spina bifida and encephalocele - springer · spina bifida and encephalocele jonathan r. ellenbogen,...
TRANSCRIPT

Spina Bifida and Encephalocele
Jonathan R. Ellenbogen, Michael D. Jenkinson, and Conor L.Mallucci
AbstractThis chapter aims to explain the range of con-genital anomies that are encompassed by theterm neural tube defects (NTD). Both cranialand spinal abnormalities occur which ariseduring embryological development due todefects in morphogenesis of the neural tube.They range from anencephaly to spina bifidaocculta. The commonest congenital nervoussystem defect encountered remains spina bifidadespite the overall incidence of neural tubedefects declining. The chapter explains theclassification, pathogenesis, assessment, man-agement, treatment, and prognosis related tothese conditions.
KeywordsHydrocephalus • Cerebrospinal fluid (CSF) •Ultrasound • Computed tomography (CT) •Magnetic resonance imaging (MRI) • Shunt •Neural tube defects (NTD) • Spina bifida •Anencephaly • Myelomeningocele • Embryol-ogy • Encephalocele • Meningocele • Serumalphafetoprotein • Counseling • In-utero sur-gery • Hydrocephalus • Arnold–Chiari type IImalformation • Craniorachischisis •Dysraphism
ContentsIntroduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
A Historical Perspective . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
Embryological Development . . . . . . . . . . . . . . . . . . . . . . . . . 3
Classification and Pathogenesis . . . . . . . . . . . . . . . . . . . . . . 4Brain . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4Spinal Cord . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5Other Defects . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
Spinal Dysraphism . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5Spina Bifida Occulta . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5Spina Bifida Cystica: Meningocele . . . . . . . . . . . . . . . . . . . . 5Spina Bifida Cystica: Myelomeningocele . . . . . . . . . . . . . . 6
Etiology . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7Genetics . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7Dietary Factors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7Diabetes and Obesity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7Teratogens . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7
Prenatal Diagnosis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8Maternal Serum Alphafetoprotein . . . . . . . . . . . . . . . . . . . . . . 8Imaging . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8Amniocentesis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
J.R. Ellenbogen (*)Department of Neurosurgery, Alder Hey Children’s NHSFoundation Trust, Liverpool, UK
Department of Neurosurgery, The Walton Centre NHSFoundation Trust, Liverpool, UKe-mail: [email protected]
M.D. JenkinsonDepartment of Neurosurgery, The Walton Centre NHSFoundation Trust, Liverpool, UKe-mail: [email protected]
C.L. MallucciDepartment of Neurosurgery, Alder Hey Children’s NHSFoundation Trust, Liverpool, UKe-mail: [email protected]
# Springer-Verlag GmbH Germany 2017P. Puri (ed.), Pediatric Surgery,https://doi.org/10.1007/978-3-642-38482-0_87-1
1

Clinical Management . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9Prenatal Assessment and Counseling . . . . . . . . . . . . . . . . . 9In-Utero Surgery . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
Postnatal Assessment . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10Inspection . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10Investigations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11Parental Counseling . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11
General Management, Operative Treatment,and Technique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12
Postoperative Management and Complications . . . 13
Long-Term Management and Outcome . . . . . . . . . . . . 15Medical Problems . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
Lifestyle Concerns . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16Intellect . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16Ambulation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16Psychosocial Problems . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
Encephalocele . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17Classification . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17Etiology . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17Pathology . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17Clinical Features . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18Differential Diagnosis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18Investigations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18Treatment . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18Prognosis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20
Conclusion and Future Directions . . . . . . . . . . . . . . . . . . 20
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20
Introduction
Neural tube defects (NTDs) arise due to aberra-tions in the embryological development of theneural tube and encompass a variety of congenitalanomalies ranging from spina bifida occultatoanencephaly, occurring in one in every 1000births. Geographic variations exist in the inci-dence of spina bifida and neural tube defects, forexample, the incidence of spina bifida cysticavaries between 0.3 per 1000 live births in Finland,and 4.5 per 1000 live births in Ireland (Leck1984). The incidence of spina bifida occulta hasa greater reported variation, which rangesbetween 1% and 50% depending upon the agegroup (Boone et al. 1985). Lower socioeconomicgroups have a higher incidence of developingspina bifida, as do Caucasians when compared toblack people (Leck 1972; Wiswell et al. 1990).There also exists a marked difference in
prevalence between ethnic groups and pregnancyorder in multiparous women, with a preponder-ance of anencephaly in girls (Carter 1974). NTDsare thought to be multifactorial in origin with bothgenetic and environmental factors playing a rolein their development.
While spina bifida remains the commonestcongenital central nervous defect, the overallincidence of NTDs is in decline (EUROCATWorking Group 1991; Kondo et al. 2009; Steven-son et al. 2000; Yen et al. 1992). Several factorsare thought to account for this change, whichinclude declining birth rates, increasing antenataldiagnosis, changing social attitudes, and improv-ing standards of living and nutrition. The mostappropriate management strategy for these patientsis a continued source of medical, ethical, and legaldebate. A multidisciplinary team approach, incor-porating neurosurgeons, pediatricians, neurolo-gists, endocrinologists, urologists, orthopedicsurgeons, physiotherapists, social workers, psy-chologists and nursing staff, must be utilized asa diagnosis of a NTD may have devastating conse-quences not only for the patient but their family.The aim being to provide the patient and familywith the minimal disability possible and thereforewith the best quality of life achievable.
A Historical Perspective
The first reports of fetuses and infants with anen-cephaly, myelomeningocele, and craniora-chischisis originate in ancient Egypt (Obladen2011). Caspar Baulinin is credited with the firstaccurate description of spina bifida in the earlyseventeenth century (Morgagni 1762). The term“spina dorsi bifida” was coined by NicholasTalpius (Tulp) in 1641 (Doran and Guthkelch1961; Tulp 1672), and Virchow introduced theterm “spina bifida occulta” in 1875 (Virchow1875). The association of hydrocephalus andspina bifida was recognized by Morgagni in1761, and he also described anencephaly andspina bifida as expressions of the same patholog-ical process and attributed bladder, rectal, andlimb abnormalities to the neuronal damage in thedefective spinal cord (Morgagni 1762).
2 J.R. Ellenbogen et al.

Aspiration of the lesion was the time-honoredmethod of management but had catastrophicconsequences. Forestus ligated the sac (1882),and sac excision was attempted by Tulp withfatal results (Tulp 1672). The Clinical Society ofLondon (1882) recommended the use of a localsclerosing technique as the preferred methodof treatment, which was initially advocated byMorton (Morton 1872). Excision of the sac wasagain popularized by Bayer (Bayer 1892) andFrazier (Frazier 1918) in the early twentiethcentury; however, the mortality rate remainedhigh. With the advent of antibiotics and introduc-tion of CSF shunts in the 1950s, the operativeresults encouraged more surgeons to introducecomprehensive, aggressive management. In 1963,Sharrad proposed emergency operative closure ofthe back lesion to decrease mortality and improvemuscle function (Sharrard et al. 1967). This pro-vided new hope for these patients. However, in thelatter part of the decade, it became evident that themortality remained high and those who survivedhad major handicaps. In 1971, Lorber reviewed524 cases of myelomeningocele treated activelyand concluded that there were four main criteriaassociated with a poor prognosis: gross hydroceph-alus, severe paraplegia, kyphosis, associated grosscongenital anomalies, or major birth injury (Lorber1971). He advocated that patients with one or acombination of these criteria should be managedconservatively as very few patients survived, andthose that did would suffer severe mental andphysically handicap. In recent years, the reliabilityand “predictive value” of these four criteria hasbeen questioned. It has been suggested that themanagement of these infants should be individual-ized and changed whenever necessary in the bestinterest of the patient and family (McCarthy 1991;Surana et al. 1991; Woodhouse 2008).
Embryological Development
NTDs are the results of an abnormality in theprocess of neurulation, conversion of the neuralplate into a neural tube by a process of folding,
which occurs in the fourth week. Failure of part ofthe neural tube to close disrupts both the differen-tiation of the central nervous system and theinduction of the vertebral arches and can resultin a number of developmental anomalies. Thesemalformations usually involve part of the cranialor caudal neuropore, the unfused rostral and cau-dal neural folds, resulting in a defect of the cranialor lower lumbar and sacral regions of the centralnervous system (CNS) respectively. These closeon day 24 and on day 26 respectively (Larsen1993). Once closure is initiated neurectodermcells reorganize to form the roof of the neuraltube, while overlying epidermal cells form theectodermal layer of the skin. Closure of theneuropores coincides with the establishment of ablood vascular circulation for the neural tube.Failure to close leads to escape of α-fetoproteinfrom the circulation into amniotic fluid. This pro-cess is called primary neurulation and it is respon-sible for establishing the brain and spinal cordregions down to the lowest sacral levels (probablyS4–5). From this level caudally, secondary neuru-lation forms the remainder of the cord. In thisphenomenon, the neural tube forms from meso-derm cells that coalesce and then epithelialize.These epithelial cells reorganize around a lumenforming the most caudal regions of the neural tubethat then becomes continuous with the remainderof the tube formed by primary neurulation (thejunction between the two processes is well belowthe site of virtually all occurrences of spina bifidaand so cannot be considered a potential factor inthe origin of the vast majority of these NTDs)(Sadler 2005).
Classification and Pathogenesis
All developmental defects of the central nervoussystem are NTDs. They can be classified intoneurulation defects (nonclosure of the neuraltube resulting in open lesions), postneurulationdefects (producing skin-covered, i.e., closedlesions), or migration abnormalities (Table 1).Some examples include:
Spina Bifida and Encephalocele 3

