review energetic graphene oxide: challenges and...
TRANSCRIPT

Nano Today (2012) 7, 137—152
Available online at www.sciencedirect.com
journa l homepage: www.e lsev ier .com/ locate /nanotoday
REVIEW
Energetic graphene oxide: Challenges andopportunities
Deepti Krishnan, Franklin Kim1, Jiayan Luo, Rodolfo Cruz-Silva2,Laura J. Cote, Hee Dong Jang3, Jiaxing Huang ∗
Department of Materials Science and Engineering, Northwestern University, Evanston, IL 60208, USA
Received 31 December 2011; received in revised form 7 February 2012; accepted 12 February 2012
KEYWORDSCombustion;Composite materials;Energetic materials;Fire risk;Graphene;Graphene oxide;Nanoparticles;Photothermal effects;Purification;Self-propagatingreactions
Summary Motivated by both its graphene-oriented applications and its own remarkable prop-erties, interest in graphene oxide (GO) has widely spread across many disciplines. In parallelto the rapid progress of research, industrial-scale production of GO has emerged. GO is highlyenergetic, thermally unstable and can readily undergo exothermic disproportionation reactionsto produce chemically modified graphene under mild heating conditions. This Review highlightsthe challenges and opportunities associated with GO’s thermal instability such as the potentialfire risk during large scale production and methods of mitigation, energy efficient way to reduceGO, photothermal patterning and sintering of graphene/polymer composites, and new synthe-ses using GO as an in situ power source to make nanoparticle decorated graphene compositesfor energy storage and catalysts.© 2012 Elsevier Ltd. All rights reserved.
Introduction: graphene oxide: new interest inan old material
Graphite oxide sheets, now called graphene oxide (GO) isthe product of chemical exfoliation of graphite that has
∗ Corresponding author.E-mail address: [email protected] (J. Huang).
1 Current address: Institute for Integrated Cell-Material Sciences(iCeMS), Kyoto University, Kyoto, 606-8501, Japan.
2 Current address: Research Center for Exotic Nano Carbon, Shin-shu University, Nagano 380-8553, Japan.
3 Current address: Rare Metals Research Center, Korea Institute ofGeoscience and Mineral Resources, Yuseong-gu, Daejeon 305-350,Republic of Korea.
been known for more than a century [1—3]. It is typi-cally synthesised by reacting graphite powders (Fig. 1a)with strong oxidizing agents such as KMnO4 in concen-trated sulfuric acid [4,5]. The oxidation of graphite breaksup the extended two-dimensional (2D) �-conjugation ofthe stacked graphene sheets into nanoscale graphitic sp2
domains surrounded by disordered, highly oxidised sp3
domains as well as defects of carbon vacancies. Theresulting GO sheets are derivatized by carboxylic acidat the edges, and phenol, hydroxyl and epoxide groupsmainly at the basal plane (Fig. 1b) [6,7]. Therefore,the sheets can readily exfoliate to form a stable, lightbrown coloured, single layer suspension in water [8,9].This severe functionalization of the conjugated networkrenders GO sheets insulating. However, conductivity maybe partially restored conveniently by thermal [10] or
1748-0132/$ — see front matter © 2012 Elsevier Ltd. All rights reserved.doi:10.1016/j.nantod.2012.02.003

138 D. Krishnan et al.
Figure 1 Synthesis, structural model and microstructures of GO sheets. (a) GO is typically synthesised by reacting graphite powderwith strongly oxidizing agents such as KMnO4 in concentrated H2SO4, followed by purification and exfoliation in water to yield acolloidal dispersion of single layers. (b) Structural model of graphene (left), GO (middle) and its reduction product r-GO. GO isinsulating due to broken conjugation in the basal plane. After reduction, r-GO becomes conductive but is still a very defectivestructure compared to graphene. (c) Colour coded high resolution TEM images showing the atomic structures of graphene (left), GO(middle) and r-GO (right). The green, purple and blue areas depict ordered, graphitic sp2 domains, disordered highly oxidised sp3
domains, and holes on the sheet, respectively.[(c) is adapted from [33]. Copyright 2010 Wiley-VCH].
chemical treatment [8], producing chemically modifiedgraphene sheets. Therefore, interest in this century-oldmaterial has resurged after 2004’s discovery on graphene[11]. The oxidization—exfoliation—reduction cycle effec-tively makes the insoluble graphite powders processable inwater, enabling many ways of using the conducting grapheneor reduced GO (r-GO) products [12,13]. Although the result-ing graphene product or r-GO is more defective and thusless conductive than pristine graphene [14], it is sufficientlyconductive for many applications such as electrodes forvarious energy devices [15—19]. Therefore, the ease ofsynthesizing GO and its solution processability have madeit a very attractive precursor for large scale productionof graphene in applications including transparent conduc-tors [16,20—22], chemical sensors [23,24], biosensors [23],polymer composites [25,26], battery and ultracapacitors
[15] as described in a number of review articles[5,14—16,21,23,25,27—30].
Apart from making graphene, GO itself has many intrigu-ing properties. Like graphene, GO sheets are characterisedby two abruptly different length scales. The measuredthickness is of typical molecular dimensions (ca. 1 nm) [8],but the lateral dimensions are those of common colloidalparticles, ranging from nanometers [31] up to hundreds ofmicrometers [32]. Therefore, GO sheets can be viewed aseither molecules or particles, depending on which lengthscale is of greater interest. On the other hand, GO can becharacterised as an unconventional soft material such as a2D polymer, anisotropic colloid, soft membrane, liquid crys-tal, or even amphiphile. A few examples are given below,where GO is treated as surfactant, diblock copolymer,and liquid crystal. An earlier structural model [6] and

Energetic graphene oxide: Challenges and opportunities 139
Figure 2 Snapshots showing the explosion of a piece of GO solid inside a petri dish a few seconds after placed on a hotplatepreheated at 300 ◦C. Before explosion, water vapour was driven out of the GO paper and condensed on the top cover as highlightedby the white broken circle.
recent high-resolution transmission electron microscopystudy [33] suggest that the basal plane of a GO sheet iscomposed of unoxidised, hydrophobic graphitic patches andheavily oxidised domains functionalised by hydroxyl andepoxide groups with hydrophilic carboxylic acid groups onthe edges. This makes GO effectively a 2D amphiphile witha hydrophilic periphery and largely hydrophobic centre[34,35]. Indeed, it has been shown that GO sheets aresurface active and can lower interfacial energies. Theycan accumulate at water surface during evaporation orby flotation [35], stabilise organic solvent in water [36]and disperse hard-to-process materials such as graphiteand carbon nanotubes in water [35,37], thus acting as asurfactant. An understanding of GO’s amphiphilicity hasled to improved knowledge of the structural—propertiesrelationship of GO monolayer thin films [38,39], all-carboncomposites for photovoltaics and energy storage [34,37],self-assembled GO windows for environmental scanningelectronic microscopy (SEM) [40], and GO/single walledcarbon nanotubes based interfacial layers in solution pro-cessed polymer solar cells [41]. On the other hand, GO canbe also viewed as a 2D random diblock copolymer: one blockis graphitic and the other heavily hydroxylated. Therefore,GO is capable of guiding material assembly through both�—� stacking and hydrogen bonding. For example, it hasbeen shown that GO can enhance both the electricalconductivity and solution viscosity of conjugate polymerpoly(3,4-ethylenedioxythiophene):poly(styrenesulfonate)(PEDOT:PSS), leading to a conducting gel that can be usedto establish mechanical and electrical interconnects inpolymer solar cells [42] and ultracapacitors. In a differentapplication, the chemically less stable, sp3-rich block can beselectively etched by hot water vapour to generate porousGO network, which has been shown to have much enhancedinteraction with gas molecules such as in chemical sensors[43]. GO has also been shown to be capable of forming liquid
crystalline phases [44—48], which has received increasinginterests in the past two years. A number of studies havebeen published to control the orientation and buckling ofthe aligned GO sheets [46], and to draw fibres from GObased liquid crystals [44]. In addition to the soft materialproperties, GO has been proposed as a chemically tunableplatform for optical applications based on its fluorescence,fluorescence quenching and nonlinear optical properties[30].
Motivated by both its graphene-oriented applications andits own remarkable properties, interest in GO has widelyspread across many disciplines. In parallel to the rapidprogress of research, industrial-scale production of GO hasemerged [49]. Therefore, it is important to take a look atthe thermal instability of GO, a property that is critical tothe large scale production and many applications of GO.This Review will highlight the challenges and opportunitiesassociated with GOs’ energetic nature. In the following sec-tions, we will first discuss the thermal instability of GO,metal salts catalysed combustion of its graphene product,and the potential fire risk during large scale production ofGO and methods of mitigation including an improved purifi-cation procedure for effective removal of the metal saltby-products. Next we will discuss energy efficient ways tocreate graphene from GO and the use GO as an in situ powersource to make nanoparticle decorated graphene compos-ites for energy storage and catalysts. A rapid photothermalreduction of GO and its applications in patterning and mak-ing polymer composites will also be introduced.
Thermal instability of GO
In recent years, thermal ‘‘reduction’’ of GO has become apopular method to produce chemically modified graphene[10]. In the so-called thermal ‘‘reduction’’, GO is
140 D. Krishnan et al.
actually undergoing disproportionation reactions, in whichsome of the partially oxidised sp3 carbon atoms become fullyoxidised to carbonaceous gases such as CO2 and the restreduced to sp2 graphene product. As a result of the dispro-portionation reactions, the graphitic domains in GO wouldgrow in size to form a percolated network, leading to partialrestoration of electrical conductivity [50]. However, the lossof gasified carbon atoms, which can account for around 10%of the total carbon atoms, would inevitably introduce car-bon vacancies in the basal plane. This has been confirmedby a recent high resolution transmission electron microscopy(TEM) study on GO and its reduction product (Fig. 1c) [33].Therefore, both GO and its reduced form are quite defectivein their basal planes, and GO would not be fully ‘‘reduced’’to perfect graphene unless these vacancies defects (andothers) are repaired. This suggests that simple annealingis unlikely to obtain high quality graphene, unless at ele-vated temperature and/or in carbonaceous atmosphere [51]to induce proper rearrangement and/or addition of car-bon atoms. This is a primary reason that the electronicproperties of r-GO are much degraded compared to pris-tine graphene. Therefore, GO derived graphene is usuallyreferred to as chemically modified graphene or reduced GO(r-GO) to distinguish from the more perfect graphene madefrom mechanical exfoliation from graphite crystals or chem-ical vapour deposition.
