selective colloid mobilization through surface-charge manipulation
TRANSCRIPT

Selective Colloid MobilizationThrough Surface-ChargeManipulationJ O H N C . S E A M A N * A N DP A U L M . B E R T S C H
Advanced Analytical Center for Environmental Sciences,Savannah River Ecology Laboratory, The University ofGeorgia, Drawer E, Aiken, South Carolina 29802
The objective of the current study was to evaluate theuse of amine hexadecyltrimethylammonium bromide (HDTMA)to enhance the mobilization and subsequent transport ofcolloidal iron oxides by selectively blocking negatively chargedsites within soil or aquifer sediments. Two materialswere used in a series of column leaching studies, a surfacesoil (Orangeburg Series) and an iron oxide-rich subsurfacesediment (Tobacco Rd. Formation) both from Aiken, SC.For comparison, the same materials were leached withsodium hexametaphosphate (Na-P) as a nonselectivedispersing agent. As a cationic surfactant, HDTMA is generallyconsidered a strong flocculent for soils because of itsability to shield the electrostatic repulsion on opposingnegatively charged clays, which was observed in leachingexperiments for the surface soil material. LeachingHDTMA solutions through the iron oxide-coated aquifersediments resulted in the selective dispersion and transportof iron oxides, relative to the more abundant kaolinite.Despite effluent colloid levels in excess of 6 g L-1, no columnplugging was observed for the HDTMA treatments. TheNa-P treatment, however, produced effluent turbidity levelsthat were less than HDTMA but induced rapid columnplugging. Thermal characterization of the Na-P-derivedcolloids indicated that they were similar to the same as thebulk clay fraction of the aquifer sediment, indicating thatdispersion was nonselective. HDTMA appears to blocknegatively charged filtration sites that limit iron oxide transport,thus enhancing colloid dispersion without inducingcolumn plugging observed for nonselective dispersants.Iron oxides have been demonstrated to be the resident phasefor many inorganic and organic contaminants withinhighly weathered, organic matter-poor systems. Thissuggests that selective mobilization of colloidal iron oxidesand their associated contaminants can potentially enhancesubsurface remediation activities via implementation ofpump-and-treat technologies.
IntroductionThe strong partitioning (i.e., precipitation, sorption, etc.) ofgroundwater contaminants to the immobile solid aquifermatrix typically limits the success of remediation efforts. Theslow release of contaminants can make it inefficient to reclaiman aquifer by simply capturing and treating the groundwater(1). Recent studies suggest that soil and groundwater colloids
mobilized as a result of chemical perturbation may activelyfacilitate the subsurface migration of sparingly solublecontaminants, such as radionuclides, transition metals,metalloids, and hydrophobic organics (2-8). Therefore,enhancing the migration of mobile colloids and theirassociated contaminants has been proposed as a means ofincreasing the efficacy of contaminant extraction systems,such as pump-and-treat, with the obvious recognition thatformation damage due to pore clogging may be an importantobstacle to overcome (2, 9). Despite such assertions, littleprogress has been made in the effective management ofcolloidal migration as a reclamation strategy.
Considerable research has focused on elucidating theprocesses controlling mobile colloid generation. Previousstudies suggest that mobile colloids may be pro-duced in the environment by a number of mechanismsincluding clay dispersion due to changes in groundwaterpH, ionic strength, and/or Na/Ca ratios (3, 6, 10-18); dissolu-tion of carbonate or iron cementing agents resulting in therelease and transport of silicate clays (19-21); and theprecipitation of colloidal particulates resulting from changesin local groundwater chemistry (22). Physical perturbationdue to increased shear velocities associated with elevatedpumping rates or bailing groundwater samples can artifac-tually mobilize colloids that would otherwise remain in place(23-25).
Colloid dispersion resulting from a change in ionicstrength, pH, or the sodium adsorption ratio (SAR) of the soilor geologic material has been the focus of most controlledlaboratory studies due to the deleterious impact of Na+ onthe physical properties of agricultural soils and geologicformations as it influences hydraulic conductivity (13, 14,26, 27). To a degree, these factors have been emphasizedbecause they are easier to experimentally control than factorssuch as redox potential or the partial pressure of CO2. Previousstudies have clearly demonstrated the importance of ironand aluminum oxides in controlling aggregation/filtrationbehavior (10, 19, 28-33). The inability to predict colloiddeposition rates under so-called “unfavorable” captureconditions has been attributed to surface charge heteroge-neities associated with the presence of iron and aluminumoxides (33-35). Thus, colloid mobility in low-carbon, oxide-coated systems has generally been thought to be quite limiteddue the favorable conditions for attachment of negativelycharged clays (33). To overcome the flocculating effect ofiron oxides, studies often resort to conditions that highlyfavor the generation and transport of negatively chargedphyllosilicates, such as the use of sodic solutions as surrogatesfor native or contaminated pore waters (3, 6, 10, 11). Ingeneral, these studies demonstrate that a high exchangeablesodium percentage (ESP) and an elevated pH, which reducespositive charge by inducing oxide charge reversal, are requiredin addition to low ionic strengths to observe significant colloidgeneration.