Brain
Anencephaly: Failure of closure of rostral-neuropore. Occurs in about 1 out of 10,000 births.Results in absence of skull, cerebral hemispheresor cerebellum, and facial abnormalities. There arethree types of anencephaly: (a) meroanencephaly,where there is rudimentary brain tissue and partialformation of the cranium; (b) holoanencephaly, themost common type, in which the brain is completelyabsent; and (c) craniorachischisis, the most severe,where area cerebrovasculosa andareamedullovasculosafill both cranial defects and the spinal column (Isadaet al. 1993; Lemire et al. 1978).
Encephalocele: Postneurulation disorder. Fail-ure of the surface ectoderm to separate from theneuroectoderm. Occurs approximately once inevery 2000 births. Results in a bony defect in thecranium (cranium bifidum) allowing herniation of
intracranial contents, most commonly in theoccipital region (75%) in the United States andWestern Europe. Approximately 90% of casesinvolve the midline. The hernia may containmeninges (meningocele), meninges and part of thebrain (meningoencephalocele), or meninges, part ofthe brain, and part of the ventricular system(meningohydroencephalocele) (Moore et al. 2011).
Spinal Cord
Meningocele: Defect postneurulationMyelomeningocele: Failure of caudal neuroporeclosure. Myelomeningoceles can occur anywherefrom cervical to lumbar spine, and numerous the-ories have been put forward to explain the precisemechanism of the defective neurulation. It iseither due to failure of neural folds to fuse or a
Table 1 Classification of the commonest neural tube defects (Copp et al. 2013)
Site Lesion Sex ratio Clinical presentation Pathology
Cranial Anencephaly Femaleexcess(3:1)
Lack of brain and cranialvault; fetal loss, or stillbirth
Failed fusion of anteriorneuropore; degeneration ofneural tissue and absence ofcranial vault formation
Encephalocoele Femaleexcess inoccipitallesions
Meningeal sac, oftencontaining brain tissue,protrudes from skull;commonly in occipital,parietal, or frontoethmoidallocations
Brain herniation throughcongenital opening of skulland covered by meninges andskin
Spinal Spina bifida cystica/aperta
(a) Myelomeningocele(b) Meningocele
Variable;roughlyequal
Open spinal cord covered bymeningeal sac or exposed;most commonlythoracolumbar, lumbar, orlumbosacral; usuallylivebirth; frequentlyassociated withhydrocephalus postnatally
Open cord defect at any pointfrom cervical to sacralDegeneration of exposedneural tissue after failedclosureMeningeal sac without neuraltissue
Spina bifida occulta Skin covered. No visibleexposure of meninges orneural tissue
Absent spinous process andvarying amounts of lamina(may be associated withlipomyelomeningocele,tethered cord, dermal sinustract, diastematomyelia, andhemangioma)
Craniospinal Craniorachischisis Femaleexcess
Anencephaly continuouswith complete open spinabifida, i.e., total dysraphism;fetal loss or stillbirth
Combined anencephaly andmyelomeningocele
4 J.R. Ellenbogen et al.

reopening of the normally fused neural tube. Thenormally fused neural tube may reopen because ofincreased intraluminal pressure or a primarydefect in the neuroepithelium (Gardner 1961).Defective neuroepithelium itself may be respon-sible for the failure of neural folds to fuse or thedefect may lie in mesoderm, which deters theclosure of neural folds (Patten 1952). AssociatedArnold Chiari malformations may be secondary tofailure of ascent of the cord within spinal columnbecause of tethering or as a result of descentsecondary to increased pressure of hydrocephalus.
Other Defects
Occur secondary to various postneurulationabnormalities involving the neural tube or meso-derm, or because of persistence of totipotent cells.These lesions are: diastematomyelia, syringomy-elia, complete anterior and posterior spina bifida,butterfly vertebrae, lipoma, hemangioma,dermoid cyst, and sacrococcygeal teratoma. Apartial duplication and separation of the noto-chord can result in herniation of the endoderm ofthe yolk sac, called split notochord syndrome. Ifthe hernia ruptures it may result in ectopic bowel,sinus, or fistula (Bentley and Smith 1960).
Spinal Dysraphism
Spina Bifida Occulta
The term “spina bifida occulta” refers to the formof spinal dysraphism not accompanied by theextrusions of the contents of the vertebral column.There is congenital loss of the spinous process andvariable amounts of lamina without exposure ofneural tissue. Without obvious external evidenceSpina bifida occultais rarely diagnosed in the new-born. The defect may be palpable, and occasion-ally patients may have external evidence of spinabifida occulta such as a small dimple, sinus, tuft ofhair, or hamartomatous lesions such as hemangi-oma, lipoma, and naevi. This incidental finding isusually of no clinical importance when it occurs
alone. Although there may be neurological deficitbecause of tethering of the cord, manifesting asurinary problems, e.g., recurrent urinary infectionor enuresis, motor deficit with pedal deformity,pelvic tilt and muscle weakness, and sensoryinvolvement in the form of trophic ulceration.These patients warrant careful examination,investigation, and follow-up. Spinal X-ray willshow evidence of spina bifida and other spinalabnormalities. Ultrasonography can be useful inthe newborn period to diagnose diastematomyelia(McConnell et al. 1986). The common clinicallyrelevant lesions are diastematomyelia, tetheredcord, lipomyelomeningocele, and dermal sinustract. All of these lesions need referral to a neuro-surgical specialist and investigation withacraniospinal Magnetic Resonance Imaging(MRI). Many patients with these lesions requireregular follow-up and surgery in the future. Theincreased incidence and treatment of these lesionsover the last 10–20 years probably reflectsincreased diagnosis due to the more widespreadavailability of MRI.
Spina Bifida Cystica: Meningocele
Ameningocele is an epithelial lined sac filled withcerebrospinal fluid (CSF), which communicateswith the spinal subarachnoid space. There is adefect in the vertebral arches with distention ofthe meninges through this but no abnormality ofneural tissue. Meningocele, which comprisesabout 5% of all spina bifida cystica cases, is usu-ally not associated with neurological deficit andhydrocephalus, and is most frequently observed inthe lumbar region.
Spina Bifida Cystica:Myelomeningocele
Myelomeningocele is one of the most commoncongenital malformations and the commonestform of NTD. There is a congenital defect in thevertebral arches with cystic dilatation of themeninges with underlying structural and func-
Spina Bifida and Encephalocele 5
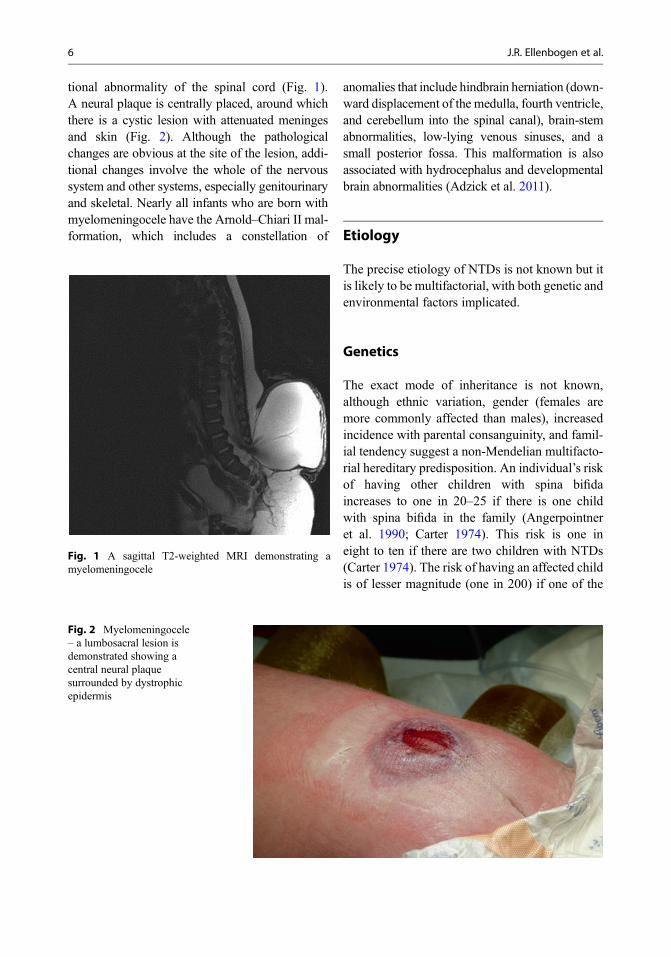
tional abnormality of the spinal cord (Fig. 1).A neural plaque is centrally placed, around whichthere is a cystic lesion with attenuated meningesand skin (Fig. 2). Although the pathologicalchanges are obvious at the site of the lesion, addi-tional changes involve the whole of the nervoussystem and other systems, especially genitourinaryand skeletal. Nearly all infants who are born withmyelomeningocele have the Arnold–Chiari II mal-formation, which includes a constellation of
anomalies that include hindbrain herniation (down-ward displacement of the medulla, fourth ventricle,and cerebellum into the spinal canal), brain-stemabnormalities, low-lying venous sinuses, and asmall posterior fossa. This malformation is alsoassociated with hydrocephalus and developmentalbrain abnormalities (Adzick et al. 2011).
Etiology
The precise etiology of NTDs is not known but itis likely to be multifactorial, with both genetic andenvironmental factors implicated.
Genetics
The exact mode of inheritance is not known,although ethnic variation, gender (females aremore commonly affected than males), increasedincidence with parental consanguinity, and famil-ial tendency suggest a non-Mendelian multifacto-rial hereditary predisposition. An individual’s riskof having other children with spina bifidaincreases to one in 20–25 if there is one childwith spina bifida in the family (Angerpointneret al. 1990; Carter 1974). This risk is one ineight to ten if there are two children with NTDs(Carter 1974). The risk of having an affected childis of lesser magnitude (one in 200) if one of the
Fig. 1 A sagittal T2-weighted MRI demonstrating amyelomeningocele
Fig. 2 Myelomeningocele– a lumbosacral lesion isdemonstrated showing acentral neural plaquesurrounded by dystrophicepidermis
6 J.R. Ellenbogen et al.

