relating carbon monoxide photoproduction to dissolved organic matter functionality
TRANSCRIPT

Relating Carbon MonoxidePhotoproduction to DissolvedOrganic Matter FunctionalityA R O N S T U B B I N S , * , † , ‡ , §
V E S P E R H U B B A R D , † G U E N T H E R U H E R , ‡
C L I F F S . L A W , § , |
R O B E R T C . U P S T I L L - G O D D A R D , ‡
G E O R G E R . A I K E N , ⊥ A N DK E N N E T H M O P P E R * , †
Department of Chemistry and Biochemistry, Old DominionUniversity, Norfolk, Virginia 23529, Marine Science andTechnology, Armstrong Building, Newcastle University, NE17RU, U.K., Plymouth Marine Laboratory, The Hoe, Plymouth,PL1 3DH, U.K., NIWA, 301 Evans Bay Parade,Wellington, 6021, New Zealand, and U.S. Geological Survey,3215 Marine Street, Boulder, Colorado 80303
Received December 3, 2007. Revised manuscript receivedFebruary 11, 2008. Accepted February 15, 2008.
Aqueous solutions of humic substances (HSs) and puremonomeric aromatics were irradiated to investigate the chemicalcontrols upon carbon monoxide (CO) photoproduction fromdissolved organic matter (DOM). HSs were isolated from lakes,rivers, marsh, and ocean. Inclusion of humic, fulvic, hydrophobicorganic, and hydrophilic organic acid fractions from theseenvironments provided samples diverse in source and isolationprotocol. In spite of these major differences, HS absorptioncoefficients (a) and photoreactivities (a bleaching and COproduction) were strongly dependent upon HS aromaticity (r2
>0.90; n ) 11), implying aromatic moieties are the principalchromophores and photoreactants within HSs, and by extension,DOM. Carbonyl carbon and CO photoproduction were notcorrelated, implying that carbonyl moieties are not quantitativelyimportant in CO photoproduction. CO photoproductionefficiency of aqueous solutions of monomeric aromaticcompounds that are common constituents of organic mattervaried with the nature of ring substituents. Specifically, electrondonating groups increased, while electron withdrawinggroupsdecreasedCOphotoproductivity,supportingourconclusionthat carbonyl substituents are not quantitatively important inCO photoproduction. Significantly, aromatic CO photoproductionefficiency spanned 3 orders of magnitude, indicating thatvariations in the CO apparent quantum yields of natural DOMmay be related to variations in aromatic DOM substituent groupchemistry.
Introduction
Carbon monoxide (CO) is quantitatively the second largestphotoproduct of dissolved organic matter (DOM) photom-
ineralization (1, 2) and a significant term in the aquatic carboncycle. Oceanic CO photoproduction mineralizes 30–90 Tg ofdissolved organic carbon annually (3, 4), driving CO emissionto the atmosphere (5) where it plays an important role inclimate regulation (6).
Low background levels and precise and accurate analyticaltechniques allow precise and accurate quantification of COphotoproduction rates. Consequently, CO is used as a proxyfor the photoproduction of dissolved inorganic carbon (DIC)(1, 7, 8) and biolabile organic carbon (9, 10), which accountfor the majority of DOM photomineralization in naturalwaters, but are considerably more difficult to measure thanCO. CO is also a key tracer for testing and tuning models ofmixed layer processes (11–14). Despite widespread interest,little is known about the chromophoric sites responsible forDOM photoreactivity (15–17). Although previous studies havehypothesized that CO is produced by direct photocleavageof carbonyl groups from DOM (18–20), the chemical controlsgoverning CO production have not been empiricallydetermined.
Humic substances (HSs) typically account for 40-90% ofthe DOM pool (21) and have photoreactivities and opticalproperties comparable to natural DOM at similar carbonand chromophore concentrations (19, 22) making themsuitable DOM surrogates in photochemical experiments (23).In addition, HS isolates are amenable to 13C nuclear magneticresonance spectroscopy (13C NMR) providing a level ofchemical characterization presently unattainable for noniso-lated DOM.
HSs can be referred to as aquatic (aHSs) or terrestrial(tHSs). tHSs prevail in freshwaters receiving significant DOMthrough soil leaching and surface runoff and contain highlevels of aromatic carbon derived from lignin and lignindegradation products (24, 25). In contrast, aHSs are producedin situ from microbial sources and are usually highly aliphaticwith lower aromaticity (24, 26, 27). They are prominent inoceans, eutrophic lakes, and lakes receiving limited terrestrialinput. The impact of DOM chemistry upon CO photopro-duction was investigated using HSs from a diverse selectionof natural waters.
In addition, sets of monomeric, structurally relatedaromatic compounds were used as proxies for DOM pho-toreaction sites. Chosen aromatics consisted only of carbon,hydrogen, and oxygen. Although DOM includes a wide varietyof biochemical residues (16, 28) and condensation products(29), our experiments focused on monomeric aromatics forthe following reasons: (a) Natural DOM and HSs areprohibitively complex for preliminary mechanistic studies.(b) The absorption spectra of many naturally occurringaromatics extend into the UV-B and UV-A, the mainwavelengths for CO photoproduction (19). (c) Syringyl,vanillyl, cinnamyl, and quinone moieties occur in marineDOM (30–32) and tHSs (33, 34). (d) Algal polyphenols aremajor constituents of DOM in coastal surface waters (35, 36)and estuarine phenol concentrations can be well correlatedwith CDOM absorbance (36). (e) Many aromatic compoundsare photoreactive in dilute aqueous solutions at environ-mentally relevant wavelengths (34, 37, 38). (f) The number,type, and location of functional groups (ring substituents)can be systematically changed, facilitating evaluation ofsubstituent impact upon photoreactivity. And (g) A largenumber of natural and synthetic aromatic compounds arecommercially available and relatively inexpensive.
