redox cycling of iron by aβ42
TRANSCRIPT

/freeradbiomed
Free Radical Biology & M
Original Contribution
Redox cycling of iron by Ah42
Ayesha Khan a, Jon P. Dobson b, Christopher Exley a,*
a Birchall Centre for Inorganic Chemistry and Materials Science, Lennard-Jones Laboratories, Keele University, Staffordshire ST5 5BG, UKb Institute for Science and Technology in Medicine, Keele University, Staffordshire ST5 5BG, UK
Received 27 June 2005; revised 31 August 2005; accepted 7 September 2005
Available online 14 October 2005
Abstract
The amyloid cascade hypothesis and oxidative damage have been inextricably linked in the neurodegeneration that is characteristic of
Alzheimer_s disease. We have investigated this link and sought to suggest a mechanism whereby the precipitation of Ah42 might contribute to the
redox cycling of iron and hence the generation of reactive oxygen species via Fenton-like chemistry. We have shown that the critical step in the
auto-oxidation of Fe(II) under the near-physiological conditions of our study involved the generation of H2O2 via O2S� and that Ah42 influenced
Fenton chemistry through aggregation state-specific binding of both Fe(II) and Fe(III). The net result of these interactions was the delayed
precipitation of kinetically redox-inactive Fe(OH)3(s) such that Fe(II)/Fe(III) were cycled in redox-active forms over a substantially longer time
period than if peptide had been absent from preparations. The addition of physiologically significant concentrations of either Cu(II) or Zn(II)
reduced the role played by Ah42 in the Fe(II)/Fe(III) redox cycle whereas a pathophysiologically significant concentration of Al(III) potentiated
the redox cycle in favour of Fe(II) whether or not Cu(II) or Zn(II) was additionally present. The results support the notion that oxidative damage in
the immediate vicinity of, for example, senile plaques, may be the result of Fenton chemistry catalysed by the codeposition of Ah42 with metals
such as Fe(II)/Fe(III) and Al(III).
D 2005 Elsevier Inc. All rights reserved.
Keywords: Amyloid; Alzheimer_s disease; h-Sheet; Fenton chemistry; Iron; Aluminum; Biological oxidation; Free radical
Introduction
Fibrillar deposits of h-amyloid (Ah) are neurotoxic in vivo
and may be involved in the pathogenesis of Alzheimer_sdisease (AD) [1]. Neurotoxicity may be mediated by fibrillar
Ah per se, prefibrillar Ah in equilibrium with precipitated
amyloid, or a combination of solution (colloidal) and ‘‘solid
phase’’ Ah. The mechanism of in vivo toxicity is unknown
though neuronal damage occurring in the immediate vicinity of
fibrillar amyloid deposits is often associated with markers for
oxidative stress [2]. In addition there is also evidence that Ahin senile plaques is itself oxidatively modified [3] and that
codeposits of iron and Ah are significant sources of reactive
oxygen species (ROS) [4]. Senile plaques are sinks for metals
which are both redox active, such as iron and copper, and redox
inactive, such as zinc and aluminum [5], and synergies between
0891-5849/$ - see front matter D 2005 Elsevier Inc. All rights reserved.
doi:10.1016/j.freeradbiomed.2005.09.013
Abbreviations: Ah, h-amyloid; ROS, reactive oxygen species; KH, Krebs-
Henseleit; ThT, Thioflavin T.
* Corresponding author.
E-mail address: [email protected] (C. Exley).
different metals codeposited with different forms of Ah might
define the neurotoxic potential of amyloid deposits in vivo [6].
Ferrous iron, Fe(II), catalyses the formation of ROS, such as
the hydroxyl radical (OHS), by its reaction with hydrogen
peroxide (H2O2). However, extremely low concentrations of
Fe2+ and competitive substrates for H2O2, such as the enzyme
catalase, would normally ensure that this reaction was not
favoured in vivo. Therefore, it was surprising to read that Fe(II)
had been identified associated with senile plaques [4] and even
more surprising to learn that the origin of such could be the
reduction of Fe(III) in the presence of Ah42 [7]. If senile
plaques were catalysts for the formation of Fe(II) then this
might explain their association with the generation of ROS and
neuronal damage in their immediate vicinities [8].
Ah42 localised to senile plaques has been shown to adopt h-pleated sheet conformation [3] and it is this fibrillar Ah in
concert with iron which is now implicated in the formation of
ROS [9]. However, several other metals are found associated
with senile plaques [5] and either alone or in tandem with iron
may influence the conformation of deposited Ah42 [10] and its
potential to catalyse the formation of ROS [11–13]. We have
edicine 40 (2006) 557 – 569
www.elsevier.com/locate

A. Khan et al. / Free Radical Biology & Medicine 40 (2006) 557–569558
investigated the role of the aggregation state of Ah42 in
promoting the redox cycling of Fe(II) and the concomitant
effects of companion metals, aluminum, copper, and zinc.
Materials and methods
Peptide preparations
Ah42 was purchased as the lyophilised salt (Bachem,
Saffron Walden, UK), dissolved in 0.010 mM NaOH in
ultrapure water (conductivity <0.067 AS/cm, Elga, UK) to
give a ca 0.200 mM stock solution. This stock was then
centrifuged (15,000 rpm for 300 s) and divided into an
appropriate number of aliquots which were immediately frozen
Fig. 1. Summary of the different preparations, their conditions of incubation, the sam
of Fe(II) following the addition of either Fe(II) or Fe(III).
and thereafter thawed and used within 3 days of storage at
�20-C. Peptide aliquots were diluted into an appropriate
volume of modified Krebs-Henseleit (KH) medium (NaCl,
123.5 mM; KCl, 4.8 mM; MgSO4, 1.2 mM; CaCl2, 1.4 mM;
glucose, 11.0 mM) which was buffered at pH 7.40 T 0.05 with
100 mM Pipes (1,4-piperazinediethanesulfonic acid) to give
nominal [Ah42] of 0.29, 1.83, and 3.52 AM. Al(III), Fe(III),
Cu(II), and Zn(II) were added to preparations as certified
stocks of their nitrate salt (Perkin-Elmer, Beaconsfield, UK)
whereas Fe(II) was added each time from freshly prepared
stocks of 1.50 mM FeCl2 in 2% v/v HCl (Riedel de Haan,
Poole, UK). A summary of the peptide preparations and their
respective peptide-free controls is shown schematically in Fig.
1. The peptide preparations are also listed in Tables 1–3.
pling to determine aggregation states, and the spectrophotometric measurement

Table 1
The influence of the concentration of Ah42 on Thioflavin T fluorescence (AU)
of preparations (a–c) incubated at 37-C for 1, 24, or 48 h (pre-Fe(II) addition)
and the Thioflavin T fluorescence (AU) and concentration (AM) of Fe(II)
([Fe2+]T) following the addition of 5.0 AM Fe(II) and incubation at 37-C for a
further 30 min (post-Fe(II) addition)
Preparation Incubation time
1 hour 24 hours 48 hours
(a) 0.29 lM Ab42 + Fe(II)
ThT fluorescence
Pre-Fe(II) 4 (0.3) 6 (1.0) 4 (0.4)
Post-Fe(II) 4 (0.4) 5 (0.7) 3 (0.4)
Fe(II) concentration
[Fe2+]T 0.84 (0.055) 1.57 (0.204) 1.35 (0.123)
(b) 1.83 lM Ab42 + Fe(II)
ThT fluorescence
Pre-Fe(II) 46 (20.7) 60 (24.8) 41 (22.5)
Post-Fe(II) 26 (14.2) 36 (11.8) 21 (15.0)
Fe(II) concentration
[Fe2+]T 1.14 (0.223) 2.09 (0.303) 1.92 (0.368)
(c) 3.52 lM Ab42 + Fe(II)
ThT fluorescence
Pre-Fe(II) 189 (31.5) 192 (18.3) 130 (23.4)
Post-Fe(II) 137 (68.6) 110 (23.6) 60 (12.0)
Fe(II) concentration
[Fe2+]T 0.98 (0.163) 3.10 (0.300) 3.14 (0.901)
Mean and SD are given, n = 15.
