safety of dextromethorphan/quinidine in patients issn ... for 12-08-2017 pharma...
TRANSCRIPT

Full Terms & Conditions of access and use can be found athttp://www.tandfonline.com/action/journalInformation?journalCode=icmo20
Download by: ["University at Buffalo Libraries"] Date: 28 November 2017, At: 19:52
Current Medical Research and Opinion
ISSN: 0300-7995 (Print) 1473-4877 (Online) Journal homepage: http://www.tandfonline.com/loi/icmo20
An open-label multicenter study to assess thesafety of dextromethorphan/quinidine in patientswith pseudobulbar affect associated with a rangeof underlying neurological conditions
Gary L. Pattee, James P. Wymer, Catherine Lomen-Hoerth, Stanley H. Appel,Andrea E. Formella & Laura E. Pope
To cite this article: Gary L. Pattee, James P. Wymer, Catherine Lomen-Hoerth, Stanley H. Appel,Andrea E. Formella & Laura E. Pope (2014) An open-label multicenter study to assess the safetyof dextromethorphan/quinidine in patients with pseudobulbar affect associated with a range ofunderlying neurological conditions, Current Medical Research and Opinion, 30:11, 2255-2265, DOI:10.1185/03007995.2014.940040
To link to this article: https://doi.org/10.1185/03007995.2014.940040
© 2014 The Author(s). Published by Taylor &Francis.
View supplementary material
Accepted author version posted online: 25Jul 2014.Published online: 28 Jul 2014.
Submit your article to this journal
Article views: 1684 View related articles
View Crossmark data Citing articles: 11 View citing articles

Current Medical Research & Opinion Vol. 30, No. 11, 2014, 2255–2265
0300-7995 Article FT-0229.R1/940040
doi:10.1185/03007995.2014.940040 All rights reserved: reproduction in whole or part not permitted
Original articleAn open-label multicenter study to assess thesafety of dextromethorphan/quinidine in patientswith pseudobulbar affect associated with arange of underlying neurological conditions
Gary L. PatteeNeurology Associates, Lincoln, NE, USA
James P. WymerThe Neurosciences Institute, Albany Medical Center,
Albany, NY, USA
Catherine Lomen-HoerthDepartment of Neurology, University of California, San
Francisco, CA, USA
Stanley H. AppelDepartment of Neurology, Methodist Neurological
Institute, The Methodist Hospital Research Institute,
The Methodist Hospital, Houston, TX, USA
Andrea E. FormellaLaura E. PopeAvanir Pharmaceuticals Inc., Aliso Viejo, CA, USA
Address for correspondence:Gary L. Pattee MD, Neurology Associates, PC, 2631
South 70th Street, Lincoln, NE 68506, USA.
Tel.: +1 402 483 7226; Fax: +1 402 483 5440;
Keywords:Dextromethorphan/quinidine – Pseudobulbar affect –
Safety – Tolerability
Accepted: 26 June 2014; published online: 28 July 2014
Citation: Curr Med Res Opin 2014; 30:2255–65
Abstract
Background:
Pseudobulbar affect (PBA) is associated with neurological disorders or injury affecting the brain, and
characterized by frequent, uncontrollable episodes of crying and/or laughing that are exaggerated or
unrelated to the patient’s emotional state. Clinical trials establishing dextromethorphan and quinidine
(DM/Q) as PBA treatment were conducted in patients with amyotrophic lateral sclerosis (ALS) or multiple
sclerosis (MS). This trial evaluated DM/Q safety in patients with PBA secondary to any neurological condition
affecting the brain.
Objective:
To evaluate the safety and tolerability of DM/Q during long-term administration to patients with PBA
associated with multiple neurological conditions.
Methods:
Fifty-two-week open-label study of DM/Q 30/30 mg twice daily. Safety measures included adverse events
(AEs), laboratory tests, electrocardiograms (ECGs), vital signs, and physical examinations.
Clinical trial registration:
#NCT00056524.
Results:
A total of 553 PBA patients with430 different neurological conditions enrolled; 296 (53.5%) completed. The
most frequently reported treatment-related AEs (TRAEs) were nausea (11.8%), dizziness (10.5%), headache
(9.9%), somnolence (7.2%), fatigue (7.1%), diarrhea (6.5%), and dry mouth (5.1%). TRAEs were mostly
mild/moderate, generally transient, and consistent with previous controlled trials. Serious AEs (SAEs) were
reported in 126 patients (22.8%), including 47 deaths, mostly due to ALS progression and respiratory
failure. No SAEs were deemed related to DM/Q treatment by investigators. ECG results suggested no
clinically meaningful effect of DM/Q on myocardial repolarization. Differences in AEs across neurological
disease groups appeared consistent with the known morbidity of the primary neurological conditions. Study
interpretation is limited by the small size of some disease groups, the lack of a specific efficacy measure and
the use of a DM/Q dose higher than the eventually approved dose.
Conclusions:
DM/Q was generally well tolerated over this 52 week trial in patients with PBA associated with a wide range
of neurological conditions.
! 2014 Informa UK Ltd www.cmrojournal.com Safety of dextromethorphan/quinidine in PBA Pattee et al. 2255
Dow
nloa
ded
by [
"Uni
vers
ity a
t Buf
falo
Lib
rari
es"]
at 1
9:52
28
Nov
embe
r 20
17

Introduction
Pseudobulbar affect (PBA) is a neurological condition thatexerts a significant health burden on patients and care-givers1. PBA is associated with a wide range of neuro-logical disorders and is characterized by frequent, sudden,uncontrollable episodes of crying and/or laughing that aregreatly exaggerated or contrary to the patient’s emotionalstate2. Available prevalence studies suggest PBA is presentin at least 5% of Parkinson’s disease (PD), 10% of multiplesclerosis (MS), stroke, traumatic brain injury (TBI), andAlzheimer’s disease (AD), 20% of progressive supranuclearpalsy, and up to 50% of amyotrophic lateral sclerosis(ALS) patients3–11.
PBA is thought to arise from disruption of corticobulbarand cerebellar/pontine pathways controlling emotionalexpression12–14. These pathways may be disrupted by mul-tiple neurological conditions, yet the ensuing clinicalmanifestations of PBA are indistinguishable, consistentwith a common etiology across disorders12–14. The com-bination of dextromethorphan and quinidine (DM/Q) isthe first pharmacotherapy approved by the US Food andDrug Administration (FDA) and European MedicinesAuthority (EMA) for treating PBA15,16.Dextromethorphan (DM) has many pharmacologicalactions, including uncompetitive N-methyl-D-aspartate(NMDA) receptor antagonism17, sigma-1 receptor agon-ism18, and serotonin reuptake inhibition, among others;the precise mechanism(s) accounting for PBA suppressionis/are unknown19. DM is co-administered with low-dosequinidine, a potent CYP2D6 inhibitor that reduces rapidfirst-pass metabolism of DM. This inhibition increases DMbioavailability and half-life20,21. The efficacy, safety, andtolerability of DM/Q as PBA treatment was established inthree controlled clinical trials lasting 422 or 1223,24 weeks,using fixed-dose combinations of twice daily DM/Q 30/30 mg22,23, 30/10 mg24, or 20/10 mg24 in patients withALS22,24 or MS23,24. The present trial was designed to pro-vide long-term safety data using the higher DM/Q 30/30 mg dose in patients with PBA, regardless of primaryneurological condition.
Methods
Study design
Study 02-AVR-107 (NCT00056524) is a multicenter, 52week, open-label safety trial. The trial began in March2003 at 44 sites internationally (39 in the US, four inIsrael, and one in Serbia and Montenegro). An extensionwas added to allow completing patients the option toremain on treatment past 1 year. The trial was terminatedby the sponsor in June 2007, following the decision topursue development of a lower DM/Q dose. Patients
were instructed to take DM/Q 30/30 mg in the eveningfor 7 days and then twice daily thereafter. Patients kept adiary of dosage times and recorded any adverse events(AEs). Clinic visits occurred at screening, baseline(Day 1), and after 1, 4, 8, and 12 months of treatment orat patient discontinuation. During months when no clinicvisit was scheduled, patients were contacted by telephoneand asked about medication compliance and AEs.
Study population
Eligible patients were 18 to 75 years of age with a clinicaldiagnosis of PBA. For the purposes of this study, PBA wasdefined as ‘a syndrome characterized by outbursts of cryingand/or laughing that occur spontaneously and inappropri-ately given the context in which they occur’. No specificthreshold for severity of PBA symptoms was required forstudy entry. Patients were required to have an electrocar-diogram (ECG) with no evidence of rate-corrected QTinterval (QTc) prolongation (�450 msec in men;�470 msec in women), heart block (isolated right bundlebranch block without clinical history of heart disease wasallowable), sinus bradycardia (550 bpm), ventriculartachycardia, multifocal ventricular ectopic beats (any fre-quency), or unifocal ventricular ectopic beats (45/min).Patients with ALS were required to have a vital capacity�50% at baseline. Patients completing prior controlledstudies of twice daily DM/Q 30/30 mg to treat PBA inMS23 or ALS22 were also eligible to participate, providedthey met all eligibility requirements at the time of enroll-ment in this study.
Exclusion criteria were myasthenia gravis; a history ofventricular tachycardia or torsades de pointes; sensitivityto quinidine or opiate drugs; major psychiatric disturbance;a history of substance abuse in the past 2 years; any majorsystemic disease that would interfere with interpretation ofstudy results (e.g., malignancy, uncontrolled diabetes,dilated cardiac myopathy, ischemic or valvular heartdisease); hypotension (systolic blood pressure [BP]5100 mmHg); a history of postural or any unexplainedsyncope; renal, hepatic, or pulmonary disease; or clinicallysignificant deviations in standard laboratory tests. Femalepatients could not be pregnant or breastfeeding; those withchild-bearing potential were required to use an establishedmethod of birth control.
Patients were allowed to continue existing medications,except for the following starting from 1 week before DM/Qinitiation: ketoconazole, voriconazole, verapamil, diltia-zem, nifedipine, sodium bicarbonate, carbonic anhydraseinhibitors, thiazide diuretics (unless urine pH�6.5 and onthiazide for �1 month at enrollment), warfarin, haloperi-dol, monoamine oxidase inhibitors, and tricyclic anti-depressants at doses 475 mg/day (420 mg/day fordesipramine; 415 mg/day for protriptyline). Other than
Current Medical Research & Opinion Volume 30, Number 11 November 2014
2256 Safety of dextromethorphan/quinidine in PBA Pattee et al. www.cmrojournal.com ! 2014 Informa UK Ltd
Dow
nloa
ded
by [
"Uni
vers
ity a
t Buf
falo
Lib
rari
es"]
at 1
9:52
28
Nov
embe
r 20
17

the study drug, medications containing dextromethorphanor quinidine were prohibited.
Safety/tolerability assessments
Safety and tolerability measures included: AEs recorded inpatient diaries and during clinic visits and telephone con-tacts (serious adverse events [SAEs], including deaths,were required to be reported through the 30 days followingfinal dose); vital signs (all visits); resting 12-lead ECG(screening, Day 29, and Final Visit at Week 52 or discon-tinuation); clinical laboratory values, including serumchemistry, hematology, and urinalysis (screening, Day29, and Final Visit); and physical examination (screeningand Final Visit).
Pharmacokinetic assessments
A blood sample was obtained on Day 29 within 12 hafter dosing to determine plasma concentrations of DM,quinidine, and the DM metabolite dextrorphan (DX).
Safety-data analyses
The safety population comprised all patients receiving atleast one DM/Q dose. The numbers of patients experi-encing AEs were summarized by body system, relation-ship to study drug, and severity. SAEs and AEs thatresulted in study discontinuation were also summarizeddescriptively. Additionally, patient demographics, dis-position, concomitant medications, and AEs were ana-lyzed by primary neurological condition. Likelihood-ratiochi-square tests were used to compare AE incidenceamong disease groups. Assignment of patients to sub-groups for analysis by primary neurological conditionwas made based on similarities among nosologicalcharacteristics and without knowledge of patient dataor safety outcome.
Clinical laboratory values were summarized descrip-tively; shifts between normal, low, or high values wereanalyzed using McNemar’s test. Changes in physical-examination findings (normal vs. abnormal) were assessedby McNemar’s test. Summary statistics of absolute valuesand percentage change from screening value were pro-vided for systolic and diastolic BP, heart rate, respiratoryrate, QT interval, QTc interval, ventricular rate, PR inter-val, and QRS duration. Clinically significant abnormal-ities were documented.
Ethics and Good Clinical Practice
The study was conducted in conformity with the standardsof Good Clinical Practices and the Declaration ofHelsinki. Eligible patients were informed of the study’s
purposes and of anticipated AEs that recipients mightexperience, and ethics committee approval was obtainedfor each study site. Before any study procedures, a signedinformed consent document was obtained.
Results
A total of 553 patients with PBA were enrolled (494 in theUS, 48 in Israel, and 11 in Serbia and Montenegro).Eighty-nine of these patients entered directly from a12 week, placebo-controlled DM/Q study to treat PBAin MS, and 56 others (9 with ALS and 47 with MS) hadbeen previously exposed to DM and/or quinidine in clin-ical studies22,23. Over 30 neurological conditions were rep-resented among the participating cohort. In addition toALS (n¼ 176) and MS (n¼ 223), 154 participants hadPBA secondary to diverse neurological conditions(Table 1). These conditions were categorized into sevendisease groups as follows: ALS/motor neuron diseases(MND) (n¼ 199), MS (n¼ 223), stroke/cerebrovasculardisorders (CBVD) (n¼ 51), TBI (n¼ 23), PD/movementdisorders (n¼ 23), AD/dementia (n¼ 17), and ‘otherPBA’ (n¼ 17), as shown in Table 1.
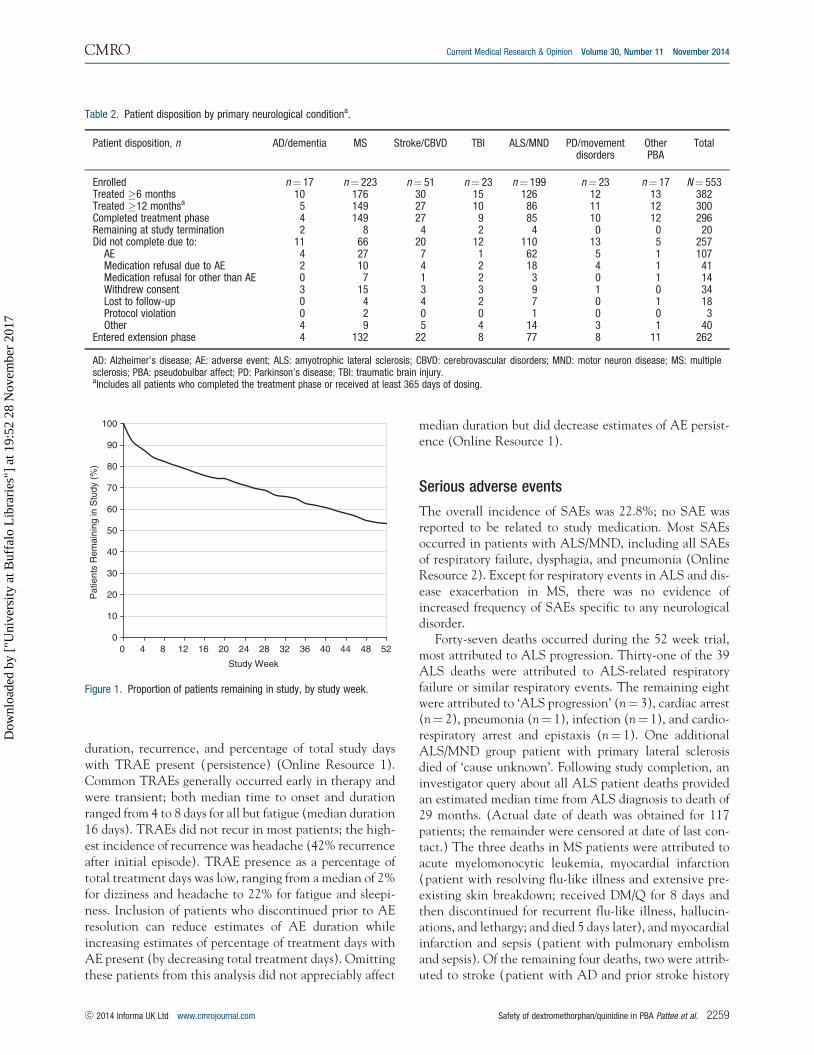
Patient disposition
Patient disposition is shown in Table 2 and duration ofstudy participation is shown in Figure 1. Of the 553patients treated with DM/Q, 382 patients (69.1%) com-pleted at least 6 months of treatment, 300 (54.2%) com-pleted 1 year, and 296 (53.5%) completed the protocol-specified visits. One hundred forty-eight (26.8%) patientsdropped out of the trial for either an AE or medicationrefusal due to an AE; 109 (19.7%) dropped out for otherreasons. Of the completers, 262 (88.5%) elected to con-tinue DM/Q in the optional extension. At trial termin-ation, 20 patients (3.6%) remained in the treatment phaseand 168 (64.1% of those entering the extension) remainedin the extension. This report provides results from the 52week treatment phase.
Patient demographics
Patient demographics by primary disease are provided inTable 3. Patients reported a median of 7.0 (range 0–420;mean 16.7� 32.6) PBA episodes per week at baseline.Some patients transitioned directly from a previous DM/Q study and therefore may have had low or no PBA epi-sodes at baseline. The median patient age was 52.0 years(range 18–86), 15.9% were�65 years old, 58.2% werefemale, and 90.8% were Caucasian.
Current Medical Research & Opinion Volume 30, Number 11 November 2014
! 2014 Informa UK Ltd www.cmrojournal.com Safety of dextromethorphan/quinidine in PBA Pattee et al. 2257
Dow
nloa
ded
by [
"Uni
vers
ity a
t Buf
falo
Lib
rari
es"]
at 1
9:52
28
Nov
embe
r 20
17

