systemic anti-vegf treatment strongly reduces skin inflammation … · systemic anti-vegf treatment...
TRANSCRIPT

Systemic anti-VEGF treatment strongly reduces skininflammation in a mouse model of psoriasisHelia B. Schonthalera,b, Reto Huggenbergerc, Stefanie K. Wculeka, Michael Detmarc, and Erwin F. Wagnera,1
aBanco Bilbao Vizcaya Argentaria (BBVA)-Foundation, Cancer Cell Biology Programme, Centro Nacional de Investigaciones Oncologicas (CNIO),28029 Madrid, Spain; bResearch Institute of Molecular Pathology (IMP), Vienna, Austria; and cInstitute of Pharmaceutical Sciences,Swiss Federal Institute of Technology, ETH Zurich, 8093 Zurich, Switzerland
Edited by Michael Karin, University of California, San Diego School of Medicine, La Jolla, CA, and approved October 22, 2009(received for review July 8, 2009)
Although, vascular remodeling is a hallmark of many chronic inflam-matory disorders such as rheumatoid arthritis, inflammatory boweldisease, and psoriasis, anti-vascular strategies to treat these condi-tions have received little attention to date. We investigated theanti-inflammatory activity of systemic blockade of VEGF-A by theinhibitory monoclonal antibody G6–31, employing a therapeutic trialin a mouse model of psoriasis. Simultaneous deletion of JunB andc-Jun (DKO*) in the epidermis of adult mice leads to a psoriasis-likephenotype with hyper- and parakeratosis and increased subepider-mal vascularization. Moreover, an inflammatory infiltrate and ele-vated levels of cytokines/chemokines including TNF�, IL-1�/�, IL-6,and the innate immune mediators IL-22, IL-23, IL-23R, and IL-12p40 aredetected. Here we show that anti-VEGF antibody treatment of micealready displaying disease symptoms resulted in an overall improve-ment of the psoriatic lesions leading to a reduction in the number ofblood vessels and a significant decrease in the size of dermal bloodand lymphatic vessels. Importantly, anti-VEGF–treated mice showeda pronounced reduction of inflammatory cells within the dermis anda normalization of epidermal differentiation. These results demon-strate that systemic blockade of VEGF by an inhibitory antibody mightbe used to treat patients who have inflammatory skin disorders suchas psoriasis.
activator protein-1 � Jun proteins � epidermis � angiogenesis �G6-31 antibody
Angiogenesis has an important role in tumor growth andmetastasis. However, vascular remodeling also occurs in many
inflammatory and autoimmune disorders (1, 2), including rheuma-toid arthritis, inflammatory bowel disease, and the chronic inflam-matory skin disease, psoriasis (3–5). Psoriasis affects �2% of theworld population and is characterized by epidermal hyperplasia,inflammatory infiltrates, enlarged, tortuous, and hyperpermeableblood vessels, and an enlargement of lymphatic vessels (6, 7).Several studies indicate a crucial role of VEGF-A (here termed‘‘VEGF’’) in the pathogenesis of psoriasis: (i) epidermis-derivedVEGF is strongly up-regulated in psoriatic skin lesions (8); (ii)VEGF serum levels are correlated with disease severity (9); (iii) agenetic predisposition caused by single-nucleotide polymorphismsof the VEGF gene may be involved in the pathogenesis of psoriasis(10); and (iv) K14-VEGF transgenic mice expressing mouseVEGF164 in the epidermis spontaneously develop a chronic pso-riasiform skin inflammation (6). Moreover, VEGF transgenic miceshow the characteristic Koebner phenomenon, because induction ofskin inflammation in transgenic mice results in the development ofchronic, psoriasis-like skin inflammation (7). Treatment of K14-VEGF transgenic mice with a VEGF receptor tyrosine-kinaseinhibitor, NVP-BAW288, reduced the number of blood and lym-phatic vessels and the infiltrating leukocytes in the skin andnormalized the epidermal architecture (11). Recently developedanti-psoriatic therapies (e.g., ustekinumab and ABT-874) predom-inantly target specific components of the immune system, such asTNF�, or molecules involved in T-cell activation (12–14); however,no cure is currently available (15). Surprisingly anti-angiogenictreatments for chronic inflammatory conditions have received little
attention thus far, although some therapies for psoriasis, such aspaclitaxel and shark fin cartilage, are thought to have an anti-angiogenic effect (16). Moreover, the prominent involvement ofangiogenesis in the pathogenesis of psoriasis and the validated useof anti-angiogenic therapy in human cancers using a monoclonalantibody directed against VEGF (bevacizumab, AvastinR) (17, 18)suggests anti-VEGF treatment as a possible therapy for patientssuffering from psoriasis. Recently, a patient was reported to expe-rience complete remission of psoriasis during bevacizumab treat-ment of metastatic colon cancer (19).
Jun proteins are members of the transcription factor complexAP-1 and are essential in establishing and maintaining skin ho-meostasis (20). Epidermal c-Jun controls keratinocyte proliferationand differentiation by regulating the expression of epidermalgrowth factor receptor and heparin-binding EGF-like growth factor(21). On the other hand, absence of epidermal JunB in mice leadsto ulcerative skin lesions and a multiorgan disease caused by thesecretion of granulocyte colony-stimulating factor and IL-6 bykeratinocytes (22).
In the present study we used a therapeutic approach in a geneticmouse model of chronic, psoriasis-like skin inflammation (20, 23,24), using the anti-VEGF antibody G6–31, which potently inhibitsboth human and murine VEGF (25). After Cre-mediated deletion,mice carrying floxed alleles for JunB and c-Jun develop a chronic,psoriasis-like skin inflammation comprising many immunologicalfeatures observed in human psoriatic patients (23). Systemic treat-ment of mutant mice with an anti-VEGF antibody strongly reducedskin inflammation within 8 days of treatment in contrast withcontrol IgG-treated animals. The mutant mice showed an overallimprovement of the psoriatic phenotype, normalization of theepidermal architecture, and a reduction in the number and size ofblood vessels. Moreover, the immune infiltrate in the skin wasreduced in antibody-treated mice.