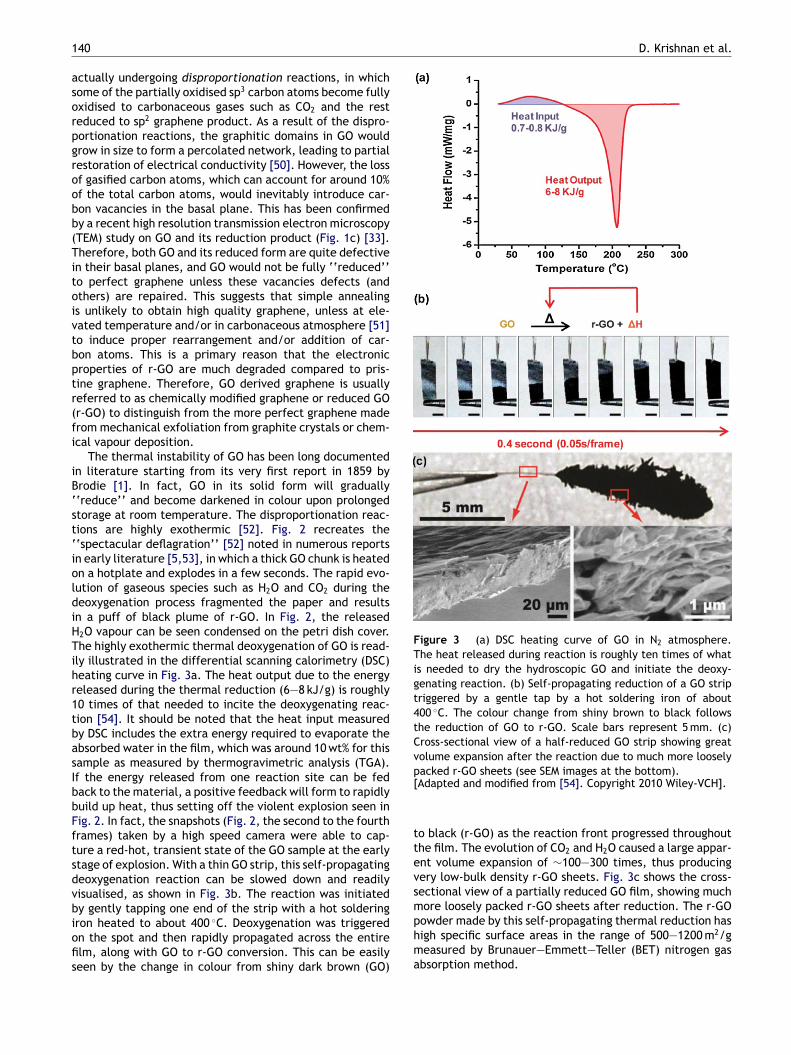
The thermal instability of GO has been long documentedin literature starting from its very first report in 1859 byBrodie [1]. In fact, GO in its solid form will gradually‘‘reduce’’ and become darkened in colour upon prolongedstorage at room temperature. The disproportionation reac-tions are highly exothermic [52]. Fig. 2 recreates the‘‘spectacular deflagration’’ [52] noted in numerous reportsin early literature [5,53], in which a thick GO chunk is heatedon a hotplate and explodes in a few seconds. The rapid evo-lution of gaseous species such as H2O and CO2 during thedeoxygenation process fragmented the paper and resultsin a puff of black plume of r-GO. In Fig. 2, the releasedH2O vapour can be seen condensed on the petri dish cover.The highly exothermic thermal deoxygenation of GO is read-ily illustrated in the differential scanning calorimetry (DSC)heating curve in Fig. 3a. The heat output due to the energyreleased during the thermal reduction (6—8 kJ/g) is roughly10 times of that needed to incite the deoxygenating reac-tion [54]. It should be noted that the heat input measuredby DSC includes the extra energy required to evaporate theabsorbed water in the film, which was around 10 wt% for thissample as measured by thermogravimetric analysis (TGA).If the energy released from one reaction site can be fedback to the material, a positive feedback will form to rapidlybuild up heat, thus setting off the violent explosion seen inFig. 2. In fact, the snapshots (Fig. 2, the second to the fourthframes) taken by a high speed camera were able to cap-ture a red-hot, transient state of the GO sample at the earlystage of explosion. With a thin GO strip, this self-propagatingdeoxygenation reaction can be slowed down and readilyvisualised, as shown in Fig. 3b. The reaction was initiatedby gently tapping one end of the strip with a hot solderingiron heated to about 400 ◦C. Deoxygenation was triggeredon the spot and then rapidly propagated across the entirefilm, along with GO to r-GO conversion. This can be easilyseen by the change in colour from shiny dark brown (GO)
Figure 3 (a) DSC heating curve of GO in N2 atmosphere.The heat released during reaction is roughly ten times of whatis needed to dry the hydroscopic GO and initiate the deoxy-genating reaction. (b) Self-propagating reduction of a GO striptriggered by a gentle tap by a hot soldering iron of about400 ◦C. The colour change from shiny brown to black followsthe reduction of GO to r-GO. Scale bars represent 5 mm. (c)Cross-sectional view of a half-reduced GO strip showing greatvolume expansion after the reaction due to much more looselypacked r-GO sheets (see SEM images at the bottom).[Adapted and modified from [54]. Copyright 2010 Wiley-VCH].
to black (r-GO) as the reaction front progressed throughoutthe film. The evolution of CO2 and H2O caused a large appar-ent volume expansion of ∼100—300 times, thus producingvery low-bulk density r-GO sheets. Fig. 3c shows the cross-sectional view of a partially reduced GO film, showing muchmore loosely packed r-GO sheets after reduction. The r-GOpowder made by this self-propagating thermal reduction hashigh specific surface areas in the range of 500—1200 m2/gmeasured by Brunauer—Emmett—Teller (BET) nitrogen gasabsorption method.

Energetic graphene oxide: Challenges and opportunities 141
Figure 4 Snapshots showing natural gas flame treatments on r-GO film with intervals of 3—4 s. No combustion occurred even aftermultiple flame treatments, clearly showing the high thermal stability of r-GO.[Adapted from [54]. Copyright 2010 Wiley-VCH].
Flammability of GO: flame retardant or firehazard?
The explosive deoxygenation shown in Fig. 2 suggests that tostore large quantities of GO, one should avoid any potentialsource of heating to prevent triggering of the exothermicreaction. Earlier literature has reported something muchmore alarming about the potential fire hazard of GO. Forexample, in his original paper published in 1859, Brodiedescribed that the solution oxidation product of graphite‘‘decomposed with ignition’’ upon heating [1]. The combus-tion graphite oxide or graphitic acid during ‘‘deflagration’’upon heating has also been noted in some other papersfrom earlier literature [5,53]. It is interesting to note thatalthough the GO solid shown in Fig. 2 exploded, it did notcombust at all even though the sample was self-heated toa red-hot state during explosion. In view of these historicalaccounts on GO’s flammability, it is quite puzzling that oneof the relatively new applications of GO and r-GO is actuallyas flame retardant adduct for polymeric materials [55—61]!Naturally one would ask are GO and r-GO flame retardantsor fuels for combustion that can lead to potential fire hazard[54,62]?
The flammability of GO and r-GO can be easily testedby burning with a natural gas flame. Flame treatment ofGO triggers the same type of self-propagating reduction asshown in Fig. 3b. However, the resulting graphene prod-uct is quite stable against combustion, even though it isalready well exfoliated and has high surface area. This canbe noticed in Fig. 4 which shows an r-GO film being recur-rently exposed to a natural gas flame. Although the exposededge turned red hot, no further combustion was observedupon removal from the flame. Prolonged flame treatmentonly caused minor material loss, suggesting the high thermalstability of the graphene sheets due to their highly stablegraphitic structure. This is also consistent with the flame-retarding behaviour of graphene nanoplatelets made fromexpandable graphite. It appears that GO and r-GO shouldnot be flammable, although as we know now, GO can beeasily reduced to r-GO by burning.
Now the question is why GO was reported to be flammablein some of the papers from the early literature? Since thesynthesis of GO requires the use of very strong oxidant,
and some of the oxidants such as KMnO4 used in the Hum-mers method [4] can readily decompose to produce oxygenupon heating, one could infer that maybe there was residueoxidant not completely removed during purification, whichmight be responsible for the reported flammability of GO.To test the thought, we deliberately contaminated neat GOsamples by soaking a strip in KMnO4 solution. After drying,the resulting thin film did become flammable. Treating itwith a natural gas flame triggered both explosion and com-bustion, burning away most of the material. However, if onefollows the synthetic protocols correctly, it is very unlikelyto find unreacted KMnO4 in the final product since excesshydrogen peroxide is added after reaction to reduce KMnO4.
