studies on genotoxicity and mutagenicity of certain pesticidesir.amu.ac.in/4769/1/ds 2504.pdf ·...
TRANSCRIPT

STUDIES ON GENOTOXICITY AND MUTAGENICITY OF CERTAIN PESTICIDES
By
SANGEETA SAXENA
Date :
Approved :
Supervisor
A DISSERTATION SUBMITTED IN PARTIAL FULFILMENT OF THE REQUIREMENTS FOR THE DEGREE OF
MASTER OF PHILOSOPHY IN BIOTECHNOLOGY OF THE ALIGARH MUSLIM UNIVERSITY, ALIGARH
1994

DS2504
;^^''i'^i>m A ijfj^
I? 3 FEB W<>

C E R T I F I C A T E
I certify that the work presented in the following
pages has been carried out by Ms Sangeeta Saxena and that it
is suitable for the award of M.Phil, degree in Biotechnology
of the Aligarh Muslim University, Aligarh.
/ ^ ^ v ^ ^ ^ (SAAD TAWAB)
Senior Lecturer Interdisciplinary Biotechnology Unit
Aligarh Muslim University Aligarh 202002 (U.P) INDIA

ACKNOWLEDGEMENT
I wish to express my deep sense of gratitude to my
supervisor Dr. Saad Tayyab for his able guidance , keen
intrest, moral support and help extended to me in the
accomplishment of this task.
I am extremely grateful to Dr. Javed flussarrat for
his invaluable guidance, constructive and intelligent
criticism and encouragement , throughout the course of
this study.
I wish to thank Prof. A. Salahuddin, Coordinator,
Interdisciplinary Biotechnology Unit, A.M.U. for
providing necessary research facilties.
I thank Dr. Arshad Rahman for the help I received
from him.
I also wish to express my heartfelt gratitude to
Dr. P.V. Sane and Dr. K.li. Srivastava, NBRI , Lucknow
for their cooperation and genuine help in the
compilation of this dissertation.
I sincerely acknowledge the cooperation which I
received from my seniors, Aftab Bhai, Sadhna apa, Zoya
apa, Rizwan Bhai, Shama apa and Pasha Bhai.
I owe a lot to my friends liani ,Fariha, Ronana,
Nishat, Sunita, Sakhra, Sadia, Moin, Mozzafar, Jalal,
Fahim, Madhvi, Vipin and Rizwan for their genuine help
and love they showered on me.
I wish to express mv deep sense of appreciation to
my friend and colleague, Ashok for his great help,
moral support and always being there at my hour of
need.
ili

I sincerely acknowledge my parents ,SORADH ,
GAIJRAV and RA.TEEV for the encouragempn t , TIP 1 p and
cooperation I received in my academic endeavours.
I also extend my thanks to Mr. Fazlur Rahman Khan,
Mr. Lai Mohammad Khan and the other non-teaching staff
of the department.
1 am also thankful to Mr. Meraj Uddin, Design
Centre, Aligarh for painstaking, and timely typing of
this manuscript.
(SANGEETA SAXENA )

CONTENTS
PAGE NO
CERTIFICATE ii
ACKNOWLEDGEMENT iii
LIST OF TABLES vi
LIST OF FIGURES vii
LIST OF ABBREVIATIONS viii
ABSTRACT ix
I. REVIEW OF LITERATURE 1
II. GENERAL MATERIALS AND METHODS 3 7
III. MUTAGENICITY ASSAY 45
IV. GENOTOXICITY ASSAY 58
V. DISCUSSION 68
VI. REFERENCES 73
VII. BIOGRAPHY 88

LIST OF TABLES
PAGE
Table I Categories of different pesticides. 5
Table II High-risk cancers in mortality 16
surveys of farmers.
Table III Types of cancer associated with 18
specific pesticide exposure.
Table IV The Salmonella typhimurium and 37
Escherichia coli strains used in
this study .
Table V Relevant genetic and biochemical 56
markers of Ames Strains.

LIST OF FIGURES
PAGE NO.
Fig. 1. Dose response curves of captan (-S9)
with Ames tester strains. 47
Fig. 2. Dose response curves of captan (+S9)
with Ames tester strains. 49
Fig. 3. Dose response curves of foltaf (-S9)
with Ames tester strains. 50
Fig. 4. Dose response curves of phosphamidon
(-S7) with Ames tester strains. 52
Fig. 5. Dose response curves of furadan (-S9)
with Ames tester strains. 53
Fig. 6. Survival of SOS defective Ej;_ col i
K-12 strains exposed to captan. 61
Fig. 7. Survival of SOS defective E^. col i
K-12 strains exposed to foltaf. 63
Fig. 8. Survival of SOS defective E^ coli
K-12 strains exposed to phosphamidon. 64
Fig. 9. Survival of SOS defective Ej_ coli
K-12 strains exposed to furadan. 66
Iviii

LIST OF ABBREVIATION
Amp Ampicillin resistant
CFU Colony forming units
g Gram
Kg Kilogram
L Litre
ug Microgram
ml Millilitre
mg Milligram
N.B. Nutrient Broth
SCE Sister chromatid exchange
STT Short term test
Str Streptomycin resistant
UV Ultra violet
V.B. Salts Vogel Bonner Salts
(vi

ABSTRACT
Among the plethora of environmental mutagens,
pesticides belong to a noxious chemical class of compounds,
posing a major threat to global environment and human
health. Many of them possess mutagenic, carcinogenic and
teratogenic properties. However, very few of them have been
tested for their mutagenic and/or carcinogenic properties.
It is generally believed that a large proportion of human
cancers are caused mainly due to the genotoxic chemicals
present in the environment. It is therefore necessary to
identify the entire spectrum of chemical carcinogens by the
short-term mutagenicity and carcinogenicity assays in
combination with epidemiological investigations.
It is against this background that we have
evaluated the genotoxic and mutagenic potential of four
pesticides viz., captan, foltaf,phosphamidon and furadan.
Our studies have provided a complete profile of the
mutagenic potential and provided insights into their
mechanism of repair in bacteria. Of the four pesticides
studied, captan showed highest mutagenic response followed
by foltaf, phosphamidon and furadan. Captan was found to be
most mutagenic in the absence of metabolic activation (-S9).
However, in the presence of S9 mix its mutagenic activity
dropped significantly. About 1.6 to 13 times higher
ix

mutagenic response of captan was observed compared to the
reported values. Foltaf, phosphamidon and furadan have been
detected as comparatively weak mutagens.
A clear role for pol^ recA and lexA genes in the
repair of the pesticide induced DNA damage is suggested by
studies on the genotoxic effect of the pesticides on DNA
repair defective mutants of E^ coli. The wild-type strain
AB1157 showed a decline in survival in the presence of
captan, foltaf and furadan. The mutants polA~, recA~ and
lexA~ also showed a significant decrease in survival on
exposure to the pesticides. Based on our repair studies, it
is suggested that inducible error-prone repair pathways
involving the recA and lexA genes are in operation in the
enzymatic removal of pesticide induced lesions in DNA.
x

CHAPTER -1
REVIEW OF LITERATURE

REVIEW OF LITERATURE
The human environment is being polluted
continuously by the ever increasing presence of toxic
pollutants, introduction of synthetic chemicals, rapid
and unplanned industrialization, deforestation, burning
of domestic and industrial fuels and many such
activities. While there are a large number of
environmental agents to which people are exposed in an
occupational setting, carcinogenic substances are also
entering our system through food, air and water(Gupta,
1989 ) .
The realization that many chemicals have a
potential of damaging the genetic material has led to
considerable scientific effort to detect, in the tiuman
environment, the noxious substances that might induce
mutations culminating in tumorigenesis. In recent years
it has become more and more substantiated that
mutagenic chemicals pose danger to the present
generation because of the high correlation between
mutagenicity and carcinogenicity (Gocke et. al.» 1981).
Many heavy metals, several food mycotoxins, pesticides
which may contaminate food , synthetic hormones and
other food additives have been found to be carcinogenic
(Ray, 1986 ; Culliney gt. al . 1992).
It has long been known that cancer can arise as a
result of exposure to a variety of agents, and studies
on the mechanism of the induction process have revealed

that pure chemicals themselves are able to cause cancer
(Miller, 1978). Damage to DNA by environmental mutagens
may be the main cause of death and disability in
advanced societies (Cairns, 1975). It is believed that
this damage, accumulating during the lifetime initiates
most human cancer and genetic defects and is quite
likely a major contributor to aging (Burnet, 1974) and
heart disease as well (Pearson et. aX> 1975).
Pesticides - Uses and Consequences :
A pesticide is defined as "any substance or
mixture of substances for destroying, preventing,
repelling or mitigating any pest, including chemicals
intended for use as a plant regulator, defoliant or
dessicant" (CFR, 1986). The purpose of a pesticide is
to control insect and animal vectors in human disease
and to increase agricultural productivity which has led
to a 307. increase in food production worldwide (Jain,
1992). It has been projected that there will be a 117.
increase in the overall share of pesticide use in
developing countries from the current 24% to 35% by
the year 2000 (Jain, 1992).
The benefits of pesticide use should also be
weighed against their harmful effects on human health,
biological interactions with non-target species,
pesticide resistance, and alterations to and /or
accumulation of pesticide in the environment. The

increased and continued use of pesticide is associated
with human health risks, adverse effect on non target
organisms and contamination of air, soil and water.
Less than 0.17. of the applied pesticides are estimated
to actually reach the target pests and therefore large
amounts are entering the environment and contaminating
soil and water resources (Young, 1987). In order to
minimize the risks associated with the use of
pesticides, Young (1987) advocated a better
understanding of pesticide pharmacology and toxicology,
pests and pest management, and potential hazards
associated with pesticide use.
All pesticides are considered to be toxic to
humans and other organisms. The degree of harmfulness
to humans and other living organisms depends on
pesticide characteristics, the amount or dosage of the
pesticide, and the duration of exposure or contact
time. Studies on mammalian toxicity including acute,
subchronic, chronic, mutagenicity and special tests
provided a data base for evaluating the hazards and
risk assessment associated with pesticides (Cardona,
1987).
Substantial amount of literature is available
indicating that the pesticide / insecticide load in the
food chain is increasing (Anon , 1973; Manske and
Johnson, 1977 ),due to their intensive, widespread and
indiscriminate use. Even the mother's milk has been

reported to contain certain level of pesticides.
Various fruits, vegetables and cereals also accumulate
biologically significant quantities of pesticides. (
Kannan et. al . 1992).
For this reason, the genotoxic and mutagenic
activities of a number of pesticides have been the
subject of extensive research. Such studies have a
predictive value for the potential of the pesticide to
cause cancer ( Innes et. ai.5l969; Ames et, al_»i*?75; Mahr
and Miltenburger, 1976; Simmon, 1978; Waters et. al ,
1980; Moriya et. al_, 1983; Rosenkranz et. al.. 1984),
Classification of Pesticides :
The term "pesticide" is a generic name, which
includes not only insecticides but also herbicides,
fungicides, rodenticides, fumigants , other growth
regulators and repellents. A list of some
representative pesticides is given in table I.

Table-I: Categories of different pesticides
(1) Insecticides
(a) Chlorinated hydrocarbons DDT, Aldrin, Dieldrin, Heptachlor, (organochlorines) Toxaphene, Chlordane, Methoxychlor
Endosulfan, BHC, Endrin.
(b) Organophosphates Malathion, Parathion, Diazinon, Guthion, Phosphamidon, Dimethoate, Monocrotophos, Dichlorvos.
(c) Carbamates Sevin, Zectran, Baygon, Temik, Furadan, Carbaryl, Isolan.
(d) Pyrethrins Allethrin and Cyclethrin
(2) Fungicides
(a) Dithiocarbamates
(b) Nitrogen containing compounds
Ferbam, Ziram and Maneb.
PhenyImercuric acid acetate, Triazines, Quinones, Heterocyclics, and some heavy metals.
(c) Organochlorine Hexachlorobenzene, Captan, Foltaf.
(3) Herbicides
(a) Phenoxy acids
(b) Aquatic herbicides
(4) Rodenticides :
(a) Inorganic
(b) Organic
2 , 4 - D and 2 , 4 , 5 - T .
E n d o t h a l and D i q u a t .
Barium carbonate.
Sodium F l u o r o a c e t a t e , W a r f a r i n ,
(5 ) Fuffligants ;

(a) Organic Methyl Bromide, CS2•
(6) Others :
(a) Plant growth regulators 1-Naphthylacetic acid.
(b) Repellents Deet, Dimethyl phthalate.
(von Rumker e^ al., 1975)

Pesticides as Carcinogens :
There are numerous reports on the mutagenicity of
captafol (Difolatan) and captan (Seller , 1973; Kada et.
al . 1974). Kada et. ai. (1974) have reported captafol to
be a weak base change mutagen. Moriya ejt al_ (1983) have
also reported the same for captafol by E.coli WP2 her
strain. Captafol, an alkylating agent, has produced
genotoxic effects in, in. vitro systems (IPCS, 1990a).
Captan, a broad spectrum fungicide is also an
alkylating agent which has demonstrated genotoxic
properties iri. vitro (IPCS, 1990b). Captan has also
been reported to be a weak base change mutagen (Siebert
et aX, 1970; Clarke, 1971; Brideges et. al_ , 1972).
Moriya et. al_ (1983) have also reported captan to be a
base-change mutagen in a test on WP2 her, TA1535,TA100,
TA1537, TA1538 and TA 98. Moriya et. al (1978) have
reported that mutagenicity of captan decreased by S9 as
also by blood and cysteine. Captan has been shown to
increase in mutagencity in TA98 by the plant extract
activation (S14), while in TA100 it decreased
(Rasquinha et. al., 1988). Captan induced mutation has
been observed with the recA assay of B. subtilis (Kada
et al. 1974) and the involvement of the error-prone
recombination - dependent repair system and the
recombination- dependent repair mechanisms in both the
£.. coli K-12 as well as E.coli WP2 repair systems
(Rashid and Ralph, 1986). A Microscreen phage-induction

assay performed by Houk and DeMarini (1787) showed that
captan was genotoxic and induced prophage. Reuber
(1989) has reviewed the carcinogenicity of captan in
mice and rats.
Carbofuran or Furadan, is a broad spectrum
carbamate and used as an insecticide and nematicide.
This insecticide has been shown to be a frame-shift
mutagen. Carbofuran has been tested for mutagenicity
and shown to be positive in frame shift type strains
(Moriya et. al_) 1983). Carbofuran and its metabolite 3-
ketocarbofuran have been reported to be acutely toxic
in the Microtox method of toxicity assessment (Kross et.
aJL, 1992).
Mutagenicity of organo-phosphorous pesticides ,
especially phosphamidon has been reported by Vishwanath
and Jamil (1986). This was found to be a weak mutagen
and caused base change mutations in the Ames Salmonella
assay. This pesticide has also been reported to be
mutagenic • in the E. coli WP2 Try-systems and
clastigenic in the human leucocyte tests system
(Patankar and Vaidya, 1980). Phosphamidon has been
shown to have mutagenic activity in. vitro and iji vivo
in mammalian systems (Georgian, 1975; Usha Rani et. al.
1980) .
It has been reported that mutagenic activation or
inactivation of the ingested chemicals can occur
through various metabolic processes in the animal body
8

(Lu et. aX» 1^72; Prins, 1978). In such transformation
of the chemicals, liver microsomal enzymes and
intestinal microflora play major roles (Prins, 1978).
Pednekar et. al_ (1987) have evaluated the mutagenic
activities of commonly used insecticides - endosulfan
(organochlorine), phosalone and malathion
(organophosphorus) and permethrin(pyrethroid) before
and after activation with fecal microbial extracts or
with liver post mitrochondrial fraction (39 fraction)
or rat in Ames test with Salmonella tvphimurium tester
strains TA97a, TA98 and TA100.
The microtitration SOS chromotest to study
genotoxicity, showed that nine pesticides were capable
of inducing SOS response in E.. col i. The SOS inducing
potency was least for captafol and captan (Xu and
Schurr, 1990). Wong et. aj. (1989) have reported that
diazinon, dimethoate, fenitrothion, malathion,
phenothoate and quinolphos were non-mutagenic in the
absence of S9, diazinon, dimethoate and quinolphos were
mutagenic in the absence of S9, while in the presence
of S9, diazinon, dimethoate and quinolphos were
mutagenic. Several new chemical mutagens in
pesticides have been detected by the rec-assay
procedures. The mutagenic specificities of Captafol;
DAPA (Dexon) and NBT (Nirit) were determined using gj.
coli WP2 and Ames Salmonella system by Kada et. al.
(1974).