ResultsSystemic Inhibition of VEGF Reduces the Psoriasis-Like Skin Inflam-mation. The effects of the monoclonal anti-VEGF antibody G6–31(25) were tested in a mouse model displaying many characteristicfeatures of psoriasis. Eight-week-old mice carrying floxed alleles forthe JunB and c-Jun locus and the K5-CreERT transgene receivedconsecutive i.p. injections of tamoxifen (1 mg/day) for a period of5 days. This treatment leads to the deletion of both genes in theepidermis by inducible Cre-recombinase activity (see also SI Ma-terials and Methods). When the mice showed the first symptoms ofa psoriatic phenotype (10–11 days after the last tamoxifen injec-
Author contributions: H.B.S., M.D., and E.F.W. designed research; H.B.S., R.H., and S.K.W.performed research; M.D. contributed new reagents/analytic tools; H.B.S., R.H., and S.K.W.analyzed data; and H.B.S., R.H., M.D., and E.F.W. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
1To whom correspondence should be addressed. E-mail: [email protected].
This article contains supporting information online at www.pnas.org/cgi/content/full/0907550106/DCSupplemental.
21264–21269 � PNAS � December 15, 2009 � vol. 106 � no. 50 www.pnas.org�cgi�doi�10.1073�pnas.0907550106

tion), they received 4 consecutive injections of the anti-VEGFantibody or an isotype control IgG every second day (Fig. 1A). Theappearance of the phenotype was determined by macroscopicallyvisible scaly areas on ears and tail. At day 23–24, the double-knockout (DKO*) mice injected with isotype control IgG (n � 8)showed prominent inflammatory, scaly skin lesions on the ears (Fig.1C). The skin of the paws and tails showed the typical signs ofpsoriasis macroscopically as well as in histological analysis [sup-porting information (SI) Fig. S1 A and B]. In contrast, Cre� miceinjected with either isotype control IgG (n � 3) or anti-VEGF (n �2) did not show any skin abnormalities (Figs. 1B and S1B).However, systemic treatment with the anti-VEGF antibody for 8days resulted in an almost complete reduction of skin inflammation,
scaliness, and swelling in 5 of 12 treated mice (referred to as strongresponders, ‘‘DKO* anti-VEGFs’’; Fig. 1E). Six of 12 treated miceshowed a moderate anti-inflammatory response in the ears (re-ferred to as moderate responders, ‘‘DKO* anti-VEGFm’’; Fig. 1D).No major response was observed in only 1 mouse. The differencesbetween strong responders (5 of 12 mice) versus moderate respond-ers (6 of 12 mice) to the anti-VEGF treatment might result from thevariability observed in biological systems or might be explained bythe complexity of the phenotype and the mixed genetic background.The anti-VEGF treatment of DKO* mice also ameliorated thepsoriasis-like symptoms in the paws and in the tails (Fig. S1 A andB). Dramatically reduced redness and swelling of the toes wasobserved in anti-VEGF–treated mice (Fig. S1A). These findingsindicate that inhibition of the angiogenic growth factor VEGF isable to reverse the psoriasis-like skin phenotype including theepidermal abnormalities.
Anti-VEGF Treatment Normalizes the Epidermal Architecture in In-flamed Skin. To characterize better the efficacy of systemic VEGFblockade in reducing psoriasis-like skin inflammation, histolog-ical analyses were performed on ear sections obtained fromDKO* mice and Cre� littermate controls treated with anti-VEGF or with isotype control IgG. After 8 days of treatment,H&E-stained sections revealed the typical histopathologicalsigns of the psoriasis-like phenotype in the IgG-treated DKO*mice, i.e., acanthosis (thickened epidermis), hyperkeratosis(thickening of the stratum corneum), parakeratosis (retention ofnuclei in the stratum corneum), epidermal rete-ridge projectionsinto the dermis, and microabscesses (Fig. 1G). In contrast,systemic inhibition of VEGF led to a noticeable reduction in thepsoriasis-like histological features (Fig. 1 H and I).
The epidermal hyperproliferation marker keratin 6 is normallyabsent from interfollicular epidermis, whereas keratin 10 is ex-pressed in the suprabasal layers of the normal epidermis. Duringpsoriatic hyperproliferation of keratinocytes, both keratin 6 andkeratin 10 display a much broader staining pattern. Treatment withthe anti-VEGF antibody for 8 days normalized the expression ofboth keratin 6 (Fig. 1 L and M) and keratin 10 (Fig. 1 P and Q) toa staining pattern much more similar to that observed in unin-flamed epidermis (Fig. 1 J and N), unlike the inflamed epidermisof isotype control-treated mice (Fig. 1 K and O). After anti-VEGFtreatment, loricrin, a marker of epidermal cornification, was re-stricted largely to the upper granular layer (Fig. 1 T and U), astypically observed in normal epidermis (Fig. 1R). By contrast,loricrin expression was increased, was more focal, and was presentin several keratinocyte layers in the ears of isotype control-treatedmice (Fig. 1S). Thus, inhibition of VEGF normalized the epidermalskin architecture in this mouse model of psoriasis.
As reported earlier, deletion of JunB and c-Jun in the epidermisleads to up-regulation of the chemotactic proteins S100A8 andS100A9 in the epidermis before the onset of disease symptoms (23).To corroborate the observation in DKO* mice, we analyzedsections for up-regulation of the S100A8 and S100A9 proteins (Fig.2 B–D and F–H). Both groups, DKO* IgG-treated and DKO*anti-VEGF–treated, displayed the characteristic up-regulation ofthese 2 proteins in the epidermis. Quantification of S100A8 levelsby Western blot showed only a slight reduction of S100A8 proteinin the epidermis of anti-VEGF–treated mice compared with DKO*IgG–treated mice (Fig. 2 S and T). Furthermore, the apparentincrease of S100A8� cells in the dermis of DKO*IgG-treated micewas region specific and was reduced in anti-VEGF–treated mice(Fig. 2 F–H).
