
Environ. Sci. Technol., Vol. 28, No. 6, 1994 999
- Table 4. General Partition Model Isotherm Parameters, 90% Confidence Limits (in parentheses), and Derived Quantities for Sorption Experiments Reported Here
soil sorbate
a fenuron monuron chlorotoluron diuron neburon
b fenuron monuron chlorotoluron diuron neburon
C fenuron monuron chlorotoluron diuron neburon
(i plR* evaluated at @1 = @l*.
In m* 1.06 (0.17) 1.79 (0.18) 2.96 (0.27) 2.61 (0.39) 4.16 (0.08) 1.26 (0.22) 2.07 (0.12) 3.06 (0.18) 2.38 (0.24) 3.64 (0.11) 1.66 (0.19) 2.16 (0.13) 3.10 (0.14) 2.49 (0.14) 3.41 (0.34)
l l(m + 1)
0.822 (0.018) 0.783 (0.021) 0.719 (0.052) 0.718 (0.056) 0.634 (0.018) 0.882 (0.026) 0.824 (0.014) 0.762 (0.032) 0.840 (0.038) 0.744 (0.027) 0.843 (0.022) 0.804 (0.015) 0.769 (0.026) 0.821 (0.022) 0.835 (0.090)
R2
0.999 0.997 0.988 0.986 0.998 0.997 0.999 0.995 0.993 0.998 0.998 0.998 0.996 0.998 0.982
m @l*
0.22 0.35 0.28 0.17 0.39 0.05 0.39 0.07 0.58 0.02 0.13 0.29 0.21 0.13 0.31 0.05 0.19 0.09 0.35 0.03 0.19 0.19 0.24 0.12 0.30 0.05 0.22 0.08 0.20 0.03
X* plR* (kJ/mol)"
0.95 1.0 1.38 2.4 2.24 5.0 1.96 4.2 3.28 7.9 1.06 1.3 1.56 3.0 2.32 5.2 1.78 3.6 2.81 6.6 1.30 2.1 1.63 3.2 2.35 5.3 1.87 3.9 2.61 6.0
._ Substituted Aromatics /
-16:-- ~-~ ~
X I 0 ' 80 120 160 200 240 5 7 9 11 13
Molar Volume (cm3/mol) In 'Y, Figure 4. Standard-state interaction parameters x" for three compound families vs molar volumes V,. Nonlinear substituted phenylurea data are reported here; linear substituted aromatics and linear PAH data are reported in ref 45. Error bars represent the mean of three soils f u"-~.
5. In-ln plot of sorbed phase concentration 4, vs aqueous- phase activity coefficient yw for substituted benzenes and five substituted phenylureas studied here at two equilibrium so,ution concentrations. Substituted benzene data were calculated using Koc
Concentration Dependence of x. The interaction parameter x may be calculated as a function of $1 from isotherm parameters or evaluated at a particular isotherm data point. Subject to VI << Vplpolymer, x is given by (45)
In y1 - (1 - X =
(1 - $J2
= In y1 - 1 << 1) (2)
The sorbed-phase activity coefficient y1 for particular $1
may be determined from isotherm parameters:
In y1 = m In c)~ + ( m + 1) In yl* (3)
For a particular data point, the choice of standard states leads to the equivalence of sorbate activities in solution and sorbed phases a t equilibrium yielding
(4)
Figure 6 illustrates the variation in x and residual partial molar free energy p1R (= RTx for $1 << 1; ref 45) with sorbed-phase concentration $1 for five of the substituted ureas on soil a. The x values decrease with concentration, corresponding to increasingly favorable residual partial
In y1 = In Xy, - In d1
and liquid solubility data from ref 3. Urea data were calculated from experimental isotherms determined on soil c. (0) Benzenes. (A) Ureas.
molar free energies with decreasing concentration. Strongly negative x values are not without precedent in the polymer literature. The work of Jones and co-workers (51,521 has been summarized in the data compilation of Orwoll (53), where values of x in the range of -4 were reported for polar compounds in cellulose nitrate polymer.
The negative interaction parameters reported here quantitatively demonstrate strong interactions in the sorbed phase and are consistent with the report by Senesi and Testini (25) of specific interactions between the ureas and humic materials. It is difficult to justify the strongly negative x values based on entropic considerations. Therefore, it appears that enthalpic effects (i.e., strong attractive specific interactions) increase with decreasing concentration. Intuitively, it seems that some lower limit for x must eventually be reached; however, no indications of limiting behavior were observed for equilibrium sorbed- phase concentrations down to - 5 pg (g of 0C)- (cor- responding to solution concentrations of ea. 100 ng mL-] in the case of fenuron).