In a series of column experiments in which coarse-textured, oxide-coated Atlantic Coastal Plain sediments wereleached with solutions containing various Na/Ca ratios atdifferent ionic strengths and pH values, Seaman et al. (12,36) observed that colloidal dispersion resulted from minorchanges in solution chemistry, many of which are consideredhighly flocculating based on current thoughts regardingcolloid generation in the subsurface environment (25). Theintroduction of dilute CaCl2 solutions resulted in colloidmobilization and a decrease in effluent pH attributed to Al3+
exchange and hydrolysis reactions as well as specific cation* Corresponding author phone: (803)725-0977; fax: (803)725-3309;
e-mail: [email protected].
Environ. Sci. Technol. 2000, 34, 3749-3755
10.1021/es001056w CCC: $19.00 2000 American Chemical Society VOL. 34, NO. 17, 2000 / ENVIRONMENTAL SCIENCE & TECHNOLOGY 9 3749Published on Web 07/29/2000

sorption reactions on hydrous oxide surfaces that can impartgreater net-positive charge (Figure 1). The resulting colloidsdisplayed positive electrophoretic mobilities, and electronmicroscopy and thermal analysis confirmed that theyconsisted primarily of iron oxides (goethite) rather than thepredominate phyllosilicate, kaolinite (12, 36, 37). The abilityto produce stable positively charged colloids has beenconfirmed at Darcy flow velocities ranging from 0.73 to 3.7m d-1 using multiple sediment samples displaying a rangeof iron oxide/kaolinite contents, with the production of ironoxide colloids generally increasing with increasing iron oxidecontent (12, 36).
According to the conceptual model presented in Figure1, such materials are at or near the zero point of net charge(ZPNC) for the combined mineral assemblage (i.e., variablecharge oxides and constant charge phyllosilicates) and thusremain flocculated regardless of the SAR or the ionic strengthof the suspending solution. This behavior is consistent withthe low ionic strength native solutions to which such materialsare typically exposed (IS < 0.5 mM, pH ≈ 5.0) (38). Reactionsthat alter pH, however, including an increase in the con-centration of polyvalent cations which lowers pH, canincrease net matrix charge.
Although the low pH mechanism of colloid generationmay be more common than previously recognized, themobilization of significant iron oxide coatings can exposenegatively charged surfaces, which then act as depositionsites. To significantly increase colloid mobility, one mustblock the negatively charged sites in a manner similar to theway previously deposited colloids inhibit the subsequentfiltration of additional particles (33, 34). On the other hand,widespread colloid dispersion can induce formation damagedue to pore clogging, which may limit further particle migra-tion. Once the hydraulic conductivity of a given aquifer regionhas been reduced, the efficacy of any remediation techniqueis compromised. Therefore, the ability to control these twodisparate processes is critical to the use of colloid mobilizationas an enhanced contaminant extraction technique.
Cationic surfactants such as the quaternary amine hexa-decyltrimethylammonium bromide (HDTMA) are generallypoor detergents because they readily flocculate clay suspen-sions at surface coverages equal to or less than the CEC byshielding negative charge sites and favoring the face-to-faceassociation of opposing clay surfaces (39, 40). At relativelyhigh surface coverages, however, the cooperative adsorptionof HDTMA in excess of the cation exchange capacity (CEC)can induce charge reversal and the eventual stabilization ofa suspension as positively charged colloids (39, 41). Theobjective of the current study was to evaluate the use ofHDTMA to enhance and control the mobilization andsubsequent transport of native colloidal iron oxides byselectively blocking negatively charged sites within the aquifermatrix.
Materials and MethodsA surface soil horizon (Ap) (Orangeburg Series; TypicPaleudult, fine-loamy, siliceous, thermic) and an aquifersediment (Tobacco Rd. Formation) were collected on theDepartment of Energy’s Savannah River Site (SRS), nearAiken, SC (Table 1). These materials are typical of the coarse-textured, highly weathered soils and sediments of the AtlanticCoastal Plain. The surface soil was collected from an exposedprofile located within a mixed deciduous/coniferous forest.The aquifer sediment was collected from a deep erosionalexposure of sediments typical of the water table aquifer(Tobacco Rd. Formation) and the first confined aquifer(Barnwell Formation). Prior to sample collection, the driedsurface crust and overburden present on the exposure wereremoved to reveal the moist homogeneous material that wasstored in a field-moist state (typically 0.5-5%) at 4 °C untilcolumn packing. Typical specimens of both materials havebeen characterized extensively and described in previousstudies (36, 42). The clay fractions of both materials areprimarily kaolinitic with the surface soil also containinghydroxy-interlayered vermiculite (HIV) and gibbsite, whilethe remaining clay fraction of the aquifer material consists
FIGURE 1. Mechanisms responsible for shifts in solution pH (A) and the generation of positive surface charge (B) resulting in iron oxidedispersion (based on data from ref 47).
