structure based pharmacophore, virtual screening...
TRANSCRIPT

Virtual Screening and Antibacterial Assay
173
STRUCTURE BASED PHARMACOPHORE, VIRTUAL SCREENING AND ANTIBACTERIAL ACTIVITY OF NOVEL MOLECULES
Among all the virtual approaches, structure-based pharmacophore (SBP) and molecular
docking based Virtual Screening (VS) are probably the most efficient methods to identify
potentially potent compounds from chemical databases containing a large amount of
molecules (Valasani et al., 2013). Virtual Screening is a knowledge driven process that
uses computational chemistry techniques to analyze large chemical databases in order to
identify possible new leads (Valasani et al., 2013). Virtual Screening is used as an initial
screen for large databases to prune the number of compounds that are to be screened
experimentally (Lyne et al., 2002; Liu et al., 2013). This process of finding ‘needles in a
haystack’ produces leads that add immense value to the early drug discovery stages. VS
protocols include ligand based screens like 1D filters (e.g. molecular weight), 2D filters
(similarity, substructure fingerprints), 3D filters (3D pharmacophore, 3D shape matching)
and structure based screens like docking (Sirois et al., 2004; Valasani et al., 2013). A key
pre-requisite is the knowledge about the spatial and energetic criteria responsible for
protein–ligand binding (Klebe et al., 2006).
Application of computational methodologies in the drug discovery process is well
established. Technologies like combinatorial chemistry and high-throughput screening
(HTS) (Jhoti et al., 2013) are being used to synthesize and screen large number of
compounds in a short period of time to boost the productivity of the drug discovery
process. In virtual screening, computational models are used to predict the biological
activity of compounds. The computational models can be generated and validated utilizing
either the 3D structure of the target or a set of active analogues specific to the target.
Computational models can also be built combining information from structure of the drug
target and a set of active analogues specific to the target (Moro et al., 2007; Liu et al.,
2013). It is identified that there are no efforts carried to enrich the structure types and find
out novel potent inhibitors against Staphylococcus aureus PheRS using VS technologies.
Hence, it is worthy to develop an effective and accurate virtual screening strategy to design
novel druggable PheRS inhibitors.

Virtual Screening and Antibacterial Assay
174
Clearly, there is a need for pharmaceutical agents that inhibit novel bacterial targets
outside the realm of established antimicrobial therapies (Woodford, 2005; Fatima et al.,
2013). The search for such targets is ongoing and has intensified both in scope and volume
in the past few years. Among the numerous enzyme families that have become the focus of
antibacterial research recently, the aminoacyl-tRNA synthetases (AaRS) are particularly
attractive (Schimmel et al., 1998; Tao et al., 2000; Hurdle et al., 2005; Yao et al., 2013).
Since ribosomal protein synthesis absolutely depends on the steady supply of charged
tRNA molecules, AaRS are essential for all living organisms. Inhibition of AaRS results in
arrest of ribosomal protein synthesis for bacteria this causes attenuation and eventual
cessation of growth in vitro and in vivo (Tao et al., 2000).
Herein, we report an effective VS model combining ensemble pharmacophore
models and cascade docking for the discovery of novel phenylalanine-tRNA synthetase
inhibitors (Figure 1). Ten SBP models are generated based on the developed homology
model. The common features among these models are reserved, makes the based model.
However, this model is too large and not sensitive to differentiate the active from inactive.
For simplification, we have used a Receptor−Ligand pharmacophore model (a model focus
on Receptor−Ligand complexes based on the LigandScout algorithm) to support selection
of critical features from the SBP models. The ensemble model is then applied for the VS of
a database containing over 680,000 compounds derived from Asinex, ZINC, National
Cancer Institute (NCI) and SPECS (www.specs.net) databases. The hit compounds are
then evaluated for their binding affinities and conformations with PheRS using a cascade
docking method. Thirty compounds are reserved and evaluated using comparative invitro
inhibitory concentration assay (MIC) against Staphylococcus aureus. Five compounds with
novel scaffolds exhibiting potent target-based and cell-based activities can be served as
promising lead compounds for further optimization.
5.1. MATERIALS AND METHODS
Biological data: For modeling studies, a data set of molecules having activities against
PheRS was selected from the literature (Xiang et al., 2004; Xiang et al., 2004; Richard et
al., 2005). Ten active compounds and ten inactive compounds were selected to validate the
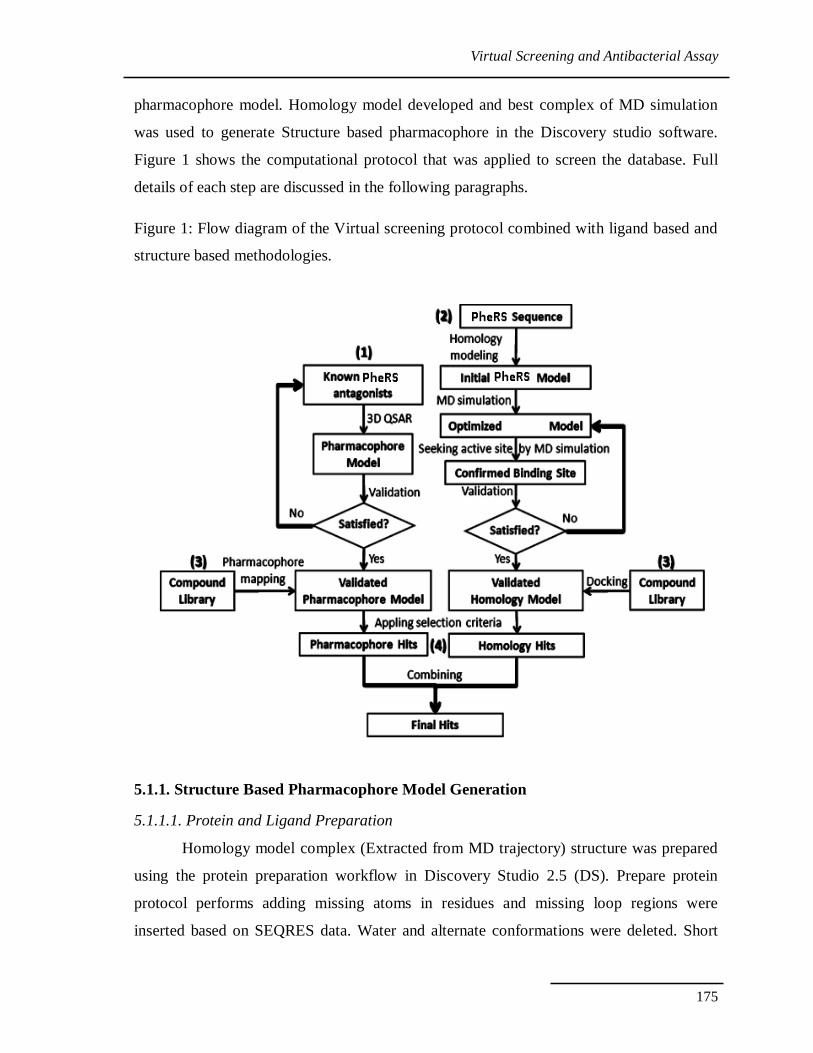
Virtual Screening and Antibacterial Assay
175
pharmacophore model. Homology model developed and best complex of MD simulation
was used to generate Structure based pharmacophore in the Discovery studio software.
Figure 1 shows the computational protocol that was applied to screen the database. Full
details of each step are discussed in the following paragraphs.
Figure 1: Flow diagram of the Virtual screening protocol combined with ligand based and
structure based methodologies.
5.1.1. Structure Based Pharmacophore Model Generation
5.1.1.1. Protein and Ligand Preparation
Homology model complex (Extracted from MD trajectory) structure was prepared
using the protein preparation workflow in Discovery Studio 2.5 (DS). Prepare protein
protocol performs adding missing atoms in residues and missing loop regions were
inserted based on SEQRES data. Water and alternate conformations were deleted. Short