The next suspects would be other water soluble potas-sium salts. It has been well known that alkali and alkalinemetal salts are quite effective catalysts for graphite com-bustion even at low loading percent [63—68]. Potassium saltcatalysed ‘‘carbon activation’’ reactions have been used toetch nanoporous structures in carbon materials [69], or forcoal gasification in oxidative atmospheres [65]. The synthe-sis of GO typically involves potassium salts such as KMnO4
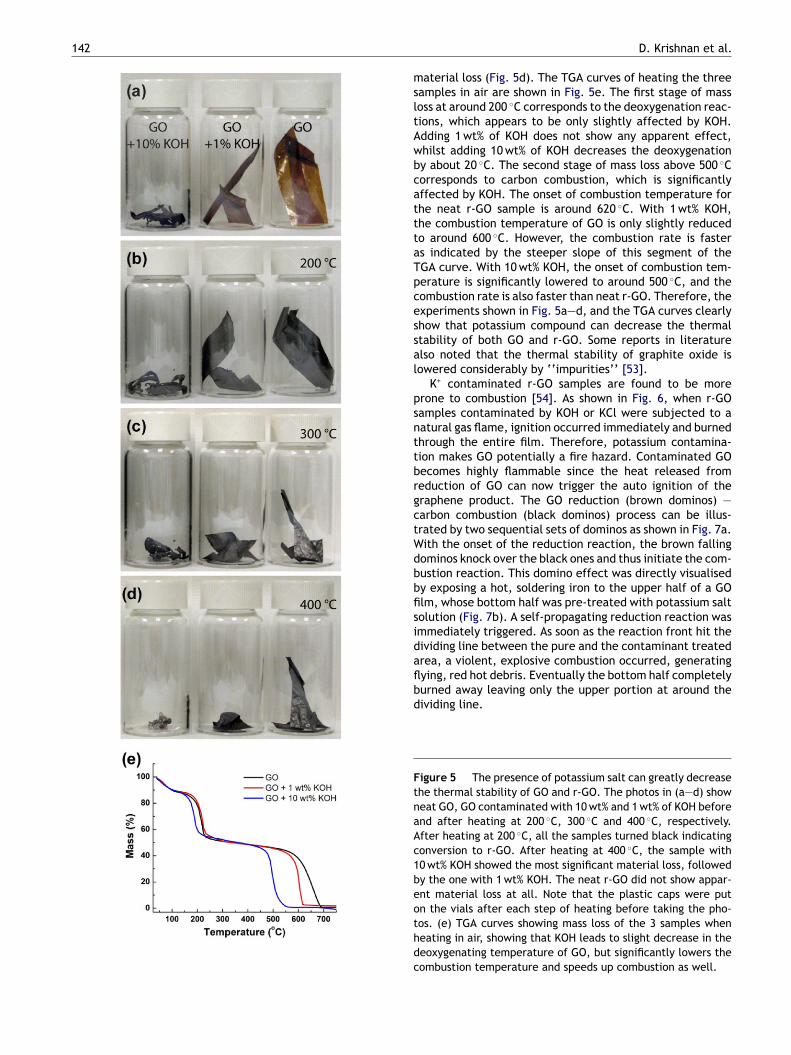
and K2S2O8, which after oxidation and subsequential washingsteps, may turn into various forms of potassium compounds,including KOH, KCl, K2SO4, KNO3, etc. Although these saltsare highly soluble in water, it is not trivial to remove themfrom GO. Filtration usually becomes very time consuming(days to weeks), since exfoliated GO tends to block the filterpapers. As noted by Brodie in his original paper, GO tends togel during washing, making purification even more tedious.All these make it difficult to completely remove the inor-ganic ions, often resulting in trace amounts of potassiumsalts. To illustrate the effect of K+ on graphene’s thermalstability, 3 samples: GO + 10 wt% KOH, GO + 1 wt% KOH andneat GO were placed in glass vial and heated in air (Fig. 5a).KOH was chosen due to its non-oxidative nature. To avoidexplosive exfoliation of GO, the samples were first heatedto 200 ◦C and maintained at the temperature overnight. Thistreatment converted all the GO samples to r-GO, as indi-cated by the change of colour from brown to black (Fig. 5b).Then the samples were heated at 300 ◦C for another 5 h,and some material loss can be seen (Fig. 5c). After heatingat 400 ◦C for another 12 h, the sample with 10 wt% of KOHhad the most significant material loss, followed by the onewith 1 wt%. The neat r-GO sample did not show any apparent
142 D. Krishnan et al.
material loss (Fig. 5d). The TGA curves of heating the threesamples in air are shown in Fig. 5e. The first stage of massloss at around 200 ◦C corresponds to the deoxygenation reac-tions, which appears to be only slightly affected by KOH.Adding 1 wt% of KOH does not show any apparent effect,whilst adding 10 wt% of KOH decreases the deoxygenationby about 20 ◦C. The second stage of mass loss above 500 ◦Ccorresponds to carbon combustion, which is significantlyaffected by KOH. The onset of combustion temperature forthe neat r-GO sample is around 620 ◦C. With 1 wt% KOH,the combustion temperature of GO is only slightly reducedto around 600 ◦C. However, the combustion rate is fasteras indicated by the steeper slope of this segment of theTGA curve. With 10 wt% KOH, the onset of combustion tem-perature is significantly lowered to around 500 ◦C, and thecombustion rate is also faster than neat r-GO. Therefore, theexperiments shown in Fig. 5a—d, and the TGA curves clearlyshow that potassium compound can decrease the thermalstability of both GO and r-GO. Some reports in literaturealso noted that the thermal stability of graphite oxide islowered considerably by ‘‘impurities’’ [53].
K+ contaminated r-GO samples are found to be moreprone to combustion [54]. As shown in Fig. 6, when r-GOsamples contaminated by KOH or KCl were subjected to anatural gas flame, ignition occurred immediately and burnedthrough the entire film. Therefore, potassium contamina-tion makes GO potentially a fire hazard. Contaminated GObecomes highly flammable since the heat released fromreduction of GO can now trigger the auto ignition of thegraphene product. The GO reduction (brown dominos) —carbon combustion (black dominos) process can be illus-trated by two sequential sets of dominos as shown in Fig. 7a.With the onset of the reduction reaction, the brown fallingdominos knock over the black ones and thus initiate the com-bustion reaction. This domino effect was directly visualisedby exposing a hot, soldering iron to the upper half of a GOfilm, whose bottom half was pre-treated with potassium saltsolution (Fig. 7b). A self-propagating reduction reaction wasimmediately triggered. As soon as the reaction front hit thedividing line between the pure and the contaminant treatedarea, a violent, explosive combustion occurred, generatingflying, red hot debris. Eventually the bottom half completelyburned away leaving only the upper portion at around thedividing line.
Figure 5 The presence of potassium salt can greatly decreasethe thermal stability of GO and r-GO. The photos in (a—d) showneat GO, GO contaminated with 10 wt% and 1 wt% of KOH beforeand after heating at 200 ◦C, 300 ◦C and 400 ◦C, respectively.After heating at 200 ◦C, all the samples turned black indicatingconversion to r-GO. After heating at 400 ◦C, the sample with10 wt% KOH showed the most significant material loss, followedby the one with 1 wt% KOH. The neat r-GO did not show appar-ent material loss at all. Note that the plastic caps were puton the vials after each step of heating before taking the pho-tos. (e) TGA curves showing mass loss of the 3 samples whenheating in air, showing that KOH leads to slight decrease in thedeoxygenating temperature of GO, but significantly lowers thecombustion temperature and speeds up combustion as well.

Energetic graphene oxide: Challenges and opportunities 143
Figure 6 Snapshots showing flame treatment on r-GO films contaminated with (a) KOH and (b) KCl, respectively (ca. 1 wt%).The flame immediately triggered the self-propagating combustion in the film, which suggests that potassium salts can significantlyreduce the thermal stability of r-GO. The intervals between the snapshots are a few seconds.[(a) is adapted from [54]. Copyright 2010 Wiley-VCH].
Now it is clear that although GO is energetic and ther-mally unstable, its clean graphene product is remarkablystable against combustion (Fig. 4). In fact, the explo-sive exfoliation shown in Fig. 2 may even be useful forextinguishing fire since it releases a puff of light weight,flame-retarding graphene powders. Combustion of GO notedin the early literature may be a result of incomplete removalof the salt by-products. Indeed, it has not been reportedas much in recent literature, presumably due to moreadvanced sample purification apparatuses available to mod-ern researchers. Other than potassium salts, we found thatsodium salts can also increase the flammability of r-GO.Studies of catalytic carbon combustion [70] implied thatthis effect could be general to alkali and alkaline salts. Infact, Brodie also noted that the complex of GO with bar-ium hydroxide tends to ‘‘explode yet with greater violence’’upon heating. Combustion catalysed by other metal com-pounds such as Fe has also been noted [71]. Higher saltcontent results in lower thermal stability as shown in Fig. 5,which also makes GO more flammable.
Fire safety of GO
Although graphite nanoplatelets, r-GO and even GO havebeen demonstrated to have flame-retarding properties, it isnow clear that GO solids containing salt by-products can behighly flammable. Since commercial scale production of GOhas emerged [49], it is time to be prepared for the potentialfire risk of GO. An accidental spark, excessive exposure tolight, or a local hot spot generated by heat accumulatedfrom slow self-reduction can trigger rapid, catastrophictotal combustion, which poses a serious fire hazard. This
is particularly alarming for the fire safety of large scaleproduction, processing, and storage of GO materials, espe-cially in solid state. In addition, the gas evolution during GOreduction could rapidly build up pressure in a sealed con-tainer. Note that in a highly moisturised environment, thealkali metal salts could also catalyse additional water—gasreactions [66—68], converting some of the graphene mate-rial into CO and H2, which poses an even greater safetyhazard. Therefore, from a chemical safety point of view,large amounts of GO should be best stored in solution. WhenGO is stored as a solid, heat sources, electrical sparks andexposure to high intensity light should be avoided. GO thinfilms and porous GO foams having effective heat dissipa-tion should be relatively safer. It is also helpful to monitorthe sample temperature regularly to watch for excess heatbuildup. To avoid combustion, it may also be helpful to stor-age GO in oxygen-free atmosphere.