To investigate the proliferative activity in the skin, the number ofKi67� proliferating epidermal keratinocytes and dermal cells wasmeasured and was found to be markedly reduced in both moderateand strong anti-VEGF responders (Fig. 2 K, L, and Q), suggestingreduced proliferative activity of keratinocytes after anti-VEGFtreatment. Staining for phosphorylation of histone H3 serine 10
Fig. 1. Systemic inhibition of VEGF alleviates the psoriasis-like phenotypeinduced by epidermal deletion of JunB and c-Jun. (A) The schematic represen-tation of the experimental procedure. (B–E) Ears of control IgG-treated DKO*mice showed prominent inflammatory, scaly skin lesions. Mice that respondedonly moderately to anti-VEGF antibody treatment (DKO* anti-VEGFm; n � 6)displayedamildpsoriaticphenotype (D),whereas theear skinappeared largelynormal in mice with a strong response to the anti-VEGF treatment (DKO*anti-VEGFs: n � 5) (E). A control IgG-injected Cre� mouse is shown for compar-ison (n � 3). (F–I) H&E staining of control IgG-treated mice (F) and DKO*IgG-treatedmice (G) reveals thetypical signsof thepsoriasis-likephenotype. (H,I)Anti-VEGF–treatedmiceshowedsignificant improvement inthesesymptoms.(Scalebars,100 �m.) (J–U)NormalizedkeratinocytedifferentiationafterG6–31treatment. Immunofluorescence analyses showed strong induction of the hy-perproliferation marker keratin 6 (K) and altered expression patterns of theepidermal differentiation-related proteins keratin 10 (O) and loricrin (S) in theisotype control IgG-treated DKO* mice, whereas after anti-VEGF treatment theexpression patterns normalized showing reduced expression of keratin 6 (L, M)and restriction of the expression of keratin 10 (P, Q) and loricrin (T, U) to thesuprabasal layers of the epidermis. A normal control mouse is shown forcomparison (J, N, R). Blood vessels (MECA-32�) are shown in red; nuclei arestained in blue (Hoechst 33342 stain). (Scale bars, 50 �m.)
Schonthaler et al. PNAS � December 15, 2009 � vol. 106 � no. 50 � 21265
IMM
UN
OLO
GY
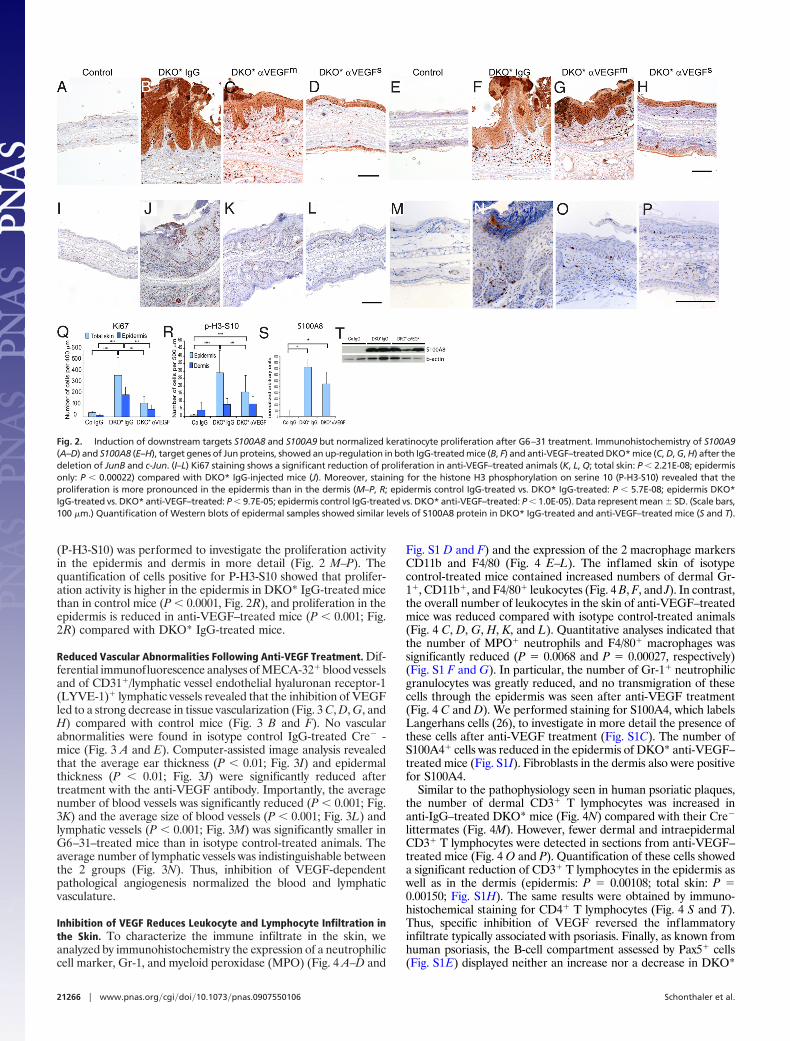
(P-H3-S10) was performed to investigate the proliferation activityin the epidermis and dermis in more detail (Fig. 2 M–P). Thequantification of cells positive for P-H3-S10 showed that prolifer-ation activity is higher in the epidermis in DKO* IgG-treated micethan in control mice (P � 0.0001, Fig. 2R), and proliferation in theepidermis is reduced in anti-VEGF–treated mice (P � 0.001; Fig.2R) compared with DKO* IgG-treated mice.