3750 9 ENVIRONMENTAL SCIENCE & TECHNOLOGY / VOL. 34, NO. 17, 2000

mainly of goethite and mica (illite). Analysis of exchangeablecations indicated that both materials have a high degree ofAl3+ saturation on the exchange complex (g80%) (12, 43).
The water-dispersible clay (WDC) content for each soilwas determined as an indicator of inherent clay dispersionpotential. Four grams of each soil was weighed into threereplicate 50-mL centrifuge tubes and 40 mL of deionizedwater (DIW) was added to each tube prior to shakingovernight. After being shaken, each soil suspension wasallowed to settle undisturbed for approximately 2 h beforethe dispersed colloidal fraction (3 mL) was sampled by slowlypipetting at a fixed, predetermined depth within the tubebelow which particles >2 µm would be expected to havesettled (44). The dispersed clay present in the sampled aliquotwas quantified by placing the pipetted suspension inpreweighed, oven-dried aluminum tins and heating at 110°C to dry the sample prior to reweighing the pan.
Column Methods. Field-moist samples were packed in10-cm-long columns with interior diameters of 5 cm to auniform bulk density of ≈ 1.5 g cm-3. Above and below thesample, 1-cm layers of Ottawa sand were included to disperseflow throughout the entire cross-section of the column andreduce the impact of turbulent flow on colloid formation.The packed columns were oriented vertically and slowlysaturated from the outlet in an upward manner with anartificial groundwater solution (AGW) based on the com-position of groundwater collected from several nonimpactedwells located on the SRS (Table 2). The saturated columnswere then oriented horizontally and leached at a constantDarcy velocity of ≈ 0.72 m d-1 with one of several treatmentsolutions: 0.001 M NaCl or 0.0005 M CaCl2 (inlet pH ∼ 6.0);acidic 0.005 M CaCl2 (adjusted to a pH of 3.0 with HCl); 0.001M Na-hexameta-polyphosphate (NA-P); and 0.00025-0.001M HDTMA. HDTMA breakthrough in the organic poorTobacco Rd. material was evaluated based on dissolvedorganic carbon in the effluent (Shimadzu, Inc.).
The pH, electrical conductivity (EC), and turbidity of thecolumn effluents were continuously monitored, and turbidsamples were collected for surface charge and chemicalcharacterization. When effluent turbidity exceeded the 100NTU linear range of the instrument (DRT 200B, Hialeah, FL),an aliquot of the turbid sample was diluted with DIW towithin the linear range. A linear regression comparing themass (g L-1) of colloids mobilized as a function of NTU basedon the diluted samples had an r2 of 0.974. Column plugging,as influenced by solution treatment, was monitored using a0-1 PSI pressure transducer (OMEGA Engineering, StamfordCT) calibrated with a piezometer tube at the column inlet.Each repacked column was used for only one leachingtreatment. The electrophoretic mobilities of suspendedcolloids produced in the column experiments were deter-mined by laser doppler velocimetry with a Delsa 440 (CoulterElectronics, Hialeah, FL).
Thermal Analysis of Column Suspensions. Suspensionsgenerated by the HDTMA and Na-P treatments were dialyzedagainst DIW to remove soluble salts, quick-frozen in liquidN2, and then freeze-dried prior to thermal analysis. Suspen-sions generated by the other treatments, such as 0.001 NCaCl2, required preconcentration prior to analysis. The dilutesuspensions were filtered through a 0.1 µm pore-size poly-carbonate filter. The filter was then placed in a 250 mLNalgene bottle, filled to half its volume with DIW, and shakento resuspend the captured particulates. The clean filter wasremoved from the bottle and the resulting suspension wasfreeze-dried as described above. Approximately 10 mg offreeze-dried clay were analyzed by thermal-gravimetricanalysis using the dynamic-rate, high-resolution mode (TGA2950, TA Instruments Inc.).
Postcolumn Flocculation Studies. Suspensions generatedby HDTMA and Na-P treatments were used in flocculationstudies as an indication of particle charge and colloidalstability. The ability to flocculate the captured suspensionsefficiently is critical for the development of a practicalgroundwater remediation scheme. Bulk suspension samplesgenerated by each treatment were split into several equivalentfractions and subjected to the following flocculation treat-ments: (1) control (i.e., no chemical adjustment); (2) pHadjustment to 7.5 with NaOH; (3) addition of CaCl2 for a finalconcentration of 0.001 M; and (4) pH adjustment to 7.5 plusaddition of CaCl2 for a final concentration of 0.001 M. Afterchemical treatment, all of the tubes were physically agitatedto resuspend the colloids and then periodically photographedas a function of settling time.
Results and DiscussionThe Orangeburg surface soil was included in the presentstudy to illustrate how the evaluated treatments affect thedispersion of materials which are similar in mineralogy (i.e.kaolinitic) and texture (≈90% sand) to the oxide-coatedsubsurface sediments, but display little positive charge dueto the higher organic matter content and the presence ofmore-negatively charge clays such as HIV. As a result, theOrangeburg soil can be easily dispersed when shaken in DIW,i.e., water-dispersible clay (WDC), in contrast with the oxide-coated Tobacco Rd. material which readily flocculates inbatch dispersion tests (Table 1).