The objective of this study was to relate the lightabsorbance and CO photoproduction of a suite of diverseHSs to source, mode of extraction, spectral properties, and
* Corresponding authors e-mail: [email protected] (K.M.),[email protected] (A.S.).
† Old Dominion University.‡ Newcastle University.§ Plymouth Marine Laboratory.| NIWA.⊥ U.S. Geological Survey.
Environ. Sci. Technol. 2008, 42, 3271–3276
10.1021/es703014q CCC: $40.75 2008 American Chemical Society VOL. 42, NO. 9, 2008 / ENVIRONMENTAL SCIENCE & TECHNOLOGY 9 3271
Published on Web 03/26/2008

chemical composition (based on 13C NMR). Subsequentexperiments with monomeric aromatics explored the rela-tionships between CO photoproduction and the substituentchemistry of chromophores potentially present within naturalDOM.
Experimental SectionHumic Substance Isolation, Characterization, and SamplePreparation. Whole water samples were collected fromterrestrial and marine environments (described below) andfiltered immediately (0.45 µm, AquaPrep 600, Pall Gelman).Samples were acidified to pH 2 with hydrochloric acid (HCl)passed through XAD-8 and XAD-4 resins (Amberlite) to extractthe HPOA (hydrophobic organic acid) and HPIA (hydrophilicorganic acid) fractions, respectively (30, 39). These fractionswere then eluted from the resins with 0.1 N sodium hydroxide.Eluates were immediately acidified with reagent-grade HClto pH 3 (minimizing sample alteration at high pH), desaltedusing H+ saturated AG-MP 50 cation exchange resin (Bio-Rad), lyophilized, and stored in a desiccator. For somesamples, the HPOA fraction was further separated into humicacid (HA) and fulvic acid (FA) fractions by acidifying theXAD-8 eluate to pH <1 using HCl. The HA precipitate wasremoved by centrifugation, and lyophilized. The supernatantcontaining FA was then desalted, hydrogen saturated,lyophilized, and stored in a desiccator.
In this study, HS isolates are categorized by the dominantsource of DOM as aquatic HS (aHS; in situ microbial sourcesincluding algal and bacterial), terrestrial HS (tHS; higher plantand soil-derived), and a mixture of both (Table 1). Theterrestrial HSs were the Suwannee River FA and HA referencematerials (riverine FA and HA obtained from the InternationalHumic Substances Society, IHSS (40)). Predominantly aquatic-derived samples included Lake Fryxell HPOA (Lake Fryxell,an ice bound Antarctic lake 26, 27), and Pacific Ocean FA(eastern equatorial Pacific Ocean near Hawaii, depth)200 m(24)). The other samples represent mixed sources, includingWilliams Lake, a seepage lake in Minnesota, the ShingobeeRiver, Minnesota, and samples from the Florida Everglades.DOM in Williams Lake is primarily from aquatic sources,with the addition of DOM from groundwater that rechargesthe lake (41). The Shingobee River, downstream of WilliamsLake, receives DOM from local lakes, wetlands, and soils(41). Other mixed samples from the northern Everglades, anarea of subtropical wetland environment containing peatsoils, cattail and saw grass vegetation, and significantmicrobial activity, include S10E Hillsborough Canal (26°27′32′′N, 80°26′54′′ W); the U3 (26°17′15′′ N, 80°24′41′′ W), and F1
(26°21′35′′ N, 80°22′14′′ W) in Water Conservation Area 2A;and 2BS (26°09′00′′ N, 80°22′30′′ W) from Water ConservationArea 2B (Table 1).
Quantitative liquid state 13C NMR spectra were obtainedfrom 100 mg of HS sample dissolved in 1 mL of H2O/D2O(1:1), adjusted to pH 7 and analyzed on a Varian 300spectrometer at 75.429 MHz using inverse gated-decouplingwith an 8-s delay (27). Quantification of the 13C NMR spectraintegrated the area under the bands (aliphatics 0–62 ppm,heteroaliphatics 62–90, anomerics 90–110, aromatics 110–160,carboxyls 190–220) to derive approximate concentrations asa percentage of total carbon (42). Total aliphatic carbon wascalculated as the sum of aliphatics, heteroaliphatics, andanomerics. Data were converted to concentrations (mg CL-1) using the weight of HS added (5 mg L-1) and HS carboncontent (Table 1) determined by elemental analyses (HuffmanLaboratories, Golden, CO; (43)).
HS isolates were added to ultrapure laboratory water (DIW;Millipore, Milli-Q) to give solutions with HS concentrationsof 5.00 mg L-1. Samples were sonicated for one hour tofacilitate dissolution, refiltered (0.45 µm GF/F, Whatman) toremove any minor particulates, and refrigerated in the darkfor <24 h before use. Solutions were irradiated in gastightquartz tubes (diameter 22 mm, length 145 mm, volume 37mL) for three hours using a solar simulator carousel (150 Wxenon arc lamp (Oriel) with a borosilicate shield, 1%transmission cutoff ∼300 nm). Samples were irradiated underoxic conditions. Dark controls were foil wrapped and placedin the irradiation carousel. The intensity of the solar simulatorwas equivalent to midsummer, solar noon at about 45° N(measured using a Biospherical PUV 2500). All samples wererun over a two-day period at Plymouth Marine Laboratory(May 1999). Photochemical rates were calculated as thedifferences in concentration between irradiated and darksamples divided by the irradiation time.