Concurrent medications
Patients were taking a median of seven additionalmedications at baseline (range 0–30) including those fortheir primary neurological condition and other conditions.The number of baseline medications was similar for studycompleters and those who discontinued for AEs.
The most frequently reported concurrent chronicmedications (taken �3 months during treatment) werenonsteroidal anti-inflammatory drugs and other anal-gesics, antidepressants, lipid-modifying agents, antithrom-botics, inhibitors of gastric acid production, vitamins,anticonvulsants, and benzodiazepines (Table 4).Antipsychotics were used chronically by 2.2% of patients.
Common disease-specific therapies included: interferonbeta, glatiramer, baclofen, and stimulants in patientswith MS; donepezil, and memantine in patients withAD; riluzole in patients with ALS; dopaminergic drugsin patients with PD; antithrombotics and cardiovascularmedications in patients with stroke; and antiepilepticsin patients with TBI (Table 5).
Adverse events
Over 90% of patients (n¼ 508; 91.8%) reported at leastone AE during the 52 week trial. The most frequentlyreported AEs (incidence�15%) were nausea, headache,dizziness, fall, and diarrhea. Most AEs were mild to mod-erate in severity (64.8% of patients with AEs). Table 6shows the incidence of commonly reported AEs acrossdisease groups.
Although chi-square tests were performed for each AE,resulting p values must be regarded as exploratory due tomultiple comparisons and the small size of some groups.Differences in AE incidence across disease groups appearedconsistent with the expected manifestations of the primaryneurological conditions. For example, respiratory failureoccurred exclusively, and dysphagia and dyspnea predom-inantly, in ALS/MND patients; fatigue was more thantwice as common in TBI patients; somnolence was mostcommon in PD/movement disorders patients; weaknesswas most common in ALS/MND and MS patients; andconstipation was most common in AD/dementia, ALS/MND, and ‘other PBA’ patients.
Because of the high morbidity associated with severalneurological conditions, it seems particularly important toalso note the frequency of AEs that investigators con-sidered possibly related to DM/Q treatment (TRAEs)(Table 7). Most TRAEs (91%) were mild-to-moderatein severity. Only seven occurred with incidence �5%:nausea (11.8%), dizziness (10.5%), headache (9.9%),somnolence (7.2%), fatigue (7.1%), diarrhea (6.5%), anddry mouth (5.1%).
To further assess the time course of these seven TRAEs,a post hoc analysis was conducted evaluating time to onset,Ta
ble
1.C
ateg
oriz
atio
nof
patie
nts’
prim
ary
neur
olog
ical
cond
ition
sin
todi
seas
egr
oups
(n).
AD
/dem
entia
(n¼
17)
MS
(n¼
223)
Str
oke/
CB
VD(n¼
51)
ALS
/MN
D(n¼
199)
TBI
(n¼
23)
PD/m
ovem
ent
diso
rder
s(n¼
23)
Oth
erPB
A(n¼
17)
AD
15M
S22
3S
trok
e46
ALS
176
TBI
21PD
11C
ereb
ella
rat
axia
3Fr
onta
llo
bede
men
tia2
Veno
usan
giom
a1
Prim
ary
late
ral
scle
rosi
s16
Hea
d trau
ma
2Pa
rkin
son
synd
rom
e1
Spi
noce
rebe
llar
atax
ia2
Pont
ine
AVM
1Pr
ogre
ssiv
ebu
lbar
pals
y5
Aty
pica
lPD
1C
ereb
ralp
alsy
2
Post
-cer
ebra
lan
eury
smsu
rger
y1
Bul
bar
mot
orne
uron
dise
ase
orde
gene
ratio
n1
Park
inso
nian
synd
rom
e1
Chr
onic
coug
h1
Bra
inan
eury
sm1
Bul
bar
pals
y1
Prog
ress
ive
supr
anuc
lear
pals
y5
Sm
all-
fiber
neur
opat
hy1
Sub
arac
hnoi
dhe
mor
rhag
e1
Hun
tingt
on’s
dise
ase
1Le
uko-
ence
phal
opat
hy1
Cho
reifo
rmdi
sord
er1
Post
-her
pes
sim
plex
viru
s1
Mov
emen
tdi
sord
er1
Sub
dura
lhem
atom
a1
Cor
ticob
asila
rde
gene
ratio
n1
Vira
lmen
ingi
o-en
ceph
aliti
s1
Hyd
roce
phal
us1
Post
-enc
epha
litis
1Fo
ster
-Ken
nedy
synd
rom
e1
Non
ekn
own
1
AD
:A
lzhe
imer
’sdi
seas
e;A
LS:
amyo
trop
hic
late
ral
scle
rosi
s;A
VM:
arte
riov
enou
sm
alfo
rmat
ion;
CB
VD:
cere
brov
ascu
lar
diso
rder
s;M
ND
:m
otor
neur
ondi
seas
e;M
S:
mul
tiple
scle
rosi
s;PB
A:
pseu
dobu
lbar
affe
ct;
PD:
Park
inso
n’s
dise
ase;
TBI:
trau
mat
icbr
ain
inju
ry.
Current Medical Research & Opinion Volume 30, Number 11 November 2014
2258 Safety of dextromethorphan/quinidine in PBA Pattee et al. www.cmrojournal.com ! 2014 Informa UK Ltd
Dow
nloa
ded
by [
"Uni
vers
ity a
t Buf
falo
Lib
rari
es"]
at 1
9:52
28
Nov
embe
r 20
17
duration, recurrence, and percentage of total study dayswith TRAE present (persistence) (Online Resource 1).Common TRAEs generally occurred early in therapy andwere transient; both median time to onset and durationranged from 4 to 8 days for all but fatigue (median duration16 days). TRAEs did not recur in most patients; the high-est incidence of recurrence was headache (42% recurrenceafter initial episode). TRAE presence as a percentage oftotal treatment days was low, ranging from a median of 2%for dizziness and headache to 22% for fatigue and sleepi-ness. Inclusion of patients who discontinued prior to AEresolution can reduce estimates of AE duration whileincreasing estimates of percentage of treatment days withAE present (by decreasing total treatment days). Omittingthese patients from this analysis did not appreciably affect
median duration but did decrease estimates of AE persist-ence (Online Resource 1).
Serious adverse events
The overall incidence of SAEs was 22.8%; no SAE wasreported to be related to study medication. Most SAEsoccurred in patients with ALS/MND, including all SAEsof respiratory failure, dysphagia, and pneumonia (OnlineResource 2). Except for respiratory events in ALS and dis-ease exacerbation in MS, there was no evidence ofincreased frequency of SAEs specific to any neurologicaldisorder.
Forty-seven deaths occurred during the 52 week trial,most attributed to ALS progression. Thirty-one of the 39ALS deaths were attributed to ALS-related respiratoryfailure or similar respiratory events. The remaining eightwere attributed to ‘ALS progression’ (n¼ 3), cardiac arrest(n¼ 2), pneumonia (n¼ 1), infection (n¼ 1), and cardio-respiratory arrest and epistaxis (n¼ 1). One additionalALS/MND group patient with primary lateral sclerosisdied of ‘cause unknown’. Following study completion, aninvestigator query about all ALS patient deaths providedan estimated median time from ALS diagnosis to death of29 months. (Actual date of death was obtained for 117patients; the remainder were censored at date of last con-tact.) The three deaths in MS patients were attributed toacute myelomonocytic leukemia, myocardial infarction(patient with resolving flu-like illness and extensive pre-existing skin breakdown; received DM/Q for 8 days andthen discontinued for recurrent flu-like illness, hallucin-ations, and lethargy; and died 5 days later), and myocardialinfarction and sepsis (patient with pulmonary embolismand sepsis). Of the remaining four deaths, two were attrib-uted to stroke (patient with AD and prior stroke history
Table 2. Patient disposition by primary neurological conditiona.
Patient disposition, n AD/dementia MS Stroke/CBVD TBI ALS/MND PD/movementdisorders
OtherPBA
Total
Enrolled n¼ 17 n¼ 223 n¼ 51 n¼ 23 n¼ 199 n¼ 23 n¼ 17 N¼ 553Treated �6 months 10 176 30 15 126 12 13 382Treated �12 monthsa 5 149 27 10 86 11 12 300Completed treatment phase 4 149 27 9 85 10 12 296Remaining at study termination 2 8 4 2 4 0 0 20Did not complete due to: 11 66 20 12 110 13 5 257
AE 4 27 7 1 62 5 1 107Medication refusal due to AE 2 10 4 2 18 4 1 41Medication refusal for other than AE 0 7 1 2 3 0 1 14Withdrew consent 3 15 3 3 9 1 0 34Lost to follow-up 0 4 4 2 7 0 1 18Protocol violation 0 2 0 0 1 0 0 3Other 4 9 5 4 14 3 1 40
Entered extension phase 4 132 22 8 77 8 11 262
AD: Alzheimer’s disease; AE: adverse event; ALS: amyotrophic lateral sclerosis; CBVD: cerebrovascular disorders; MND: motor neuron disease; MS: multiplesclerosis; PBA: pseudobulbar affect; PD: Parkinson’s disease; TBI: traumatic brain injury.aIncludes all patients who completed the treatment phase or received at least 365 days of dosing.
100
90
80
70
60
50
40
30
20
10
00 4 8 12 16 20 24 28 32 36 40 44 48 52
Pat
ient
s R
emai
ning
in S
tudy
(%
)
Study Week
Figure 1. Proportion of patients remaining in study, by study week.
Current Medical Research & Opinion Volume 30, Number 11 November 2014
! 2014 Informa UK Ltd www.cmrojournal.com Safety of dextromethorphan/quinidine in PBA Pattee et al. 2259
Dow
nloa
ded
by [
"Uni
vers
ity a
t Buf
falo
Lib
rari
es"]
at 1
9:52
28
Nov
embe
r 20
17
who died on Day 217, and a patient with prior strokehistory who died on Day 41); one was attributed to cardiacarrest (patient with AD and history of acute coronarysyndrome and hypertension); and one patient committedsuicide on Day 104 (patient with spinocerebellar ataxia
and reactive depression). In all cases, death was consideredunlikely related to DM/Q treatment. All deaths occurringduring the complete DM/Q clinical development programwere additionally reviewed by a group of consulting cardi-ologists and neurologists as part of the FDA review process
Table 4. Common concurrent medicationsa by primary neurological condition.
Description, % AD/dementia(n¼ 17)
MS(n¼ 223)
Stroke/CBVD(n¼ 51)
TBI(n¼ 23)
ALS/MND(n¼ 199)
PD/movementdisorders(n¼ 23)
Other PBA(n¼ 17)
Total(N¼ 553)
NSAIDs 23.5 36.3 19.6 30.4 40.2 17.4 5.9 33.8Antidepressants 23.5 23.3 31.4 30.4 29.1 34.8 23.5 26.9
Tricyclic antidepressants 0 4.9 2.0 13.0 13.1 4.3 0 7.6SSRIs 5.9 15.2 21.6 17.4 10.6 13.0 5.9 13.6Otherb 17.6 8.1 7.8 8.7 10.1 17.4 17.6 9.8
Lipid modifiers 35.3 22.4 35.3 21.7 20.6 13.0 23.5 23.0Antithrombotics 35.3 12.1 52.9 17.4 24.6 21.7 5.9 21.5Analgesics (e.g., acetaminophen) 35.3 25.6 19.6 21.7 14.1 13.0 5.9 19.9Drugs to inhibit gastric acid
production23.5 15.7 23.5 21.7 22.6 17.4 23.5 19.7
Vitamin E 11.8 7.6 3.9 4.3 37.7 8.7 29.4 18.8Antiepileptics 17.6 23.8 25.5 43.5 9.0 8.7 29.4 18.8Multivitamins, plain 5.9 22.4 7.8 8.7 20.1 0 11.8 17.9Anxiolytics 23.5 16.6 11.8 13.0 20.1 21.7 11.8 17.5Vitamin C 11.8 12.6 0 4.3 30.7 8.7 0 17.0Antihistamines for systemic use 17.6 14.8 7.8 26.1 18.1 4.3 17.6 15.6Vitamin B12 and folic acid 11.8 9.9 7.8 17.4 20.1 4.3 11.8 13.6Calcium 5.9 20.2 7.8 8.7 7.5 17.4 5.9 13.0Hypnotics and sedatives 17.6 8.5 3.9 4.3 20.6 17.4 0 12.7Opioids 11.8 11.7 5.9 8.7 15.1 4.3 11.8 11.9Laxatives 11.8 9.4 5.9 4.3 17.1 0 11.8 11.4Misc. herbal 0 13.0 2.0 0 15.1 0 0 10.8Beta-blocking agents 11.8 6.7 13.7 4.3 13.1 17.4 5.9 10.1Thyroid preparations 17.6 9.4 9.8 17.4 7.5 13.0 5.9 9.4ACE inhibitors, plain 17.6 5.8 19.6 13.0 9.0 4.3 11.8 9.0
ACE: angiotensin-converting enzyme; AD: Alzheimer’s disease; ALS: amyotrophic lateral sclerosis; CBVD: cerebrovascular disorders; MND: motor neuron disease;MS: multiple sclerosis; NSAIDs: nonsteroidal anti-inflammatory drugs; PBA: pseudobulbar affect; PD: Parkinson’s disease; SSRI: selective serotonin reuptakeinhibitor; TBI: traumatic brain injury.aUsed for �3 months and taken during the study; drug use represents minimum use in each category and may not include some combination products such asantihistamine cold products, combinations of vitamins and herbals, etc.bOther includes: venlafaxine, 3.6%; bupropion, 3.3%; trazodone, 2.5%; mirtazepine, 1.3%; duloxetine, mianserin, nefazodone, reboxetine, and hydroxytryptophan,0.2% (1 patient) each.
Table 3. Patient demographics by primary neurological condition.
Category AD/dementia(n¼ 17)
MS(n¼ 223)
Stroke/CBVD(n¼ 51)
TBI(n¼ 23)
ALS/MND(n¼ 199)
PD/movementdisorders(n¼ 23)
Other PBA(n¼ 17)
Age (years)Mean 63.5 45.7 54.8 45.6 55.6 64.9 49.1SD 11.20 10.23 10.93 13.81 11.06 8.00 13.71Median 66.0 47.0 56.0 44.0 57.0 66.0 52.0Min, max 38, 79 18, 71 30, 80 18, 74 18, 80 50, 86 22, 67
Gender, n (%)Male 10 (58.8) 49 (22.0) 24 (47.1) 10 (43.5) 116 (58.3) 15 (65.2) 7 (41.2)Female 7 (41.2) 174 (78.0) 27 (52.9) 13 (56.5) 83 (41.7) 8 (34.8) 10 (58.8)
Ethnicity, n (%)Caucasian 16 (94.1) 208 (93.3) 43 (84.3) 18 (78.3) 179 (89.9) 22 (95.7) 16 (94.1)Black 1 (5.9) 5 (2.2) 5 (9.8) 1 (4.3) 5 (2.5) 0 (0) 0 (0)Asian 0 (0) 1 (0.4) 1 (2.0) 0 (0) 1 (0.5) 0 (0) 0 (0)Hispanic 0 (0) 8 (3.6) 2 (3.9) 3 (13.0) 9 (4.5) 1 (4.3) 1 (5.9)Other 0 (0) 1 (0.4) 0 (0) 1 (4.3) 5 (2.5) 0 (0) 0 (0)
AD: Alzheimer’s disease; ALS: amyotrophic lateral sclerosis; CBVD: cerebrovascular disorders; MND: motor neuron disease; MS: multiple sclerosis; PBA:pseudobulbar affect; PD: Parkinson’s disease; SD: standard deviation; TBI: traumatic brain injury.
Current Medical Research & Opinion Volume 30, Number 11 November 2014
2260 Safety of dextromethorphan/quinidine in PBA Pattee et al. www.cmrojournal.com ! 2014 Informa UK Ltd
Dow
nloa
ded
by [
"Uni
vers
ity a
t Buf
falo
Lib
rari
es"]
at 1
9:52
28
Nov
embe
r 20
17

for the DM/Q application for treatment of PBA. Theirfindings corroborated the investigators’ initial assessmentthat the deaths were not related to treatment but to pro-gression of underlying neurological or other medicalconditions.
AEs leading to discontinuation
One hundred forty-nine patients (26.9%) withdrew fromthe trial or refused further study medication due to AEs(Online Resource 3). This number includes one discon-tinuation during the extension for an AE beginning in thetreatment phase. AE dropout was more frequent in ALS/MND (n¼ 80; 40.2% of ALS/MND patients), PD/move-ment disorders (n¼ 9; 39.1% of PD/movement disorderspatients), and AD/dementia (n¼ 6; 35.3% of AD/demen-tia patients), and less frequent in stroke/CBVD (n¼ 11;21.5%), MS (n¼ 37; 16.6%), TBI (n¼ 3; 13.0%), and‘other PBA’ (n¼ 2; 11.8%). The most common AEs lead-ing to discontinuation were respiratory failure in ALS/MND patients (14.1% of ALS/MND group; 5.1% oftotal), nausea (3.3%), dizziness (2.9%), headache(2.5%), and diarrhea (1.8%). Other than respiratoryfailure and nausea in the ALS/MND group and dizzinessand headache in the PD/movement disorders group,discontinuation due to AEs appeared similar regardlessof primary neurological disorder. Approximately half ofthe AE-related dropouts, (n¼ 74 or 13.5% of totalpatients) included AEs that were judged to be at leastpossibly related to treatment. In these patients, themedian time to discontinuation was 16.5 days (mean31.4; range 1–243).
Clinical laboratory values and vital signs
Laboratory test results remained stable during the trial.Shift tables showed no changes of clinical relevance.There were no clinically relevant changes in mean systolicand diastolic BP, heart rate, respiratory rate, or body tem-perature at any visit. Increased BP was recorded as an AEin 11 patients (2.0%), and increased systolic BP wasrecorded as an AE in one patient; however, these wereconsidered ‘not related’ or ‘unlikely related’ to DM/Qtreatment in all but one patient (‘possibly related’).
Physical examinations
No abnormal physical findings related to study-drug treat-ment were observed.
Electrocardiography
Mean ECG values showed no clinically relevant post-base-line changes (Online Resource 4). QTc intervals showed a
Table 5. Common concurrent disease-specific medicationsa by primaryneurological condition.
AD/dementia (n¼ 17) %Anticholinesterase (e.g., donepezil) 47.1Memantine 29.4
MS (n¼ 223) %Interferon beta 45.3Glatiramer 27.4Centrally acting muscle relaxants (e.g., baclofen,
tizanidine, orphenadrine)33.2
Urinary antispasmodics (e.g., oxybutynin, tolterodine) 25.6Psychostimulants (e.g., amphetamine derivatives,
modafinil, pemoline)18.8
Glucocorticoids 5.8Immunosuppressants (e.g., methotrexate,
mycophenolate, azathioprine)5.8
Mitoxantrone 4.04-Aminopyridine 2.2
Stroke/CBVD (n¼ 51) %Aspirin 43.1Clopidogrel 19.6Calcium channel blockers (e.g., dihydropyridine) 11.8
TBI (n¼ 23) %Topiramate 13.0Carbamazepine/oxcarbazepine 13.0Thyroid preparations 17.4Psychostimulants (e.g., amphetamine derivatives,modafinil, pemoline)
13.0
Antimigraine (e.g., triptans, selective 5HT1 agonists) 13.0Glucocorticoids 8.7
ALS/MND (n¼ 199) %Riluzole 47.7Centrally acting muscle relaxants (e.g., baclofen,
tizanidine, orphenadrine)31.7
Coenzyme Q10 31.2Tetracycline antibiotics (e.g., minocycline, doxycycline) 17.6Creatine 15.6Beta-2-adrenergic agonists, inhalants 14.6Beta carotene/vitamin A 13.1Urinary antispasmodics (e.g., oxybutynin, tolterodine) 11.1Quinine 10.6Expectorants and mucolytics (e.g., guaifenesin,
n-acetylcysteine)9.0
Belladonna alkaloids (e.g., atropine, hyocyamine,scopolamine)
9.0
Glycopyrrolate 7.5
PD/movement disorders (n¼ 23) %Dopamine derivatives (� carbidopa, benserazide,
or entacapone)65.2
Dopamine agonists 30.4Amantadine 17.4Selegiline 8.7Trihexyphenidyl 0.5
Other PBA (n¼ 17) %Phenytoin 11.8Tiagabine 11.8
5HT1: type 1 5-hydroxytryptamine (serotonin) receptor; AD: Alzheimer’sdisease; ALS: amyotrophic lateral sclerosis; CBVD: cerebrovascular dis-orders; MND: motor neuron disease; MS: multiple sclerosis; PBA: pseudo-bulbar affect; PD: Parkinson’s disease; TBI: traumatic brain injury.aUsed for �3 months and taken during the study.
Current Medical Research & Opinion Volume 30, Number 11 November 2014
! 2014 Informa UK Ltd www.cmrojournal.com Safety of dextromethorphan/quinidine in PBA Pattee et al. 2261
Dow
nloa
ded
by [
"Uni
vers
ity a
t Buf
falo
Lib
rari
es"]
at 1
9:52
28
Nov
embe
r 20
17
Table 6. Incidence of adverse events reported by �5% and at least two patients in any primary-neurological-condition category (safety population).
Preferredterm, % (n)
AD/dementia(n¼ 17)
MS(n¼ 223)
Stroke/CBVD
(n¼ 51)
TBI(n¼ 23)
ALS/MND(n¼ 199)
PD/movementdisorders(n¼ 23)
Other PBA(n¼ 17)
p Valuea Total(N¼ 553)
Any AE 70.6 (12) 89.7 (200) 90.2 (46) 82.6 (19) 98.0 (195) 91.3 (21) 88.2 (15) 0.0004 91.9 (508)Nausea 11.8 (2) 23.8 (53) 19.6 (10) 13.0 (3) 31.7 (63) 8.7 (2) 23.5 (4) 0.0544 24.8 (137)Headache 5.9 (1) 25.1 (56) 15.7 (8) 26.1 (6) 22.1 (44) 26.1 (6) 29.4 (5) 0.4613 22.8 (126)Dizziness 5.9 (1) 21.5 (48) 19.6 (10) 21.7 (5) 17.1 (34) 30.4 (7) 17.6 (3) 0.5201 19.5 (108)Fall 17.6 (3) 13.5 (30) 7.8 (4) 13.0 (3) 22.6 (45) 21.7 (5) 5.9 (1) 0.0674 16.5 (91)Diarrhea 23.5 (4) 15.7 (35) 11.8 (6) 13.0 (3) 19.1 (38) 0 (0) 23.5 (4) 0.2426 16.3 (90)Fatigue 17.6 (3) 16.6 (37) 7.8 (4) 39.1 (9) 11.1 (22) 13.0 (3) 17.6 (3) 0.0141 14.6 (81)Weakness 0 (0) 14.8 (33) 3.9 (2) 4.3 (1) 18.6 (37) 4.3 (1) 11.8 (2) 0.0245 13.7 (76)Nasopharyngitis 0 (0) 16.1 (36) 9.8 (5) 13.0 (3) 9.5 (19) 4.3 (1) 11.8 (2) 0.1944 11.9 (66)Somnolence 5.9 (1) 4.9 (11) 15.7 (8) 4.3 (1) 15.6 (31) 26.1 (6) 11.8 (2) 0.0019 10.8 (60)Arthralgia 0 (0) 14.3 (32) 5.9 (3) 0 (0) 10.1 (20) 13.0 (3) 5.9 (1) 0.1334 10.7 (59)Cough 5.9 (1) 10.3 (23) 3.9 (2) 13.0 (3) 14.6 (29) 4.3 (1) 0 (0) 0.1633 10.7 (59)Ecchymosis 5.9 (1) 8.1 (18) 5.9 (3) 4.3 (1) 12.6 (25) 13.0 (3) 17.6 (3) 0.4343 9.8 (54)Pain in limb 0 (0) 13.0 (29) 11.8 (6) 0 (0) 8.5 (17) 0 (0) 5.9 (1) 0.1056 9.6 (53)Constipation 11.8 (2) 5.4 (12) 5.9 (3) 0 (0) 15.1 (30) 4.3 (1) 11.8 (2) 0.0124 9.0 (50)Insomnia 0 (0) 6.7 (15) 7.8 (4) 8.7 (2) 13.6 (27) 0 (0) 11.8 (2) 0.1004 9.0 (50)Back pain 5.9 (1) 9.9 (22) 5.9 (3) 0 (0) 9.0 (18) 4.3 (1) 5.9 (1) 0.6684 8.3 (46)Dysphagia 0 (0) 1.8 (4) 3.9 (2) 0 (0) 20.1 (40) 0 (0) 0 (0) 50.0001 8.3 (46)Dry mouth 5.9 (1) 5.8 (13) 9.8 (5) 0 (0) 8.5 (17) 13.0 (3) 0 (0) 0.4119 7.1 (39)Edema lower limb 5.9 (1) 3.6 (8) 7.8 (4) 8.7 (2) 9.5 (19) 8.7 (2) 11.8 (2) 0.3244 6.9 (38)Respiratory failure 0 (0) 0 (0) 0 (0) 0 (0) 19.1 (38) 0 (0) 0 (0) 50.0001 6.9 (38)Sore throat 5.9 (1) 9.0 (20) 3.9 (2) 4.3 (1) 7.0 (14) 0.4887 6.9 (38)Urinary tract
infection0 (0) 9.9 (22) 5.9 (3) 4.3 (1) 4.5 (9) 4.3 (1) 11.8 (2) 0.2970 6.9 (38)
Dyspnea 0 (0) 3.6 (8) 3.9 (2) 8.7 (2) 11.6 (23) 4.3 (1) 5.9 (1) 0.0399 6.7 (37)Fatigue aggravated 0 (0) 9.4 (21) 3.9 (2) 0 (0) 5.0 (10) 0 (0) 5.9 (1) 0.1663 6.1 (34)Laceration 0 (0) 5.8 (13) 5.9 (3) 0 (0) 8.0 (16) 4.3 (1) 5.9 (1) 0.6724 6.1 (34)MS aggravated 0 (0) 14.3 (32) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 50.0001 5.8 (32)Pyrexia 5.9 (1) 7.6 (17) 3.9 (2) 5.0 (10) 4.3 (1) 5.9 (1) 0.7549 5.8 (32)Vomiting 5.9 (1) 7.6 (17) 2.0 (1) 8.7 (2) 5.0 (10) 4.3 (1) 0 (0) 0.6161 5.8 (32)Muscle spasms 0 (0) 6.7 (15) 0 (0) 8.7 (2) 6.0 (12) 0 (0) 5.9 (1) 0.3610 5.4 (30)Nasal congestion 0 (0) 6.3 (14) 3.9 (2) 4.3 (1) 6.0 (12) 0 (0) 0 (0) 0.6634 5.2 (29)Anxiety 0 (0) 3.6 (8) 0 (0) 4.3 (1) 7.5 (15) 13.0 5.9 (1) 0.1138 5.1 (28)Joint stiffness 0 (0) 5.4 (12) 2.0 (1) 0 (0) 6.5 (13) 0 (0) 11.8 (2) 0.3292 5.1 (28)
AD: Alzheimer’s disease; AE: adverse event; ALS: amyotrophic lateral sclerosis; CBVD: cerebrovascular disorders; MND: motor neuron disease; MS: multiplesclerosis; PBA: pseudobulbar affect; PD: Parkinson’s disease; TBI: traumatic brain injury.aChi-square test.
Table 7. Incidence of treatment-relateda adverse events by primary neurological condition (safety population).
Preferred term, % AD/dementia(n¼ 17)
MS(n¼ 223)
Stroke/CBVD(n¼ 51)
TBI(n¼ 23)
ALS/MND(n¼ 199)
PD/movementdisorders(n¼ 23)
Other PBA(n¼ 17)
Total(N¼ 553)
Any TRAE 23.5 39.9 51.0 47.8 59.3 60.9 52.9 49.0Nausea 0 8.1 7.8 4.3 19.1 4.3 17.6 11.8Dizziness (excluding vertigo) 0 10.3 7.8 13.0 10.1 21.7 17.6 10.5Headache NOS 0 9.4 5.9 8.7 11.6 21.7 5.9 9.9Somnolence 0 2.7 5.9 0 13.1 13.0 11.8 7.2Fatigue 5.9 5.8 2.0 26.1 7.5 4.3 11.8 7.1Diarrhea NOS 11.8 4.0 3.9 4.3 9.0 0 23.5 6.5Dry mouth 0 3.1 5.9 0 8.0 8.7 0 5.1
Additional TRAEs that occurred in at least 5% of patients and in two or more patients in any disease category were stroke/CBVD: insomnia (not elsewhereclassified), n¼ 3 (5.9%); TBI: lethargy, n¼ 3 (13.0%), abnormal dreams, n¼ 2 (8.7%); ALS/MND: constipation, n¼ 11 (5.5%); PD/movement disorders: confusion,n¼ 2 (8.7%); other PBA: joint stiffness, n¼ 2 (11.8%); flatulence, n¼ 2 (11.8%); dyspepsia, n¼ 2 (11.8%).AD: Alzheimer’s disease; ALS: amyotrophic lateral sclerosis; CBVD: cerebrovascular disorders; MND: motor neuron disease; MS: multiple sclerosis; NOS: nototherwise specified; PBA: pseudobulbar affect; PD: Parkinson’s disease; TRAE: treatment-related adverse event.aTRAEs that occurred in at least 5% of total patients; treatment-related categories included possible, probable, highly probable, and missing.
Current Medical Research & Opinion Volume 30, Number 11 November 2014
2262 Safety of dextromethorphan/quinidine in PBA Pattee et al. www.cmrojournal.com ! 2014 Informa UK Ltd
Dow
nloa
ded
by [
"Uni
vers
ity a
t Buf
falo
Lib
rari
es"]
at 1
9:52
28
Nov
embe
r 20
17