Column Results. Initial column effluents from theOrangeburg soil display extremely high turbidities thatdepend on inlet solution flow rate, regardless of the com-position of the treatment solution. As the higher ionicstrength treatment solution breaks through, however, theresponse in terms of mobile colloid generation is consistentwith numerous studies in that Ca2+-rich or acidic treat-ments tend to inhibit colloid transport compared to high-pH, sodic solutions (data not shown). In fact, the 0.001 MHDTMA solution was more effective than Na+ and even Ca2+
in decreasing effluent turbidity during leaching. A significant
TABLE 1. Chemical and Physical Characteristics of SamplesUsed in Column and Batch Studies.
Tobacco Rd.sedimenta
OrangeburgSeriesb
pHwater 5.30 5.52pHKCl 4.98 4.43TOC (g/100 g)c 0.02 0.76CDB Fe (g/100 g)d 0.19 0.74PSDe g 100 g-1 g 100 g-1
sand 87.2 85.5silt 4.5 7.8clay 8.3 6.6
WDC f BDL 4.5clay mineralogyg k, goe, m k, HIV, gibba 2:1 solution/soil ratio in DIW. b 2:1 solution/soil ratio in 1 M KCl.
c TOC, total organic carbon, dry combustion method (55). d CDB, citrate-dithionite-bicarbonate extraction (54). e PSD, particle size distribution(44). f WDC, water-dispersible clay determined without the aid of adispersing agent (i.e., sodium hexametaphosphate) (44). BDL, belowdetection limits. g Clay mineralogy determined by X-ray diffraction: k,kaolinite; HIV, hydroxy-interlayered vermiculite; gibb, gibbsite; goe,goethite; m, mica (illite).
TABLE 2. Composition of Artificial Groundwater Based onRoutine Monitoring Data from the Department of Energy’sSavannah River Site (38)a
component concn (mg L-1) component concn (mg L-1)
Ca2+ 1.00 Na+ 1.40Mg2+ 0.37 SO4
2- 0.73K+ 0.21 pH 5.2
a Na2SO4 was used to make up the sulfate component, and thenchloride salts were used for the remaining Na and the other cations aswell. The pH was adjusted using HCl.
VOL. 34, NO. 17, 2000 / ENVIRONMENTAL SCIENCE & TECHNOLOGY 9 3751
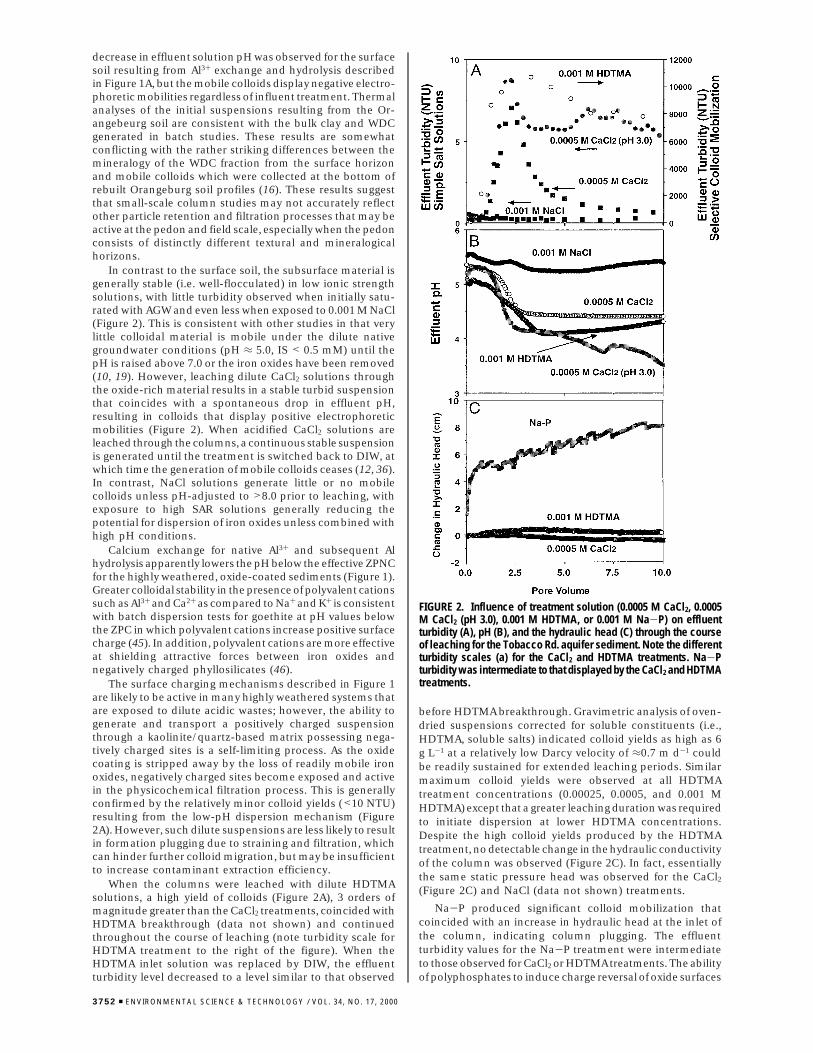
decrease in effluent solution pH was observed for the surfacesoil resulting from Al3+ exchange and hydrolysis describedin Figure 1A, but the mobile colloids display negative electro-phoretic mobilities regardless of influent treatment. Thermalanalyses of the initial suspensions resulting from the Or-angebeurg soil are consistent with the bulk clay and WDCgenerated in batch studies. These results are somewhatconflicting with the rather striking differences between themineralogy of the WDC fraction from the surface horizonand mobile colloids which were collected at the bottom ofrebuilt Orangeburg soil profiles (16). These results suggestthat small-scale column studies may not accurately reflectother particle retention and filtration processes that may beactive at the pedon and field scale, especially when the pedonconsists of distinctly different textural and mineralogicalhorizons.
