the drug price competition and patent term restoration act of 1984: the basics of the waxman-hatch...
TRANSCRIPT

THE DRUG PRICE COMPETITION AND PATENT TERM RESTORATION ACT OF 1984: THE BASICS OF THE
WAXMAN-HATCH ACT
Michael A. Swit, Esq.Vice President
Center for Professional AdvancementGeneric Drug Approvals Course

Why The 1984 Law Was Created
Difficulty in obtaining an ANDA for post-1962 drugs
Erosion of patent protection for pioneer drugs due to lengthy FDA approval process
Increased pressure for competitive pricing
Federal government’s role as major drug purchaser

Statute Sections - NDAs
FDCA § 505(b)(1)
“Any person may file with the Secretary [FDA] an application with respect to any [new] drug . . .” {21 U.S.C. § 355(b)(1)}
FDCA § 505(b)(2)
“An application . . . for a [new] drug for which the [safety and effectiveness] investigations . . . relied upon by the applicant for approval of the application were not conducted by or for the applicant and for which the applicant has not obtained a right of reference or use from the person by or for whom the investigations were conducted . . .” {21 U.S.C. § 355(b)(2)}

Post-1984 NDA Requirements Preclinical and Clinical Data For New Chemical Entity, New
Indication, or New Formulation Submission of Patent Information Eligibility for Patent Extension or Market
Exclusivity Pediatric Use Assessment

User Fees & Review Schedule – FY2009
Application with Clinical Data = $1,247,200 Application without Clinical Data/
Supplement with Clinical Data = $623,600 60 Days – FDA Determines if NDA is
Accepted for Filing 180 Days – FDA Presents all Major
Deficiencies 10 Months – 1st Target Date for Approval 12 Months – 2nd Target Date for Approval

Post-1984 ANDA Requirements Based on a “Reference Listed Drug”
(RLD) Bioequivalence Data Manufacturing Information Approval is Limited by Patent Extension
and/or Market Exclusivity Limited Eligibility for Market Exclusivity
– 180 Days No User Fees

Safe Harbor for ANDA R&D on Patent-Protected RLD
History: Court held “patent infringement” includes precommercial testing of product (flurazepam/Dalmane®) (Roche v. Bolar, Federal Circuit, 1984)
Waxman Hatch Act -- Overturned the court decision, specifically declaring that precommercial testing is NOT an infringement (35 USC Section 271(e)(1))
Statute Language: Person may make, use or sell a patented drug during the patent life if solely for uses reasonably related to the development and submission of information under a federal law which regulates the manufacture, use or sale of drugs

Safe Harbor for ANDA R&D on Patent-Protected RLD
Safe Harbor -- applies to innovator and generic firms (Bristol-Myers Squibb v. Rhone-Poulenc Rorer, S.D.N.Y. 2001, aff’d, 3rd Circuit, 2003)
“Reasonably Related” -- means having a decent prospect that the “use” would generate the kind of information relevant to FDA approval requirements Construed:
Intermedics v. Ventritex, N.D.Ca. 1991, aff’d, Federal Circuit, 1993 – clinical trials on intermediates, yes
Medtronic v. Lohr, U.S. Supreme Court, 1996 – applies to medical devices
Integra Life Sciences v. Merck, U.S. Supreme Court, 2005 – preclinical research to identify future candidate, yes

Basic Requirements for Securing ANDA Approval
“Listed Drug” Conditions of use Active ingredient(s) Route of administration, dosage form
and strength Bioequivalent Labeling CMC Certification to patents May be delayed by RLD’s market
exclusivity

The Concept of the Listed Drug
Required for ANDA Approval Generic must be the same as the RLD Before 1984 – Federal Register Notices
Declared DESI Drugs to be “Effective” Now Appear in FDA’s Orange Book,
updated monthly

Electronic Orange Book
Approved Drug Products with
Therapeutic Equivalence EvaluationsCurrent through March 2008
PrefaceFAQ
Search by Active Ingredient Search by Applicant Holder Search by Proprietary Name Search by Application No.
The products in this list have been approved under section 505 of the Federal Food, Drug, and Cosmetic Act.
Drug question email: [email protected]
U.S. Department of Health and Human ServicesFood and Drug Administration
Center for Drug Evaluation and ResearchOffice of Pharmaceutical Science
Office of Generic Drugs
Updated: May 1, 2008