parents had spina bifida than if a sibling had spinabifida (Angerpointner et al. 1990).
Dietary Factors
Substantial data have been accumulated to sug-gest that myelomeningocele and other neuraldefects may be reduced by improved maternalnutritional status. Mothers of spina bifida patientswere found to have an increased incidence offolate metabolism abnormalities and it has sincebeen suggested that folic acid might be involved(Smithells and Chinn 1965). Several studies havereported a beneficial role of folic acid and othervitamins (Atta et al. 2016; Laurence et al. 1981;Olney and Mulinare 2002; Willett 1992). TheMedical Research Council conducted a random-ized double-blind prevention trial with factorialdesign at 33 centers in seven countries to deter-mine whether supplementation with folic acid ora mixture of seven other vitamins (A, D, B1, B2,B6, C, and nicotinamide) around the time of con-ception could prevent NTDs (Group 1991). Thewomen at risk were randomly allocated to variousgroups including a control group to avoid bias.This study found a significant reduction in thenumber of children born with NTDs to high-riskmothers who had taken folic acid in the peri-conceptional period. Periconceptional use offolic acid has been recommended to all womenwith or without risk. A concern that large dosesof folic acid may delay the diagnosis of perniciousanemia has led to the fortification recommenda-tions being limited to a level that may addon average, only about 0.1 mg folic acid/day.However, others feel that a daily dose of 0.4 mg/day should be continued (Wald and Bower 1994).Indeed in recent studies published from Canada,United States, and Australia have shown a declinein neural tube defects especially in high-riskgroups not only with folic supplementation butalso with food fortification (Bower et al. 2009;Stevenson et al. 2000; De Wals et al. 2007).Despite folic acid fortification, the incidenceof myelomeningocele has stabilized at 3.4 per10,000 live births in the United States (Bouletet al. 2008).
Diabetes and Obesity
Mothers with preexisting diabetes and obesityhave each independently been found to have anincreased risk of having a child with a NTD. Arecent paper has demonstrated a 0.7% prevalenceof diabetes mellitus in mothers of children withNTD, as compared to 0.4% of controls (adjustedodds ratio 1.84). Nineteen percent of mothers ofchildren with NTD were obese (Body Mass Index(BMI) �30) as compared with 10.8% of control(adjusted odds ratio 1.97) (Parker et al. 2013).
Teratogens
Many agents have been blamed as possible terato-gens responsible for the occurrence of NTDs.Exposure to the antiepileptic drugs valproate andcarbamazepine in utero carries a 1.2% risk of fetalNTDs, which have been reported to be moresevere open defects with a high incidence ofhydrocephalus (Lindhout et al. 1992; Rosa 1991;1988; 1983). Certain viruses and hyperthermiahave also been hypothesized to cause NTDs,and exposure to heat in the form of a hot tub,sauna, or fever in the first trimester of pregnancyhas also been associated with an increased riskof NTDs (Janerich 1971; Layde et al. 1980;Milunsky et al. 1992; Sandford et al. 1992).
Prenatal Diagnosis
Prenatal diagnosis of myelomeningocele allowsfor parental informed decision to continue thepregnancy or terminate if desired, and alsoimproved obstetric and neonatal care of theaffected infant (White-Van Mourik et al. 1990).
Maternal Serum Alphafetoprotein
Alphafetoprotein (AFP) can be detected in mater-nal serum in open NTDs. It is a 70 kd glycoproteinproduced initially by the yolk sac but by the end ofthe third trimester solely by the fetal liver. AFP isexcreted in the urine by the fetal kidneys and thus
Spina Bifida and Encephalocele 7
into the amniotic fluid. It is an effective methodfor mass screening to identify pregnancies requir-ing further evaluation. Elevated levels, �2 multi-ples of the median for the appropriate week ofgestation, between 15 and 20 weeks gestationcarries a relative risk of 224 for neural tube defects(Milunsky et al. 1989). If positive, the test isrepeated a week later to confirm the presence orabsence of NTDs. The sensitivity of this test isabout 97% for anencephaly and 72% for spinabifida (Wald et al. 1977). This second test requiresfurther confirmation by amniocentesis and prena-tal ultrasonography after appropriate counseling.Since maternal serum AFP rises during normalpregnancy, an underestimate of gestational agemay cause a normal level to be interpreted aselevated, and an overestimate of gestation agemay lead to an elevated AFP interpreted as nor-mal. Maternal serum AFP may be elevated inmany other fetal abnormalities, including thoseof the abdominal and urological systems, suchas omphalocele, gastroschisis, cloacal exstrophy,esophageal atresia, and renal agenesis. Therefore,the diagnosis should be confirmed using othertests.
Imaging
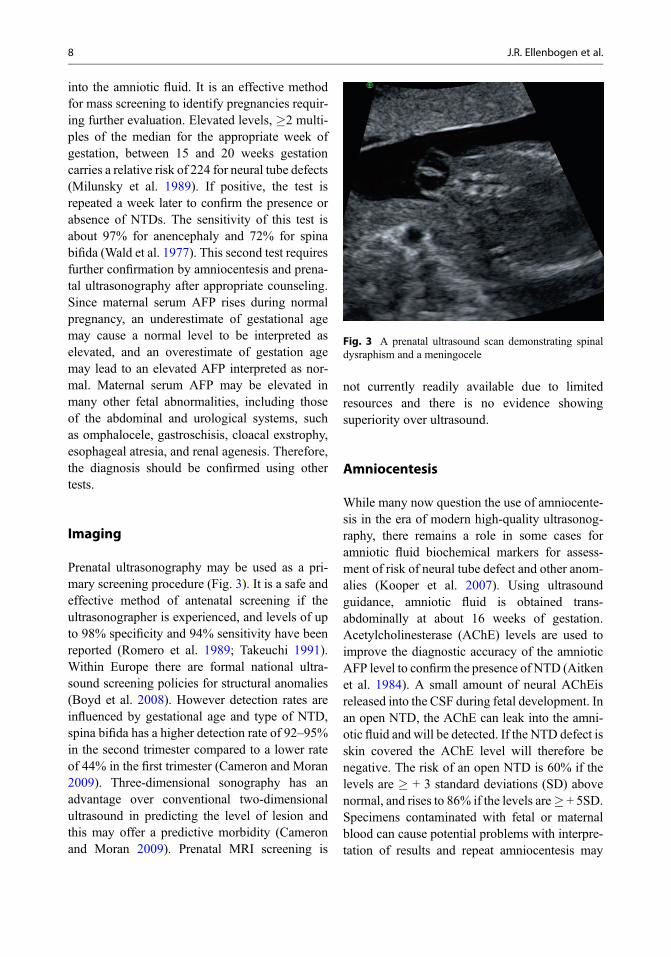
Prenatal ultrasonography may be used as a pri-mary screening procedure (Fig. 3). It is a safe andeffective method of antenatal screening if theultrasonographer is experienced, and levels of upto 98% specificity and 94% sensitivity have beenreported (Romero et al. 1989; Takeuchi 1991).Within Europe there are formal national ultra-sound screening policies for structural anomalies(Boyd et al. 2008). However detection rates areinfluenced by gestational age and type of NTD,spina bifida has a higher detection rate of 92–95%in the second trimester compared to a lower rateof 44% in the first trimester (Cameron and Moran2009). Three-dimensional sonography has anadvantage over conventional two-dimensionalultrasound in predicting the level of lesion andthis may offer a predictive morbidity (Cameronand Moran 2009). Prenatal MRI screening is
not currently readily available due to limitedresources and there is no evidence showingsuperiority over ultrasound.
Amniocentesis
While many now question the use of amniocente-sis in the era of modern high-quality ultrasonog-raphy, there remains a role in some cases foramniotic fluid biochemical markers for assess-ment of risk of neural tube defect and other anom-alies (Kooper et al. 2007). Using ultrasoundguidance, amniotic fluid is obtained trans-abdominally at about 16 weeks of gestation.Acetylcholinesterase (AChE) levels are used toimprove the diagnostic accuracy of the amnioticAFP level to confirm the presence of NTD (Aitkenet al. 1984). A small amount of neural AChEisreleased into the CSF during fetal development. Inan open NTD, the AChE can leak into the amni-otic fluid and will be detected. If the NTD defect isskin covered the AChE level will therefore benegative. The risk of an open NTD is 60% if thelevels are � + 3 standard deviations (SD) abovenormal, and rises to 86% if the levels are� + 5SD.Specimens contaminated with fetal or maternalblood can cause potential problems with interpre-tation of results and repeat amniocentesis may
Fig. 3 A prenatal ultrasound scan demonstrating spinaldysraphism and a meningocele
8 J.R. Ellenbogen et al.

be required. There is approximately a 6% risk offetal loss in this population with this procedure(Greenberg 2010).
Clinical Management
This section relates primarily to the managementof myelomeningocele as this is the commonestNTD that presents to pediatric neurosurgicalpractice.
Prenatal Assessment and Counseling
Once a prenatal diagnosis of myelomeningocelehas been made, further detailed imaging isperformed to determine the level and extent ofthe defect (Fig. 4). This allows the neurosurgeonto predict the likely degree of neurological deficitand facilitates prenatal counseling. Antenatalcounseling is undertaken in specialist clinics andinvolves fetal medicine specialists, obstetricians,neurosurgeons, and radiologists. Parents need tobe given realistic expectations regarding the prog-nosis for intellectual development, ambulation,and survival, as well as information on hydro-cephalus and neurogenic bowel and bladder. Inthe UK, a review process is underway with a viewto setting up national standard.
It still remains controversial whether cesareansection should be carried out if the prenatal diag-nosis of myelomeningocele has been made. Someauthors report that cesarean section offers advan-tages to those born by this route as compared tothose born by vaginal delivery, although otherauthors do not support this (Luthy et al. 1991;Sakala and Andree 1990). Luthy et al. reportedthat delivery by cesarean section for the fetus withuncomplicated myelomeningocele before onset oflabor may result in better subsequent motor func-tion than vaginal delivery or cesarean section afterlabor has commenced (Luthy et al. 1991). Whilethere is no clear evidence, either way, cesareansection facilitates a timed delivery to allow plan-ning for surgical closure of the myelomeningocele(Thompson 2009).
In-Utero Surgery
There is a potential role for fetal surgery as neuraltube defects are diagnosed prenatally. Techniquesusing endoscopy, percutaneous fetoscopic patchcoverage, and via hysterotomy have beendescribed. Human prenatal myelomeningocelerepair by hysterotomy was first performed in1997, and by 2003, more than 200 fetuses hadundergone the procedure (Adzick et al. 2011).However, until recently little evidence as to thebenefit of fetal surgery existed (Sutton 2008).The Management of Myelomeningocele Study(MOMS) was a three-center randomized prospec-tive trial comparing prenatal surgery before26 weeks of gestation or standard postnatal repairof myelomeningocele based in the USA. Out-comes assessed include death, the need for cere-brospinal fluid shunting by one year of life, andneurological function at 30 months of age. Theyfound that prenatal surgery for myelomeningocelereduced the need for CSF shunting (40% in theprenatal-surgery group and 82% in the postnatal-surgery group) and improved the composite scorefor mental development and motor function at
Fig. 4 A sagittal inter-uterine fetal MRI demonstrating aChiari malformation
Spina Bifida and Encephalocele 9