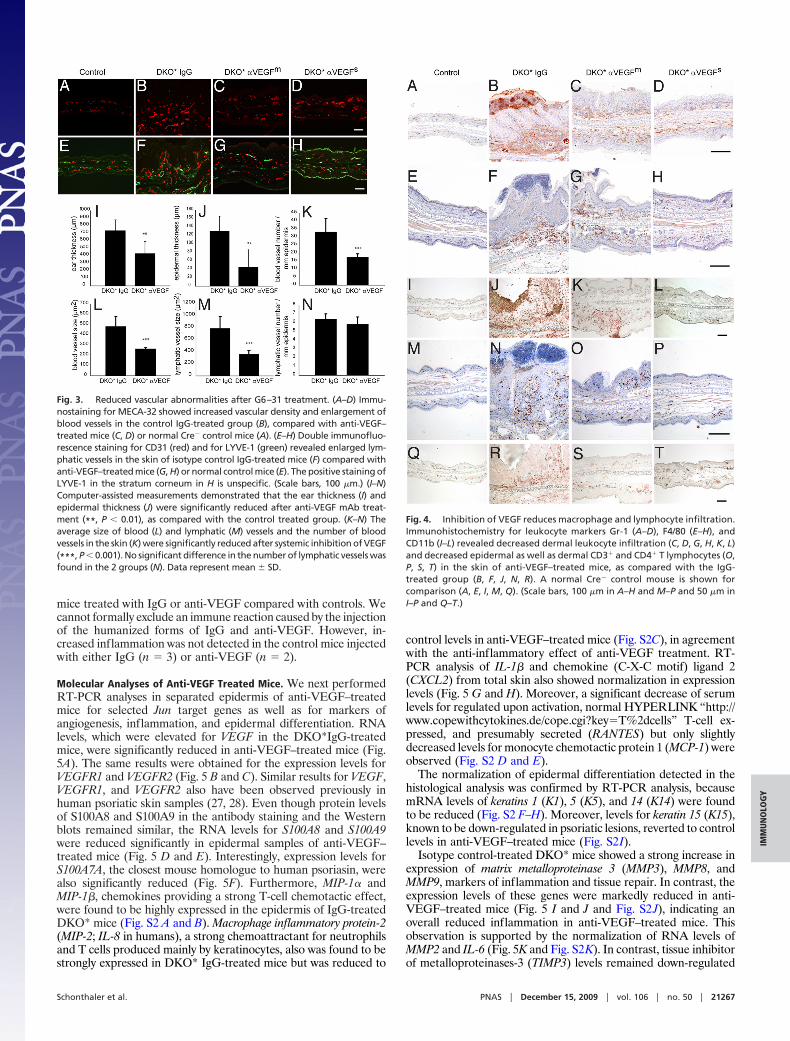
Reduced Vascular Abnormalities Following Anti-VEGF Treatment. Dif-ferential immunofluorescence analyses of MECA-32� blood vesselsand of CD31�/lymphatic vessel endothelial hyaluronan receptor-1(LYVE-1)� lymphatic vessels revealed that the inhibition of VEGFled to a strong decrease in tissue vascularization (Fig. 3 C, D, G, andH) compared with control mice (Fig. 3 B and F). No vascularabnormalities were found in isotype control IgG-treated Cre� -mice (Fig. 3 A and E). Computer-assisted image analysis revealedthat the average ear thickness (P � 0.01; Fig. 3I) and epidermalthickness (P � 0.01; Fig. 3J) were significantly reduced aftertreatment with the anti-VEGF antibody. Importantly, the averagenumber of blood vessels was significantly reduced (P � 0.001; Fig.3K) and the average size of blood vessels (P � 0.001; Fig. 3L) andlymphatic vessels (P � 0.001; Fig. 3M) was significantly smaller inG6–31–treated mice than in isotype control-treated animals. Theaverage number of lymphatic vessels was indistinguishable betweenthe 2 groups (Fig. 3N). Thus, inhibition of VEGF-dependentpathological angiogenesis normalized the blood and lymphaticvasculature.
Inhibition of VEGF Reduces Leukocyte and Lymphocyte Infiltration inthe Skin. To characterize the immune infiltrate in the skin, weanalyzed by immunohistochemistry the expression of a neutrophiliccell marker, Gr-1, and myeloid peroxidase (MPO) (Fig. 4 A–D and
Fig. S1 D and F) and the expression of the 2 macrophage markersCD11b and F4/80 (Fig. 4 E–L). The inflamed skin of isotypecontrol-treated mice contained increased numbers of dermal Gr-1�, CD11b�, and F4/80� leukocytes (Fig. 4 B, F, and J). In contrast,the overall number of leukocytes in the skin of anti-VEGF–treatedmice was reduced compared with isotype control-treated animals(Fig. 4 C, D, G, H, K, and L). Quantitative analyses indicated thatthe number of MPO� neutrophils and F4/80� macrophages wassignificantly reduced (P � 0.0068 and P � 0.00027, respectively)(Fig. S1 F and G). In particular, the number of Gr-1� neutrophilicgranulocytes was greatly reduced, and no transmigration of thesecells through the epidermis was seen after anti-VEGF treatment(Fig. 4 C and D). We performed staining for S100A4, which labelsLangerhans cells (26), to investigate in more detail the presence ofthese cells after anti-VEGF treatment (Fig. S1C). The number ofS100A4� cells was reduced in the epidermis of DKO* anti-VEGF–treated mice (Fig. S1I). Fibroblasts in the dermis also were positivefor S100A4.
Similar to the pathophysiology seen in human psoriatic plaques,the number of dermal CD3� T lymphocytes was increased inanti-IgG–treated DKO* mice (Fig. 4N) compared with their Cre�
littermates (Fig. 4M). However, fewer dermal and intraepidermalCD3� T lymphocytes were detected in sections from anti-VEGF–treated mice (Fig. 4 O and P). Quantification of these cells showeda significant reduction of CD3� T lymphocytes in the epidermis aswell as in the dermis (epidermis: P � 0.00108; total skin: P �0.00150; Fig. S1H). The same results were obtained by immuno-histochemical staining for CD4� T lymphocytes (Fig. 4 S and T).Thus, specific inhibition of VEGF reversed the inflammatoryinfiltrate typically associated with psoriasis. Finally, as known fromhuman psoriasis, the B-cell compartment assessed by Pax5� cells(Fig. S1E) displayed neither an increase nor a decrease in DKO*
Fig. 2. Induction of downstream targets S100A8 and S100A9 but normalized keratinocyte proliferation after G6–31 treatment. Immunohistochemistry of S100A9(A–D) and S100A8 (E–H), target genes of Jun proteins, showed an up-regulation in both IgG-treated mice (B, F) and anti-VEGF–treated DKO* mice (C, D, G, H) after thedeletion of JunB and c-Jun. (I–L) Ki67 staining shows a significant reduction of proliferation in anti-VEGF–treated animals (K, L, Q; total skin: P � 2.21E-08; epidermisonly: P � 0.00022) compared with DKO* IgG-injected mice (J). Moreover, staining for the histone H3 phosphorylation on serine 10 (P-H3-S10) revealed that theproliferation is more pronounced in the epidermis than in the dermis (M–P, R; epidermis control IgG-treated vs. DKO* IgG-treated: P � 5.7E-08; epidermis DKO*IgG-treated vs. DKO* anti-VEGF–treated: P � 9.7E-05; epidermis control IgG-treated vs. DKO* anti-VEGF–treated: P � 1.0E-05). Data represent mean � SD. (Scale bars,100 �m.) Quantification of Western blots of epidermal samples showed similar levels of S100A8 protein in DKO* IgG-treated and anti-VEGF–treated mice (S and T).