Fig. 2. The auto-oxidation for 150 min of 5.0 AM Fe(II) added in modified KH
medium buffered at pH 7.40 T 0.05 and 37-C compared to the same conditions
but including either 8 mM sodium azide or 10 AM ascorbic acid. Mean and SD
are plotted, n = 5.
A. Khan et al. / Free Radical Biology & Medicine 40 (2006) 557–569 559
Spectrophotometric determination of Fe(II)
The reaction of Fe(II) with 1,10-phenanthroline to form a
coloured complex with an absorption maximum at around 510
nm is a tried and tested method for the determination of Fe(II) in
aqueous solutions at near-neutral pH [14]. The application of
the Beer-Lambert law and the extinction coefficient for the
Fe(II)–phenanthroline complex (11,110 M�1 cm�1) allows for
the precise determination of Fe(II) in the submicromolar range
and, providing that 1,10-phenanthroline is present to excess, the
method is free of interferences from other metals (e.g., Fe(III),
Al(III), Cu(II), Zn(II)) bound by this ligand. We tested the
possible interference of each and combinations of these metals
on the determination of Fe(II) and confirmed the lack of any
interferences under the conditions of our study. We optimised
the application of this method to our assay conditions by
measuring the auto-oxidation of added Fe(II) over time and
included either 8 mM sodium azide (NaN3) as an accelerant or
10 AM ascorbic acid as an inhibitor of auto-oxidation (Fig. 2).
We used these data to choose 30 min as an appropriate time
period for auto-oxidation for all further auto-oxidation assays.
Thioflavin T fluorescence
Thioflavin T (ThT) is a benzothiazole dye which undergoes
a characteristic spectral alteration upon being bound by h-pleated sheet structures [15]. It is not bound by monomers or
oligomers of Ah42 nor is it bound by amorphous precipitates
of the peptide and in this study it was used to identify and
loosely quantify the formation of h-pleated fibrils of Ah42.
The assay has been described in detail elsewhere [16] but
briefly 100 AL of peptide (or control) preparation was sampled
as per Fig. 1 and mixed with 10 AL of ThT such that the
concentration of the latter was 10 AM and significantly in
excess of the concentration of peptide. The ThT fluorescence
of each sample was then determined at 37-C over a 300 s
period (Perkin-Elmer LS50B Luminescence Spectrometer; Ex,
450 nm, 5 nm bandpass; and Em, 482 nm, 10 nm bandpass),
the latter ensuring that a stable ThT fluorescence had been
reached before the reading was taken. Fluorescence readings
for peptide-free preparations, though usually very low, less
than 5 fluorescence units (<5 AU), were substracted from the
appropriate peptide preparation.
Transmission electron microscopy
Samples for TEM were taken according to the protocol in
Fig. 1, mounted on formvar-coated copper grids, negatively
stained with 2% uranyl acetate, and viewed using a JEOL JEM-
1230 electron microscope.
Statistical analyses
Statistical analyses were carried out using the Mann-
Whitney test (Minitab, version 14). This performs a hypothesis
test of the equality of two population medians and calculates
the confidence level and was used as a nonparametric
alternative to the two-sample t test due to data not being
normally distributed. Confidence levels > p = 0.05 were
rejected as not being statistically significant.
Results
To determine how solution chemistry in the absence of Ah42
influenced the auto-oxidation of Fe(II) all peptide preparations
were compared with peptide-free preparations. In this way it
was possible to determine how the presence of peptide
contributed to the auto-oxidation of Fe(II) under each set of

A. Khan et al. / Free Radical Biology & Medicine 40 (2006) 557–569560
experimental conditions. In addition this allowed computation
of the peptide-attributable [Fe2+] which was defined as the
difference between the [Fe2+] measured in the presence of
peptide and the [Fe2+] measured in the corresponding peptide-
free preparation:
½Fe2þ�PA
¼ Fe2þ� �
T� Fe2þ� �
C:
How did [Ab42] and its time-dependent formation of b-pleatedsheets affect [Fe2+]
At the lowest [Ah42] of 0.29 AM and following only a
1-h incubation at 37-C the presence of h-pleated conformers of
peptide was indicated by ThT fluorescence. The measured
values of 3–6 AU were close to the limit of detection of this
application of the assay and did not change significantly ( p >
0.05) following incubation of this concentration of peptide for a
further 24 and 48h (Table 1). This [Ah42] increased [Fe2+]Trelative to peptide-free controls at 1 h ( p = 0.003), 24 h ( p <
0.001), and 48 h ( p < 0.001) though the increases were not time
dependent with the highest [Fe2+]T (1.57 T 0.204 AM ) which
represented ca 30% of added Fe(II), being measured in peptide
incubated for 24 h. A sixfold increase in [Ah42] to 1.83 AMproduced significantly higher ThT fluorescence (20–80 AU)
though the extent of formation of h-pleated conformers was not
significantly influenced ( p > 0.05) by ageing peptide solutions
for a further 24 or 48 h (Table 1). Once again, the presence of
peptide significantly increased [Fe2+]T relative to peptide-free
controls at 1, 24, and 48 h ( p < 0.001) and the highest [Fe2+]T(2.09 T 0.303 AM), measured at 24 h, represented ca 40% of the
added Fe(II). At 3.52 AM Ah42 the formation of h-pleatedconformers was extensive after only a 1-h incubation (189 T31.5 AU), remained unchanged over the next 24 h (193 T 18.3
AU), and fell significantly ( p < 0.001) by 48 h (130 T 23.4 AU)(Table 1). At 1 h incubation [Fe2+]T was not significantly
increased with respect to the peptide-free control ( p = 0.054)
whereas the increases were highly significant at both 24 and
48 h ( p < 0.001). Ageing of the peptide solutions resulted in
significant increases in [Fe2+]T from 0.98 T 0.163 AM at 1 h to
3.10 T 0.300 AM at 24 h ( p < 0.001) and a further
insignificant increase ( p = 1.000) from 24 to 48 h (3.14 T0.901 AM). The latter was equivalent to ca 65% of added
Fe(II). Ah42 was clearly influential in determining [Fe2+]Tthough there was not a clear relationship between the extent
of formation of h-pleated conformers of Ah42 and [Fe2+]T.
For example, at 3.52 AM Ah42 after 1 h incubation the ThT
fluorescence was 189 T 31.5 AU and the corresponding
[Fe2+]T was 0.98 T 0.163 AM whereas after 24 h incubation of
the peptide the ThT fluorescence was unchanged at 193 T18.3 AU while the [Fe2+]T was 3.10 T 0.300 AM. The
apparent lack of an association between ThT fluorescence and
[Fe2+]T was supported by an insignificant correlation coeffi-
cient (r2 = 0.113) when ThT fluorescence prior to Fe(II)
addition for all [Ah42] was plotted against their corresponding
[Fe2+]T. However, when the same analyses were carried out
using the control-subtracted data, [Fe2+]PA, a much more
positive correlation (r2 = 0.267) suggested, if only weakly,
that higher pre-Fe(II) addition ThT fluorescence resulted in
higher [Fe2+]PA (Fig. 3a). When this analysis was broken
down to show how the correlation between pre-Fe(II) addition
ThT fluorescence and [Fe2+]PA was influenced by the three
different [Ah42] it was found to be strongest for the
intermediate [Ah42]; the r2 were 0.078, 0.279, and 0.027
for 0.29, 1.83, and 3.52 AM Ah42, respectively (Figs. 3b–d).