Avoid flammable GO: improved purificationmethod
Due to the explosive and flammable nature of GO in thepresence of metal salt contaminants, it is necessary tofurther improve the purification process of GO, especiallyin scaled-up reactions. Recent work showed that salt by-product can also interfere with how GO sheets assemble insolution [44]. Although the inorganic salt by-products fromGO synthesis are quite water soluble, removing them is nottrivial, since GO dispersions tend to undergo gelation as thepH value increases during washing. This was also noted inBrodie’s original paper in 1859. Gelation hinders washingof the GO product and makes commonly used purification

144 D. Krishnan et al.
Figure 7 Reduction—combustion reactions in potassium compound contaminated GO. (a) Schematic illustration of self-propagating, reduction—combustion domino-like reactions in GO. The two sets of dominos represent GO reduction (grey) and carboncombustion (black), respectively. (b) Reactions were triggered by a hot soldering iron from the untreated side. Reduction startedfrom the upper half and rapidly propagated over the boundary (dotted line), which then triggered violent reduction—combustionreactions in the KOH treated bottom half area. The heat released from reduction is capable of igniting r-GO contaminated withpotassium salt, which is an even more exothermic reaction as represented by the taller dominos in (a). The bottom half completelyburned away whilst the upper part stayed stable.[Adapted from [54]. Copyright 2010 Wiley-VCH].
techniques, such as centrifugation and, especially, filtration,very tedious tasks. In fact, in some reported purification pro-cedures, GO needs to be filtered for days and/or dialysed forweeks. For example, filtration of GO is a self-limiting pro-cess as GO sheets tend to block the filter paper — in fact,this is how GO papers are made [9,20]. In our own experi-ence, after the formation of the first ten microns of GO filtercake, filtration essentially stops and evaporation becomesmore effective for removing water. In addition, evaporationis also hindered by GO’s amphiphilicity as they tend to forma dense film at the air—water interface [35,40]. All thesedifficulties not only make it tedious to purify GO, but alsoincrease the likelihood of trapping metal salt by-products inGO.
Salts and acids are two major types of inorganic by-products in GO syntheses. Although both are well solublein water, if the as synthesised product is rinsed with waterdirectly, volume expansion and gelation would start to occur,rendering filtration or centrifugation quite difficult (Fig. 8,top). We have found that gelation of GO can be greatlysuppressed in strong acids or acetone. Therefore, we havedeveloped a two-step, acid—acetone washing procedure to
avoid gelation and speed up the purification process [54].In the first step, excess amount of strong acid is used toremove the salts, followed by acetone to remove the acid.In a typical experiment, 3.4 wt% HCl is used for the first stepof rinsing (Fig. 8). The pH was measured to be around 0,which can suppress the gelation of GO. More importantly,HCl solution is quite effective in removing ions from theproducts. HCl does not leave any solid residue, either. Afterrinsing 3—4 times with HCl the product is fully dried in air orunder vacuum, and re-dispersed in acetone. The dispersionis then filtered and rinsed using more acetone. Acetone canremove the remaining HCl effectively without causing gela-tion, and readily evaporates without leaving solid residue.In addition, GO sheets can disperse temporarily to allow suf-ficient washing, but will aggregate later to avoid forming asmooth and dense filter cake, thus making filtration muchfaster. This two-step method can greatly speed up filtrationor centrifugation of GO, facilitating the purification of GO.
We developed and tested the two-step procedure inde-pendently, and included the results in Ref. [54]. However,when we were writing the manuscript, we found that a 1974paper published in Russian Journal of Inorganic Chemistry by

Energetic graphene oxide: Challenges and opportunities 145
Figure 8 Improved purification method of GO for effective removal of metal ions. (a) The left vial contains as-synthesised GO,which greatly expands and gelates upon washing by water, making filtration or centrifugation quite tedious. However, gelation canbe overcome by a two-step, acid—acetone washing procedure. First, the sample is rinsed with HCl (pH 0) to remove metal ionswithout exfoliating GO sheets. Next, acetone is used to remove the excess acid (b). Finally, GO water dispersion can be obtainedthrough solvent-exchange or by directly dispersing dried GO solid product.[Adapted and modified from [54]. Copyright 2010 Wiley-VCH].
Nikolaev et al. had already reported purifying GO by washingwith strong acid and acetone [72]. Unfortunately this paperseems to have been largely unnoticed in the past 20 yearsbased on its citation history: according to Web of Sciencerecord, it was cited 4 times between 1986 and 1993 but hasnot been cited again until by us in Ref. [54] in 2010. Never-theless, this coincidence suggests that the two-step washingmethod has already been proven effective by at least twoindependent studies. Using this method, large quantitiesof GO solid can be obtained by filtration and vacuum dry-ing. GO dispersion in various solvents can then be obtainedthrough solvent exchange after the final acetone-washingstep (Fig. 8), or by directly dispersing the dried solid inthe solvent of choice. The potassium content in GO samplesrinsed with HCl and acetone 3—4 times was undetectable byenergy-dispersive X-ray spectroscopy (EDX), which indicatesthe concentration should be lower than 0.1 wt%. Such puri-fied GO samples produces graphene products that are verystable against combustion, like those shown in Fig. 4.
Flash photothermal reduction of GO
Instantaneous reduction of GO by a camera flash
The most popular methods for reducing GO include chemicaltreatment using strong reducing agents such as hydrazineor its derivatives, and thermal treatment in various inertor reducing atmospheres. However, applying such methodscould be challenging if GO is to be blended with othermaterials, such as in composites since the polymer compo-nent may prevent the reducing agent from fully reactingwith GO, or it may be unstable at the annealing tempera-ture. Alternative reducing methods that do not rely on theuse of chemicals or high temperature would maximise theadvantage of GO’s excellent solvent processability. Fig. 3asuggests that GO can be easily reduced by absorbing a smallamount of thermal energy (on the order of 0.6 kJ/g), which is
also demonstrated by the self-propagating reduction shownin Fig. 3b. This allows great flexibility in designing reductionmethods for GO other than direct heating or chemical reduc-tion. This section will introduce a flash reduction processwhere GO can be photothermally reduced upon exposure toa pulsed Xenon flash at ambient conditions [73]. Flash reduc-tion is rapid, clean, and versatile and can be done with aconsumer camera flash unit. It consumes a lot less energycompared to conventional heating treatment in furnaces.
A close camera flash provides sufficient energy to pho-tothermally heat up GO to r-GO (Fig. 9a). Fig. 9b is theabsorption spectrum of a 1 �m thick GO film. It can be cal-culated from the spectrum that the film absorbs about 63%of the incident photon energy in the visible range between400 nm and 800 nm. Common camera flash units as thoseequipped with digital cameras can deliver 0.1—2 J/cm2 ofenergy per pulse at close distances (<2 mm) as measured bypyroelectric energy meter. For the sake of simplicity, herewe assume the pulse energy is 1 J/cm2. Then 630 mJ/cm2
of photon energy will be absorbed by the GO film. Sincethere is no significant alternative pathways to convert thephoton energy for GO, most of the absorbed light energyshould be converted to heat. The DSC heating curve of GOin Fig. 3a suggests that it takes up to 0.8 kJ/g of heat to trig-ger the deoxygenating reaction of GO. Since the density ofthe 1 �m thick GO paper is calculated to be 0.187 mg/cm2,the mass-based energy input measured in DSC can be con-verted to area-based energy input. The energy needed toreduce GO 0.8 kJ/g would be equal to 150 J/cm2. Therefore,a pulse of camera flash can deliver more than enough energy(630 mJ/cm2) to photothermally reduce GO. Note that dur-ing the conversion from GO to r-GO, the light absorptionefficiency dramatically increases. For example, based onFig. 9b, assuming there is no electronic coupling betweenthe stacked GO sheets, a single layer of GO (roughly 1000sheets in a 1 �m thick film) should absorb about 0.063% ofwhite light. A single layer graphene absorbs 2.3% of whitelight [74], which is more than 30 times greater than GO.

146 D. Krishnan et al.
Figure 9 Flash reduction of GO. (a) GO solid can be photothermally reduce to r-GO. (b) UV/vis spectrum of a 1 �m thick GOfilm. The average absorption over the visible range (400—800 nm) is 63%. A GO paper (c) was instantaneously reduced (d) uponexposure to a photographic camera flash. The grids in the background are 1 mm × 1 mm. The flash reduction of GO was evident bythe dramatic changes in colour (c, before; d, after) and electrical conductivity as shown in (e, before; f, after).[Adapted and modified from [73]. Copyright 2010 American Chemical Society].
Therefore, formation of r-GO during reduction will greatlyintensify the photothermal heating effect. Indeed, a closecamera flash can turn the brown, transparent GO film shownin Fig. 9c to a black and opaque (Fig. 9d) r-GO film alongwith a loud pop sound. For thicker, translucent GO films,flash irradiation can heat up the surface and trigger self-propagating reduction throughout the whole material. Theconductivity of flash reduced r-GO was measured to bearound 1000 S/m (or 10 S/cm), which is comparable to thosemade from common chemical or thermal treatments. Fig. 9eand f shows that a camera flash instantaneously convert theinsulating GO film to conductive r-GO, thus completing thecircuit and lighting up the bulb.