21266 � www.pnas.org�cgi�doi�10.1073�pnas.0907550106 Schonthaler et al.
mice treated with IgG or anti-VEGF compared with controls. Wecannot formally exclude an immune reaction caused by the injectionof the humanized forms of IgG and anti-VEGF. However, in-creased inflammation was not detected in the control mice injectedwith either IgG (n � 3) or anti-VEGF (n � 2).
Molecular Analyses of Anti-VEGF Treated Mice. We next performedRT-PCR analyses in separated epidermis of anti-VEGF–treatedmice for selected Jun target genes as well as for markers ofangiogenesis, inflammation, and epidermal differentiation. RNAlevels, which were elevated for VEGF in the DKO*IgG-treatedmice, were significantly reduced in anti-VEGF–treated mice (Fig.5A). The same results were obtained for the expression levels forVEGFR1 and VEGFR2 (Fig. 5 B and C). Similar results for VEGF,VEGFR1, and VEGFR2 also have been observed previously inhuman psoriatic skin samples (27, 28). Even though protein levelsof S100A8 and S100A9 in the antibody staining and the Westernblots remained similar, the RNA levels for S100A8 and S100A9were reduced significantly in epidermal samples of anti-VEGF–treated mice (Fig. 5 D and E). Interestingly, expression levels forS100A7A, the closest mouse homologue to human psoriasin, werealso significantly reduced (Fig. 5F). Furthermore, MIP-1� andMIP-1�, chemokines providing a strong T-cell chemotactic effect,were found to be highly expressed in the epidermis of IgG-treatedDKO* mice (Fig. S2 A and B). Macrophage inflammatory protein-2(MIP-2; IL-8 in humans), a strong chemoattractant for neutrophilsand T cells produced mainly by keratinocytes, also was found to bestrongly expressed in DKO* IgG-treated mice but was reduced to
control levels in anti-VEGF–treated mice (Fig. S2C), in agreementwith the anti-inflammatory effect of anti-VEGF treatment. RT-PCR analysis of IL-1� and chemokine (C-X-C motif) ligand 2(CXCL2) from total skin also showed normalization in expressionlevels (Fig. 5 G and H). Moreover, a significant decrease of serumlevels for regulated upon activation, normal HYPERLINK “http://www.copewithcytokines.de/cope.cgi?key�T%2dcells” T-cell ex-pressed, and presumably secreted (RANTES) but only slightlydecreased levels for monocyte chemotactic protein 1 (MCP-1) wereobserved (Fig. S2 D and E).
The normalization of epidermal differentiation detected in thehistological analysis was confirmed by RT-PCR analysis, becausemRNA levels of keratins 1 (K1), 5 (K5), and 14 (K14) were foundto be reduced (Fig. S2 F–H). Moreover, levels for keratin 15 (K15),known to be down-regulated in psoriatic lesions, reverted to controllevels in anti-VEGF–treated mice (Fig. S2I).
Isotype control-treated DKO* mice showed a strong increase inexpression of matrix metalloproteinase 3 (MMP3), MMP8, andMMP9, markers of inflammation and tissue repair. In contrast, theexpression levels of these genes were markedly reduced in anti-VEGF–treated mice (Fig. 5 I and J and Fig. S2 J), indicating anoverall reduced inflammation in anti-VEGF–treated mice. Thisobservation is supported by the normalization of RNA levels ofMMP2 and IL-6 (Fig. 5K and Fig. S2K). In contrast, tissue inhibitorof metalloproteinases-3 (TIMP3) levels remained down-regulated
Fig. 3. Reduced vascular abnormalities after G6–31 treatment. (A–D) Immu-nostaining for MECA-32 showed increased vascular density and enlargement ofblood vessels in the control IgG-treated group (B), compared with anti-VEGF–treated mice (C, D) or normal Cre� control mice (A). (E–H) Double immunofluo-rescence staining for CD31 (red) and for LYVE-1 (green) revealed enlarged lym-phatic vessels in the skin of isotype control IgG-treated mice (F) compared withanti-VEGF–treated mice (G, H) or normal control mice (E). The positive staining ofLYVE-1 in the stratum corneum in H is unspecific. (Scale bars, 100 �m.) (I–N)Computer-assisted measurements demonstrated that the ear thickness (I) andepidermal thickness (J) were significantly reduced after anti-VEGF mAb treat-ment (**, P � 0.01), as compared with the control treated group. (K–N) Theaverage size of blood (L) and lymphatic (M) vessels and the number of bloodvessels in the skin (K) were significantly reduced after systemic inhibition of VEGF(***, P � 0.001). No significant difference in the number of lymphatic vessels wasfound in the 2 groups (N). Data represent mean � SD.
Fig. 4. Inhibition of VEGF reduces macrophage and lymphocyte infiltration.Immunohistochemistry for leukocyte markers Gr-1 (A–D), F4/80 (E–H), andCD11b (I–L) revealed decreased dermal leukocyte infiltration (C, D, G, H, K, L)and decreased epidermal as well as dermal CD3� and CD4� T lymphocytes (O,P, S, T) in the skin of anti-VEGF–treated mice, as compared with the IgG-treated group (B, F, J, N, R). A normal Cre� control mouse is shown forcomparison (A, E, I, M, Q). (Scale bars, 100 �m in A–H and M–P and 50 �m inI–P and Q–T.)