Virtual Screening and Antibacterial Assay
176
and medium size loop regions were optimized with the LOOPER algorithm (Spassov et al.,
2008) and the remaining loop regions were minimized in the CHARMm force field. All
structures’ pKa were calculated and protonated. (Spassov et al., 2008).
5.1.1.2. Receptor-Ligand Pharmacophore Generation
Receptor-Ligand Pharmacophore Generation protocol is available in DS 3.1
(www.accelrys.com) was used to investigate the essential amino acids that participate in
the binding of ligands with PheRS enzyme. This protocol acts by generating selective
pharmacophore models by employing Genetic Function Approximation (GFA) technique.
The pharmacophore hypotheses were constructed using the LigandScout algorithm
(Wolber et al., 2004). The LigandScout algorithm allowed the automatic construction of
the pharmacophore model from the structural data of the protein−ligand complex. The
resulting pharmacophore model is based on receptor-ligand interactions and adds certain
features such as Hydrogen bond acceptor (HBA), Hydrogen bond donor (HBD),
Hydrophobic (HY), Negative ionizable (NI), Positive ionizable (PI) and Ring aromatic
(RA). The essential amino acids will be identified in which the ligand interacts with and
these amino acids will be considered later when the final pharmacophore model is being
built using Interaction Generation protocol. This protocol extracts pharmacophore query
from the Ludi interaction map which is created inside the active site sphere and only
assigns three main features namely HBA, HBD and HY. All the parameters within this
protocol were left as their default values (Fei et al., 2013).
5.1.1.3. Pharmacophore Validation
How to modify a pharmacophore model based on protein structures is always a
perplexing issue. Researchers used a test cluster of active and inactive inhibitors to validate
and modify the pharmacophore model. So while generating the pharmacophore to validate
the pharmacophore we have given 10 actives and 10 inactive molecules as positive and
negative controls. This will aid us to do the different validations to the developed SBP
model like ROC curve, selectivity and sensitivity tables. Herein we use a pharmacophore
model based on Receptor-Ligand which focuses more attention on ligands clusters. The
pharmacophore model built on the Receptor-Ligand complex reflects more features of the
interaction between PheRS and ligands than which built on ligands. The pharmacophore

Virtual Screening and Antibacterial Assay
177
model built on receptor structures can factually represent the energy contributed by critical
points in the process of interacting between protein and ligands. However, these
hypotheses were usually used to design inhibitors.
5.1.1.4. Enrichment Factor Validation
For validating the reliability of the constructed pharmacophore models, the
enrichment factor (EF) (Halgren et al., 2004; Anighoro et al., 2013) was calculated using a
decoy database of 1000 molecules. The EF was calculated using the following formula.
Where n = total number of hits, a = the total number of active molecules in the n hits, N =
total number of molecules in database and A = the total number of actives in the database.
The decoy database was built by mixing the 10 active ligands (IC50 <1 μ M) with
990 compounds (selected randomly from ZINC database). Total 5% actives were
considered in the dataset. All the compounds were converted to 3D structures and multiple
conformers were generated using the Diverse Conformation Generation module in DS
running with the Best conformations option. The energy of each compound was minimized
using CHARMm (Chemistry at HARvard Macromolecular Mechanics) force field.
5.2. CHEMICAL LIBRARY DESIGN
In view of the structural diversity and availability at the time of study, among the
numerous commercial and academic compound databases, the Asinex, NCI Plated 2007
Database and SPECS Natural Database were selected for the screening purpose. The total
databases with 680,000 compounds were filtered by well-known Lipinski’s rule of five
(Lipinski et al., 2004) which states, in general; an orally active drug has no more than one
violation of the following criteria:
Not more than 5 hydrogen bond donors (nitrogen or oxygen atoms with one
or more hydrogen atoms).
Not more than 10 hydrogen bond acceptors (nitrogen or oxygen atoms).

Virtual Screening and Antibacterial Assay
178
A molecular mass less than 500 Daltons.
An octanol-water partition coefficient log P not greater than 5.
Molecules which are not fitting in the Lipinski rule were eliminated from the hits
list. Then the database was calculated to predict Absorption, Distribution, Metabolism,
Excretion and Toxicity (ADMET) properties by Pipeline Pilot (PP) 8.0. The molecules
which had unacceptable ADMET properties were removed. Those compounds that passed
all of the screening experiments were retained for further study. The filtered database was
built of multi conformers by using the ‘Build 3D Database’ module in DS (best method,
maximum number of conformers = 255).
5.3. CASCADE DOCKING
Native-Docking: The output Molecular dynamics protein ligand complex is used to
conduct Native-Docking. Complex ligand was docked back into protein structure using DS
2.5 CDOCKER (Wu et al., 2003) and Schrodinger Glide (Glide Version 3.5, Schrodinger,
L.L.C., New York. 2006). The docking results were evaluated through comparison of the
best docked ligands binding modes with the input pose. The root-mean-square deviation
(RMSD) was used to compare differences between the atomic distances of the docked
poses and the real input ligand pose to measure docking reliability. The docking software
with the smallest RMSD was selected to perform Docking.
Docking with Decoy: A decoy set was used to validate the docking efficiency of the
programs. Docking with decoy set (http://dud.docking.org) benefits to determine the
percentage of the ranked compounds that we should select in the docking. The parameters
of the docking function were determined by Native-Docking (Thilagavathi et al., 2010;
Tubert-Brohman et al., 2013). Enrichment factor (EF) in the top 5%, 10% and 25% was
calculated.

Virtual Screening and Antibacterial Assay
179
5.4. VIRTUAL SCREENING
5.4.1. Pharmacophore Screening The chemical database with about 860,827 molecules was screened employing the
ensemble pharmacophore model using the Search 3D Database module with the fast
flexible search method in DS. The index ‘Fit value’ was calculated to rank the screened
molecules. The molecules with the Fit value greater than 3 were retained. Conformers
belonged to the same molecule were also ranked and the best conformer with the highest
Fit value remained.
5.4.2. Docking Screening All the molecules which passed the pharmacophore screening were aligned in
Schrödinger 2009 and processed the cascade docking with the parameter discussed above.
Then compounds were evaluated by consistency scoring functions in Glide and Gold 5.0
using several algorithms: Ludi, Goldscore, Chemscore, ASP, CHEMPLP, LigScore1,
LigScore2, Jain and Ludi Energy Estimate 1 (Jain, 2006). The consistent score was
calculated and ranked. 2% of molecules with a ranked consistent score were retained and
clustered to 10 sets by their similarity using Tanimoto in DS. Finally, one or two
compounds with the highest consistent score were picked out from each set subject to the
bioassay.
5.5. BIOLOGICAL ASSAY
The potency (activity) of an antibiotic product is expressed as the ratio of the dose
that inhibits the growth of a suitable susceptible microorganism to the dose of an
International Biological Standard, an International Biological Reference Preparation, or an
International Chemical Reference Substance of that antibiotic that produces similar
inhibition. Properly validated secondary reference materials may also be utilized in the
assay. To carry out the assay a comparison is made between the inhibition of the growth of
microorganisms produced by known concentrations of the reference material and that
produced by measured dilutions of the test substance. This response is measured by the
disk diffusion method (Negi et al., 2012; Krishnamurthy et al., 2013; Oksuz et al., 2013;
Nagarajan et al., 2013).

Virtual Screening and Antibacterial Assay
180
5.5.1. Assay Procedure
Petri dishes are filled to a depth of 3-4 mm, with a culture medium that has
previously been inoculated with a suitable inoculum of a susceptible test organism. The
nutrient agar (Antibiotic assay medium) composed based on standard concentrations of the
ingredients (Nagarajan et al., 2013). The concentration of the inoculum was selected that
the sharpest zones of inhibition and suitable dose response at different concentrations of
the standard are obtained. When using the inoculum, an inoculated medium containing 1
ml of inoculum per 100 ml of the culture medium is selected. When the inoculum consists
of vegetative organisms, the temperature of the molten agar medium was not exceeding to
48-50 °C. The dishes were specially selected with flat bottoms. During the filling they are
placed on a flat, horizontal surface so as to ensure that the layer of the medium will be of a
uniform thickness.
For the application of the test solution, previously sterilized borer 8-10 mm in
diameter was bored on the surface of the inoculated medium to make the holes. The holes
arrangement on the plate was made such that overlapping of zones is avoided.
Solutions of the reference material of known concentration and corresponding
dilutions of the test substance at the same concentration are prepared in a sterile buffer of a
suitable pH value. To assess the validity of the assay 3 different doses of the reference
material is used together with an equal number of doses of the test substance. The dose
levels were used in the geometric progression. Once the relationship between the logarithm
of concentration of the antibiotic and the diameter of the zone of inhibition has been shown
to be approximately rectilinear, routine assays were carried out using only 2 concentrations
of the reference material and 2 dilutions of the test substance.
The solutions of the reference material and the test substance were arranged on
each dish so that the solutions of the reference material and those of the test substance
alternate around the dish and are placed in such a manner that the highest concentrations of
the reference material and of the test substance are not adjacent. The solutions are placed in
the holes by means of a pipette that delivers a uniform volume of liquid. Delivered
volumes were sufficient to fill holes almost completely.