30 months. Prenatal surgery also resulted inimprovement in hindbrain herniation by12 months, and ambulation by 30 months. How-ever, prenatal surgery was associated withincreased fetal risks and an increased risk of pre-term delivery and uterine dehiscence at delivery(Adzick et al. 2011; Johnson et al. 2016). Whilstthis is positive, fetal surgery is a very complexprocedure. The centers performing fetal surgeryneed to be very well organized with diagnosis,counseling, intervention, follow-up, and supportperformed by a highly skilled and trained multi-disciplinary team. All fetal surgical interventionsthat involve low volume and highly complex careshould be restricted to dedicated centers withstrict regulations and a high level of quality con-trol (van Lith et al. 2013).
Postnatal Assessment
Inspection
In about 80% of infants with myelomeningocele,the defect includes the lumbar region because thisis the last region of the neural tube to close.Occasionally more than one lesion can be foundand there is often marked kyphosis or scoliosis pre-sent (Thompson 2009). Most myelomeningocelescontain an enlarged subarachnoid space ventrally,with the neural tissue displaced dorsally; in combina-tion this creates a herniated sac on the infant’s back.
Motor AssessmentMotor function is assessed when the infant is atrest. Sharp stimulation above the level of themeningocele is administered with careful obser-vation of voluntary movement below the affectedlevel. Varying degrees of paralysis below the levelof the lesion are common, except in rarer cervicaland upper thoracic lesions, which are usuallyspared. The paralysis is usually flaccid, indicatinga complete neural lesion. It must be borne in mindthat there are some abnormal reflex activities inthe lower extremities that have no bearing onvolitional motor function.
Sensory AssessmentSensory loss is determined by pinprick test fromdistal to proximal, looking for an upper limb orfacial response characteristic of those experienc-ing a pain sensation. The level at which anesthesiastarts indicates the level of the lesion and predictsthe degree of disability. Proper care is necessary toavoid trophic changes in anesthetic areas.
Assessment of Bladder and BowelFunctionOver 90% of patients with myelomeningocelehave a form of neurogenic bladder. The vastmajority of these patients have disturbances ofdetrusor and sphincter balance resulting in a large,trabeculated bladder with urinary stasis. The analexternal sphincter and puborectalis are ofteninvolved, resulting in patulous anus and sometimesrectal prolapse. It is difficult to ascertain bladderinvolvement in the newborn but steps should betaken to ensure the bladder is kept empty by inter-mittent catheterization. The upper urinary tract isusually normal but some affected patients willexperience changes in the upper urinary tract atbirth (Greig et al. 1991). These patients needcareful urological and renal follow-up.
HydrocephalusHydrocephalus develops in 65–85% of patientswith myelomeningocele, the majority beforethe age of 6 months, and approximately 5–10%have overt hydrocephalus at birth (Stein andSchut 1979). Assessment of the anterior fonta-nelle and occipitofrontal circumference are impor-tant to determine the timing of any CSF diversionprocedures. In those infants with clear evidenceof hydrocephalus, early CSF diversion is indi-cated. Delayed placement of a ventriculo-peritoneal shunt is not associated with a lowerinfection rate compared to placement at the timeof myelomeningocele closure (Chadduck andReding 1988; Parent and McMillan 1995).It may reduce the risk of myelomeningocele repairbreakdown previously seen in the interval beforeshunting (Greenberg 2010). Myelomeningocelerepair may actually convert a latent hydrocephalus
10 J.R. Ellenbogen et al.

to active as the route of CSF egress is eliminated.Almost half of meningocele patients shunted atbirth require shunt revision in the first year of life,mostly due to mechanical failure (Caldarelli et al.1996). Endoscopic third ventriculosomy (ETV)has also been used in some patients althoughthe failure rate is high when performed as aprimary procedure (Fritsch et al. 2005; O’Brienet al. 2005). In patients presenting with shuntmalfunction, secondary ETV has a better successrate and is therefore best reserved for latermanagement (O’Brien et al. 2005).
Arnold–Chiari Type II MalformationArnold–Chiari II malformation is invariably asso-ciated with myelomeningocele, often accompa-nied by hydrocephalus. The caudally displacedcervicomedullary junction, pons, fourth ventricleand medulla together with a small posterior fossaand tectal “beaking” are demonstrated well onMRI. Between 25 and 33% of patients are symp-tomatic from the Chiari II malformation causedby brainstem dysfunction and lower cranial nervepalsies. Stevenson et al. reported that apneicepisodes, stridor (vocal cord paresis), andswallowing difficulties in infancy were associatedwith a 15% mortality rate in those affected(Stevenson 2004). Whether early decompressionimproves symptoms remain controversial as sur-gery related morbidity and mortality is high ininfants (up to 15–20%) and will not correct theintrinsic brainstem abnormalities (Pollack et al.1992; Vandertop et al. 1992). From a practicalpoint of view, symptomatic Chiari II are rare ininfants as long as the hydrocephalus is correctlymanaged. Chiari II can become a secondary problemin adult life with delayed deterioration.
Skeletal AbnormalitiesTalipes deformity is the most commonly occur-ring abnormality with spina bifida. Other defor-mities include hip dysplasia, genu recurvatum,and kyphoscoliosis. Orthopedic assessment andspecialist physiotherapy services are required tomanage these problems and minimize the extentof deformity.
Investigations
A full blood count is obtained and blood is cross-matched with maternal serum. A plain X-ray ofthe spine will reveal the extent of the bony lesionand associated kyphoscoliosis. An ultrasoundscan of the head, renal tract, and hips are carriedout as baseline investigations. MRI of the entirecraniospinal axis will determine the presence ofassociated congenital abnormalities. An assess-ment of congenital pituitary insufficiency viaanterior pituitary function tests should also beperformed.
Parental Counseling
Babies born with NTDs deserve an ethical,humane program of management based on accu-rate background data involving the parents fully inthe decision-making process (Charney 1990;Surana et al. 1991). The management of eachchild should be individualized and reviewed reg-ularly. It has been observed that in spite of earlyclosure and application of all measures available,there is still a significant rate of disability andmortality (Fitzgerald and Healy 1974; Hunt1990; Hunt and Oakeshott 2003). Historically,Lorber outlined some adverse criteria for conser-vative management of these patients, includinggross paraplegia, hydrocephalus exceeding theNinetieth centile by 2 cm or more, severe kypho-sis, thoracolumbar lesions, and other associatedcongenital anomalies (Lorber 1971). Thesecriteria are based on the belief that these patientswill die early in infancy; this type of managementwill also give some time for discussion with theparents, enabling them to make rational decisions.These severely affected babies were often man-aged conservatively with demand feeding andsedation. In the past decade with improved ante-natal diagnosis and prenatal counseling, many ofthe severe defects are avoided with early termina-tion and therefore the trend in the western world isfor active management as outcomes are improv-ing. Parents are usually aware of the expected
Spina Bifida and Encephalocele 11