Schonthaler et al. PNAS � December 15, 2009 � vol. 106 � no. 50 � 21267
IMM
UN
OLO
GY

in DKO* anti-VEGF–treated mice (Fig. S2L), as expected becauseof the deletion of c-Jun and JunB. Most importantly, RNA levels ofcytokines such as TNF� and IL-23, known from human data to behighly important in the pathophysiology of psoriasis (29), weresignificantly reduced in anti-VEGF–treated mice but were stronglyincreased in DKO* anti-IgG–treated mice. Notably, TNF�, IL-23,IL-23R, IL-22, and IL-12p40 were reduced to normal levels afteranti-VEGF treatment (Fig. 5 L–P). Therefore, anti-VEGF treat-ment of mutant mice led to the normalization of the IL-23/T-helper-17 (Th17) axis and consequently to an amelioration of thephenotype.
DiscussionPsoriasis is a common inflammatory skin disease that is heteroge-neous, and its pathogenesis still is not well understood. Importantly,therapeutic interventions currently are limited and are restricted tothe treatment of symptoms rather than targeting the mechanisms
underlying the disease. Our study, employing a therapeutical design,reveals that systemic administration of an anti-VEGF antibodyeffectively inhibited pathological angiogenesis. Characteristicpsoriasis-like symptoms observed in the skin lesions of these mice,such as epidermal hyperplasia with altered differentiation, inflam-matory cell infiltration, vascular abnormalities, and elevated levelsof IL-23/Th17-related cytokines, were markedly reduced followingtreatment with anti-VEGF antibody.
In cutaneous lesions and in the serum of psoriatic patients,elevated levels of IL-23 and Th17-related cytokines were detected,and the development and maintenance of Th17 cells has beenshown to be linked to IL-23, a key cytokine in the development ofautoimmunity (30, 31). Injection of IL-23 in mice leads to erythema,mixed dermal infiltrate, and epidermal hyperplasia associated withparakeratosis comprising histopathological features resemblingpsoriasis. These responses were shown to be mediated by TNF� andIL-20R2 (32). Moreover, variants of the IL-23R gene are associatedwith psoriasis (33), demonstrating the relevance of IL-23/IL-23Rsignaling in psoriasis. The common subunit IL-12/IL-23, IL12p40,and IL-23p19 were highly expressed in psoriatic skin lesions ascompared with uninvolved skin (34). Moreover, ectopic expressionof IL12p40 from the keratin 14 promoter led to enhanced expres-sion of IL-23p19 in mouse keratinocytes (35).
A strong increase of TNF� is observed following deletion of JunBand c-Jun in epidermal tissues during pre- and postnatal develop-ment (Fig. 5L and refs. 23 and 36). Because TNF� can regulate IL-6as well as MMP9 levels (37–39), the increase in TNF� could beresponsible for the increased VEGF levels in the DKO* mice. Inmice, the embryonic fibroblasts JunB and c-Jun are required forhypoglycemia-induced expression of VEGF (40). However, wefavor the hypothesis that induction of VEGF in keratinocytes ismediated instead by a cytokine-dependant mechanism.
Besides cytokines, chemoattractant molecules such as S100A8and S100A9 are increased in DKO* mice at the protein level (Fig.2 B, F, S, and T). Interestingly, RNA levels of S100A8 and S100A9appear to be decreased in anti-VEGF–treated DKO* mice (Fig. 5D and E); this reduction might be caused by a posttranslationalmechanism or by an unknown negative feedback loop.
Our findings strongly indicate that therapeutic intervention at thelevel of the vasculature can be sufficient to reduce the immune-mediated and epidermal components of the disease. We speculatethat the anti-inflammatory effects of anti-VEGF antibody treat-ment block VEGF effects in endothelial cells and also in other celltypes in the skin. Besides its role in angiogenesis, VEGF is awell-documented regulator of various aspects of the inflammatoryresponse, wound healing, and tumor growth (41). VEGF induceshyperpermeability of blood vessels, leading to tissue edema duringinflammation (42). Furthermore, chronic overexpression of VEGFin the skin of K14-VEGF transgenic mice promoted leukocyterolling and adhesion in skin microvessels, probably resulting fromthe increased expression of adhesion molecules such as E- andP-selectin (43). In addition to endothelial cells, monocytes andmacrophages also express VEGFR-1, and VEGF can chemoattractthese cells (44). Thus, the inhibition of these additional activities ofVEGF related to attracting and activating immune cells by systemictreatment with an anti-VEGF antibody probably contributed to thediminished inflammatory cell infiltration observed in the skin.
Recent evidence indicates that, lymphangiogenesis, as well asangiogenesis, is involved in certain inflammatory and autoimmuneconditions. It is proposed that lymphatic vessel formation andremodeling participates in the regulation of the immune responseby affecting the transport of antigens and leukocytes to draininglymph nodes (45). It was recently shown that VEGF can promotelymphangiogenesis associated with tumor formation, tissue repair,and inflammation (7, 46, 47). However, the exact role of lymphaticvessels in inflammation remains to be established.
Whether anti-angiogenic therapy for benign diseases such aspsoriasis or rheumatoid arthritis is a viable option will require
Fig. 5. Expression analyses of genes involved in inflammation and epidermaldifferentiation. RT-PCR analysis of mRNA expression levels revealed strong ex-pression of VEGF A, VEGFR1, and VEGFR2 in DKO* IgG-treated mice which wasreduced in DKO* anti-VEGF–treated mice (A–C). S100A8 (D) and A9 (E) wereincreased in DKO* mice and decreased in anti-VEGF–injected mice, perhapsbecause of a posttranslational mechanism or a negative feedback loop. Theexpression levels of epidermal samples of the following genes (especially cyto-kines related to the IL-23/Th17 axis and the related receptors) reflect a normal-ization of the inflammatory process in anti-VEGF–injected mice: S100A7A (F),MMP3 (I), MMP9 (J), MMP2 (K), TNF� (L), IL-23 (M), IL-23R (N), IL-22 (O), andIL-12p40 (P).RNAexpression levelsof total skinfor IL-1� (G)andCXCL2 (H)alsoarereduced back to control levels in anti-VEGF–injected mice. Controls are set to 1,and the y axis shows relative expression levels. Control IgG-treated: n � 3; DKO*IgG-treated: n � 3–4; DKO* anti-VEGF–treated: n � 4–5. D–F represent data(mean � SD) from control IgG-treated: n � 3; DKO* IgG-treated: n � 5–6; DKO*anti-VEGF–treated: n � 10–12.