Genotoxic and Non-genotoxic Carcinogens.
The classification of carcinogens into two
categories, genotoxic and non-genotoxic has been
proposed to give a logical foundation on which cancer
risk assessment is reasonably based. The term
"genotoxic carcinogen' indicates a chemical capable of
producing cancer by directly altering the genetic
material of target cells, while "non-genotoxic
carcinogen" represents a chemical capable of producing
cancer by some secondary mechanism not related to
direct gene damage. Most classical carcinogens such as
polycyclic aromatic hydrocarbons, nitroso-compounds,
aromatic amines and azo dyes belong to the genotoxic
category. However, sex hormones like estrogen and
progesterone can be raised as typical examples of non-
genotoxic carcinogns (Hayashi, 1972). While this
classification can not be applied, at present to all
instances due to insufficiencies in necessary
information or limited current knowledge on
carcinogenesis, the genotoxic versus non-genotoxic
carcinogen concept can still contribute to a basic
understanding of carcinogenic mechanisms and
establishment of strategies for cancer risk evaluation
from exposure to the environmental chemicals.
Carcinogenesis is a multi-step evolutionary
process occuring both in. vivo and in cell culture
(Newbold,1983; Boone et. art, 1992). The concept of
multistage carcinogenesis was initially developed
10

decades ago in rodent skin models (Rous and Kidd, 1941;
Mottram, 1744) and has since been shown to apply more
generally to cancers of many species and cell types. It
is now beleived that experimental carcinogenesis
proceeds through at least 3 distinct stages viz.
initiation, promotion and progression. The first stage
of carcinogenesis, initiation, results from
irreversible genetic alterations, most likely one or
more simple mutations, transversions, transitions
and/or small deletions in DNA. The reversible stage of
promotion does not involve changes in the structure of
DNA but rather in the expression of the genome mediated
through promoter—receptor interactions. The final
irreversible stage of progression is characterized by
karyotypic instability and malignant growth (Pitot,
1993). Oncogene research has provided new insight into
the cellular signal pathways, as well as genetic
alterations leading to neoplastic growth. The current
understanding of the processes regulating tumor
initiation and progression is based on the observation
that genes found in tumorigenic retroviruses, genes
from spontaneus human tumors that could transform
recipient murine or rat cells, and genes located at
sites of chromosomal translocations in human cancers
have similar origins (Bishop, 1991; Boyle, 1992;
Rozengurt, 1992) . These genes commonly called as
oncogenes (Garrett, 1986) are mutated forms of normal
11

cellular genes, designated protooncagenes (Temin, 1974)
which are components of the pathways regulating normal
cellular proliferation and differentiation. (Schmandt
and Mills, 1993). There are principally two sets of
oncogenes in the cell- those acting in the nucleus, and
those acting in the cytoplasm. The most thoroughly
studied cytoplasmic oncogene is the family of ras,
which is probably involved in over 307. of human cancer
(Ramel, 1991). The product of ras (p21 ) functions as
a GTPase in combination with other proteins, such as
GTPase-activating proteins, GAP. This process, implies
an amplification of the trans-membrane signals that
control cell proliferation and differentiation. Mutant
ras interrupts the GTP-GDP cycle, causing a
constitutive signal output (McCormick, 1989).
Activating mutations in ras are provided bv base
substitutions in DNA, located mainly in codons 12, 13
and 61 which coincide with regions for binding between
p21 and GAP (Khosravi-Far and Der, 1994). Nuclear
proto-oncogenes on the other hand are activated by
genetic alterations affecting the expression of the
genes. A continuous expression of the oncogene mvc has
been demonstrated in Burkitt's lymphoma as a result of
translocations of mvc to the vicinity of the promoter
of immunoglobulin genes (Ramel, 1991). Also, nuclear
oncogenes are often activated by gene amplification
causing increased output of the gene product.
12

Another critical molecular target during the
stages of carcinogenesis are tumor suppressor genes
(Marshall, 1991). In this instance, the mutational
alteration of the gene leads to a complete loss of
function or to a change in function such that its
activity in suppressing the neoplastic phenotype is
drastically altered. In the human, the best
documentation that a tissue -specific neoplasm arises
as a result of deletion or mutation of both copies of
the tumor supressor gene is the retinoblastoma
(Knudsen, 1971). Mutations in p53 tumor suppressor gene
have now turned out to be the most common genetic
alterations yet identified in human cancers (Ryberg et.
al , 1994; Deng et. al., 1994). The genetic alteration of
a single copy of a tumor suppressor gene may be a
critical event for the stage of initiation, with
alteration of the other allele occurring as the
determinant in the transition from the stage of
promotion to that of progression.
Although other potential critical molecular
targets exist, such as those involved in the cell cycle
(Nishimoto et. aX ,1992; Kaufmann and Kaufman,
1993),whose alterations result in neoplasia. What is
not clear, with few exceptions, are the ultimate number
and characteristics of critical molecuar alterations
that absolutely define the malignancy of a cell. Hence,
the molecular nature of neoplasia and its development
stand as a continuing challenge to the human intellect.
13

Carcinogenic Effects of Pesticides
In recent years there has been increasing concern
that exposure to pesticides may pose a potentially
serious cancer risk to the general population. The
currently available data are insufficient to estimate a
pesticide-related cancer rate for the general
population. However, a variety of pesticides are known
to cause cancer in laboratory animals (Blair et. al ,
1990; Hooever and Blair, 1991). In a recent survey of
47 pesticides evaluated for carcinogenecity, it was
found that six pesticides (13'/.) were positive in both
sexes in mice and rats, ten pesticides (217.) were
positive in both sexes of one species and six
pesticides (13"/.) were positive in one sex of atleast
one species (Hooever and Blair, 1991). Of 16 chemicals
that were positive in both sexes of atleast one
species, included organochlorine and organophosphate
insecticides, herbicides, fungicides and fumigants,
suggesting that no chemical class of pesticides can be
considered problem free. Similarly, the International
Agency for Research on Cancer recently reported that
43'/. of a wide variety of pesticides were carcinogenic
in humans (Blair et. al., 1990). With pesticides of all
the variant types showing some evidence of
carcinogenicity, the concern about human exposure seems
to be well founded. With regard to mechanisms of
carcinogenesis, a survey of the genotoxic activities of
65 pesticides by Garrett et. al_ (1987) revealed that 33
14

of the pesticides (54'/.) tested positive, including
organophosphates, thiocarbamate insecticides,
pyrethroid insecticides, and a variety of herbicides
and fungicides. However, some non-genotoxic pesticides
also may be involved in carcinogenesis via their
epigenetic properties, such as tumor promotion,
inhibition of intercellular communication, or induction
of peroxisome proliferations.
Substantial amount of epidemiologic data is
available on the carcinogenicity of pesticides based on
the persons employed in agricultural or related
occupation (Weisenburger, 1793). Although farmers
typically perform many tasks resulting in a variety of
exposures it is the pesticides that have received the
most attention. Table II shows the cancers that have
been associated with occupational exposures to specific
pesticides or class of pesticides (Blair and Zahm,
1991). The epidemiologic data linking specific
pesticide exposures to hematopeitic cancer (i.e., non-
Hodgkin's lymphoma, leukemia and multiple myeloma) are
the strongest compared to the data on soft tissue
sarcoma and Hodgkin's disease. Very little is known
about specific pesticides and cancer of the brain,
although ovarian carcinoma has been shown to be
associated with exposure to triazine herbicides (Donna
et al. 19B9) Neverthless, the leukemia and brain cancer
in children have also been reported to be associated
15
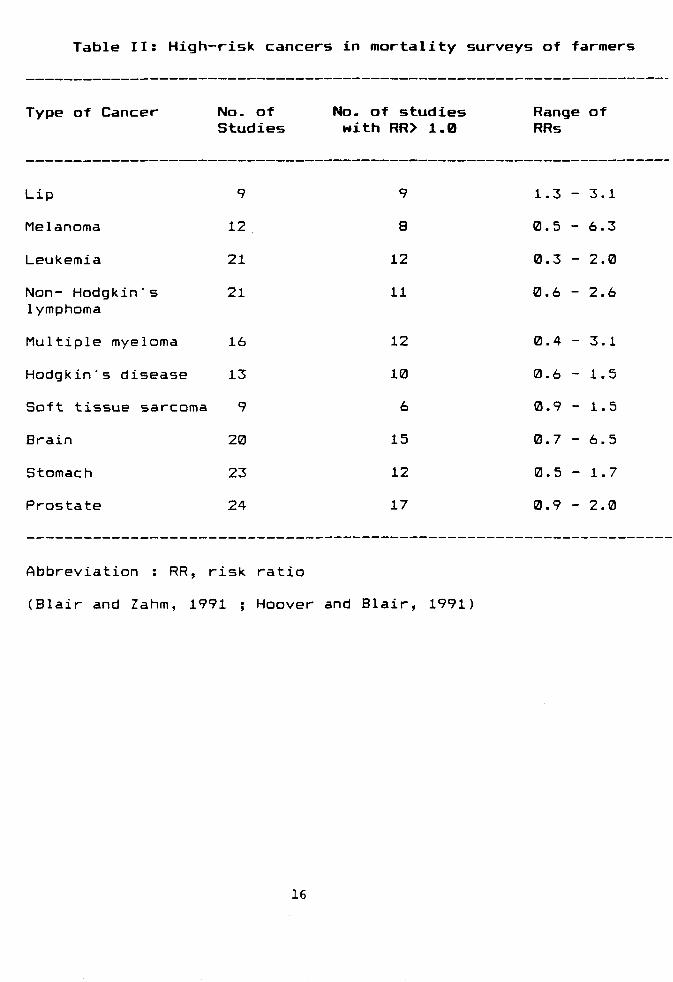
Table II: High-risk cancers in mortality surveys of farmers
Type of Cancer No. of Studies
No. of studies with RR> 1.0
Range of RRs
Lip
Melanoma
Leukemia
Non- Hodgkin's lymphoma
Multiple myeloma
Hodgkin's disease
Soft tissue sarcoma
Brain
Stomach
Prostate
9
12
21
21
16
13
9
20
23
24
9
8
12
11
12
10
6
15
12
17
1.3 - 3.1
0.5 - 6.3
0.3 - 2.0
0.6 - 2.6
0.4 - 3.1
0.6 - 1.5
0.9 - 1.5
0.7 - 6.5
0.5 - 1.7
0.9 - 2.0
Abbreviation : RR, risk ratio
(Blair and Zahm, 1991 ; Hoover and Blair, 1991
16

with parental employment in agriculture and exposure to
pesticides (Lowengart et. aj., 1987; WilkinS and sinks,
(1990). Table III shows a list of different types of
cancer associated with specific pesticide exposures.
17

Table -III Types of cancer associated with specific pesticide exposures.
Cancer Pesticides
Soft tissue sarcoma and lymphoma
Non-Hodgkin's lymphoma
Soft tissue sarcoma
Leukemia
Lung Carcinoma
Phenoxyacetic acid herbicides (2,4,D, 2,4,5-T), DDT
Organophosphate and Organochlorine insecticides, fumigants
Chlorophenols, animal insecticides
Dichlorvos, crotoxyphos, famphur, methoxychlor, pyrethrins, DDT, chlordane, heptachlor, lindane, ethylene oxide
Organochlorine insecticide (DDT), arsenicals
Ovarian Carcinoma Triazines
18

Immunologic Effects of Pesticides:
Although there is little evidence to suggest that
pesticides exposure compromise human health through
interference with immune system, however, Thomas et. ai.
(1990) reported that pesticides or pesticide
contaminants can modulate the immune response in
experimental animals. Sharma and Kour (1990) suggested
that contact dermatitis is a common disease caused by
pesticides and also the asthma-type reactions can be
triggered by pesticide exposures. Although altered
immunoglobulin and competent levels and changes in T-
cell populations have been reported in human exposed to
pesticides (Thomas et. aX> 1990), no adverse health
effects have been noted. However, impaired neutrophil
chemotaxis accompanied by an increase in respiratory
tract infections was correlated with the length of
occupational exposure to organophosphate pesticides
(Hermanowicz and Kossman, 1984). Further studies on the
immunological effects of pesticides in humans are
needed since depressed immunity is known to predispose
humans to a variety of cancers, including non-Hodgkin' s
lymphoma.
Carcinogenicity Testing System :
It has become increasingly apparent that the
traditional methods for identifying carcinogens by
using long term studies in rodents are unable to meet
the demands for a quick, sure and inexpensive
19

identification of environmental carcinogens. This led
to an intensive search for appropriate test systems and
over the last few decades more than one hundred short-
term assays for mutagenicity have been developed (
Ramel , 1991).
Despite the fact that long-term tests are
expensive and time consuming they provide considerable
evidence in estimating the potency of a carcinogen
(Farmer, 1982). However, there is a problem of
predicting the response of humans to the carcinogens
because of the perplexing differential responses
exhibited by the animal model systems. Therefore, the
extrapolation of the risk factor from rodents to humans
could be quite contentious (Ames et_ aX 1987). Short-
term tests (STTs) •'or genotoxicity testing have
therefore been developed to study the mechanism of
chemically induced DNA damage and to assess the
potential genetic hazard of chemicals to humans. The
most commonly employed short-term tests are chromosome
aberrations, sister chromatid exchange (SCE),
mutagenicity assay using mouse lymphoma cells and the
Ames-microsome test. The chromosomal abberations are
the manifestation of the deleterious effects that occur
anywhere in the genome. Because of the difference in
size between a genome and a gene, the cytogenetic
effects manifested as gross chromosome aberration occur
at a much greater frequency than do mutations observed
at a single locus. This test has provided a very
20

sensitive method for determining, whether or not
environmental agents can interact with the genetic
material (Wolff, 1984). SCE method has been widely used
to detect whether or not a chemical is potentially
dangerous and which of the metabolites of a
premutagenic and precarcinogenic agent could be a
potential candidate to interact with DNA resulting in
adduction (Wolff, 1984).
Ashby and Tennant (1988) reported that no
combination of the STTs is any better than the
Salmonella test, on its own for flagging probable
carcinogens. The significance of STTs has increased
tremendously because of observations that many rodent
carcinogens have been found to be genotoxic in in, vitro
STTs (Ames, 1979). The in. vitro STTs have the advantage
that they can be conducted relatively quickly and
inexpensively compared to long- term carcinogenicity
assays with rodents. These short-term tests have
significantly contributed to the identification of many
natural mutagens and carcinogens in the human diet
(Kapadia, 1982). There is a range of applications in
which STTs have been successfully used for the
identification of mutagenic fractions in complex
mixtures such as cooked meat (Hatch et. al_f 1984;
Sugimura, 1985) or air pollutants (Schuetzle and
Lewtas, 1986). Nevertheless, there are two impediments
to a thorough evaluation of the ability of these tests
21

to predict carcinogenicity . For most STTs there is a
dearth of positive results for documented
noncarcinogens (Shelby and Stasiewiez, 1784; Kier et.
aJL, 1786; Ray et. aj., 1787) and statistically
insignificant number of chemicals had been tested in
multiple STTs to provide meaningful comparisons of the
ability of different STTs and STT combinations to
predict carcinogens. Even with a battery of assays, not
all rodent carcinogens have been found to be in. vitro
mutagens nor are all in. vitro mutagens proven as
rodent carcinogens. Even then STTs do, contribute to
offer an economical, rapid and dependable means to
detect genotoxic chemicals (Tennant et. al_, 1787).
Ames Testing System :
The Salmonel la mutagenicity test (Ames et. al ,
1775), has been extensively used to survey a variety of
substances in the environment for their mutagenic
activities (Ames, 1784). The assay measures back
mutation in several specially constructed mutants of
Salmonella typhimurium strains. The method has been
adapted for use in detecting chemicals which are
potential human carcinogens or mutagens supplementing
homogenates of rat liver (S7 fraction) directly to the
petri-plates, as an approximation of mammalian
metabolism (Ames et. al_> 1773). The compounds are tested
on petri-plates with several specially constructed
mutants of Salmonella typhimurium LT2 type selected
22

for sensitivity and specificity in being reverted from
a histidine requirement back to prototrophy by a wide
variety of mutagens (Ames et. al., 1973). These histidine
requiring mutants have GC base pairs at the critical
site for reversion e.g. -C-C-C in the base pair
substitution strain, TA100 (Barnes et. aj^, 1982), -C-C-
C-C-C-C in the frameshift tester strain, TA97a (Levin
et al, 1982b) and -C-B-C-G-C-G-C-G in the frame shift
tester strain, TA98 (Isono and Yourno, 1974). However,
TA102 and TA104 have AT base pairs at the critical site
for reversion. These strains detect a variety of
oxidants and other agents as mutagens. (Levin ez_ al .
1984).
A new mutagenicity testing system to test a
chemical with more than one strain simultaneously (the
"SIMULTEST') has been develped . It involves the jse of
different Salmonella typhimurium tester strains with
and without R-plasmid, in combination on the same
plate. This approach may be useful in reducing the
workload associated with mutagenicity testing with
Salmonel la (Nestman et. al., 1987).
A great deal of controversy is over the popular
view that synthetic carcinogens are posing a higher
risk than the natural ones. According to Ames e^ al_
(1987) the carcinogenic hazards from current levels of
pesticide residues or water pollution are likely to be
of minimal concern relative to the background level of
natural substances.
23