Electronic Orange Book Search in 3 Databases
Rx OTC Discontinued
Request FDA Determination that RLD Was Not Withdrawn from the Market for Reasons of Safety or Efficacy
FDA Answers: Approved Discontinued Drug Products Safety and Effectiveness Determinations

“Same As” RLD Conditions of Use, Route of
Administration, Dosage Form and Strength
Exception: Suitability Petition Requests Approval of Differences
FDA Must Approve Before ANDA Is Filed Examples Advantages/Disadvantages Effect of Pediatric Study Rule – Denial or
Waiver

Bioequivalence Clinical Comparison of Generic and RLD No Significant Difference in the Rate and
Extent to which the Active Ingredient Becomes Available at the Site of Drug Action
Details in Next Lecture

Labeling Same as RLD Except for Certain Differences:
Changes Permitted Under Suitability Petition Based on Different Manufacturer Based on Patents or Market Exclusivity
Electronic Version Required As Of June 8, 2004 (21 CFR 314.94(d); 68 FR 69009 (12/11/03))
Problem Areas Copyright -- Trade Name Trademark/Trade Dress -- USP Specs Indication “Carve Out” -- “Same”?

Chemistry, Manufacturing & Controls
Include List of Articles Used as Components of the Drug
Full Statement of Composition of the Drug
Full Description of Methods, Facilities, and Controls Used in Manufacturing, Processing, and Packaging the Drug
Samples of the Drug and Components, If Requested

Inactive Ingredients
Listed on the Product Label and Labeling Need Not Be the Same as the RLD Exceptions: Parenterals, Ophthalmics, Otics,
Topicals ANDA may be delayed if formulation contains
inactives never before included in an approved drug of that route of administration (Qualitative) – Q1
Or inactives at a higher concentration (Quantitative) – Q2
If different as to Q1 or Q2, applicant must submit information to demonstrate the difference has no effect on the drug’s safety
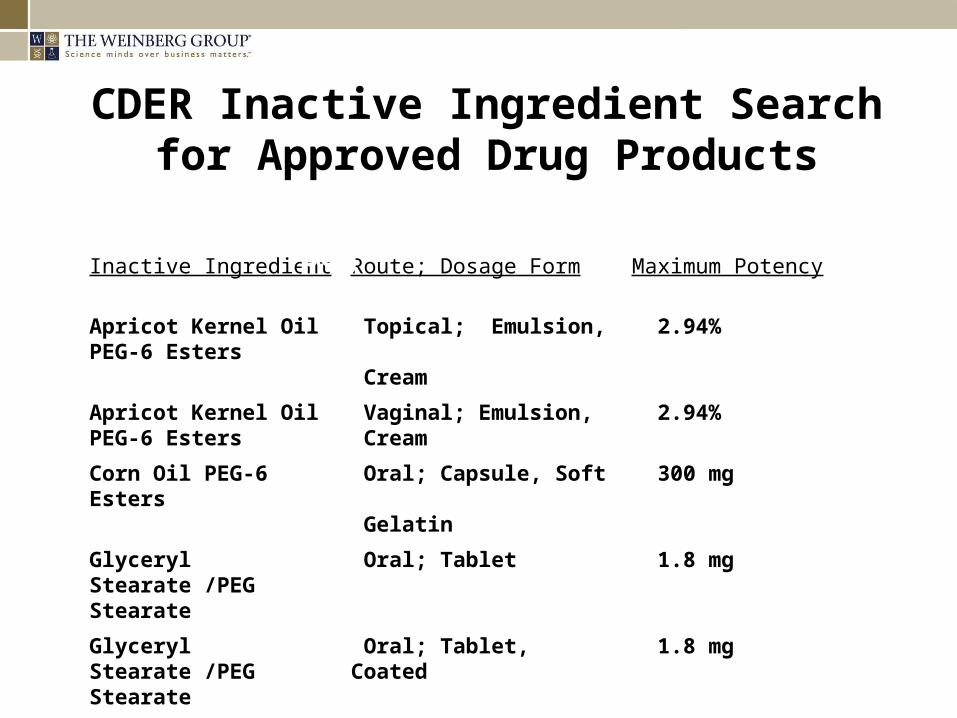
CDER Inactive Ingredient Search for Approved Drug Products
Inactive Ingredient Route; Dosage Form Maximum Potency
Apricot Kernel Oil PEG-6 Esters
Topical; Emulsion, Cream
2.94%
Apricot Kernel Oil PEG-6 Esters
Vaginal; Emulsion, Cream
2.94%
Corn Oil PEG-6 Esters
Oral; Capsule, Soft Gelatin
300 mg
Glyceryl Stearate /PEG Stearate
Oral; Tablet 1.8 mg
Glyceryl Stearate /PEG Stearate
Oral; Tablet, Coated 1.8 mg
Glyceryl Stearate /PEG Stearate
Rectal; Suppository 36.85 mg
Search Results for “peg”