This, along with the observation that a 12-fold increase in
[Ah42] (i.e., 0.29 to 3.52 AM) only resulted in a 2-fold
increase in [Fe2+]PA, may have suggested that some aspect of
the aggregation state of Ah42 was more important than its
absolute concentration in determining [Fe2+]T.
A consistent effect which was measured for each of the
[Ah42] (though not a statistically significant effect for 0.29 AMAh42) was that the addition of Fe(II) and the subsequent
incubation for 30 min always resulted in a significant fall in
ThT fluorescence over this short period. This remarkable effect
was loosely associated with [Fe2+]T in that proportional
reductions in ThT fluorescence were consistently greater in
those treatments in which [Fe2+]T were highest. For example,
for 3.52 AM Ah42 incubated for 1 h the 28% reduction in ThT
fluorescence which followed the addition of 5.0 AM Fe(II) was
associated with an [Fe2+]T of 0.98 T 0.163 AM whereas for the
same peptide incubated for 48 h the 54% reduction in ThT
fluorescence which followed the addition of 5.0 AM Fe(II) was
associated with an [Fe2+]T of 3.14 T 0.901 AM.
How was the influence of 3.52 lM Ab42 on [Fe2+] affected by
the additional presence of Al(III), Cu(II), and Zn(II)
Peptide-free preparations
The auto-oxidation at 37-C over 30 min of 5.0 AM Fe(II)
added to peptide-free preparations which had previously been
incubated at 37-C for 1, 24, and 48 h resulted in each case in ca
85% of the available Fe(II) being oxidised to Fe(III) (Table 2).
For example, for the 1 h preparation, [Fe2+]C fell from a
maximum possible concentration of 4.75 T 0.206 to 0.83 T0.105 AM. When the peptide-free preparations also included
8 mM NaN3, a known scavenger of OHS, the auto-oxidation
was accelerated significantly ( p < 0.001) such that more than
95% of the available Fe(II) was oxidised (Table 2). For
example, for the 1 h preparation, [Fe2+]C fell from a maximum
possible concentration of 4.75 T 0.206 to 0.24 T 0.075 AM. The
opposite effect was found for peptide-free preparations which
additionally included 5.0 AM Al(III), a known pro-oxidant,
such that [Fe2+]C in these preparations was only reduced by
50% (Table 2). For example, for the 1 h preparation, [Fe2+]Cfell from a maximum possible concentration of 4.13 T 0.374 to
2.15 T 0.155 AM. When 5.0 AM Fe(III) was added to peptide-
free preparations instead of Fe(II) the resultant [Fe2+]C were
very low, for example, 0.14 T 0.050 AM for the 1 h preparation,
and suggested that the peptide-free preparations were not
themselves capable of reducing Fe(III) to Fe(II). The additional
presence of 5.0 AM Al(III) in these Fe(III) preparations did not
influence the measured [Fe2+]C and this showed that Al(III) per
se could not reduce Fe(III) to Fe(II) (Table 2). The additional
presence of 10.0 AM Cu(II), a redox active metal, accelerated
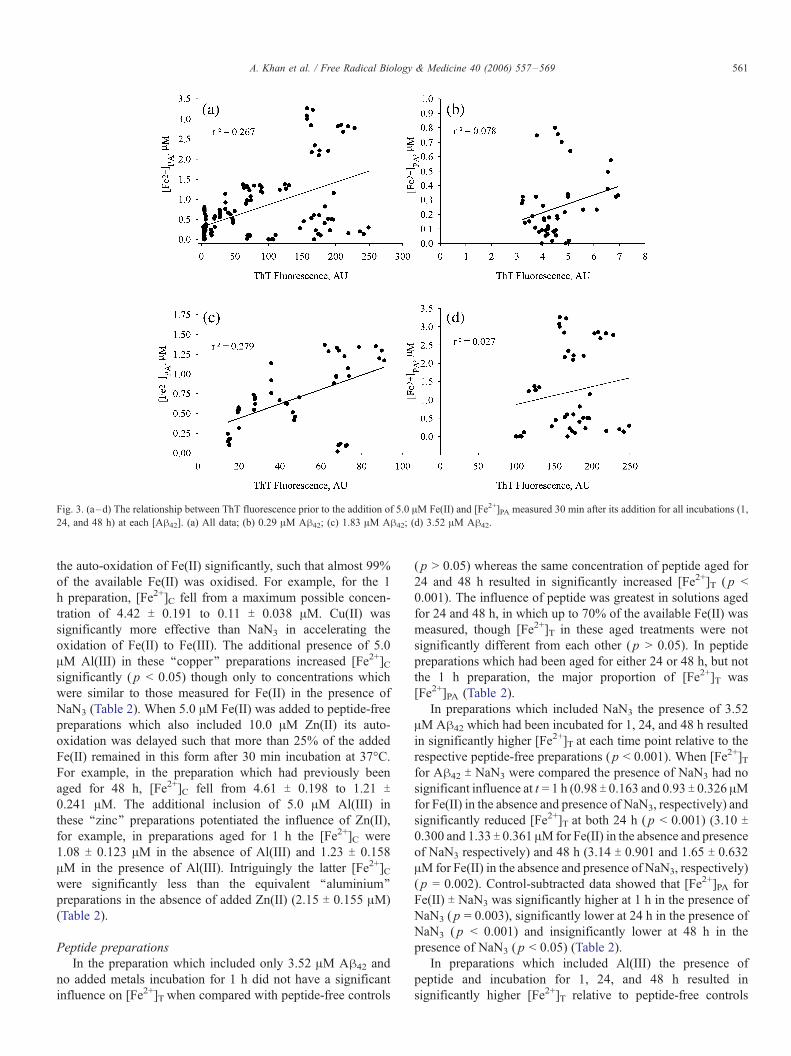
Fig. 3. (a–d) The relationship between ThT fluorescence prior to the addition of 5.0 AM Fe(II) and [Fe2+]PA measured 30 min after its addition for all incubations (1,
24, and 48 h) at each [Ah42]. (a) All data; (b) 0.29 AM Ah42; (c) 1.83 AM Ah42; (d) 3.52 AM Ah42.
A. Khan et al. / Free Radical Biology & Medicine 40 (2006) 557–569 561
the auto-oxidation of Fe(II) significantly, such that almost 99%
of the available Fe(II) was oxidised. For example, for the 1
h preparation, [Fe2+]C fell from a maximum possible concen-
tration of 4.42 T 0.191 to 0.11 T 0.038 AM. Cu(II) was
significantly more effective than NaN3 in accelerating the
oxidation of Fe(II) to Fe(III). The additional presence of 5.0
AM Al(III) in these ‘‘copper’’ preparations increased [Fe2+]Csignificantly ( p < 0.05) though only to concentrations which
were similar to those measured for Fe(II) in the presence of
NaN3 (Table 2). When 5.0 AM Fe(II) was added to peptide-free
preparations which also included 10.0 AM Zn(II) its auto-
oxidation was delayed such that more than 25% of the added
Fe(II) remained in this form after 30 min incubation at 37-C.For example, in the preparation which had previously been
aged for 48 h, [Fe2+]C fell from 4.61 T 0.198 to 1.21 T0.241 AM. The additional inclusion of 5.0 AM Al(III) in
these ‘‘zinc’’ preparations potentiated the influence of Zn(II),
for example, in preparations aged for 1 h the [Fe2+]C were
1.08 T 0.123 AM in the absence of Al(III) and 1.23 T 0.158
AM in the presence of Al(III). Intriguingly the latter [Fe2+]Cwere significantly less than the equivalent ‘‘aluminium’’
preparations in the absence of added Zn(II) (2.15 T 0.155 AM)
(Table 2).