Degree of Nonlinearity, m. The degree of nonlinearity describes the variation in sorbate excess partial molar free energy (in units of RT, given by In 71) with sorbed-phase
1000 Environ. Sci. Technol., Vol. 28, No. 6, 1994

3 r i 6 Neburon
-21
4 % L
E O E -
0,
75 2 n
-2 - ! -4 -1-6
-3 I I 1 I 0 0.002 0.004 0.006 0.008
- volume fraction sorbed
Flgure 6. Flory interaction parameter x and residual free energy (plR. kJ/mol) vs sorbed-phase concentration 41 for five substituted ureas on soil a. Lines were calculated from isotherm data; points are from individual isotherm data points (eqs 3-5). Residual free energies were calculated from plR = xRT(1 - 4 ~ ) ~ (45, 46) and assuming (1 - $1)'
= 1. Note strong attractive free energies at low sorbed loadings.
loading 41:
On a molecular level, specific interactions are by definition site-directed, i.e., directional in nature. For organic carbon- based sorption, values of m therefore reflect the complex interplay between various sorbate properties (size, shape, and polarity) and the physical (steric) and electronic (polar) environment about the ensemble of specific interaction sites within the heterogeneous (amorphous) soil humic phase.
In contrast to the water/l-octanol system, sorption of the substituted phenylureas in natural soils is strongly influenced by specific interactions. Based on past research findings and this study, nonlinear sorption of the sub- stituted phenylureas may be interpreted in terms of specific interactions between the urea linkage and the organic carbon phase of the soil (subject to crn/OC < -50 and total OC > 0.3 % ). In treating the process as nonlinear partitioning, specific interactions are thermodynamically characterized by decreasing values of the Flory interaction parameter with decreasing sorbed-phase concentration. The results show that specific interactions contribute a proportionally lesser amount (relative to hydrophobicity and van der Waals forces in the humic phase) to sorption with increasing concentration, with the variable nature of these sorbed-phase interactions resulting in isotherm nonlinearity.
Acknowledgments The authors thank an anonymous reviewer for a detailed
and thorough review. This work supported by USDA- CSRS Grant 92-34214-7350 and a University of California Jastro-Shields Research Fellowship.
Glossary
C
Y1 sorbate solution-phase concentration (pg mL-1) volume fraction-based sorbate activity coefficient
in sorbed phase
Y1*
YW
kF Koc
Kow m N PIR 4i
41*
R
T S
V l
X* X
XH xs X
upper theoretical limiting sorbate activity coef-
mole fraction-based sorbate aqueous activity coef-
Freundlich constant (pgl-N mLN gm-l) organic carbon-normalized partition coefficient
[mL (g of OC)-l] octanol/water partition coefficient degree of nonlinearity Freundlich exponent residual chemical potential (J mol-l) volume fraction concentration of species i (cm3
~ m - ~ ) theoretical limiting sorbate volume fractional
solubility in sorbed phase corrected for crystal energy, if any (cm3 cm-3)
ficient in sorbed phase
ficient
gas constant (J K-' mol-') sorbed-phase concentration [pg (g of soil)-lI temperature (K) sorbate molar volume (cm3 mol-') Flory interaction parameter (dimensionless) limiting Flory interaction parameter, evaluated at
enthalpic component of x (dimensionless) entropic component of x (dimensionless) sorbate mole fraction in solution
41 = +I* (dimensionless)
Literature Cited
Karickhoff, S. W. Chemosphere 1981, 10, 833. Karickhoff, S. W. J . Hydraul. Eng. 1984, 110, 707. Chiou, C. T. In Reactions and Movement of Organic Chemicals in Soil; Sawhney, B. L., Brown, K., Eds.; SSSA Special Publication 22; American Society of Agronomy: Madison, WI, 1989; pp 1-29. Lyman, W. J. In Handbook of Chemical Property Estima- tion Methods; Lyman, W. J., Rheel, W. F., Rosenblatt, D. H., Eds.; American Chemical Society: Washington, DC, 1982, Chapter 4. Minglegrin, U.; Gerstl, Z. J. Environ. Qual. 1983, 12, 1. Weber, W. J., Jr.; McGinley, P. M.; Katz, L. E. Enuiron. Sei. Technol. 1992, 26, 1955. Karickhoff, S. W.; Brown, D. S.; Scott, T. A. Water Res. 1979, 13, 241. Chin, Y. P.; Weber, W. J., Jr. Environ. Sci. Technol. 1989, 23, 978. McCall, P. J.; Agin, G. L. Environ. Toxicol. Chem. 1985,4, 37. Brusseau, M. L.; Rao, P. S. C. Environ. Sei. Technol. 1991, 25, 903. Rao, P. S. C.; Davidson, J. M. In Environmental Impact of Non-point Source Pollution; Overcash, M. R., Davidson, J. M., Eds.; Ann Arbor Science: Ann Arbor, MI, 1980; pp 23- 67. Hance, R. J. Can. J. Soil Sei. 1969, 49, 357. Bailey, G. W.; White, J. L.; Rothberg, T. Soil Sei. SOC. Am. Proc. 1968, 32, 222. Chiou, C. T.; Porter, P. E.; Shoup, T. D. Environ. Sei. Technol. 1984, 18, 295. Lee, L. S.; Rao, P. S. C.; Nkedi-Kizza, P.; Delfino, D. D. Environ. Sci. Technol. 1990, 24, 654. Pennell, K. D.; Rhue, R. D.; Rao, P. S. C.; Johnston, C. T. Environ. Sei. Technol. 1992, 26, 756. Smith, J. A.; Jaffe, P. R. Environ. Sci. Technol. 1991, 25, 2054. Mills, A. C. Ph.D. Dissertation, University of California at Davis, 1968. Murphy, E. M.; Zachara, J. M.; Smith, S. C. Environ. Sci. Technol. 1990,24, 1507.