Animal models on the other hand provided valuable
information related to the mechanism of carcinogenesis
(Harris, 1985; Hay, 1988). Extrapolation of this
information from experimental animals to humans however
remains a problematic endeavour. Most scientific
workers consider the qualitative extrapolation to be
accurate, i.e., a chemical , carcinogenic in
experimental animals is likely to be carcinogenic in
humans. Although there is a positive association
between adduct levels and tumor initiating potency in
many but not all studies using animal models (Wogan and
Gorlick, 1985), it is not known whether such
association exists in human carcingenesis (Harris,
1985). Lifetime carcinogenicity studies will still be
needed for those non- genotoxic carcinogens that are
tissue, sex, species specific in contrast to the
genotoxic carcinogens that cause tumors in different
species at multiple sites. The latter will need only a
limited bioassay (Hay, 1988). A study on mutagenic
and non-mutagenic rodent carcinogens shows that the
tested carcinogens in rats are potentially mutagenic
in S_i_ typhimurium compared to their non-mutagenic
counterparts (Rosenkranz, and Ennever, 1990).
DNA Repair in Prokaryotes
DNA is the primary carrier of genetic information
and the structural integrity of DNA is a prerequisite
for gene expression (Modak, 1972). It is a known fact
24

that the primary structure of DNA is dynamic and
subject to a constant change (Friedberg, 1785).
Ionizing and UV-radiations as well as a multitude of
other chemical agents upset the genetic and metabolic
machinery of the living system. That would perhaps
render our planet barren were it not subjected to the
constant cellular monitoring and repair. Moreover, the
contemporary global environment also posed a continual
threat to the hereditary material. The living systems
have therefore evolved repair processes to maintain
structural and functional fidelity of DNA against a
large range of insult (Friedberg, 1985). The molecular
mechanisms involved in repair of damaged portions of
DNA and their restriction into functionally intact
informational units is fundamental to the maintenance
of the genomic integrity (Modak, 1972). Defective DNA
repair could cause an accumulation of lesions or
mutations which might be either lethal, lead to an
altered phenotype, or neoplastic transformation. Repair
at the cellular and macromolecular level is multiple in
its form and varies as a function of the cell cycle
(Hart et. al, 1979) .
A great deal of research has been directed towards
gaining new insights into the mechanistic regulation of
repair machinery in Escherichia coli. The physiological
studies on the recovery from DNA damage have
established the existence of DNA repair pathways.
Moveover, the genetic studies have identified a large
25

number of genes participating in the repair of damaged
DNA (Walker, 1985).
The earliest suggestion on recovery of bacteia
after exposure to ultraviolet (UV) light was made by
Hollaender and Curtis in 1935. Later, Kelner (1949)
observed that exposure of UV-irradiated bacteria to
visible light reversed the killing and mutagenic effect
of UV light and coined the term, 'photo-reactivation'.
The isolation of an E.coli by Hill (1958) provided the
first evidence on genetic control of radiation
sensitivity . Setlow and Carrier (1964) and Boyce and
Howard-Flanders (1964) independently demonstrated that
UV-induced thymine dimers in bacterial DNA were not
excised in a UV-sensitive strain but were excised in
the wild type strain. This suggested that excision of
thymine dimers from bacterial DNA may be important for
cell survival and that it is genetically controlled.
Further, the same repair system has been shown to
operate on the damage induced in DNA by carcinogens,
mutagens and other hazardous chemicals ( Seeberg, 1981;
Friedberg, 1985). The foregoing developments paved the
way for the systematic study of DNA repair mechanism in
prokaryotes and thus led to the discovery of additional
repair systems. The following repair systems have been
shown to be existing in bacteria.
26

Photoreactivation :
The simplest class of repair pathway is
photoreactivation that directly rectifies the cyclo-
butane type pyrimidine dimers in UV-irradiated DNA
without the formation of new phosphodiester bonds
(Walker ejt al_, 1985). This type of recovery process
requires the exposure of cell to visible light. It was
the first system to be observed iri. vitro (Rupert et. al.
1958) and was the first to be characterized with regard
to mechanism (Rupert, 1962).
Photoreactivation is a universal phenomenon
because it is known to occur in E.. col i , yeast and
possibly in higher animals and plants. (Schild et_ al,
1984).
Excision Repair :
An important mechanism for cell survival after UV-
irradiation depends upon the release or excision of
pyrimidine dimers from the DNA by excision enzymes, and
the subsequent reconstruction of the double helix by
repair enzymes that trjake use of the intact opposite
strand as template. Excision repair appears to be a
significant source of DNA repair virtually in all
organisms (Walker et. al_, 1985). UV-induced damage in
cellular DNA is also repaired in the dark by excision
repair system. Mechanism of repair of DNA by excision
has been studied extensively using mutants of E. coli
sensitive to ultraviolet radiations (Setlow and
27

Carrier, 1964; Boyce and Howard-Flanders, 1964). Two
basic types of excision repair have been described in
bacteria (van Houten, 1990). One system, base excision
repair, removes smaller types of base damage, such
lesions caused by ionizing radiation or monofunctional
alkylating agents. Another system, nucleotide excision
repair, removes bulky DNA adducts of which the UV-
induced pyrimidine dimer is the prototype (Bohr, 1991).
The basic steps involved in excision repair includes
(i) pre-incision recognition of DNA damage, (ii)
incision of the damaged DNA strand near the defect,
(iii) excision of the defective site with concomitant
degradation of the excised DNA strand (iv) replication
of a repair patch to replace the excised strand with
the opposite intact strand being used as a template and
(v) ligation of the repair patch at its 3" end. The
resulting patch in nucleotide excision repair is much
larger (20-100 bases) than in base excision repair (1-5
bases) (Bohr, 1991).
Although some of the enzymes involved in
prokaryotic DNA repair have been characterized, their
mammalian counterparts are less well described. Two
prokaryotic enzymes, T4 endonuclease V and uvrABC
excinuc lease, play roles in the excision pathways of E..
coli and have proven particularly useful in the study
of gene-specific repair. The T4 endonuclease V, which
recognizes cyclobutane pyrimidine dimers, has both DNA
glycosylase and Ap endonuclease activities resulting in
28

a single-stranded nick in DNA at the site of a
pyrimidine dimer (Bohr et. al., 1985). The E_; coli ABC
excinuclease, encoded by the uvrA, uvrB and uvrC genes,
recognizes a wide range of bulky DNA adducts. These
proteins act in concert to accomplish the first three
steps of excision repair i.e. recognition, incision and
excision. This complex enzyme has also been used in
gene specific repair studies because the damage formed
by several environmental and chemotherapeutic agents,
including cisplatin, polycylic aromatic hydrocarbons,
mitomycin C and psoralen, can be recognized by the
enzyme (Thomas et. al^, 1988).
Post Replication Recombinational Repair :
The DNA lesions, especially UV-induced pyrimidine
dimers that are neither split photoenzymatically nor
removed from DNA by excision repair, block the
continuous progress of the DNA replication fork.
However, they do not prevent the re-initiation of DNA
synthesis at a point beyond the dimer (Rupp and Howard-
Flanders, 1968). As a result, gaps are produced in the
daughter strand opposite the lesions. The continuity of
daughter strand is interrupted by gaps of about 1000
nucleotides (Howard-Flanders, 1968; Iyer and Rupp,
1971; Benbow et. al_, 1974). This type of enzymatic DNA
repair by which the molecular weight of the newly
synthesized strand increases is called post replication
repair. This was first demonstrated in E. coli by Rupp
29

and Howard-Flanders (1768). In excision deficient uvr
mutants that are otherwise normal, the major mechanism
of post replication repair is recombinational ,
requiring the activity of rec genes.( Howard-Flanders
et al, 1781). The most fascinating property of the RecA
protein of Escherichia coli is its ability to form
filaments with nucleic acid (Flory and Radding, 1782;
Dunn et. aX, 1782). These filaments interact with linear
DNA molecules and exhibit end-joining activity
(Register and Griffith, 1786). The RecA protein also
plays an important role in SOS induced targeted
mutagenesis in addition to its recombinations and
regulatory roles (Walker, 1784).
Recently, Lu ejt aJL. (1786) have proposed a model
for targeted mutagenesis. According to this model (i)
RecA protein should bind more effectively to UV-
irradiated double stranded DNA than to un-irradiated
DNA (ii) RecA should inhibit the 3'-5' activity of the
isolated £ subunit of DNA polymerase III.
Inducible Error Prone (SOS) Repair :
The term 'SOS' (international distress signal)
implied to an error prone repair, is induced under
enormously stressed condition of growth as a last
resort to the survival of cells. The existence of
'SOS'network was clearly postulated by Defais et. ai.
(1771) and was further developed and amplified by
Radman (1774, 1775) and Witkin (1776). The exposure of
30

E. coli to the agents that damage DNA or interfere with
DNA replication results in the induction of a diverse
set of physiological responses called "SOS' response.
+ +
It requires recA and lexA genotypes of host (Muira
and Tomizawa, 1968; Defais et. ai., 1971).'SOS' repair is
a highly integrated and sophisticated regulatory
network that requires de. novo protein synthesis for
expression (Kovel, 1986). It is an inducible repair
process and is believed to be responsible for common
mutagenic pathways (Radman, 1974, Witkin, 1976; Walker,
1985). In response to DNA damage calling for the SOS
repair, DNA repair systems in Ej_ coli are activated,
cell division is altered, integrated viruses ars
induced and respiration is blocked (Witkin, 1976;
Little and Mount, 1982).
Several bacterial genes have been identified which
coordinately function in 'SOS' repair. These are uvrA
and uvrB (DNA repair), umuC (mutagenesis), sfiA
(filamentation), himA (site specific recombination) and
several din genes with known functions in additon to
the recA and lexA genes (Witkin, 1976;Kenyon, 1983).
Role of recBC and recN genes has also been suggested in
'SOS' induction (Chaudhury and Smith, 1985; Finch et.
al, 1985).
The recA and lexA genes are the regulators which
control "SOS" response. Under normal conditons, LexA
protein represses the subordinate genes of the system
31

and RecA protein derepresses these loci in response to
DNA damage (Kenyon, 1983). LexA protein is a self
repressor and also binds to similar operator sequences
in each gene (Little et. BI_, 1981; Sancar et. aj., 1982;
Little, 1984). During 'SOS' induction, the LexA
repressor is cleaved between ala-gly bond by the second
regulator, the RecA protein (Little 1984). RecA protein
if acquires a specific protease activty to form RecA when
it interacts with intracellular molecule that results
from certain type of DNA damage and therefore, RecA
protein is considered to play a key role in the
induction of 'SOS' response (Radman, 1975; Takahashi et_
al , 1986) .
The "SOS' response is transient and thus following
DNA repair and removal of inducing stimulus (i) RecA
protein loses its protease activity, (ii) level of LexA
repressor rises and (iii) repression of the SOS genes
resume (Brent and Ptashna, 1981; Little et, al., 1981;
Sancar et. aX, 1982).
The accumulation of certain deoxynucleotide
monophosphates and a high level induction of recA gene
expression have been suggested to stimulate the "SOS'
repair (Gottesman, 1981). Moreover, Salles et. al_ (1983)
reported the full amplification of RecA protein without
any amplification of other single strand binding
proteins under 'SOS' inducing conditions. Bebenek and
Janion (1985) proposed the possible role of mismatch
repair also in the induction of 'SOS'.
32

Mutagenesis resulting from'SOS' processing of
damaged DNA template is targeted and is not due to the
induction of some random mutator activity (niller,
1783; Walker, 1984). Radman (1974, 1975) suggested that
the appearance of mutation during "SOS' processing may
be due to the inducible infidelity of DNA replication,
because pyrimidine dimers were found to block the chain
elongation by purified DNA polymerase I and III on UV-
irradiated DNA template. Subsequent studies revealed
the involvement of inducible inhibition of proof
reading (3'-5' exonuclease) activity of DNA polymerases
.(Radman et. ai, 1977; Boiteux et. aj^, 1978; Villani et.
al. 1978). Moreover, the lexA dependent conversion
involving the £ subunit of DNA polymerase III
holoenzyme to 'SOS' polymerase has also been postulated
(Scheuermann et. aX, 1963; Piechocki et. aJL, 1986). Lu eX.
al (1986) infact, have demonstrated the recA mediated
inhibition of 3'-5' nuclease activity of isolated £
subunit of DNA polymerase III and was suggested to be
responsible for targeted mutagenesis.
Gene Specific DNA Repair:
Recent studies demonstrated that in several
biological systems, the DNA repair processes are
heterogeneous within the genome (Bohr, 1991). It has
been reported that active genes are preferentially
repaired in hamster (Bchr et. aJL ,1985) and human cells
(Mellon et. al_. 1986) after exposure to UV radiation.
33

This heterogeneity is further extended to a strand bias
of DNA repair after UV exposure and has been
experimentally proved that the transcribed strand is
repaired much more efficiently than the nontranscribed
strand (Mellon et. al, 1987; Stevnsner and Bohr, 1993).
The first study of gene specific repair in heritable
disorder Xeroderma pigmentosum (XP) group 6 cells
revealed 10-307. repair in the dihydrofolate reductase
(DHFR) gene in XP4R0 strain (Bohr et. aJL, 19B6a).
Recently, Venema et. al_ (1990) reported that adenosine
deaminase and DHFR genes were preferentially repaired
in XP-C fibroblasts from individuals XPITE and XP
21R0. The strand bias of the repair in these genes in
XP-C (XPITE) cells has also been analyzed (Venema et.
al, 1991).
Preferential DNA repair is a general feature in
many biological systems. It has been observed in
various mammalian cell lines, in fish, yeast (Terlleth
et al , 1989) and in E^. coli (Mellon and Hanawalt,
1989). Its occurrence in bacteria suggests that the
preferential DNA repair of active genes is not
dependent upon chromatin structure. Studies on the fine
structure of DNA repair of UV damage in the hamster
DHFR gene suggest that the region of the preferential
repair could be larger than the size of the gene.
Characterization of a DNA repair domain containing the
DHFR gene in CHO cells indicated the size of the domain
in the range of 60 -80 Kb (Bohr et. JLL. I' BA.b) . Since
34

this is the size of the higher -order structures in
chromatin, it is possible that DNA repair is regulated
within such chromatin loops. One parameter to consider
is the degree of genomic demethylation. There are very
few studies on the relation of DNA repair to genomic
methylation patterns, at the gene level showing that
genomic demethylation could affect the pattern repair
in the hamster DHFR domain .
Several studies have demonstrated that the
efficiency of repair in a gene also correlates to its
transcriptional activity, as in the case of the hamster
and human metallothionein genes and can be blocked by
the transcription inhibitors . Moreover, with respect
to repair, different genomic regions have been
categorized from the lowest to the highest repair
efficiency as follows (i) the inactive genomic DNA ,
(ii) the inactive genes that are turned off early in
development, (iii) the structural genes that are
capable of transcription at certain times in
metabolism, (iv) the essential genes that are always
active but at a low level of transcription and finally
(v) an active gene transcribed at a high rate. This
categorization is obviously speculative and needs to be
explored (Bohr, 1991).
In view of the present literature we have
undertaken this study ta determine the mutagenic and
genotoxic potential of four pesticides viz captan,
35

foltaf, phosphamidon and furadan.
The major objectives were as follows:-
(1) Screening of the pesticides for mutagenicity by
means of Ames testing system. (Plate incorporation
assay) .
(2) The effect of the pesticides on the survival of
SOS repair defective strains of E.. col i K-12.
36

CHAPTER - II
GENERAL MATERIALS AND METHODS

Table IV: The Salmonella typhimurium and Escherichia coli strains
used in this study have been tabulated as under :
Strain designation
Relevant genetic markers Source
Ames Strains TA97a
TA98
TA100
TA102
TA104
uvrB. hisD6610, bio, rfa. R-factor plasmid-pkMlBi
urvB. hi5D5052. bio, rfa. R-factor plasmid-pkH101
uvrB. hi5G46. bio, rfa. R-factor plasmid-pkM101
uvrB. hi5G428. bio, rfa R-factor plasmid-pkM101 multicopy plasmid-pAQl
uvrB, hi5G42B. rfa. R-factor plasmid-pkM101
Ames, B.N.
Ames, B.N.
Ames, B.N.
Ames, B.N.
Ames, B.N.
E. coli K-i2 strains
AB1157
AB2463
AB2494
AB2470
KL400
KL 403
MM159
thi-1. arqE3. thr-1. 1euB6. ProA2. hisG4. lacYl. F-, Str' . A'^
recA13. thi-1. argE3» thr—1. leuBd). proA2. hisG4. F-, StrPr~^
lexA. thi-1. thi-1. leuB6. proA2. hisG4, metB. lacYl. F-, Str-, A"'
thr-1. leuB6. lacYl. ^, bisG4. recB21. arqE—. thi-1.
thi-1. leuB6. proC32. hi5FB60. thvA54. lacZ36. 5trA109. A .
leuB6. lacZ36. DrDC32. ^ , hisF860. thvA54. polAl. thi-i
qalK2./f-. raaL200. ijvrAi57. IN (rrnD-rrnE)l
Howard-Flanders,P,
Howard-F1anders,P.
Howard-F1anders,P.
Howard-Flanders,P.
K.B.Law laboratory,
K.B.Low laboratory
Meselson, M.
37

METHODS
Maintenance and growth of bacteria : Each strain of
Salmonella typhimurium was streaked over master plate.
A single colony was picked up, grown in minimal medium
and repurified by streaking over fresh master plate.
Likewise each strain of E.coli was streaked over
nutrient agar plates. A single colony was picked up and
repurified by streaking over agar plates. The culture
was tested on the basis of associated genetic markers
raising it from a single colony from the master plate.
Having satisfied with the test clone the culture was
raised and streaked over minimal and nutrient agar
slants. It was then allowed to grow over night at Zl'C
and stored at 4°C. Every month, cultures were
transferred over fresh slants with TA102 strain as an
exception. It was transferred after every 15 days.
Stabs were prepared for longer storage.
Overnight culture of S_ typhimurium strains were
used as such for experiments. Overnight culture of
E.coli strains were raised in nutrient broth at 37'C
The culture was diluted fifty times in fresh broth
followed by shaking at 37'C till the cell density
8 ~1 reached to about 2 x 10 viable counts ml . Such
exponential cultures were used in all the experiments.
38