Flash photothermal lithography of GO and r-GO
Being a light based method, flash reduction has a greatadvantage over the conventional reducing processes in that
it readily allows photo patterning. Using a photomask ordirect writing with a high intensity beam, conducting r-GO patterns can be generated on the insulating GO film[73]. Note that this works best for substrate supported thinfilms, as the self-propagating reduction along lateral direc-tions (see Fig. 3b) is limited due to efficient dissipation ofreleased heat. By flashing through a photomask with a hol-low letter ‘‘N’’ (Fig. 10a, left), a black ‘‘N’’ shaped r-GOdomain was created in a GO film supported on filter paper(Fig. 10a, centre). When flashed further with a flash lampof higher intensity, the reduced r-GO area can be ablateddue to rapid degassing and air expansion, leaving an etched‘‘N’’-shaped mark in the GO film (Fig. 10a, right). There-fore, tuning the intensity of the flash irradiations, which canbe done by adjusting either the energy output of the flashlamp or its distance to the sample, enables both pattern-ing and etching to be performed in a single experimentalsetup.

Energetic graphene oxide: Challenges and opportunities 147
Figure 10 Flash patterning of GO. (a) Dry flash lithography of GO. Left: a shadow mask defining an exposed area with ‘‘N’’ shapewas taped onto a GO film supported by filter paper. Centre: the black, N-shaped, r-GO pattern obtained after flash. Right: theexposed area was ablated by higher power flashes, leaving an etched ‘‘N’’ pattern on the film. (b) Fuel ignition by flashing GO. Aseries of photographs showing a GO foam sample before (a), during (b) and after (c) flash ignition with the time interval labelled inmilliseconds (ms). By placing a GO foam sample onto a paper soaked with ethanol and flashing, the GO foam is capable of ignitingthe ethanol vapour as depicted in the centre photograph. [(a) is adapted from [73].Copyright 2010 American Chemical Society; (b) is adapted from [75]. Copyright 2010 Wiley-VCH].
GO assisted photothermal fuel ignition
Since the exothermic decomposition of GO can be eas-ily triggered by a flash, the additional heat released fromGO can be conducted to a fuel system to aid its ignitionfor more efficient combustion. Gilje et al., demonstratedsuccessful ignition of ethanol fuels using GO as a pho-tothermal initiator [75]. Fig. 10b shows a photo of a GOfoam sample placed on a paper soaked with ethanol. Uponflashing, the ethanol vapour readily combusts. After theethanol fuel is consumed, the graphene product can beseen glowing bright red as a result of the combustion reac-tion, again demonstrating excellent stability of r-GO (ifnot contaminated with salt) against combustion. Pyrom-eter readings of this ignition process indicate that byflashing GO achieves temperatures of 400—500 ◦C withina few milliseconds. Thus, GO can be used to tune thephoto ignition behaviour of a fuel for a more controlledand distributed ignition. However, extending the appli-cation to more aliphatic fuels may be limited beforesolving the poor dispersibility of GO in these non-polar sol-vents.
Flash sintering and patterning of GO/polymercomposite thin films
The excellent solution processability of GO in polar sol-vents makes it an ideal precursor for the preparationof graphene/polymer composites. However, after blendingwith the polymer component, chemical reduction meth-ods may not be feasible as GO is now protected by thesurrounding polymer. Direct heating could be a problem if
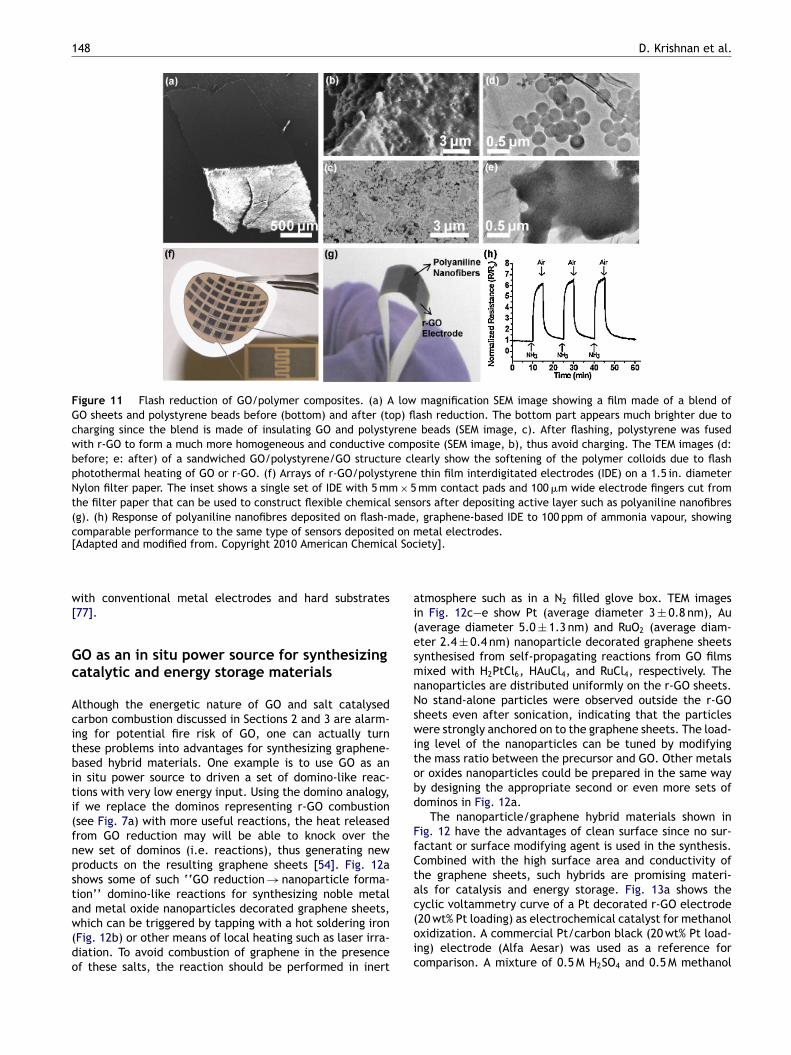
the polymer is unstable upon annealing. Flash photother-mal irradiation can offer transient, selective heating ofGO, if the polymer component is not strongly absorbing.And the excess heat from either direct flash heating ordeoxygenation can sinter the r-GO sheets and polymer par-ticles to form more continuous structures [73]. Fig. 11a—eshows that GO can be flash-reduced and welded withpolystyrene beads. Before flash, the film of GO/polystyrene,made by filtration from their colloidal solutions, is brit-tle and cracks easily as shown in the SEM image inFig. 11a (bottom). Flash irradiation can reduce GO andmelt the surrounding polystyrene particles (Fig. 11b andc), leading to a continuous, conducting film (Fig. 11a,top). The brightness contrast between the bottom (beforeflash) and top (after flash) of the film shown in Fig. 11areflects significant improvement in electrical conductivitybefore and after flash irradiation. The melting of poly-mer colloids can be clearly observed by TEM (Fig. 11d ande). Since polystyrene is transparent in the visible range,they act as a heat sink through melting to drain awayexcess heat, which is particularly beneficial for pattern-ing applications as it avoids overexposure due to lateralself-propagating reduction of GO. Fig. 11f shows arrays ofinterdigitated r-GO/polystyrene electrodes fabricated on aflexible Nylon filter paper by flash patterning. The exposedareas became conducting as the sheet resistance droppedsignificantly from 2 × 105 k�/square to 9.5 k�/square. Whendeposited with conducting polymer such as polyanilinenanofibres [76], these flexible electrodes (Fig. 11g) canbe used as chemical vapour sensors. Fig. 11h shows theresponse of one of such all-organic, flexible sensors to100 ppm of NH3 vapour. The sensitivity and time responseare comparable to polyaniline nanofibres sensors made
148 D. Krishnan et al.
Figure 11 Flash reduction of GO/polymer composites. (a) A low magnification SEM image showing a film made of a blend ofGO sheets and polystyrene beads before (bottom) and after (top) flash reduction. The bottom part appears much brighter due tocharging since the blend is made of insulating GO and polystyrene beads (SEM image, c). After flashing, polystyrene was fusedwith r-GO to form a much more homogeneous and conductive composite (SEM image, b), thus avoid charging. The TEM images (d:before; e: after) of a sandwiched GO/polystyrene/GO structure clearly show the softening of the polymer colloids due to flashphotothermal heating of GO or r-GO. (f) Arrays of r-GO/polystyrene thin film interdigitated electrodes (IDE) on a 1.5 in. diameterNylon filter paper. The inset shows a single set of IDE with 5 mm × 5 mm contact pads and 100 �m wide electrode fingers cut fromthe filter paper that can be used to construct flexible chemical sensors after depositing active layer such as polyaniline nanofibres(g). (h) Response of polyaniline nanofibres deposited on flash-made, graphene-based IDE to 100 ppm of ammonia vapour, showingcomparable performance to the same type of sensors deposited on metal electrodes.[Adapted and modified from. Copyright 2010 American Chemical Society].
with conventional metal electrodes and hard substrates[77].