Model Aromatic Sample Preparation and Irradiation.Pure solutions (10.0 mM) of 27 (Table 2) reagent grade,monomeric aromatic compounds were prepared in DIW.Aromatic compounds were added to DIW, stirred in foil-covered glassware (to prevent light exposure) overnight toensure dissolution of aromatics, and filtered (0.45 µm GF/F,Whatman) to check for particles. Samples and dark controls(foil wrapped) were transferred to gastight quartz tubes andirradiated for 3 h on a rotating Table (1 rpm) under 12 UVA-340 bulbs (integrated irradiance ∼25 W m-2; Q-Panel), whichprovide a spectral shape and flux closely approximatingnatural sunlight from 295 to 365 nm, the main wavelengthrange for environmental photochemical reactions involving
TABLE 1. Humic Substance Source, Isolation Protocol, Percent (%) Carbon, Aromatic and Ketonic Carbon (mgC L-1) by 13C NMR,Absorption Coefficient (a350), Absorption Photobleaching (δa350), and CO Photoproduction Rates (CO) (All Samples 5 mg L-1
Humic Substance in Ultrapure Water)
sample source%
carbonaliphatic
(mg C L-1)aromatic
(mg C L-1)ketonic
(mg C L-1)a350
(m-1)δa350
(m-1 hr-1)CO
(nmol L-1 hr-1)
Pacific Ocean HPOAa aHSe 57.5 2.06 0.21 0.05 0.65 0.03 5.0Lake Fryxell HPOA aHS 55 1.80 0.42 0.00 2.48 0.07 14.6Williams Lake HPOA mixed aHSf 53.9 1.94 0.34 0.05 1.43 0.04 9.3F1 Everglades HPOA mixed tHSg 51.9 1.76 0.47 0.07 6.07 0.10 22.2F1 Everglades HPIAb mixed tHS 47.7 1.68 0.31 0.04 2.48 0.04 15.12BS Everglades HPOA mixed tHS 51.7 1.24 0.36 0.22 3.84 0.06 14.22BS Everglades HPIA mixed tHS 47.2 1.64 0.30 0.05 1.65 0.03 8.1U3 Everglades HPOA mixed tHS 54.7 1.71 0.49 0.11 5.03 0.09 16.7S10E Everglades HPOA mixed tHS 54.5 1.78 0.47 0.09 5.50 0.10 21.0IHSS Suwannee River HAc tHSh 53.4 0.93 1.12 0.19 12.83 0.19 43.4IHSS Suwannee River FAd tHS 53.5 1.25 0.75 0.16 6.36 0.12 24.3
a HPOA ) Hydrophobic organic acid. b HPIA ) Hydrophilic organic acid. c HA ) Humic acid. d FA ) Fulvic Acid. e aHS )Aquatic humic substance derived from aquatic microbial sources. f Mixed aHS ) Predominantly aHS with some tHS inputs.g Mixed tHS ) Predominantly tHS with some aHS inputs. h tHS ) Terrestrially derived humic substance.
3272 9 ENVIRONMENTAL SCIENCE & TECHNOLOGY / VOL. 42, NO. 9, 2008

DOM (8). Simulated sunlight was chosen above naturalsunlight due to the need for reproducible irradiation condi-tions between sample sets. Samples were run over a 2-monthperiod (Summer 2005) at Old Dominion University (VA)during which time the solar simulator output was monitored(Biospherical PUV 2500) and remained stable. Samples wereirradiated under oxic conditions. CO quantum yields werenot measured; however, CO production rates were dividedby the absorbed light (the cross-product of the aromaticsolution optical density spectrum (OD nm-1) and solarsimulator spectral irradiance (W m-2 nm-1)) to account fordifferences in molar absorption coefficients, yielding COphotoproduction efficiencies (Table 2).
Carbon Monoxide. For HSs, 60-mL crimp top vials wereflushed and filled with sample immediately after irradiation,sealed with Teflon faced butyl septa (Supelco), and analyzedfor CO. CO scrubbed (Hopcalite, Supelco) carrier gas (zerograde air) was introduced through the vial septa to create a25-mL headspace. Following 30-min dark equilibration usinga wrist action shaker, 15 mL of headspace gas was collectedin a gastight syringe (Hamilton). For aromatic compounds,sample was drawn directly from the gastight flasks into 100-mL gastight glass syringes (Hamilton), ensuring neitheratmospheric contact nor bubbling occurred. Syringes werefilled with 70 mL of sample. Then, 30 mL of CO-free headspacegas was added to bring the total volume to 100 mL. Sampleswere equilibrated for 30 min using a wrist action shaker beforethe headspace was injected directly onto the GC drier.
CO in sample gaseous headspace was measured using areduction gas detector (RGD2) with UV-photometer (HSs:Trace Analytical; aromatics: SRI) following separation by gaschromatography (4, 5). For all samples, atmospheric pressureand aqueous sample temperature were recorded for use incalculating CO solubility.
Absorption Coefficient Spectra. Samples were stored inirradiation flasks, kept in the dark, and allowed to reach roomtemperature before processing. Absorbance spectra wereobtained using a scanning UV–visible, double-beam, spec-trophotometer (Kontron, Uvicon 923) with DIW as a referencebeam blank. Samples were measured in 1-cm quartz cuvettes,cleaned with HCl (0.1 M), copious DIW, and then triple rinsedwith sample before use. Wavelengths from 250 to 800 nmwere measured and corrected for offsets due to scattering,particulate absorbance, and instrument drift by subtractingthe average absorbance between 700 and 800 nm. Data outputfrom the spectrophotometer were in the form of dimension-less absorbance or optical density (OD) at wavelength λ. ODwas converted to the absorption coefficient, a, defined as aλ
) (ODλ × ln10)/l where l is the optical path length of thecuvette (44).