In contrast to the surface soil, the subsurface material isgenerally stable (i.e. well-flocculated) in low ionic strengthsolutions, with little turbidity observed when initially satu-rated with AGW and even less when exposed to 0.001 M NaCl(Figure 2). This is consistent with other studies in that verylittle colloidal material is mobile under the dilute nativegroundwater conditions (pH ≈ 5.0, IS < 0.5 mM) until thepH is raised above 7.0 or the iron oxides have been removed(10, 19). However, leaching dilute CaCl2 solutions throughthe oxide-rich material results in a stable turbid suspensionthat coincides with a spontaneous drop in effluent pH,resulting in colloids that display positive electrophoreticmobilities (Figure 2). When acidified CaCl2 solutions areleached through the columns, a continuous stable suspensionis generated until the treatment is switched back to DIW, atwhich time the generation of mobile colloids ceases (12, 36).In contrast, NaCl solutions generate little or no mobilecolloids unless pH-adjusted to >8.0 prior to leaching, withexposure to high SAR solutions generally reducing thepotential for dispersion of iron oxides unless combined withhigh pH conditions.
Calcium exchange for native Al3+ and subsequent Alhydrolysis apparently lowers the pH below the effective ZPNCfor the highly weathered, oxide-coated sediments (Figure 1).Greater colloidal stability in the presence of polyvalent cationssuch as Al3+ and Ca2+ as compared to Na+ and K+ is consistentwith batch dispersion tests for goethite at pH values belowthe ZPC in which polyvalent cations increase positive surfacecharge (45). In addition, polyvalent cations are more effectiveat shielding attractive forces between iron oxides andnegatively charged phyllosilicates (46).
The surface charging mechanisms described in Figure 1are likely to be active in many highly weathered systems thatare exposed to dilute acidic wastes; however, the ability togenerate and transport a positively charged suspensionthrough a kaolinite/quartz-based matrix possessing nega-tively charged sites is a self-limiting process. As the oxidecoating is stripped away by the loss of readily mobile ironoxides, negatively charged sites become exposed and activein the physicochemical filtration process. This is generallyconfirmed by the relatively minor colloid yields (<10 NTU)resulting from the low-pH dispersion mechanism (Figure2A). However, such dilute suspensions are less likely to resultin formation plugging due to straining and filtration, whichcan hinder further colloid migration, but may be insufficientto increase contaminant extraction efficiency.
When the columns were leached with dilute HDTMAsolutions, a high yield of colloids (Figure 2A), 3 orders ofmagnitude greater than the CaCl2 treatments, coincided withHDTMA breakthrough (data not shown) and continuedthroughout the course of leaching (note turbidity scale forHDTMA treatment to the right of the figure). When theHDTMA inlet solution was replaced by DIW, the effluentturbidity level decreased to a level similar to that observed
before HDTMA breakthrough. Gravimetric analysis of oven-dried suspensions corrected for soluble constituents (i.e.,HDTMA, soluble salts) indicated colloid yields as high as 6g L-1 at a relatively low Darcy velocity of ≈0.7 m d-1 couldbe readily sustained for extended leaching periods. Similarmaximum colloid yields were observed at all HDTMAtreatment concentrations (0.00025, 0.0005, and 0.001 MHDTMA) except that a greater leaching duration was requiredto initiate dispersion at lower HDTMA concentrations.Despite the high colloid yields produced by the HDTMAtreatment, no detectable change in the hydraulic conductivityof the column was observed (Figure 2C). In fact, essentiallythe same static pressure head was observed for the CaCl2
(Figure 2C) and NaCl (data not shown) treatments.
Na-P produced significant colloid mobilization thatcoincided with an increase in hydraulic head at the inlet ofthe column, indicating column plugging. The effluentturbidity values for the Na-P treatment were intermediateto those observed for CaCl2 or HDTMA treatments. The abilityof polyphosphates to induce charge reversal of oxide surfaces
FIGURE 2. Influence of treatment solution (0.0005 M CaCl2, 0.0005M CaCl2 (pH 3.0), 0.001 M HDTMA, or 0.001 M Na-P) on effluentturbidity (A), pH (B), and the hydraulic head (C) through the courseof leaching for the Tobacco Rd. aquifer sediment. Note the differentturbidity scales (a) for the CaCl2 and HDTMA treatments. Na-Pturbidity was intermediate to that displayed by the CaCl2 and HDTMAtreatments.