prognosis for intellectual development, ambula-tion, and survival after antenatal counseling.
General Management, OperativeTreatment, and Technique
The initial management is aimed at stabilizing theinfant with the aim that all actively treated spinabifida patients should undergo closure of thedefect within 24–48 h of delivery. It was previ-ously thought that early closure resulted inimproved neurological outcome, but this has notbeen supported by more recent studies (Charneyet al. 1985; Frazier 1918; Robards et al. 1975).Early defect closure prevents infection and furtherdamage to exposed neural tissue – the aim beingto preserve existing function. The infant should bekept euvolemic, normothermic with latex precau-tions in place, as 73% of these infants will beallergic to proteins present in latex. Themyelomeningocele should be covered in sterilesaline-moistened gauze and wrapped in plasticcling film to minimize dehydration and to preventcontamination. The infant is nursed lateral orprone to ensure no pressure is placed on the neuralplacode.
The principle of surgery is to reconstruct thespinal cord by five-layer closure of the pia andarachnoid, lumbar fascia, subcutaneous tissue,and skin. The vascular supply to the neuroplaqueis maintained and unnecessary neural injury isavoided. The patient is placed in the prone positionwith a soft roll under the hips and shoulder, andwith the head turned to the right through 90�
(Figs. 5 and 8a). The table is in the Trendelenbergposition to minimize CSF drainage from the spinaldefect. Swabs are taken from the lesion for micro-bial examination and culture. An antiseptic soak isplaced over the anus. The lesion is covered with awarm swab and the surrounding skin is cleanedand draped. The skin is incised at the junction ofthe neural placode arachnoid membrane and thedystrophic epidermis (Fig. 8b). The membranebetween the edge of the skin defect and neuralplaque is removed carefully to avoid inclusioncysts (dermoid/lipoma) (Fig. 8c). The dura isfreed laterally and then superiorly and inferiorly
to normal intact dura (Fig. 8d). Dura is then suturedin the midline with continuous mono-filamentabsorbable sutures (Figs. 6 and 8e). A suctiondrain may be placed extradurally though this isnot mandatory. The lumbodorsal fascia is incisedlaterally and dissected free from the posterior lilaccrest. These are folded medially and sutured overthe dorsal dural layer. The subcutaneous tissue isclosed with absorbable interrupted sutures andthen the skin is closed with interrupted nylonstitches (Figs. 7 and 8f). The infant is nursed in aprone position. A plastic drape is positionedbetween the wound and the anus to prevent con-tamination of the wound. Feeding is commencedonce the bowel starts working. The wound isperiodically inspected. The suction drain isremoved 24–48 h after operation.
Postoperative Managementand Complications
Adequate nutrition is vital to allow wound healingto take place and supplementary nutrition may benecessary. Wound dehiscence in the commonestcomplication and is usually secondary to unduetension on the skin edges or skin necrosis(Rickwood 1976). Infection occurs with an inci-dence of 1–1.5% and usually occurs 5–7 dayspostsurgery (Pang 1995). Treatment is withappropriate antibiotics and local drainage. If theinvolvement is deep to the lumbodorsal fascia, itmay cause meningitis or ventriculitis. These arevigorously treated with local dressings, systemicantibiotics, and external ventricular drainage, ifhydrocephalus is present and CSF leak occurs.With meticulous closure of the dura, CSF fistulashould be rare, however if it does occur, immedi-ate repair is preferred rather than conservativetreatment. Hydrocephalus may be present inabout 15% of patients with myelomeningocele atbirth, while it eventually develops in 85% of thesepatients. The exact etiology of this is not knownbut it is likely to be multifactorial. It may be aresult of aqueductal stenosis or fourth ventricularoutflow obstruction, or due to underdevelopmentof the arachnoid granulations. Close observationfor signs of raised intracranial pressure should
12 J.R. Ellenbogen et al.
occur in the first few days followingmyelomeningocele repair. At the first signs ofraised intracranial pressure, a CSF diversion pro-cedure should take place to avoid stress on themyelomeningocele wound. From a practical pointof view, after neurosurgical closure and shuntplacement, if indicated, referral and managementis continued by pediatric orthopedic surgeons,urologists, and physiotherapist, thusencompassing a multidisciplinary team approach.
Long-TermManagement andOutcome
Parents who choose to continue the pregnancy ofa fetus with spina bifida must prepare for a childwith significant care needs. The results of
Fig. 6 Dura is then suturedin the midline and skin isundermined to allowmobilization and closure
Fig. 7 Skin closure
Fig. 5 Patient position onoperating table
Spina Bifida and Encephalocele 13
myelomeningocele operations vary considerablybecause of differences in approach to manage-ment. Historically, in the units where a highlyselective approach was taken, all 100% conserva-tively managed patients died, while only 14.3% ofactively managed patients died (Lorber andSalfield 1981). In centers where patients weremanaged unselectively, a 41% mortality rate hasbeen reported (Hunt 1990). More recently despiteaggressive intervention, nearly 14% of all spinabifida neonates do not survive past 5 years of age,with the mortality rising to 35% in those withsymptoms of brainstem dysfunction secondary tothe Arnold–Chiari malformation. While 70% ofpatients have an IQ above 80, only half are able to
live independently as adults, even with adaptedaccommodations (Oakeshott and Hunt 2003). Theemotional and financial impact on the family andcommunity are enormous. No recent data areavailable, but in 1994 the cost of care exceeded$500 million per year (in 1992 dollars) in theUnited States alone (Adzick 2013).
Medical Problems
Urinary IncontinenceOnly 10% of myelomeningocele patients have anormal bladder, the remainder have a neurogenicbladder. The introduction of clean intermittent
Fig. 8 Myelomeningoceleclosure. (a) Patient positionon operating table;(b) elliptical incision atjunction of arachnoid andskin; (c) membrane excisedand neural plaque freed;(d) dura dissected free fromunderlying muscle; (e) durais closed with absorbablesuture and adjacent skin isundermined to allowmobilization; (f) skinclosure with nylon suture(# Puri P, editor. NewbornSurgery. 3rd ed. London,Hodder Arnold; 2011)
14 J.R. Ellenbogen et al.

self-catheterization, pharmacological agents (e.g.,anticholinergics), external devices, biofeedback,and innovative surgical procedures for the neuro-genic bladder have enabled patients to developsocial relationships while preserving renal func-tion – up to 75% of these patients can be sociallycontinent of urine (Rudy and Woodside 1991).Urodynamic studies in the newborn period areuseful in identifying “at-risk” children with highbladder pressure and detrusor instability(Kasabian et al. 1992; Stoneking et al. 2001;Snow-Lisy et al. 2015).
HydrocephalusShunt placement has a significant impact for thefuture life of the child and parents. More than 80%of spina bifida patients require placement ofshunts to prevent the neurologic and intellectualcompromise that accompanies significantventriculomegaly, and 46% have complicationsof shunts within the first year of placement(Caldarelli et al. 1996). Shunt malfunction andcomplications (e.g., infection, hemorrhage) inmyelomeningocele patients has a cognitiveimpact as well as being related to long-term sur-vival (Barf et al. 2004; Davis et al. 2005; Huntand Oakeshott 2003; Tuli et al. 2003). If shuntmalfunction occurs in later childhood then ETVhas a 89% success rate and should be consideredbefore shunt revision (Jenkinson et al. 2009).
Chiari II Malformation and HindbrainDysfunctionOlder children and young adults with Chiari IImalformation may complain of symptoms relatedto foramen magnum impaction such as headache,neck pain, and upper limb sensory disturbance.Brainstem dysfunction and lower cranial nervepalsies may also be present. Surgical treatmentmay be indicated in symptomatic patients, onceadequate CSF diversion by either shunting orendoscopic third ventriculostomy has beenestablished (Jenkinson et al. 2009). In a series of21 patients with myelomeningocele surviving intoadulthood, eight patients underwent foramenmagnum decompression for late deterioration,which stabilized symptom progression (Jenkinsonet al. 2008).
Tethered CordSpinal cord tethering is fixation of the spinal cordsecondary to adhesions between the previouslyexposed neural elements and the surrounding tis-sues, leading to tension on the neural axis andsecondary neurological injury due to ischemia.As many as 70% of myelomeningocele patientshave radiologically confirmed cord tethering butonly a minority are symptomatic, usually identi-fied after a patient develops progressive worsen-ing of neurologic function (Greenberg 2010).While surgical release can limit further damagein some patients, the functional decline may beirreversible in others (Adzick 2013).
Lifestyle Concerns
Whether patients are managed conservatively oractively, the quality of life is an important factor inthose who survive. In the long-term follow-upstudy by Oakshott and Hunt of 117 patientstreated unselectively, who were followed up for16–20 years, it was found that 41% of patients haddied before 16-years of age (Hunt 1990; Hunt andOakeshott 2003). Of the survivors, almost 31%were mentally delayed and 48% were unable tolive without help or supervision, while only aboutone-quarter of the survivors were capable of com-petitive employment (Hunt 1990; Hunt andOakeshott 2003). Although a more recent studyshowed that 38% of adult survivors were in activeemployment (Valtonen et al. 2006).
Intellect
Patients who have associated hydrocephalus andepisodes of shunt malfunction (blockage andinfection) have lower intelligence than thosewho only have myelomeningocele, and are lesslikely to walk, live independently, and drive acar (Barf et al. 2004; Oakeshott et al. 2012). Twostudies have demonstrated significant cognitiveimpairment in myelomeningocele patients in thepediatric population that have been attributed tothe structural defects associated with the Chiari IImalformation, rather than repeated episodes of
Spina Bifida and Encephalocele 15