Results and DiscussionRelating Chemical and Absorption Characteristics to HumicSubstance Source and Isolation Protocol. Terrestrial humicsubstances (tHSs; Suwannee River HA and FA) containedhighest levels of aromaticity (28–42% by carbon; Table 1).Aquatic humic substances (aHSs) had lower aromatic carboncontents (7–15% by carbon) due to their dominantly microbialsources (Table 1). HPOA and HA isolates had higher aromaticcarbon levels than HPIA and FA extracts from the same waters(Table 1).
HS absorption coefficient (a) spectra (Figure 1) exhibitedapproximately exponential increases with decreasing wave-length, in agreement with previous studies (45). The absorp-tion coefficient at 350 nm (a350) ranged from 0.65 to 12.83m-1 (Table 1; Figure 2a), compared to ranges of 0.5 to 1.0 a350
m-1 in coastal seawaters and 20 to 104 a350 m-1 in freshwaters(45–47). As HS concentrations were identical for all samples(5.0 mg L-1), large differences in a350 (Table 1) are attributedto differences in aromatic carbon concentration (mg ArCL-1) with which HS a350 was linearly related (r2 ) 0.90, p )7.13 × 10-6, n ) 11; Figure 2a). These results agree withprevious studies that demonstrated the dominant role ofaromatic chromophores in colored dissolved organic matter(CDOM) (48, 49). The nonzero x-intercept of the a350 versusaromatic carbon linear regression (∼1.67( 0.74 mg aromaticC L-1; Figure 2a) may be due to unconjugated olefin moieties,which absorb in the same region of the 13C NMR spectrumas aromatic carbons (110–160 ppm) but do not absorb lightat wavelengths >250 nm (49). Alternatively, this could beattributable to more extensive charge transfer for HS withgreater aromatic composition or increased resonancethroughout the pi-electron system at higher aromatic con-
TABLE 2. Carbon Monoxide (CO) Photoproduction Efficiency fora Suite of 27 Monomeric Aromatic Compounds Dissolved inUltrapure Water (Aromatic Concentrations 10 mM) andIrradiated under Simulated Sunlight in Order of Increasing COPhotoproduction Efficiency
monomeric aromaticcompound
CO production efficiency(production/light absorbed)
1-(2-hydroxy-phenyl)-ethanone 0.011-(3-hydroxy-phenyl)-ethanone 0.112-hydroxybenzaldehyde 0.11benzaldehyde 0.113-phenyl-propenal 0.113-hydroxybenzaldehyde 0.134-hydroxybenzaldehyde 0.213-phenyl-acrylic acid 0.28ethoxybenzene 0.391-(4-hydroxy-phenyl)-ethanone 0.504-hydroxybenzoic acid 0.57methoxybenzene 0.83benzoic acid 0.893-methoxybenzaldehyde 1.08phenol 1.244-methoxybenzaldehyde 2.57acetophenone 3.222-methoxybenzaldehyde 3.461-(4-hydroxy-3-methoxy-phenyl)-
ethanone3.58
benzene-1,3-diol 4.55benzoquinone 5.502-methoxyphenol 11.164-methoxyphenol 15.803-methoxyphenol 17.054-ethoxyphenol 19.122-methoxy-4-methylphenol 20.012-ethoxyphenol 34.67
FIGURE 1. Ultraviolet–visible absorption coefficient spectra forhumic substances dissolved in deionized water.
VOL. 42, NO. 9, 2008 / ENVIRONMENTAL SCIENCE & TECHNOLOGY 9 3273
centrations (e.g., unsubstituted dimeric aromatics havegreater absorption coefficients >300 nm than monomericanalogs).
The tHS samples had higher a350 per unit carbon thanaHSs, and HPOA and HA samples had higher a350 per unitcarbon than corresponding HPIA and FA extracts from thesame water samples (Table 1) indicating that source andisolation procedure affect a350. However, the strong, linearregression between a350 and aromatic carbon for all samples(Figure 2a) suggests that HS a350 is controlled primarily bythe concentration of aromatic carbon in the sample and thatHS source, isolation protocol, and chemistry have onlysecondary impact. The statistical relation between aromaticcarbon and a350 (mg aromatic carbon L-1 ) 0.0710 × a350 +0.1646; r2 ) 0.90, p ) 7.13 × 10-6, n ) 11) can be used topredict aromatic carbon concentrations from absorptioncoefficients of natural waters (note that in this regressiona350 is the independent variable as the slope is designed toallow prediction of aromatic carbon from DOM absorption).
No relation was found between a350 and ketonic, het-eroaliphatic, anomeric, or carboxylic carbon levels (datareported in ref (23)). However, aliphatic and aromatic carbons(Table 1) were negatively correlated (r ) -0.91, p ) 6.53 ×10-6, n ) 11), resulting in a negative correlation between a350
and aliphatic carbon (r ) -0.87, n ) 11).Humic Substance Photoreactivity. CO photoproduction
rates ranged from 5.0 to 43.4 nmol L-1 hr-1 (Table 1; Figure2b) and HS photobleaching rates ranged from 0.03 to 0.19a350 m-1 h-1 (Table 1; Figure 2c). As previously reported forDOM samples (19, 20, 50), CO photoproduction and HS aphotobleaching were related to initial a. CO productionrelated most strongly with initial a at 350 nm, where a linear
regression best described the relationship (r2 ) 0.97, p )4.30 × 10-8, n ) 11).
Both CO production and photobleaching rates were higherfor tHSs than aHSs, while the rates for HPOA and HA sampleswere higher than those of corresponding HPIA and FAsamples (Table 1). However, linear regressions of initial a350
(Figure 2a), CO production (Figure 2b; r2 ) 0.91, p ) 4.30 ×10-6, n ) 11) and photobleaching at a350 (Figure 2c; r2 ) 0.93,p ) 2.27 × 10-6, n ) 11) versus aromaticity indicate that lightabsorption, CO production, and CDOM photobleaching aredetermined by aromatic chromophore concentrations.