3752 9 ENVIRONMENTAL SCIENCE & TECHNOLOGY / VOL. 34, NO. 17, 2000

is a major factor in their use as dispersing agents for manyprocesses. A similar role in charge reversal has beenimplicated in previous field and column studies using anionicsurfactants and natural organic matter (10, 16, 19). The lackof reduced hydraulic conductivity observed for the HDTMAtreatments, despite the high colloid yields, is thought to reflectthe smaller particle size of the mobilized iron oxide phasesas compared to the coarser clay fraction mobilized by Na-Ptreatment (47). The effluent pH for the HDTMA treatmentwas similar to that observed for the CaCl2 treatments despitethe much higher colloid yield, again suggesting that thesurfactant is effective in exchanging Al3+ and blockingnegatively charged sites that inhibit transport of the ironoxide suspension (Figure 2B).
Thermal Analysis of the Resulting Suspensions. Thermalanalysis confirmed that the mobile colloids resulting fromCa2+ treatment solutions consisted almost exclusively ofgoethite, with far lesser amounts of other clay-sized minerals(Figure 3A). Suspensions generated from the HDTMA treat-ment (Figure 3C) were highly enriched in iron oxides ascompared to suspensions by the Na-P treatment (Figure3B). The TGA pattern for the Na-P treatment was essentiallythe same as that observed for the bulk clay from the TobaccoRd sediment (bulk clay data not shown). The thermal peakon the low-temperature side of kaolinite for Figure 3C hasbeen attributed to crandallite (CaAl3(PO4)2(OH)5‚H2O), amember of the plumbogummite mineral group (36, 48).
Post-capture Flocculation Studies. Suspensions gener-ated by the Na-P and HDTMA treatments were exposed tovarious post-collection treatments to evaluate the ease inwhich the colloids could be separated from the bulk solution,an important factor in the application of colloid mobilizationas a remediation technology. Response to such treatmentsis also indicative of colloidal stability and surface charge.The oxide suspensions generated using either HDTMA orNa-P were stable (Figure 4A), with a significant portion failingto flocculate and settle after several days of storage (Figure4C). The addition of CaCl2 (final concentration 0.001 M) failedto induce significant flocculation for the HDTMA-derivedsuspension (Figure 4B), but caused complete flocculationfor the Na-P suspension in accordance with the Schulze-Hardy rule for the coagulation of negatively charged colloids.However, adjusting the pH to neutrality (pH ≈ 7.5), a basicrequirement for the reclamation of acidic metal-contami-nated groundwater, induced rapid complete flocculation byreducing positive charge such that the HDTMA facilitatesflocculation. After settling, a clear supernatant results thatcan then be recycled for use as the injection solution. Incontrast, adjustment of the pH to 7.5 for the Na-P-derivedsuspension had little effect on flocculation and settling, withthe addition of CaCl2 required to ensure efficient phaseseparation.
Post-column flocculation results indicate that pH adjust-ment after suspension collection may be the only necessarywater treatment prior to recycling the HDTMA back througha pump-and-treat system to mobilize additional colloids(Figure 4B). The pH adjustment can potentially increase thesorption by the iron oxide suspension of any solublecontaminant metals that were extracted with the suspension.Therefore, the presence of the iron oxides may reduce theneed for additional iron salts or Al-based flocculating agentsto ensure sorption/precipitation, flocculation, and settlingof contaminant metals prior to re-injection or disposal ofthe treated water.
Despite previous assertions that colloid mobilization maybe an effective means of enhancing subsurface remediation(9), the current study is the first to demonstrate the controlledselective mobilization of an important sorptive phase formany contaminants, i.e., iron oxides. The application of this
method, known as Selective Colloid Mobilization (SCM), hasbeen awarded a U.S. Patent (49). HDTMA blocks cationexchange sites on phyllosilicate clays that act as capture sites,thus enhancing the mobilization of colloidal iron oxides, thesize of which are less likely to induce pore clogging comparedto the bulk clay fraction (37). Iron oxides tend to be theresident phase for many contaminants within these highlyweathered, organic matter poor systems (50, 51). In additionto enhancing the mobility of colloid-associated contaminants,HDTMA can increase the migration of cationic metalcontaminants through simple competitive exchange pro-cesses (52) and maintain a lower solution pH favoring reducedmetal sorption. Sorbed contaminants remain in a fairly inertstate (i.e., sorbed) throughout the extraction and treatment
FIGURE 3. Derivative weight loss as a function of temperature forthe mobile Tobacco Rd. colloids resulting from the 0.0005 M CaCl2
(A), 0.001 M Na-P (B), and 0.001 M HDTMA (C) treatment solutions.
VOL. 34, NO. 17, 2000 / ENVIRONMENTAL SCIENCE & TECHNOLOGY 9 3753

process and, therefore, pose less of a hazard as compared toother enhanced extraction technologies (i.e., chelates).