Peptide preparations
In the preparation which included only 3.52 AM Ah42 and
no added metals incubation for 1 h did not have a significant
influence on [Fe2+]T when compared with peptide-free controls
( p > 0.05) whereas the same concentration of peptide aged for
24 and 48 h resulted in significantly increased [Fe2+]T ( p <
0.001). The influence of peptide was greatest in solutions aged
for 24 and 48 h, in which up to 70% of the available Fe(II) was
measured, though [Fe2+]T in these aged treatments were not
significantly different from each other ( p > 0.05). In peptide
preparations which had been aged for either 24 or 48 h, but not
the 1 h preparation, the major proportion of [Fe2+]T was
[Fe2+]PA (Table 2).
In preparations which included NaN3 the presence of 3.52
AM Ah42 which had been incubated for 1, 24, and 48 h resulted
in significantly higher [Fe2+]T at each time point relative to the
respective peptide-free preparations ( p < 0.001). When [Fe2+]Tfor Ah42 T NaN3 were compared the presence of NaN3 had no
significant influence at t = 1 h (0.98 T 0.163 and 0.93 T 0.326 AMfor Fe(II) in the absence and presence of NaN3, respectively) and
significantly reduced [Fe2+]T at both 24 h ( p < 0.001) (3.10 T0.300 and 1.33 T 0.361 AMfor Fe(II) in the absence and presence
of NaN3 respectively) and 48 h (3.14 T 0.901 and 1.65 T 0.632
AM for Fe(II) in the absence and presence of NaN3, respectively)
( p = 0.002). Control-subtracted data showed that [Fe2+]PA for
Fe(II) T NaN3 was significantly higher at 1 h in the presence of
NaN3 ( p = 0.003), significantly lower at 24 h in the presence of
NaN3 ( p < 0.001) and insignificantly lower at 48 h in the
presence of NaN3 ( p < 0.05) (Table 2).
In preparations which included Al(III) the presence of
peptide and incubation for 1, 24, and 48 h resulted in
significantly higher [Fe2+]T relative to peptide-free controls
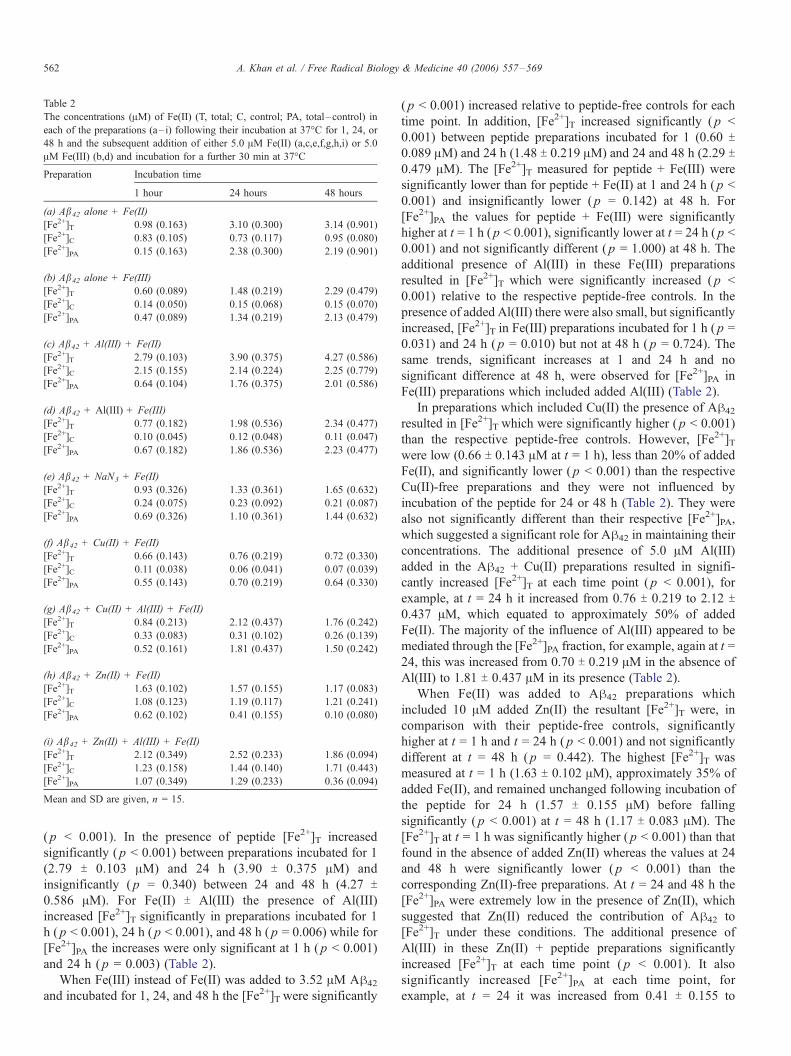
Table 2
The concentrations (AM) of Fe(II) (T, total; C, control; PA, total–control) in
each of the preparations (a– i) following their incubation at 37-C for 1, 24, or
48 h and the subsequent addition of either 5.0 AM Fe(II) (a,c,e,f,g,h,i) or 5.0
AM Fe(III) (b,d) and incubation for a further 30 min at 37-C
Preparation Incubation time
1 hour 24 hours 48 hours
(a) Ab42 alone + Fe(II)
[Fe2+]T 0.98 (0.163) 3.10 (0.300) 3.14 (0.901)
[Fe2+]C 0.83 (0.105) 0.73 (0.117) 0.95 (0.080)
[Fe2+]PA 0.15 (0.163) 2.38 (0.300) 2.19 (0.901)
(b) Ab42 alone + Fe(III)
[Fe2+]T 0.60 (0.089) 1.48 (0.219) 2.29 (0.479)
[Fe2+]C 0.14 (0.050) 0.15 (0.068) 0.15 (0.070)
[Fe2+]PA 0.47 (0.089) 1.34 (0.219) 2.13 (0.479)
(c) Ab42 + Al(III) + Fe(II)
[Fe2+]T 2.79 (0.103) 3.90 (0.375) 4.27 (0.586)
[Fe2+]C 2.15 (0.155) 2.14 (0.224) 2.25 (0.779)
[Fe2+]PA 0.64 (0.104) 1.76 (0.375) 2.01 (0.586)
(d) Ab42 + Al(III) + Fe(III)
[Fe2+]T 0.77 (0.182) 1.98 (0.536) 2.34 (0.477)
[Fe2+]C 0.10 (0.045) 0.12 (0.048) 0.11 (0.047)
[Fe2+]PA 0.67 (0.182) 1.86 (0.536) 2.23 (0.477)
(e) Ab42 + NaN3 + Fe(II)
[Fe2+]T 0.93 (0.326) 1.33 (0.361) 1.65 (0.632)
[Fe2+]C 0.24 (0.075) 0.23 (0.092) 0.21 (0.087)
[Fe2+]PA 0.69 (0.326) 1.10 (0.361) 1.44 (0.632)
(f) Ab42 + Cu(II) + Fe(II)
[Fe2+]T 0.66 (0.143) 0.76 (0.219) 0.72 (0.330)
[Fe2+]C 0.11 (0.038) 0.06 (0.041) 0.07 (0.039)
[Fe2+]PA 0.55 (0.143) 0.70 (0.219) 0.64 (0.330)
(g) Ab42 + Cu(II) + Al(III) + Fe(II)
[Fe2+]T 0.84 (0.213) 2.12 (0.437) 1.76 (0.242)
[Fe2+]C 0.33 (0.083) 0.31 (0.102) 0.26 (0.139)
[Fe2+]PA 0.52 (0.161) 1.81 (0.437) 1.50 (0.242)
(h) Ab42 + Zn(II) + Fe(II)
[Fe2+]T 1.63 (0.102) 1.57 (0.155) 1.17 (0.083)
[Fe2+]C 1.08 (0.123) 1.19 (0.117) 1.21 (0.241)
[Fe2+]PA 0.62 (0.102) 0.41 (0.155) 0.10 (0.080)
(i) Ab42 + Zn(II) + Al(III) + Fe(II)
[Fe2+]T 2.12 (0.349) 2.52 (0.233) 1.86 (0.094)
[Fe2+]C 1.23 (0.158) 1.44 (0.140) 1.71 (0.443)
[Fe2+]PA 1.07 (0.349) 1.29 (0.233) 0.36 (0.094)
Mean and SD are given, n = 15.