While it has been proposed that CO is photoproducedthrough photolytic cleavage of carbonyl groups from DOM(18, 51), we observed no relation between total ketonic carbonand CO photoproduction (Table 1), indicating that carbonylmoieties are apparently not important in controlling the rateof CO photoproduction. This conclusion is supported byexperiments with model aromatic chromophores (below).
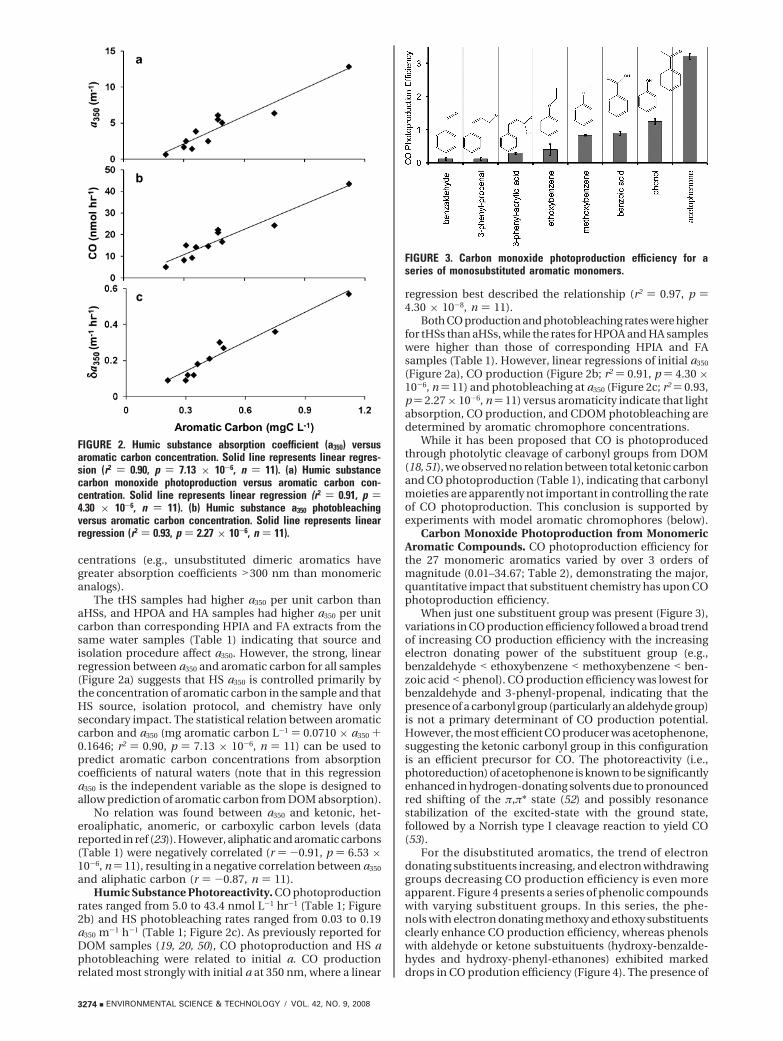
Carbon Monoxide Photoproduction from MonomericAromatic Compounds. CO photoproduction efficiency forthe 27 monomeric aromatics varied by over 3 orders ofmagnitude (0.01–34.67; Table 2), demonstrating the major,quantitative impact that substituent chemistry has upon COphotoproduction efficiency.
When just one substituent group was present (Figure 3),variations in CO production efficiency followed a broad trendof increasing CO production efficiency with the increasingelectron donating power of the substituent group (e.g.,benzaldehyde < ethoxybenzene < methoxybenzene < ben-zoic acid < phenol). CO production efficiency was lowest forbenzaldehyde and 3-phenyl-propenal, indicating that thepresence of a carbonyl group (particularly an aldehyde group)is not a primary determinant of CO production potential.However, the most efficient CO producer was acetophenone,suggesting the ketonic carbonyl group in this configurationis an efficient precursor for CO. The photoreactivity (i.e.,photoreduction) of acetophenone is known to be significantlyenhanced in hydrogen-donating solvents due to pronouncedred shifting of the π,π* state (52) and possibly resonancestabilization of the excited-state with the ground state,followed by a Norrish type I cleavage reaction to yield CO(53).
For the disubstituted aromatics, the trend of electrondonating substituents increasing, and electron withdrawinggroups decreasing CO production efficiency is even moreapparent. Figure 4 presents a series of phenolic compoundswith varying substituent groups. In this series, the phe-nols with electron donating methoxy and ethoxy substituentsclearly enhance CO production efficiency, whereas phenolswith aldehyde or ketone substuituents (hydroxy-benzalde-hydes and hydroxy-phenyl-ethanones) exhibited markeddrops in CO prodution efficiency (Figure 4). The presence of
FIGURE 2. Humic substance absorption coefficient (a350) versusaromatic carbon concentration. Solid line represents linear regres-sion (r2 ) 0.90, p ) 7.13 × 10-6, n ) 11). (a) Humic substancecarbon monoxide photoproduction versus aromatic carbon con-centration. Solid line represents linear regression (r2 ) 0.91, p )4.30 × 10-6, n ) 11). (b) Humic substance a350 photobleachingversus aromatic carbon concentration. Solid line represents linearregression (r2 ) 0.93, p ) 2.27 × 10-6, n ) 11).
FIGURE 3. Carbon monoxide photoproduction efficiency for aseries of monosubstituted aromatic monomers.