small mean increase (QTcF 3.2; QTcB 4.6 msec) frombaseline to final measurement. Five patients had QTcincreases �60 msec; however, all values remained5500 msec during the 52 week treatment phase.
Pharmacokinetics
Blood samples were provided by 202 patients on Day 29.Mean (standard deviation) plasma concentrations wereDM: 92.7 (45.6) ng/mL, DX: 78.0 (43.4) ng/mL, and quini-dine: 0.15 (0.09) mg/mL. Mean DM concentration wassimilar to that reported in previous placebo-controlledtrials using this dosage22,23.
Discussion
Long-term administration of DM/Q was well tolerated inthis large, open-label study of patients with PBA associatedwith a wide range of neurological conditions. These resultsprovide important safety data for DM/Q, the onlyapproved medication for PBA, a common and distressingdisorder experienced by patients with neurologic disease orinjury. The low incidence of treatment-emergent AEs withDM/Q used at a higher dose than the approved dose, andoccurring in a diverse, clinic-based, ‘real-world’ popula-tion, provides reassuring clarity for physicians on therisks of this treatment. The overall incidence of reportedAEs is consistent with the trial length and chronic andoften progressive nature of the underlying neurologicalconditions. Although the majority (90%) of patientsreported at least one AE, only 49% reported AEs possiblyrelated to DM/Q treatment, and only 13.4% discontinuedfor TRAEs. The most frequently reported AEs were con-sistent with those reported at higher incidence than con-trols in shorter, double-blind DM/Q 30/30 mg trials inPBA patients with ALS or MS22,23. In a 4 week study inALS patients22, the most commonly reported AEs (inci-dence �15% and more than controls) among DM/Qrecipients (n¼ 70) were nausea, dizziness, fatigue, diar-rhea, and headache. In a 12 week study in MS patients23,the most commonly reported AEs (incidence�15% andmore than placebo) among the DM/Q recipients (n¼ 76)were dizziness, nausea, and headache.
This patient population is susceptible to falls. Falls werereported by 16.5% of patients in this study; however, onlysix patients (1.1%) experienced a fall that was deemed byinvestigators as possibly related to treatment. In double-blind clinical trials falls were reported with similar inci-dence in DM/Q and placebo recipients.
Several types of AEs, particularly SAEs, wereattributed to primary neurological conditions, for examplerespiratory depression, dyspnea, and dysphagia in ALSand disease aggravation in MS. Underlying diseasecontribution to AEs is supported by the AE analysis by
disease group, AE data from DM/Q double-blind trials,and literature-reported mortality patterns in ALSpatients22–26. Overall, the 39/176 ALS patient deaths con-stitute a mortality rate of 22.2%, which is lower than thedeath rate reported in prospective trials. In large-scaleALS-treatment trials, 1 year mortality rates ranged fromapproximately 26% to 35% in treatment arms and as highas 46% in placebo arms27,28. Furthermore, epidemiologicalstudies generally suggest median survival after ALS diag-nosis is approximately 15 to 19 months29–34, although atleast one study has reported median survival post-diagnosisof 30 months35. The median survival after ALS diagnosisin the current study was estimated at 29 months (n¼ 176);however, inferences regarding potential effect of DM/Q onsurvival are not possible without controlled studies.
The DM/Q 30/30 mg twice daily dosage administered inthis study is greater than the DM/Q 20/10 mg twice dailydosage approved by the FDA and EMA for PBA treatmentand also greater than the 30/10 mg twice daily dosageapproved by the EMA. Given that the incidence of someTRAEs, such as dizziness and nausea, appear dose-relatedin controlled trials22–24, it is likely that the AE incidencereported in this long-term study would be greater thanobserved with the lower approved doses. Indeed, amongpatients treated with DM/Q 20/10 mg for 12 weeks duringthe pivotal trial (n¼ 107) the percentages reporting head-ache, dizziness, and nausea were 14.0%, 10.3%, and 7.5%24
versus 22.8%, 19.5%, and 24.8%, respectively, in the pre-sent study.
Quinidine is a potent and reliable inhibitor of CYP2D6,and the dose in the approved DM/Q formulation (10 mgtwice daily) is sufficient to increase DM exposure approxi-mately 20-fold24. Other medications metabolized byCYP2D6 include antidepressants, antipsychotics, andbeta-blockers. While concentrations of these medicationsmay be increased in the presence of quinidine, potentialdrug interactions can be managed with simple dose adjust-ments. The overall number and type of concomitant medi-cations taken in this study appear to reflect those seen intypical clinical practice. Patients who withdrew from thestudy for AEs did not appear to have a higher medicationburden than those who completed the trial.
While quinidine is a well established type 1a antiar-rhythmic drug with potential to cause dose-relatedQT-interval prolongation36, ventricular arrhythmia, andtorsades de pointes37–40, no clinically significant arrhyth-mias were reported in controlled trials of DM/Q for PBAutilizing either the 30 or 10 mg doses of quinidine22–24. Nopatient had QTc increase4500 msec during the treatmentphase. Two patients (both with MS) had a QTc4500 msecduring the elective treatment extension; one had QTcB/QTcF¼ 493/505 msec and was withdrawn from the trial,and the other had prolonged QTc on two occasions (high-est QTcB/QTcF¼ 526/506 msec on Day 1183) andremained in treatment until study termination. Overall
Current Medical Research & Opinion Volume 30, Number 11 November 2014
! 2014 Informa UK Ltd www.cmrojournal.com Safety of dextromethorphan/quinidine in PBA Pattee et al. 2263
Dow
nloa
ded
by [
"Uni
vers
ity a
t Buf
falo
Lib
rari
es"]
at 1
9:52
28
Nov
embe
r 20
17

ECG results suggested no clinically meaningful effect ofDM/Q on myocardial repolarization or any ECG variable.
In summary, no significant ECG changes were observedon DM/Q during this 1 year study. The observed TRAEsoccurred early in the treatment course, were largely mild-to-moderate, predominantly transient, and typically didnot result in discontinuation. The safety and tolerabilityprofile of DM/Q in this study was consistent with thatobserved in previous DM/Q trials, and the outcomes forpatients receiving DM/Q followed the anticipated clinicalcourse of the underlying neurological conditions.
Key points
� Pseudobulbar Affect (PBA) is an uncontrollable dis-order of emotional expression, occurring in a broadrange of neurological diseases or injuries affecting thebrain.
� Dextromethorphan/quinidine is the only medicationapproved by the US Food and Drug Administrationand European Medicines Authority for the treatmentof PBA.
� This 52 week open-label study of 553 patients demon-strates the long-term safety and tolerability of dextro-methorphan and quinidine in patients with PBAsecondary to a wide variety of neurological conditions.
TransparencyDeclaration of fundingThis work was supported by Avanir Pharmaceuticals Inc.
Declaration of financial/other relationshipsG.L.P. has disclosed that he has served on the speakers bureau forand received research support from Avanir Pharmaceuticals Inc.J.P.W. has disclosed that he has served on the speakers bureau forand received research support from Avanir Pharmaceuticals Inc.C.L.-H. has disclosed that she has no significant relationshipswith or financial interests in any commercial companies relatedto this study or article. S.H.A. has disclosed that he serves on ascientific advisory board for Neuraltus Pharmaceuticals Inc.; hasreceived a speaker honorarium from Avanir PharmaceuticalsInc.; receives research support from the National Institutes ofHealth and the Muscular Dystrophy Association; and hasserved as an expert consultant in a medicolegal case. A.E.F.and L.E.P. have disclosed that they are employees of AvanirPharmaceuticals Inc.
The CMRO peer reviewer on this manuscript has received anhonorarium from CMRO for review work, but has no relevantfinancial or other relationships to disclose.
AcknowledgmentsThe authors thank Laura Truskowski of INC Research for statis-tical analysis of the data; INC Research for data collection and
management; Adrian Hepner MD, previously of AvanirPharmaceuticals Inc., for contributing to the analysis of thestudy and the early stages of this manuscript; Joao Siffert MDof Avanir Pharmaceuticals Inc. and Randall E. Kaye MD, previ-ously of Avanir Pharmaceuticals Inc., for critical review of thedata and manuscript; and The Curry Rockefeller Group LLC forhelp in preparing the manuscript for publication.
References1. Colamonico J, Formella A, Bradley W. Pseudobulbar affect: burden of illness
in the USA. Adv Ther 2012;29:775-98
2. Wortzel HS, Oster TJ, Anderson CA, Arciniegas DB. Pathological laughing
and crying: epidemiology, pathophysiology and treatment. CNS Drugs
2008;22:531-45
3. Parvizi J, Arciniegas DB, Bernardini GL, et al. Diagnosis and management of
pathological laughter and crying. Mayo Clin Proc 2006;81:1482-6
4. Work S, Colamonico JA, Bradley WG, Kaye RE. Pseudobulbar affect: an
under-recognized and undertreated neurological disorder. Adv Ther
2011;28:586-601
5. Brooks BR, Crumpacker D, Fellus J, et al. PRISM: a novel research tool to
assess the prevalence of pseudobulbar affect symptoms across neurological
conditions. PLoS One 2013;8:1-8
6. Lopez OL, Gonzalez MP, Becker JT, et al. Symptoms of depression and
psychosis in Alzheimer’s disease and frontotemporal dementia: exploration
of underlying mechanisms. Neuropsychiatry Neuropsychol Behav Neurol
1996;9:154-61
7. Feinstein A, Feinstein K, Gray T, O’Connor P. Prevalence and neurobehavioral
correlates of pathological laughing and crying in multiple sclerosis.
Arch Neurol 1997;54:1116-21
8. Siddiqui MS, Fernandez HH, Garvan CW, et al. Inappropriate crying and
laughing in Parkinson disease and movement disorders. World J Biol
Psychiatry 2009;10:234-40
9. House A, Dennis M, Molyneux A, et al. Emotionalism after stroke. BMJ
1989;298:991-4
10. Tateno A, Jorge RE, Robinson RG. Pathological laughing and crying following
traumatic brain injury. J Neuropsychiatry Clin Neurosci 2004;16:426-34
11. Gallagher JP. Pathologic laughter and crying in ALS: a search for their origin.
Acta Neurol Scand 1989;80:114-17
12. Parvizi J, Anderson SW, Martin CO, et al. Pathological laughter and crying: a
link to the cerebellum. Brain 2001;124:1708-19
13. Parvizi J, Joseph J, Press DZ, Schmahmann JD. Pathological laughing and
crying in patients with multiple system atrophy-cerebellar type. Mov Disord
2007;22:798-803
14. Parvizi J, Coburn KL, Shillcutt SD, et al. Neuroanatomy of pathological
laughing and crying: a report of the American Neuropsychiatric
Association Committee on Research. J Neuropsychiatry Clin Neurosci
2009;21:75-87
15. Nuedexta (dextromethorphan hydrobromide and quinidine sulfate) capsules.
Full prescribing information. Available at: https://www.nuedexta.com/sites/
default/files/pdf/Prescribing_Information.pdf [Last accessed 14 March 2014]
16. European Medicines Agency. Nuedexta. Summary of product characteristics.
Available at: http://www.ema.europa.eu/docs/en_GB/document_library/
EPAR_-_Product_Information/human/002560/WC500145050.pdf [Last
accessed 14 March 2014]
17. Choi DW, Peters S, Viseskul V. Dextrorphan and levorphanol selectively block
N-Methyl-D-aspartate receptor-mediated neurotoxicity on cortical neurons.
J Pharmacol Exp Ther 1987;242:713-20
18. Musacchio JM, Klein M, Canoll PD. Dextromethorphan and sigma ligands:
common sites but diverse effects. Life Sci 1989;45:1721-32
19. Werling LL, Keller A, Frank JG, Nuwayhid SJ. A comparison of the binding
profiles of dextromethorphan, memantine, fluoxetine and amitriptyline:
Current Medical Research & Opinion Volume 30, Number 11 November 2014
2264 Safety of dextromethorphan/quinidine in PBA Pattee et al. www.cmrojournal.com ! 2014 Informa UK Ltd
Dow
nloa
ded
by [
"Uni
vers
ity a
t Buf
falo
Lib
rari
es"]
at 1
9:52
28
Nov
embe
r 20
17

treatment of involuntary emotional expression disorder. Exp Neurol 2007;207:
248-57
20. Zhang Y, Britto MR, Valderhaug KL, et al. Dextromethorphan:
enhancing its systemic availability by way of low-dose quinidine-
mediated inhibition of cytochrome P4502D6. Clin Pharmacol Ther
1992;51:647-55
21. Pope LE, Khalil MH, Berg JE, et al. Pharmacokinetics of dextromethorphan
after single or multiple dosing in combination with quinidine in extensive and
poor metabolizers. J Clin Pharmacol 2004;44:1132-42
22. Brooks BR, Thisted RA, Appel SH, et al.; AVP-923 ALS Study Group.
Treatment of pseudobulbar affect in ALS with dextromethorphan/quinidine:
a randomized trial. Neurology 2004;63:1364-70
23. Panitch HS, Thisted RA, Smith RA, et al.; Pseudobulbar Affect in Multiple
Sclerosis Study Group. Randomized, controlled trial of dextromethorphan/
quinidine for pseudobulbar affect in multiple sclerosis. Ann Neurol
2006;59:780-7
24. Pioro EP, Brooks BR, Cummings J, et al.; Safety, Tolerability, and Efficacy
Results Trial of AVP-923 in PBA Investigators. Dextromethorphan plus
ultra low-dose quinidine reduces pseudobulbar affect. Ann Neurol 2010;
68:693-702
25. Gil J, Funalot B, Verschueren A, et al. Causes of death amongst French
patients with amyotrophic lateral sclerosis: a prospective study. Eur J
Neurol 2008;15:1245-51
26. Spataro R, Lo Re M, Piccoli T, et al. Causes and place of death in Italian
patients with amyotrophic lateral sclerosis. Acta Neurol Scand 2010;
122:217-23
27. Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in
amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med
1994;330:585-91
28. Louwerse ES, Weverling GJ, Bossuyt PM, et al. Randomized, double-blind,
controlled trial of acetylcysteine in amyotrophic lateral sclerosis. Arch Neurol
1995;52:559-64
29. Rooney J, Byrne S, Heverin M, et al. Survival analysis of Irish amyotrophic
lateral sclerosis patients diagnosed from 1995–2010. PLoS One 2013;8:
e74733
30. Zoccolella S, Beghi E, Palagano G, et al.; SLAP Registry. Analysis of survival
and prognostic factors in amyotrophic lateral sclerosis: a population based
study. J Neurol Neurosurg Psychiatry 2008;79:33-7
31. Murphy M, Quinn S, Young J, et al. Increasing incidence of ALS in Canterbury,
New Zealand: a 22-year study. Neurology 2008;71:1889-95
32. del Aquila MA, Longstreth Jr WT, McGuire V, et al. Prognosis in amyotrophic
lateral sclerosis: a population-based study. Neurology 2003;60:813-19
33. Chio A, Mora G, Leone M, et al.; Piemonte and Valle d’Aosta Register for
ALS (PARALS). Early symptom progression rate is related to ALS outcome: a
prospective population-based study. Neurology 2002;59:99-103
34. Logroscino G, Traynor BJ, Hardiman O, et al.; EURALS. Descriptive epidemi-
ology of amyotrophic lateral sclerosis: new evidence and unsolved issues.
J Neurol Neurosurg Psychiatry 2008;79:6-11
35. Millul A, Beghi E, Logroscino G, et al. Survival of patients with amyotrophic
lateral sclerosis in a population-based registry. Neuroepidemiology
2005;25:114-19
36. Quinidine Sulfate Tablets USP Revised: June 2005 Rx only. Available at:
http://pi.watson.com/data_stream.asp?product_group¼1306&p¼pi [Last
accessed 14 March 2014]
37. Holford NH, Coates PE, Guentert TW, et al. The effect of quinidine and its
metabolites on the electrocardiogram and systolic time intervals: concentra-
tion–effect relationships. Br J Clin Pharmacol 1981;11:187-95
38. Coplen SE, Antman EM, Berlin JA, et al. Efficacy and safety of quinidine
therapy for maintenance of sinus rhythm after cardioversion. A meta-analysis
of randomized control trials. Circulation 1990;82:1106-16
39. Morganroth J, Goin JE. Quinidine-related mortality in the short-to-medium-
term treatment of ventricular arrhythmias. A meta-analysis. Circulation
1991;84:1977-83
40. Grace AA, Camm AJ. Quinidine. N Engl J Med 1998;338:35-45
Current Medical Research & Opinion Volume 30, Number 11 November 2014
! 2014 Informa UK Ltd www.cmrojournal.com Safety of dextromethorphan/quinidine in PBA Pattee et al. 2265
Dow
nloa
ded
by [
"Uni
vers
ity a
t Buf
falo
Lib
rari
es"]
at 1
9:52
28
Nov
embe
r 20
17

ORIGINAL ARTICLE
Dextromethorphan Plus Ultra Low-DoseQuinidine Reduces Pseudobulbar Affect
Erik P. Pioro, MD, PhD,1 Benjamin Rix Brooks, MD,2 Jeffrey Cummings, MD,3
Randolph Schiffer, MD,1 Ronald A. Thisted, PhD,4 Daniel Wynn, MD,5
Adrian Hepner, MD,6 and Randall Kaye, MD6 for the Safety, Tolerability, and Efficacy
Results Trial of AVP-923 in PBA Investigators
Objective: To evaluate dextromethorphan combined with ultra low-dose quinidine (DMq) for treating pseudobulbaraffect (PBA) in patients with amyotrophic lateral sclerosis (ALS) or multiple sclerosis (MS).Methods: In a 12-week randomized, double-blind trial, ALS and MS patients with clinically significant PBA (a baselinescore �13 on the Center for Neurologic Studies–Lability Scale [CNS-LS]) were maintained, twice daily, on placebo,DMq at 30/10mg (DMq-30), or DMq at 20/10mg (DMq-20).Results: In 326 randomized patients (of whom 283, or 86.8%, completed the study), the PBA-episode daily rate was46.9% (p < 0.0001) lower for DMq-30 than for placebo and 49.0% (p < 0.0001) lower for DMq-20 than for placeboby longitudinal negative binomial regression, the prespecified primary analysis. Mean CNS-LS scores decreased by8.2 points for DMq-30 and 8.2 for DMq-20, vs 5.7 for placebo (p¼ 0.0002 and p¼ 0.0113, respectively). Otherendpoints showing statistically significant DMq benefit included, for both dosage levels, the likelihood of PBAremission during the final 14 days and, for the higher dosage, improvement on measures of social functioning andmental health. Both dosages were safe and well tolerated.Interpretation: DMq markedly reduced PBA frequency and severity, decreasing the condition’s detrimental impacton a patient’s life, with satisfactory safety and high tolerability. The findings expand the clinical evidence that DMqmay be an important treatment for patients suffering from the socially debilitating symptoms of PBA.
ANN NEUROL 2010;68:693–702
Introduction
Pseudobulbar affect (PBA) is a neurologic condition
characterized by involuntary outbursts of laughing
and/or crying incongruous or disproportionate to the
patient’s emotional state.1 The condition, hypothesized to
arise from disconnection of brainstem structures from
cortical inhibition, is associated with underlying central
nervous system disorders, including stroke,2 traumatic
brain injury,3 Alzheimer disease,4 amyotrophic lateral scle-
rosis (ALS),5–7 and multiple sclerosis (MS).8 Prevalence
studies have reported that it affects 11% of patients 1 year
after a stroke,2 11% of patients during the first year after
traumatic brain injury,9 18% of patients with Alzheimer
disease,4 10% of patients with MS,8 and 49% of patients
with ALS.10 In addition to the effects of the underlying
disorder, PBA can have a severe impact on well-being and
social functioning and can be highly disabling, owing in
part to the stigma attached to loss of emotional control.11
Yet even with such a significant burden of illness, PBA
appears to be poorly recognized and consequently is
undertreated.11,12
In settings of ALS or MS, dextromethorphan plus
quinidine (DMQ) has been found to be beneficial in reduc-
ing PBA.13,14 Dextromethorphan (DM) is known to be a
low-affinity, noncompetitive antagonist of the N-methyl-d-
aspartate glutamate receptor,15 and also a sigma-receptor
agonist.16 To block its first-pass metabolism, it was originally
coadministered with low-dose quinidine (Q), a potent cyto-
chrome P450 2D6 inhibitor,17 at DMQ dosage of 30/30mg
in a capsule taken twice daily. Without such blockade, DM
blood levels in some ALS patients have been undetectably
low even following DM dosage as high as 750mg/day.17 In
View this article online at wileyonlinelibrary.com. DOI: 10.1002/ana.22093
Received Feb 24, 2010, and in revised form Apr 30, 2010. Accepted for publication May 20, 2010.
Address correspondence to Dr Pioro, Director, Section of ALS and Related Disorders, Department of Neurology, Neurological Institute, Cleveland
Clinic, Desk S90, 9500 Euclid Avenue, Cleveland, OH 44195. E-mail: [email protected]
From the 1Cleveland Clinic, Cleveland, OH; 2Carolinas Medical Center, Charlotte, NC; 3David Geffen School of Medicine at University of California at Los
Angeles, Los Angeles, CA; 4University of Chicago, Chicago IL; 5Consultants in Neurology, Northbrook, IL; and 6Avanir Pharmaceuticals, Aliso Viejo, CA.
VC 2010 American Neurological Association 693