Media
Media far Ames Strains :
Medium for master plates and slants : The composition of the
medium for Ames tester strains to prepare master plates and
slants is as under:
Sterile 50X VB Salts 20 ml
Sterile agar 15 g per 910 ml
Sterile 407. glucose 50 ml
Sterile histidine HCI.H2O 10 ml
(2 g per 400 ml H2O)
Sterile 0.5mM biotin 6 ml
Sterile ampicillin solution 3.15 ml
(a mg/ml 0.02 N NaOH)
Sterile tetracycline solution 0.25 ml
(8 mg/ml 0.02 N HCl)
The above components were mixed with the molten agar to
prepare the plates. Tetracycline was added only for use with
TA102 which is tetracycline resistant.
Stock solution of VB salts (IX) was prepared using the
following ingredients :
MgSD4.7H20 0.2 g/1
Citric acid monohydrate 0.2 g/1
K2HPO4 (anhydrous) 10.0 g/1
NaHNH4Pa4.4H20 3.4 g/1
39

The salts were added in the order indicated to warm
distilled water and each salt was allowed to dissolve
completely before adding the next. The solution was then
autoclaved for 20 min. at 121"C.
Minimal glucose plates for mutagenicity assay :
Sterile VB salts 20 ml
Sterile 407, glucose 50 ml
Sterile agar 15 g/930 ml distilled water
The above components were mixed with the molten agar
and poured atleast 30 ml in each plate.
Top agar for mutagenicity assay : The top agar contained
0.67. Difco agar and 0.57. NaCl. At the time of experiment
i0ml of sterile solution of 0.5 mM histidine.HCl and 0.5 mM
biotin was added to the molten agar and mixed thoroughly by
swirling.
0.5 mM histine/biotin solution for mutagenicity assay :
D-Biotin 30.9 mg
L-Histidine HCl 24.0 mg
Distilled water 250 ml
Dissolved biotin by heating the water to the boiling
point and mixed histidine to it. Autoclaved for 20 minutes
at 121'C.
40

Media far E.coli K-12 strains :
Nutrient broth (13g/l): Nutrient broth had the following
composition.
Peptone 5 g/1
NaCl 5 g/1
Beef extract 1.5 g/1
Yeast extract 1.5 g/1
pH (approx.) 7.4 + 0.2
Nutrient agar (Hard agar) :
Nutrient broth 13 g/1
(Hi media)
Agar powder 15 g/1
(Hi media)
Buffers
MgS04.7H20 solution (B.BIM) : For all dilution, 0.31M MgS04
solution was used.
B.2 M sodium phosphate buffer, pH 7.4 :
NaH2P04.H20 13.8 g Na2HP04 14.2 g Distilled water 500 ml
Adjusted the pH to 7.4 and then sterilized it at 121'C
for 20 minutes.
41

CHEMICALS
The fallowing chemicals were used :
Chemicals
Agar Powder
Ampicil1 in
Biotin
Chloramphenicol
Dimethyl sulphoxide
Dipotassium hydrogen phosphate
Disodium hydrogen phosphate
D-glucose
Histidine monohydrochloride
Hydrochloric acid
Magnesium sulphate
Nutrient agar
Sodium ammonium phosphate
Sodium chloride
Sodium hydoxide
Tetracycline
Source
Hi-Media, India
Ranbaxy, India
Hi-Media, India
Biochem Pharmaceutical Industries, India
s.d. fine chemicals, India
Qualigens, India
Sarabhai Chemicals, India
s.d. fine chemicals, India
CDH, India
s.d. fine chemicals, India
s.d. fine chemicals, India
Hi-Media, India
E. Merck, Germany
s.d. fine chemicals, India
Qualigens, India
IDPL, India
42
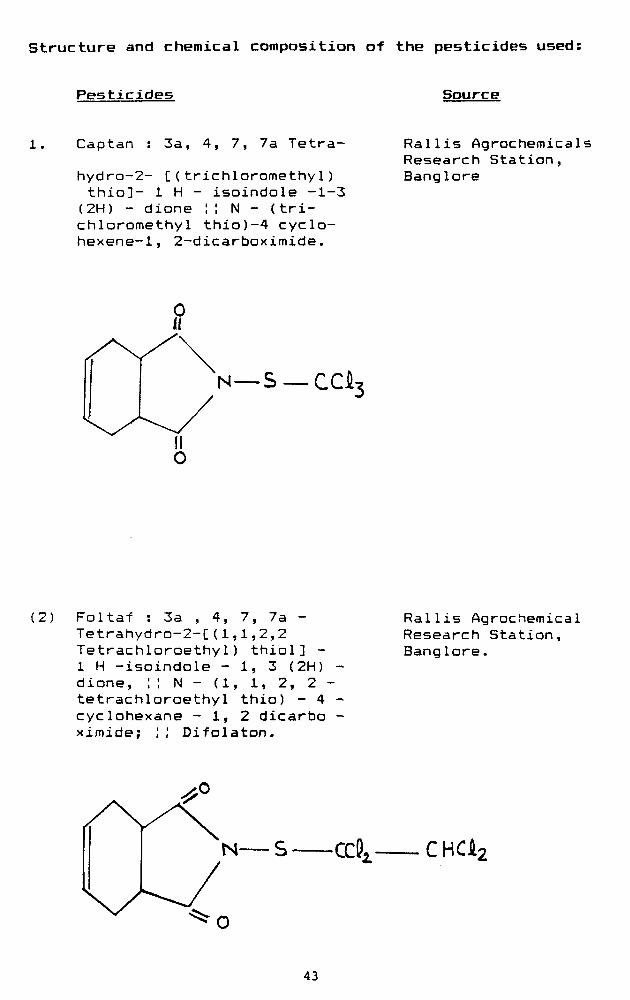
structure and chemical composition of the pesticides used:
Pesticides Source
Captan 3a, 4, 7, 7a Tetra-
hydra-2- C(trichloromethyl) thio]- 1 H - isoindole -1-3 (2H) - dione !I N - (trichloromethyl thio)-4 cyclo-hexene-1, 2-dicarboximide.
Rallis Agrochemicals Research Station, Banglore
—S —CCi
(2) Foltaf : 3a , 4, 7, 7a -Tetrahydro-2-C(1,1,2,2 Tetrachloroethy1) thiol] -1 H -isoindole - 1, 3 (2H) -dione, ;! N - (1, 1, 2, 2 -tetrachloroethyl thio) - 4 -cyclohexane - 1, 2 dicarbo -ximide; ;; Difolaton.
Rallis Agrochemical Research Station, Banglore.
CHCk:
43
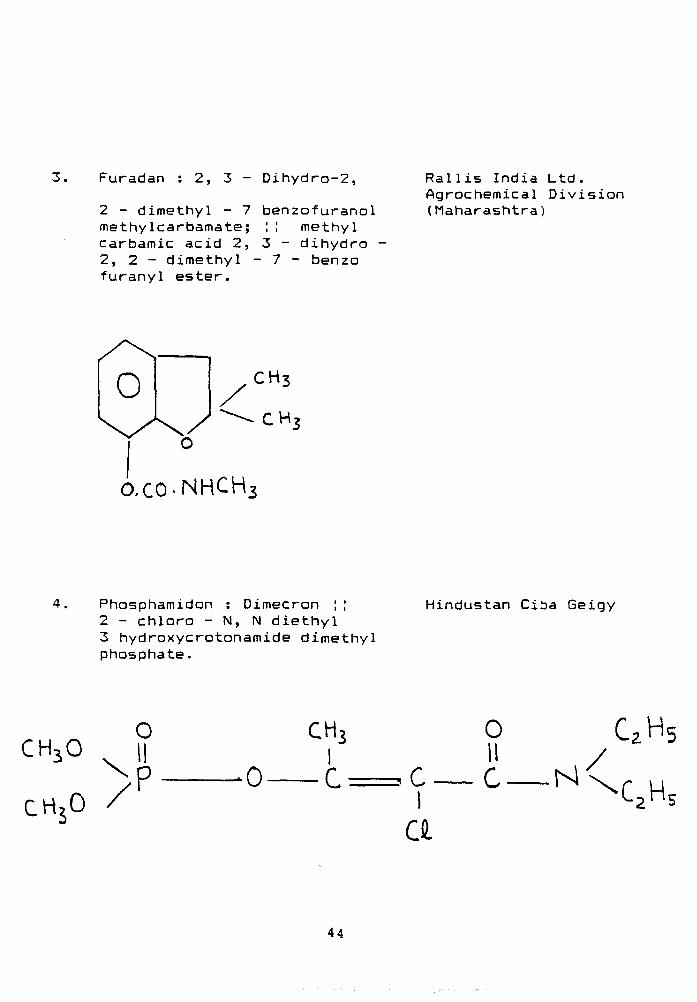
3. Furadan : 2, 3 - Dihydro-2,
2 - dimethyl - 7 methylcarbamate; carbamic acid 2, 2, 2 - dimethyl -furanyl ester.
benzofuranol :! methyl 3 - dihydro • 7 - benzo
Rallis India Ltd. Agrochemical Division (Maharashtra)
O.CO-NHCH
Phosphamidon : Dimecron ; ; 2 - chloro - N, N diethyl 3 hydroxycrotonamide dimethyl phosphate.
Hindustan Ciba Geigy
o
CHjO II
CHjO /
0
CH3 I
I a
o 11 c N
CzHs /
44

CHAPTER - lil
MUTAGENICITY ASSAY

MUTAGENICITY ASSAY
INTRODUCTION
There is considerable evidence that a large
proportion of human cancer may be caused by exposure to
toxic chemicals in the environment, very few of which
have been tested for carcinogenicity or mutagenicity. A
program of cancer prevention aimed at identifying and
eliminating human exposure to hazardous chemicals
requires the development of rapid, inexpensive, long-
term animal tests, to pinpoint dangerous chemicals
among the thousands to which humans are exposed.
The Salmonella/microsome mutagenicity test,
relating mutagenicity to the number of revertant
colonies, has been sufficiently developed and validated
to be seriously considered for widespread use in this
way (Ames, 1971; Ames et. al, 1973; McCann et al_, 1975;
van der Hoeven e . ajL., 1990). The evidence accumulated
so far indicates that with a few exceptions,
carcinogens are mutagens (Ames et. a_l.j 1973; Flessel et.
al. 1987; Zeiger, 1987). It thus supports the
desirability of using this type of rapid and economical
test system as a screening technique (Ames, 1971; Ames
et al. 1973). The work embodied in this chapter was
therefore, designed to screen the available pesticides
for their potential mutagenic and carcinogenic
behaviour employing the Ames testing system.
45

Methods
Bacteria, pesticides and media are listed in
Chapter II. All the media were freshly prepared. Plate
incorporation assay was routinely performed for testing
the mutagenicity of the pesticides.
Plate incorporation assay : This test was
performed by combining the pesticide, the bacterial
tester strain and 0.2M phosphate buffer (pH 7.4) in
soft agar which was poured onto a minimal agar plate.
Positive and negative controls were also included in
each assay. After incubation at ZVC for 48 hours,
revertant colonies were counted and the mutation
frequency calculated by subtracting the spontaneous
reversions from the induced reversions.
Routine examination of the bacterial background
lawn resulting from the trace amount of histidine added
to the top agar aided in determining the toxicity of
the test chemicals and in the interpretation of
results.
RESULTS
Assessment of the mutagenic potential of certain
pesticides
Mutagenicity of captan:
Mutagenicity of four different pesticides viz
captan, foltaf, phosphamidon and furadan have been
evaluated employing Ames salmonella tester strains.
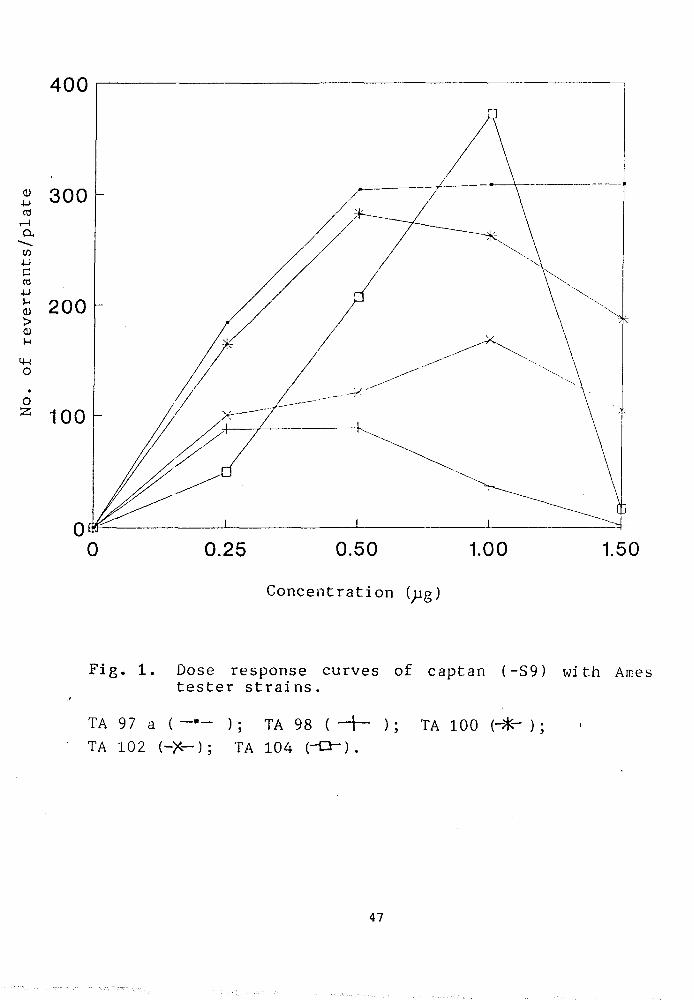
Fig. 1 shows a dose dependent increase in the mutagenic
46
400
d) •U rt
1-1
a tn 4-1 C rt 4-1
(1) > OJ
u 14-1 O
o 2
'^00 \ j \ j \ j
200
100
0 0 0.25 0.50 1.00 1.50
Concentration (;jg)
Fig. 1. Dose response curves of captan (-S9) with Ames tester strains.
TA 97 a ( ); TA 98 ( - j — ); TA 100 (-- ) ; TA 102 (-X-); TA 104 (-Q") .
47

response of captan with five different histidine
mutants. At concentration range of 0- 1.5^g/plate, a
considerable increase in the number of his revertants
was observed with a certain degree of variability
depending upon the sensitivity of the strain. The total
number of induced revertants as shown in fig. 1 have
been obtained after subtracting the spontaneous
revertants. The results indicate that the tester strain
TA104, TA97a and TA100 are maximally sensitive compared
to TA102 and TA78 at a concentration of 1.0;jg/plate. A
higher dose of captan i.e. 1.5_^g/plate was invariably
found to be toxic to all the tested strains. At this
concentration, a sharp decline in the dose response
curve of TA104, corresponding to 987. decrease in the
number of revertants, was clearly observed. The maximum
mutagenic response was seen at a plate concentration of
l.B^g/plate captan, in the following order of
sensitivity: TA104> TA77a> TA100 > TA102 and TA98.
Interestingly, a 2-4 fold decrease in the mutagenic
response of his mutants was noticed in the similar
concentration range in presence of S9 mix (Fig. 2).
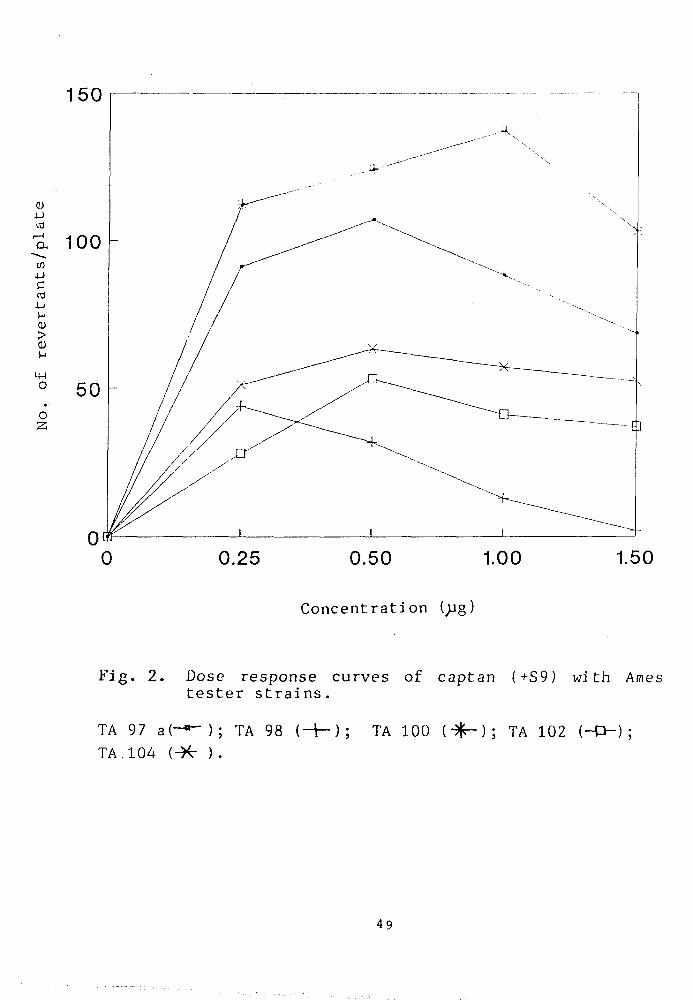
Mutagenicity of foltaf:
The mutagenic response of foltaf, evaluated in
concentration range of 0-2.5 g/ plate, is shown in
Fig. 3. The results show that the maximum number of
revertants were produced with TA102 and TA77a at a dose
of 1.0jjg/ plate. Any further increase in the dose of
48
150
Q) 4J
a CO
c u
>
o 2;
100
1.50
Concentration ( g)
Fig. 2. Dose response curves of captan (+S9) with Ames tester strains.
TA 97 a(-*-); TA 98 (-H); TA 100 {^); TA 102 (-D-) ; TA.104 (-H- ).
49
400
Concentration (>Jg)
Fig. 3. Dose response curves of foltaf (~S9) with Ames tester strains.
TA 97 a(-»-); TA 100 (-1—); TA 102 (-O-) ; TA 104 (- -] .
TA 98 (Not mutagenic).
50