A. Khan et al. / Free Radical Biology & Medicine 40 (2006) 557–569562
( p < 0.001). In the presence of peptide [Fe2+]T increased
significantly ( p < 0.001) between preparations incubated for 1
(2.79 T 0.103 AM) and 24 h (3.90 T 0.375 AM) and
insignificantly ( p = 0.340) between 24 and 48 h (4.27 T0.586 AM). For Fe(II) T Al(III) the presence of Al(III)
increased [Fe2+]T significantly in preparations incubated for 1
h ( p < 0.001), 24 h ( p < 0.001), and 48 h ( p = 0.006) while for
[Fe2+]PA the increases were only significant at 1 h ( p < 0.001)
and 24 h ( p = 0.003) (Table 2).
When Fe(III) instead of Fe(II) was added to 3.52 AM Ah42
and incubated for 1, 24, and 48 h the [Fe2+]T were significantly
( p < 0.001) increased relative to peptide-free controls for each
time point. In addition, [Fe2+]T increased significantly ( p <
0.001) between peptide preparations incubated for 1 (0.60 T0.089 AM) and 24 h (1.48 T 0.219 AM) and 24 and 48 h (2.29 T0.479 AM). The [Fe2+]T measured for peptide + Fe(III) were
significantly lower than for peptide + Fe(II) at 1 and 24 h ( p <
0.001) and insignificantly lower ( p = 0.142) at 48 h. For
[Fe2+]PA the values for peptide + Fe(III) were significantly
higher at t = 1 h ( p < 0.001), significantly lower at t = 24 h ( p <
0.001) and not significantly different ( p = 1.000) at 48 h. The
additional presence of Al(III) in these Fe(III) preparations
resulted in [Fe2+]T which were significantly increased ( p <
0.001) relative to the respective peptide-free controls. In the
presence of added Al(III) there were also small, but significantly
increased, [Fe2+]T in Fe(III) preparations incubated for 1 h ( p =
0.031) and 24 h ( p = 0.010) but not at 48 h ( p = 0.724). The
same trends, significant increases at 1 and 24 h and no
significant difference at 48 h, were observed for [Fe2+]PA in
Fe(III) preparations which included added Al(III) (Table 2).
In preparations which included Cu(II) the presence of Ah42
resulted in [Fe2+]T which were significantly higher ( p < 0.001)
than the respective peptide-free controls. However, [Fe2+]Twere low (0.66 T 0.143 AM at t = 1 h), less than 20% of added
Fe(II), and significantly lower ( p < 0.001) than the respective
Cu(II)-free preparations and they were not influenced by
incubation of the peptide for 24 or 48 h (Table 2). They were
also not significantly different than their respective [Fe2+]PA,
which suggested a significant role for Ah42 in maintaining their
concentrations. The additional presence of 5.0 AM Al(III)
added in the Ah42 + Cu(II) preparations resulted in signifi-
cantly increased [Fe2+]T at each time point ( p < 0.001), for
example, at t = 24 h it increased from 0.76 T 0.219 to 2.12 T0.437 AM, which equated to approximately 50% of added
Fe(II). The majority of the influence of Al(III) appeared to be
mediated through the [Fe2+]PA fraction, for example, again at t =
24, this was increased from 0.70 T 0.219 AM in the absence of
Al(III) to 1.81 T 0.437 AM in its presence (Table 2).
When Fe(II) was added to Ah42 preparations which
included 10 AM added Zn(II) the resultant [Fe2+]T were, in
comparison with their peptide-free controls, significantly
higher at t = 1 h and t = 24 h ( p < 0.001) and not significantly
different at t = 48 h ( p = 0.442). The highest [Fe2+]T was
measured at t = 1 h (1.63 T 0.102 AM), approximately 35% of
added Fe(II), and remained unchanged following incubation of
the peptide for 24 h (1.57 T 0.155 AM) before falling
significantly ( p < 0.001) at t = 48 h (1.17 T 0.083 AM). The
[Fe2+]T at t = 1 h was significantly higher ( p < 0.001) than that
found in the absence of added Zn(II) whereas the values at 24
and 48 h were significantly lower ( p < 0.001) than the
corresponding Zn(II)-free preparations. At t = 24 and 48 h the
[Fe2+]PA were extremely low in the presence of Zn(II), which
suggested that Zn(II) reduced the contribution of Ah42 to
[Fe2+]T under these conditions. The additional presence of
Al(III) in these Zn(II) + peptide preparations significantly
increased [Fe2+]T at each time point ( p < 0.001). It also
significantly increased [Fe2+]PA at each time point, for
example, at t = 24 it was increased from 0.41 T 0.155 to
A. Khan et al. / Free Radical Biology & Medicine 40 (2006) 557–569 563
1.29 T 0.233 AM, though it did not prevent significant
reductions in both [Fe2+]T and [Fe2+]PA between 24 and
48 h (Table 2).