Virtual Screening and Antibacterial Assay
181
The plates are incubated at a suitable temperature, the selected temperature (33°C)
being controlled at ±0.5 °C, for approximately 16 hours and the diameters or areas of the
zones of inhibition produced by the varied concentrations of the standard and of the test
substance are measured accurately, by using a suitable measuring device. From the results,
the potency of the tested substance was calculated. Conditions for the assay of individual
antibiotics and suitable test organisms were considered according to Indian pharmacopeia
and WHO guidelines.
Figure 2: Mother culture of S. aureus NCIM 5345.
5.5.2. Culture Media Preparation Culture medium was prepared according to Indian pharmacopeia standard
antibiotic assay method media preparation section (Table 1). The described raw materials,
reagents and test solutions were used. Mother cultures obtained from National Collection
of Industrial Microorganisms (NCIM), Pune (Figure 2) was used to prepare culture slants
(Figure 3).

Virtual Screening and Antibacterial Assay
182
Table 1: Reference antibiotic assay conditions for Staphylococcus aureus as test organism.
Antibiotic Test organism
Culture medium
aPhosphate buffer, sterile,
pHa, TS
bConcentration (weight or
International Units per ml)
Incubation temperature (°C) final pH
Cefalexin Staphylococcus aureus cCm1; 6 10-40 μg 32-35 NCTC 6571 6.5-6.6 NCIM 5345 Staphylococcus aureus Cm1; 6 10-40 μg 32-35 ATCC 6538-P 6.5-6.6
a Phosphate buffers, sterile, of suitable pH. Buffers designated as TS, TS1, or TS2 may be used. b Range within which suitable concentrations may be found. cCm: Culture media.
Figure 3: Aseptically prepared cultures of Staphylococcus aureus.
5.5.3. Preparation of Inoculums
Mother cultures of Staphylococcus aureus are obtained from National Collection of
Industrial Microorganisms (NCIM), Pune, INDIA. The strain NCIM5345 is chosen as test
organism as it has close resembles with many other stains. The test organism is grown
overnight on culture medium Cm1 (pH 6.5-6.6 after sterilization) at a temperature of 35-37
°C. A suspension was prepared by washing off the growth with saline test solution and
diluting to a suitable opacity, such that a 1-cm layer transmits 50% of the incident light
when examined at 650 nm.

Virtual Screening and Antibacterial Assay
183
5.5.4. Calculation of Results
Inhibition zone diameters measured to compare the inhibition of the tested
substance with the reference molecule. Standard methods were used to carry out the
statistical evaluation of the microbiological assay of antibiotics (Bliss et al., 1952).
5.6. RESULTS AND DISCUSSION
5.6.1. Proteins and Ligands Preparation
As initial input is highly validated and minimized homology model complex there
is no missing side chain atoms, alternate conformations and loops to build. Bonds and
bond orders have been checked and corrected. Only some side chains movement is
occurred due to CHARMm minimization process. Minimized protein complex is
considered for further Receptor based pharmacophore generation.
5.6.2. Structure Based Pharmacophore Model Generation
A Receptor-Ligand Pharmacophore Generation protocol in the DS2.1 software was
used to develop ten pharmacophore models ranking them according to their selectivity
score, the higher the better. According to the results, ten structure based pharmacophore
models (SBP) that scored selectivity scores from 2.75 to 1.11 were generated from features
that matched the receptor-ligand interactions HBD, HBD, HY, HY, PI (Table 2). These
interactions revealed the important amino acids that are helpful to choose the final
structure-based pharmacophore model.
The active site of PheRS enzyme has three major binding areas (Figure 4). The first
and the most important area is the Polar pocket 1; which is formed by Met314 and Glu216.
Second, Two hydrophobic pockets formed by Phe212, Arg318 and Phe254 respectively.
Hydrophobic pocket 2 is inserted deeply inside the active site, which can accommodate up
to two aromatic rings. Finally polar pocket 2 which is composed of Val261 and Ser252.
The hydrophobic pocket was filled by one or two HY features in all ten generated
pharmacophores. On the other hand, the key interactions of the active site represented by
HBD, PI features correlated to the polar amino acids that are present.

Virtual Screening and Antibacterial Assay
184
Table 2: The generated pharmacophore models with their selectivity scores based on
PheRS enzyme bound to its ligand.
Figure 4: A 3D view representing the three main areas that forms the active site of PheRS
enzyme. Ligands are represented in tube and the active site residues are represented in
sticks. Mapped ligands in grey colour.