hydrocephalus (Vachha et al. 2006; Vinck et al.2006). Fiber tract anomalies in the limbic systemwere correlated with memory deficits (Vachhaet al. 2006). A recent study has confirmed this tobe the case in adults with spina bifida and a ChiariII malformation, as repeated episodes of shuntmalfunction or foramen magnum decompressionwere not associated with a worse cognitive func-tion (Jenkinson et al. 2011). In a small adult cohortundergoing in depth neuropsychological testing,all myelomeningocele patients had elements ofcognitive impairment, notably attention and mem-ory function and visuospatial construction, whichinvariably influences social integration andemployment prospects (Jenkinson et al. 2008,2011). Nevertheless, patients though severelyphysically disabled but who have relatively mildcognitive impairment can be self-supporting andin competitive employment.
Ambulation
Ambulatory potential and capacity are related tointelligence, orthopedic deformity, level of lesion,obesity, and motivation (Dicianno et al. 2015).Most of the patients with lesions below L5 areambulators, those with lesions at L4 are functionalambulators, and those with lesions above L3 arewheelchair-bound (Asher and Olson 1983;Oakeshott et al. 2012; Seitzberg et al. 2008). Theproportion of those patients who retain ambulantstatus into adulthood gradually decreases due tocombinatorial factors including increasing weight,spinal and foot deformity, and respiratory compro-mise. Long-term follow-up studies have reportedthat only 30% of patients remain ambulated at30 years of age (Oakeshott and Hunt 2003).A wheelchair is certainly a more energy efficientmeans of mobility (Bruinings et al. 2007).
Psychosocial Problems
Educational mainstreaming, special counseling,improved understanding of the patient’s potential,and increased public awareness of spina bifidacontribute to a reduction in stress through
psychosocial problems originating within them-selves, their families, and society.
Encephalocele
Encephalocele, a cranial neural tube defect, ismuch less common than the spinal equivalentand constitutes 10–20% of all NTDs. A spectrumof “cranial dysraphism” exists from a cranial der-mal sinus, a persistent ectodermal connectionbetween the skin and the meninges, to trueencephaloceles, were components of all intracra-nial contents herniate through a defect in the skull(Fig. 9). When only meninges and CSF protrude itis called a meningocele. An atretic meningocele isa small, nodular, noncystic lesions, commonly atthe vertex (Thompson 2009). Encephaloceleshave a prevalence of between 1 in 2000 and 1 in5000 live births, but there is a significant geo-graphical variation with a higher incidence inAsia than in the West (EUROCAT WorkingGroup 1991; Gorlin et al. 2001).
Classification
Several classifications of encephalocele exist.They can be classified according to the contentsof the swelling, the location of the swelling, andthe location of cranial defect (Choux et al. 1999).Location can be classified as occipital, parietal,frontal, temporal, nasopharyngeal, nasal,frontoethmoid (sincipital), basal encephalocele,and posterior fossa (Suwanwela and Suwanwela1972). The location also varies with geographicallocation with 80–90% of all encephaloceles beingoccipital in the Western Hemisphere but anterioror sincipital encephaloceles occurring 9.5 timesmore frequently in Thailand, Indonesia, northernIndia, and Africa (Choux et al. 1999).
Etiology
There is no definitive explanation for themaldevelopment of the neural tube at the cephalicend leading to the formation of an encephalocele.
16 J.R. Ellenbogen et al.
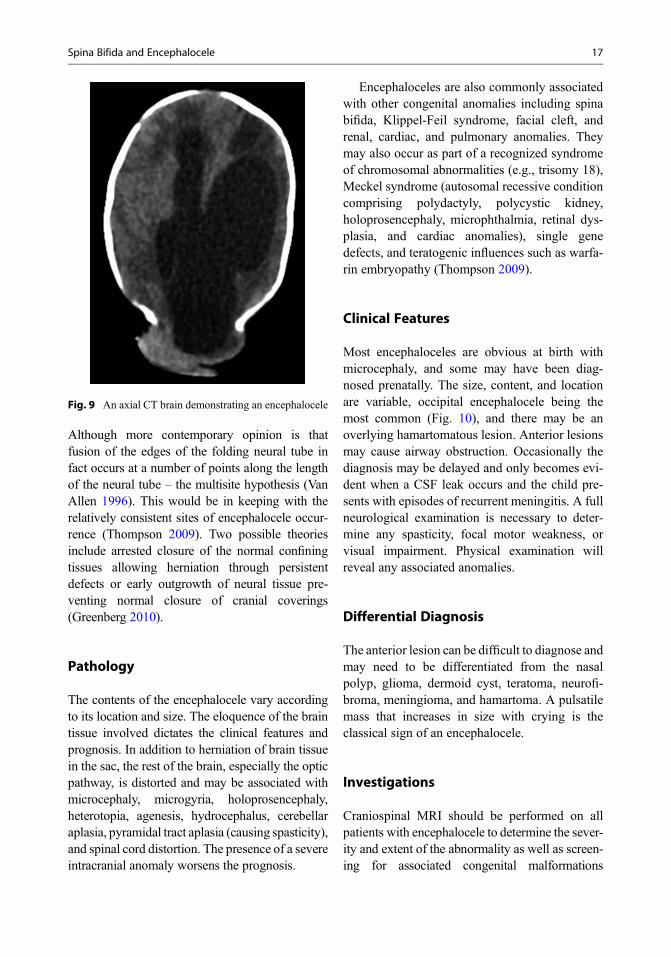
Although more contemporary opinion is thatfusion of the edges of the folding neural tube infact occurs at a number of points along the lengthof the neural tube – the multisite hypothesis (VanAllen 1996). This would be in keeping with therelatively consistent sites of encephalocele occur-rence (Thompson 2009). Two possible theoriesinclude arrested closure of the normal confiningtissues allowing herniation through persistentdefects or early outgrowth of neural tissue pre-venting normal closure of cranial coverings(Greenberg 2010).
Pathology
The contents of the encephalocele vary accordingto its location and size. The eloquence of the braintissue involved dictates the clinical features andprognosis. In addition to herniation of brain tissuein the sac, the rest of the brain, especially the opticpathway, is distorted and may be associated withmicrocephaly, microgyria, holoprosencephaly,heterotopia, agenesis, hydrocephalus, cerebellaraplasia, pyramidal tract aplasia (causing spasticity),and spinal cord distortion. The presence of a severeintracranial anomaly worsens the prognosis.
Encephaloceles are also commonly associatedwith other congenital anomalies including spinabifida, Klippel-Feil syndrome, facial cleft, andrenal, cardiac, and pulmonary anomalies. Theymay also occur as part of a recognized syndromeof chromosomal abnormalities (e.g., trisomy 18),Meckel syndrome (autosomal recessive conditioncomprising polydactyly, polycystic kidney,holoprosencephaly, microphthalmia, retinal dys-plasia, and cardiac anomalies), single genedefects, and teratogenic influences such as warfa-rin embryopathy (Thompson 2009).
Clinical Features
Most encephaloceles are obvious at birth withmicrocephaly, and some may have been diag-nosed prenatally. The size, content, and locationare variable, occipital encephalocele being themost common (Fig. 10), and there may be anoverlying hamartomatous lesion. Anterior lesionsmay cause airway obstruction. Occasionally thediagnosis may be delayed and only becomes evi-dent when a CSF leak occurs and the child pre-sents with episodes of recurrent meningitis. A fullneurological examination is necessary to deter-mine any spasticity, focal motor weakness, orvisual impairment. Physical examination willreveal any associated anomalies.
Differential Diagnosis
The anterior lesion can be difficult to diagnose andmay need to be differentiated from the nasalpolyp, glioma, dermoid cyst, teratoma, neurofi-broma, meningioma, and hamartoma. A pulsatilemass that increases in size with crying is theclassical sign of an encephalocele.
Investigations
Craniospinal MRI should be performed on allpatients with encephalocele to determine the sever-ity and extent of the abnormality as well as screen-ing for associated congenital malformations
Fig. 9 An axial CT brain demonstrating an encephalocele
Spina Bifida and Encephalocele 17

(Fig. 10). Visual evoked response will establish thepresence of the occipital cortex within the sac,which may be helpful in surgical planning.
Treatment
Conservative treatment may be justified forpatients with microcephaly and large amounts ofbrain within the encephalocele, where death isinevitable. Most of the patients are treated bysurgical repair of the encephalocele. The aims ofthe surgery are excision of the extracranial non-functioning brain tissue, water-tight dural closureat the level of the cranium, and restoration ofcranial contour with good skin coverage. Takingthese steps to treatment also helps to preventinfection and preserve function.
Operative Treatment and TechniqueUnder general anesthesia the patient is positionedwith the encephalocele uppermost (e.g., prone foran occipital lesion). The lesion is prepped anddraped while ensuring adequate support for theencephalocele. Large lesions may be aspirated tofacilitate handling and dissection. A sample ofCSF is sent for microbiological examination andcultures. Usually a transverse ellipse incision ismade near the base of the lesion, planned so as toenable closing it without causing undue tension(Fig. 11b, c). The incision is then deepened untilthe dura is seen, which is traced up to the bonydefect. The sac is opened where the cerebral tissueis not adhered. If the cerebral tissue is too large,necrotic and if no visual evoked potentials aredemonstrated, it is excised. Every effort shouldbe made to preserve brain tissue without causingan acute rise in intracranial pressure. Bony defectsmay sometimes need to be enlarged. Whiledissecting the dura and brain tissue, care must betaken to avoid abnormal venous sinuses andvenous connections. The distal sac is then excised.The dura is closed with continuous monofilamentabsorbable sutures and a dural graft may berequired (pericranium is ideal). Meticulous hemo-stasis is achieved with the closure of all layers. Asmall suction drain is occasionally used. Subcuta-neous tissue is approximated with fine, absorbable
interrupted sutures, and the skin is closed withinterrupted nylon or continuous absorbable sub-cuticular stitches (Fig. 11d). A dressing and ban-dage is applied.
Postoperative Care and ComplicationThe infant is nursed opposite to site to minimizepressure in the wound and closely observed forsigns of increasing intracranial pressure or devel-opment of hydrocephalus. Meningitis is commonwith anterior encephalocele repair, where contam-ination is more likely because of the proximity tothe nose, mouth, and air sinuses. A CSF leak canoccur despite meticulous dural closure. A com-bined intracranial and transnasal approach mayhelp to avoid this. Once hydrocephalus has beenexcluded with a CT scan, CSF leaks are managedwith additional skin sutures and a lumbar drain.If a CSF leak persists then wound revision may beindicated. Most infants with encephalocele repairdevelop hydrocephalus which should be treatedwith a CSF shunt.
Prognosis
The prognosis for encephaloceles is largely dic-tated by three variables: anatomical site, volume
Fig. 10 A sagittal T1-weighted MRI demonstrating anoccipital encephalocele
18 J.R. Ellenbogen et al.

of neural contents, and the presence of coexistingcerebral and extracerebral malformations. Formeningoceles, atretic meningoceles, and cranialdermal sinuses, the underlying brain is usuallystructurally normal, the prognosis for these lesionsis extremely favorable and normal or near normaldevelopment can be anticipated (Thompson 2009).Encephalocele carries a high rate of mortality butsimple isolated encephaloceles without associatedabnormalities have a better prognosis than largerlesions. The prognosis in children with anteriorencephaloceles is better than those with occipitalencephaloceles. Deaths may be due to cerebralanomalies, associated congenital abnormalities,hydrocephalus/shunt malfunction, and complica-tions. The presence of hydrocephalus has beendemonstrated to correlate with poor outcome, ashas the presence of additional congenital
abnormalities (Brown and Sheridan-Pereira 1992;Bui et al. 2007; Date et al. 1993).
Conclusion and Future Directions
These congenital defects of the central nervoussystem have devastating consequences both forthe child and their family. Appropriate manage-ment aims to recognize and reduce the pre-disposing factors to such congenital defects, toidentify early and appropriately counsel thosepregnancies with defects, to ensure timely surgi-cal management of exposed spinal cord and asso-ciated hydrocephalus, and maintain a holisticmultidisciplinary approach to the care of thepatients and their family.
Further research with the aim of expandingour understanding of the pathophysiology of
Fig. 11 Encephaloceleclosure. (a) Diagrammaticalrepresentation of anoccipital encephalocele;(b, c) incision around baseof encephalocele; (d) skinclosure (#Puri P, editor.Newborn Surgery.3rd ed. London, HodderArnold; 2011)
Spina Bifida and Encephalocele 19