atrial fibrillation and flutter, Q is utilized for conversion as
well as reduction in frequency of relapse. However, Q doses
are often in excess of 1,000 to 1,600mg/day and may affect
cardiac function in ways that include prolongation of the
QTc interval,18 which in turn may be associated with risk of
ventricular arrhythmias.19 In the treatment of PBA, a formal
pharmacokinetic/pharmacodynamic analysis has predicted
that the Q dosage can be reduced to 10mg per capsule (ultra
low dosage, q), as a treatment referred to as DMq, with
maintained efficacy and a decreased potential for proarrhyth-
mic risk.20 The present 12-week trial was designed to evalu-
ate DMq at 30/10mg and at 20/10mg twice daily versus pla-
cebo for treating PBA in patients with ALS or MS. An
additional objective was to determine the pharmacokinetic
parameters of each DMq formulation in a subset of the
study population, as will be reported separately.
Patients and Methods
DesignThis was a 12-week, randomized, double-blind, placebo-con-
trolled, 3-arm, parallel-group study conducted at 60 centers in the
United States and South America between December 2007 and
March 2009. Patients completed screening procedures 1 to 4
weeks before their baseline visit. At the screening visit, those meet-
ing all inclusion/exclusion criteria (see below) were randomized
(1:1:1) to receive placebo, DM 30mg þ Q 10mg (DMq-30), or
DM 20mg þ Q 10mg (DMq-20). For the first treatment week,
patients took a single capsule of study drug in the morning. Dur-
ing weeks 2 through 12, they took study drug once in the morn-
ing and once in the evening. Follow-up visits occurred at 2, 4, 8,
and 12 weeks. In addition, for 1 week prior to baseline and
throughout the trial, patients were required to maintain a diary
recording the daily number of laughing and/or crying episodes
experienced, the medications they took, and any adverse experien-
ces. Patients completing the study were eligible to continue treat-
ment in a 12-week open-label phase with DMq-30 twice daily.
The study protocol was approved by local institutional
review boards or independent ethics committees and was con-
ducted in accordance with Good Clinical Practice Consolidated
Guidance, as approved by the International Conference on Har-
monization (1997), and also with local or national laws and
regulations. Prior to entry, study procedures and risks were
explained to each subject, and written informed consent was
obtained. The study’s randomization code (blocked by center
and by underlying neurological disorder) was computer-gener-
ated, and study drug was supplied in blister packs of identical-
looking capsules. The sponsor, all patients, and all investigators
were blind to treatment identification and allocation.
PatientsFor entry, men or women 18 to 80 years old were required to
have clinically significant PBA, with a score �13 on the Center
for Neurologic Study–Lability Scale (CNS-LS),21 and a diagno-
sis either of ALS (by El Escorial criteria22) within the past 30
months or of MS or probable MS (by McDonald criteria23).
Patients were excluded for any evidence of clinically significant
abnormality on screening electrocardiogram, a family history of
congenital QT-interval prolongation syndrome, a resting respi-
ratory rate outside the range of 12 to 20/min, or a resting diur-
nal oxygen saturation <95%. Patients were also excluded for
any presence or history of major psychiatric disturbance, includ-
ing current symptoms of a depressive disorder (or a score >19
on the Beck Depression Inventory–II [BDI-II]24); major sys-
temic disease or organ dysfunction capable of interfering with
study assessments or putting the patient at risk; and exacerba-
tion of the patient’s underlying ALS or MS within the previous
2 months. Women with childbearing potential were required to
use a medically acceptable form of birth control; pregnant or
lactating women were excluded.
Efficacy AssessmentsThe primary efficacy outcome was a patient’s change from base-
line in the number of PBA episodes (laughing and/or crying)
per day, as recorded in the patient’s diary. Diary data also
yielded, as secondary outcomes, a responder analysis (the pro-
portion of patients with an improvement from baseline PBA
rate, assessed across all degrees of improvement); number of
episode-free days; and occurrence of remission from PBA
(defined by absence of episodes during the study’s final 14
days). Additional secondary outcomes were a patient’s change
from baseline on CNS-LS, which was administered at baseline
and at each follow-up visit, and on BDI-II, the Neuropsychiat-
ric Inventory (NPI),25 and the Medical Outcomes Study 36-
Item Short-Form Health Survey Version 1.0 (SF-36),26 which
were administered at baseline and at 12 weeks.
The CNS-LS is a 7-item self-assessment of PBA severity,
validated for measuring PBA in ALS21 and MS.27 Total scores
range from 7 to 35; a score �13 is the instrument’s range for
clinical PBA. The BDI-II is a 21-item self-assessment of symp-
toms of depression. A total score of 14–19 is considered mild,
20–28 is moderate, and 29–63 is severe. The NPI is a question-
naire covering 12 neuropsychiatric symptom domains; it
FIGURE 1: Subjects’ disposition. DMq-305 dextromethorphancombined with ultra low-dose quinidine at 30/10mg; DMq-205DMq at 20/10mg.
ANNALS of Neurology
694 Volume 68, No. 5
provides a brief, informant-based assessment of neuropsychiatric
symptoms and caregiver distress. The SF-36 is a 36-item
health-status assessment, with subdomains for Physical Func-
tioning, Role-Physical, Bodily Pain, General Health, Vitality,
Social Functioning, Role-Emotional, and Mental Health. Each
of 2 summary scores (Mental Component and Physical Com-
ponent) is standardized so that 50 represents the US general
population norm (for 1998).
Safety/Tolerability AssessmentsAt all visits, vital signs were measured, 12-lead electrocardiography
was performed, and reports of adverse events (AEs) were obtained.
Serious AEs were defined as fatal, life-threatening, significantly
disabling, or requiring hospitalization. Resting diurnal oxygen sat-
uration and nocturnal oxygen saturation were measured (with
pulse oximetry) at screening and at 2 weeks. Resting diurnal oxy-
gen saturation was also measured at 12 weeks. Clinical laboratory
testing was performed at screening and at 4 and 12 weeks.
Statistical AnalysesIn the intent-to-treat population, comprising all randomized
patients, change from baseline in laughing/crying episode rate
was analyzed using longitudinal negative binomial regression,28
with adjustment for baseline rate, diagnosis, and study site. As
a sensitivity analysis, change in episode rate was also assessed by
a nonlongitudinal negative binomial model. In addition, 12-
week change in episode rate was analyzed using the Wilcoxon
rank sum test. For number of episode-free days, a 2-sample t
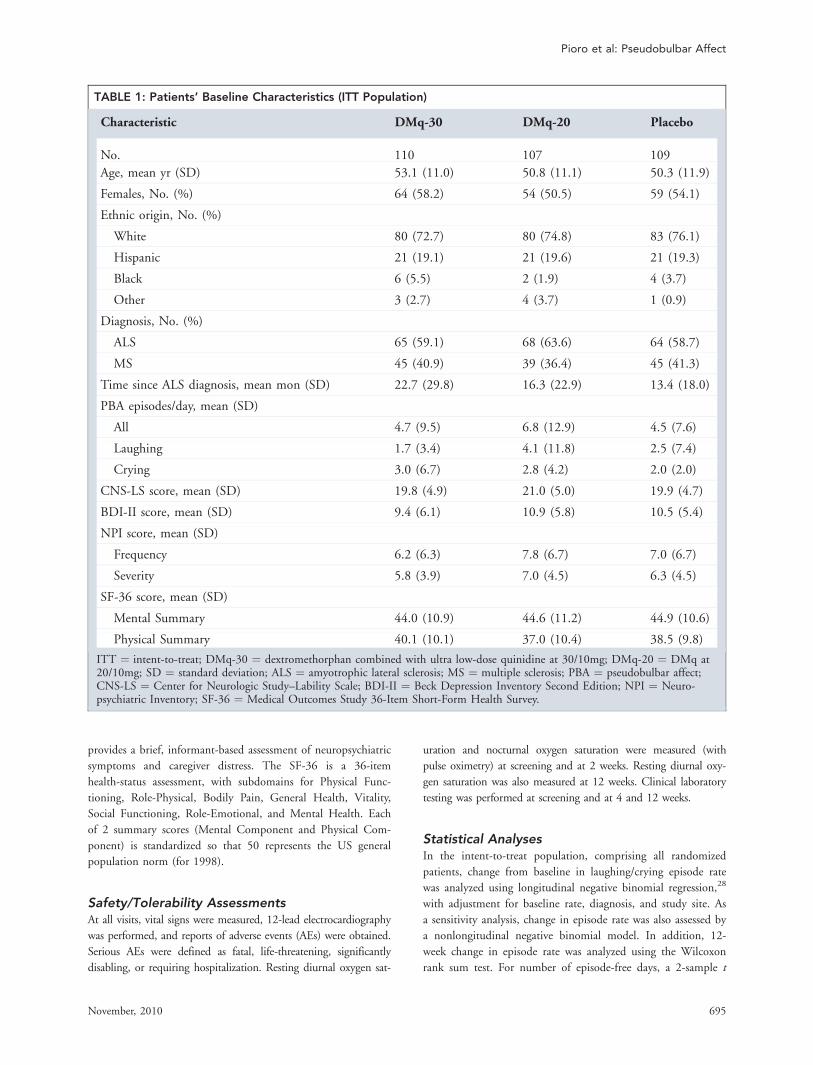
TABLE 1: Patients’ Baseline Characteristics (ITT Population)
Characteristic DMq-30 DMq-20 Placebo
No. 110 107 109
Age, mean yr (SD) 53.1 (11.0) 50.8 (11.1) 50.3 (11.9)
Females, No. (%) 64 (58.2) 54 (50.5) 59 (54.1)
Ethnic origin, No. (%)
White 80 (72.7) 80 (74.8) 83 (76.1)
Hispanic 21 (19.1) 21 (19.6) 21 (19.3)
Black 6 (5.5) 2 (1.9) 4 (3.7)
Other 3 (2.7) 4 (3.7) 1 (0.9)
Diagnosis, No. (%)
ALS 65 (59.1) 68 (63.6) 64 (58.7)
MS 45 (40.9) 39 (36.4) 45 (41.3)
Time since ALS diagnosis, mean mon (SD) 22.7 (29.8) 16.3 (22.9) 13.4 (18.0)
PBA episodes/day, mean (SD)
All 4.7 (9.5) 6.8 (12.9) 4.5 (7.6)
Laughing 1.7 (3.4) 4.1 (11.8) 2.5 (7.4)
Crying 3.0 (6.7) 2.8 (4.2) 2.0 (2.0)
CNS-LS score, mean (SD) 19.8 (4.9) 21.0 (5.0) 19.9 (4.7)
BDI-II score, mean (SD) 9.4 (6.1) 10.9 (5.8) 10.5 (5.4)
NPI score, mean (SD)
Frequency 6.2 (6.3) 7.8 (6.7) 7.0 (6.7)
Severity 5.8 (3.9) 7.0 (4.5) 6.3 (4.5)
SF-36 score, mean (SD)
Mental Summary 44.0 (10.9) 44.6 (11.2) 44.9 (10.6)
Physical Summary 40.1 (10.1) 37.0 (10.4) 38.5 (9.8)
ITT ¼ intent-to-treat; DMq-30 ¼ dextromethorphan combined with ultra low-dose quinidine at 30/10mg; DMq-20 ¼ DMq at20/10mg; SD ¼ standard deviation; ALS ¼ amyotrophic lateral sclerosis; MS ¼ multiple sclerosis; PBA ¼ pseudobulbar affect;CNS-LS ¼ Center for Neurologic Study–Lability Scale; BDI-II ¼ Beck Depression Inventory Second Edition; NPI ¼ Neuro-psychiatric Inventory; SF-36 ¼ Medical Outcomes Study 36-Item Short-Form Health Survey.
Pioro et al: Pseudobulbar Affect
November, 2010 695
test was used. Changes on CNS-LS, SF-36, BDI-II, and NPI
were analyzed with analysis of covariance, using the method of
Frison and Pocock.29 Baseline value, study site, and diagnosis
were covariates. Observed cases were used in the sensitivity
analyses, with no imputation for missing data. All analyses were
2-sided hypothesis tests at the 0.05 significance level.
The safety population comprised all patients who took at
least 1 dose of study medication. Their AE rates, for types reported
by �5% of patients in any treatment group, were compared among
groups, and mean change in resting nocturnal oxygen saturation
(from baseline to day 15) was assessed by 2-sample t test.
Sample Size CalculationBased on PBA episode rates in previous studies of DMq for
PBA in ALS13 and in MS,14 a sample size of approximately 90
patients (60 with ALS and 30 with MS) per treatment group
was planned. This size was expected to be sufficient to detect a
36% reduction in mean episode rate for DMq-30 vs placebo
with at least 90% power. The study was not powered to test a
difference between DMq-30 and DMq-20.
Results
SubjectsIn all, 332 patients were screened, and among them 326
were randomized, 110 to DMq-30, 107 to DMq-20, and
109 to placebo (Fig 1). The main reasons for screening
failure were unwillingness to discontinue disallowed medi-
cations, CNS-LS score <13, and BDI-II score >19. In all,
283 patients (86.8% of 326) completed the study, includ-
ing 101 (91.8% of 110) in the DMq-30 group, 88
(82.2% of 107) in the DMq-20 group, and 94 (86.2% of
109) in the placebo group. Demographically and in base-
line PBA characteristics, the treatment groups were well
matched (Table 1), except for a higher baseline PBA epi-
sode rate in the DMq-20 group than in the other groups,
and a longer time since ALS diagnosis in the DMq-30
group. At entry, no patient had clinical depression.
EfficacyOver the course of the study, all 3 groups showed sub-
stantial reduction in daily PBA episode rates relative to
baseline. However, the reduction in daily PBA episode
rate was significantly greater in each of the DMq groups
than in the placebo group. By longitudinal negative bino-
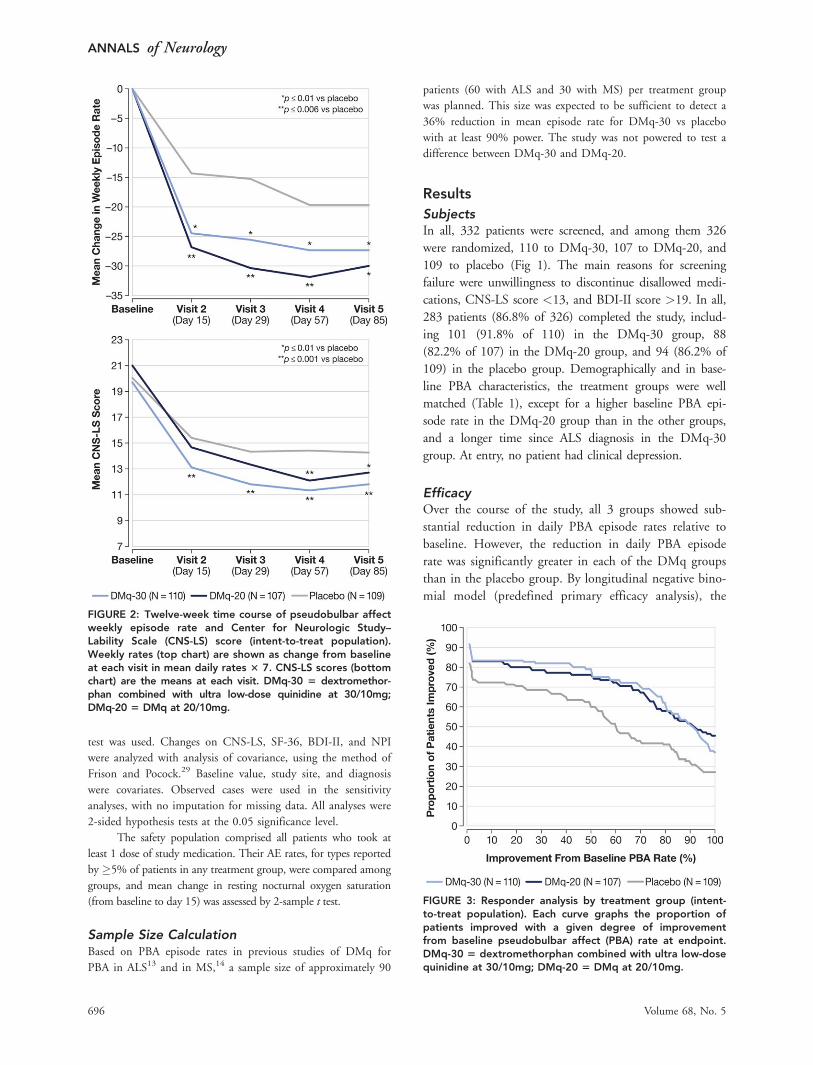
mial model (predefined primary efficacy analysis), the
FIGURE 2: Twelve-week time course of pseudobulbar affectweekly episode rate and Center for Neurologic Study–Lability Scale (CNS-LS) score (intent-to-treat population).Weekly rates (top chart) are shown as change from baselineat each visit in mean daily rates 3 7. CNS-LS scores (bottomchart) are the means at each visit. DMq-30 5 dextromethor-phan combined with ultra low-dose quinidine at 30/10mg;DMq-20 5 DMq at 20/10mg.
FIGURE 3: Responder analysis by treatment group (intent-to-treat population). Each curve graphs the proportion ofpatients improved with a given degree of improvementfrom baseline pseudobulbar affect (PBA) rate at endpoint.DMq-30 5 dextromethorphan combined with ultra low-dosequinidine at 30/10mg; DMq-20 5 DMq at 20/10mg.
ANNALS of Neurology
696 Volume 68, No. 5
treatment effect in each DMq group over that seen in
the placebo group was an incremental reduction in PBA
episode rate of 46.9% (p < 0.0001) for DMq-30 com-
pared to placebo and 49.0% (p < 0.0001) for DMq-20
compared to placebo. By nonlongitudinal negative bino-
mial model with constant dispersion (predefined efficacy
sensitivity analysis), the additional improvement over pla-
cebo at both dosage levels was also statistically significant
(p < 0.0001 and p¼ 0.0370, respectively). The 12-week
mean change in daily episode rate was �4.1 for DMq-30
and �3.9 for DMq-20, vs �3.0 for placebo (p¼ 0.0099
and p¼ 0.0048, respectively). Weekly rates (daily rates �7) showed significant decrease at all time points assessed,
beginning with day 15 (Fig 2, top).
Among secondary outcomes, the 12-week mean
reduction from baseline CNS-LS score was significantly
greater at both DMq dosage levels than for placebo
(Table 2), and for DMq-30, the mean reduction was sig-
nificant at all time points assessed, beginning with day
15 (see Fig 2, bottom). Among secondary outcomes
derived from diary data, the proportion of patients with
an improvement from their baseline PBA rate was higher
for both DMq-30 and DMq-20 than for placebo, across
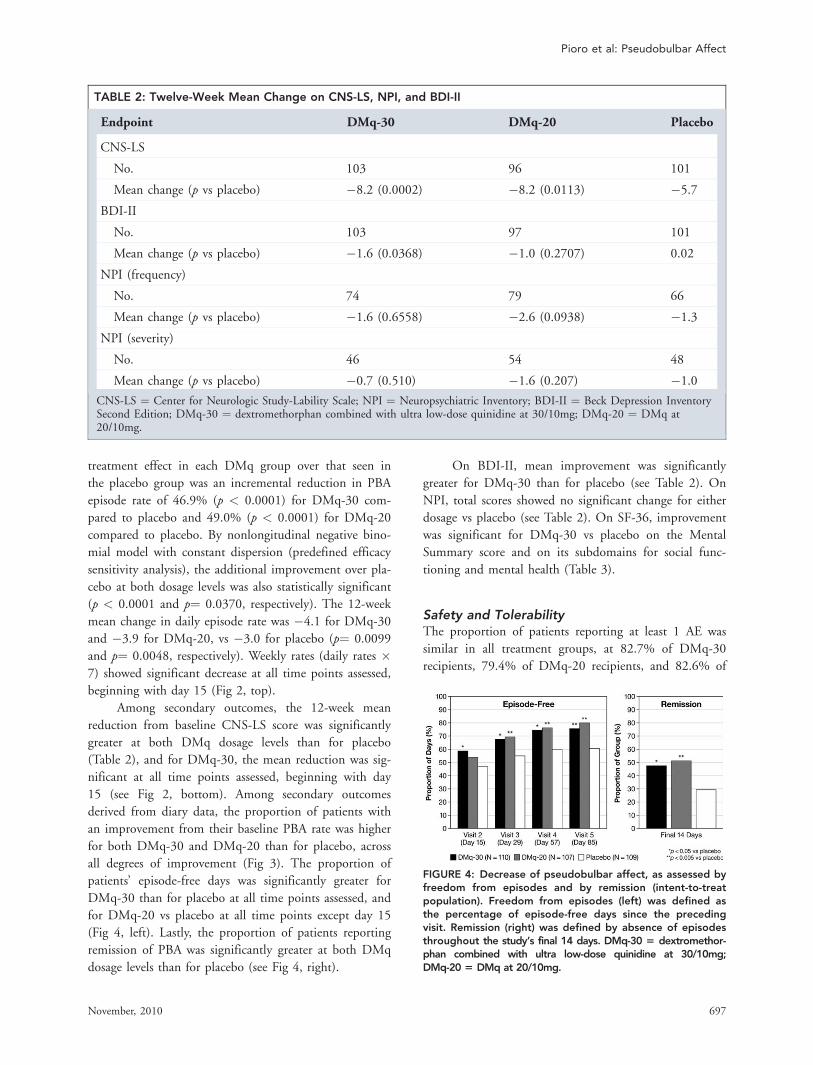
all degrees of improvement (Fig 3). The proportion of
patients’ episode-free days was significantly greater for
DMq-30 than for placebo at all time points assessed, and
for DMq-20 vs placebo at all time points except day 15
(Fig 4, left). Lastly, the proportion of patients reporting
remission of PBA was significantly greater at both DMq
dosage levels than for placebo (see Fig 4, right).
On BDI-II, mean improvement was significantly
greater for DMq-30 than for placebo (see Table 2). On
NPI, total scores showed no significant change for either
dosage vs placebo (see Table 2). On SF-36, improvement
was significant for DMq-30 vs placebo on the Mental
Summary score and on its subdomains for social func-
tioning and mental health (Table 3).
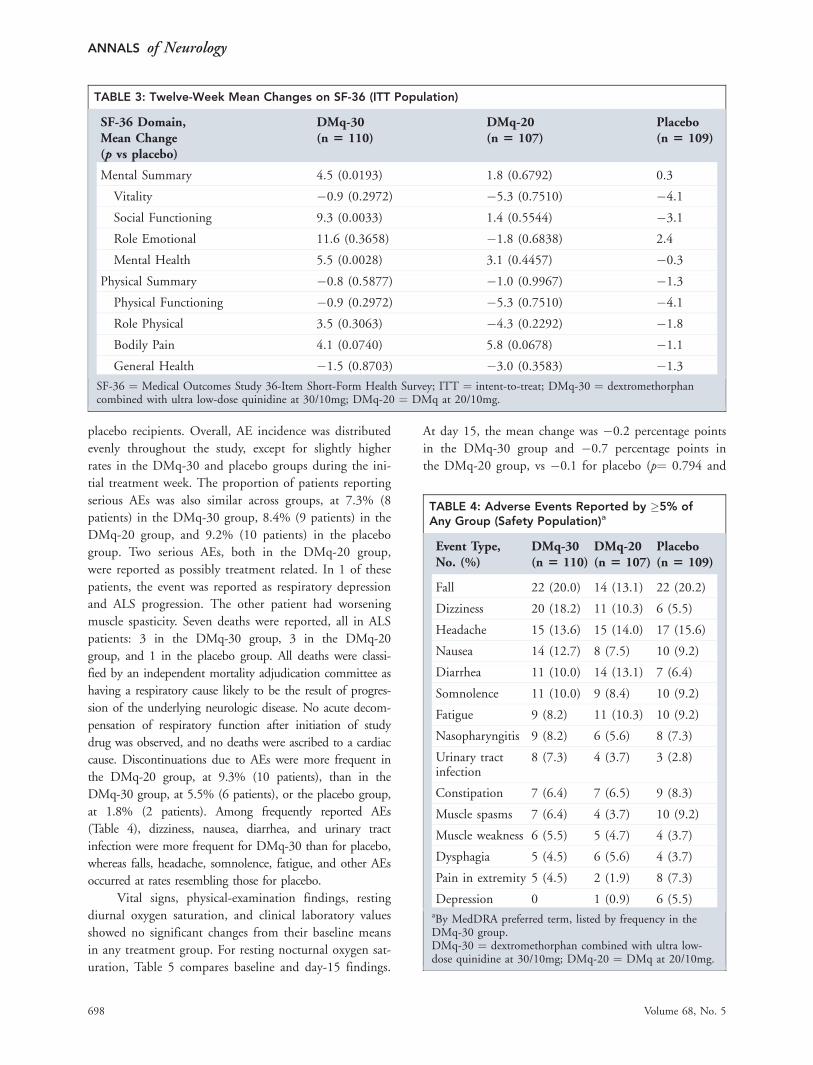
Safety and TolerabilityThe proportion of patients reporting at least 1 AE was
similar in all treatment groups, at 82.7% of DMq-30
recipients, 79.4% of DMq-20 recipients, and 82.6% of
TABLE 2: Twelve-Week Mean Change on CNS-LS, NPI, and BDI-II
Endpoint DMq-30 DMq-20 Placebo
CNS-LS
No. 103 96 101
Mean change (p vs placebo) �8.2 (0.0002) �8.2 (0.0113) �5.7BDI-II
No. 103 97 101
Mean change (p vs placebo) �1.6 (0.0368) �1.0 (0.2707) 0.02
NPI (frequency)
No. 74 79 66
Mean change (p vs placebo) �1.6 (0.6558) �2.6 (0.0938) �1.3NPI (severity)
No. 46 54 48
Mean change (p vs placebo) �0.7 (0.510) �1.6 (0.207) �1.0CNS-LS ¼ Center for Neurologic Study-Lability Scale; NPI ¼ Neuropsychiatric Inventory; BDI-II ¼ Beck Depression InventorySecond Edition; DMq-30 ¼ dextromethorphan combined with ultra low-dose quinidine at 30/10mg; DMq-20 ¼ DMq at20/10mg.
FIGURE 4: Decrease of pseudobulbar affect, as assessed byfreedom from episodes and by remission (intent-to-treatpopulation). Freedom from episodes (left) was defined asthe percentage of episode-free days since the precedingvisit. Remission (right) was defined by absence of episodesthroughout the study’s final 14 days. DMq-30 5 dextromethor-phan combined with ultra low-dose quinidine at 30/10mg;DMq-20 5 DMq at 20/10mg.
Pioro et al: Pseudobulbar Affect
November, 2010 697
placebo recipients. Overall, AE incidence was distributed
evenly throughout the study, except for slightly higher
rates in the DMq-30 and placebo groups during the ini-
tial treatment week. The proportion of patients reporting
serious AEs was also similar across groups, at 7.3% (8
patients) in the DMq-30 group, 8.4% (9 patients) in the
DMq-20 group, and 9.2% (10 patients) in the placebo
group. Two serious AEs, both in the DMq-20 group,
were reported as possibly treatment related. In 1 of these
patients, the event was reported as respiratory depression
and ALS progression. The other patient had worsening
muscle spasticity. Seven deaths were reported, all in ALS
patients: 3 in the DMq-30 group, 3 in the DMq-20
group, and 1 in the placebo group. All deaths were classi-
fied by an independent mortality adjudication committee as
having a respiratory cause likely to be the result of progres-
sion of the underlying neurologic disease. No acute decom-
pensation of respiratory function after initiation of study
drug was observed, and no deaths were ascribed to a cardiac
cause. Discontinuations due to AEs were more frequent in
the DMq-20 group, at 9.3% (10 patients), than in the
DMq-30 group, at 5.5% (6 patients), or the placebo group,
at 1.8% (2 patients). Among frequently reported AEs
(Table 4), dizziness, nausea, diarrhea, and urinary tract
infection were more frequent for DMq-30 than for placebo,
whereas falls, headache, somnolence, fatigue, and other AEs
occurred at rates resembling those for placebo.
Vital signs, physical-examination findings, resting
diurnal oxygen saturation, and clinical laboratory values
showed no significant changes from their baseline means
in any treatment group. For resting nocturnal oxygen sat-
uration, Table 5 compares baseline and day-15 findings.
At day 15, the mean change was �0.2 percentage points
in the DMq-30 group and �0.7 percentage points in
the DMq-20 group, vs �0.1 for placebo (p¼ 0.794 and
TABLE 3: Twelve-Week Mean Changes on SF-36 (ITT Population)
SF-36 Domain,Mean Change(p vs placebo)
DMq-30(n 5 110)
DMq-20(n 5 107)
Placebo(n 5 109)
Mental Summary 4.5 (0.0193) 1.8 (0.6792) 0.3
Vitality �0.9 (0.2972) �5.3 (0.7510) �4.1Social Functioning 9.3 (0.0033) 1.4 (0.5544) �3.1Role Emotional 11.6 (0.3658) �1.8 (0.6838) 2.4
Mental Health 5.5 (0.0028) 3.1 (0.4457) �0.3Physical Summary �0.8 (0.5877) �1.0 (0.9967) �1.3Physical Functioning �0.9 (0.2972) �5.3 (0.7510) �4.1Role Physical 3.5 (0.3063) �4.3 (0.2292) �1.8Bodily Pain 4.1 (0.0740) 5.8 (0.0678) �1.1General Health �1.5 (0.8703) �3.0 (0.3583) �1.3
SF-36 ¼ Medical Outcomes Study 36-Item Short-Form Health Survey; ITT ¼ intent-to-treat; DMq-30 ¼ dextromethorphancombined with ultra low-dose quinidine at 30/10mg; DMq-20 ¼ DMq at 20/10mg.
TABLE 4: Adverse Events Reported by �5% ofAny Group (Safety Population)a
Event Type,No. (%)
DMq-30(n 5 110)
DMq-20(n 5 107)
Placebo(n 5 109)
Fall 22 (20.0) 14 (13.1) 22 (20.2)
Dizziness 20 (18.2) 11 (10.3) 6 (5.5)
Headache 15 (13.6) 15 (14.0) 17 (15.6)
Nausea 14 (12.7) 8 (7.5) 10 (9.2)
Diarrhea 11 (10.0) 14 (13.1) 7 (6.4)
Somnolence 11 (10.0) 9 (8.4) 10 (9.2)
Fatigue 9 (8.2) 11 (10.3) 10 (9.2)
Nasopharyngitis 9 (8.2) 6 (5.6) 8 (7.3)
Urinary tractinfection
8 (7.3) 4 (3.7) 3 (2.8)
Constipation 7 (6.4) 7 (6.5) 9 (8.3)
Muscle spasms 7 (6.4) 4 (3.7) 10 (9.2)
Muscle weakness 6 (5.5) 5 (4.7) 4 (3.7)
Dysphagia 5 (4.5) 6 (5.6) 4 (3.7)
Pain in extremity 5 (4.5) 2 (1.9) 8 (7.3)
Depression 0 1 (0.9) 6 (5.5)aBy MedDRA preferred term, listed by frequency in theDMq-30 group.DMq-30 ¼ dextromethorphan combined with ultra low-dose quinidine at 30/10mg; DMq-20 ¼ DMq at 20/10mg.
ANNALS of Neurology
698 Volume 68, No. 5
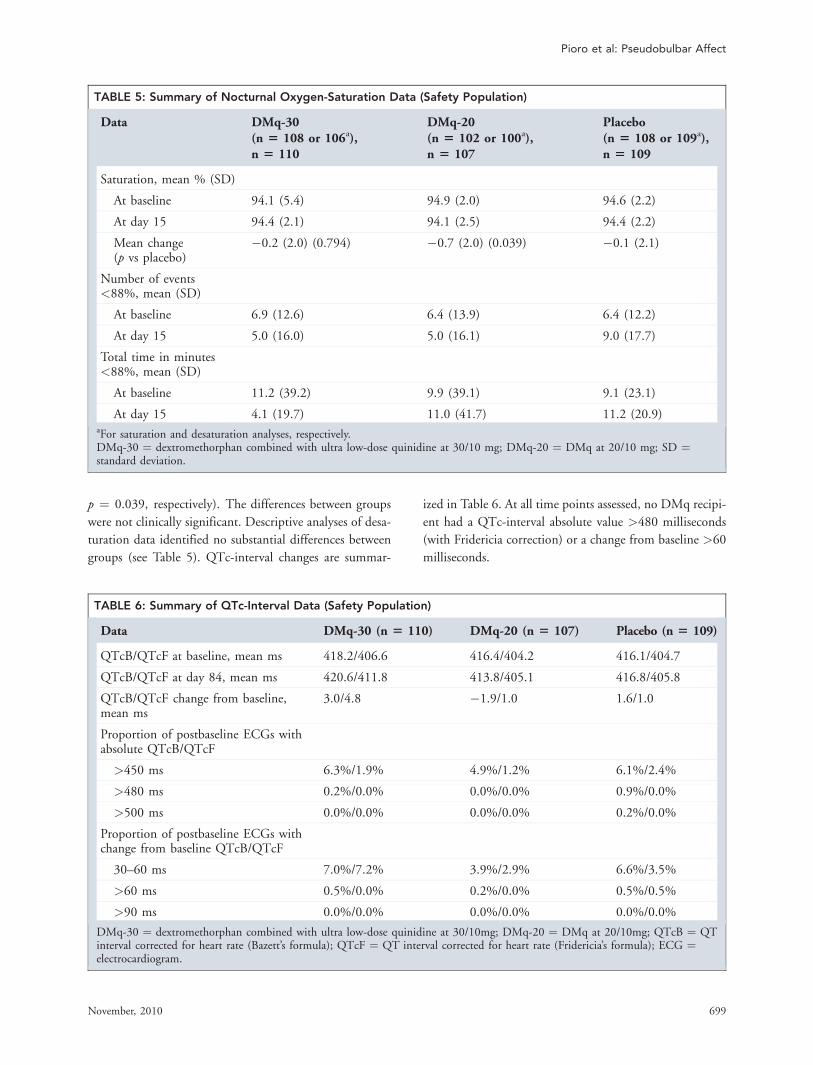
p ¼ 0.039, respectively). The differences between groups
were not clinically significant. Descriptive analyses of desa-
turation data identified no substantial differences between
groups (see Table 5). QTc-interval changes are summar-
ized in Table 6. At all time points assessed, no DMq recipi-
ent had a QTc-interval absolute value >480 milliseconds
(with Fridericia correction) or a change from baseline >60
milliseconds.
TABLE 5: Summary of Nocturnal Oxygen-Saturation Data (Safety Population)
Data DMq-30(n 5 108 or 106a),n 5 110
DMq-20(n 5 102 or 100a),n 5 107
Placebo(n 5 108 or 109a),n 5 109
Saturation, mean % (SD)
At baseline 94.1 (5.4) 94.9 (2.0) 94.6 (2.2)
At day 15 94.4 (2.1) 94.1 (2.5) 94.4 (2.2)
Mean change(p vs placebo)
�0.2 (2.0) (0.794) �0.7 (2.0) (0.039) �0.1 (2.1)
Number of events<88%, mean (SD)
At baseline 6.9 (12.6) 6.4 (13.9) 6.4 (12.2)
At day 15 5.0 (16.0) 5.0 (16.1) 9.0 (17.7)
Total time in minutes<88%, mean (SD)
At baseline 11.2 (39.2) 9.9 (39.1) 9.1 (23.1)
At day 15 4.1 (19.7) 11.0 (41.7) 11.2 (20.9)aFor saturation and desaturation analyses, respectively.DMq-30 ¼ dextromethorphan combined with ultra low-dose quinidine at 30/10 mg; DMq-20 ¼ DMq at 20/10 mg; SD ¼standard deviation.
TABLE 6: Summary of QTc-Interval Data (Safety Population)
Data DMq-30 (n 5 110) DMq-20 (n 5 107) Placebo (n 5 109)
QTcB/QTcF at baseline, mean ms 418.2/406.6 416.4/404.2 416.1/404.7
QTcB/QTcF at day 84, mean ms 420.6/411.8 413.8/405.1 416.8/405.8
QTcB/QTcF change from baseline,mean ms
3.0/4.8 �1.9/1.0 1.6/1.0
Proportion of postbaseline ECGs withabsolute QTcB/QTcF
>450 ms 6.3%/1.9% 4.9%/1.2% 6.1%/2.4%
>480 ms 0.2%/0.0% 0.0%/0.0% 0.9%/0.0%
>500 ms 0.0%/0.0% 0.0%/0.0% 0.2%/0.0%
Proportion of postbaseline ECGs withchange from baseline QTcB/QTcF
30–60 ms 7.0%/7.2% 3.9%/2.9% 6.6%/3.5%
>60 ms 0.5%/0.0% 0.2%/0.0% 0.5%/0.5%
>90 ms 0.0%/0.0% 0.0%/0.0% 0.0%/0.0%
DMq-30 ¼ dextromethorphan combined with ultra low-dose quinidine at 30/10mg; DMq-20 ¼ DMq at 20/10mg; QTcB ¼ QTinterval corrected for heart rate (Bazett’s formula); QTcF ¼ QT interval corrected for heart rate (Fridericia’s formula); ECG ¼electrocardiogram.
Pioro et al: Pseudobulbar Affect
November, 2010 699