Virtual Screening and Antibacterial Assay
185
The results have shown a similar trend to the Ligand based pharmacophore
generation protocol by indicating the presence of two hydrophobic pockets and the
Hydrogen bond donors and one Positive ionization feature points. Pharmacophore mapped
with the best active ligand showed in the figure 5.
5.6.3. Pharmacophore validation
While generating the pharmacophore a test cluster of active and inactive inhibitors
were added to validate and modify the pharmacophore model. Cluster of 10 actives and 10
inactive molecules as positive and negative controls were used. This helps in validating the
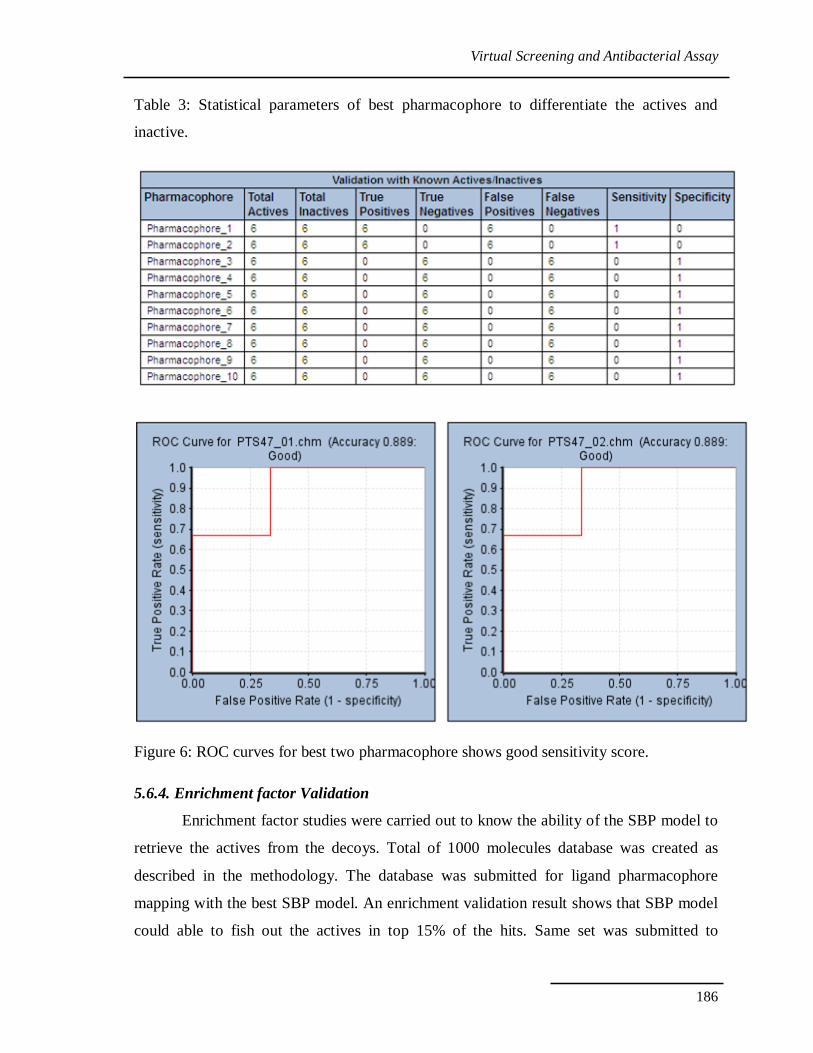
generated models by generating ROC curve, selectivity and sensitivity tables. The
pharmacophore model built on the Receptor −Ligand complex reflects features of the
interactions between PheRS and ligands. The best Structure based pharmacophore (SBP)
model poses good sensitivity towards actives and inactive. It could able separate actives
from inactive with less/no number of true negatives (Table 3). Sensitivity table along with
ROC curve for best two pharmacophore with accuracy rate 0.899 gives confidence on the
model to use for virtual screening (Figure 6).
Figure 5: Structure-based pharmacophore of the active site of PheRS. 3D representation of
this pharmacophore; HY: cyan; HBD: magenta; PI: Red. Mapped ligand in green colour.
Virtual Screening and Antibacterial Assay
186
Table 3: Statistical parameters of best pharmacophore to differentiate the actives and
inactive.
Figure 6: ROC curves for best two pharmacophore shows good sensitivity score.
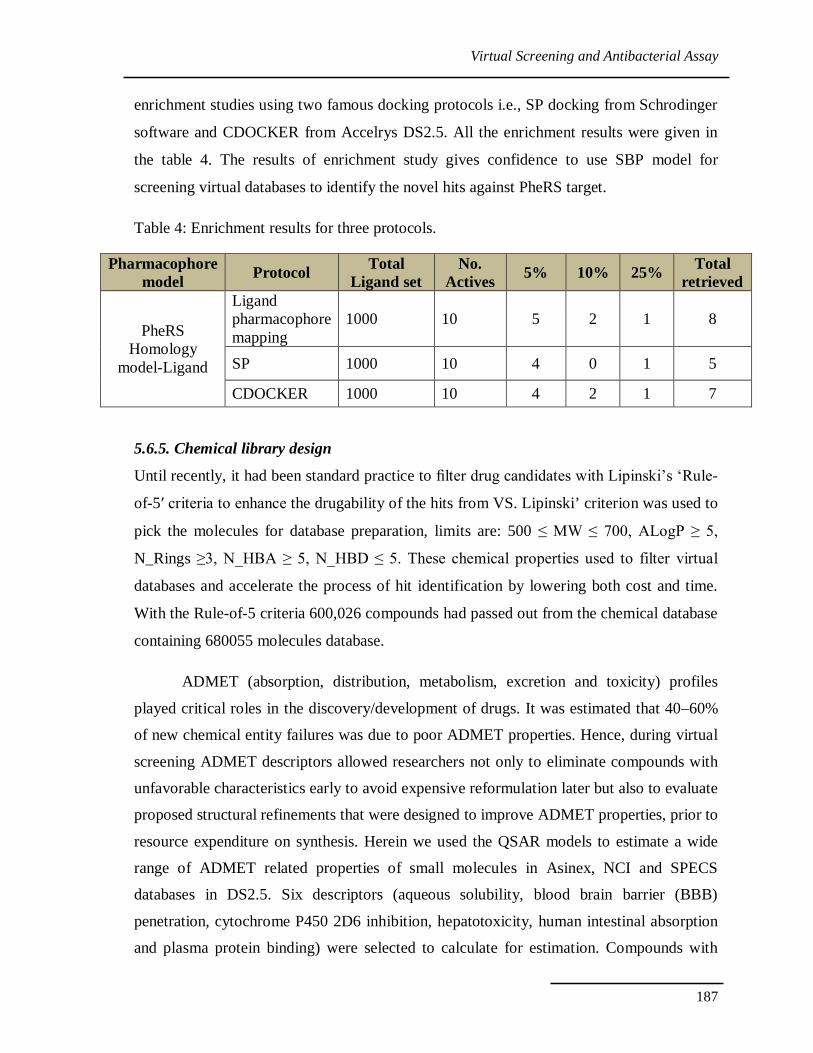
5.6.4. Enrichment factor Validation
Enrichment factor studies were carried out to know the ability of the SBP model to
retrieve the actives from the decoys. Total of 1000 molecules database was created as
described in the methodology. The database was submitted for ligand pharmacophore
mapping with the best SBP model. An enrichment validation result shows that SBP model
could able to fish out the actives in top 15% of the hits. Same set was submitted to
Virtual Screening and Antibacterial Assay
187
enrichment studies using two famous docking protocols i.e., SP docking from Schrodinger
software and CDOCKER from Accelrys DS2.5. All the enrichment results were given in
the table 4. The results of enrichment study gives confidence to use SBP model for
screening virtual databases to identify the novel hits against PheRS target.
Table 4: Enrichment results for three protocols.
Pharmacophore model Protocol Total
Ligand set No.
Actives 5% 10% 25% Total retrieved
PheRS Homology
model-Ligand
Ligand pharmacophore mapping
1000 10 5 2 1 8
SP 1000 10 4 0 1 5
CDOCKER 1000 10 4 2 1 7
5.6.5. Chemical library design
Until recently, it had been standard practice to filter drug candidates with Lipinski’s ‘Rule-
of-5′ criteria to enhance the drugability of the hits from VS. Lipinski’ criterion was used to
pick the molecules for database preparation, limits are: 500 ≤ MW ≤ 700, ALogP ≥ 5,
N_Rings ≥3, N_HBA ≥ 5, N_HBD ≤ 5. These chemical properties used to filter virtual
databases and accelerate the process of hit identification by lowering both cost and time.
With the Rule-of-5 criteria 600,026 compounds had passed out from the chemical database
containing 680055 molecules database.
ADMET (absorption, distribution, metabolism, excretion and toxicity) profiles
played critical roles in the discovery/development of drugs. It was estimated that 40–60%
of new chemical entity failures was due to poor ADMET properties. Hence, during virtual
screening ADMET descriptors allowed researchers not only to eliminate compounds with
unfavorable characteristics early to avoid expensive reformulation later but also to evaluate
proposed structural refinements that were designed to improve ADMET properties, prior to
resource expenditure on synthesis. Herein we used the QSAR models to estimate a wide
range of ADMET related properties of small molecules in Asinex, NCI and SPECS
databases in DS2.5. Six descriptors (aqueous solubility, blood brain barrier (BBB)
penetration, cytochrome P450 2D6 inhibition, hepatotoxicity, human intestinal absorption
and plasma protein binding) were selected to calculate for estimation. Compounds with

Virtual Screening and Antibacterial Assay
188
good absorption level (level 0 according to DS), optimal solubility (level 3 or 4), low BBB
penetrability (level 3), CYP450 2D6 non-inhibition and non-hepatotoxic properties were
selected as druggable compounds. Finally, 21,827 molecules were selected from 82026
molecules filtered with ‘Rule-of-5-for-PheRS’ criteria.
5.6.6. Docking of known inhibitors
According to the results of Native-Docking of modeled protein to its native ligand,
docking using the Glide program (Schrodinger, 2009) had the smallest average RMSD and
standard deviation. Although docking using CDOCKER had an approximate std (0.18), its
average RMSD was not acceptable. The reproducibility of the Glide program was higher
than CDOOKER programs. Therefore, we have used Glide program to dock protein with
inhibitors. Ideally it’s better to select the protein after conducting a cross docking study
with its available crystal structures, in this case we don’t have the luxury of multiple
crystal structures. For this reason, some known actives and inactives to PheRS target were
selected to perform Glide docking study. This study enables to understand the activity
difference between actives and inactives. Understanding the activity difference in protein
and interactions point of view is very crucial to pick best hits from the virtual screening.
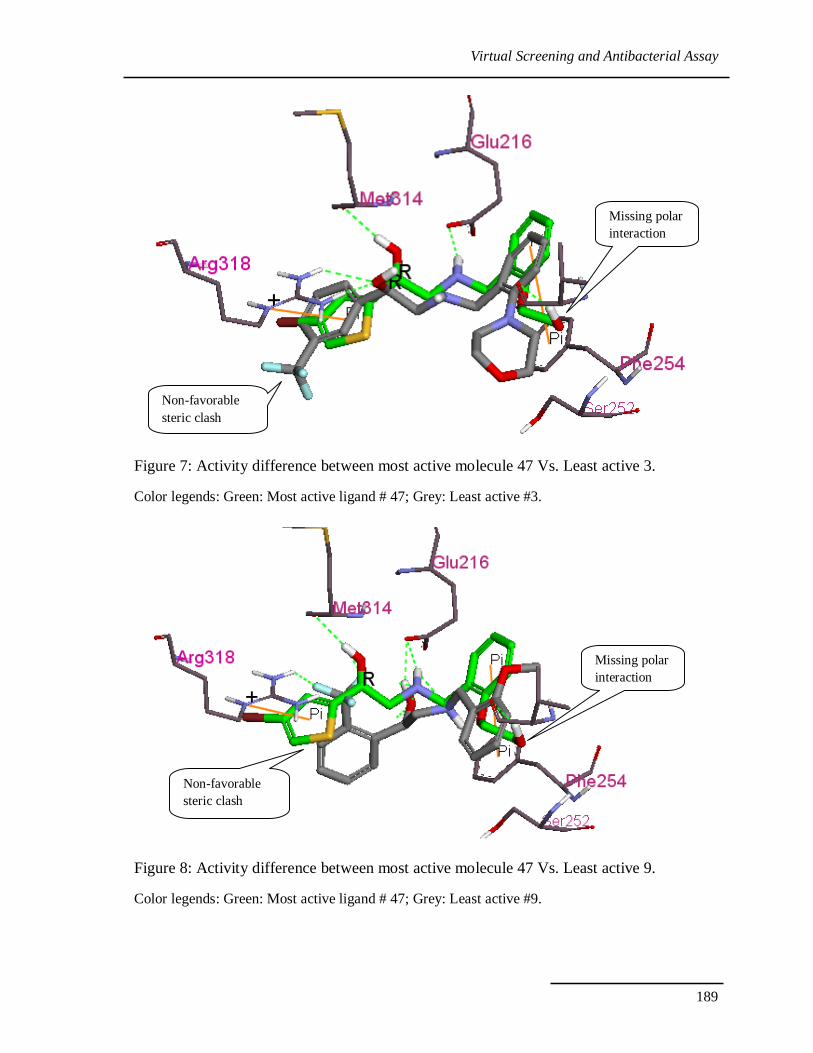
5.6.7. Understanding the activity difference
A molecular dynamics study expedited to understand the binding mode of the most
active molecule in the Chapter IV. In the process of virtual screening it is important to
understand the inactive molecules and their features to reduce the false negatives in the
VS. To do so we have compared the docking pose of best active molecule 47 with two
least active molecules 3 and 9.
In most active compound 47 binding pose, the ligand NH of amine forms an ionic
and hydrogen bond with Glu216 and forms key interaction with Met314. Ligand Phenyl
moiety and extended alkyl chain occupies a relatively wide hydrophobic pocket (P1)
formed by Val261, Phe254, Thr257, Phe312, Gly217 and Gly290. In addition to the
essential interactions between PheRS antagonists and Met314 ligand has the cation –π
interactions with Arg318. Where as in compound 3 and 9 molecules has steric clash with
Arg318 and Phe212, also they are missing donor interactions with Val261 and Ala289.
Binding modes of the both molecule can be observed in Figure 7 and 8.
Virtual Screening and Antibacterial Assay
189
Figure 7: Activity difference between most active molecule 47 Vs. Least active 3.
Color legends: Green: Most active ligand # 47; Grey: Least active #3.
Figure 8: Activity difference between most active molecule 47 Vs. Least active 9.
Color legends: Green: Most active ligand # 47; Grey: Least active #9.
Missing polar interaction
Non-favorable steric clash
Non-favorable steric clash
Missing polar interaction