MMC, together with the development of surgicaltechnique and procedures, will improve our abil-ity to prevent and treat these conditions. Futurestudies will aim to evaluate the long-term impactof in-utero intervention on maternal and fetal out-come as compared to postnatal intervention withfollow-up of those patients in the MOMS trial(MOMS II).
References
Adzick NS. Fetal surgery for spina bifida: past, present,future. Semin Pediatr Surg. 2013;22(1):10–7.
Adzick NS, Thom EA, Spong CY, Brock JW, Burrows PK,JohnsonMP, et al. A randomized trial of prenatal versuspostnatal repair of myelomeningocele. N Engl J Med.2011;364(11):993–1004.
Aitken DA, Morrison NM, Ferguson-Smith MA. Predic-tive value of amniotic acetylcholinesterase analysis inthe diagnosis of fetal abnormality in 3700 pregnancies.Prenat Diagn. 1984;4(5):329–40.
Atta CA, Fiest KM, Frolkis AD, et al. Global birth preva-lence of spina bifida by folic acid fortification status: asystematic review and meta-analysis. Am J PublicHealth. 2016;106(1):e24–34.
Van Allen MI. Multisite neural tube closure in humans.Birth Defects Orig Artic Ser. 1996;30(1):203–25.
Angerpointner TA, Pockrandt L, Schroer K. Course ofpregnancy, family history and genetics in childrenwith spina bifida. Z Kinderchir. 1990;45(2):72–7.
Asher M, Olson J. Factors affecting the ambulatory statusof patients with spina bifida cystica. J Bone Joint SurgAm. 1983;65(3):350–6.
Barf HA, Verhoef M, Post MW, Jennekens-Schinkel A,Gooskens RH, Mullaart RA, et al. Educational careerand predictors of type of education in young adults withspina bifida. Int J Rehabil Res. 2004;27(1):45–52.
Bayer C. Zur technik der operation der spina bifida andencephalocoele. Prag Med Wochenschr. 1892;17:317.
Bentley JF, Smith JR. Developmental posterior entericremnants and spinal malformations: the split notochordsyndrome. Arch Dis Child. 1960;35:76–86.
Boone D, Parsons D, Lachmann SM, Sherwood T. Spinabifida occulta: lesion or anomaly? Clin Radiol.1985;36(2):159–61.
Boulet SL, Yang Q, Mai C, Kirby RS, Collins JS,Robbins JM, et al. Trends in the postfortificationprevalence of spina bifida and anencephaly in theUnited States. Birth Defects Res A Clin Mol Teratol.2008;82(7):527–32.
Bower C, D’Antoine H, Stanley FJ. Neural tube defects inAustralia: trends in encephaloceles and other neuraltube defects before and after promotion of folic acidsupplementation and voluntary food fortification. BirthDefects Res A Clin Mol Teratol. 2009;85(4):269–73.
Boyd PA, Devigan C, Khoshnood B, Loane M, Garne E,Dolk H. Survey of prenatal screening policies inEurope for structural malformations and chromosomeanomalies, and their impact on detection and termina-tion rates for neural tube defects and Down’s syndrome.BJOG. 2008;115(6):689–96.
BrownMS, Sheridan-Pereira M. Outlook for the child witha cephalocele. Pediatrics. 1992;90(6):914–9.
Bruinings AL, van den Berg-Emons HJ, Buffart LM, vander Heijden-Maessen HC, Roebroeck ME, Stam HJ.Energy cost and physical strain of daily activities inadolescents and young adults with myelomeningocele.Dev Med Child Neurol. 2007;49(9):672–7.
Bui CJ, Tubbs RS, Shannon CN, Acakpo-Satchivi L,Wellons JC, Blount JP, et al. Institutional experiencewith cranial vault encephaloceles. J Neurosurg.2007;107(1 Suppl):22–5.
Caldarelli M, Di Rocco C, La Marca F. Shunt complica-tions in the first postoperative year in children withmyelomeningocoele. Childs Nerv Syst. 1996;12(12):748–54.
Cameron M, Moran P. Prenatal screening and diagnosis ofneural tube defects. Prenat Diagn. 2009;29(4):402–11.
Carter CO. Clues to the aetiology of neural tubemalformations. Dev Med Child Neurol. 1974;16(6 Suppl 32):3–15.
Centers for Disease Control (CDC). MMWRMorb MortalWkly Rep. 1983;32(33):438–9. Valproate: a new causeof birth defects–report from Italy and follow-up fromFrance.
ChadduckWM, Reding DL. Experience with simultaneousventriculo-peritoneal shunt placement and myelo-meningocele repair. J Pediatr Surg. 1988;23(10):913–6.
Charney EB. Parental attitudes toward management ofnewborns with myelomeningocele. Dev Med ChildNeurol. 1990;32(1):14–9.
Charney EB, Weller SC, Sutton LN, Bruce DA, Schut LB.Management of the newborn with myelomeningocele:time for a decision-making process. Pediatrics.1985;75(1):58–64.
Choux M, Di Rocco C, Hockley AD, Walker ML, editors.Pediatric neurosurgery. 1st ed. London: ChurchillLivingstone; 1999. p. 875.
Copp AJ, Stanier P, De GN. Neural tube defects: recentadvances, unsolved questions, and controversies.Lancet Neurol. 2013;4422(13):1–12.
Date I, Yagyu Y, Asari S, Ohmoto T. Long-term outcome insurgically treated encephalocele. Surg Neurol.1993;40(2):125–30.
Davis BE, Daley CM, Shurtleff DB, Duguay S, Seidel K,Loeser JD, et al. Long-term survival of individuals withmyelomeningocele. Pediatr Neurosurg. 2005;41(4):186–91.
Dicianno BE, Karmarkar A, Houtrow A, et al. Factorsassociated with mobility outcomes in a National SpinaBifida Patient Registry. Am J Phys Med Rehabil.2015;94(12):1015–25.
Doran PA, Guthkelch AN. Studies in spina bifida cystica I:general surgery and reassessment of the problem.J Neurol Neurosurg Psychiatry. 1961;24:331–45.
20 J.R. Ellenbogen et al.

EUROCAT Working Group. Prevalence of neural tubedefects in 20 regions of Europe and the impact ofprenatal diagnosis, 1980–1986. J Epidemiol Commu-nity Health. 1991;45(1):52–8.
Fitzgerald RJ, Healy B. The spina bifida problem: a longerterm review with special reference to the quality ofsurvival. Ir Med J. 1974;67(21):565–7.
Folate supplements prevent recurrence of neural tubedefects. Nutr Rev. 1992;50:22–24. doi:10.1111/j.1753-4887.1992.tb02459.x.
Frazier CH. Surgery of the spine and spinal cord.New York: Appleton & Co.; 1918.
Fritsch MJ, Kienke S, Ankermann T, Padoin M,Mehdorn HM. Endoscopic third ventriculostomy ininfants. J Neurosurg. 2005;56(6):1271–8.
Gardner WJ. Rupture of the neural tube. Arch Neurol.1961;4:1–7.
Gorlin RJ, Cohen MM, Hennekam RCM. Syndromes ofthe head and neck. NewYork: Oxford University Press;2001.
Greenberg M. Handbook of neurosurgery. 7th ed.New York: Thieme; 2010. p. 1352.
Greig JD, Young DG, Azmy AF. Follow-up of spina bifidachildren with and without upper renal tract changes atbirth. Eur J Pediatr Surg. 1991;1(1):5–9.
Group MRCVSR. Prevention of neural tube defects:results of the Medical Research Council vitamin studyMRC vitamin study research group. Lancet.1991;338(8760):131–7.
Hunt GM. Open spina bifida: outcome for a completecohort treated unselectively and followed into adult-hood. Dev Med Child Neurol. 1990;32(2):108–18.
Hunt GM, Oakeshott P. Outcome in people with open spinabifida at age 35: prospective community based cohortstudy. BMJ. 2003;326(7403):1365–6.
Isada NB, Qureshi F, Jacques SM, Holzgreve W, Tout MJ,Johnson MP, et al. Meroanencephaly: pathology andprenatal diagnosis. Fetal Diagn Ther. 1993;8(6):423–8.
Janerich DT. Influenza and neural-tube defects. Lancet.1971;2(7723):551–2.
Jenkinson MD, Campbell S, Hayhurst C, Clark S,Kandasamy J, Lee MK, et al. Cognitive and functionaloutcome in spina bifida-Chiari II malformation. ChildsNerv Syst. 2011;27(6):967–74.
Jenkinson MD, Hayhurst C, Al-Jumaily M, Kandasamy J,Clark S, Mallucci CL. The role of endoscopic thirdventriculostomy in adult patients with hydrocephalus.J Neurosurg. 2009;110(5):861–6.
Jenkinson MD, Hayhurst C, Clark S, Campbell S,Murphy P, Mallucci CL. Long-term functional andneuropsychological outcome in Chiari II malformation.Childs Nerv Syst. 2008;24(10):1270.
Johnson MP, Bennett KA, Rand L, et al. Management ofmyelomeningocele study investigators. The managementof myelomeningocele study: obstetrical outcomes and riskfactors for obstetrical complications following prenatalsurgery. Am J Obstet Gynecol. 2016;215(6):778.e1–9.
Kasabian NG, Bauer SB, Dyro FM, Colodny AH,Mandell J, Retik AB. The prophylactic value of clean
intermittent catheterization and anticholinergic medica-tion in newborns and infants with myelodysplasia atrisk of developing urinary tract deterioration. Am J DisChild. 1992;146(7):840–3.
Kondo A, Kamihira O, Ozawa H. Neural tube defects:prevalence, etiology and prevention. Int J Urol.2009;16(1):49–57.
Kooper AJ, de Bruijn D, van Ravenwaaij-Arts CM,Faas BH, Creemers JW, Thomas CM, et al. Fetal anom-aly scan potentially will replace routine AFAFP assaysfor the detection of neural tube defects. Prenat Diagn.2007;27(1):29–33.
Larsen W. Human Embryology. 1st ed. New York: Chur-chill Livingstone; 1993. p. 479.
Laurence KM, James N, Miller MH, Tennant GB,Campbell H. Double-blind randomised controlled trialof folate treatment before conception to prevent recur-rence of neural-tube defects. Br Med J (Clin Res Ed).1981;282(6275):1509–11.
Layde PM, Edmonds LD, Erickson JD. Maternal fever andneural tube defects. Teratology. 1980;21(1):105–8.
Leck I. The etiology of human malformations: insightsfrom epidemiology. Teratology. 1972;5(3):303–14.
Leck I. The geographical distribution of neural tube defectsand oral clefts. Br Med Bull. 1984;40(4):390–5.
Lemire RJ, Beckwith JB, Warkny J. Anencephaly.New York: Raven Press; 1978.
Lindhout D, Omtzigt JG, Cornel MC. Spectrum of neural-tube defects in 34 infants prenatally exposed to anti-epileptic drugs. Neurology. 1992;42(4 Suppl 5):111–8.
Van Lith JMM, Johnson MP, Wilson RD. Current contro-versies in prenatal diagnosis 3: fetal surgery afterMOMS: is fetal therapy better than neonatal? PrenatDiagn. 2013;33(1):13–6.
Lorber J. Results of treatment of myelomeningocele. Ananalysis of 524 unselected cases, with special referenceto possible selection for treatment. Dev Med ChildNeurol. 1971;13(3):279–303.
Lorber J, Salfield SA. Results of selective treatment of spinabifida cystica. Arch Dis Child. 1981;56(11):822–30.
Luthy DA, Wardinsky T, Shurtleff DB, Hollenbach KA,Hickok DE, Nyberg DA, et al. Cesarean section beforethe onset of labor and subsequent motor function ininfants with meningomyelocele diagnosed antenatally.N Engl J Med. 1991;324(10):662–6.
McCarthy GT. Treating children with spina bifida. BMJ.1991;302(6768):65–6.
McConnell JR, Holder JC, Menick JR, Alexander JE. Theradiology of neural tube defects. Curr Probl DiagnRadiol. 1986;15(4):244–76.
Milunsky A, Jick SS, Bruell CL, MacLaughlin DS,Tsung YK, Jick H, et al. Predictive values, relativerisks, and overall benefits of high and low maternalserum alpha-fetoprotein screening in singleton preg-nancies: new epidemiologic data. Am J ObstetGynecol. 1989;161(2):291–7.
Milunsky A, Ulcickas M, Rothman KJ, Willett W, Jick SS,Jick H. Maternal heat exposure and neural tube defects.JAMA. 1992;268(7):882–5.
Spina Bifida and Encephalocele 21