Discussion
In this large, double-blind, placebo-controlled study,
both dosage levels of DMq were significantly superior to
placebo for reducing PBA episode frequency among
patients with underlying ALS or MS, as assessed by lon-
gitudinal and nonlongitudinal statistical models and also
by mean change in daily PBA episode rate. At both dos-
age levels, DMq also significantly reduced the severity of
PBA, as represented by CNS-LS score. For reduction in
episode rate, a prespecified responder analysis showed, at
both dosage levels, a substantial difference from placebo
across all degrees of improvement, despite a strong pla-
cebo effect (resembling those seen in previous studies13).
The differences between DMq and placebo included, at
both dosage levels, a significantly higher likelihood of
PBA remission on DMq than on placebo, suggesting that
for large proportions of patients, the active treatment’s
amelioration of PBA may be marked.
Numerically, the responses to the higher DMq dos-
age were more robust than those to the lower dosage in
several ways, including an earlier improvement in CNS-
LS score, an earlier time to significant difference vs pla-
cebo in number of episode-free days, and a slightly
greater 12-week mean change vs placebo in PBA daily
episode rate. At the higher dosage, DMq was also associ-
ated with significant improvement in mental-health
measures, by BDI-II and SF-36. Because none of the
subjects in this study was clinically depressed, and
because PBA can result in substantial reduction of quality
of life,11 this improvement may have been in well-being.
Specific improvements on SF-36 subdomains for social
functioning and mental health are further evidence that
the social and psychological disability associated with
PBA may have been reduced. However, the possibility
that DMq may have direct antidepressant properties can-
not be excluded, and would require further study. Over-
all, the efficacy reported for DMq containing Q at ultra
low dosage—10mg per capsule—resembled the benefits
reported by measures including CNS-LS scores and PBA
episode counts in studies of DMq in its original formula-
tion, in which the Q content per capsule was 30mg.13,14
In the present study, both dosage levels were safe.
In particular, cardiovascular safety was satisfactory, with
mild QTc prolongation and no proarrhythmic events.
Respiratory findings appeared to be consistent with ALS
progression. However, physicians should always exercise
caution in managing a patient population that has com-
promised respiratory function. Both dosage levels were
also well tolerated, with only 13% of DMq recipients
discontinuing during 12 weeks of double-blind treatment.
The overall discontinuation rate was lower for DMq-30, at
8%, than for DMq-20, at 18%. In studies of DMQ as
originally formulated, with DM at 30mg per capsule and
Q at 30mg, the 12-week discontinuation rate had been
much higher, at 28% in ALS patients13 and 25% in MS
patients.14 The implication is that the improved tolerability
demonstrated in the present study may reflect ultra low
dosing of Q. Usage of dose escalation (with once-daily
dosing during week 1) may also have contributed.
A body of published evidence suggests that PBA
may be ameliorated pharmacologically,1 but the trials
assessing current agents, all of which are being utilized
off-label, have limitations. In 1979, dopaminergic treat-
ment, specifically L-dopa, was reported to be effective for
‘‘emotional incontinence,’’30 but in a follow-up uncon-
trolled study of L-dopa or amantadine, only 10 of 25
recipients responded.31 Since then, reports have centered
on antidepressants, notably tricyclic agents (eg, amitripty-
line32 or nortriptyline33) and selective serotonin reuptake
inhibitors (eg, fluoxetine,34 citalopram,35,36 paroxetine,36
or sertraline37). Overall, the trials have been hampered
by small size (12 to 28 subjects, among those referenced
above) and by methodological problems, such as their
definitions of PBA improvement. Substantial placebo
effects, as demonstrated in the present study, make
uncontrolled findings all the more difficult to interpret.
In brief, well-controlled data to support current options
are scarce, and no option is currently approved by the
US Food and Drug Administration. In addition, antide-
pressants are associated with incompletely elucidated AE
profiles (including QT-interval prolongation19).
In interpreting the present study’s findings, the trial’s
limitations should be taken into account. Because its subjects
were carefully selected, the findings should be generalized to
a broader spectrum of PBA with caution. The study
required, for instance, a baseline CNS-LS score of at least
13. Accordingly, the effects of DMq-30 or DMq-20 on
milder forms of PBA are unknown. In addition, the study
enrolled only patients with underlying ALS or MS. Because
the pathophysiologic mechanisms causing PBA are probably
similar regardless of the underlying CNS pathology, DMq
will likely be effective in reducing symptoms of PBA arising
in various brain disease or injury states, much in the same
way that antispastic medication reduces spasticity, irrespective
of the underlying condition. Even so, additional studies of
the effect of DMq on PBA in various neurological disorders
could provide enhanced safety, efficacy, and health outcome
insight. Hence, further clinical studies of PBA are warranted.
Nevertheless, the present study represents the largest
and longest double-blind, randomized, placebo-controlled
trial of DMq conducted to date in PBA, and also the
first to test DMq in PBA patients at ultra low Q dosage.
Its findings expand the clinical evidence that with
ANNALS of Neurology
700 Volume 68, No. 5

satisfactory safety and high tolerability, DMq markedly
reduces PBA frequency and severity, decreasing the con-
dition’s detrimental impact on a patient’s life.
Acknowledgment
This study was supported by Avanir Pharmaceuticals.
Potential Conflicts of Interest
E.P.P. has received research support and compensation for
consulting from Avanir Pharmaceuticals. B.R.B. has
received compensation for consulting from Avanir Phar-
maceuticals, Bayer Healthcare Pharmaceuticals, Biogen
Idec, Genentech, and Teva Neuroscience, and has received
research support from Avanir Pharmaceuticals, Biogen
Idec, National Institutes of Neurological Disorders and
Stroke Clinical Research Consortium, Novartis, and Teva
Neuroscience. J.C. has received compensation for consult-
ing from Abbott, Acadia, Accera, ADAMAS, Astellas,
Avanir Pharmaceuticals, Bristol-Myers Squibb, CoMentis,
Eisai, Elan, EnVivo, Forest, GlaxoSmithKline, Janssen,
Lilly, Lundbeck, Medivation, Merck, Merz, Myriad,
Neuren, Novartis, Pfizer, Prana, Schering Plough, Sonexa,
Takeda, Toyama, and Wyeth, and owns the copyright of
the Neuropsychiatric Inventory. A.H. and R.K. are
employees of and own stock options in Avanir Pharma-
ceuticals. R.S. has received compensation for consulting
from Teva and Avanir Pharmaceuticals and owns 300
shares of common stock in Johnson & Johnson Corpora-
tion. R.A.T. is a statistical consultant to Avanir
Pharmaceuticals, and has been an expert witness on behalf
of Eli Lilly, Forest Laboratories, Wyeth, Otsuka, Glaxo-
SmithKline, and Hoffmann-LaRoche. D.W. has received
compensation for consulting from Teva Neurosciences, Pfizer,
Serono, Acorda Therapeutics, GlaxoSmithKline, Avanir
Pharmaceuticals, and Eli Lilly, and has received research
support from Biogen Idec, Serono, Pfizer, Teva, UCB
Pharma, PDL BioPharma, BioMS, Sanofi-Aventis, Opexa
Therapeutics, Genzyme, GlaxoSmithKline, XenoPort, Avanir
Pharmaceuticals, Eli Lilly, and Shire Laboratories.
Appendix
AVP-923 in PBA Trial Investigators
USA. Carmel Armon, MD (Baystate Medical Center);
Richard Bedlack, MD (Duke University); Kevin Boylan,
MD (Mayo Clinic Jacksonville); Elena Bravver, MD
(Carolinas Medical Center); Andrea Corse, MD (Johns
Hopkins University); Merit E. Cudkowicz, MD, MSc
(Massachusetts General Hospital); Dennis Dietrich, MD
(Advanced Neurology Specialists); Peter Donofrio, MD
(Vanderbilt Neurology, Medical Center North); David
Ginsburg, MD (University of Nevada School of Medi-
cine); Jonathan Glass, MD (Emory University); Michael
Graves, MD (University of California at Los Angeles
School of Medicine); Laurie Gutmann, MD (West Vir-
ginia University School of Medicine); Bianca Weinstock-
Guttman, MD (Buffalo General Hospital); Ghazala
Hayat, MD (Saint Louis University); Daragh Heitzman,
MD (Texas Neurology, PA); Catherine Lomen-Hoerth,
MD (Amyotrophic Lateral Sclerosis Center at University
of California at San Francisco); Carlayne E. Jackson, MD
(University of Texas Health Science Center); Edward
Kasarskis, MD (University of Kentucky); Jason Kellogg,
MD (South Coast Clinical Trials, Inc.); Jonathan Licht,
MD (Coordinated Clinical Research); Ann Little, MD
(University of Michigan Health System); Jau-Shin Lou,
MD (Oregon Health Science University); Catherine
Madison, MD (California Pacific Medical Center); Leo
McCluskey, MD (University of Pennsylvania Health Sys-
tem); April McVey, MD (University of Kansas Medical
Center, Landon Center on Aging); Hiroshi Mitsumoto,
MD (Columbia Presbyterian Center); Tahseen Mozaffar,
MD (University of California at Irvine); Steven Nash,
MD (Ohio State University Medical Center); Daniel
Newman, MD (Henry Ford Hospital); Oliver Ni, MD
(Dean Foundation); Gary Pattee, MD (Neurology Asso-
ciates, Inc.); Terry Heiman-Patterson, MD (Drexel Uni-
versity College of Medicine); Erik Pioro, MD, PhD
(Cleveland Clinic Foundation); Yvonne Rollins, MD,
PhD (University of Colorado, Denver)(replacing Dr Bjorn
Oskarsson); Jiong Shi, MD (Barrow Neurological Institute
of St. Joseph’s Hospital and Medical Center) (replacing Dr
Timothy Vollmer); Ericka Simpson, MD (Methodist Hos-
pital Research Institute); Mark Sivak, MD (Mount Sinai
Medical Center)(replacing Dr Dale Lang); Kumaraswamy
Sivakumar, MD (Neuromuscular Research Center); Brian
Steingo, MD (Neurological Associates); Robert Sufit, MD
(Northwestern University); Rup Tandan, MD (University
of Vermont, College of Medicine); Alberto Vasquez, MD
(Suncoast Neuroscience Associates, Inc.); Ashok Verma,
MD (University of Miami); Joseph Weissman, MD (Neu-
rology Specialists of Decatur Research Center); Ben Wil-
liams, MD (Texas Tech University)(replacing Dr Randolph
Schiffer); James Wymer, MD (Upstate Clinical Research,
LLC); Daniel Wynn, MD (Consultants in Neurology, Ltd.).
SOUTH AMERICA. Alberto Francisco Rodriguez
Alfici, MD (Instituto Medico Rodriguez Alfici); Dago-
berto Callegaro, MD (Faculdade de Medicina da Uni-
versidade Sao Paulo); Adriana Josefa Carra, MD
(Hospital Britanico Buenos Aires); Edgardo Cristiano,
MD (Hospital Italiano de Buenos Aires); Jefferson
Gomes Fernandes, MD (Hospital Moinhos de Vento);
Pioro et al: Pseudobulbar Affect
November, 2010 701