3274 9 ENVIRONMENTAL SCIENCE & TECHNOLOGY / VOL. 42, NO. 9, 2008

electron donating (e.g., hydroxy, methoxy, and ethoxy) andwithdrawing (e.g., aldehyde, ketone) substituents have beenshown to cause similar increases and decreases in e-aq andOH production efficiency relative to phenol (39, 54). Gross-weiner and Joschek (54) found that compounds with lowgas-phase photoionization potentials have the greatest e-aq
yields, and concluded that monosubstituted benzene de-rivatives with electron donating substituents in the groundstate favor photoionization in solution, while the presenceof electron withdrawing substituents inhibit it. Thus, it seemslikely that the trends for CO photoproduction have a similarbasis. Possible explanations of these trends include theincreased acidity of electron donating groups in the excitedsinglet state (55) and/or the increased stability of the excitedtriplet state and reaction intermediates imparted by electrondonating groups, particularly alkoxy groups (56). Surprisingly,the position of substituent groups (ortho, meta, or para) didnot cause significant or consistent trends in CO productionefficiency (Figure 4), indicating that, for the current data set,steric effects have only secondary influence upon COphotoproduction.
Atmospheric studies have shown that a variety of volatileorganic compounds are readily mineralized through director photosensitized degradation, with CO as an end product(57). Therefore, observed high CO production by methoxy-aromatics could result from the reaction of formaldehydewith hydroxyl radicals (57), as both are photoproducts ofaqueous solutions of methoxy-phenolic compounds (38) andDOM (8).
CO photoproduction efficiencies for studied monomericaromatics vary by more than 3 orders of magnitude (Table2). Therefore, although the exact mechanisms of CO pro-duction are not apparent, it is clear that variations in aromaticsubstituent chemistry have a profound influence upon COphotoproduction efficiencies. CO photoproduction efficien-cies (apparent quantum yields) for natural DOM vary by overan order of magnitude at a given wavelength depending uponsource, environment and previous light exposure (3, 5, 14, 19).Our results suggest that these natural variations in DOMapparent quantum yields may be related to variations in thesubstituent chemistry of aromatic chromophores within theDOM pool. For instance, the rapid photodegradation orrearrangement of aromatics with substituent chemistrieswhich confer high photoreactivity may be responsible forthe rapid decreases in CO apparent quantum yields withincreasing absorbed photon dose (58). Continued work withsimple aromatics is required to further explore the mech-
anisms and chemical controls upon the efficiencies of COphotoformation and other photoreactions.
AcknowledgmentsWe thank Dr. Vassilis Kitidis, Malcolm Woodward and JohnR. Helms for help running the irradiation system and thethree anonymous ES&T reviewers for their valuable com-ments. This work was funded by the UK National Environ-mental Research Council (grant GR3 11665), the PlymouthMarine Laboratory, the Marine Biological Association of theUK via a Ray Lankester Investigatorship to K.M., the U.S.National Science Foundation (OCE0196220, OCE0327446),and the U.S. Geological Survey National Research Program.Opinions, findings, conclusions, and recommendationsexpressed are those of the authors and do not necessarilyreflect those of the funding bodies. Brand names are foridentification purposes only and do not imply endorsement.
Literature Cited(1) Miller, W. L.; Zepp, R. G. Photochemical production of dissolved
inorganic carbon from terrestrial organic-matter - significanceto the oceanic organic-carbon cycle. Geophys. Res. Lett. 1995,22, 417–420.
(2) Mopper, K.; Kieber, D. J. Photochemistry and the cycling ofcarbon, sulfur, nitrogen and phosphorus. In Biogeochemistry ofMarine Dissolved Organic Matter; Hansell, D., Carlson, C., Eds.;Academic Press: San Diego, CA, 2002.
(3) Zafiriou, M. C.; Andrews, S. S.; Wang, W. Concordant estimatesof oceanic carbon monoxide source and sink processes in thePacific yield a balanced global “blue-water” CO budget. GlobalBiogeochem. Cycles 2003, DOI: 10.1029/2001GB001638.
(4) Stubbins, A.; Uher, G.; Kitidis, K.; Law, C. S.; Upstill-Goddard,R. C.; Woodward, E. M. S. The Open Ocean Source ofAtmospheric Carbon Monoxide. Deep Sea Res.: II 2006, 53, 1685–1694.
(5) Stubbins, A.; Uher, G.; Law, C. S.; Mopper, K.; Robinson, C.;Upstill-Goddard, R. C. Open ocean carbon monoxide photo-production. Deep Sea Res.: II 2006, 53, 1695–1705.
(6) Thompson, A. M.; Cicerone, R. J. Atmospheric CH4, CO and OHfrom 1860 to 1985. Nature 1986, 321, 148–150.
(7) Johannessen, S.; Ziolkowski, L.; Miller, W. Comparison ofphotochemical production rates of carbon monoxide anddissolved inorganic carbon in the ocean.; presented at AmericanChemical Society Pacifichem 2000, Honolulu, Hawaii, 2000.
(8) Mopper, K.; Kieber, D. J.; Marine photochemistry and its impacton carbon cycling. In The Effects of UV Radiation in the MarineEnvironment; d. Mora, S. J., Demers, S., Vernet, M., Eds.;Cambridge University Press: New York, 2000.
(9) Kieber, D. J.; McDaniel, J. A.; Mopper, K. Photochemical sourceof biological substrates in seawater: Implications for carboncycling. Nature 1989, 341, 637–639.
(10) Miller, W. L.; Moran, M. A.; Sheldon, W. M.; Zepp, R. G.; Opsahl,S. Determination of apparent quantum yield spectra for theformation of biologically labile photoproducts. Limnol. Ocean-ogr. 2002, 47, 343–352.
(11) Doney, S. C.; Najjar, R. G.; Stewart, S. Photochemistry, mixingand diurnal cycles in the upper ocean. J. Mar. Res. 1995, 53,341–369.