foltaf resulted in decreased survival of the bacteria
except strain TA104 which has exhibited a concentration
dependent increase in the number of revertants upto the
level of i. 5)jg/plate. There after, a sharp decline in
the mutagenicity curve was observed, most probably due
to the toxic effect of the pesticide. No significant
induction in the mutagenic response in TA78 was
noticed. The maximum number of revertants, induced by
foltaf at a concentration of Ijjg/plate in different
tester strains were in the fallowing sequence: TA102 >
TA97a> TA104> TA10IZ) and TA78.
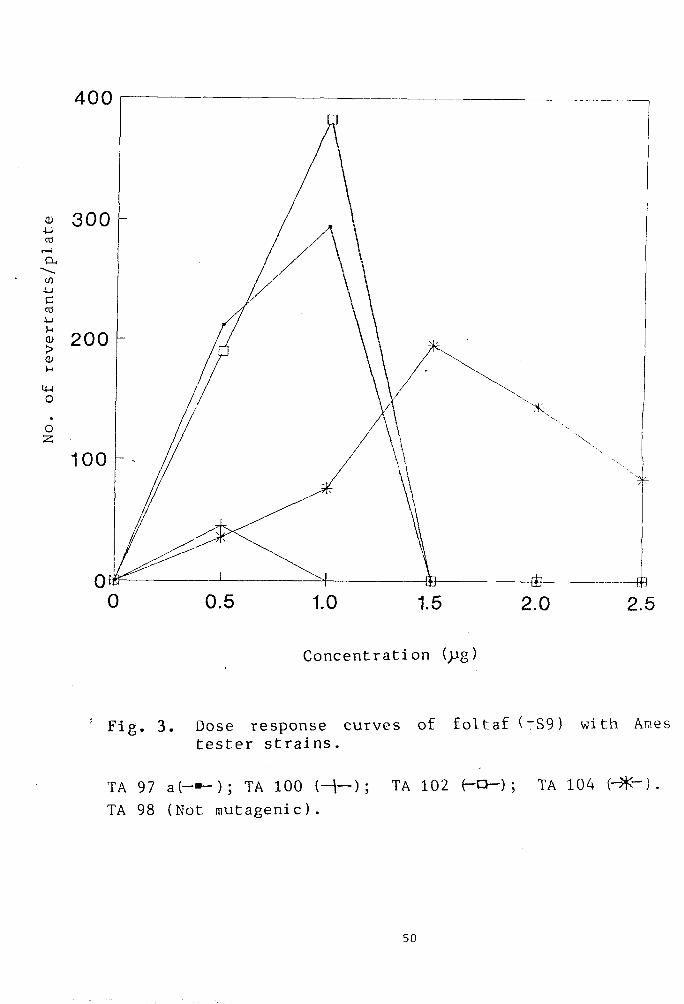
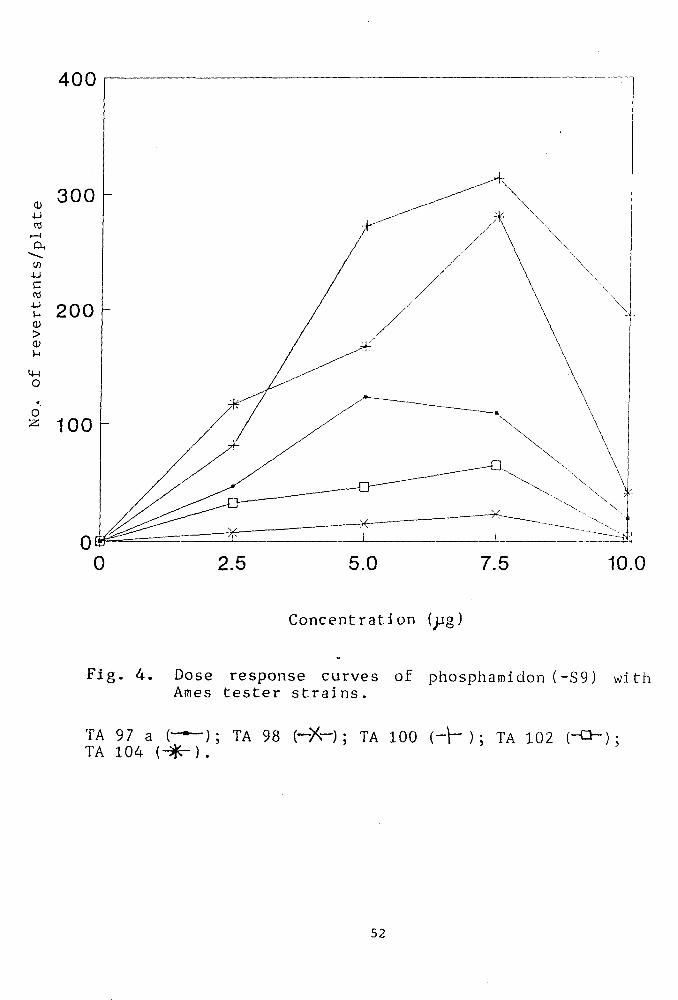
Mutagenicity of phosphamidon
Fig.4 shows the mutagenicity profile of
phosphamidon in a concentration range of 0-i0^g/plate.
A linear dose dependent mutagenic response was observed
upto a concentration of 7.5 ^pg/plate in the following
order of sensitivity;TA100> TA104 > TA97a > TA102 and
TA98. Further increase in dosage to 10^g/plate resulted
in reduced survival of the bacterial strains. The
strain TA9a was found to be least sensitive and
produced the lowest number of revertants even at the
optimum plate concentration of 7.5^g which is causing
maximum induction of mutagenicity in other tester
strains.
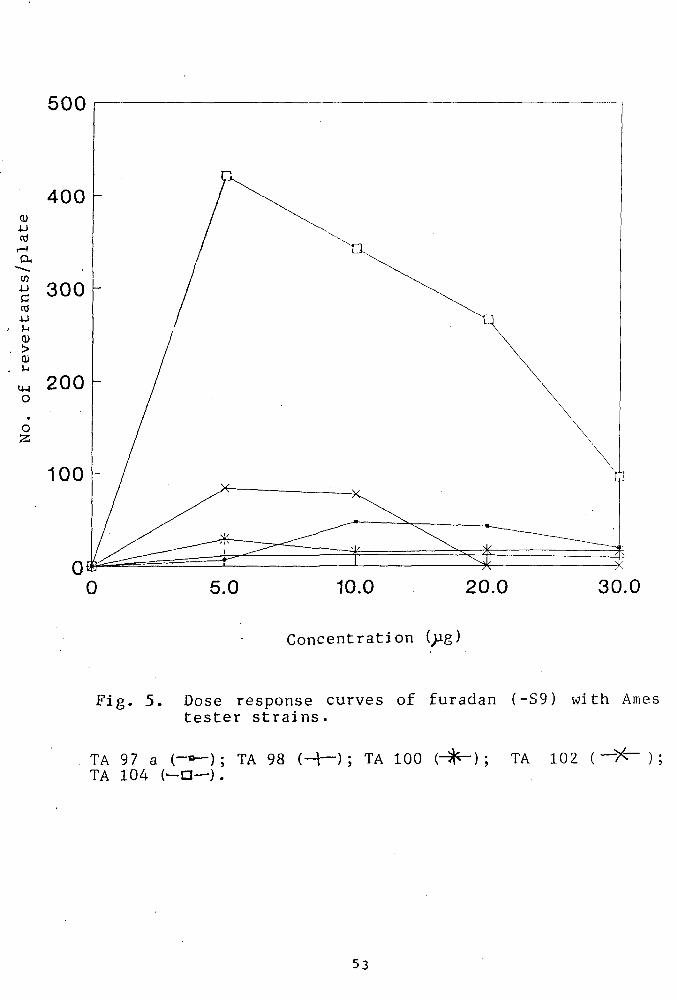
Mutagenicity of furadan
Fig. 5 shows the mutagenic response exhibited by
furadan at a concentration range of 0-30pg/plate. A
51
400
10.0
Concentration ( g)
Fig. 4. Dose response curves of phosphamidon (-S9) with Ames tester strains.
TA 97 a ( — ) ; TA 98 (-X-) ; TA 100 {-\-) ; TA 102 (-O-): TA 104 (^^) .
52
500
400
cd
a
^ 300 CO
> CD
O
O 2;
200
100
30.0
Concentration ( g)
Fig. 5. Dose response curves of furadan (-S9) with Ames tester strains.
TA 97 a (-»-); TA 98 (-+-); TA 100 (- ) ; TA 102 ( — > ^ ); TA 104 (—a—).
53

significantly high level of mutagenic response was seen
in strain TA1(Z)4 producing the highest number, 422
revertants/plate at concentration of 5;jg/plate followed
by TA102 (85 revertants/plate).Other strains including
TA97a, TA100 and TA98 did not exhibit any significant
increase in the reversion frequency. The results also
indicate that an increase in amount of furadan beyond
5ijg resulted in a remarkable decrease in the viability
of the bacterial strains.
Discussion
The Salmonella test was first validated in a study
of 300 chemicals most of which were known carcinogens
(McCann ejt aJL. 1975; licCann and Ames, 1976; McCann and
Ames 1977). It was subsequently employed in other
studies by the Imperial Chemical Industries (Purchase
et al. 1976), and the International Agency for Research
on Cancer (Bartsch et. ai., 1980). Nearly 90"/. of the
carcinogens tested were found to be mutagenic in these
studies, but there was considerable overlapping of
chemicals tested. It is customary to define the
compound as negative if none of the tester strains
responds with and without metabolic activation upto the
limit of toxicity not exceeding to 5ng/plate
concentration (deSerres and Ashby, 1981; Dyrby and
Ingvardsen, 1983). An increase in the number of
colonies (over the number of spontaneous background
revertants) indicates that the chemical is mutagenic

while the number of revertant colonies provides an
index of the mutagenic activity of the sample (deSerres
and Ashby, 1981; Levin et. aj., 1984; Flessel e^ al,
1987). Our results indicated an invariable increase in
number of revertant colonies with all the pesticides
viz. captan, furadan, foltaf and phosphamidon. Moreover
all the pesticides exhibited a remarkable degree of
mutagenicity with TA97a, TA1(Z)0, TA104 and TA102.
Table V, shows the relevant genetic and
biochemical markers of the Ames tester strains. All the
strains carry the pKM101 plasmid which is believed to
enhance the error—prone repair process (Shanabruch and
Walker, 1980; Levin et. aJL. 1982a). TA97a strain has
been recommended for detecting frameshift mutagens
(Levin et. al, 1982a), whereas TA102 is known to detect
the oxidative mutagens (Levin et. al, 1984). TA97a, TA98
and TA100 tester strains contain G-C base pairs at the
critical site for reversion (Isono and Yourno, 1974;
Barnes et. al., 1982; Levin et, al_, 1982a). With regard to
the individual difference among the G-C specific
mutants, TA97a, TA98 and TA100, the first two strains
are the frameshift type while TA100 is the base-pair
substitution mutant (Isono and Yourno, 1974, Barnes et.
al. 1982). The tester strain, TA102, is proficient in
the excision repair machinery but carries a mutation in
his G428 gene on the multicopy plasmid, pAQl. On the
contrary, TA104 carries the same mutation on the
55
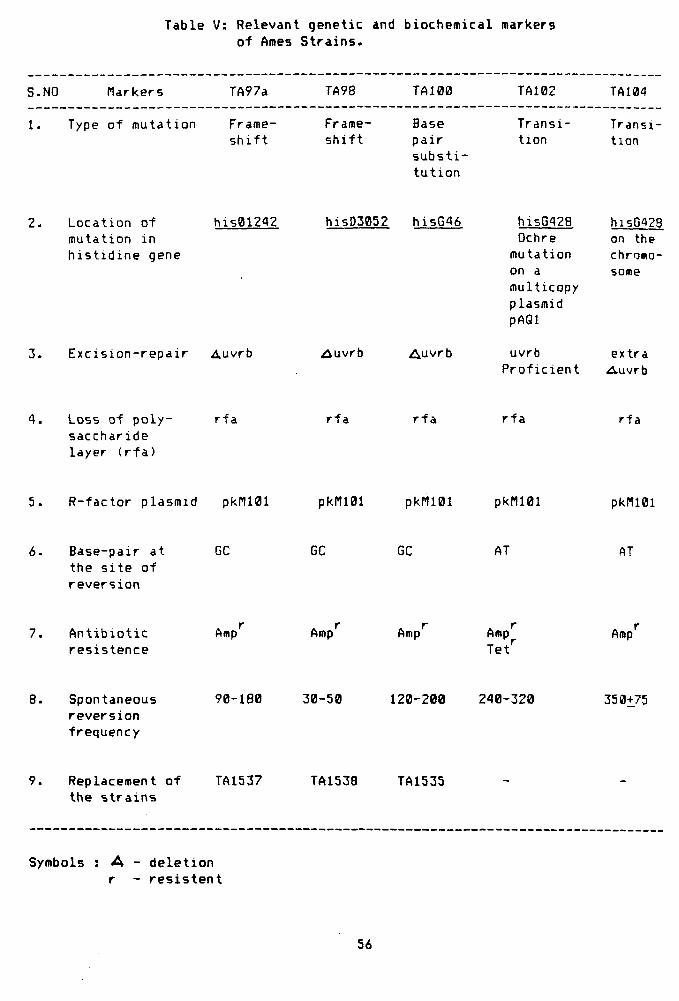
Table V: Relevant genetic and biochemical markers of Ames Strains.
S.NO Markers TA97a TA98 TA100 TA102 TA104
1. Type of mutation Frame-shift
Frame-shift
Base pair substi tution
Transition
Transition
2. Location of mutation in histidine gene
his012A2
3. Excision-repair Auvrb
hi5D3052 hi5G46
^uvrb Auvrb
hisG^28 Ochre mutation on a multicopy plasmid pAQl
uvrb Proficient
hisG428 on the chroao-some
extra Auvrb
4 . Loss of p o l y sacchar ide layer ( r f a )
r f a r f a r f a r f a r f a
5. R- fac tor p lasmid pkri l01 pkni01 pkr i l01 pkn iB l pkI1101
Base-pai r a t the s i t e o f r eve rs i on
GC GC GC AT AT
7. Antibiotic resistence
Amp Amp Amp Amp Tet''
Amp
Spontaneous reversion frequency
90-180 30-50 120-200 240-320 350-^75
Replacement of the strains
TA1537 TA1538 TA1535
Symbols : A - deletion r - resistent
56

chromosome but carries a defective excision repair
gene, uvrB which obviously diverts the repair towards
the errorprone side and thus changes the mutagenic
activity of the compound (Levin e^ al_» 1982b). In view
of our findings, it is noteworthy that the pesticides
act on the A-T base pair mutants TAi04 and TA102 in
addition to TA97a and TA100 having G-C base pairs at
the site of mutation. However, the pesticide, captan
seems to revert the frameshift tester strain, TA97a,
more efficiently as compared to base-pair and
transition mutants.
We have then, concentrated our studies on four
pesticides. Quantitative Ames testing revealed that the
tester strains, TA97a, TAi00, TA102, TA104 were
responsive (Fig. 1,2,3,4 and 5). TA104 happened to be
the most responsive. In view of the present findings it
is clear that the pesticide treatment brought about a
transition mutation owing to the error prone repair of
the damaged cell. This idea gains further support by
the relatively strong mutagenic response of the test
compound on the uvrB defective TA 104 strain.
57

CHAPTER - IV
GEiNOTOXICITY ASSAY

Genotoxocity Assay
Introduction
Various physical and chemical environmental agents
have been shown to damage cellular DNA in. vivo. The
capacity of a system to repair such damage is basic to
the ability of an organism to withstand the biological
consequences of environmental stress. Correctly
repaired, genetic damage has little effect on the
biological function of a system. Studies on
microorganisms, mammmalian and plant cells have shown
that DNA damage results in change in the physiological
processes such as growth, division, transcription of
DNA, cell death, mutation and induction of
transformation (Elkind and Whitmore, 1967; Price and
Makinodon, 1973; Lohman and Bootsma, 1974).
E.coli responds to DNA damage with the expression
of a set of functions usually termed as SOS response.
This includes the induction of a transitory mutagenic
DNA repair system, the activation of inducible prophage
and of several other functions involved in cell
division and DNA metabolism (Radman, 1974; Witkin,
1976).
The work embodied in this chapter was therefore
planned to estimate the extent of injury produced, to
understand the mutagenic behaviour and the mode of
interaction of pesticides with DNA.
58