GO as an in situ power source for synthesizingcatalytic and energy storage materials
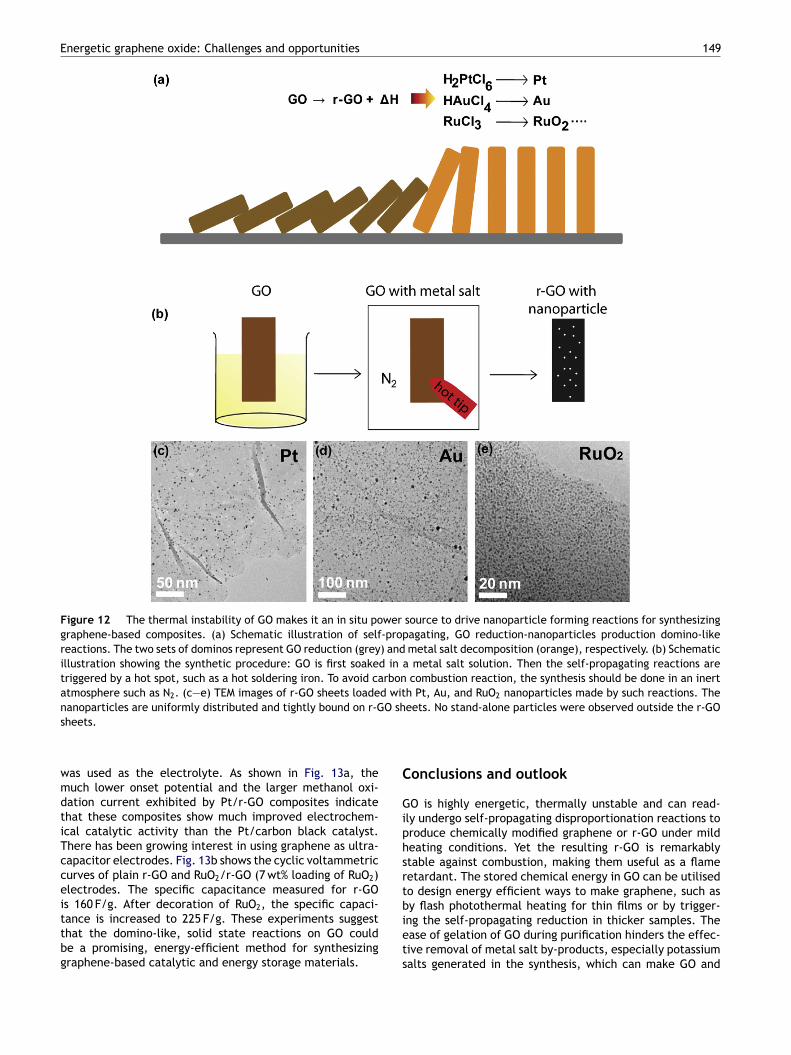
Although the energetic nature of GO and salt catalysedcarbon combustion discussed in Sections 2 and 3 are alarm-ing for potential fire risk of GO, one can actually turnthese problems into advantages for synthesizing graphene-based hybrid materials. One example is to use GO as anin situ power source to driven a set of domino-like reac-tions with very low energy input. Using the domino analogy,if we replace the dominos representing r-GO combustion(see Fig. 7a) with more useful reactions, the heat releasedfrom GO reduction may will be able to knock over thenew set of dominos (i.e. reactions), thus generating newproducts on the resulting graphene sheets [54]. Fig. 12ashows some of such ‘‘GO reduction → nanoparticle forma-tion’’ domino-like reactions for synthesizing noble metaland metal oxide nanoparticles decorated graphene sheets,which can be triggered by tapping with a hot soldering iron(Fig. 12b) or other means of local heating such as laser irra-diation. To avoid combustion of graphene in the presenceof these salts, the reaction should be performed in inert
atmosphere such as in a N2 filled glove box. TEM imagesin Fig. 12c—e show Pt (average diameter 3 ± 0.8 nm), Au(average diameter 5.0 ± 1.3 nm) and RuO2 (average diam-eter 2.4 ± 0.4 nm) nanoparticle decorated graphene sheetssynthesised from self-propagating reactions from GO filmsmixed with H2PtCl6, HAuCl4, and RuCl4, respectively. Thenanoparticles are distributed uniformly on the r-GO sheets.No stand-alone particles were observed outside the r-GOsheets even after sonication, indicating that the particleswere strongly anchored on to the graphene sheets. The load-ing level of the nanoparticles can be tuned by modifyingthe mass ratio between the precursor and GO. Other metalsor oxides nanoparticles could be prepared in the same wayby designing the appropriate second or even more sets ofdominos in Fig. 12a.
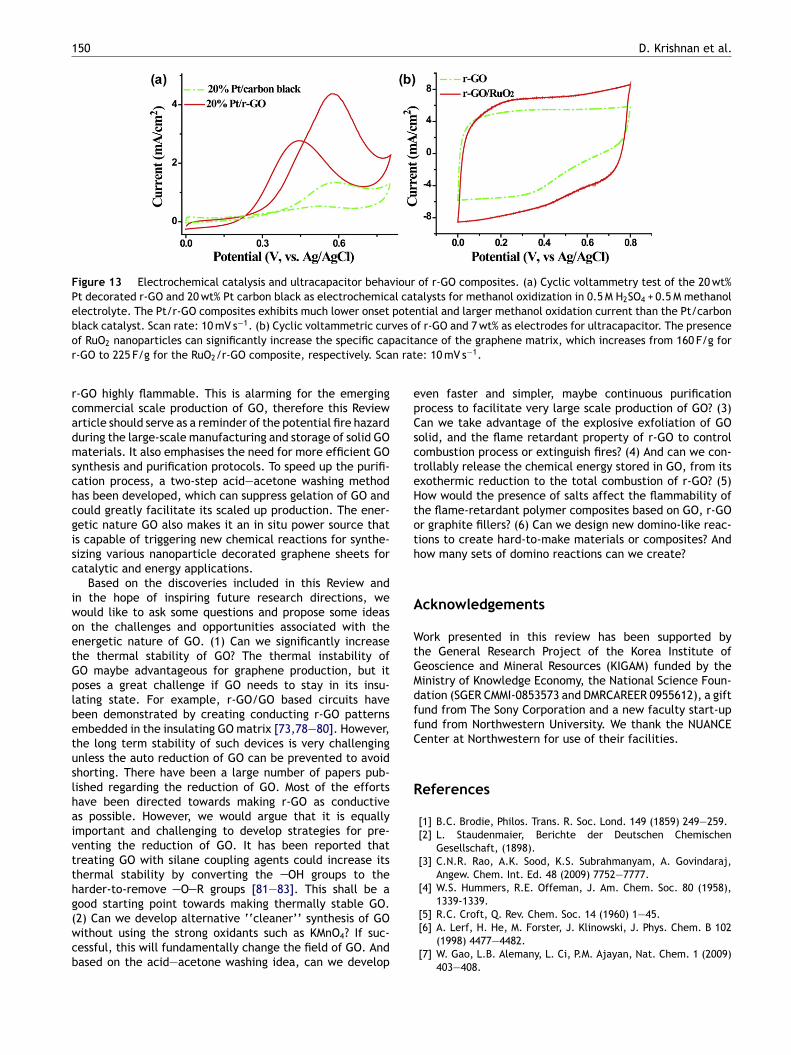
The nanoparticle/graphene hybrid materials shown inFig. 12 have the advantages of clean surface since no sur-factant or surface modifying agent is used in the synthesis.Combined with the high surface area and conductivity ofthe graphene sheets, such hybrids are promising materi-als for catalysis and energy storage. Fig. 13a shows thecyclic voltammetry curve of a Pt decorated r-GO electrode(20 wt% Pt loading) as electrochemical catalyst for methanoloxidization. A commercial Pt/carbon black (20 wt% Pt load-ing) electrode (Alfa Aesar) was used as a reference forcomparison. A mixture of 0.5 M H2SO4 and 0.5 M methanol
Energetic graphene oxide: Challenges and opportunities 149
Figure 12 The thermal instability of GO makes it an in situ power source to drive nanoparticle forming reactions for synthesizinggraphene-based composites. (a) Schematic illustration of self-propagating, GO reduction-nanoparticles production domino-likereactions. The two sets of dominos represent GO reduction (grey) and metal salt decomposition (orange), respectively. (b) Schematicillustration showing the synthetic procedure: GO is first soaked in a metal salt solution. Then the self-propagating reactions aretriggered by a hot spot, such as a hot soldering iron. To avoid carbon combustion reaction, the synthesis should be done in an inertatmosphere such as N2. (c—e) TEM images of r-GO sheets loaded with Pt, Au, and RuO2 nanoparticles made by such reactions. Thenanoparticles are uniformly distributed and tightly bound on r-GO sheets. No stand-alone particles were observed outside the r-GOsheets.
was used as the electrolyte. As shown in Fig. 13a, themuch lower onset potential and the larger methanol oxi-dation current exhibited by Pt/r-GO composites indicatethat these composites show much improved electrochem-ical catalytic activity than the Pt/carbon black catalyst.There has been growing interest in using graphene as ultra-capacitor electrodes. Fig. 13b shows the cyclic voltammetriccurves of plain r-GO and RuO2/r-GO (7 wt% loading of RuO2)electrodes. The specific capacitance measured for r-GOis 160 F/g. After decoration of RuO2, the specific capaci-tance is increased to 225 F/g. These experiments suggestthat the domino-like, solid state reactions on GO couldbe a promising, energy-efficient method for synthesizinggraphene-based catalytic and energy storage materials.
Conclusions and outlook
GO is highly energetic, thermally unstable and can read-ily undergo self-propagating disproportionation reactions toproduce chemically modified graphene or r-GO under mildheating conditions. Yet the resulting r-GO is remarkablystable against combustion, making them useful as a flameretardant. The stored chemical energy in GO can be utilisedto design energy efficient ways to make graphene, such asby flash photothermal heating for thin films or by trigger-ing the self-propagating reduction in thicker samples. Theease of gelation of GO during purification hinders the effec-tive removal of metal salt by-products, especially potassiumsalts generated in the synthesis, which can make GO and
150 D. Krishnan et al.
Figure 13 Electrochemical catalysis and ultracapacitor behaviour of r-GO composites. (a) Cyclic voltammetry test of the 20 wt%Pt decorated r-GO and 20 wt% Pt carbon black as electrochemical catalysts for methanol oxidization in 0.5 M H2SO4 + 0.5 M methanolelectrolyte. The Pt/r-GO composites exhibits much lower onset potential and larger methanol oxidation current than the Pt/carbonblack catalyst. Scan rate: 10 mV s−1. (b) Cyclic voltammetric curves of r-GO and 7 wt% as electrodes for ultracapacitor. The presenceof RuO2 nanoparticles can significantly increase the specific capacitance of the graphene matrix, which increases from 160 F/g forr-GO to 225 F/g for the RuO2/r-GO composite, respectively. Scan rate: 10 mV s−1.
r-GO highly flammable. This is alarming for the emergingcommercial scale production of GO, therefore this Reviewarticle should serve as a reminder of the potential fire hazardduring the large-scale manufacturing and storage of solid GOmaterials. It also emphasises the need for more efficient GOsynthesis and purification protocols. To speed up the purifi-cation process, a two-step acid—acetone washing methodhas been developed, which can suppress gelation of GO andcould greatly facilitate its scaled up production. The ener-getic nature GO also makes it an in situ power source thatis capable of triggering new chemical reactions for synthe-sizing various nanoparticle decorated graphene sheets forcatalytic and energy applications.