Virtual Screening and Antibacterial Assay
190
5.7. VIRTUAL SCREENING RESULTS
5.7.1. Pharmacophore Screening The database constructed using Build database module in Discovery studio with 82,000
molecules which were filtered out from the Lipinski rule filter. The constructed database
was used to screen against final validated structure-based pharmacophore (SBP) model.
At this step, it is important to find candidate structures that share similarity with
active molecules reported in the literature and fulfill the requirements of being able to fit in
the generated structure-based pharmacophore model which is a reflection of the active site
geometry. Consequently, both 2D and 3D criteria will be attained. The Screen Library
protocol was run for database consists of 82,686 compounds molecules which are filtered
out from the Lipinski rule filter. Database was generated by applying Best Conformation
generation of 255 conformers for each structure with 20 kcal energy threshold. This
screening step has returned 2,373 compounds able to match the structure-based
pharmacophore with a minimum of three features out of five being satisfied and every
compound should map the critical binding feature and the HBD and PI feature adjacent to
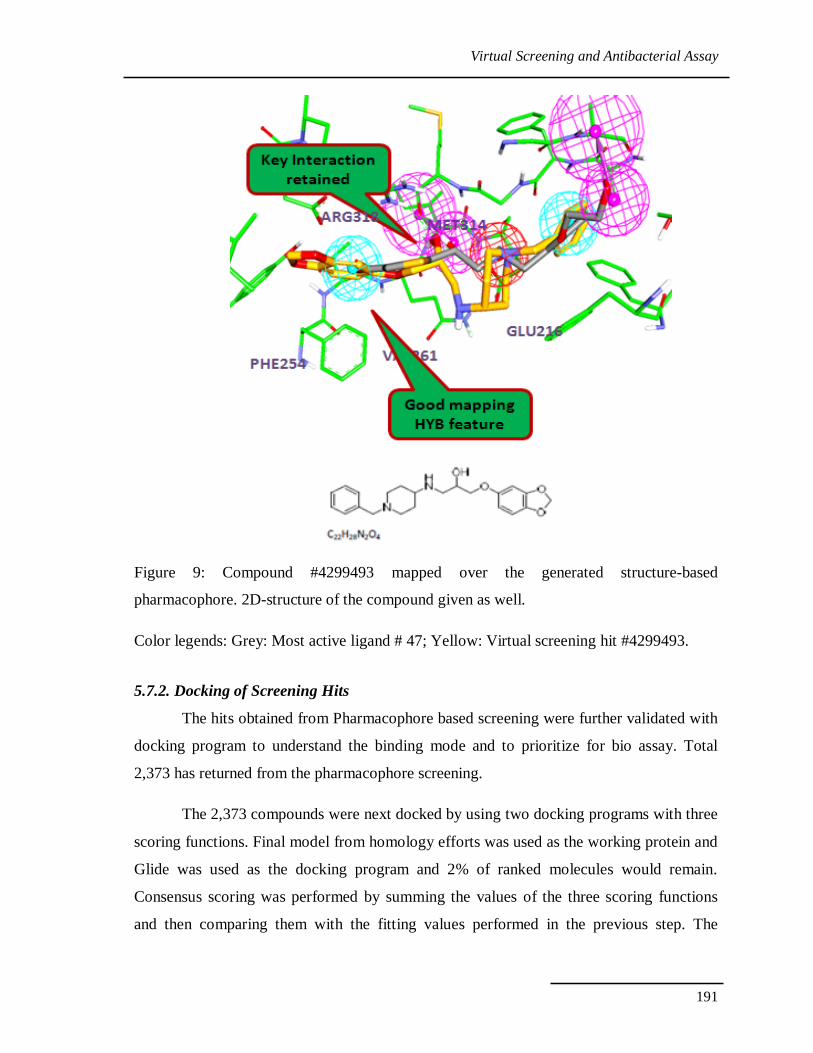
it. SBP model mapping of compound 4299493 is depicted in figure 9. Total five features
were fulfilled by the compound; the benzo[1,3]dioxole ring fits the HY, the NH and OH
groups nicely mapped with HBD and PI features, hydrophobic part of the piperdine group
maps with second HY features and finally benzyl group try to manage in the polar HBD
region. Compound has good fit value 3.89, while the one of the feature was not mapped
well still protein has the some hydrophobic region to accommodate the hydrophobic part of
the ligand.
For the final selection of candidate inhibitors of PheRS, the fit values of these
compounds will be compared with the consensus scoring of the molecular docking.
Virtual Screening and Antibacterial Assay
191
Figure 9: Compound #4299493 mapped over the generated structure-based
pharmacophore. 2D-structure of the compound given as well.
Color legends: Grey: Most active ligand # 47; Yellow: Virtual screening hit #4299493.
5.7.2. Docking of Screening Hits
The hits obtained from Pharmacophore based screening were further validated with
docking program to understand the binding mode and to prioritize for bio assay. Total
2,373 has returned from the pharmacophore screening.
The 2,373 compounds were next docked by using two docking programs with three
scoring functions. Final model from homology efforts was used as the working protein and
Glide was used as the docking program and 2% of ranked molecules would remain.
Consensus scoring was performed by summing the values of the three scoring functions
and then comparing them with the fitting values performed in the previous step. The