Organizational Structure of OGD
Office of the Director Division of Bioequivalence Divisions of Chemistry (3) Division of Labeling and Program Support Review Queue – 1st In, 1st Reviewed, But
For First generic products for which there are no
blocking patents or exclusivities on the RLD Clusters of ANDAs for same active ingredient Expertise of Reviewers

Statutory Exclusivities Under Waxman-Hatch
New Chemical Entity (NCE) Exclusivity Prohibits the filing of an ANDA (or 505(b)(2)
NDA) for a product that contains the NCE for 5 years after approval of the first NDA. (4 years if ANDA includes a Paragraph IV
challenge to listed patent) NCE: "a drug that contains no active moiety
that has been approved by FDA in any other [NDA]."

Statutory Exclusivities …
3-Year Exclusivity Available for NDAs which contain:
Reports of "new" "clinical trials" That were "essential to approval" of the NDA Conducted or sponsored by the applicant
FDA may not approve an ANDA or 505(b)(2) NDA for 3 years after approval
Applies for new indications, Rx OTC switch, new dosing regimen, and some other labeling changes.

Statutory Exclusivities -- Other
Orphan Drug Exclusivity 7 year exclusivity Drugs for rare conditions (<200,000 people in
U.S.) Pediatric Exclusivity
6-month extension of existing patent or Waxman-Hatch exclusivity
180-day generic (ANDA) exclusivity

“180-Day” or “ANDA” Exclusivity
Basics: First person to file an ANDA with a Paragraph IV
certification gets 180 days during which no other ANDA can be approved for that drug
Must either (a) not be sued by brand co. in 45-day period or (b) prevail in litigation (or get favorable settlement)
180 days starts from earlier of: Date of first commercial marketing (changed in 2003;
used to peg to a court decision as well)

Present Goals of OGD
Decrease Time to Approval & Review Backlogs Now called GIVE - the Generic Initiative for Value and
Efficiency Use existing resources to help modernize &
streamline Hire and train new reviewers Enhanced use of electronic programs for handling
submissions and internal documents Identify low-risk manufacturing changes that do not
require an intense review Educate consumers on safety and low-cost of generics Timely Foreign Inspections (APIs/DMFs) Implement changes under FDAAA and MMA

FDA Amendments Act of 2007 (FDAAA)
Post Labeling of RLD 21 days after approval Post Approval Package 30 days after approval Database For Authorized Generic Drugs
FDA must publish a complete list on its Internet site of all authorized generic drugs, updated quarterly
Drug trade name, brand manufacturer, and date the authorized generic drug entered the market
Drugs marketed, sold, or distributed directly or indirectly to retail class of trade with either labeling, packaging, product code, labeler code, trade name, or trade mark that differs from that of the RLD