Maria Lucia Brito Ferreira, MD (Hospital da Restaura-
cao, Secretaria Estadual da Saude); Soniza Vieira Alves
Leon, MD (Hospital Universitario Clementino Fraga
Filho); Geraldine Luetic, MD (Instituto Neurociencias
Rosario); Antonio Pereira Gomes Neto, MD (Santa
Casa de Misericordia de Belo Horizonte); Martin Alejan-
dro Nogues, MD (Fundacion para la Lucha de las Enfer-
medades Neurologicas de la Infancia); Miguel Angel
Pagano, MD (Fundacion contra las Enfermedades Neuro-
logicas del Envejecimiento); Gustavo Martin Petracca,
MD (Instituto de Neurociencias Buenos Aires); Roberto
Daniel Rey, MD (Instituto Argentino Investigacion Neu-
rologica); Rosana Herminia Scola, MD (Hospital de
Clınicas, UFPR); Adriana Nora Tarulla, MD (Policlınico
Bancario); Andres Marıa Villa, MD (Hospital General de
Agudos Ramos Mejıa); Carlos Alejandro Vrech, MD
(Hospital Militar Regional de Cordoba).
References1. Dark FL, McGrath JJ, Ron MA. Pathological laughing and crying.
Aust N Z J Psychiatry 1996;30:472–479.
2. House A, Dennis M, Molyneux A, et al. Emotionalism after stroke.BMJ 1989;298:991–994.
3. Brooks N. Personality change after severe head injury. Acta Neu-rochir Suppl (Wien) 1988;44:59–64.
4. Starkstein SE, Migliorelli R, Teson A, et al. Prevalence and clinicalcorrelates of pathological affective display in Alzheimer’s disease.J Neurol Neurosurg Psychiatry 1995;59:55–60.
5. Jackson CE, Bryan WW. Amyotrophic lateral sclerosis. Semin Neu-rol 1998;18:27–39.
6. Miller RG, Rosenberg JA, Gelinas DF, et al. Practice parameter:the care of the patient with amyotrophic lateral sclerosis (an evi-dence-based review): report of the Quality Standards Subcommit-tee of the American Academy of Neurology: ALS PracticeParameters Task Force. Neurology 1999;52:1311–1323.
7. Miller RG, Jackson CE, Kasarskis EJ, et al. Practice parameterupdate: The care of the patient with amyotrophic lateral sclerosis:multidisciplinary care, symptom management, and cognitive/be-havioral impairment (an evidence-based review): report of theQuality Standards Subcommittee of the American Academy ofNeurology. Neurology 2009;73:1227–1233.
8. Feinstein A, Feinstein K, Gray T, O’Connor P. Prevalence and neu-robehavioral correlates of pathological laughing and crying in mul-tiple sclerosis. Arch Neurol 1997;54:1116–1121.
9. Tateno A, Jorge RE, Robinson RG. Pathological laughing and cry-ing following traumatic brain injury. J Neuropsychiatry Clin Neuro-sci 2004;16:426–434.
10. Gallagher JP. Pathologic laughter and crying in ALS: a search fortheir origin. Acta Neurol Scand 1989;80:114–117.
11. Schiffer R, Pope LE. Review of pseudobulbar affect including a noveland potential therapy. J Neuropsychiatry Clin Neurosci 2005;17:447–454.
12. Parvizi J, Arciniegas DB, Bernardini GL, et al. Diagnosis and managementof pathological laughter and crying. Mayo Clin Proc 2006;81:1482–1486.
13. Brooks BR, Thisted RA, Appel SH, et al. Treatment of pseudobul-bar affect in ALS with dextromethorphan/quinidine: a randomizedtrial. Neurology 2004;63:1364–1370.
14. Panitch HS, Thisted RA, Smith RA, et al. Randomized, controlledtrial of dextromethorphan/quinidine for pseudobulbar affect inmultiple sclerosis. Ann Neurol 2006;59:780–787.
15. Choi DW. Dextrorphan and dextromethorphan attenuate gluta-mate neurotoxicity. Brain Res 1987;403:333–336.
16. Musacchio JM, Klein M, Paturzo JJ. Effects of dextromethorphan siteligands and allosteric modifiers on the binding of (þ)-[3H]3-(-3-hydroxyphenyl)-N-(1-propyl)piperidine. Mol Pharmacol 1989;35:1–5.
17. Zhang Y, Britto MR, Valderhaug KL, et al. Dextromethorphan:enhancing its systemic availability by way of low-dose quinidine-mediated inhibition of cytochrome P4502D6. Clin Pharmacol Ther1992;51:647–655.
18. Holford NH, Coates PE, Guentert TW, et al. The effect of quini-dine and its metabolites on the electrocardiogram and systolictime intervals: concentration-effect relationships. Br J Clin Pharma-col 1981;11:187–195.
19. De Ponti F, Poluzzi E, Montanaro N. QT-interval prolongation bynon-cardiac drugs: lessons to be learned from recent experience.Eur J Clin Pharmacol 2000;56:1–18.
20. Brooks BR, Cummings J, Pioro EP, et al. Pharmacokinetic/pharma-codynamic modeling of dextromethorphan/quinidine for a studyin pseudobulbar affect. Presented at: American NeurologicalAssociation’s 133rd Annual Meeting; September 21–24, 2008; SaltLake City, Utah [abstract T 172].
21. Moore SR, Gresham LS, Bromberg MB, et al. A self report measureof affective lability. J Neurol Neurosurg Psychiatry 1997;63:89–93.
22. Brooks BR, Miller RG, Swash M, et al. El Escorial revisited: revisedcriteria for the diagnosis of amyotrophic lateral sclerosis. Amyo-troph Lateral Scler Other Motor Neuron Disord 2000;1:293–299.
23. McDonald WI, Compston A, Edan G, et al. Recommended diagnosticcriteria for multiple sclerosis: guidelines from the International Panelon the diagnosis of multiple sclerosis. Ann Neurol 2001;50:121–127.
24. Beck AT, Steer RA, Brown GK. Manual for the Beck Depression In-ventory—II. San Antonio, TX: Psychological Corporation, 1996.
25. Kaufer DI, Cummings JL, Ketchel P, et al. Validation of the NPI-Q,a brief clinical form of the Neuropsychiatric Inventory. J Neuropsy-chiatry Clin Neurosci 2000;12:233–239.
26. Ware JE Jr, Sherbourne CD. The MOS 36-item short-form healthsurvey (SF-36). I. Conceptual framework and item selection. MedCare 1992;30:473–483.
27. Smith RA, Berg JE, Pope LE, et al. Validation of the CNS emo-tional lability scale for pseudobulbar affect (pathological laughingand crying) in multiple sclerosis patients. Mult Scler 2004;10:679–685.
28. Hausman J, Hall BH, Griliches Z. Econometric models for countdata with an application to the patents-R&D relationship. Econo-metrica 1984;52:909–938.
29. Frison L, Pocock SJ. Repeated measures in clinical trials: analysisusing mean summary statistics and its implications for design. StatMed 1992;11:1685–1704.
30. Wolf JK, Santana HB, Thorpy M. Treatment of ‘‘emotional inconti-nence’’ with levodopa. Neurology 1979;29:1435–1436.
31. Udaka F, Yamao S, Nagata H, et al. Pathologic laughing and cry-ing treated with levodopa. Arch Neurol 1984;41:1095–1096.
32. Schiffer RB, Herndon RM, Rudick RA. Treatment of pathologic laughingand weeping with amitriptyline. N Engl J Med 1985;312:1480–1482.
33. Robinson RG, Parikh RM, Lipsey JR, et al. Pathological laughing andcrying following stroke: validation of a measurement scale and adouble-blind treatment study. Am J Psychiatry 1993;150:286–293.
34. Seliger GM, Hornstein A, Flax J, et al. Fluoxetine improves emo-tional incontinence. Brain Inj 1992;6:267–270.
35. Andersen G, Vestergaard K, Riis JO. Citalopram for post-strokepathological crying. Lancet 1993;342:837–839.
36. Muller U, Murai T, Bauer-Wittmund T, von Cramon DY. Paroxetineversus citalopram treatment of pathological crying after braininjury. Brain Inj 1999;13:805–811.
37. Burns A, Russell E, Stratton-Powell H, et al. Sertraline in stroke-asso-ciated lability of mood. Int J Geriatr Psychiatry 1999;14:681–685.
ANNALS of Neurology
702 Volume 68, No. 5

RESEARCH ARTICLE Open Access
PRISM II: an open-label study to assesseffectiveness of dextromethorphan/quinidine for pseudobulbar affect inpatients with dementia, stroke or traumaticbrain injuryFlora M. Hammond1*, David N. Alexander2, Andrew J. Cutler3, Stephen D’Amico4, Rachelle S. Doody5, William Sauve6,Richard D. Zorowitz7, Charles S. Davis8, Paul Shin9, Fred Ledon9, Charles Yonan9, Andrea E. Formella9 and Joao Siffert9
Abstract
Background: Phase 3 trials supporting dextromethorphan/quinidine (DM/Q) use as a treatment for pseudobulbaraffect (PBA) were conducted in patients with amyotrophic lateral sclerosis (ALS) or multiple sclerosis (MS). The PRISM IIstudy provides additional DM/Q experience with PBA secondary to dementia, stroke, or traumatic brain injury (TBI).
Methods: Participants in this open-label, multicenter, 90-day trial received DM/Q 20/10 mg twice daily. The primaryoutcome was the Center for Neurologic Study-Lability Scale (CNS-LS), assessing change in PBA episode frequency andseverity. The CNS-LS final visit score was compared to baseline (primary analysis) and to the response in a previouslyconducted placebo-controlled trial with DM/Q in patients with ALS or MS. Secondary outcomes included change inPBA episode count and Clinical Global Impression of Change with respect to PBA as rated by a clinician (CGI-C) and bythe patient or caregiver (PGI-C).
Results: The study enrolled 367 participants with PBA secondary to dementia, stroke, or TBI. Mean (standard deviation[SD]) CNS-LS score improved significantly from 20.4 (4.4) at baseline to 12.8 (5.0) at Day 90/Final Visit (change, −7.7 [6.1]; P< .001, 95 % CI: −8.4, −7.0). This magnitude of improvement was consistent with DM/Q improvement in the earlierphase-3, placebo-controlled trial (mean [95 % CI] change from baseline, −8.2 [−9.4, −7.0]) and numerically exceeds theimprovement seen with placebo in that study (−5.7 [−6.8, −4.7]). Reduction in PBA episode count was 72.3 % at Day90/Final Visit compared with baseline (P < .001). Scores on CGI-C and PGI-C showed that 76.6 and 72.4 % of participants,respectively, were “much” or ”very much” improved with respect to PBA. The most frequently occurring adverse events(AEs) were diarrhea (5.4 %), headache (4.1 %), urinary tract infection (2.7 %), and dizziness (2.5 %); 9.8 % had AEs that led todiscontinuation. Serious AEs were reported in 6.3 %; however, none were considered treatment related.
Conclusions: DM/Q was shown to be an effective and well-tolerated treatment for PBA secondary to dementia, stroke, orTBI. The magnitude of PBA improvement was similar to that reported in patients with PBA secondary to ALS or MS, andthe adverse event profile was consistent with the known safety profile of DM/Q.(Continued on next page)
* Correspondence: [email protected] Yonan and Joao Siffert are former employees of AvanirPharmaceuticals, Inc.1Physical Medicine and Rehabilitation, Indiana University School of Medicine,Rehabilitation Hospital of Indiana, 4141 Shore Drive, Indianapolis, IN 46254, USAFull list of author information is available at the end of the article
© 2016 The Author(s). Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, andreproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link tothe Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver(http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
Hammond et al. BMC Neurology (2016) 16:89 DOI 10.1186/s12883-016-0609-0

(Continued from previous page)
Trial registration: Clinicaltrials.gov, NCT01799941, registered on 25 February 2013
Keywords: Dextromethorphan, Quinidine, Pseudobulbar affect, Dementia, Brain injuries, Stroke, Neuropsychiatricsymptoms, Center for neurologic study-lability scale
BackgroundAffective lability is a commonly described sequela ofmultiple neurologic disorders and brain injury. Pseudo-bulbar affect (PBA) is a type of affect lability character-ized by sudden, involuntary, and distressing outbursts oflaughing and/or crying that are often exaggerated or dis-connected from mood state or social context [1–3]. PBAepisodes tend to be stereotypical, can last from secondsto several minutes, and often occur multiple times perday. PBA is thought to occur as a result of injury or dis-ease that disrupts pathways regulating emotional expres-sion, or affect, including the corticobulbar tracts andbasal ganglia [4, 5]. PBA is diagnosed clinically through amedical and neurologic evaluation and a careful assess-ment of symptoms in order to distinguish it from featuresof other psychiatric or neurobehavioral conditions such asdepression, anxiety disorders, post-traumatic stress dis-order, dementia-related agitation and other conditionsthat can occur separately or comorbidly with PBA in per-sons with neurologic diseases or brain injuries [2].PBA episodes can be highly disruptive to everyday life,
causing embarrassment, social isolation and, in somecases, impacting the ability to work [6]. Prevalence esti-mates vary by rigor of diagnostic requirements, screen-ing methodology, and causative neurological disorder;however a registry sample of 5,290 clinic patients with 1of 6 neurologic conditions known to be associated withPBA—Alzheimer’s disease (AD), amyotrophic lateralsclerosis (ALS), multiple sclerosis (MS), Parkinson’s dis-ease (PD), stroke, and traumatic brain injury (TBI)—found 36.7 % had a Center for Neurologic Study-LabilityScale (CNS-LS) score ≥13, a score that predicted neur-ologist diagnosis of PBA in validation studies for 82 % ofpatients with ALS and 78 % with MS; 9.3 % of this regis-try sample had a CNS-LS ≥21 [5]. Other studies reportPBA symptom prevalence between 7 and 39 % amongparticipants with dementia [5, 7, 8], 5 to 48 % amongthose with TBI [7, 9, 10], and 11 to 34 % following stroke[11–13]. However, in everyday practice, PBA symp-toms are generally under-recognized due to lack ofroutine screening, limited awareness of the conditionand confusion with other neuropsychiatric conditions[3, 7, 14]. Various drugs have been studied as treat-ments for PBA, including tricyclic antidepressants andselective serotonin reuptake inhibitors, though noagents in these drug classes have been approved forthis use [15].
Dextromethorphan hydrobromide and quinidine sul-fate in the fixed combination NUEDEXTA® (DM/Q;commercially available since 2010 from Avanir Pharma-ceuticals, Inc., Aliso Viejo, CA, USA) is currently theonly pharmaceutical agent approved by the US Food andDrug Administration for the treatment of PBA [16].DM, the CNS-active component of the medication, is aweak, uncompetitive N-methyl-D-aspartate (NMDA)receptor antagonist, a sigma-1 receptor agonist, a sero-tonin and norepinephrine reuptake inhibitor, and anα3β4 neuronal nicotinic receptor antagonist [17–19].However, the mechanism whereby DM exerts its clinicaleffects is not fully elucidated. Normally, DM is rapidly me-tabolized through the cytochrome P450 2D6 (CYP2D6)isoenzyme to dextrorphan, a metabolite that is rapidly glu-curonidated, limiting its CNS bioavailability when DM isadministered at approved doses. When administeredalone, DM achieves only minimal plasma exposure. Low-dose quinidine (10 mg) is a potent inhibitor of cytochromeP450 2D6 that, when combined with DM, substantially in-creases DM bioavailability and enables greater central ner-vous system exposure. Well-controlled Phase 3 studieshave shown that the DM/Q combination is efficacious forthe treatment of PBA [20–22], and is significantly moreefficacious than DM alone or Q alone [22].The DM/Q combination was approved in the United
States for the treatment of PBA, irrespective of the typeof neurologic etiology, as a result of phase 3 trials in par-ticipants with PBA secondary to ALS or MS [20–22].Across the phase 3 trials in patients with MS or ALS,DM/Q was generally safe and well tolerated [20–22]. Inaddition, DM/Q was found to be generally well toler-ated in a long-term, 52-week safety trial that enrolledparticipants with PBA associated with a wide range ofunderlying neurological conditions [23]. To date, therehave been only limited clinical trial data evaluatingthe safety and efficacy of DM/Q for PBA in specificand well-defined patient populations beyond MS andALS.The purpose of the present study (referred to as the
Pseudobulbar Affect Registry Investigating SymptomManagement II [PRISM II] trial) was to provide data onthe effectiveness of DM/Q for the treatment of PBA sec-ondary to dementia, stroke, and TBI. This article de-scribes the findings for outcomes common to the overallstudy population. Outcomes in individual neurologicdisease cohorts are reported separately [24–26].
Hammond et al. BMC Neurology (2016) 16:89 Page 2 of 12

MethodsStudy designThe study measured effectiveness by evaluating efficacy,safety/tolerability and other patient outcomes when usedunder usual conditions in patients with dementia, stroke,or TBI. Adults with PBA secondary to dementia, stroke,or TBI and a CNS-LS score ≥13 (scale range, 7 [nosymptoms] to 35 [maximum]) were enrolled in an open-label, 90-day study of DM/Q 20/10 mg twice daily (oncedaily during Week 1). Clinical assessments were per-formed at baseline, Day 30, and Day 90, with a telephonecontact at Day 60. This study was conducted in theUnited States at 74 enrolling sites, from 26Feb2013 to30Apr2015, according to Good Clinical Practice and theDeclaration of Helsinki. The protocol and study mate-rials were approved by the institutional review board ateach site and registered on www.clinicaltrials.gov. (iden-tifier: NCT01799941).
ParticipantsParticipants were eligible for enrollment if they had aclinical diagnosis of PBA based on published criteria anda clinical diagnosis of dementia, stroke, or TBI [2]. PBAdiagnostic criteria were (1) involuntary or exaggeratedepisodes of emotional expression (i.e., laughing or cry-ing); (2) development of symptoms that represent achange from the person's usual emotional reactivity oc-curring subsequent to a specified brain disorder; (3) epi-sodes are incongruent with or out of proportion to theindividual's mood state and independent to or in excessof a provoking stimulus; and (4) symptoms are not bet-ter accounted for by another disorder, substance abuse,or medication use. Patients were also required to have aCNS-LS score ≥13 [27, 28], the same score required forentry into phase 3 trials of DM/Q for PBA [20–22].Eligible participants had documented diagnoses of one
of the following: dementia, (including Alzheimer’s, vas-cular, Lewy body, or frontotemporal dementia; ischemicor hemorrhagic stroke; or mild, moderate, or severenon-penetrating TBI. There were no restrictions ontypes of allowed concomitant medications with the ex-ception of those contraindicated by the US PrescribingInformation [16], specifically monoamine oxidase inhibi-tors (MAOIs), and drugs that both significantly prolongQT interval and are primarily metabolized by CYP2D6(e.g., thioridazine). Other medications typically used bypatients with the studied neurologic conditions (includ-ing those that are metabolized by CYP2D6) were allowedwith the stipulation that medications for management ofdementia, such as memantine or acetylcholinesterase in-hibitors should be stable for at least 6 weeks and otherneuropsychiatric medications such as anticonvulsants,antidepressants, antipsychotics, anxiolytics, and sedative/hypnotics should be stable for at least 2 months prior to
baseline; any medication changes, if deemed necessary,were recorded. Potential participants were excluded ifthey had severe dementia (Mini-Mental State Examin-ation [MMSE] score <10); stroke within 3 months ofstudy enrollment; penetrating TBI; severe depressive dis-order; history or current symptoms of schizophrenia (in-cluding psychosis), schizoaffective disorder or bipolardisorder; substance/alcohol abuse in the preceding3 years; systemic disease, neurologic condition or braininjury that was unstable or rapidly changing within the3 months prior to enrollment; life expectancy ≤6 months;contraindication to DM/Q use (including known QTinterval prolongation); DM/Q use during the previous6 months; or interventional clinical study participationwithin the preceding 30 days. Written informed consentwas obtained from all participants or from legally autho-rized representatives.
Outcome measuresPrimary measureThe primary outcome was the change in CNS-LS scorefrom baseline to Day 90 (or final visit). The CNS-LS wascompleted by the patient or the caregiver acting as a pa-tient proxy at Day 1 (baseline), Day 30 (visit 1), and Day90 or early withdrawal (final visit). The CNS-LS is a 7-item (4 laughing items; 3 crying items), self-report ratingscale measure of PBA episode frequency and severitythat was validated in persons with ALS and persons withMS and is sensitive to change over time and treatmenteffects [20–22].
Secondary measuresSecondary measures included PBA episode count for the7 days preceding each study visit as well as the ClinicalGlobal Impression of Change (CGI-C) and Patient (orCaregiver) Global Impression of Change (PGI-C; 7-pointscales ranging from 1 [very much improved] to 7 [verymuch worse]) based on overall change in the patient’scondition with respect to PBA. In addition, a quality-of-life visual analog scale (QOL-VAS) assessed the degreeto which PBA episodes affected the patient’s overallquality of life (11-point scale ranging from 0 [“not atall”] to 10 [“significantly”]) during the past week and a5-point Likert-type scale rated patient satisfaction withtreatment from 1 [very dissatisfied] to 5 [very satisfied].The Folstein Mini-Mental State Examination (MMSE;11-item assessment of orientation, memory, attention,and language scored from 0 to 30) was included as acognitive assessment [29]. As an additional measure, thePatient Health Questionnaire (PHQ-9; a 9-item assess-ment of depression symptoms with each item rated 0[not at all] to 3 [nearly every day] based on frequency ofoccurrence over the past 2 weeks, for a total possiblescore of 27) was included [30]. Disease-specific
Hammond et al. BMC Neurology (2016) 16:89 Page 3 of 12

functional measures, namely the Neurobehavioral Func-tioning Inventory and Stroke Impact Scale were assessedin the TBI and stroke cohorts, respectively, and will bereported separately. Caregivers were allowed to complete
all assessments except the MMSE for any patients whowere unable to do so. The timing of assessments is illus-trated in Fig. 1.
SafetySafety measures included reporting of adverse events (AEs)occurring at any time between study enrollment and up to30 days after the last dose of DM/Q in the study, as well asvital signs and concomitant medication use.
Statistical analysisThe safety analysis set included all participants who re-ceived at least 1 dose of DM/Q. The effectiveness ana-lysis set comprised all participants in the safety set whoalso met all study eligibility criteria, and completed atleast 1 post-baseline CNS-LS assessment. The MedicalDictionary for Regulatory Activities (version 15.1) wasused for coding, categorizing, and reporting AEs.All data were analyzed descriptively. Changes from
baseline in ratings at Day 30 and 90 were also analyzedinferentially using one-sample t-tests for rating scalemeasures (CNS-LS, QOL-VAS, MMSE, and PHQ-9) anda mixed-effects Poisson regression model using numberof PBA episodes in the past 7 days as the dependentvariable to estimate change in PBA episode counts. Themixed-effects Poisson regression model incorporatedage, gender, and time (Day 30 and Day 90) as fixed ef-fects while allowing for individual differences in baselinerate (random-subject effects). The percentage change
Fig. 1 Schedule of study assessments. Caregivers completed ratings asproxies for patients who were unable (except for the MMSE). AE =adverse event; CGI-C = Clinical Global Impression of Change; CNS-LS =Center for Neurologic Study–Lability Scale; MMSE =Mini-Mental StateExam; NFI = Neurobehavioral Functioning Inventory; PBA = pseudobulbaraffect; PGI-C = patient/caregiver Global Impression of Change; PHQ-9 = 9-item Patient Health Questionnaire; QOL-VAS = quality-of-life visual analogscale; SIS = Stroke Impact Scale
Fig. 2 Consort diagram for PRISM II cohort. CNS-LS = center for neurologic study–lability scale
Hammond et al. BMC Neurology (2016) 16:89 Page 4 of 12