Virtual Screening and Antibacterial Assay
192
candidate molecules were selected based on the ability of the chemical structures to map
the generated structure-based pharmacophore, getting the highest ranks in the consensus
scoring tactic and visually examining the binding pattern of the docked poses.
Consequently, 30 compounds were chosen to be potential inhibitors which contain various
functional groups performing binding with critical amino acids Met214 and Glu216 at the
active site (Table 5). For example, 13658686 is a 1,3-diphenylthiourea based structure in
which the two NH donor groups forming critical interactions with Met314 and Glu216.
Two dimethyl substituted morpholine groups occupying the hydrophobic pockets on both
sides which formed by Phe212 and Phe312, Phe256 respectively. (Figure 10).
Figure 10: Top VS hit Compound #13658686 mapped over the generated structure-based
pharmacophore. 2D- structure of the compound given as well.
Color legends: Grey: Most active ligand # 47; Yellow: Virtual screening hit #13658686.
Virtual Screening and Antibacterial Assay
193
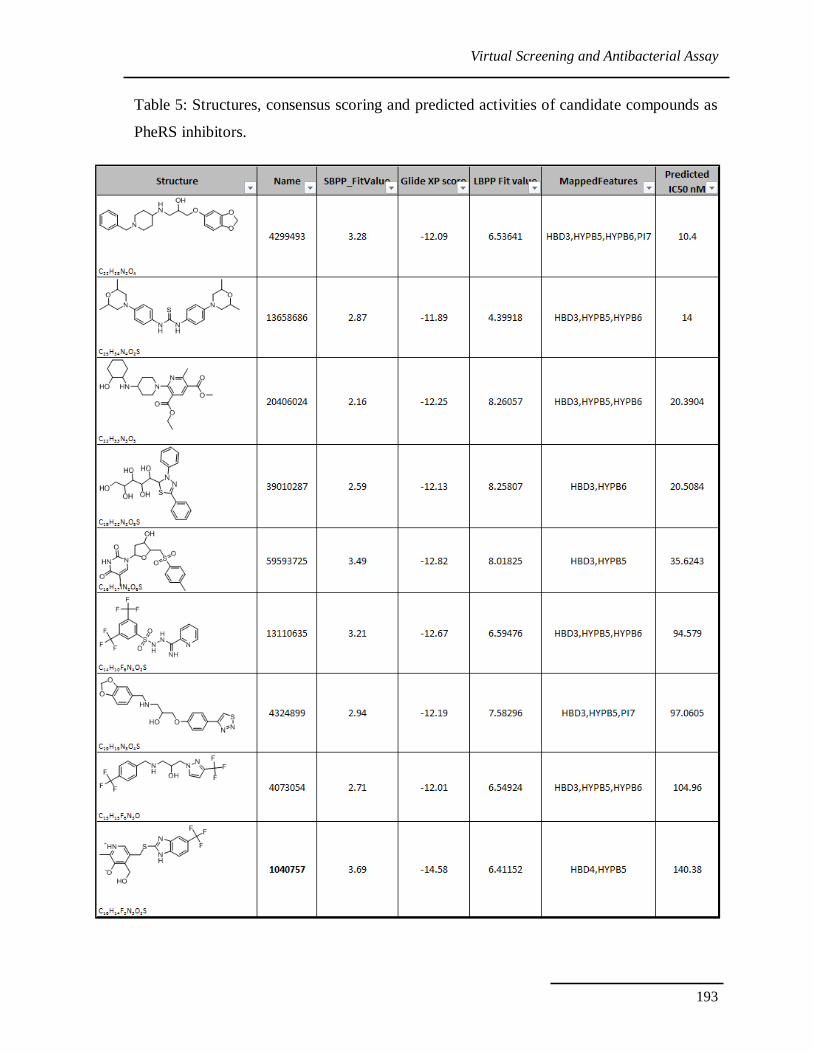
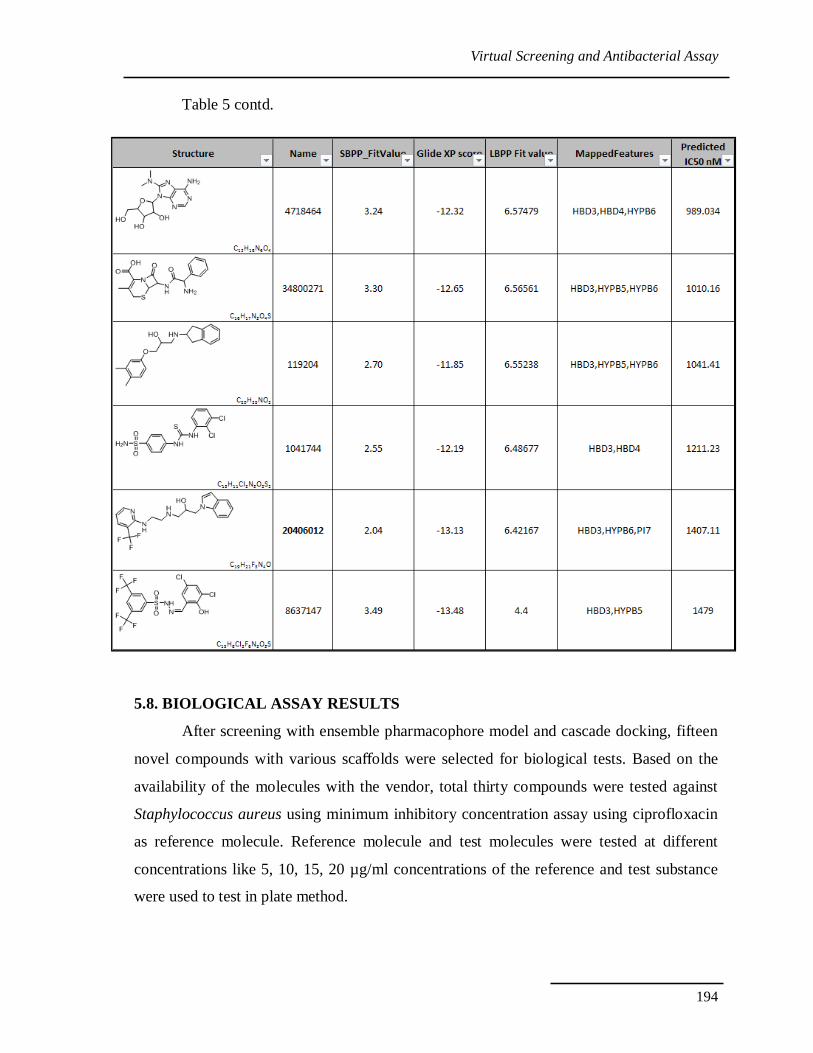
Table 5: Structures, consensus scoring and predicted activities of candidate compounds as
PheRS inhibitors.
Virtual Screening and Antibacterial Assay
194
Table 5 contd.
5.8. BIOLOGICAL ASSAY RESULTS
After screening with ensemble pharmacophore model and cascade docking, fifteen
novel compounds with various scaffolds were selected for biological tests. Based on the
availability of the molecules with the vendor, total thirty compounds were tested against
Staphylococcus aureus using minimum inhibitory concentration assay using ciprofloxacin
as reference molecule. Reference molecule and test molecules were tested at different
concentrations like 5, 10, 15, 20 µg/ml concentrations of the reference and test substance
were used to test in plate method.

Virtual Screening and Antibacterial Assay
195
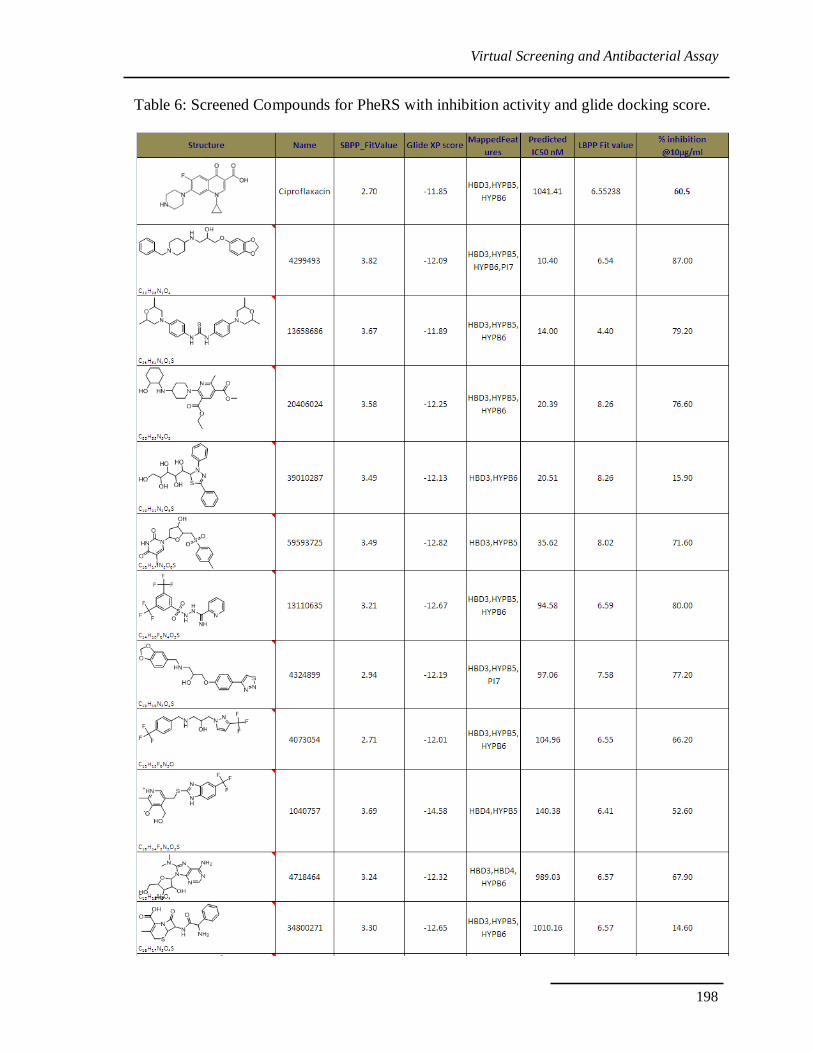
Interestingly 10 out of 30 molecules showed good inhibition against
Staphylococcus aureus. Five novel scaffolds showed better activity than existing drug
ciprofloxacin. Results were given in table 6.
5.8.1. Assay results for Compound 4299493
Figure 11: Top VS hit Compound #4299493 inhibition zones against
Staphylococcus aureus in petri dish MIC assay. Both standard compound and sample are
tested at concentrations 5, 10, 20 and 30 µg/ml.
Most scored virtual screen hit 4299493 was tested for inhibitory activity against
Staphylococcus aureus. MIC results show that tested compound has better clear zones of
Inhibition zone