The inability to uniformly deliver the HDTMA treatmentsolutions to the contaminated zone of interest may limit theeffectiveness of colloid mobilization as a remediation tech-nique (9), especially in physically heterogeneous materialsthat are typical of many contaminated sites. However, similarlimitations impede any enhanced chemical extraction tech-nique (i.e., chelates, surfactants, etc.). In addition, colloid
mobilization may increase the preferential flow throughcoarse-textured regions of the aquifer as they becomestripped of colloidal fines. Diffusion of contaminants out offiner textured aquifer zones limits the rate of contaminantremoval when such zones are bypassed by the recirculatingfluid, a major problem that already reduces the efficiency ofcurrent pump-and-treat systems (1). As demonstrated bythe inability of HDTMA to enhance colloidal transport forthe Orangeburg surface soil, however, its effectiveness in
FIGURE 4. Post-column flocculation studies for colloids generated from the Tobacco Rd. sediments in response to the 0.0005 M HDTMAor 0.001 M Na-P: (A) immediately after shaking; (B) 24 h after shaking; and (C) 72 h after shaking. Flocculation treatments include thefollowing from left to right: (1) HDTMA control, (2) HDTMA adjusted to pH 7.5 with NaOH, (3) HDTMA in 0.001 M CaCl2, (4) Na-P control,(5) Na-P adjusted to pH 7.5 with NaOH, (6) Na-P in 0.001 M CaCl2, and (7) Na-P in 0.001 M CaCl2 and adjusted to pH with NaOH.
3754 9 ENVIRONMENTAL SCIENCE & TECHNOLOGY / VOL. 34, NO. 17, 2000

selectively mobilizing iron oxides is limited to highly weath-ered oxide-coated sediments. The clear incompatibility ofthe cationic surfactants with the highly weathered aquifermaterial is an important consideration in the use of suchcompounds to develop in situ reactive barriers, as proposedin previous studies (52, 53).
AcknowledgmentsThis research was supported by Financial Assistance AwardDE-FC09-96SR18546 from the U.S. Department of Energy tothe University of Georgia Research Foundation. The authorsacknowledge the thoughtful comments of Drs. C. Strojanand B. Jackson on an early version of the manuscript.
Literature Cited(1) National Research Council. Alternatives for ground water
cleanup; National Academy Press: Washington, DC, 1994.(2) McCarthy, J. F.; Zachara, J. M. Environ. Sci. Technol. 1989, 23,
496-502.(3) Grolimund, D.; Borkevec, M.; Barmettler, K.; Sticher, H. Environ.
Sci. Technol. 1996, 30, 3118-3123.(4) Puls, R. W.; Powell, R. M. Environ. Sci. Technol. 1992, 26, 614-
621.(5) Seta, A. K.; Karathanasis, A. D. Soil Sci. Soc. Am. J. 1997, 61,
612-617.(6) Roy, S. B.; Dzombak, D. A. Environ. Sci. Technol. 1997, 31, 656-
664.(7) Newman, M. E.; Elzerman, A. W.; Looney, B. B. J. Contam. Hydrol.
1993, 14, 233-246.(8) Sairs, J. E.; Hornberger, G. M. Water Resour. Res. 1996, 32, 33-
41.(9) McCarthy, J. F.; Wobber, F. J. Manipulation of Groundwater
Colloids for Environmental Restoration; Lewis Publishers: AnnArbor, MI, 1993.
(10) Ryan, J. N.; Gschwend, P. M. Environ. Sci. Technol. 1994, 28,1717-1726.
(11) Roy, S. B.; Dzombak, D. A. Colloids Surf. A 1996, 107, 245-262.(12) Seaman, J. C.; Bertsch, P. M.; Miller, W. P. Environ. Sci. Technol.
1995, 29, 1808-1814.(13) Kia, S. F.; Fogler, H. S.; Reed, M. G. J. Colloid Interface Sci. 1987,
118, 158-168.(14) Khilar, K. C.; Fogler, H. S. J. Colloid Interface Sci. 1984, 101,
214-224.(15) Kaplan, D. I.; Sumner, M. E.; Bertsch, P. B.; Adriano, D. C. Soil
Sci. Soc. Am. J. 1996, 60, 269-274.(16) Kaplan, D. I.; Bertsch, P. M.; Adriano, D. C.; Miller, W. P. Environ.
Sci. Technol. 1993, 27, 1193-1200.(17) Kaplan, D. I.; Bertsch, P. M.; Adriano, D. C. Soil Sci. Soc. Am.
J. 1997, 61, 641-649.(18) Ryan, J. N.; Gschwend, P. M. J. Colloid Interface Sci. 1994, 164,
21-34.(19) Ryan, J. N.; Gschwend, P. M. Water Resour. Res. 1990, 26, 307-
322.(20) Ronen, D.; Magaritz, M.; Weber, U.; Amiel, A. J.; Klein, E. Water
Resour. Res. 1992, 28, 1279-1291.(21) Gschwend, P. M.; Backhus, D. A.; MacFarlane, J. K.; Page, A. L.