21268 � www.pnas.org�cgi�doi�10.1073�pnas.0907550106 Schonthaler et al.

additional experiments in other model systems. Moreover, topicalapplication might provide a valuable alternative to overcomeunwanted side effects. Nevertheless, these findings provide a proofof concept for the proposition that inflammatory diseases can betreated by anti-angiogenic therapies.
Materials and MethodsMouse Strains and Treatment. Mice carrying JunB and c-Jun alleles flanked byloxP sites and the K5-CreERT allele were used in this study (23). DKO* mice wereinjected i.p. with 1 mg tamoxifen (Sigma) for 5 consecutive days to induce thedeletion of the 2 genes in the epidermis. Mice showing the first signs of thepsoriasis-like phenotype were injected every 48 h with 4 doses of the monoclonalantibody G6–31 (25) or a human control IgG. All mouse experiments wereperformed in accordance with local and institutional regulations and licenses.
Immunohistochemistry and Immunofluorescence. Mice were killed, and earswere collected and cut in half. One half of each ear was processed for paraffinsections. The other half was embedded in optimal cutting temperature (OCT)compound, and 7-�m cryostat sections were cut. Immunohistological analyseswere performed as described previously (7, 48).
For paraffin embedding, tissues were fixed in 3.7% paraformaldehyde inPBS at 4 °C overnight. For H&E staining, 3-�m sections of formalin-fixedsamples were processed according to standard procedures. Staining for MPO(Dako) and immunohistochemistry with antibodies against CD3 (Vector Lab-oratories), S100A8, and S100A9 (both Santa Cruz) was performed as describedpreviously (23, 49, 50) or according to the manufacturer’s protocol.
Additional Information. For information on computer-assisted morphometricanalyses, statistical analysis, and quantitative RT-PCR, see SI Materials andMethods.
ACKNOWLEDGMENTS. We thank Maria Helena Idarraga-Amado, Jessica Rubio,and Jeannette Scholl for their excellent technical support and Drs. Latifa Bakiri,Aline Bozec, Juan Guinea Viniegra, Mirna Perez-Moreno, Maria Sibilia, and ErwinTschachler for discussions and for critically reading the manuscript. This work wassupported by National Institutes of Health Grant CA69184, Swiss National FundGrant 3100A0–108207, Austrian Science Foundation Grant NFN S94-SP11, theCommissionoftheEuropeanCommunitiesGrantLSHC-CT-2005–518178(toM.D.)and the Banco Bilbao Vizcaya Argentaria (BBVA)-Foundation. The ResearchInstitute of Molecular Pathology is supported by Boehringer Ingelheim Inc.
1. Carmeliet P (2003) Angiogenesis in health and disease. Nat Med 9(6):653–660.2. Pober JS, Sessa WC (2007) Evolving functions of endothelial cells in inflammation.
Nature Reviews Immunology 7(10):803–815.3. Leong TT, Fearon U, Veale DJ (2005) Angiogenesis in psoriasis and psoriatic arthritis:
Clues to disease pathogenesis. Current Rheumatology Reports 7(4):325–329.4. Danese S, et al. (2006) Angiogenesis as a novel component of inflammatory bowel
disease pathogenesis. Gastroenterology 130(7):2060–2073.5. Bainbridge J, Sivakumar B, Paleolog E (2006) Angiogenesis as a therapeutic target in
arthritis: Lessons from oncology. Current Pharmaceutical Design 12(21):2631–2644.6. Xia YP, et al. (2003) Transgenic delivery of VEGF to mouse skin leads to an inflammatory
condition resembling human psoriasis. Blood 102(1):161–168.7. Kunstfeld R, et al. (2004) Induction of cutaneous delayed-type hypersensitivity reac-
tions in VEGF-A transgenic mice results in chronic skin inflammation associated withpersistent lymphatic hyperplasia. Blood 104(4):1048–1057.
8. Detmar M, et al. (1994) Overexpression of vascular permeability factor/vascular endo-thelial growth factor and its receptors in psoriasis. J Exp Med 180(3):1141–1146.
9. Nielsen HJ, et al. (2002) Elevated plasma levels of vascular endothelial growth factorand plasminogen activator inhibitor-1 decrease during improvement of psoriasis.Inflammation Research 51(11):563–567.
10. Young HS, Summers AM, Bhushan M, Brenchley PE, Griffiths CE (2004) Single-nucleotide polymorphisms of vascular endothelial growth factor in psoriasis of earlyonset. J Invest Dermatol 122(1):209–215.
11. Halin C, et al. (2008) Inhibition of chronic and acute skin inflammation by treatmentwith a vascular endothelial growth factor receptor tyrosine kinase inhibitor. Am JPathol 173(1):265–277.
12. Krueger GG, et al. (2007) A human interleukin-12/23 monoclonal antibody for thetreatment of psoriasis. N Engl J Med 356(6):580–592.
13. Ding C, Xu J, Li J (2008) ABT-874, a fully human monoclonal anti-IL-12/IL-23 antibodyfor the potential treatment of autoimmune diseases. Current Opinion in Investiga-tional Drugs 9(5):515–522.
14. Leonardi CL, et al. (2008) Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a random-ised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet 371(9625):1665–1674.
15. Menter A, Griffiths CE (2007) Current and future management of psoriasis. Lancet370(9583):272–284.
16. Halverstam CP, Lebwohl M (2008) Nonstandard and off-label therapies for psoriasis.Clinics in Dermatology 26(5):546–553.
17. Kerr DJ (2004) Targeting angiogenesis in cancer: Clinical development of bevacizumab.Nature Clinical Practice Oncology 1(1):39–43.