(12) Najjar, R. G.; Erickson, D. J., III; Madronich, S. Modeling theair-sea fluxes of gases formed from the decomposition ofdissolved organic matter: Carbonyl sulfide and carbon mon-oxide. In The Role of Nonliving Organic Matter in the Earth’sCarbon Cycle: Report of the Dahlem Workshop on the Role ofNonliving Organic Matter in the Earth’s Carbon Cycle; Zepp,R. G., Sonntag, C., Eds.; John Wiley: New York, 1995.
(13) Gnanadesikan, A. Modeling the diurnal cycle of carbon mon-oxide: Sensitivity to physics, chemistry, biology, and optics. J.Geophys. Res. 1996, 101 (C5), 12,177–12,191.
(14) Kettle, A. J. Diurnal cycling of carbon monoxide (CO) in theupper ocean near Bermuda. Ocean Modell. 2005, 8, 337–367.
(15) Zika, R. G. Advances in marine photochemistry l983–l987. Rev.Geophys. 1987, 25, 1390–1394.
(16) Zafiriou, O. C.; Joussot-Dubien, J.; Zepp, R. G.; Zika, R. G.Photochemistry of natural waters. Environ. Sci. Technol. 1984,18, 358A–371A.
(17) Zepp, R. G. Environmental photoprocesses involving naturalorganic matter. In Humic Substances and Their Role in the
FIGURE 4. Carbon monoxide photoproduction efficiency forphenol and a series of monosubstituted phenols. Photoproduc-tion efficiency was calculated as the cross-product of thespectral aromatic solution optical density (absorbance) per nm(OD nm-1) and solar simulator spectral irradiance per nm (Wm-2 nm-1).
VOL. 42, NO. 9, 2008 / ENVIRONMENTAL SCIENCE & TECHNOLOGY 9 3275

Environment; Frimmel, F. H., Christman, R. H., Eds.; Wiley:New York, 1988.
(18) Redden, G. D. Characteristics of photochemical production ofcarbon monoxide in seawater. M.Sc. Thesis, Oregon StateUniversity, 1983.
(19) Valentine, R. L.; Zepp, R. G. Formation of Carbon Monoxidefrom the Photodegradation of Terrestrial Dissolved OrganicCarbon in Natural Waters. Environ. Sci. Technol. 1993, 27, 409–412.
(20) Pos, W. H. On the Processes and Mechanisms Affecting CarbonylSulfide and Carbon Monoxide Photoproduction in NaturalWaters. Ph.D. Thesis, Florida, 1997.
(21) Amador, J. A.; Milne, P. J.; Moore, C. A.; Zika, R. G. Extractionof chromophoric humic substances from seawater. Mar. Chem.1990, 29, 1–17.
(22) Kablitz, K.; Geyer, S.; Geyer, W. A comparative characterizationof dissolved organic matter by means of original aqueoussamples and isolated humic substances. Chemosphere 2000,40, 1305–1312.
(23) Anesio, A. M.; Granéli, W.; Aiken, G.; Kieber, D. J.; Mopper, K.Effect of humic substance photodegradation on bacterial growthand respiration in lake water. Appl. Environ. Microbiol. 2005,71, 6267–6275.
(24) Malcolm, R. L. The uniqueness of humic substances in each ofsoil, stream and marine environments. Anal. Chim. Acta 1990,232, 19–30.
(25) Ertel, J. R.; Hedges, J. I.; Perdue, E. M. The lignin componentof humic substances: Distribution among soil and sedimentaryhumic, fulvic and base-insoluble fractions. Geochem. Cosmo-chem. Acta 1984, 48, 2065.
(26) McKnight, D. M.; Aiken, G. R.; Smith, R. L. Aquatic Fulvic-Acidsin Microbially Based Ecosystems - Results From 2 Desert Lakesin Antarctica. Limnol. Oceanogr. 1991, 36, 998–1006.
(27) Aiken, G. R.; McKnight, D.; Harnish, R.; Wershaw, R. Geochem-istry of aquatic humic substances in the Lake Fryxell Basin,Antarctica. Biogeochemistry 1996, 34, 157–188.
(28) Benner, R. Chemical composition and reactivity. In Bio-geochemistry of Marine Dissolved Organic Matter; Hansell, D.,Carlson, C., Eds.; Academic Press: San Diego, CA, 2002.
(29) Ikan, R.; Ioselis, P.; Rubinsztain, Y.; Aizenshtat, Z.; Miloslavsky,I.; Yariv, S.; Pugmire, R.; Anderson, L. L.; Woolfenden, W. R.; etal. Chemical, isotopic, spectroscopic and geochemical aspectsof natural and synthetic humic substances. Sci. Total Environ.1992, 117/118, 1–12.
(30) Rashid, M. A. Quinone content of humic compounds isolatedfrom the marine environment. Soil Sci. 1972, 113, 181–188.
(31) Meyers-Schulte, K. J.; Hedges, J. I. Molecular evidence for aterrestrial component of organic matter dissolved in oceanwater. Nature 1986, 321, 61–63.
(32) Benner, R.; Opsahl, S. Molecular indicators of the sources andtransformations of dissolved organic matter in the Mississippiriver plume. Org. Geochem. 2001, 32, 597–611.
(33) Schulten, H.-R.; Plage, B.; Schnitzer, M. A chemical structurefor humic substances. Naturwissenschaften 1991, 78, 311–312.
(34) Choudhry, G. G. Humic substances. Part II: Photophysical,photochemical and free radical characteristics. Toxicol. Environ.Chem. 1981, 4, 261–295.
(35) Sieburth, J. M.; Jensen, A. Studies on algal substances in the sea.II. The formation of Gelbstoff (humic material) by exudates ofphaeophyta. J. Exp. Mar. Biol. Ecol. 1969, 3, 275–289.