J. Contam. Hydrol. 1990, 6, 307-320.(22) Gschwend, P. M.; Reynolds, M. D. J. Contam. Hydrol. 1987, 1,
309-327.(23) Backhus, D. A.; Ryan, J. N.; Groher, D. M.; MacFarlane, J. K.;
Gschwend, P. M. Ground Water 1993, 31, 466-479.(24) Puls, R. W. Nucl. Saf. 1990, 31, 58-65.(25) Ryan, J. N.; Elimelech, M. Colloids Surf. A 1996, 107, 1-56.(26) Shainberg, I.; Rhoades, J. D.; Prather, R. J. Soil Sci. Soc. Am. J.
1981, 45, 273-277.
(27) Frenkel, H.; Goertzen, J. O.; Rhoades, J. D. Soil Sci. Soc. Am. J.1978, 42, 32-39.
(28) Shainberg, I.; Singer, M. J.; Janitzky, P. Soil Sci. Soc. Am. J. 1987,51, 1283-1287.
(29) Goldberg, S.; Glaubig, R. A. Clays Clay Miner. 1987, 35, 220-227.
(30) Goldberg, S.; Kapoor, B. S.; Rhoades, J. D. Soil Sci. 1990, 150,588-593.
(31) Kretzschmar, R.; Robarge, W. P.; Weeds, S. B. Soil Sci. Soc. Am.J. 1993, 57, 1277-1283.
(32) Ryan, J. N.; Gschwend, P. M. Geochim. Cosmochim. Acta 1994,56, 1507-1521.
(33) Johnson, P. R.; Sun, N.; Elimelech, M. Environ. Sci. Technol.1996, 30, 3284-3293.
(34) Liu, D.; Johnson, P. R.; Elimelech, M. Environ. Sci. Technol.1995, 29, 2963-2973.
(35) McDowell-Boyer, L. M. Environ. Sci. Technol. 1992, 26, 586-593.
(36) Seaman, J. C.; Bertsch, P. M.; Strom, R. N. Environ. Sci. Technol.1997, 31, 2782-2790.
(37) Seaman, J. C. Environ. Sci. Technol. 2000, 34, 187-191.(38) Strom, R. N.; Kaback, D. S. SRP Baseline Hydrogeologic Inves-
tigation: Aquifer Characterization Groundwater Geochemistryof the Savannah River Site and Vicinity (U); WestinghouseSavannah River Company, Environmental Sciences Section:1992.
(39) Xu, S.; Boyd, S. A. Soil Sci. Soc. Am. J 1994, 58, 1382-1391.(40) Stapleton, M. G.; Sparks, D. L.; Dentel, S. K. Environ. Sci. Technol.
1994, 28, 2330-2335.(41) Haggerty, G. M.; Bowman, R. S. Environ. Sci. Technol. 1994, 28,
452-458.(42) Seaman, J. C.; Bertsch, P. M.; Korom, S. F.; Miller, W. P. Ground
Water 1996, 34, 778-783.(43) Seaman, J. C. Dissertation, University of Georgia, 1994.(44) Miller, W. P.; Miller, D. M. Commun. Soil Plant Anal. 1987, 18,
1-15.(45) Herrera-Ramos, A. C.; McBride, M. B. Clays Clay Miner. 1996,
44, 286-296.(46) Peng, F. F.; Di, P. J. Colloid. Interface Sci. 1994, 164, 229-237.(47) Bertsch, P. M.; Seaman, J. C. Proc. Natl. Acad. Sci. 1999, 96,
3350-3357.(48) Brindley, G. W.; Brown, G. Crystal structures of clay minerals
and their identification by X-ray diffraction; MineralogicalSociety: London, 1980.
(49) Seaman, J. C.; Bertsch, P. U.S. Patent No. 5,846,434, 1998.(50) Zachara, J. M.; Smith, S. C.; Kuzel, L. S. Geochim. Cosmochim.
Acta 1995, 59, 4825-4844.(51) Coston, J. A.; Fuller, C. C.; Davis, J. A. Geochim. Cosmochim.
Acta 1995, 59, 3525-3547.(52) Wagner, J.; Chen, H.; Brownawell, B. J.; Westall, J. C. Environ.
Sci. Technol. 1994, 28, 231-237.(53) Hayworth, J. S.; Burris, D. R. Environ. Sci. Technol. 1997, 31,
1277-1283.(54) Jackson, M. L.; Lin, C. H.; Zelazny, L. W. In Methods of Soil
Analysis: Part 1, Physical and Mineralogical Methods; Klute, A.,Ed.; American Society of Agronomy, Inc.: Madison, WI, 1986;pp 101-150.
(55) Nelson, D. W.; Sommers, L. E. In Methods of Soil Analysis; Page,A. L., Miller, R. H., Keeney, D. R., Eds.; American Society ofAgronomy, Inc.: Madison, WI, 1982; Vol. 2.
Received for review February 28, 2000. Revised manuscriptreceived June 7, 2000. Accepted June 16, 2000.
ES001056W
VOL. 34, NO. 17, 2000 / ENVIRONMENTAL SCIENCE & TECHNOLOGY 9 3755