18. Folkman J (2006) Angiogenesis. Annu Rev Med 57 S1–18.19. Akman A, Yilmaz E, Mutlu H, Ozdogan M (2008) Complete remission of psoriasis
following bevacizumab therapy for colon cancer. Clin Exp Dermatol. 34(5):e202–204.20. Zenz R, Wagner EF (2006) Jun signalling in the epidermis: From developmental defects
to psoriasis and skin tumors. Int J Biochem Cell Biol 38(7):1043–1049.21. Zenz R, et al. (2003) c-Jun regulates eyelid closure and skin tumor development
through EGFR signaling. Dev Cell 4(6):879–889.22. Meixner A, et al. (2008) Epidermal JunB represses G-CSF transcription and affects
haematopoiesis and bone formation. Nat Cell Biol 10(8):1003–1011.23. Zenz R, et al. (2005) Psoriasis-like skin disease and arthritis caused by inducible
epidermal deletion of Jun proteins. Nature 437(7057):369–375.24. Zenz R, et al. (2008) Activator protein 1 (Fos/Jun) functions in inflammatory bone and
skin disease. Arthritis Research and Therapy10(1):201.25. Liang WC, et al. (2006) Cross-species vascular endothelial growth factor (VEGF)-
blocking antibodies completely inhibit the growth of human tumor xenografts andmeasure the contribution of stromal VEGF. J Biol Chem 281(2):951–961.
26. Zibert JR, Skov L, Thyssen JP, Jacobsen GK, Grigorian M (July 30, 2009) Significance ofthe S100A4 protein in psoriasis. J Invest Dermatol, 10.1038/jid.2009.206.
27. Man XY, Yang XH, Cai SQ, Yao YG, Zheng M (2006) Immunolocalization and expressionof vascular endothelial growth factor receptors (VEGFRs) and neuropilins (NRPs) onkeratinocytes in human epidermis. Molecular Medicine 12(7–8):127–136.
28. Man XY, Yang XH, Cai SQ, Bu ZY, Zheng M (2008) Overexpression of vascular endothelialgrowth factor (VEGF) receptors on keratinocytes in psoriasis: Regulated by calcium indepen-dent of VEGF. Journal of Cellular and Molecular Medicine 12(2):649–660.
29. Di Cesare A, Di Meglio P, Nestle FO (2009) The IL-23/Th17 axis in the immunopatho-genesis of psoriasis. J Invest Dermatol 129(6):1339–1350.
30. Bettelli E, et al. (2006) Reciprocal developmental pathways for the generation ofpathogenic effector TH17 and regulatory T cells. Nature 441(7090):235–238.
31. Kastelein RA, Hunter CA, Cua DJ (2007) Discovery and biology of IL-23 and IL-27: Related butfunctionally distinct regulators of inflammation. Annu Rev Immunol 25:221–242.
32. Chan JR, et al. (2006) IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2-dependent mechanisms with implications for psoriasis pathogenesis. J Exp Med203(12):2577–2587.
33. Nair RP, et al. (2008) Polymorphisms of the IL12B and IL23R genes are associated withpsoriasis. J Invest Dermatol 128(7):1653–1661.
34. Lee E, et al. (2004) Increased expression of interleukin 23 p19 and p40 in lesional skinof patients with psoriasis vulgaris. J Exp Med 199(1):125–130.
35. Kopp T, et al. (2003) IL-23 production by cosecretion of endogenous p19 and transgenicp40 in keratin 14/p40 transgenic mice: Evidence for enhanced cutaneous immunity.J Immunol 170(11):5438–5444.
36. Guinea-Viniegra J, et al. (2009) TNFalpha shedding and epidermal inflammation arecontrolled by Jun proteins. Genes Dev 23:2663–2674.
37. Scott KA, et al. (2004) TNF-alpha regulates epithelial expression of MMP-9 and integrinalphavbeta6 during tumour promotion. A role for TNF-alpha in keratinocyte migra-tion? Oncogene 23(41):6954–6966.
38. Rega G, et al. (2007) Vascular endothelial growth factor is induced by the inflammatorycytokines interleukin-6 and oncostatin m in human adipose tissue in vitro and inmurine adipose tissue in vivo. Arterioscler Thromb Vasc Biol 27(7):1587–1595.
39. Bergers G, et al. (2000) Matrix metalloproteinase-9 triggers the angiogenic switchduring carcinogenesis. Nat Cell Biol 2(10):737–744.
40. Textor B, Sator-Schmitt M, Richter KH, Angel P, Schorpp-Kistner M (2006) c-Jun and JunBare essential for hypoglycemia-mediated VEGF induction. Ann N Y Acad Sci 1091:310–318.
41. Rossiter H, et al. (2004) Loss of vascular endothelial growth factor a activity in murineepidermal keratinocytes delays wound healing and inhibits tumor formation. CancerRes 64(10):3508–3516.
42. Ferrara N, Gerber HP, LeCouter J (2003) The biology of VEGF and its receptors. Nat Med9(6):669–676.
43. Detmar M, et al. (1998) Increased microvascular density and enhanced leukocyterolling and adhesion in the skin of VEGF transgenic mice. J Invest Dermatol 111(1):1–6.
44. Sawano A, et al. (2001) Flt-1, vascular endothelial growth factor receptor 1, is a novelcell surface marker for the lineage of monocyte-macrophages in humans. Blood97(3):785–791.
45. Angeli V, et al. (2006) B cell-driven lymphangiogenesis in inflamed lymph nodesenhances dendritic cell mobilization. Immunity 24(2):203–215.
46. Hirakawa S, et al. (2005) VEGF-A induces tumor and sentinel lymph node lymphangio-genesis and promotes lymphatic metastasis. J Exp Med 201(7):1089–1099.
47. Hong YK, et al. (2004) VEGF-A promotes tissue repair-associated lymphatic vesselformation via VEGFR-2 and the alpha1beta1 and alpha2beta1 integrins. FASEB J18(10):1111–1113.
48. Traxler P, et al. (2004) AEE788: A dual family epidermal growth factor receptor/ErbB2and vascular endothelial growth factor receptor tyrosine kinase inhibitor with anti-tumor and antiangiogenic activity. Cancer Res 64(14):4931–4941.
49. Gebhardt C, et al. (2008) RAGE signaling sustains inflammation and promotes tumordevelopment. J Exp Med 205(2):275–285.
50. Hui L, Zatloukal K, Scheuch H, Stepniak E, Wagner EF (2008) Proliferation of humanHCC cells and chemically induced mouse liver cancers requires JNK1-dependent p21downregulation. J Clin Invest 118(12):3943–3953.
Schonthaler et al. PNAS � December 15, 2009 � vol. 106 � no. 50 � 21269
IMM
UN
OLO
GY