Virtual Screening and Antibacterial Assay
196
inhibition than the reference compound see figure 11. This gives confidence on the ligand
and structure based methods, the hints gained from the analysis.
5.8.2. Assay results for Compound 13658686
One of the best scored virtual screen hit no. 13658686 was tested for inhibitory
activity against Staphylococcus aureus. MIC results show that tested compound has better
and clear zones of inhibition than the reference compound see figure 12. This gives
confidence on the ligand and structure based methods, the hints gained from the analysis.
Figure 12: Top VS hit Compound #13658686 inhibition zones against Staphylococcus
aureus in petri dish MIC assay. Both standard compound and sample are tested at
concentrations 5, 10, 20 and 30 µg/ml.
5.8.3. Assay results for Compound 20406024
Virtual screen hit no. 20406024 was tested for inhibitory activity against Staphylococcus
aureus. MIC results show that tested compound has better and clear zones of inhibition
Inhibition zone

Virtual Screening and Antibacterial Assay
197
than the reference compound see figure 13. This gives confidence on the ligand and
structure based methods, the hints gained from the analysis.
Figure 13: Top VS hit Compound #20406024 inhibition zones against Staphylococcus
aureus in petri dish MIC assay. Both standard compound and sample are tested at
concentrations 5, 10, 20 and 30 µg/ml.
Total thirty candidate compounds were purchased from Asinex and were later
tested for minimum inhibitory concentration at Chandra labs, an ISO certified company for
microbiology services, Hyderabad, India. Following the ligand based virtual screening and
finally validated by docking with the PheRS model, docking score, fit value and estimated
activity of top 30 molecules screened and suggested as inhibitors of PheRS. The selected
compounds with Asinex id is given in the table 6.
Inhibition zone
Virtual Screening and Antibacterial Assay
198
Table 6: Screened Compounds for PheRS with inhibition activity and glide docking score.

Virtual Screening and Antibacterial Assay
199
Contd..
In our study, we successfully demonstrated an efficient virtual screening model
using ligand protein based combined pharmacophore based screening model to remove
false positives. Initially, pharmacophore model was developed and validated using known
PheRS inhibitors to retrieve the crucial features. The validated pharmacophore model was
used as a query to screen database of compounds and top 2726 hits were selected based
pharmacophore fit value. Subsequently, feature & docking studies developed using the
predicted bound conformation of the highest active compound and successfully validated
based on enrichment factor.
Virtual screening has been established as a powerful alternative complementary to
high throughput screening (Morya et al., 2012). When virtual screening is performed
optimally, impressive hit rates have been reported. Though few Virtual screening studies
were reported in the literature on different receptors of the Staphylococcus (Lee JY et al.,
2012; Ananthula et al., 2012; Sabatini et al., 2013), there are no report on virtual screening
tools to identify novel compounds specifically for PheRS receptors. Virtual screening with
a receptor model is hindered by at least three factors: the model reliability, the absence of a
good docking score and the low throughput. Many scoring functions for ligand-protein
docking have been developed in the past decade and several successful examples have

Virtual Screening and Antibacterial Assay
200
been reported (Jain, 2006; Tubert-Brohman et al., 2013) but they are still far from perfect
and have yet to be proven adequate for predicting binding affinities. Docking is relatively
slow and currently, virtual screening of a 33 lakhs compounds takes extensive
computational resources (Hongwu et al., 2008). To overcome these uncertainties scientists
using combined ligand and structure-based virtual screening as a tool for detecting novel
lead compounds.( Mladenovic et al., 2013; Zhou et al., 2013; Wang et al., 2014).
The present study established that combined ligand and structure-based virtual
screening is a useful tool for detecting novel lead compounds for the antibiotics such as the
Staphylococcus aureus PheRS receptors. The hit molecule serves as a lead structure for
novel PheRS antagonists that are intended for therapeutic use. We have reported the
development and validation of common feature-based pharmacophore models for PheRS
receptor ligands. In addition, the present study also demonstrated that refined PheRS
receptor homology model provides a relevant structural basis for rationalizing the PheRS
receptor binding.
The established insilico receptor-based screening approach and consensus scoring
functions (Jain, 2006). Previously developed, studied homology model of PheRS is
currently used in 3D database searches to identify new chemical scaffolds for PheRS
antagonist. This virtual screening workflow resulted in the success of 10 out of 30
compounds in biological testing against Staphylococcus aureus. Five novel and diverse
compounds tested in vitro biology assays for Staphylococcus aureus can be subjected to
medicinal chemistry optimization in order to obtain new leads for subsequent
pharmacological evaluation. This study demonstrated the power of virtual screening with a
pharmacophore model and using the modeling techniques in performance was essential.
CONCLUSIONS
Successful approaches should optimize each procedure in virtual screening to
distinguish actives from false positives. From chemical database to pharmacophore model,
docking screening and result validation, our work has made much effort to optimize the VS
strategy special for PheRS. Comparing to classic VS strategy, our VS strategy combined
an ensemble pharmacophore model with cascade docking. The chemical database with
appropriate chemical space was built for screening drug-like molecules. Cascade docking

Virtual Screening and Antibacterial Assay
201
was used to ensure that the protein, the docking program and evaluation were optimal for
this VS process. We constructed a pharmacophore model, which ensemble a Receptor–
Ligand complex-based pharmacophore model to simply the SBP model, with the proper
size and shape for screening nonpeptide inhibitors. With the effective VS strategy, five
novel scaffolds for inhibiting the PheRS were identified from a modest database of
commercially available compounds. In addition to the success with chemical diversity, the
hit rate was nearly 30%. This demonstrated the ability of our VS strategy to broadly and
effectively search and identify more diverse inhibitors.

Virtual Screening and Antibacterial Assay
202
REFERENCES
Ananthula RS, Ravi kumar M, Mahmood SK and Kumar MN (2012). Insights from ligand
and structure based methods in virtual screening of selective Ni-peptide
deformylase inhibitors. J Mol Model. 18: 693-708.
Anighoro A and Rastelli, G (2013). Enrichment factor analyses on G-protein coupled
receptors with known crystal structure. J. Chem. Inf. Model. 53: 739-743.
Bharatham N, Bharatham K and Lee KW (2007). Pharmacophore identification and virtual
screening for methionyl-tRNA synthetase inhibitors. J. Mol. Graph. Model. 25:
813-823.
Bohm HJ (1992). The computer program LUDI: A new method for the de novo design of
enzyme inhibitors. J. Comput. Aided. Mol. Des. 6: 61-78.
Desiraju GR, Gopalakrishnan B, Jetti RK, Nagaraju A, Raveendra JD, Sharma A, Sobhia
ME and Thilagavathi R (2002). Computer-aided design of selective COX-2
inhibitors: comparative molecular field analysis, comparative molecular similarity
indices analysis and docking studies of some 1,2-diarylimidazole derivatives. J.
Med. Chem. 45: 4847-4857.
Dmitrieva NF, Kliachko NL, Bondarenko VM, Shabanova NA, Eshchina AS, Filatova LIu,
Morozova NI, Timofeev IuM, Kabanov AV and Briko NI. (2011). Development of
Streptococcus pyogenes cells inactivation for turbidimetric determination of
bacteriolytic activity of phage-associated enzyme. Zh. Mikrobiol. Epidemiol.
Immunobiol. 6: 14-19.
Evdokimov AG, Mekel M, Hutchings K, Narasimhan L, Holler T, McGrath T, Beattie B,
Fauman E, Yan C, Heaslet H, Walter R, Finzel B, Ohren J, McConnell P, Braden
T, Sun F, Spessard C, Banotai C, Al-Kassim L, Ma W, Wengender P, Kole D,
Garceau N, Toogood P and Liu J (2008). Rational protein engineering in action: the
first crystal structure of a phenylalanine tRNA synthetase from Staphylococcus
haemolyticus. J. Struct. Biol. 162: 152-169.
Fatima A, Shyum-Naqvi SB, Khaliq SA, Perveen S, Yousuf RI and Saeed R (2013).
Staphylococcal resistance against five groups of life saving antibiotics in the year
2003-2005. Pak. J. Pharm. Sci. 26: 1137-40.