Envlron. Sci. Technoi., Vol. 28, No. 6, 1994 1001

(20) Kahn, A.; Hassett, J. J.; Banwart, W. L.; Means, J. C.; Wood,
(21) Hassett, J. J.; Banwart, W. L.; Wood, S. G.; Means, J. C.
(22) Grover, R. Can. J . Soil Sci. 1975, 55, 127. (23) Farmer, W. J.; Alrichs, J. L. Soil Sci. SOC. Am. Proc. 1969,
33, 254. (24) Bohn, H. L.; McNeal, B. L.; O'Connor, G. A. Soil Chemistry;
Wiley-Interscience: New York, 1979; p 82. (25) Senesi, N.; Testini, C. Pestic. Sci. 1983, 14, 79. (26) Wershaw, R. L.; Burcar, P. J.; Goldberg, P. J. Environ. Sci.
(27) Li, G. C.; Felbeck, G. T. Soil Sci. 1972, 116, 140. (28) Kahn, S. U. Can. J . Soil Sci. 1973, 53, 429. (29) Kahn, S. U.; Mazurkewhich, R. Soil Sci. 1974, 118, 339. (30) Haque, R.; Schmedding, D. J. Enuiron. Sci. HealthB 1976,
(31) Moreale, A.; Gallez, A.; van Bladel, R. Weed Res. 1977,17,
(32) Kahn, S. U. Can. J . Soil Sci. 1977, 57, 9. (33) Hance, R. J. Weed Res. 1965, 5, 98. (34) Upchurch, R. P. Weeds 1958, 6, 161. (35) Upchurch, R. P.; Mason, D. D. Weeds 1962, 10, 9. (36) Savage, K. E.; Wauchope, R. D. Weed Sci. 1974,22, 106. (37) Peck, D. E.; Corwin, D. L.; Farmer, W. J. J. Environ. Qual.
(38) Nkedi-Kizza, P.; Rao, P. S. C.; Johnson, J. W. J. Environ.
(39) Hance, R. J. Weed Res. 1965, 5, 108. (40) Spurlock, F. C. Ph.D. Dissertation, University of California
S. G. Soil Sci. 1979, 128, 297.
Soil Sci. SOC. Am. J . 1981, 45, 38.
Technol. 1969, 3, 271.
2, 129.
349.
1980, 9, 101.
Qual. 1983, 12, 195.
a t Davis, 1992.
(41) Smith, A. E.; Grover, R. In Analysis of Pesticides in Water; Chau, A. S. Y., Afghan, B. K., Robinson, J. W., Eds.; CRC Press: Boca Raton, FL, 1982; Vol. 111, pp 183-212.
(42) Mackay, D.; Shiu, W. Y.; Maijanen, A.; Feenstra, S. J . Contam. Hydrol. 1991, 8, 23.
(43) Tsonopoulos, C.; Prausnitz, J. M. Ind. Eng. Chem. Fundam. 1971, 10, 593.
(44) Day, P. R. Soil Sci. SOC. Am. Proc. 1956, 20, 167. (45) Spurlock, F. C.; Biggar, J. W. Enuiron. Sci. Technol. 1994,
(46) Flory, P. J. Discuss Faraday SOC. 1970, 49, 7. (47) Joesten, M. D.; Schaad, L. J. Hydrogen Bonding; Marcel-
Dekker: New York, 1974; pp 247-251. (48) Gur'yanova, E. N.; Gol'dshtein, I. P.; Romm, I. P. Donor-
Acceptor Bond; Wiley: New York, 1975; pp 348-353. (49) Curtis, G. P.; Reinhard, M.; Roberts, P. V. In Geochemical
Processes at Mineral Surfaces; Davis, J. A., Hayes, K. F., Eds.; American Chemical Society: Washington, DC, 1986;
(50) Rutherford, D. W.; Chiou, C. T.; Kile, D. E. Enuiron. Sci.
(51) Jones, A. L. Trans. Faraday SOC. 1956, 52, 1408. (52) Baughan, E. C.; Jones, A. L.; Stewart, K. Proc. R. SOC.
(53) Orwoll, R. A. Rubber Chem. Technol. 1977,50, 451.
preceding paper in this issue.
pp 192-216.
Technol. 1992, 26, 336.
London, Ser. A 1954,225,478.
Received for review January 21, 1993. Revised manuscript received August 6, 1993. Accepted February 25, 1994."
@Abstract published in Aduance ACS Abstracts, April 1, 1994.
1002 Environ. Sci. Technol.. Vol. 28, No. 6, 1994