Our contention was that SOS defective strains of
E.coli K-12 might serve as a convenient model for this
purpose because even a slight change in environment
could be reflected in its survival pattern.
Methods
Bacteria, media, pesticides and buffers are listed
in Chapter II. Relevant genetic markers associated with
the strains is given in (Table IV ). Survival patterns
of E•coli strains have been studied in the presence of
pesticides.
The SOS defective E.coli strains were treated
with that concentration of pesticides at which the
highest peak was observed in the Ames test . Except
otherwise stated^ the E.coli cells were treated for 6
hours with pesticide. All the media were freshly
prepared.
Pesticide treatment to bacteria :
The SOS defective recA. recB. lexA. polA and uvrA
mutants of E.coli K-12 as well as the isogenic wild
type strain were harvested by centrifugation from g
exponentially growing cultures (1-3 x 10 viable counts
ml ). The pellets so obtained were suspended directly
in 0.01M MgS04 solution and treated with the pesticides
at various concentrations. Samples were withdrawn after
the treatment period of 6 hours, suitably diluted and
plated to assay the colony forming ability. Plates were
incubated over night at 37*C. Treated controls were
59

also run simultaneously.
RESULTS
Evaluation of the qenotoxic effect of the pesticides
Genotoxicity of captan
The genotoxicity of captan has been asssessed by
determining the colony forming ability of the bacterial
strains treated with varying doses of captan. The
relationship between the percent survival and the dose
of captan is shown in Fig. 6.The trend of the survival
curves of DNA repair defective mutants, recA , recB ,
lexA , and pol campared to their isogenic wild types
ABii57 and pol A clearly indicates the DNA damaging
activity of the captan. The mutants particularly pol ,
recA , and lexA exhibited a significant decrease in
the colony farming ability within a concentration range
of 2 to IB^g^itJesticide. Among all mutants, pol was
found to be the most sensitive and its survival reached
to the baseline, indicating 99.3X loss in viability
campared to the pol strain. The mutants recA and lexA
have also shown decrease in the colony forming
ability compared to their wild-type counterparts. It
was noticed, that a higher concentration i.e. IQiuq/fni of
captan, the wildtype cells also exhibited a decrease in
the survival with a proportional reduction in the
survival of mutant strains.
60
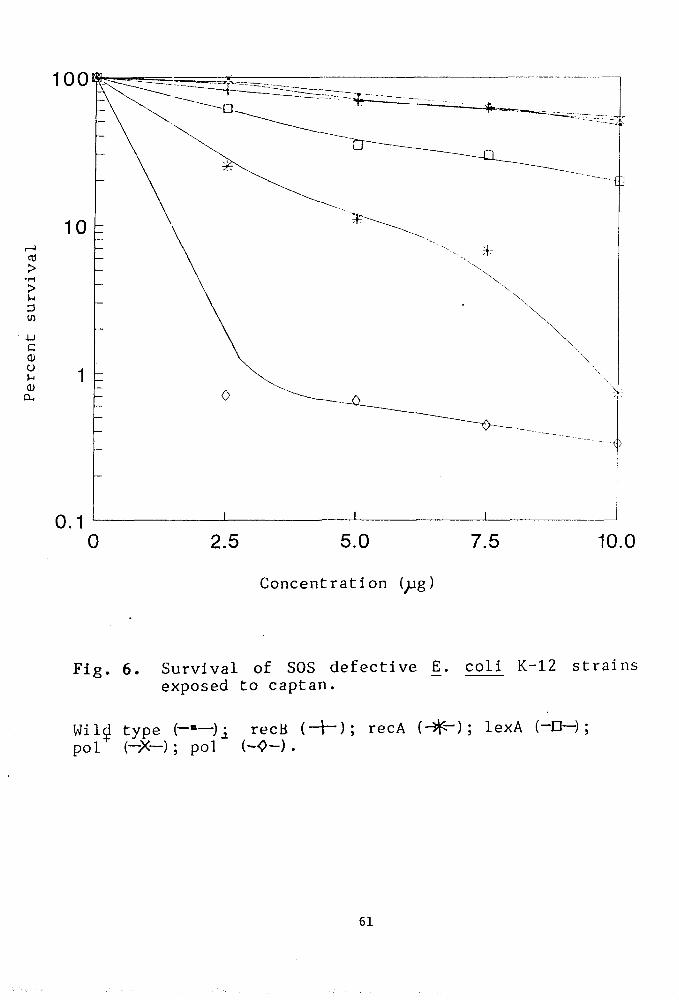
100
>
>
3 w
c 0) o >-l
a> 0-,
10.0
Concentration ( g)
Fip. 6. Survival of SOS defective E. coli K-12 strains exposed to captan
Wild type (——)i recB (-H); recA (- ) ; lexA (-D—) ; pol (-X-); pol {-<>-) .
61

Genotoxicity of foltaf
A relatively higher amount of foltaf was used for
the genotoxicity assay. Fig. 7 shows the comparision of
the survival curves of various DNA repair defective
mutants with the repair proficient strains in a
concentration range of 0 to 15pqWof foltaf. At a sub
lethal concentration of 5^g, a noticeable change in the
survival pattern of the mutants was observed. This
effect became more pronounced at a higher concentration
of i5^g foltaf resulting in percent reduction in the
colony forming ability of pol and recA mutants,
respectively. The highest toxic dose was found to be
25ug which causes significant loss in the viability of
all the strains and no differential response was
noticed at this concentration .
Genotoxicity of phosphamidon
Fig.8 shows the survival curves of recA , recB ,
uvrA, lexA and polA mutants upon treatment with
phosphamidon in a broad concentration range of 0-100u^W-
Relatively weak genotoxic activities were noticed even
at a higher concentration of 100 cj^,Hawever, certain
mutants including polA , recA and lexA were found to
be much more sensitive compared to uvrA. recB and wild
type strains. An almost linear dose dependent decrease
in the colony forming ability of polA mutant was
observed. On the other hand , the mutant recA~ and
lexA exhibited resistance against the higher
62
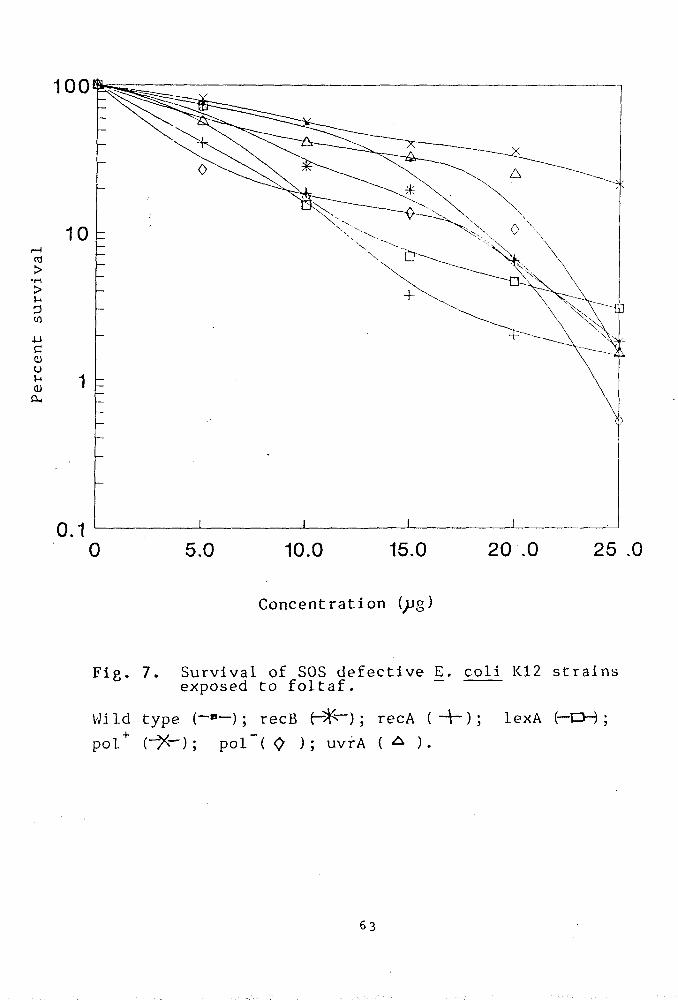
100
> •r-l
> O
C (1) o 0)
a.
20 .0 25 .0
Concentration ( g)
Fig. 7. Survival of SOS defective E. coli K12 strains exposed to foltaf.
Wild type (-"—); recB H ^ ) ; recA (-+"); lexA (—CH ;
pol^ (-^-); pol~{Q ) ; uvrA (^ ).
63
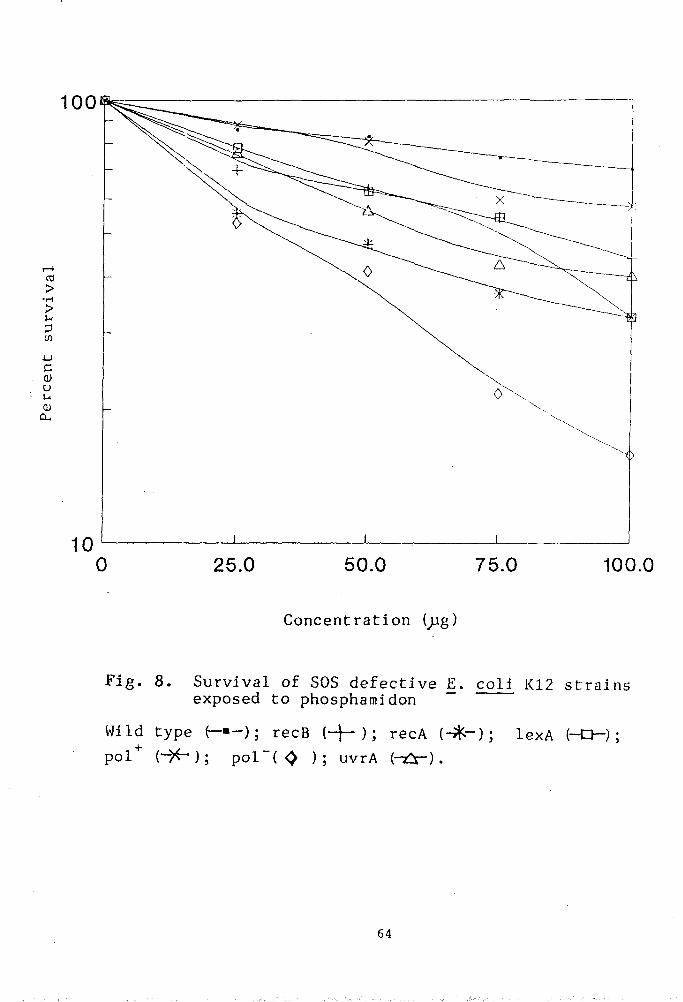
100
CO >
>
to
u c u QJ CL,
25.0 100.0
Concentration (;jg)
Fig. 8. Survival of SOS defective E. coli K12 strains exposed to phosphamidon
Wild type e-«-); recB {-\--) \ recA (- -) ; lexA (-D-) ;
pol^ (->^); pol"( <> ); uvrA {r^:^-).
64

concentration of phosphamidon. The wildtype polA-<- and
AB1157 also showed almost similar response in the test
range.
Genotoxicity of furadan
The genotoxicity of furadan was evaluated at a
much higher concentration range, ranging form 0-300 ygftnt.
As shown in Fig. 9 , the relationship between the
percent survival and the dose of furadan represents a
weak genotoxic ability of furadan. Even the higest
concentration of 300^g was found to be insufficient to
induce significant DNA damage which could result in the
loss of viability of the bacterial strains.
Nevertheless, the differential pattern of the survival
curve, as observed at 100pg concentration of furdan,
indicates that pol , recA and lexA ars relatively
more sensitive strains as compared to the recB mutant
and the wiIdtype-strains.
Discussion
Bacterial injury caused by UV and ionizing
radiations has been well documented (Auerbach, 1976;
Howard-Flanders, 1968; Witkin, 1976; Friedberg, 1985).
Cellular DNA damage occurs following exposure to
ultraviolet light, ionizing radiation, various
environmental chemicals and reactive oxygen species. If
left uncorrected, it can result in cell death, mutation
and neoplastic transformation. Prokaryotes and
eukaryotes have evolved systems for reversing DNA
65
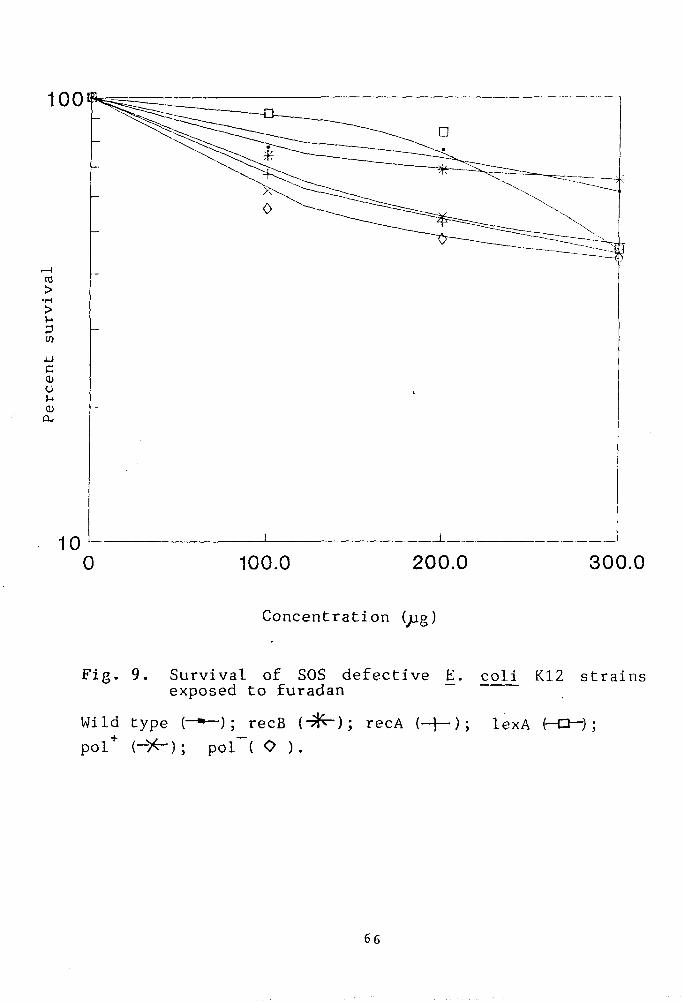
100.0 200.0 300.0
Concentration (jag)
Fig. 9. Survival of SOS defective E. coli K12 strains exposed to furadan
Wild type { — ) ; recB (- i ); recA (-H); lexA (-O-) ; pol* (-><-); pol~( O ).
66

damage, and several classes of DNA repair enzymes have
been identified (Friedberg and Bridges, 1983).
The pesticides which were detected to be mutagenic
in the Ames testing system were also tested with SOS
repair defective recA , recB , lexA , uvrA and polA
mutants of E.coli K-12. It is postulated that the
inducible error—prone repair pathway could potentially
operate on all types of lesions in DNA whether produced
by radiation or by other agents (Walker 1785). These
mutants were found to be highly sensitive to the
pesticides (Figures 6, 7, 8 and 9) suggesting thereby,
the damage to the DNA of the exposed cells as well as
the role of polA, recA, recB. uvrA and lexA genes to
cope with the hazardous effect of the pesticide. polA
mutant was found to be most sensitive to all the four
pesticides. The role of recA and lexA gene in
initiating the error prone repair in E.coli is well
documented (Witkin, 1976; Walker, 1985)This idea gains
further support by Ames testing studies (Figures 1, 2,
3 ,4 and 5) with Salmonella strains triggering the
error prone SOS response.
67

CHAPTER-V
DISCUSSION

DISCUSSION
Our studies on the mutagenicity and genotoxicity
of the pesticides viz captan, foltaf, phosphamidon and
furadan have provided, a complete profile of the
mutagenic potential as well as insights into their
mechanism of repair in bacteria. It is generally
believed that a large proportion of human cancer is
mainly due to the exposure to genotoxic chemicals
present in the environment. Ironically, very few of
them have been tested for their carcinogenic and /or
mutangenic activities. It is, therefore, emphasized to
identify the entire spectrum of chemical carcinogens by
performing the short-term mutagenicity and
carcinogenicity assays in combination with
epidemiological investigations. Among the plethora of
environmental mutagens, pesticides are considered to
belong to a noxious chemical class of compounds, posing
a major threat to the global environment, A majority of
them have been reported to be mutagenic, carcinogenic
and teratogenic (Lucier and Rambush, 1983; M£tzler,
1984; Dunkel et. al., 1985). Unfortunately due to lack of
a national database, no accurate or current statistics
are available regarding the incidence of acute
pesticide poisoning in this country. However, in the
United States atleast 30 to 40 persons die of acute
poisoning each year and approximately 20,000 persons
Are taken to emergency room each year for actual or
68

suspected pesticides poisoning (Weisenburger, 1993). In
fact acute poisoning is thought to be a major global
health problem with approximately 25 million
occupational poisoning per year (Jeyaratnam, 1990).
Most of these occur in third-world countries where
pesticide education and monitoring and the use of
safety equipments are largely non-existent.
The epidemiological data have indicated that most
of the cancer is due to the environmental factors
(Ramel, 1991). Consequently, the identification of
carcinogens in the environment is of fundamental
importance in cancer prevention. This has actually
prompted us to evaluate the genotoxic and mutagenic
potential of certain pesticides. The four selected
pesticides tested in this study have shown a
differential mutagenic response in the following order,
capton > foltaf > phosphamidon > furadan. As indicated,
captan, a highly effective broad spectrum antimicrobial
agent (Dillwith and Lewis, 1982a) was found to be most
mutagenic among all the pesticides tested. However, its
mutagenic activity was observed to be decreased in
presence of S9 mix suggesting that captan could be a
direct acting mutagen and does not require any
metabolic activation. In fact, its catabolism resulted
in the production of relatively less mutagenic
intermediates as evident in Fig. 2. This attenuated
mutagenic response of captan in presence of S9 mix is
in accordance with the earlier reported results (
69