Virtual Screening and Antibacterial Assay
203
Fersht AR (1977). Editing mechanisms in protein synthesis. Rejection of valine by the
isoleucyl-tRNA synthetase. Biochemistry. 16:1025-1030.
Finn J, Stidham M, Hilgers M and GCK (2008). Identification of novel inhibitors of
methionyl-tRNA synthetase (MetRS) by virtual screening. Bioorg. Med. Chem.
Lett. 18: 3932-3937.
Gao Q, Yang L and Zhu Y (2010). Pharmacophore based drug design approach as a
practical process in drug discovery. Curr Comput Aided Drug Des. 6: 37-49.
Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT and Banks JL
(2004). Glide: a new approach for rapid, accurate docking and scoring, Enrichment
factors in database screening, J. Med. Chem. 47: 1750-8.
Jain AN (2006). Scoring functions for protein-ligand docking. Curr Protein Pept Sci. 7:
407-420.
Jhoti H, Rees S and Solari R (2013). High-throughput screening and structure-based
approaches to hit discovery: is there a clear winner? Expert Opin Drug Discov.
[Epub ahead of print]
Jiang YR, Yang YY, Chen YL and Liang ZJ (2013). CoMFA, CoMSIA and HQSAR
studies of acetylcholinesterase inhibitors. Curr. Comput. Aided Drug Des. 9:385-
95.
Kim SY and Lee J (2003). 3-D-QSAR study and molecular docking of methionyl-tRNA
synthetase inhibitors. Bioorg. Med. Chem. 11: 5325-5231.
Klebe G (2006). Virtual ligand screening: strategies, perspectives and limitations Drug
Discov. Today. 11: 580-594.
Krishnamurthy V, GSV, Kumar MS, HVP, RP and ERN (2013). Phenotypic and
Genotypic Methods for Detection of Extended Spectrum β Lactamase Producing
Escherichia coli and Klebsiella pneumoniae Isolated from Ventilator Associated
Pneumonia. J Clin Diagn Res.7: 1975-1978.
Lee JY, Jeong KW, Shin S, Lee JU and Kim Y (2012). Discovery of novel selective
inhibitors of Staphylococcus aureus β-ketoacyl acyl carrier protein synthase III.
Eur. J. Med. Chem. 47: 261-269.

Virtual Screening and Antibacterial Assay
204
Lipinski CA (2004). Lead- and drug-like compounds: the rule-of-five revolution. Drug
Discovery Today: Technologies 1: 337-341.
Liu C, He G, Jiang Q, Han B and Peng C (2013). Novel hybrid virtual screening protocol
based on molecular docking and structure-based pharmacophore for discovery of
methionyl-tRNA synthetase inhibitors as antibacterial agents. Int. J. Mol. Sci.
14:14225-14339.
Lv PC and Zhu HL (2012). Aminoacyl-tRNA synthetase inhibitors as potent antibacterials.
Curr. Med. Chem. 9:3550-3563.
Lyne PD (2002). Structure-based virtual screening: an overview. Drug Discov. Today. 7:
1047-1055.
Mermershtain I, Finarov I, Klipcan L, Kessler N, Rozenberg H and Safro MG (2011).
Idiosyncrasy and identity in the prokaryotic Phe-system: crystal structure of E. coli
phenylalanyl-tRNA synthetase complexed with phenylalanine and AMP. Protein
Sci. 20:160-167.
Mermershtain I, Finarov I, Klipcan L, Kessler N, Rozenberg H and Safro MG (2011).
Idiosyncrasy and identity in the prokaryotic Phe-system: crystal structure of E. coli
phenylalanyl-tRNA synthetase complexed with phenylalanine and AMP. Protein
Sci. 20:160-167.
Mladenovic M, Matic S, Stanic S, Solujic S, Mihailovic V, Stankovic N and Katanic J
(2013). Combining molecular docking and 3-D pharmacophore generation to
enclose the in vivo antigenotoxic activity of naturally occurring aromatic
compounds: Myricetin, quercetin, rutin and rosmarinic acid. Biochem Pharmacol.
86:1376-1396.
Moro S, Bacilieri M and Deflorian F (2007). Combining ligand-based and structure-based
drug design in the virtual screening arena. Expert. Opin. Drug Discov. 2: 37-49.
Morya VK, Dewaker V and Kim EK (2012). Insilico study and validation of
phosphotransacetylase (PTA) as a putative drug target for Staphylococcus aureus
by homology-based modelling and virtual screening. Appl .Biochem. Biotechnol.
168:1792-805.

Virtual Screening and Antibacterial Assay
205
Nagarajan N, Vanitha G, Ananth DA, Rameshkumar A, Sivasudha T and Renganathan R
(2013). Bioimaging, antibacterial and antifungal properties of imidazole-pyridine
fluorophores: Synthesis, characterization and solvatochromism. J. Photochem.
Photobiol. B. 127: 212-22.
Negi BS, Dave BP and Agarwal YK (2012). Evaluation of antimicrobial activity of
bauhinia purpurea leaves under invitro conditions. Indian J. Microbiol. 52: 360-
365.
Oksuz L, Dupieux C, Tristan A, Bes M, Etienne J and Gurler N (2013). The high diversity
of MRSA clones detected in a university hospital in istanbul. Int. J. Med. Sci. 10:
1740-1745.
Phosrithong N and Ungwitayatorn J (2013). Ligand-based CoMFA and CoMSIA studies
on chromone derivatives as radical scavengers. Bioorg Chem. 49: 9-15.
Sabatini S, Gosetto F, Iraci N, Barreca ML, Massari S, Sancineto L, Manfroni G, Tabarrini
O, Dimovska M, Kaatz GW and Cecchetti V (2013). Re-evolution of the 2-
phenylquinolines: ligand-based design, synthesis and biological evaluation of a
potent new class of Staphylococcus aureus NorA efflux pump inhibitors to combat
antimicrobial resistance. J. Med. Chem. 56: 4975-4989.
Sirois S, Wei DQ, Du Q and Chou KC (2004). Virtual screening for SARS-CoV protease
based on KZ7088 pharmacophore points. J. Chem. Inf. Comput. Sci. 44: 1111-
1122.
Spassov VZ and Yan LA (2008). Fast and accurate computational approach to protein
ionization. Protein Sci. 17: 1955-1970.
Spassov VZ, Flook PK and Yan L (2008). LOOPER: a molecular mechanics-based
algorithm for protein loop prediction. Protein Eng. Des. Sel. 21: 91-100.
Teh CH, Nazni WA, Lee HL, Fairuz A, Tan SB and Sofian-Azirun M (2013). In vitro
antibacterial activity and physicochemical properties of a crude methanol extract of
the larvae of the blow fly Lucilia cuprina. Med. Vet. Entomol.27: 414-420.
Thilagavathi R and Mancera RL (2010). Ligand-protein cross-docking with water
molecules. J. Chem. Inf. Model. 50: 415-421.

Virtual Screening and Antibacterial Assay
206
Tubert-Brohman I, Sherman W, Repasky M and Beuming T (2013). Improved docking of
polypeptides with glide. J. Chem. Inf. Model. 53: 1689-1699.
Valasani KR, Chaney MO, Day VW and Shidu YS (2013). Acetylcholinesterase inhibitors:
structure based design, synthesis, pharmacophore modeling and virtual screening. J.
Chem. Inf. Model. 53: 2033-2046.
Wang X, Ren Z, He Y, Xiang Y, Zhang Y and Qiao Y (2014). A combination of
pharmacophore modeling, molecular docking and virtual screening for iNOS
inhibitors from Chinese herbs. Biomed Mater Eng. 24:1315-1322.
Wolber G and Langer T (2004). LigandScout: 3-D pharmacophores derived from protein-
bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 45:
160-9.
Wu G, Robertson DH, Brooks CL and Vieth M (2003). Detailed Analysis of Grid-Based
Molecular Docking: A Case Study of CDOCKER - A CHARMm-Based MD
Docking Algorithm. J. Comp. Chem. 24: 1549-1562.
Yao P and Fox PL (2013). Aminoacyl-tRNA synthetases in medicine and disease. EMBO
Mol Med. 5: 332-343.
Zhao Y, Wang Q, Meng Q, Ding D, Yang H, Gao G, Li D, Zhu W and Zhou H (2012).
Identification of Trypanosoma brucei leucyl-tRNA synthetase inhibitors by
pharmacophore and docking-based virtual screening and synthesis. Bioorg. Med.
Chem. 20: 1240-1250.
Zhou ZL, Liu HL, Wu JW, Tsao CW, Chen WH, Liu KT and Ho Y (2013). Combining
structure-based pharmacophore and insilico approaches to discover novel selective
serotonin reuptake inhibitors. Chem Biol Drug Des. [Epub ahead of print].