(36) Carlson, D. J.; Mayer, L. M. Relative influences of riverine andmacroalgal phenolic materials on UV abosrbance in temperatecoastal waters. Can. J. Fish. Aquat. Sci. 1983, 40, 1258–l263.
(37) Ononye, A. I.; Bolton, J. R. Mechanism of the photochemistryof p-benzoquinone in aqueous solutions. 2. Optical flashphotolysis. J. Phys. Chem. 1986, 90, 6270–6274.
(38) Qian, J.-G. Photochemical Production and Formation Mech-anisms of Hydroxyl Radical and Formaldehyde in AqueousSystems. M.Sc. Thesis, Pullman, Washington State University,1996.
(39) Thurman, E. M.; Malcolm, R. L. Preparative isolation of aquatichumic substances. Environ. Sci. Technol. 1981, 15, 463–466.
(40) Aiken, G. R.; Malcolm, R. Molecular weight of aquatic fulvicacids by vapor pressure osmometry. Geochim. Cosmochim. Acta1987, 51, 2177–2184.
(41) Aiken, G. R.; McKnight, D. The influence of hydrological factorson the nature of organic matter in the Williams and ShingobeeLake Systems. In Interdisciplinary research initiative: Hydrologi-cal and biogeochemical research in the Shingobee River head-waters area, north-central Minnesota; Winter, T. C., Ed.; U. S.Geological Survey Water Supply, 1997.
(42) Wilson, M. A.; Barron, P. F.; Gillam, A. H. The Structure of Fresh-Water Humic Substances As Revealed By C-13-NMR Spectros-copy. Geochim. Cosmochim. Acta 1981, 45, 1743–1750.
(43) Huffman, E. W. D.; Stuber, H. Analytical methodology forelemental analysis of humic substances. In Humic substancesin soil, sediment and water: Geochemistry, isolation, andcharacterization; Aiken, G. R., McKnight, D. M., Wershaw, R. L.,McCarthy, P., Eds.; John Wiley and Sons: New York, 1985.
(44) Hu, C.; Muller-Karger, F. E.; Zepp, R. G. Absorbance, absorptioncoefficient, and apparent quantum yield: A comment oncommon ambiguity in the use of these optical concepts. Limnol.Oceanogr. 2002, 47, 1261–1267.
(45) Blough, N. V.; Green, S. A. Spectroscopic characterization andremote sensing of NLOM. In The Role of Non-Living OrganicMatter in the Earth’s Carbon Cycle; Zepp, R. G., Sonntag, C.,Eds.; John Wiley and Sons: New York, 1995; pp 23–45.
(46) Uher, G.; Hughes, C.; Henry, G.; Upstill-Goddard, R. C. Non-conservative mixing behavior of colored dissolved organic matterin a humic-rich, turbid estuary. Geophys. Res. Lett. 2001, 28,3309–3312.
(47) Helms, J. R.; Stubbins, A.; Ritchie, J. D.; Minor, E. C.; Kieber,D. J.; Mopper, K. Absorbance spectral slopes and slope ratiosas indicators of molecular weight and sources of chromophoricdissolved organic matter. Limnol. Oceanogr. 2008, 53, 955–969.
(48) Chin, Y. P.; Aiken, G.; Oloughlin, E. Molecular-Weight, Poly-dispersity, and Spectroscopic Properties of Aquatic HumicSubstances. Environ. Sci. Technol. 1994, 28, 1853–1858.
(49) Weishaar, J. L.; Aiken, G. R.; Bergamaschi, B. A.; Fram, M. S.;Fujii, R.; Mopper, K. Evaluation of specific ultraviolet absorbanceas an indicator of the chemical composition and reactivity ofdissolved organic carbon. Environ. Sci. Technol. 2003, 37, 4702–4708.
(50) Mopper, K.; Zhou, X. L.; Kieber, R. J.; Kieber, D. J.; Sikorski, R. J.;Jones, R. D. Photochemical degradation of dissolved organic-carbon and its impact on the oceanic carbon-cycle. Nature 1991,353, 60–62.
(51) Pos, W. H.; Riemer, D. D.; Zika, R. G. Carbonyl sulfide (OCS) andcarbon monoxide (CO) in natural waters: evidence of a coupledproduction pathway. Mar. Chem. 1998, 62, 89–101.
(52) Turro, N. J. Modern Molecular Photochemistry; UniversityScience Books: Sausalito, CA, 1991.
(53) Cowan, D. O.; Drisko, R. L. Elements of Organic Photochemistry;Plenum Press: New York, 1976.
(54) Grossweiner, L. I.; Joschek, H.-I. Optical Generation of HydratedElectrons from Aromatic Compounds; Advances In ChemistrySeries, No. 50; American Chemical Society: Washington DC,1965.
(55) Wan, P.; Shukla, D. Utility of acid-base behavior of excited statesof organic molecules. Chem. Rev. 1993, 93, 571–584.
(56) Lathioor, E. C.; Leigh, W. J.; St. Pierre, M. J. Geometrical effectson intramolecular quenching of aromatic ketone (ð,ð*) tripletsby remote phenolic hydrogen abstraction. J. Am. Chem. Soc.1999, 121, 11984–11992.
(57) Saunders, S. M.; Jenkin, M. E.; Derwent, R. G.; Pilling, M. J.Protocol for the development of the Master Chemical Mech-anism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds. Atmos. Chem. Phys. 2003,3, 161–180.
(58) Zhang, Y.; Xie, H.; Chen, G. Factors affecting the efficiency ofcarbon monoxide photoproduction in the St. Lawrence estuarinesystem (Canada). Environ. Sci. Technol. 2006, 40, 7771–7777.
ES703014Q
3276 9 ENVIRONMENTAL SCIENCE & TECHNOLOGY / VOL. 42, NO. 9, 2008