episode count from baseline to a given visit is 1 minusthe appropriate time parameter (λ).There was no imputation method for missing data;
however, if the patient had a Final Visit, that visit was in-cluded as the Day 90 Visit. If there was no Final Visit,the Day 30 visit was not carried forward as the subject’sFinal Visit.The primary analysis tested the null hypothesis that the
mean change in CNS-LS score from baseline to the Day90/Final Visit was equal to zero; the 95 % confidenceinterval (CI) was also reported to enable a descriptivecomparison with the CNS-LS change seen in the pivotalphase 3 registration trial (STAR trial) that led to the USapproval of DM/Q for PBA [20]. Pearson correlation coef-ficients were calculated to assess the correlation of CNS-LS scores with other endpoints at baseline, Day 30, andDay 90/Final Visit, as well as correlation of change frombaseline in CNS-LS with changes in other measures.Tests of significance were 2-tailed and carried out at the
α = .05 level of significance; all analyses were completedusing either SAS v9.2 (SAS Institute Inc., Cary, NC) orStata v12 (StataCorp, College Station, TX). All patientswith data for the given comparison were included.A power calculation was based on mean (SD) CNS-LS
change observed from the pivotal phase 3 trial for thesame dose of DM/Q (20/10 mg twice daily): −8.2 (6.1)points vs. placebo: −5.7 (5.3) points. Based on these re-sults, it was determined that a sample size of 100 pa-tients per disease group would provide 80 % power todetect a CNS-LS mean change of −7.45 points (increaseof 1.75 points over assumed true placebo mean changeof −5.7), or 90 % power to detect a CNS-LS meanchange of −7.7 points (increase of 2.0 points over as-sumed true placebo mean change of −5.7). An interimanalysis, conducted after the first 100 patients
Table 1 Baseline demographics and clinical characteristics—safetyanalysis set
Characteristic (N = 367)
Age, mean (SD), y 59.4 (16.5)
Age category, n (%)
≥ 65 years 152 (41.4)
≥ 75 years 75 (20.4)
Gender, n (%)
Male 165 (45.0)
Female 202 (55.0)
Race, n (%)
White/Caucasian 304 (82.8)
Black/African American 50 (13.6)
Asian 3 (0.8)
Othera 4 (1.1)
Unknown 6 (1.6)
Ethnicity, n (%)
Hispanic/Latino 71 (19.4)
Presence of a caregiver, n (%) 166 (45.2)
Place of Residence, n (%)
Home 303 (82.6)
Assisted living 35 (9.5)
Skilled nursing facility 29 (7.9)
Primary diagnosis, n (%)
Dementia 134 (36.5)
Stroke 113 (30.8)
TBI 120 (32.7)
Concomitant medications at baseline(total no. of medications)
Mean 7.7
Median (min, max) 7.0 (0, 27)
Psychopharmacologic medication use,b n (%) 260 (70.8)
Any antidepressant 178 (48.5)
SSRIs 103 (28.1)
Other 83 (22.6)
Non-selective (tricyclic) 14 (3.8)
Sedative/hypnotics/anxiolytics 124 (33.8)
Any benzodiazepinec 109 (29.7)
Antipsychoticsd 66 (18.0)
Anticonvulsants 92 (25.1)
CNS-LS scoree,f
Mean (SD) 20.5 (4.4)
Median (min, max) 20 (13, 34)
PBA episode count(over 7 days prior to baseline)f
Mean (SD) 21.4 (25.0)
Median (min, max) 12 (0, 240)
Table 1 Baseline demographics and clinical characteristics—safetyanalysis set (Continued)
MMSE score, mean (SD)f 23.9 (5.9)
QOL-VAS, mean (SD) 5.9 (2.6)
PHQ-9,g mean (SD) 13.5 (5.9)
CNS-LS Center for neurologic study-lability scale, MMSE mini-mental stateexamination, PBA pseudobulbar affect, PHQ-9 patient health questionnaire-9,QOL-VAS quality-of-life visual analog scale, SD standard deviation, SSRI selectiveserotonin reuptake inhibitoraOther includes American Indian, Alaskan Native, Native Hawaiian, or otherPacific IslanderbPsychopharmacologic medications included anticonvulsants, antipsychotics,antidepressants, sedatives/hypnotics or anxiolytics, and benzodiazepinecIncludes benzodiazepines as sedatives/hypnotics plus clonazepam asan anticonvulsantdTypical antipsychotic use in 1.9 %, and atypical antipsychotic use in 16.6 %;categories were not mutually exclusiveeThe CNS-LS scale ranges from 7 to 35, with higher scores indicating increasedfrequency and severity of PBA episodesfEffectiveness analysis set (n = 298)gPHQ-9 scores range from 0 to 27, with higher scores indicating increasedseverity of depression
Hammond et al. BMC Neurology (2016) 16:89 Page 5 of 12
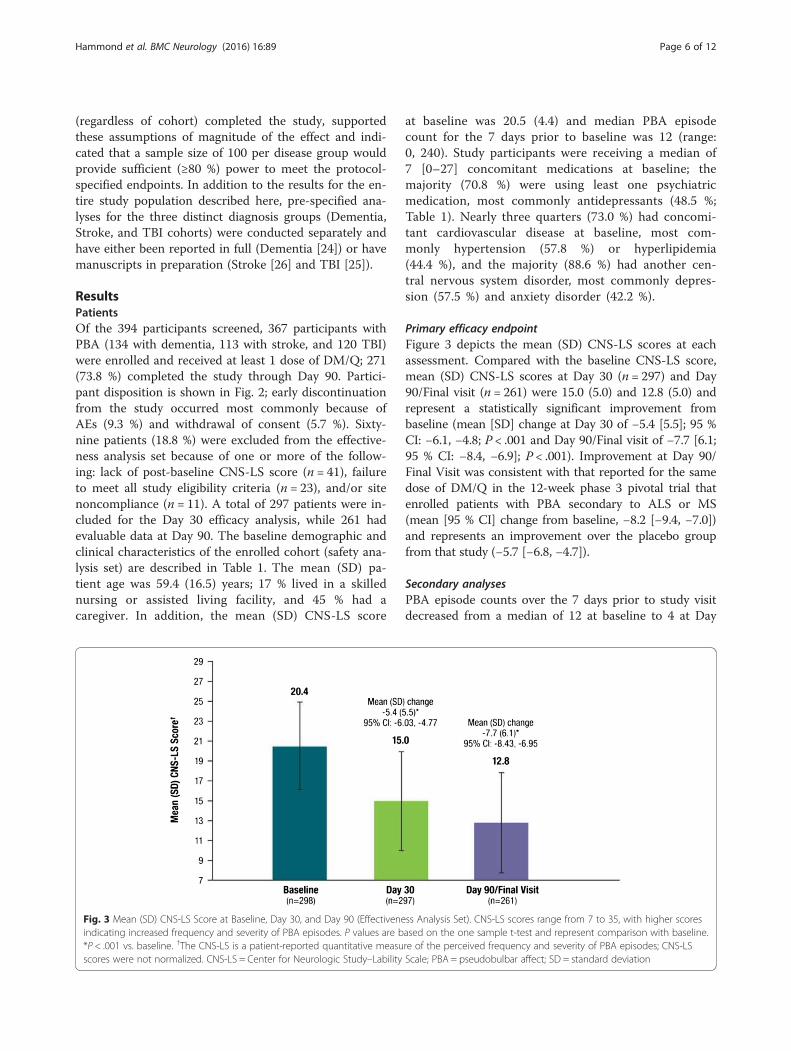
(regardless of cohort) completed the study, supportedthese assumptions of magnitude of the effect and indi-cated that a sample size of 100 per disease group wouldprovide sufficient (≥80 %) power to meet the protocol-specified endpoints. In addition to the results for the en-tire study population described here, pre-specified ana-lyses for the three distinct diagnosis groups (Dementia,Stroke, and TBI cohorts) were conducted separately andhave either been reported in full (Dementia [24]) or havemanuscripts in preparation (Stroke [26] and TBI [25]).
ResultsPatientsOf the 394 participants screened, 367 participants withPBA (134 with dementia, 113 with stroke, and 120 TBI)were enrolled and received at least 1 dose of DM/Q; 271(73.8 %) completed the study through Day 90. Partici-pant disposition is shown in Fig. 2; early discontinuationfrom the study occurred most commonly because ofAEs (9.3 %) and withdrawal of consent (5.7 %). Sixty-nine patients (18.8 %) were excluded from the effective-ness analysis set because of one or more of the follow-ing: lack of post-baseline CNS-LS score (n = 41), failureto meet all study eligibility criteria (n = 23), and/or sitenoncompliance (n = 11). A total of 297 patients were in-cluded for the Day 30 efficacy analysis, while 261 hadevaluable data at Day 90. The baseline demographic andclinical characteristics of the enrolled cohort (safety ana-lysis set) are described in Table 1. The mean (SD) pa-tient age was 59.4 (16.5) years; 17 % lived in a skillednursing or assisted living facility, and 45 % had acaregiver. In addition, the mean (SD) CNS-LS score
at baseline was 20.5 (4.4) and median PBA episodecount for the 7 days prior to baseline was 12 (range:0, 240). Study participants were receiving a median of7 [0–27] concomitant medications at baseline; themajority (70.8 %) were using least one psychiatricmedication, most commonly antidepressants (48.5 %;Table 1). Nearly three quarters (73.0 %) had concomi-tant cardiovascular disease at baseline, most com-monly hypertension (57.8 %) or hyperlipidemia(44.4 %), and the majority (88.6 %) had another cen-tral nervous system disorder, most commonly depres-sion (57.5 %) and anxiety disorder (42.2 %).
Primary efficacy endpointFigure 3 depicts the mean (SD) CNS-LS scores at eachassessment. Compared with the baseline CNS-LS score,mean (SD) CNS-LS scores at Day 30 (n = 297) and Day90/Final visit (n = 261) were 15.0 (5.0) and 12.8 (5.0) andrepresent a statistically significant improvement frombaseline (mean [SD] change at Day 30 of −5.4 [5.5]; 95 %CI: −6.1, −4.8; P < .001 and Day 90/Final visit of −7.7 [6.1;95 % CI: −8.4, −6.9]; P < .001). Improvement at Day 90/Final Visit was consistent with that reported for the samedose of DM/Q in the 12-week phase 3 pivotal trial thatenrolled patients with PBA secondary to ALS or MS(mean [95 % CI] change from baseline, −8.2 [−9.4, −7.0])and represents an improvement over the placebo groupfrom that study (−5.7 [−6.8, −4.7]).
Secondary analysesPBA episode counts over the 7 days prior to study visitdecreased from a median of 12 at baseline to 4 at Day
Fig. 3 Mean (SD) CNS-LS Score at Baseline, Day 30, and Day 90 (Effectiveness Analysis Set). CNS-LS scores range from 7 to 35, with higher scoresindicating increased frequency and severity of PBA episodes. P values are based on the one sample t-test and represent comparison with baseline.*P < .001 vs. baseline. †The CNS-LS is a patient-reported quantitative measure of the perceived frequency and severity of PBA episodes; CNS-LSscores were not normalized. CNS-LS = Center for Neurologic Study–Lability Scale; PBA = pseudobulbar affect; SD = standard deviation
Hammond et al. BMC Neurology (2016) 16:89 Page 6 of 12
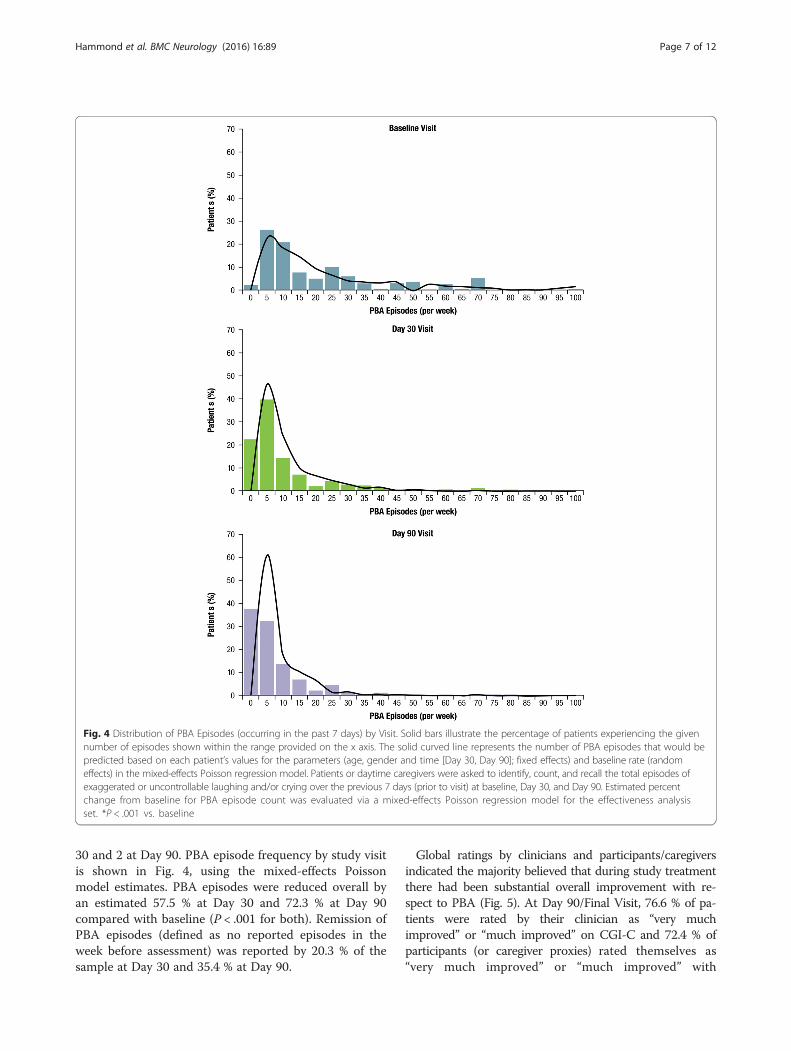
30 and 2 at Day 90. PBA episode frequency by study visitis shown in Fig. 4, using the mixed-effects Poissonmodel estimates. PBA episodes were reduced overall byan estimated 57.5 % at Day 30 and 72.3 % at Day 90compared with baseline (P < .001 for both). Remission ofPBA episodes (defined as no reported episodes in theweek before assessment) was reported by 20.3 % of thesample at Day 30 and 35.4 % at Day 90.
Global ratings by clinicians and participants/caregiversindicated the majority believed that during study treatmentthere had been substantial overall improvement with re-spect to PBA (Fig. 5). At Day 90/Final Visit, 76.6 % of pa-tients were rated by their clinician as “very muchimproved” or “much improved” on CGI-C and 72.4 % ofparticipants (or caregiver proxies) rated themselves as“very much improved” or “much improved” with
Fig. 4 Distribution of PBA Episodes (occurring in the past 7 days) by Visit. Solid bars illustrate the percentage of patients experiencing the givennumber of episodes shown within the range provided on the x axis. The solid curved line represents the number of PBA episodes that would bepredicted based on each patient’s values for the parameters (age, gender and time [Day 30, Day 90]; fixed effects) and baseline rate (randomeffects) in the mixed-effects Poisson regression model. Patients or daytime caregivers were asked to identify, count, and recall the total episodes ofexaggerated or uncontrollable laughing and/or crying over the previous 7 days (prior to visit) at baseline, Day 30, and Day 90. Estimated percentchange from baseline for PBA episode count was evaluated via a mixed-effects Poisson regression model for the effectiveness analysisset. *P < .001 vs. baseline
Hammond et al. BMC Neurology (2016) 16:89 Page 7 of 12
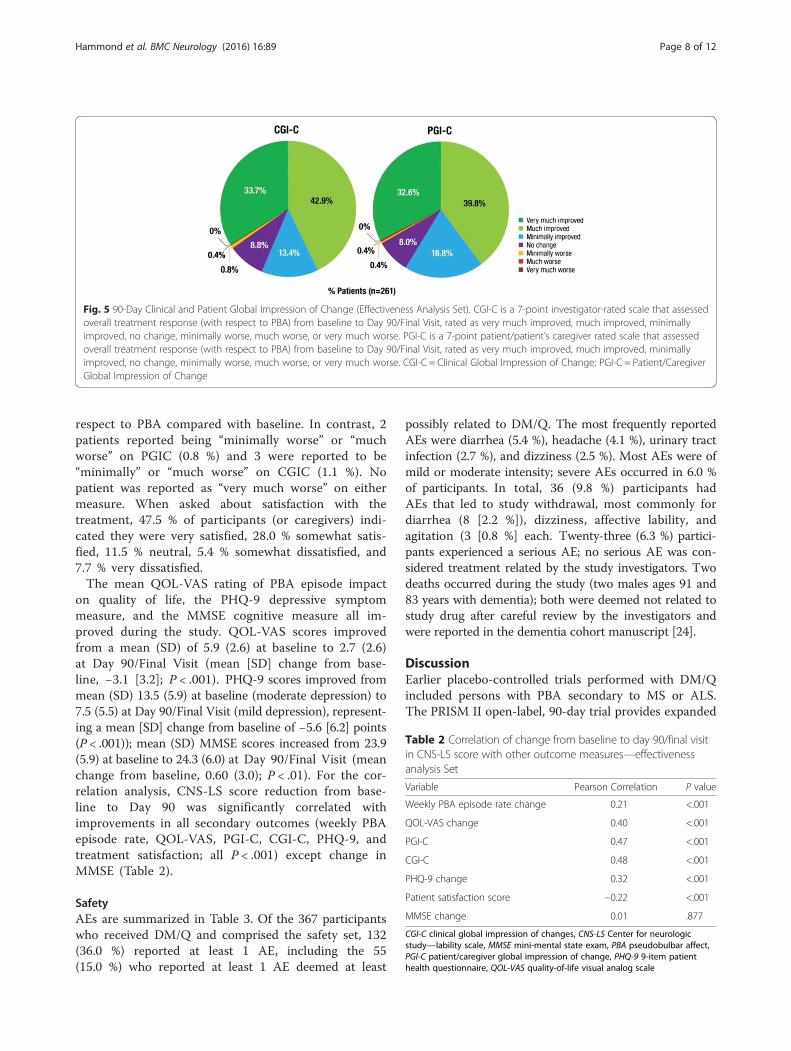
respect to PBA compared with baseline. In contrast, 2patients reported being “minimally worse” or “muchworse” on PGIC (0.8 %) and 3 were reported to be“minimally” or “much worse” on CGIC (1.1 %). Nopatient was reported as “very much worse” on eithermeasure. When asked about satisfaction with thetreatment, 47.5 % of participants (or caregivers) indi-cated they were very satisfied, 28.0 % somewhat satis-fied, 11.5 % neutral, 5.4 % somewhat dissatisfied, and7.7 % very dissatisfied.The mean QOL-VAS rating of PBA episode impact
on quality of life, the PHQ-9 depressive symptommeasure, and the MMSE cognitive measure all im-proved during the study. QOL-VAS scores improvedfrom a mean (SD) of 5.9 (2.6) at baseline to 2.7 (2.6)at Day 90/Final Visit (mean [SD] change from base-line, −3.1 [3.2]; P < .001). PHQ-9 scores improved frommean (SD) 13.5 (5.9) at baseline (moderate depression) to7.5 (5.5) at Day 90/Final Visit (mild depression), represent-ing a mean [SD] change from baseline of −5.6 [6.2] points(P < .001)); mean (SD) MMSE scores increased from 23.9(5.9) at baseline to 24.3 (6.0) at Day 90/Final Visit (meanchange from baseline, 0.60 (3.0); P < .01). For the cor-relation analysis, CNS-LS score reduction from base-line to Day 90 was significantly correlated withimprovements in all secondary outcomes (weekly PBAepisode rate, QOL-VAS, PGI-C, CGI-C, PHQ-9, andtreatment satisfaction; all P < .001) except change inMMSE (Table 2).
SafetyAEs are summarized in Table 3. Of the 367 participantswho received DM/Q and comprised the safety set, 132(36.0 %) reported at least 1 AE, including the 55(15.0 %) who reported at least 1 AE deemed at least
possibly related to DM/Q. The most frequently reportedAEs were diarrhea (5.4 %), headache (4.1 %), urinary tractinfection (2.7 %), and dizziness (2.5 %). Most AEs were ofmild or moderate intensity; severe AEs occurred in 6.0 %of participants. In total, 36 (9.8 %) participants hadAEs that led to study withdrawal, most commonly fordiarrhea (8 [2.2 %]), dizziness, affective lability, andagitation (3 [0.8 %] each. Twenty-three (6.3 %) partici-pants experienced a serious AE; no serious AE was con-sidered treatment related by the study investigators. Twodeaths occurred during the study (two males ages 91 and83 years with dementia); both were deemed not related tostudy drug after careful review by the investigators andwere reported in the dementia cohort manuscript [24].
DiscussionEarlier placebo-controlled trials performed with DM/Qincluded persons with PBA secondary to MS or ALS.The PRISM II open-label, 90-day trial provides expanded
Fig. 5 90-Day Clinical and Patient Global Impression of Change (Effectiveness Analysis Set). CGI-C is a 7-point investigator-rated scale that assessedoverall treatment response (with respect to PBA) from baseline to Day 90/Final Visit, rated as very much improved, much improved, minimallyimproved, no change, minimally worse, much worse, or very much worse. PGI-C is a 7-point patient/patient’s caregiver rated scale that assessedoverall treatment response (with respect to PBA) from baseline to Day 90/Final Visit, rated as very much improved, much improved, minimallyimproved, no change, minimally worse, much worse, or very much worse. CGI-C = Clinical Global Impression of Change; PGI-C = Patient/CaregiverGlobal Impression of Change
Table 2 Correlation of change from baseline to day 90/final visitin CNS-LS score with other outcome measures—effectivenessanalysis Set
Variable Pearson Correlation P value
Weekly PBA episode rate change 0.21 <.001
QOL-VAS change 0.40 <.001
PGI-C 0.47 <.001
CGI-C 0.48 <.001
PHQ-9 change 0.32 <.001
Patient satisfaction score −0.22 <.001
MMSE change 0.01 .877
CGI-C clinical global impression of changes, CNS-LS Center for neurologicstudy—lability scale, MMSE mini-mental state exam, PBA pseudobulbar affect,PGI-C patient/caregiver global impression of change, PHQ-9 9-item patienthealth questionnaire, QOL-VAS quality-of-life visual analog scale
Hammond et al. BMC Neurology (2016) 16:89 Page 8 of 12