FDA Amendments Act of 2007 (FDAAA)
Clinical Trial Registry Databank Not required for blood-level bio studies May be required for clinical bio studies (e.g.,
topical dosage forms) - controversial Citizen Petitions
Shall not delay ANDA approvals unless necessary for public health
If delay, 30-day notice to ANDA applicant Denial permitted based on Petitioner’s intent
to delay Certify that Info became known on XX date
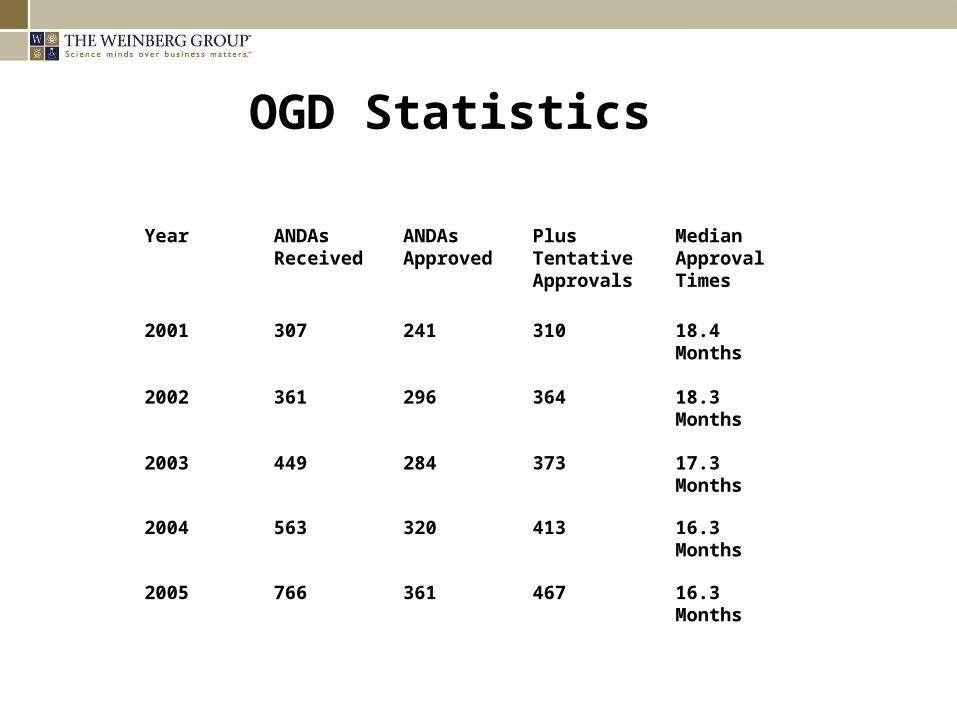
OGD Statistics
Year ANDAs Received
ANDAs Approved
Plus Tentative Approvals
Median Approval Times
2001 307 241 310 18.4 Months
2002 361 296 364 18.3 Months
2003 449 284 373 17.3 Months
2004 563 320 413 16.3 Months
2005 766 361 467 16.3 Months

What’s in a Name?
Drug Price Competition and Patent Term Restoration Act of 1984
“Waxman-Hatch Act” – universally called that until 1994
1994 – Republicans take control of Congress – “Hatch-Waxman Act”
2006 – Democrats take control of Congress So what do you call it now? – well, the first
part of the statutory name – Drug Price Competition – relates to Waxman’s role; and the second part – Patent Term Restoration – refers to Hatch’s role in pushing the 1984 compromise. What do you think?

Numerous FDA Guidance Documents
CDER Guidance Documents on Generic Drugs www.fda.gov/cder/guidance/index.htm
OGD Guidance Documents for ANDAs www.fda.gov/cder/regulatory/applications/anda
.htm
CDER Manual of Policies and Procedures (MAPP) www.fda.gov/cder/mapp.htm

Call, e-mail, fax or write:
Michael A. Swit, Esq.Vice President
The Weinberg Group Inc.336 North Coast Hwy. 101
Suite CEncinitas, CA 92024
Phone 760.633.3343Fax 760.454.2979Cell 760.815.4762
Questions?

About your speaker…Michael A. Swit, Esq., is a Vice President at The Weinberg Group Inc., where he develops and ensures the execution of a broad array of regulatory and other services to drug and biologics clients seeking to market products in the United States. His expertise includes FDA development strategies, compliance and enforcement initiatives, recalls and crisis management, submissions and related traditional FDA regulatory activities, labeling and advertising, and clinical research efforts.
Mr. Swit has been addressing critical FDA legal and regulatory issues since 1984. His multi-faceted experience includes serving for three and a half years as corporate vice president, general counsel and secretary of Par Pharmaceutical, a prominent, publicly-traded, generic drug company and, thus, he brings an industry and commercial perspective to his work with FDA-regulated companies. Mr. Swit then served for over four years as CEO of FDANews.com, a premier publisher of FDA regulatory newsletters and other specialty information products for the FDA-regulated community. His private FDA regulatory law practice has included service as Special Counsel in the FDA Law Practice Group in the San Diego office of Heller Ehrman White & McAuliffe and with the Food & Drug Law practice at McKenna & Cuneo, both in the firm’s Washington office and later in San Diego. He first practiced FDA regulatory law with the D.C. office of Burditt & Radzius.
Mr. Swit has taught and written on a wide variety of subjects relating to FDA law, regulation and related commercial activities, including, since 1989, co-directing a three-day intensive course on the generic drug approval process and editing a guide to the generic drug approval process, Getting Your Generic Drug Approved. A former member of the Food & Drug Law Journal Editorial Board, he also has been a prominent speaker at numerous conferences sponsored by such organizations as RAPS, FDLI, and DIA. A magna cum laude graduate of Bowdoin College, with high honors in history, he received his law degree from Emory University Law School and is a member of the California, D.C. and Virginia bars.