Based on the discoveries included in this Review andin the hope of inspiring future research directions, wewould like to ask some questions and propose some ideason the challenges and opportunities associated with theenergetic nature of GO. (1) Can we significantly increasethe thermal stability of GO? The thermal instability ofGO maybe advantageous for graphene production, but itposes a great challenge if GO needs to stay in its insu-lating state. For example, r-GO/GO based circuits havebeen demonstrated by creating conducting r-GO patternsembedded in the insulating GO matrix [73,78—80]. However,the long term stability of such devices is very challengingunless the auto reduction of GO can be prevented to avoidshorting. There have been a large number of papers pub-lished regarding the reduction of GO. Most of the effortshave been directed towards making r-GO as conductiveas possible. However, we would argue that it is equallyimportant and challenging to develop strategies for pre-venting the reduction of GO. It has been reported thattreating GO with silane coupling agents could increase itsthermal stability by converting the OH groups to theharder-to-remove O R groups [81—83]. This shall be agood starting point towards making thermally stable GO.(2) Can we develop alternative ‘‘cleaner’’ synthesis of GOwithout using the strong oxidants such as KMnO4? If suc-cessful, this will fundamentally change the field of GO. Andbased on the acid—acetone washing idea, can we develop
even faster and simpler, maybe continuous purificationprocess to facilitate very large scale production of GO? (3)Can we take advantage of the explosive exfoliation of GOsolid, and the flame retardant property of r-GO to controlcombustion process or extinguish fires? (4) And can we con-trollably release the chemical energy stored in GO, from itsexothermic reduction to the total combustion of r-GO? (5)How would the presence of salts affect the flammability ofthe flame-retardant polymer composites based on GO, r-GOor graphite fillers? (6) Can we design new domino-like reac-tions to create hard-to-make materials or composites? Andhow many sets of domino reactions can we create?
Acknowledgements
Work presented in this review has been supported bythe General Research Project of the Korea Institute ofGeoscience and Mineral Resources (KIGAM) funded by theMinistry of Knowledge Economy, the National Science Foun-dation (SGER CMMI-0853573 and DMRCAREER 0955612), a giftfund from The Sony Corporation and a new faculty start-upfund from Northwestern University. We thank the NUANCECenter at Northwestern for use of their facilities.
References
[1] B.C. Brodie, Philos. Trans. R. Soc. Lond. 149 (1859) 249—259.[2] L. Staudenmaier, Berichte der Deutschen Chemischen
Gesellschaft, (1898).[3] C.N.R. Rao, A.K. Sood, K.S. Subrahmanyam, A. Govindaraj,
Angew. Chem. Int. Ed. 48 (2009) 7752—7777.[4] W.S. Hummers, R.E. Offeman, J. Am. Chem. Soc. 80 (1958),
1339-1339.[5] R.C. Croft, Q. Rev. Chem. Soc. 14 (1960) 1—45.[6] A. Lerf, H. He, M. Forster, J. Klinowski, J. Phys. Chem. B 102
(1998) 4477—4482.[7] W. Gao, L.B. Alemany, L. Ci, P.M. Ajayan, Nat. Chem. 1 (2009)
403—408.

Energetic graphene oxide: Challenges and opportunities 151
[8] S. Stankovich, D.A. Dikin, R.D. Piner, K.A. Kohlhaas, A. Klein-hammes, Y. Jia, Y. Wu, S.T. Nguyen, R.S. Ruoff, Carbon 45(2007) 1558—1565.
[9] D.A. Dikin, S. Stankovich, E.J. Zimney, R.D. Piner, G.H.B. Dom-mett, G. Evmenenko, S.T. Nguyen, R.S. Ruoff, Nature 448(2007) 457—460.
[10] H.C. Schniepp, J.L. Li, M.J. McAllister, H. Sai, M. Herrera-Alonso, D.H. Adamson, R.K. Prud’homme, R. Car, D.A. Saville,I.A. Aksay, J. Phys. Chem. B 110 (2006) 8535—8539.
[11] K.S. Novoselov, A.K. Geim, S.V. Morozov, D. Jiang, Y. Zhang,S.V. Dubonos, I.V. Grigorieva, A.A. Firsov, Science 306 (2004)666—669.
[12] D. Li, R.B. Kaner, Science 320 (2008) 1170—1171.[13] S. Park, R.S. Ruoff, Nat. Nanotechnol. 4 (2009) 217—224.[14] G. Eda, M. Chhowalla, Adv. Mater. 22 (2010) 2392—2415.[15] Y.Q. Sun, Q.O. Wu, G.Q. Shi, Energy Environ. Sci. 4 (2011)
1113—1132.[16] Y.H. Hu, H. Wang, B. Hu, Chemsuschem 3 (2010) 782—796.[17] S.R.C. Vivekchand, C.S. Rout, K.S. Subrahmanyam, A. Govin-
daraj, C.N.R. Rao, J. Chem. Sci. 120 (2008) 9—13.[18] M.D. Stoller, S. Park, Y. Zhu, J. An, R.S. Ruoff, Nano Lett. 8
(2008) 3498—3502.[19] Z.Y. Yin, S.Y. Sun, T. Salim, S.X. Wu, X.A. Huang, Q.Y. He, Y.M.
Lam, H. Zhang, ACS Nano 4 (2010) 5263—5268.[20] G. Eda, G. Fanchini, M. Chhowalla, Nat. Nanotechnol. 3 (2008)
270—274.[21] O.C. Compton, S.T. Nguyen, Small 6 (2010) 711—723.[22] S.X. Wu, Z.Y. Yin, Q.Y. He, X.A. Huang, X.Z. Zhou, H. Zhang, J.
Phys. Chem. C 114 (2010) 11816—11821.[23] X. Huang, Z.Y. Yin, S.X. Wu, X.Y. Qi, Q.Y. He, Q.C. Zhang, Q.Y.
Yan, F. Boey, H. Zhang, Small 7 (2011) 1876—1902.[24] Z.J. Wang, X.Z. Zhou, J. Zhang, F. Boey, H. Zhang, J. Phys.
Chem. C 113 (2009) 14071—14075.[25] R. Sengupta, M. Bhattacharya, S. Bandyopadhyay, A.K.
Bhowmick, Prog. Polym. Sci. 36 (2011) 638—670.[26] X.Y. Qi, K.Y. Pu, H. Li, X.Z. Zhou, S.X. Wu, Q.L. Fan, B. Liu,
F. Boey, W. Huang, H. Zhang, Angew. Chem. Int. Ed. 49 (2010)9426—9429.
[27] M.J. Allen, V.C. Tung, R.B. Kaner, Chem. Rev. 110 (2010)132—145.
[28] C. Burda, X. Chen, R. Narayanan, M.A. El-Sayed, Chem. Rev.105 (2005) 1025—1102.
[29] D. Chen, L.H. Tang, J.H. Li, Chem. Soc. Rev. 39 (2010)3157—3180.
[30] K.P. Loh, Q.L. Bao, G. Eda, M. Chhowalla, Nat. Chem. 2 (2010)1015—1024.
[31] J.Y. Luo, L.J. Cote, V.C. Tung, A.T.L. Tan, P.E. Goins, J.S. Wu,J.X. Huang, J. Am. Chem. Soc. 132 (2010) 17667—17669.
[32] C.Y. Su, Y.P. Xu, W.J. Zhang, J.W. Zhao, X.H. Tang, C.H. Tsai,L.J. Li, Chem. Mater. 21 (2009) 5674—5680.
[33] K. Erickson, R. Erni, Z. Lee, N. Alem, W. Gannett, A. Zettl, Adv.Mater. (2010), doi:10.1002/adma.201000732.
[34] L.J. Cote, J. Kim, V.C. Tung, J.Y. Luo, F. Kim, J.X. Huang, PureAppl. Chem. 83 (2011) 95—110.
[35] J. Kim, L.J. Cote, F. Kim, W. Yuan, K.R. Shull, J.X. Huang, J.Am. Chem. Soc. 132 (2010) 8180—8186.
[36] F. Kim, L.J. Cote, J.X. Huang, Adv. Mater. 22 (2010) 1954—1958.[37] V.C. Tung, J.-H. Huang, I. Tevis, F. Kim, J. Kim, C.-W. Chu, S.I.
Stupp, J. Huang, J. Am. Chem. Soc. 133 (2011) 4940—4947.[38] L.J. Cote, J. Kim, Z. Zhang, C. Sun, J.X. Huang, Soft Matter 6
(2010) 6096—6101.[39] L.J. Cote, F. Kim, J.X. Huang, J. Am. Chem. Soc. 131 (2009)
1043—1049.[40] M. Krueger, S. Berg, D.A. Stone, E. Strelcov, D.A. Dikin, J.
Kim, L.J. Cote, J. Huang, A. Kolmakov, ACS Nano 5 (2011)10047—10054.
[41] J. Kim, V.C. Tung, J.X. Huang, Adv. Energy Mater. 1 (2011)1052—1057.
[42] V.C. Tung, J. Kim, L.J. Cote, J.X. Huang, J. Am. Chem. Soc.133 (2011) 9262—9265.
[43] T.H. Han, Y.K. Huang, A.T.L. Tan, V.P. Dravid, J.X. Huang, J.Am. Chem. Soc. 133 (2011) 15264—15267.