safety and efficacy data for DM/Q in a broader patientpopulation and is the first prospectively conducted, sys-tematic study to evaluate DM/Q effectiveness for PBAoccurring subsequent to dementia, stroke, or TBI. Theinclusion criteria defined within this trial allowed for apatient population that more closely resembles “real-life”clinical circumstances (e.g., participants in this trial weretaking a median of 7 concomitant medications, abouthalf were on antidepressants and nearly 20 % were in anassisted living or skilled nursing facility).Substantial improvement in clinical symptoms of PBA
were seen at both post-baseline assessments with a −7.7point (SD 6.1) reduction in CNS-LS score and a 72.3 %reduction in PBA episode frequency at Day 90/Finalvisit, both of which represented statistically significantdifferences compared with baseline. Improvements were
consistent across the three cohorts including patientswith stroke, dementia and TBI. The mean (12.8) CNS-LS score at study endpoint was below the minimumthreshold required for study inclusion. Since this studyallowed caregivers to complete assessments for patientswho were unable to do so, the planned statistical ana-lyses allowed for an evaluation of caregiver-completedvs. patient-completed outcomes. This was done for thedementia cohort, and as previously described, caregiversgenerally reported greater PBA symptom change thanpatients, but between group differences were only statis-tically significant (P < .001) for the estimated PBA epi-sode count reduction (57.7 % for patient-reported vs.77.2 % for caregiver-reported ratings) [24].Improvement was also observed in all other second-
ary endpoints, including a clinical meaningful reduc-tion in PHQ-9 scores (minimal clinically importantdifference for the PHQ-9 has been estimated to be a5 point change [31]; mean change observed in thistrial was −5.6). In addition, DM/Q was well tolerated,with 9.8 % of participants having AEs leading to dis-continuation; the most frequent AEs were diarrhea(5.4 %), headache (4.1 %), urinary tract infection(2.7 %), and dizziness (2.5 %).
Clinical implicationsAlthough the present study was open label, results ap-pear to be clinically meaningful, consistent across studycohorts, and consistent with those seen in well-controlled, phase 3 trials of DM/Q. The clinical rele-vance of CNS-LS score reductions from baseline to Day90/Final Visit is reflected in corresponding reductions inPBA episodes and improvements on other secondarymeasures, including clinician and patient global impres-sions with respect to PBA and satisfaction with treat-ment. CNS-LS reduction was moderately butsignificantly correlated with improvements in all second-ary outcomes (weekly PBA episode rate, QOL-VAS,PGI-C, CGI-C, PHQ-9, and treatment satisfaction; all P< .001) with the exception of the MMSE. While the mea-sured outcomes are directionally related, the lack ofstrong correlation (all Pearson’s coefficients were < .5)suggests that changes in CNS-LS and other outcomesare not solely reflective of changes in PBA episode num-ber and may reflect other aspects of PBA episodes, suchas episode intensity, subjective perception, functioningrelated to these episodes, or other factors not readily ap-parent from these data.A pre-specified analysis for this trial assessed the 95 %
CI for mean change from baseline in CNS-LS scores inorder to enable descriptive comparisons with the earlierplacebo-controlled Phase 3 trial in patients with PBAsecondary to ALS or MS [20]. Although drug perform-ance cannot be directly compared across different
Table 3 Summary of adverse events—safety analysis set
AE Summary, n (%) (N = 367)
Any AE 132 (36.0)
AE intensity
Mild 67 (18.3)
Moderate 72 (19.6)
Severe 22 (6.0)
Unknown 7 (1.9)
Treatment-related AEs 55 (15.0)
Treatment-related AE intensity
Mild 23 (6.3)
Moderate 28 (7.6)
Severe 7 (1.9)
Unknown 3 (0.8)
Serious AEs 23 (6.3)
Treatment-related serious AEs 0
AEs leading to discontinuation 36 (9.8)
Frequency of AEs by preferred term(occurring in >1 % of patients)
Diarrhea 20 (5.4)
Headache 15 (4.1)
Urinary tract infection 10 (2.7)
Dizziness 9 (2.5)
Nausea 6 (1.6)
Fall 6 (1.6)
Fatigue 5 (1.4)
Somnolence 5 (1.4)
Dry mouth 4 (1.1)
Gastroesophageal reflux disease 4 (1.1)
Agitation 4 (1.1)
Peripheral edema 4 (1.1)
AE adverse event
Hammond et al. BMC Neurology (2016) 16:89 Page 9 of 12
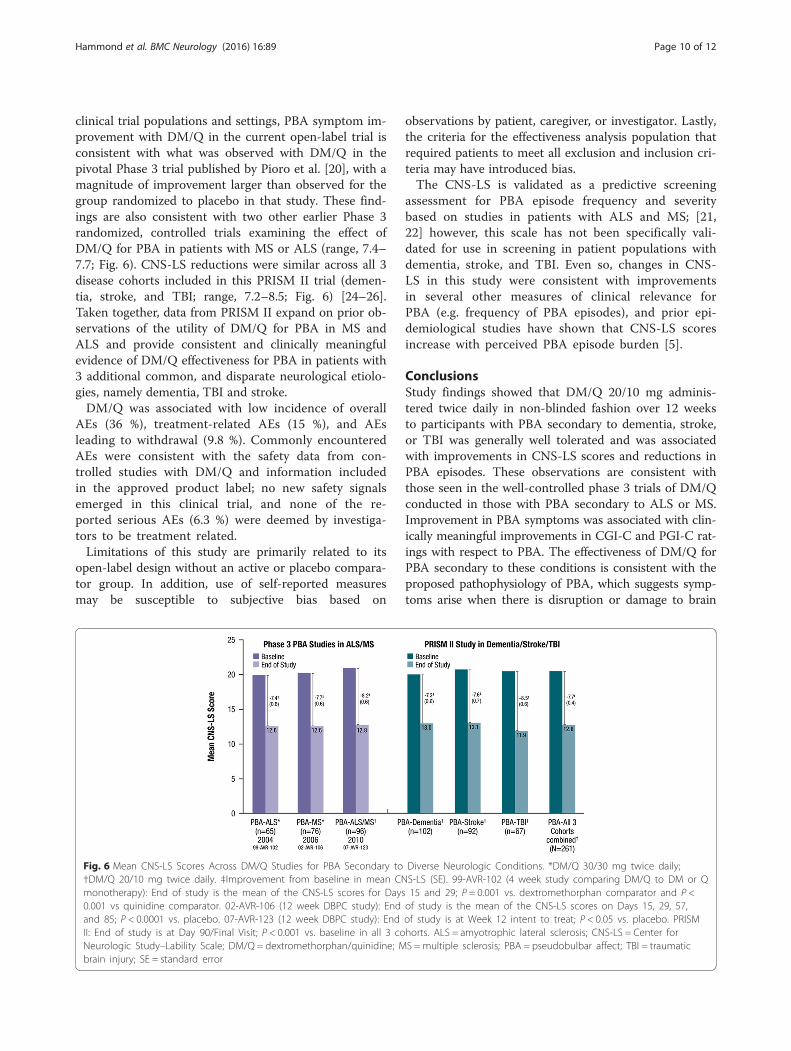
clinical trial populations and settings, PBA symptom im-provement with DM/Q in the current open-label trial isconsistent with what was observed with DM/Q in thepivotal Phase 3 trial published by Pioro et al. [20], with amagnitude of improvement larger than observed for thegroup randomized to placebo in that study. These find-ings are also consistent with two other earlier Phase 3randomized, controlled trials examining the effect ofDM/Q for PBA in patients with MS or ALS (range, 7.4–7.7; Fig. 6). CNS-LS reductions were similar across all 3disease cohorts included in this PRISM II trial (demen-tia, stroke, and TBI; range, 7.2–8.5; Fig. 6) [24–26].Taken together, data from PRISM II expand on prior ob-servations of the utility of DM/Q for PBA in MS andALS and provide consistent and clinically meaningfulevidence of DM/Q effectiveness for PBA in patients with3 additional common, and disparate neurological etiolo-gies, namely dementia, TBI and stroke.DM/Q was associated with low incidence of overall
AEs (36 %), treatment-related AEs (15 %), and AEsleading to withdrawal (9.8 %). Commonly encounteredAEs were consistent with the safety data from con-trolled studies with DM/Q and information includedin the approved product label; no new safety signalsemerged in this clinical trial, and none of the re-ported serious AEs (6.3 %) were deemed by investiga-tors to be treatment related.Limitations of this study are primarily related to its
open-label design without an active or placebo compara-tor group. In addition, use of self-reported measuresmay be susceptible to subjective bias based on
observations by patient, caregiver, or investigator. Lastly,the criteria for the effectiveness analysis population thatrequired patients to meet all exclusion and inclusion cri-teria may have introduced bias.The CNS-LS is validated as a predictive screening
assessment for PBA episode frequency and severitybased on studies in patients with ALS and MS; [21,22] however, this scale has not been specifically vali-dated for use in screening in patient populations withdementia, stroke, and TBI. Even so, changes in CNS-LS in this study were consistent with improvementsin several other measures of clinical relevance forPBA (e.g. frequency of PBA episodes), and prior epi-demiological studies have shown that CNS-LS scoresincrease with perceived PBA episode burden [5].
ConclusionsStudy findings showed that DM/Q 20/10 mg adminis-tered twice daily in non-blinded fashion over 12 weeksto participants with PBA secondary to dementia, stroke,or TBI was generally well tolerated and was associatedwith improvements in CNS-LS scores and reductions inPBA episodes. These observations are consistent withthose seen in the well-controlled phase 3 trials of DM/Qconducted in those with PBA secondary to ALS or MS.Improvement in PBA symptoms was associated with clin-ically meaningful improvements in CGI-C and PGI-C rat-ings with respect to PBA. The effectiveness of DM/Q forPBA secondary to these conditions is consistent with theproposed pathophysiology of PBA, which suggests symp-toms arise when there is disruption or damage to brain
Fig. 6 Mean CNS-LS Scores Across DM/Q Studies for PBA Secondary to Diverse Neurologic Conditions. *DM/Q 30/30 mg twice daily;†DM/Q 20/10 mg twice daily. ‡Improvement from baseline in mean CNS-LS (SE). 99-AVR-102 (4 week study comparing DM/Q to DM or Qmonotherapy): End of study is the mean of the CNS-LS scores for Days 15 and 29; P = 0.001 vs. dextromethorphan comparator and P <0.001 vs quinidine comparator. 02-AVR-106 (12 week DBPC study): End of study is the mean of the CNS-LS scores on Days 15, 29, 57,and 85; P < 0.0001 vs. placebo. 07-AVR-123 (12 week DBPC study): End of study is at Week 12 intent to treat; P < 0.05 vs. placebo. PRISMII: End of study is at Day 90/Final Visit; P < 0.001 vs. baseline in all 3 cohorts. ALS = amyotrophic lateral sclerosis; CNS-LS = Center forNeurologic Study–Lability Scale; DM/Q = dextromethorphan/quinidine; MS = multiple sclerosis; PBA = pseudobulbar affect; TBI = traumaticbrain injury; SE = standard error
Hammond et al. BMC Neurology (2016) 16:89 Page 10 of 12

pathways regulating emotional expression, regardless ofthe specific underlying neurologic condition.
Additional files
Additional file 1: Institutional Review Boards. File contains a listinglocation and members of Institutional Review Boards approving the studyprotocol. (PDF 30 kb)
AbbreviationsAD, Alzheimer’s disease; AE, adverse event; ALS, amyotrophic lateral sclerosis;CGI-C, Clinical Global Impression of Change; CI, confidence interval; CNS-LS,Center for Neurologic Study–Lability Scale; CYP2D6, cytochrome P450 2D6; DM/Q, dextromethorphan/quinidine; MMSE, Mini-Mental State Examination; MS,multiple sclerosis; NFI, neurobehavioral functioning inventory; NMDA, N-methyl-D-aspartate; PBA, pseudobulbar affect; PD, Parkinson’s disease; PGI-C, Patient/Caregiver Global impression of change; PHQ-9, patient health questionnaire-9;PRISM, Pseudobulbar affect registry investigating symptom management; QOL-VAS, quality-of-life visual analog scale; SD, standard deviation; SE, standard error;SIS, stroke impact scale; SSRI, selective serotonin reuptake inhibitor; TBI,traumatic brain injury.
AcknowledgementsWe acknowledge Shereen McIntyre, MBA (clinical data management anddata analysis); Shelby Woods, Tracy Maines and Yim Ang (projectmanagement); Jennifer Lee (CRA); Randall Kaye, MD (study concept design);Rachel Halpern, PhD, and Mike Johnson, MS (statistical analysis) for theircontributions. Medical writing and editing assistance provided by PelotonAdvantage, LLC (Parsippany, NJ) and Prescott Medical CommunicationsGroup (Chicago, IL). The authors thank all study investigators, participants,and care partners who assisted with this trial.
FundingThis study and medical writing and editing services were funded by AvanirPharmaceuticals, Inc.
Availability of data and materialsData that have been used to draw conclusions within this manuscript arereported in full. Data also will be included at www.clinicaltrials.gov usingaccession number NCT01799941.
Authors’ contributionsAll authors had full access to the study data and had final responsibility forthe decision to submit for publication. All authors provided direction onmanuscript content and data presentation. Flora Hammond, MD, wrote thefirst draft of the manuscript. All authors provided feedback and approved thefinal version. All authors read and approved the final manuscript.
Authors’ informationCharles Yonan and Joao Siffert are former employees of AvanirPharmaceuticals, Inc.
Competing interestsFlora Hammond has consulted within the past 12 months for: AvanirPharmaceuticals, Inc. as a member of the PRISM II Steering Committee; hasstock ownership in health care companies including: Abbvie Inc SHS, Eli Lilly& Co, GlaxoSmithKline PLC ADR, Exchange Traded Funds, and Mutual Funds;and her academic institution receives funding from the National Institute onDisability Independent Living and Rehabilitation Research. David Alexanderhas consulted within the past 12 months for: Avanir Pharmaceuticals, Inc. asa member of the PRISM II Steering Committee. Andrew Cutler has served asa consultant for, received research grants from, and served as a speaker forAbbott, AstraZeneca, Avanir Pharmaceuticals, Inc., Bristol-Myers Squibb,Forest, GlaxoSmithKline, Lilly, Merck, Novartis, Ortho-McNeil-Janssen, Otsuka,Pamlab, Pfizer, Shire, Sunovion, Takeda, and Vanda. Stephen D’Amico hasreceived honoraria as a consultant and speaker for Avanir Pharmaceuticals,Inc. He has been a consultant and received research grants from Sanofi,Merck, AstraZeneca, Bristol-Myers, Novartis, and Takeda Pharmaceuticals.Rachelle Doody has consulted within the past 12 months for AC Immune,
Avanir Pharmaceuticals, Inc., Axovant, AZ Therapies, Biogen, Cerespir, Forum,Genentech, Hoffman LaRoche, Shanghai Green Valley, Suven, Transition, vTv,and Takeda; holds stock options in AZ Therapies and QR Pharma, and hasfunding from the National Institutes of Aging and State of Texas. WilliamSauve is a faculty member of the Neuroscience Education Institute, is anadvisor to Avanir Pharmaceuticals, Inc., and has served as a speaker forAvanir, Pharmaceuticals, Inc., Otsuka, and Sunovion. Richard Zorowitz, hasconsulted within the past 12 months for: Avanir Pharmaceuticals, Inc. as amember of the PRISM II Steering Committee; and has stock ownership inhealth care companies including: Various Mutual Funds. He is a consultantfor Allergan, Inc., and served on data safety monitoring boards for researchprojects sponsored by SPR Therapeutics and NexStim. Charles Davis is aconsultant to Avanir Pharmaceuticals, Inc. Paul Shin, Fred Ledon, CharlesYonan, Andrea Formella and João Siffert are employees of AvanirPharmaceuticals, Inc.
Consent for publicationNot applicable.
Ethics approval and consent to participateAn ethics committee reviewed and approved the protocol. Written informedconsent was obtained from all participants or from legally authorizedrepresentatives. Additional file 1 provides a list of all IRBs.
Author details1Physical Medicine and Rehabilitation, Indiana University School of Medicine,Rehabilitation Hospital of Indiana, 4141 Shore Drive, Indianapolis, IN 46254, USA.2University of California, Los Angeles, CA, USA. 3Florida Clinical Research Center,LLC, Bradenton, FL, USA. 4Cornerstone Medical Group, Franklin, TN, USA. 5BaylorCollege of Medicine, Houston, TX, USA. 6TMS NeuroHealth Centers, Richmond,VA, USA. 7MedStar National Rehabilitation Network, Washington, DC, USA. 8CSDBiostatistics, Inc., Tucson, AZ, USA. 9Avanir Pharmaceuticals, Inc., Aliso Viejo, CA,USA.
Received: 22 December 2015 Accepted: 23 May 2016
References1. Schiffer R, Pope LE. Review of pseudobulbar affect including a novel and
potential therapy. J Neuropsychiatry Clin Neurosci. 2005;17:447–54.2. Cummings JL, Arciniegas DB, Brooks BR, Herndon RM, Lauterbach EC, Pioro
EP, et al. Defining and diagnosing involuntary emotional expressiondisorder. CNS Spectr. 2006;11:1–7.
3. Wortzel HS, Oster TJ, Anderson CA, Arciniegas DB. Pathological laughingand crying : epidemiology, pathophysiology and treatment. CNS Drugs.2008;22:531–45.
4. Parvizi J, Coburn KL, Shillcutt SD, Coffey CE, Lauterbach EC, Mendez MF.Neuroanatomy of pathological laughing and crying: a report of theAmerican Neuropsychiatric Association Committee on Research.J Neuropsychiatry Clin Neurosci. 2009;21:75–87.
5. Brooks BR, Crumpacker D, Fellus J, Kantor D, Kaye RE. PRISM: a novelresearch tool to assess the prevalence of pseudobulbar affect symptomsacross neurological conditions. PLoS One. 2013;8, e72232.
6. Colamonico J, Formella A, Bradley W. Pseudobulbar affect: burden of illnessin the USA. Adv Ther. 2012;29:775–98.
7. Work SS, Colamonico JA, Bradley WG, Kaye RE. Pseudobulbar affect: anunder-recognized and under-treated neurological disorder. Adv Ther.2011;28:586–601.
8. Starkstein SE, Migliorelli R, Teson A, Petracca G, Chemerinsky E, Manes F,et al. Prevalence and clinical correlates of pathological affective display inAlzheimer's disease. J Neurol Neurosurg Psychiatry. 1995;59:55–60.
9. Tateno A, Jorge RE, Robinson RG. Pathological laughing and cryingfollowing traumatic brain injury. J Neuropsychiatry Clin Neurosci. 2004;16:426–34.
10. Zeilig G, Drubach DA, Katz-Zeilig M, Karatinos J. Pathological laughterand crying in patients with closed traumatic brain injury. Brain Inj.1996;10:591–7.
11. House A, Dennis M, Molyneux A, Warlow C, Hawton K. Emotionalism afterstroke. BMJ. 1989;298:991–4.
12. Morris PL, Robinson RG, Raphael B. Emotional lability after stroke. Aust N Z JPsychiatry. 1993;27:601–5.
Hammond et al. BMC Neurology (2016) 16:89 Page 11 of 12

13. Kim JS, Choi-Kwon S. Poststroke depression and emotional incontinence:correlation with lesion location. Neurology. 2000;54:1805–10.
14. Engelman W, Hammond FM, Malec JF. Diagnosing pseudobulbar affect intraumatic brain injury. Neuropsychiatr Dis Treat. 2014;10:1903–10.
15. Pioro EP. Current concepts in the pharmacotherapy of pseudobulbar affect.Drugs. 2011;71:1193–207.
16. Avanir Pharmaceuticals Inc. Nuedexta [Prescribing Information. Aliso Viejo:Avanir Pharmaceuticals Inc; 2015.
17. Hernandez SC, Bertolino M, Xiao Y, Pringle KE, Caruso FS, Kellar KJ.Dextromethorphan and its metabolite dextrorphan block alpha3beta4neuronal nicotinic receptors. J Pharmacol Exp Ther. 2000;293:962–7.
18. Codd EE, Shank RP, Schupsky JJ, Raffa RB. Serotonin andnorepinephrine uptake inhibiting activity of centrally acting analgesics:structural determinants and role in antinociception. J Pharmacol ExpTher. 1995;274:1263–70.
19. Werling LL, Keller A, Frank JG, Nuwayhid SJ. A comparison of the binding profilesof dextromethorphan, memantine, fluoxetine and amitriptyline: treatment ofinvoluntary emotional expression disorder. Exp Neurol. 2007;207:248–57.
20. Pioro EP, Brooks BR, Cummings J, Schiffer R, Thisted RA, Wynn D, et al.Dextromethorphan plus ultra low-dose quinidine reduces pseudobulbaraffect. Ann Neurol. 2010;68:693–702.
21. Panitch HS, Thisted RA, Smith RA, Wynn DR, Wymer JP, Achiron A, et al.Randomized, controlled trial of dextromethorphan/quinidine forpseudobulbar affect in multiple sclerosis. Ann Neurol. 2006;59:780–7.
22. Brooks BR, Thisted RA, Appel SH, Bradley WG, Olney RK, Berg JE, et al.Treatment of pseudobulbar affect in ALS with dextromethorphan/quinidine:a randomized trial. Neurology. 2004;63:1364–70.
23. Pattee GL, Wymer JP, Lomen-Hoerth C, Appel SH, Formella AE, Pope LE. Anopen-label multicenter study to assess the safety of dextromethorphan/quinidine in patients with pseudobulbar affect associated with a range ofunderlying neurological conditions. Curr Med Res Opin. 2014;30:2255–65.
24. Doody RS, D'Amico S, Cutler AJ, Davis CS, Shin P, Ledon F, et al. An open-labelstudy to assess safety, tolerability, and effectiveness of dextromethorphan/quinidine for pseudobulbar affect in dementia: PRISM II results. CNS Spectr.2015;1–10. PMID:26471212.
25. Hammond F, Sauve W, Shin P, Ledon F, Davis CS, Yonan C, et al. Safety,tolerability, and effectiveness of dextromethorphan/quinidine forpseudobulbar affect in patients with traumatic brain injury: PRISM-II.Presented at the 33rd Annual Symposium of the National NeurotraumaSociety. June 28-July 1, 2015, Santa Fe, New Mexico.
26. Zorowitz RD, Alexander DN, Shin P, Ledon F, Davis CS, Yonan C et al.Safety, tolerability, and effectiveness of dextromethorphan/quinidine forpseudobulbar affect in patients with stroke: PRISM-II. Presented at the 92ndAnnual Conference of the American Congress of Rehabilitation Medicine,October 25-30, 2015, Dallas, Texas.
27. Moore SR, Gresham LS, Bromberg MB, Kasarkis EJ, Smith RA. A self reportmeasure of affective lability. J Neurol Neurosurg Psychiatry. 1997;63:89–93.
28. Smith RA, Berg JE, Pope LE, Callahan JD, Wynn D, Thisted RA. Validation of theCNS emotional lability scale for pseudobulbar affect (pathological laughingand crying) in multiple sclerosis patients. Mult Scler. 2004;10:679–85.
29. Folstein MF, Folstein SE, McHugh PR. "Mini-mental state". A practicalmethod for grading the cognitive state of patients for the clinician.J Psychiatr Res. 1975;12:189–98.
30. Kroenke K, Spitzer RL, Williams JB. The PHQ-9: validity of a brief depressionseverity measure. J Gen Intern Med. 2001;16:606–13.
31. Lowe B, Unutzer J, Callahan CM, Perkins AJ, Kroenke K. Monitoringdepression treatment outcomes with the patient health questionnaire-9.Med Care. 2004;42:1194–201.
• We accept pre-submission inquiries
• Our selector tool helps you to find the most relevant journal
• We provide round the clock customer support
• Convenient online submission
• Thorough peer review
• Inclusion in PubMed and all major indexing services
• Maximum visibility for your research
Submit your manuscript atwww.biomedcentral.com/submit
Submit your next manuscript to BioMed Central and we will help you at every step:
Hammond et al. BMC Neurology (2016) 16:89 Page 12 of 12

Hammond et al. BMC Neurology (2016) 16:160 DOI 10.1186/s12883-016-0679-z
ERRATUM Open Access
Erratum to: PRISM II: an open-label study toassess effectiveness of dextromethorphan/quinidine for pseudobulbar affect inpatients with dementia, stroke or traumaticbrain injury
Flora M. Hammond1*, David N. Alexander2, Andrew J. Cutler3, Stephen D’Amico4, Rachelle S. Doody5,William Sauve6, Richard D. Zorowitz7, Charles S. Davis8, Paul Shin9, Fred Ledon9, Charles Yonan9,Andrea E. Formella9 and Joao Siffert9ErratumAfter publication of the original article [1], the authorsnoticed that there were errors in the caption of Fig. 3,and the y-axis of Fig. 6 itself.The following statement should not have been in-
cluded in the caption of Fig. 3: “CNS-LS scores were notnormalized.” The CNS-LS is a rank-order scale, and isnot normalized. This statement was included errone-ously and the authors intended on removing it prior toresubmission, but this was unfortunately overlooked.Similarly, the y-axis within Fig. 6 was mislabelled. The
CNS-LS scale ranges from 7 to 35, so the y-axis for Fig.6 should start at a base score of 7 and not zero. The cor-rect and updated version of Fig. 6, in which the data pre-sented remain accurate and are unchanged, is publishedin this erratum.
Author details1Physical Medicine and Rehabilitation, Indiana University School of Medicine,Rehabilitation Hospital of Indiana, 4141 Shore Drive, Indianapolis, IN 46254,USA. 2University of California, Los Angeles, CA, USA. 3Florida Clinical ResearchCenter, LLC, Bradenton, FL, USA. 4Cornerstone Medical Group, Franklin, TN,USA. 5Baylor College of Medicine, Houston, TX, USA. 6TMS NeuroHealthCenters, Richmond, VA, USA. 7MedStar National Rehabilitation Network,Washington, DC, USA. 8CSD Biostatistics, Inc., Tucson, AZ, USA. 9AvanirPharmaceuticals, Inc., Aliso Viejo, CA, USA.
* Correspondence: [email protected] Medicine and Rehabilitation, Indiana University School of Medicine,Rehabilitation Hospital of Indiana, 4141 Shore Drive, Indianapolis, IN 46254,USA
© 2016 The Author(s). Open Access This articInternational License (http://creativecommonsreproduction in any medium, provided you gthe Creative Commons license, and indicate if(http://creativecommons.org/publicdomain/ze
Reference1. Hammond FM, Alexander DN, Cutler AJ, D’Amico S, Doody RS, Sauve W,
et al. PRISM II: an open-label study to assess effectiveness ofdextromethorphan/quinidine for pseudobulbar affect in patients withdementia, stroke or traumatic brain injury. BMC Neurol. 2016;16:89.doi:10.1186/s12883-016-0609-0.
le is distributed under the terms of the Creative Commons Attribution 4.0.org/licenses/by/4.0/), which permits unrestricted use, distribution, andive appropriate credit to the original author(s) and the source, provide a link tochanges were made. The Creative Commons Public Domain Dedication waiverro/1.0/) applies to the data made available in this article, unless otherwise stated.
Fig. 6 Mean CNS-LS Scores Across DM/Q Studies for PBA Secondary to Diverse Neurologic Conditions. *DM/Q 30/30 mg twice daily;†DM/Q 20/10 mg twice daily. ‡Improvement from baseline in mean CNS-LS (SE). 99-AVR-102 (4 week study comparing DM/Q to DM or Qmonotherapy): End of study is the mean of the CNS-LS scores for Days 15 and 29; P = 0.001 vs. dextromethorphan comparator and P <0.001 vs quinidine comparator. 02-AVR-106 (12 week DBPC study): End of study is the mean of the CNS-LS scores on Days 15, 29, 57,and 85; P < 0.0001 vs. placebo. 07-AVR-123 (12 week DBPC study): End of study is at Week 12 intent to treat; P < 0.05 vs. placebo. PRISMII: End of study is at Day 90/Final Visit; P < 0.001 vs. baseline in all 3 cohorts. ALS = amyotrophic lateral sclerosis; CNS-LS = Center forNeurologic Study–Lability Scale; DM/Q = dextromethorphan/quinidine; MS = multiple sclerosis; PBA = pseudobulbar affect; TBI = traumaticbrain injury; SE = standard error
Hammond et al. BMC Neurology (2016) 16:160 Page 2 of 2