We have used ThT fluorescence to determine the extent to
which preparations which included 3.52 AM Ah42 incubated at
37-C for 1, 24, and 48 h formed h-pleated conformers and how
aggregation status subsequently changed following the addition
of 5.0 AM Fe(II) and incubation for a further 30 min. The data
were used to identify if the propensity for Ah42 to influence
[Fe2+]T was dependent upon its prior assembly into h-pleatedsheets of amyloid (Table 3). Significant ThT fluorescence
(>100 AU) was measured after 1, 24, and 48 h in preparations
which included Ah42 alone and Ah42 + Al(III). For the former,
ThT fluorescence was only weakly correlated with either
[Fe2+]T (r2 = 0.006) or [Fe2+]PA (r2 = 0.027) whereas for
Ah42 + Al(III) r2 values of 0.190 and 0.179 for [Fe2+]T and
[Fe2+]PA respectively indicated a closer relationship between
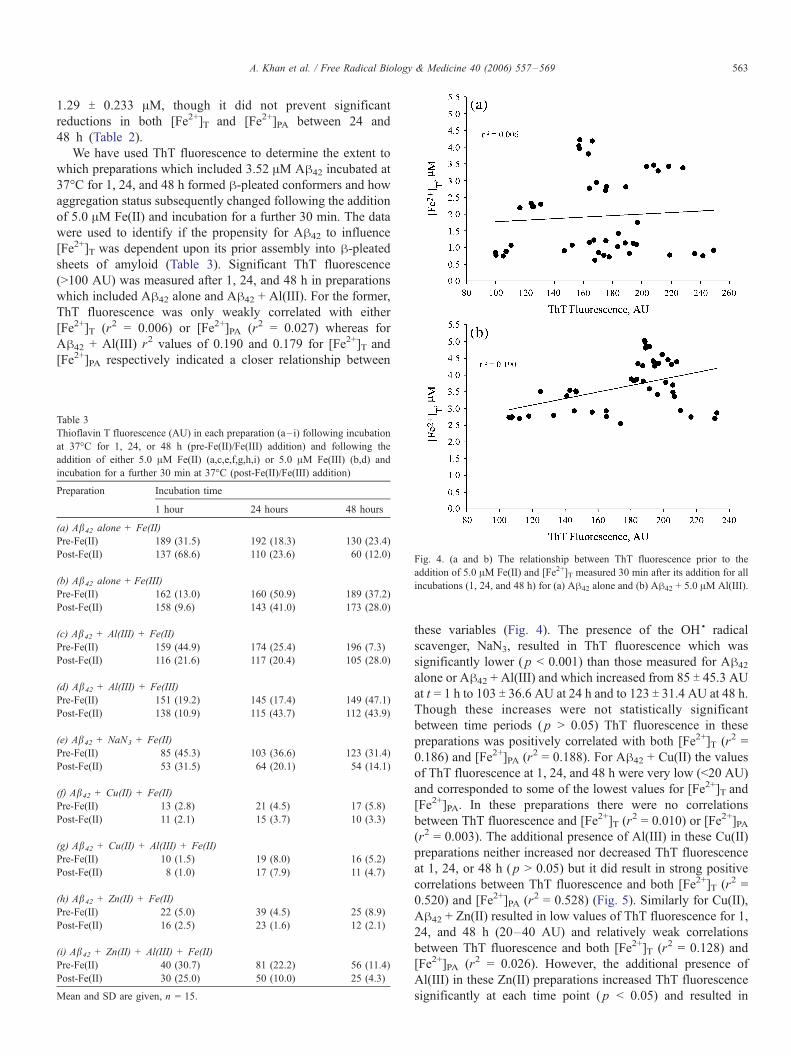
Table 3
Thioflavin T fluorescence (AU) in each preparation (a– i) following incubation
at 37-C for 1, 24, or 48 h (pre-Fe(II)/Fe(III) addition) and following the
addition of either 5.0 AM Fe(II) (a,c,e,f,g,h,i) or 5.0 AM Fe(III) (b,d) and
incubation for a further 30 min at 37-C (post-Fe(II)/Fe(III) addition)
Preparation Incubation time
1 hour 24 hours 48 hours
(a) Ab42 alone + Fe(II)
Pre-Fe(II) 189 (31.5) 192 (18.3) 130 (23.4)
Post-Fe(II) 137 (68.6) 110 (23.6) 60 (12.0)
(b) Ab42 alone + Fe(III)
Pre-Fe(II) 162 (13.0) 160 (50.9) 189 (37.2)
Post-Fe(II) 158 (9.6) 143 (41.0) 173 (28.0)
(c) Ab42 + Al(III) + Fe(II)
Pre-Fe(II) 159 (44.9) 174 (25.4) 196 (7.3)
Post-Fe(II) 116 (21.6) 117 (20.4) 105 (28.0)
(d) Ab42 + Al(III) + Fe(III)
Pre-Fe(II) 151 (19.2) 145 (17.4) 149 (47.1)
Post-Fe(II) 138 (10.9) 115 (43.7) 112 (43.9)
(e) Ab42 + NaN3 + Fe(II)
Pre-Fe(II) 85 (45.3) 103 (36.6) 123 (31.4)
Post-Fe(II) 53 (31.5) 64 (20.1) 54 (14.1)
(f) Ab42 + Cu(II) + Fe(II)
Pre-Fe(II) 13 (2.8) 21 (4.5) 17 (5.8)
Post-Fe(II) 11 (2.1) 15 (3.7) 10 (3.3)
(g) Ab42 + Cu(II) + Al(III) + Fe(II)
Pre-Fe(II) 10 (1.5) 19 (8.0) 16 (5.2)
Post-Fe(II) 8 (1.0) 17 (7.9) 11 (4.7)
(h) Ab42 + Zn(II) + Fe(II)
Pre-Fe(II) 22 (5.0) 39 (4.5) 25 (8.9)
Post-Fe(II) 16 (2.5) 23 (1.6) 12 (2.1)
(i) Ab42 + Zn(II) + Al(III) + Fe(II)
Pre-Fe(II) 40 (30.7) 81 (22.2) 56 (11.4)
Post-Fe(II) 30 (25.0) 50 (10.0) 25 (4.3)
Mean and SD are given, n = 15.
Fig. 4. (a and b) The relationship between ThT fluorescence prior to the
addition of 5.0 AM Fe(II) and [Fe2+]T measured 30 min after its addition for all
incubations (1, 24, and 48 h) for (a) Ah42 alone and (b) Ah42 + 5.0 AM Al(III).
these variables (Fig. 4). The presence of the OHSradical
scavenger, NaN3, resulted in ThT fluorescence which was
significantly lower ( p < 0.001) than those measured for Ah42
alone or Ah42 + Al(III) and which increased from 85 T 45.3 AUat t = 1 h to 103 T 36.6 AU at 24 h and to 123 T 31.4 AU at 48 h.
Though these increases were not statistically significant
between time periods ( p > 0.05) ThT fluorescence in these
preparations was positively correlated with both [Fe2+]T (r2 =
0.186) and [Fe2+]PA (r2 = 0.188). For Ah42 + Cu(II) the values
of ThT fluorescence at 1, 24, and 48 h were very low (<20 AU)
and corresponded to some of the lowest values for [Fe2+]T and
[Fe2+]PA. In these preparations there were no correlations
between ThT fluorescence and [Fe2+]T (r2 = 0.010) or [Fe2+]PA(r2 = 0.003). The additional presence of Al(III) in these Cu(II)