Moriya et. aj_ 1978). Captan was reported to be mutagenic
with the strains that detect the mutagens damaging
G > C base pairs (Bridges et, al_j 1972; Xu and
Schurr 1990). It is now clear from our results that
captan also exhibits positive mutagenic response with
the strain TA104, which suggests that it could also
induce A > T base pair substitution. We have also
noticed about 1.6 to 13 times higher mutagenic response
of captan with different tester strains under our
experimental conditions; compared to the reported
values . As expected captan was found to be relatively
less mutagenic in strain TA102, the strain with intact
excision repair system, suggesting that the captan
induced DNA lesions are repairable by the enzymatic
repair mechanism.
Other pesticides including foltaf, phosphamidon,
and furadan have been identified as comparatively weak
mutagens. Although, all of them have exhibited
mutagenic response to a certain extent in strains TA104
and TA102, none of them were significantly responsive
with strain TA100. Comoarison of the test pesticides on
the basis of their structure-activity relationship
revealed that the tricnloromethy1thio moiety of captan,
is primarily responsible for the formation of adducts
with DNA. Although, foltaf has a reactive group similar
to captan, but the substitution of its terminal CHCI2
group seems to be more difficult than in captan.
70

However, the mutagenic profile of foltaf suggests its
broad specificity for inducing both the transition and
frameshift mutations in DNA. Our studies revealed that
the most sensitive loci were his6428 and hi5(ai242 in
the multi-copy plasmid pkMi01 inside the tester strains
TA102 and TA97a respectively. Since Foltaf exhibited
maximum mutagenic response in excision repair
proficient strain TA102, it could possibly be due to
the partial removal of lesions by uvrABC excinuclease
and might have involved the error prone type of
mechanisms in repairing the damage at the expense of
mutations at the repaired sites.
Similarly, phosphamidon contains only one chlorine
2
atom a t t a c h e d t o sp - c a r b o n ( v i n y l i c c h l o r i d e ) and
t h e r e f o r e , i t s s u b s t i t u t i o n w i l l be even more d i f f i c u l t
than in f o l t a f . Moreover , t h e p l a u s i b l e r e a s o n fo r l e s s
mutagenic a c t i v i t y in c a s e of fu radan c o u l d be t h e
absence of hydrogen m o e i t y . A p p a r e n t l y , t h e on ly
r e p l a c e a b l e g r o u p i s -NHCH3, which i s supposed t o be a
poor l e a v i n g g r o u p . The a v a i l a b i l i t y of an e a s i l y
r e p l a c e a b l e e l e c t r o p h i l i c f u n c t i o n a l g r o u p in a
c a r c i n o g e n i s t h e r e f o r e an i m p o r t a n t f a c t o r in
n u c l e o p h i l i c s u b s t i t u t i o n r e a c t i o n w i t h DNA. Thus u s i n g
a s e t of S a l m o n e l l a typhimur ium s t r a i n s we have ,
e s t a b l i s h e d t h e m u t a t i o n a l s p e c i f i c i t y of t h e t e s t
p e s t i c i d e s w i t h o u t i n v o l v i n g any o t h e r complementary
a s s a y .
71

Our studies on the genotDxic effect of the
pesticides on DNA repair defective mutants of E . coli.
have also clearly suggested the role of pol. recA and
lexft genes in the repair of the pesticide induced DNA
damage . The mutants polA , recA and lexA have shown
a significant decrease in their survival upon exposure
to the pesticides. In the case of captan, foltaf and
furadan, the wild-type AB1157 has also exhibited a
decline in the survival curve particularly at a higher
concentration range which could be due to the
inhibition of the cellular metabolic machinery. There
a.re reports indicating that captan inhibits the
activity of DNA polymerase I (Dillwith and Lewis,
1982b), It also inhibits both the DNA and RNA synthesis
(Dillwith and Lewis, 1982b) by blocking the binding of
polymerase to the DNA template. The reduced survival of
wild type strain upon treatment with foltaf, furadan
and phosphamidon could be due to similar reasons. Since
the role of recA and lexA genes in initiating the
error—prone repair in E^. col i is well documented
(Witkin, 1976; Walker, 1985), the involvement of these
genes in our repair study led us to suggest the role of
inducible error—prone repair pathways in the enzymatic
removal of pesticides induced lesions in DNA.
72

CHAPTER-VI
REFERENCES

REFERENCES
Ames, B.N. (1971)- In"Chemical Mutagens: Principles and methods for thier detection". Hollaender, A; Plenum Press. N.Y. 267.
Ames, B.N.; Lee, F.D. and Durston, W.E. (1973)- Proc. Natl. Acad. Sci.(U.S.A.) 72, 2423.
Ames, B.N.; McCann, J. and Yamasaki, E. (1975)- Mutat. Res. 31, 347.
Ames, B.N. (1979)-Science, 204. 587.
Ames , B.N. (1984) -Citation Classic Curr. Cont. 27 No. 12., 18.
Ames, B.N.; liagaw, R. and Gold, L.S. (1987 )-Science, 236. 271.
Ames, B.N. (1989)- ACS Symp. Ser. 414. 223.
Anon (1973)- FAG Agricultural Series, No.90, P47.
Ashby, J. and Tennant, R.W. (1988)- Mutat. Res. 204. 17.
Auerbach, C. (1976)- In "Mutation Research." John Wiley and Sons, N.Y.
Barnes, W.; Tuley, E. and Eisenstadt, E. (1982)- In "13th Annual Meeting of the Environmental Mutagen Society Prog, and Abstracts. No. Aa - 1, 59.
Bartsch, H. ; MalaveiUe, C ; Camus, R.A. M.; Martel-Planche, G.; Brun, G.; Hantefeuille. A.; Sabadie, N.; Barbin, A.; Kuroki, T.; Drevon, C ; Picco^fi, C. and Montesano, R. (1980)- Mutat Res. 76., 1.
Bebenek, K. and Janion, C. (1985) - Mol. Gen. Genet. 201. 519.
Benbow, R.; Zuccarelli, A. and Sinsheimer, R.L. (1974) - J Mol. Biol. 88, 629.
Bishop, M. (1991)- Cel. 64.: 235.
73

Blair, A.; Axelson, 0. ; Franklin, C. et, AI_. (1990)-Carcinogenic effect of pesticides in Baker, S. R, Wilkinson, C.F (eds): The effect of pesticides on Human Health, Advances in Modern Environmental Toxicology, Princeton, NS, Princeton Scientific, 18.: 201.
Blair, A. and Zahm, S.H. (1991)- Cancer among farmers in Cordis, D.H.; Rea, D.F. (eds) Health hazards of farming. Dccupatinal Medicine: State of Art Reviews. b_; 335.
Bohr, V.A.; Smith, C.A.; Okumoto, D.S. and Hanawalt, P.C. (1985)- cell. 40, 359.
Bohr ,V.A.; Okumoto, D.S. and Hanawalt, P.C. (19e6a)-Proc. Natl. Acad. Sci. (USA). 83., 3830.
Bohr, V.A.; Okumoto, D.S.; Ho, L. and Hanawalt, P.C. (1986b)- J. Biol. Chem. 262. 16666.
Bohr, V. A. (1991)- Carcinogenesis. 12., 1983.
Boiteux, S.; Villani, G.; Spadari, S.; Zambrano, F.and Radman, M. (1978) - In "DNA Repair Mechanisms" Symp. Mol. Cell. Biol. (eds). P.C. Hanawalt; E.C. Friedberg and C.F. Fox Academic Press. N.Y. =•• 73.
Boone, C.W.; Kelloff, G.J. and Steele, V.E. (1992)-Cancer Res. 52. , 1651.
Boveri, T. (1914) - Zur. Frage. der. Entstehung. Malign. Tumoreu. Jena. Gustav Fisher.
Bowman, M.C. (ed.-1982)- In "Handbook of Carcinogens and Hazardous Substances". Marcel Dekker, Inc. New York and Basel.
Boyce, R.P. and Howard - Flanders, P. (1964) - Proc. Natl. Acad. Sci. (U.S.A.) 51., 293.
Boyle, W.J. (1992)- Curr. Opin. Oncol. 4, 156.
Brent, R. and Ptashna, M. (1981) - Proc. Natl. Acad. Sci. (U.S.A.) 78 , 4204.
Bridges, B. A.; Moltershead, R.P. ; Anne Rothwell, A.M. and Green, M.H.L.(1972) - Chem. Biol. Interact. 5, 77
74

Burnet, M. (1974) - In "Intrinsic Mutagenesis" Medical and Technical Publ. Co. Ltd. Lancaster, U.K.
Cairns, J. (1975) - Sci. Am. 233. 64.
Cardona, R.A. (1987)- In "Pesticides-Minimizing the Risks". ACS Symp. Ser. 336. Amer. Chem. Sac., Washington, D.C.
Carere, A. and Mospurgo, 6.(1981) - Prog. Mutat. Res. 2, 87.
CFR 40 162.3 (1986) - Protection of Envircnment, Office of the Federal Register, National Archives and Records Administration, U.S. Govt. P-inting Office, Washington, D.C.
Chaudhury, A.M. and Smith, G.R. (1985)- Mol. Gen. Genet. 201,525.
Clarke, C.H. (1971) - Mutat. Res. 11., 247.
Cohn, S.M. (1983) - Diss. Abstr. Int. B. 43. , 27^3.
Cooper, 6.M. (1982) - Science, 217, 801.
Crawford, B.D. (1985) - In "Advances in Modern
Environmental Toxicology" (eds.) W.6. Flames and R.J. Lorentzen. Princeton Scientific. Prirceton, N.J. p.13-39.
CuUiney, T.W. ; Pimental, D. and Pimentel , M.H. (1992) - Agric. Ecosystems Environ. 41_ , 297.
Defais, M. ; Foquet, P. ; Radman, M, and Erre-^a, M. (1971) - Virology, 43 , 495.
Deng, G. ; Chen, Ling-chun; Schott, D.R.; Thor, A. ; Bhargava, V.; Ljung, B.M.; Chew, K and Smit", H.S. (1994)- Cancer Research. 54 : 499.
deSerres, F.J. & Ashby, J. (eds.).(1981 . In "Evaluation of short-term tests for carcinogens." Reports of the International collabcrative Program. Elsevier/North-Holland, Amsterdam.
Devoret, R. (1979) - Sci. Am. 241 ,20.
Dillwith, J.W and Lewis, R.A. (1982a)- Biochem. Biophys. Acta. 696. 245.
75

Dillwith, J.W. and Lewis, R.A. (1782b)- Biochem. J. 201. 145.
Doll, R.and Peto, R. (1981) - J. Natl. Cancer Inst. 66. 1129.
Donna, A. ; Crosignani, P. and Robutti, F. (1989)-Scand. J. Work Environ. Health. 1_5., 47.
Dunn, K.; Chrysogelos, S. and Briffith, J. (1982) Cell, 28., 575.
Dunkel, V.C.; Zieger, E; Brusick, D; Mccoy, E; McGregor, D; Mortelmans, K; Rosenkranz, H. S. and Simmon, V.F. (1985)- Environ Mutagen 1 , 1.
Durstan, W.E. ; Yamasaki, E. and Lee, F.D. (1973) Proc. Natl. Acad. Sci. (U.S.A.). p 2281.
Dyrby, T. and Ingvardsen, P. (1983) - Mutat. Res. 123 , 47.
Elkind, M.M. and Whitmore, 6.F. (1967)- In "The Radiobiology of Cultural Mammalin cells. Gordan and Braden Pub., N.Y.
Farmer, P.B. (1982) - Chemistry in Britain, 18. , 790.
Finch, D.W.; Chambers, P. and Emmerson, P.T. (1985) J. Bacterial. 164 ,653.
Fink, L. (1984) - Bioscience, 34. , 75.
Flessel, P.Y. ; Wang, Y. ; Kuo In Chang and Wesolowski, J.J. (1987) - J. Chem. Education, 64 , 391 .
Florey, H. (1962) - General Pathology, 3rd eaition. London .
Flory, J. and Padding, C M . (1982) - Cell, 28, 7^7.
Friedberg, E.C. and Bridges, B (eds.) (1983)- Cellular Response to DNA Damages. Liss, New York.
Friedberg, E.C. (1985) - "DNA Repair", p 614.
Garrett, C.T. (1986)- Clin. Chim. Acta. 156. 1.
76

Barrett, N.E.; Sandhu,S.S.; Slack, H.F.and Waters, M,D. (1987) - U.S. Environ. Prat. Agency Res. Dev. EPA . EPA/600 D-87/153, 24 PP.
Georgian, L. (1975) - Mutat. Res. 31., 103.
Gocke, E. ; King, N.T.; Eckhardt, K. and Wild, D. (1981) - Mutat. Res. 90. , 91.
Sottesman, S.(1981) - Cell, 23, 1.
Gupta, B.N. (1989) - Indian J. Environ. Prot. 9, 801.
Hansen, M.F. and Cancenee, W.K. (1988) - Trends Genet. 4, 125.
Harris, C.C. (1985) - Environ. Health Perspect. 6^ , 185.
Hart, R.W.; Steven, M.; O'Ambrosio ; Kwokei, J.N.G.5 Sohan, P. and Modak, S.P. (1979) - Mechanisms of Aging and Development, 9 , 203.
Hatch, F.T.; Felton, J.S. ; Stuermer, D.H. and Bjeldanes, L.F. (1984) - In "Chemical Mutagen". Principles and Methods for their Detection (ed.) F.D. de Serres. (Plenum, N.Y.) 9, 111.
Hay, A. (1988) - Nature, 322. , 782.
Hayashi, Y. (1992)- Exp. Toxic. Pathol. 44., 465.
Hermanowicz, A. and Kossman, S. (1984)- Clin. Immunol. Immunopathol . 33., 13.
Hollstein, M.J.; Mc Cann, J.; Angelosanto, F.A. and Nichols, W.W. (1979)- Mutat. Res. 69., 173.
Heyneker, H.L. and Klenow, H.(1975) - In 'Molecular Mechanisms for Repair of DNA" (eds.) P.C. Hanawalt and R.B. Setlow. Part A, Plenum Press, MY, P219.
Hill, R.F. (1958) - Biochim. Biophys. Acta, 32., 636.
Hollaender, A. and Curtis, J.T. (1935) - Froc. Soc. Exp. Biol. Med., 33^, 61.
Hollstein, M. ; fIcCann, J. ; Angelosanto, F.A. and Nichols, W.W. (1979) - Mutat. Res. 65, 133.
77 r Ace No. V

Hoover, R.N. and Blair, A. (1991)- Pesticides and Cancer, in Devita, V. ; Helmans, Rosenberg, S. (eds.). Cancer Prevention, P.A. 1.
Houk, V.S.and De Marini, D.M. (1987) - Mutat. Res., 182 , 193.
Howard-Flanders, P.; Boyce, R.P. and Theriot, L. (1966) - Genetics, 53., 1119.
Howard - Flanders, P. (1968) - Ann. Rev. Biochem. 37. 175.
Howard - Flanders, P.; Cassuto, E. and Ross, , P.(1981) -Genet. Kongr. i£, 148.
Husain, I.; van Houten, B.; Thomas, D.C. ; Abdel-Monem, M. and Sancar, A.(1985) - Proc. Natl. Acad. Sci. 82. , 6774.
lARC Monographs on the Evaluation of Carcinogenic Risk of Chemicals to Man (1972-1984) vols. 1-35.
Innes, J.R.M.; Ulland, B. M.; Valerio, M.G.; Petrucelli, L. ; Fishbein, L.; Hart, E.R. ; Pallota, A.J.; Bates, R.R. ; Falk, H. L.; Gart, J. T.; Klein, M. ; Mitchell, I. and Peters, J. (1969) - J. Natl. Cancer. Inst. 42., 1101.
IPCS (1990a) - International Programme on Chemical Safety. Health and Safety Guide No.49 - CAPTAFOL. Pub. WHO for the IPCS, Geneva.
IPCS (1990b) - International Programme on Chemical Safety. Health and Safety Guide No. 50 -CAPTAN, Pub. WHO for the IPCS, Geneva.
Ishi, V. and Kondo, S. (1974) - Mutat. Res. ZL^ 27.
Isono, K. and Yourno, J. (1974) - Proc. Natl. Acad. Sci. /ll, 1612.
lyaniwara, T.T. (1991) - Rev. Environ. Health, 9 , 47.
Iyer , V.N. and Rupp, W.D.(1971) - Biochim. Biophys. Acta, 228., 117.
Jain, V. (1992) - Environ. Sci. Technol . 26., 226.
78