[44] J.E. Kim, T.H. Han, S.H. Lee, J.Y. Kim, C.W. Ahn, J.M. Yun, S.O.Kim, Angew. Chem. Int. Ed. 50 (2011) 3043—3047.
[45] S.H. Aboutalebi, M.M. Gudarzi, Q.B. Zheng, J.K. Kim, Adv.Funct. Mater. 21 (2011) 2978—2988.
[46] F. Guo, F. Kim, T.H. Han, V.B. Shenoy, J.X. Huang, R.H. Hurt,ACS Nano 5 (2011) 8019—8025.
[47] Z. Xu, C. Gao, ACS Nano 5 (2011) 2908—2915.[48] B. Dan, N. Behabtu, A. Martinez, J.S. Evans, D.V. Kosynkin,
J.M. Tour, M. Pasquali, I.I. Smalyukh, Soft Matter 7 (2011)11154—11159.
[49] M. Segal, Nat. Nanotechnol. 4 (2009) 612—614.[50] C. Mattevi, G. Eda, S. Agnoli, S. Miller, K.A. Mkhoyan, O. Celik,
D. Mostrogiovanni, G. Granozzi, E. Garfunkel, M. Chhowalla,Adv. Funct. Mater. 19 (2009) 2577—2583.
[51] C.Y. Su, Y.P. Xu, W.J. Zhang, J.W. Zhao, A.P. Liu, X.H. Tang,C.H. Tsai, Y.Z. Huang, L.J. Li, ACS Nano 4 (2010) 5285—5292.
[52] P.S.V. Jimenez, Mater. Res. Bull. 22 (1987) 601—608.[53] H.P. Boehm, W. Scholz, Z. Anorg. Allg. Chem. 335 (1965) 74—79.[54] F. Kim, J.Y. Luo, R. Cruz-Silva, L.J. Cote, K. Sohn, J.X. Huang,
Adv. Funct. Mater. 20 (2010) 2867—2873.[55] P. Bajaj, Bull. Mater. Sci. 15 (1992) 67—76.[56] W.G. Cui, F. Guo, J.F. Chen, Polym. Compos. 28 (2007)
551—559.[57] A. Dasari, Z.Z. Yu, Y.W. Mai, G.P. Cai, H.H. Song, Polymer 50
(2009) 1577—1587.[58] A.L. Higginbotham, J.R. Lomeda, A.B. Morgan, J.M. Tour, ACS
Appl. Mater. Interfaces 1 (2009) 2256—2261.[59] R. Zhang, Y. Hu, J.Y. Xu, W.C. Fan, Z.Y. Chen, Polym. Degrad.
Stab. 85 (2004) 583—588.[60] R. Zhang, Y. Hu, J.Y. Xu, W.C. Fan, Z.Y. Chen, Q.N. Wang,
Macromol. Mater. Eng. 289 (2004) 355—359.[61] Y.R. Lee, S.C. Kim, H.I. Lee, H.M. Jeong, A.V. Raghu, K.R.
Reddy, B.K. Kim, Macromol. Res. 19 (2011) 66—71.[62] Y.M. Shi, L.J. Li, J. Mater. Chem. 21 (2011) 3277—3279.[63] M.J. Veraa, A.T. Bell, Fuel 57 (1978) 194—200.[64] X.X. Wu, L.R. Radovic, Carbon 43 (2005) 333—344.[65] J. Lahaye, P. Ehrburger, Fundamental Issues in Control of
Carbon Gasification Reactivity, Kluwer Academic Publishers,Dordrecht, Boston, 1991.
[66] R.T. Yang, K.L. Yang, Carbon 23 (1985) 537—547.[67] Necatioz.M., H.J. Nordwall, Nucl. Sci. Eng. 44 (1971) 310—319.[68] L.D. Smoot, P.J. Smith, Coal Combustion and Gasification,
Plenum Press, New York, 1985.[69] H. Marsh, F. Rodríguez-Reinoso, Activated Carbon, Elsevier,
Amsterdam, London, 2006.[70] Heuchamp.C., X. Duval, Carbon 4 (1966) 243—253.[71] T. Szabo, A. Bakandritsos, V. Tzitzios, E. Devlin, D. Petridis, I.
Dekany, J. Phys. Chem. B 112 (2008) 14461—14469.[72] A.V. Nikolaev, A.S. Nazarov, V.V. Lisitsa, Russ. J. Inorg. Chem.
19 (1974) 3396—3397.[73] L.J. Cote, R. Cruz-Silva, J. Huang, J. Am. Chem. Soc. 131 (2009)
11027—11032.[74] R.R. Nair, P. Blake, A.N. Grigorenko, K.S. Novoselov, T.J. Booth,
T. Stauber, N.M.R. Peres, A.K. Geim, Science 320 (2008),1308—1308.
[75] S. Gilje, S. Dubin, A. Badakhshan, J. Farrar, S.A. Danczyk, R.B.Kaner, Adv. Mater. 22 (2010) 419—423.
[76] J.X. Huang, S. Virji, B.H. Weiller, R.B. Kaner, J. Am. Chem. Soc.125 (2003) 314—315.
[77] J.X. Huang, Pure Appl. Chem. 78 (2006) 15—27.[78] M. Hirata, T. Gotou, M. Ohba, Carbon 43 (2005) 503—510.[79] W. Gao, N. Singh, L. Song, Z. Liu, A.L.M. Reddy, L.J. Ci, R.
Vajtai, Q. Zhang, B.Q. Wei, P.M. Ajayan, Nat. Nanotechnol. 6(2011) 496—500.

152 D. Krishnan et al.
[80] Z.Q. Wei, D.B. Wang, S. Kim, S.Y. Kim, Y.K. Hu, M.K. Yakes, A.R.Laracuente, Z.T. Dai, S.R. Marder, C. Berger, W.P. King, W.A. deHeer, P.E. Sheehan, E. Riedo, Science 328 (2010) 1373—1376.
[81] R.F. de Farias, C. Airoldi, J. Serb. Chem. Soc. 75 (2010)497—504.
[82] Y. Matsuo, T. Fukunaga, T. Fukutsuka, Y. Sugie, Carbon 42 (2004)2117—2119.
[83] Y. Matsuo, T. Tabata, T. Fukunaga, T. Fukutsuka, Y. Sugie, Car-bon 43 (2005) 2875—2882.
Deepti Krishnan received her B. Tech. degreein Chemical Engineering in 2010 from theNational Institute of Technology, India. Afterher undergraduate degree, she worked asa research associate at the Indian Instituteof Technology, Madras. She is currently pur-suing her doctoral studies as a member ofthe research group of Prof. Jiaxing Huang inthe Materials Science and Engineering depart-ment at Northwestern University.
Franklin Kim received his Ph.D. in Chemistryfrom UC Berkeley in 2005. After graduation,he worked as a postdoctoral researcher atthe Department of Bioengineering, UC Berke-ley, and later at the Department of MaterialsScience and Engineering, Northwestern Uni-versity. In 2011, he moved to the Institutefor Integrated Cell Material Sciences (iCeMS),Kyoto University as an iCeMS Kyoto Fel-low (independent assistant professor), and iscurrently exploring the integration of nano-
materials to cell biological studies.
Jiayan Luo got his BS and MS in Chemistryfrom Fudan University. He is now a Ph.D.candidate in Prof. Huang’s group at North-western. His research relates to graphenebased materials and energy storage. Hisresearch has been recognized and partiallysupported by a 3M Graduate Fellowship anda Ryan Fellowship in Nanoscience at North-western.
Rodolfo Cruz-Silva received his bachelor’sdegree in chemical engineering from CoahuilaState University and his Ph.D. degree inpolymer science from the Applied Chem-istry Research Center, in Mexico. In 2005 hejoined the Morelos State University, where heworked on the enzymatic synthesis of con-ductive polymers. In 2008 he spent one yearas research fellow working on graphite oxidein Huang’s research group at NorthwesternUniversity. Currently, he is working at the
Research Center for Exotic Nanocarbons, at Shinshu University, inNagano Japan. His research interests are carbon nanocompositesand chemistry of nanocarbons.
Laura J. Cote received her BS degree inMaterials Science and Engineering from theUniversity of Illinois at Urbana-Champaignin 2007. She is currently an NSF GraduateResearch Fellow at Northwestern Universityin the materials science and engineeringdepartment. Her Ph.D. work in the labora-tory of Prof. Jiaxing Huang is focused oncontrolling the assembly of graphene-basedsheets to better understand the material’sstructure—property relationships. Her work
has been recognised with a Materials Research Society Graduate Stu-dent Silver Award, P.E.O. Scholar award and Josephine de KarmanFellowship.
Hee Dong Jang is currently a distinguishedprincipal researcher and a director of RareMetals Research Center of Korea Institute ofGeoscience and Mineral Resources. His areaof expertise is the synthesis of nanostructuredmaterials by aerosol self assembly. His currentinterests involve synthesis of Graphene basedcomposite materials and their application forenergy, environmental and biomaterials. Hereceived two Ph.D. from Sogang Universityin Korea (1993) and Hiroshima University in
Japan (2005). He has been a postdoctoral researcher at the Univer-sity of California at Los Angeles (1996—1997) and a visiting scholarat the Northwestern University (2009—2010).
Jiaxing Huang is an Assistant Professor ofMaterials Science and Engineering and theMorris E. Fine Junior Professor in Materialsand Manufacturing at Northwestern Univer-sity. He received a BS in Chemical Physics fromUniversity of Science and Technology of China(USTC) and a Ph.D. in chemistry from the Uni-versity of California, Los Angeles (UCLA) in2004, and became a Miller Research Fellow atthe University of California, Berkeley beforejoining Northwestern in 2007. He has been a
recipient of the Alfred P. Sloan Research Fellowship, the NSF CAREERAward and the inaugural ISEN Early Career Investigator Award atNorthwestern.