preparations neither increased nor decreased ThT fluorescence
at 1, 24, or 48 h ( p > 0.05) but it did result in strong positive
correlations between ThT fluorescence and both [Fe2+]T (r2 =
0.520) and [Fe2+]PA (r2 = 0.528) (Fig. 5). Similarly for Cu(II),
Ah42 + Zn(II) resulted in low values of ThT fluorescence for 1,
24, and 48 h (20–40 AU) and relatively weak correlations
between ThT fluorescence and both [Fe2+]T (r2 = 0.128) and
[Fe2+]PA (r2 = 0.026). However, the additional presence of
Al(III) in these Zn(II) preparations increased ThT fluorescence
significantly at each time point ( p < 0.05) and resulted in
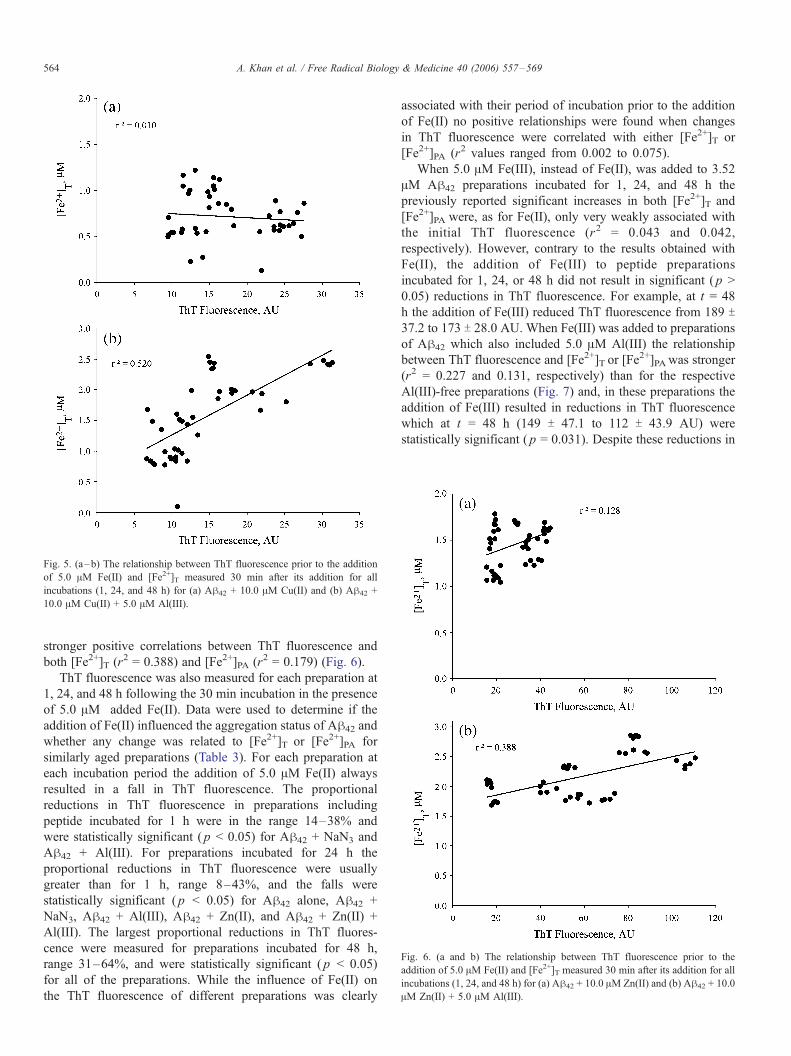
Fig. 5. (a–b) The relationship between ThT fluorescence prior to the addition
of 5.0 AM Fe(II) and [Fe2+]T measured 30 min after its addition for all
incubations (1, 24, and 48 h) for (a) Ah42 + 10.0 AM Cu(II) and (b) Ah42 +
10.0 AM Cu(II) + 5.0 AM Al(III).
Fig. 6. (a and b) The relationship between ThT fluorescence prior to the
addition of 5.0 AM Fe(II) and [Fe2+]T measured 30 min after its addition for al
incubations (1, 24, and 48 h) for (a) Ah42 + 10.0 AM Zn(II) and (b) Ah42 + 10.0
AM Zn(II) + 5.0 AM Al(III).
A. Khan et al. / Free Radical Biology & Medicine 40 (2006) 557–569564
stronger positive correlations between ThT fluorescence and
both [Fe2+]T (r2 = 0.388) and [Fe2+]PA (r2 = 0.179) (Fig. 6).
ThT fluorescence was also measured for each preparation at
1, 24, and 48 h following the 30 min incubation in the presence
of 5.0 AM added Fe(II). Data were used to determine if the
addition of Fe(II) influenced the aggregation status of Ah42 and
whether any change was related to [Fe2+]T or [Fe2+]PA for
similarly aged preparations (Table 3). For each preparation at
each incubation period the addition of 5.0 AM Fe(II) always
resulted in a fall in ThT fluorescence. The proportional
reductions in ThT fluorescence in preparations including
peptide incubated for 1 h were in the range 14–38% and
were statistically significant ( p < 0.05) for Ah42 + NaN3 and
Ah42 + Al(III). For preparations incubated for 24 h the
proportional reductions in ThT fluorescence were usually
greater than for 1 h, range 8–43%, and the falls were
statistically significant ( p < 0.05) for Ah42 alone, Ah42 +
NaN3, Ah42 + Al(III), Ah42 + Zn(II), and Ah42 + Zn(II) +
Al(III). The largest proportional reductions in ThT fluores-
cence were measured for preparations incubated for 48 h,
range 31–64%, and were statistically significant ( p < 0.05)
for all of the preparations. While the influence of Fe(II) on
the ThT fluorescence of different preparations was clearly
associated with their period of incubation prior to the addition
of Fe(II) no positive relationships were found when changes
in ThT fluorescence were correlated with either [Fe2+]T or
[Fe2+]PA (r2 values ranged from 0.002 to 0.075).
When 5.0 AM Fe(III), instead of Fe(II), was added to 3.52
AM Ah42 preparations incubated for 1, 24, and 48 h the
previously reported significant increases in both [Fe2+]T and
[Fe2+]PA were, as for Fe(II), only very weakly associated with
the initial ThT fluorescence (r2 = 0.043 and 0.042,
respectively). However, contrary to the results obtained with
Fe(II), the addition of Fe(III) to peptide preparations
incubated for 1, 24, or 48 h did not result in significant ( p >
0.05) reductions in ThT fluorescence. For example, at t = 48
h the addition of Fe(III) reduced ThT fluorescence from 189 T37.2 to 173 T 28.0 AU. When Fe(III) was added to preparations
of Ah42 which also included 5.0 AM Al(III) the relationship
between ThT fluorescence and [Fe2+]T or [Fe2+]PAwas stronger
(r2 = 0.227 and 0.131, respectively) than for the respective
Al(III)-free preparations (Fig. 7) and, in these preparations the
addition of Fe(III) resulted in reductions in ThT fluorescence
which at t = 48 h (149 T 47.1 to 112 T 43.9 AU) were
statistically significant ( p = 0.031). Despite these reductions in
l
Fig. 7. (a and b) The relationship between ThT fluorescence prior to the
addition of 5.0 AM Fe(III) and [Fe2+]T measured 30 min after its addition for all
incubations (1, 24, and 48 h) for (a) Ah42 alone and (b) Ah42 + 5.0 AM Al(III).
Fig. 8. Oligomers of Ah42 formed after only 1-h incubation, and prior to the
addition of 5.0 AM Fe(II), in the preparation which included 3.52 AM Ah42 +
10.0 AM Cu(II) + 5.0 AM Al(III). Magnification �50K.
A. Khan et al. / Free Radical Biology & Medicine 40 (2006) 557–569 565
ThT fluorescence there were no positive correlations between
the changes in ThT fluorescence and either [Fe2+]T or [Fe2+]PA.