Jeyaratnam, T. (1990)- World Health Stat Q. 43, 139.
Kada, T.; Moriya, M. and Shirasu, Y. (1974) - Mutat. Res. Z6, 243.
Kannan, K.; Tanabe, S.; Ramesh, A.; Subramanian, A. and Tatsukawa, R. (1992) - J. Agric. Food Chem. 40. 518.
Kapadia, G.J.(1982) - Oncology overview on naturally occurring dietary carcinogens of plant origin. International Cancer Research Data Bank Prog. Natl. Cancer Inst., Bethesda, Maryland.
Kaufmann, W.K. and Kaufman, D.G. (1993)- The FASEB Journal. 7, 1188.
Kelner, A. (1949) - J. Bacteriol. 58, 551.
Kenyan, C.J. (1983) - TIBS, 8, 84.
Khosravi- Far, R. and Der, C.J. (1994)- Cancer and Metastasis Reviews . 1^, 67.
Kier, L.D.; Brusick, D.J. ; Auletta, A.E.; von Halle, E.S.; Brown, M.M.; Simmon, V.F.; Dunkel, V.; McCann, J.; Mortelmans, K.; Prival, M.; Rao, T.K. and Ray, V.(1986) - Mutat. Res. 168, 69.
Knudson, A.G, (1971)- Proc. Natl. Acad. Sci. (USA). 68., 820.
Kondo, S.; Ichikawa, H.; Iwok and Kato, T.(l?70) Genetics, 66., 187.
Kovel, T.M. (1986) - Mutat, Res. 173., 291.
Kross, B.C.; Vergara,A. and Raue, L.E. (1992) - J. Toxicol. Environ. Health. 37., 149.
Levin, D.E.; Yamasaki, E.and Ames, B.N. (1982a) Mutat. Res. 94., 315.
Levin, D.E.; Hollstein M. ; Christman, M.; Schwier, E.A. and Ames, B. N. (1982b)- Proc. Natl. Acad. Sci. (USA) 79_. 7445.
Levin, D.E.; Hollstein, M.; Christman, M.F. and Ames, B.N. (1984) - Methods in Enzymol., 105. 249.
79

Little, J.W.; Mount, D.W. and Yanisch - Perron, C.R. (1981) - Proc. Natl. Acad. Sci (U.S.A). 78, 4199.
Little, J.W. and Mount, D.W. (1982) - Cell, 29, 11.
Little, J.W. (1984) - Proc. Natl. Acad. Sci. (U.S.A.) 81. 1375.
Lloyd-Lude; Trosko, J.E. and Chang, C.C. (1978) Photochem. Photobiol . Rev. Z_, 135.
Lohman, P. H. M and Bootsma, D. (1974)- Mutat. Res. 23. 147.
Lowengart, R.A.; Peters, J.M. and Cicioni, C. (1987)-J. Natl. Cancer Inst. 79., 39.
Lu, A.Y.H.; Kuntzman, R.; West, S.; Jacobson, M. and Conney, A.H. (1972) - J. Biol. Chem. 247. 1727.
Lu, C ; Scheuermann, R.H. and Echols, H. (1986) Proc. Natl. Acad. Sci. (U.S.A.) 83., 619.
Lucier, 6.W. and Rambush, R.C. (1983)- lARC. Sci. Publ . 51. 49.
Mahr, U. and Miltenburger, H.G. (1976) - Mutat. Res. 40. 107.
Manske, D. D. and Johnson, R.D. (1977) - Pestic. Monit. J. 10., 134.
Maron, D.M. and Ames, B.N. (1983)- Mutat. Res. 113. 173.
Marshall, C.J. (1991)- Cell. 61, 313.
McCann, J.; Choi, E.; Yamasaki, E. and Ames, B.N. (1975) - Proc. Natl. Acad. Sci. (U.S.A.) 72, 5135.
McCann, J. and Ames, B.N. (1976) - Proc. Natl. Acad. Sci. (U.S.A.) - 73, 950.
McCann J. and Ames, B.N.(1977). In "Origins of Human Cancer".(eds.) Hiott, H.H.; Watson, J.D. and Winsten, J. A; Cold Spring Harbor Lab. N.Y. 1431.
McCormick, A. G. (1989)- Cell, 56., 5.
80

Mellon, I.; Bohr, V.A.; Smith, C.A. and Hanawalt, P.C. (1986)-Proc. Natl. Acad. Sci (USA) 83, 8878.
Mellon, I.; Spivak, G. and Hanawalt, P.C. (1987)- Cell. 51, 241.
Mellon, I. and Hanawalt, P.C. (1787)-Nature. 342. 75.
Metzler, M. (1784). Arch. Toxocol . 55., 104.
Miller, E.C. (1778) - Cancer. Res. 38., 1477.
Miller, J.A. and Miller, E.C. (1777) - In "Origins of Human Cancer", Cold Spring Harbor, N.Y. 605.
Miller, J.M. and Heflich, R.H. (1782) - Chem. Biol. Interact. 37., 45.
Miller, J.M. (1783) - Ann. Rev. Genet. 12., 215.
Modak, S.P. (1772) - Cell Differentiation (eds. ) R. Harris; P. Allin and D. Viza. p 437-442.
Moriya, M.; Kato, K. and Shirasu, Y. (1778) - Mutat. Res. 5Z, 257.
Moriya, M.; Ohta, T.; Watanabe, K. ; Miyazawa, T.; Kato, K. and Shirasu, Y. (1783) - Mutat. Res. 116. 185.
Mosbaugh, D.W. and Linn, S. (1782) - J. Biol. chem. 257. 575.
Miura, A. and Tomizawa, J. (1768) - Mol.Gen. Genet. 103. 1
Mottram, J.C. (1744)- J. Pathol. Bacterial. 56., 181.
Nestman, E.R.; Renata , L.B.; Marcia, P.; McPherson and Luria, K.M. (1787) - Environ. Mol. Mutagen. 10. 167.
Newbold, R.F. (1785)-, Multistep malignant transformation of mammalian cells by carcinogens, induction of immortality as a key event. In carcinogenests (Barrettt, J.C. and Tennant, R.W., (eds) Vol. 7, 17 Raven press, N. York.
Nishimoto, T.; Uzawa, S. and Schlegel, R. (1972)- Curr. Opin. Cell. Biol. 4, 174.
81

Patankar, N. and Vaidya, V.6. (1980) - Indian J. Exp. Biol, ra, 1145.
Pearson, A.; Wang, A.; Solez, K. and Haplinstall, R.H.(1975) -Am. J. Pathol. 81., 379.
Pednekar, M.D.; Bandhi, S.R. and Netrawali, M.S. (1987) - Bull. Environ. Contam. Toxicol.38,, 925.
Perera., F. (1988) - Science, 239. 1227.
Piechocki, R.; Kupper, D.; Quinones, A. and Langhammer, R. (1986) - Mol. Gen. Genet. 202, 162.
Pitot, H.C. (1993)- Cancer suppl . 72. , 962.
Ponder, B. (1988) - Nature, 335, 578.
Price, G.B. & Makinodan, T. (1973)- Gerontologia. 19. 58.
Prins, R.A. (1978) -In "Nutrition and Drug Interrelations", (eds.) J.N. Hathcock and J. Coon. Academic Press, New York P. 189.
Purchase, I.F.H.; Longstaff, E.; Ashby, J.; Styles, J.A.; Anderson, D.; Lefevre, P.A. and Westwood, F.R. (1976) - Nature, 264. 624.
Radman, M. (1974) - In "Molecular and Enviramental Aspects of Mutagenesis" (eds.) L. Prokash; F. Sherman; M. Millor; C. Lawrence and H.W. Tacor. p 128.
Radman, M. (1975) - In "Molecular Mechanisms for Repair of DNA" (eds.) P.C. Hanawalt and R.B. Setlow Part A. Plenum Press. N.Y. p. 355.
Radman, M. ; Villani, G.; Boiteux, S.; Defais, H.; Caillet, F.P. and Spadari, S. (1977) - In "Origins of Human Cancer" (eds.) H. Hiatt; J.D. Watson and J.A. Winsten. Book B. N.Y. p 903.
Ramel, C. (1991)- Reviews in Oncology. 4, 27.
Rashid, A.K. and Ralph, O.M. (1986) - J. Environ. Sci. Health. B21. 319.
82

Rasquinha, I.A.; Wildeman, A.G. and Nazar, R.N. (1988) - Mutat. Res. 177. 261.
Rastogi, S.K.; Gupta, B.N.; Husain, T. (1989)- Am. J. Indust. Med. 16., 529.
Ray, P.K. (1986) - Environmental Pollution and Cancer. The Prof. S.C. Roy Commemoration Medal Lecture delivered at the Life Sciences Centre, University of Calcutta.
Ray, V.A. et. aJL, (1987) - ibid, 185. 197.
Register III, J.C. and Griffith, J. (1986) - Proc. Natl. Acad. Sci. (U.S.A.) 83., 624.
Reuber, M.D. (1989) - J. Environ. Pathol. Tcxicol. Oncol. 9, 127.
Rosenkranz, H.S.; Klopman, G.; Chankong, V.; Pet, J. and Haimes, Y.Y. (1984) - Environ. Mutagen. 3., 231,
Rosenkranz, H.S. and Ennever, F.K. (1990) - Mutat. Res. 244. 61.
Rous, P. and Kidd, J.G. (1941)- J. Exp. Med. 73., 365.
Rozengurt, E. (1992)- Curr. Opin. Cell. Biol. 4, 161.
Rupert, C.S.; Goodgal, S.H. and Herriott, P.M. (1=58) -J. Gen. Physiol. 41_, 451.
Rupert, C.S. (1962) - J. Gen. Physiol. 45., 724.
Rupp, W.D. and Howard - Flanders, P. (1968) - J. Mol . Biol. 31, 291.
Ryberg, D. ; Kure, E.; Lystad, S.; Skang, V.; Etange land, L.; Mercy, I. ; Borresen, A.L. and Hangen, A. (1994) Cancer Res. 5£, 1551.
Saffhill, R.; Margison, G.P. and O'Connor, P.J. .1985) - Biochim. Biophys. Acta, 823. 111.
Salles, B.; Paoletti, C. and ViUani, G. (1983) - Mol. Gen. Genet. 189. 175.
Salles, B. and Defais, M. (1984)- Mutat. Res. 13., 53.
83

Sancar, G.B.; Sancar, A.; Little, J.W. and Rupp, W.D. (1982) - Cell, 28., 523.
Scheuermann, R.; Tam, S.; Bergers, P.M.J.} Lu, C. and Echols, H. (1983) - Proc. Natl. Acad. Sci. (U.S.A.) 80, 7085.
Schild, D.; Johnson, J.; Chang, C. and Mortimer, R.K. (1984) - Mol. Cell. Biol., 4, 1864.
Schmandt, R.and Mills, G.B. (1993)- Clin. Chem. 39, 2375.
Schuetzle, D. and Lewtas, J. (1986) - J. Anal. Chem. 58. 1060.
Seeberg, E. (1981) - Prog. Nucleic Acid Res. Mol. Biol., 26., 217.
Seibert, D.; Zimmermann, F.K. and Lemperle, E. (1970) -Mutat. Res. 10., 533.
Seller, J.P. (1973) - Experientia, 29., 622.
Setlow, R.B. and Carrier, W.L. (1964) - Proc. Natl. Acad. Sci. (U.S.A.) 5J , 226.
Shanabruch, W.G. and Walker, B.C. (1980)- Mol. Gen. Genet. 179., 327.
Sharma, V.K. and Kour, S. (1990)- Contact Sensitization by pesticides in farmers. Contact Dermatilis. 23., 77.
Shelby, M.D. and Stasiewiez, S. (1984) - Environ. Mutagen., 6., 871.
Shirasu, Y.; Moriya, M.; Kato, K.; Furuhashi, A. and Kada, T. (1976) - Mutat. Res. 40., 19.
Shu H e r , D.E.G. (1987) - Nature. 329.
Simmon, V.F. (1978) - In vivo and in vitrao mutagenicity assays of selected pesticides. In "A Rational Evaluation of Pesticidal Vs. Mutagenic/Carcinogenic Action." (Eds.) W. Hart.; H.F. Kraybill and F.J. de Serres. DHEW Publ. No. (NIH) 78 -1306.
84

Sobels, F.H. (1984) - In "Genetics: New Frontiers", Proceed-ings of the XV International Congress of Genetics. (eds.) V.L. Chopra; B.C. Joshi; R.P. Sharma and H.C. Bansal, 3., 63.
Stevnsner, T. and Bohr, V.A. (1993)- Carcinogenesis. 14. 1841.
Straus, D.S. (1981) - J. Natl. Cancer, Inst. 67., 233.
Sugimura, T.; Matsushima, Y.; Takauchi, M. and Kawachi, T. (1976) - In "Fundamentals in Cancer Prevention" (eds.) P.N. Magee et al. University of Tokyo, Press, Tokyo.
Sugimura, T. (1985) - Mutat. Res. 150, 33.
Sweasy, Joann B.; Wilkin, Evelyn M.; Sinha, N.; Reogner Maniscalco, V. (1990)- J. Bacterial 172. 3030.
Szent -Gyorgyi, A. (1977) - Proc. Natl. Acad. Sci. (U.S.A.) 74, 2844.
Takahashi, M.; Daune, n, and Schnarr, M. (1986) - FEBS Letters, 196. 215.
Tassignon, J.P. (1985) - Food Chem. Toxicol. 23., 5.
Temin, H.M. (1974)- Ann. Rev. Genet. 8., 155.
Tennant, R.W.; Margolin, B.H.; Shelby, M.D,; Zeiger, E.; Haseman, J.K.; Spalding, J.; Caspary, W.; Resnick, M. Stasiewicz, S.; Anderson, 3. and Minor, R. (1987) - Science, 236., 933.
Terlleth, C. ; van Sluis, C.A. and van de Putte, P. (1989)- Nucleic Acids Res. 17., 4433.
Thomas, D.C.; Morton, A.G.; Bohr, V.A. and Sancar, A. (1988)- Proc. Natl. Acad. Sci (USA) 85., 3723.
Thomas, P.T.; Busse, W.W. and Kerkvliet, N. I. (1990)-Immunologic effects of pesticides, in Baker, SR, Wilkinson, C.F.(eds): The effects of pesticides on Human Health. Advances in modern environmental Toxicology. 18., 261.
Umorasaite - Rancelience, V.; Pinto, R.I.; Barenfeld, L.S. and Cieminis, K.(1986) - Tositologiya, 2B., 86.
85

Usha Rani, M.V.; Reddi, O.S. and Reddy, P.P. (1980) -Bull. Environ. Contam. Toxicol. 25., 277.
van Houten, B. (1990)- Microbiological Rev. 54_, 18.
van der Hoeven, N.; Kooijman, S.A. L.M. and De Raat, W.K. (1990)- Mutat Res. 254. 289.
Venema, J. ; Mullenders, L.F. ; Natrajan, A.T.; van Zeeland, A.A. and Mayne, L.V. (1990)- Proc. Natl. Acad. Sci (USA) 87., 4707.
Venema, J.; van Hoffen, A. ; Natrajan, A.T.; van Zeeland, A.A. and Mullenders, L.H.F. (1991)- Mol. Cell. Biol. 11, 4128.
Villani, G.; Boiteux, 3. and Radman, M. (1978, - Proc. Natl. Acad. Sci. (U.S.A.) 7 ., 3037.
Vishwanath, R. and Ja.-nil, K. (1986) - Indian J. Exp. Biol. 2A_ 305.
Vogelstein, B. et. al (1988) - J. Med. 319. 525.
von Rumker, R.; Lawless, E.W.; Meiners, A.G.; Lawr-ence, K.A.; Kelso, 6. L. and Moray, F. (1975) - E=A 540/1-74-001. Washington, D.C., U.S.A.
Walker, B.C. (1984) - Microbiol. Rev. 48., 60.
Walker, G.C. (1985) - Ann. Rev. Biochem. 5£, -2-25.
Walker, G . C ; Marsh, L, and Dodson, L.A. (19S5) - Ann. Rev. Genet. i9_, 133.
Waters, M.D.; Simmon, V.F.; Mitchell, A.D.; Jorcenson, T.A. and Valencia. R (1980) - J. Environ. Sci. Health, B15, 867.
Waters, M.D. and Auletta, A.(1981) - J. Chem. Inf. Comput. Sci. 21, 35.
Weisenburger, D. D. (1993)- Perspectives in Pathology. 21, 571.
Wilkins, J.R. and Sinks, T. (1990) - Am.J. Epidemiol 132. 275.
Witkin, E.M. (1976). Bacteriol. Rev. 40, 869.
86

Wogan, G.N. and Gorlick, N.J. (1985) - Environ. Health. Perspect. 62., 5.
Wolff, P.S. (1984) - In"6enetics : New Frontiers". Proceedings of the XV International Congress of Genetics. (Eds.) V.L. Chopra; B.C. Joshi; P.P. Sharma and H.C. Bansal. Oxford and IBH Publ. Co. New Delhi, 3, 3.
Wong, P.K.; Wai, C.C. and Liong, E. (1989) Chemosphere, 18., 2413.
Xu, H.H. and Schurr, K.M.(1990) - Toxic. Assess. 5., 1.
Young, A.L. (1987) - In "Pesticides : Minimizing the Risks (ACS Symposium Series 336). American Chemical Society, Washington, D.C.
Younos, T.M. and Weigmann, D.L. (1988)- Journal WPCF. 60: 1199.
Zeiger, E. (1987)- Cancer Res. 47., 1287.
87

B I O G R A P H Y
Ms. Sangeeta Saxena was born at Dehradoon (Uttar
Pradesh) on March 15,1968. She passed her High School
Examination in 1983 from A.S.S. Inter College, Hardwar.
She received her B.Sc. (Hons.) degree in 1988 and
Masters degree in Microbiology in 1990 from Aligarh
Muslim University, Aligarh. She was enrolled as a Ph.D.
student in July, 1992 in the Interdisciplinary
Biotechnology Unit, Aligarh Muslim University, Aligarh.
She is a member of the Society of Biological Chemists
(India).
88