Moore K, Persaud T, Torchia M. The developing human.9th ed. Saint Louis: Elsevier; 2011. p. 562.
Morgagni JB. Je sedibus et causis morborum per indagatis.Naples: Typographia Simoniana; 1762. p. 1962.
Morton J. Case of spina bifida cured by injection. BrMed J.1872;1:364.
O’Brien DF, Javadpour M, Collins DR, Spennato P,Mallucci CL. Endoscopic third ventriculostomy: anoutcome analysis of primary cases and proceduresperformed after ventriculoperitoneal shunt malfunc-tion. J Neurosurg. 2005;103(5 Suppl):393–400.
Oakeshott P, Hunt GM. Long-term outcome in open spinabifida. Br J Gen Pract. 2003;53(493):632–6.
Oakeshott P, Hunt GM, Poulton A, Reid F. Open spinabifida: birth findings predict long-term outcome. ArchDis Child. 2012;97(5):474–6.
Obladen M. Cats, frogs, and snakes: early concepts of neuraltube defects. J Child Neurol. 2011;26(11):1452–61.
Olney RS, Mulinare J. Trends in neural tube defect preva-lence, folic acid fortification, and vitamin supplementuse. Semin Perinatol. 2002;26(4):277–85.
Pang D. Surgical complications of open spinal dysraphism.Neurosurg Clin N Am. 1995;6(2):243–57.
Parent AD, McMillan T. Contemporaneous shunting withrepair of myelomeningocele. Pediatr Neurosurg.1995;22(3):132–5. discussion 136
Parker SE, Yazdy MM, Tinker SC, Mitchell AA,Werler MM. The impact of folic acid intake on theassociation among diabetes mellitus, obesity, and spinabifida. Am J Obstet Gynecol. 2013;209(3):239.e1–8.
Patten BM. Overgrowth of the neural tube in young humanembryos. Anat Rec. 1952;113(4):381–93.
Pollack IF, Pang D, Albright AL, Krieger D. Outcomefollowing hindbrain decompression of symptomaticChiari malformations in children previously treatedwith myelomeningocele closure and shunts.J Neurosurg. 1992;77(6):881–8.
Report of a committee of the society nominated to investi-gate spina bifida. Trans Chir Soc. London; 1882 p. 339.
Rickwood AMK. Infective problems encountered in neo-natal closure of the neural tube defects. Dev Med ChildNeurol. 1976;18:164–5.
Robards MF, Thomas GG, Rosenbloom L. Survival ofinfants with unoperated myeloceles. Br Med J.1975;4(5987):12–3.
Romero R, Mathisen JM, Ghidini A, Sirtori M,Hobbins JC. Accuracy of ultrasound in the prenatal diag-nosis of spinal anomalies. Am J Perinatol. 1989;6(3):320–3.
Rosa FW. Spina bifida in infants of women treated withcarbamazepine during pregnancy. N Engl J Med.1991;324(10):674–7.
Rudy DC, Woodside JR. The incontinent myelodysplasticpatient. Urol Clin North Am. 1991;18(2):295–308.
Sadler TW. Embryology of neural tube development. Am JMed Genet C Semin Med Genet. 2005;135C(1):2–8.
Sakala EP, Andree I. Optimal route of deliveryfor meningomyelocele. Obstet Gynecol Surv.1990;45(4):209–12.
Sandford MK, Kissling GE, Joubert PE. Neural tube defectetiology: new evidence concerning maternal hyperther-mia, health and diet. Dev Med Child Neurol.1992;34(8):661–75.
Seitzberg A, Lind M, Biering-Sorensen F. Ambulation inadults with myelomeningocele. Is it possible to predictthe level of ambulation in early life? Childs Nerv Syst.2008;24(2):231–7.
Sharrard WJ, Zachary RB, Lorber J. Survival and paralysisin openmyelomeningocele with special reference to thetime of repair of the spinal lesion. Dev Med ChildNeurol. 1967;Suppl 13:35–50.
Smithells RW, Chinn ER. Spina bifida in Liverpool. DevMed Child Neurol. 1965;7:258–68.
Snow-Lisy DC, Yerkes EB, Cheng EY. Update on urologi-cal management of spina bifida from prenatal diagnosisto adulthood. J Urol. 2015;194(2):288–96.
Stein SC, Schut L. Hydrocephalus in myelomeningocele.Childs Brain. 1979;5(4):413–9.
Stevenson KL. Chiari type II malformation: past, present,and future. Neurosurg Focus. 2004;16(2):E5.
Stevenson RE, Allen WP, Pai GS, Best R, Seaver LH,Dean J, et al. Decline in prevalence of neural tubedefects in a high-risk region of the United States. Pedi-atrics. 2000;106(4):677–83.
Stoneking BJ, Brock JW, Pope JC, Adams MC. Earlyevolution of bladder emptying after myelomeningoceleclosure. Urology. 2001;58(5):767–71.
Surana RH, Quinn FM, Guiney EJ, Fitzgerald RJ. Are theselection criteria for the conservative management inspina bifida still applicable? Eur J Pediatr Surg.1991;1(Suppl 1):35–7.
Sutton LN. Fetal surgery for neural tube defects. Best PractRes Clin Obstet Gynaecol. 2008;22(1):175–88.
Suwanwela C, Suwanwela N. A morphological classifica-tion of sincipital encephalomeningoceles. J Neurosurg.1972;36(2):201–11.
Takeuchi H. Prenatal ultrasound diagnosis of central nervoussystem anomalies. No To Hattatsu. 1991;23(2):183–8.
Thompson DN. Postnatal management and outcome forneural tube defects including spina bifida andencephalocoeles. Prenat Diagn. 2009;29(4):412–9.
Tuli S, Drake J, Lamberti-Pasculli M. Long-term outcomeof hydrocephalus management in myelomeningoceles.Childs Nerv Syst. 2003;19(5–6):286–91.
Tulp N. Observationes medicae. Amsterdam: Elsevier;1672.
Vachha B, Adams RC, Rollins NK. Limbic tract anomaliesin pediatric myelomeningocele and Chiari II malforma-tion: anatomic correlations with memory and learning:initial investigation. Radiology. 2006;240(1):194–202.
Valproate: a new cause of birth defects–report from Italyand follow-up from France. MMWR Morb MortalWkly Rep. 1983;32(33):438–9.
Valproate, spina bifida, and birth defect registries. Lancet.1988;332(8625):1404–5.
Valtonen K, Karlsson A-K, Alaranta H, Viikari-Juntura E.Work participation among persons with traumatic
22 J.R. Ellenbogen et al.

spinal cord injury and meningomyelocele. J RehabilMed. 2006;38(3):192–200.
Vandertop WP, Asai A, Hoffman HJ, Drake JM,Humphreys RP, Rutka JT, et al. Surgical decompres-sion for symptomatic Chiari II malformation inneonates with myelomeningocele. J Neurosurg.1992;77(4):541–4.
Vinck A, Maassen B, Mullaart R, Rotteveel J. Arnold-Chiari-II malformation and cognitive functioning in spinabifida. J Neurol Neurosurg Psychiatry. 2006;77(9):1083–6.
Virchow R. Ein fall von hypertrichosis circumscriptamediana kornbiniert mit spina bifida. Ztschaz Ethnol.1875;7:279.
Wald NJ, Bower C. Folic acid, pernicious anaemia, and pre-vention of neural tube defects. Lancet. 1994;343(8893):307.
Wald NJ, Cuckle H, Brock JH, Peto R, Polani PE,Woodford FP. Maternal serum-alpha-fetoproteinmeasurement in antenatal screening for anencephalyand spina bifida in early pregnancy. Report ofU.K. collaborative study on alpha-fetoprotein in
relation to neural-tube defects. Lancet. 1977;1(8026):1323–32.
De Wals P, Tairou F, Van Allen MI, Uh SH, Lowry RB,Sibbald B, et al. Reduction in neural-tube defects afterfolic acid fortification in Canada. N Engl J Med.2007;357(2):135–42.
White-Van Mourik MC, Connor JM, Ferguson-Smith MA.Patient care before and after termination of pregnancyfor neural tube defects. PrenatDiagn. 1990;10(8):497–505.
Willett WC. Folic acid and neural tube defect: can’t we cometo closure? Am J Public Health. 1992;82(5):666–8.
Wiswell TE, Tuttle DJ, Northam RS, Simonds GR. Majorcongenital neurologic malformations: a 17-year survey.Am J Dis Child. 1990;144(1):61–7.
Woodhouse CR. Myelomeningocele: neglected aspects.Pediatr Nephrol. 2008;23(8):1223–31.
Yen IH, Khoury MJ, Erickson JD, James LM, Waters GD,Berry RJ. The changing epidemiology of neural tubedefects. United States, 1968–1989. Am J Dis Child.1992;146(7):857–61.
Spina Bifida and Encephalocele 23