Discussion
Auto-oxidation at 37 -C of 5.0 AM added Fe(II) in KH
medium buffered at pH 7.40 was >80% complete within 30
min and independent of preincubation for 1, 24, or 48 h. The
inclusion of 0.29, 1.83, or 3.52 AM Ah42 inhibited auto-
oxidation. For example, auto-oxidation was only 25%
complete in media which included 3.52 AM Ah42 which
had been preincubated for 48 h. [Ah42] were chosen to
encompass three saturation states of peptide, sub (0.29 AM),
near (1.83 AM), and super (3.52 AM) [17], distinguished by
their propensities to self-aggregate and form h-pleated sheet
conformers. The auto-oxidation of added Fe(II) was influ-
enced by synergies between peptide concentration and
incubation period though the formation of h-pleated sheets
was only part of any synergy. For example, the strongest
correlation between ThT fluorescence and [Fe2+]PA was found
for the near-saturation (1.83 AM) and not the supersaturation
(3.52 AM) concentration of Ah42 (Figs. 3a–d), indicating that
the form of peptide which influenced the auto-oxidation of
added Fe(II) was probably intermediate in structure between
the random turn/coil conformation of monomeric Ah42 and
the h-pleated sheets which bind ThT. To understand this
further the auto-oxidation of Fe(II) was determined in
preparations in which Ah42 was precipitated in the presence
of Cu(II), Zn(II), or Al(III). Cu(II) inhibited the formation of
h-pleated sheets and precipitated Ah42 as amorphous aggre-
gates which had no influence on the auto-oxidation of added
Fe(II). No correlation was found between ThT fluorescence
and [Fe2+]T (Fig. 5a). Zn(II) reduced the formation of h-pleated conformers of Ah42 and, concomitantly, the role of
Ah42 in influencing the auto-oxidation of added Fe(II). There
was a weak correlation between ThT fluorescence and [Fe2+]T(Fig. 6a). Al(III) reduced neither the formation of h-pleatedconformers nor the influence of Ah42 on the auto-oxidation of
added Fe(II). The latter was accentuated by Al(III) and
resulted in an improved correlation between ThT fluorescence
and [Fe2+]T (Fig. 4b). The additional presence of Al(III) also
resulted in a significantly improved positive correlation
between ThT fluorescence and [Fe2+]T in Ah42 + Cu(II)
preparations (Fig. 5b). Though not reflected in statistically
significant changes in ThT fluorescence this influence of
Al(III) was accompanied by the presence of oligomers of
Ah42 at t = 1 h (Fig. 8) and the disappearance of these
oligomers and the appearance of distinctly fibrillar structures
at t = 24 and 48 h. The additional presence of Al(III) in Ah42 +
Zn(II) preparations resulted in similar effects except that the
improved correlation between ThT fluorescence and [Fe2+]T(Fig. 6b) was also accompanied by significant increases in ThT
fluorescence. These observations suggested that Al(III) slowly
reversed the inhibitive influences of Cu(II) and Zn(II) on both
the formation of h-pleated conformers of Ah42 and the
propensity for Ah42 to influence the auto-oxidation of added
Fe(II). That the improved correlations between ThT fluores-
cence and [Fe2+]T occurred concomitantly with the transforma-
tion between non-ThT-reactive oligomeric forms of Ah42 and
ThT-reactive h-pleated conformers supported the previous
notion that either the conformational transformation per se or
an intermediate involved in the transformation between

A. Khan et al. / Free Radical Biology & Medicine 40 (2006) 557–569566
conformers was active in influencing the auto-oxidation of
added Fe(II).
With the notable exception of preparations which included
Cu(II), Ah42 increased [Fe2+]T relative to preparations which
included neither peptide nor added metal. This may be due
to binding of Fe(II) by particular conformers or polymers of
Ah42 which, upon addition of 1,10-phenanthroline, released
the metal to form the characteristic 1,10-phenanthroline–
Fe(II) complex or it may indicate a role for Ah42 in the
redox cycling of iron. That the addition of Fe(III) to Ah42
resulted in the formation of Fe(II) supports the latter though
does not discount an additional role for peptide binding of
Fe(II).
To discriminate between these two mechanisms it is
necessary to identify the redox chemistry which in the absence
of Ah42 is occuring under the physiological-like conditions of
the assay. These redox reactions ((1)–(13)) and some of their
rate constants [18] are shown in Table 4. The oxidation of
Fe(II) by molecular oxygen (O2) is slow (1). However, it is
facilitated by the subsequent formation of the superoxide
radical anion (O2S�) which acts as an accelerant both directly
(3) and indirectly (2) via the formation of hydrogen peroxide
(H2O2) (4). Anything that influences this cascade of reactions
will impact significantly upon [Fe2+]C. These reactions involve
soluble forms of iron and their individual contributions to
redox cycling will be heavily influenced by factors affecting
iron solubility. Some reactions (e.g.,(6)) are not favoured due to
the extreme insolubility of iron (III) hydroxides (Ksp ca 10�37).
The OHS
scavenger NaN3 (7) accelerated auto-oxidation
(Table 2) which suggested that the reaction of Fe(II) with
H2O2 (4) was a significant rate-determining step in its auto-
oxidation. Cu(II) was more efficient than NaN3 in accelerating
the auto-oxidation of Fe(II) (Table 2) and this was almost
certainly due to its known influence upon the dismutation of
O2S� (8 and 9). There is controversy as to whether O2
S� acts as
a reductant or an oxidant ((10) and (11)) in the first instance
[19]. However, either way Cu(II) resulted in the lowest [Fe2+]C,
most probably through its acceleration of the formation of both
H2O2 and O2. The influence of Cu(II) pointed toward the
reaction of Fe(II) with H2O2 (4) as being of most importance in
Table 4
Redox reactions and, where appropriate, their rate constant
Reaction Rate constant k (M�1 s�1)
1. Fe(II) + O2 Y Fe(III) + O2S� 13
2. 2O2S� + 2H2O Y H2O2 + 2OH� 1.0 � 105
3. Fe(II) + O2S� + 2H+ Y Fe(III) + H2O2 1.0 � 107
4. Fe(II) + H2O2 Y Fe(III) + OHS+ OH� 3.1 � 104
5. Fe(II) + OHS Y Fe(III) + OH� 5.0 � 108
6. Fe(III) + O2S� Y Fe(II) + O2 1.5 � 108
7. N3� +SOH Y SN3 + OH�
8. Cu(II) + O2S� Y Cu(I) + O2 6.6 � 108
9. Cu(I) + O2S� + 2H+ Y Cu(II) + H2O2 2.0 � 109
10. Cu(II) + O2S� + 2H+ Y Cu(III) + H2O2
11. Cu(III) + O2S� Y Cu(II) + O2
12. H+/Fe2+/Al 3+ + O2S� Y AlO2
S2+ + Fe2+ + H+
13. Fe 3+ + AlO2S2+ Y Fe2+ + O2 + Al3+
All rate constants taken from [18].
the auto-oxidation of Fe(II). The highest [Fe2+]C were
measured in the presence of Al(III) and despite the fact that
these solutions were saturated with respect to aluminum
hydroxide and therefore during 48 h incubations would be
prone to considerable changes in their colloidal chemistry the
measured [Fe2+]C were independent of whether the prepara-
tions had been aged for 1, 24, or 48 h. It has been suggested
that previously noted effects of Al(III) on the auto-oxidation of
Fe(II) were due to Fe(II) being bound by particulate aluminum
such that it was protected from oxidation but still available for
binding by 1,10-phenanthroline [13]. The lack of influence of
the colloidal chemistry of Al(III) on the auto-oxidation of
Fe(II) combined with the observation that Al(III) per se did not
reduce Fe(III) to Fe(II) may suggest an alternative mechanism
involving the formation of a strong and fairly stable complex
between Al(III) and a reactant involved in the auto-oxidation of
Fe(II). Such a reaction and complex, the binding of Al3+ by
O2S� to form the semireduced radical cation AlO2
S2+, have
already been postulated [20] and would increase [Fe2+]C by
limiting the formation of H2O2 via reactions involving O2S�
(12) and accelerating the reduction of Fe(III) (13). Which of
these mechanisms was responsible for the remarkable influence
of Al(III) on the auto-oxidation of Fe(II) remains to be
determined. Zn(II) also increased [Fe2+]C and, like Al(III),
the explanation may be an interaction between O2S� and Zn2+.
Zn(II) as part of a Zn(II)/Cu(II) complex in superoxide
dismutase (SOD) is known to bind O2S� and binding is
thought to stabilise O2S� prior to it being ‘‘handed-on’’ to Cu(II)
for dismutation [21]. In this way Zn(II) would delay the
formation of H2O2 and thus, for this study, the critical step in
the oxidation of Fe(II). That Al(III) potentiated this activity of
Zn(II) or, looking at it in reverse, Zn(II) inhibited the
potentiation of [Fe2+]C due to Al(III), suggested a common
mechanism of effect and competition for either metal cation to
be bound by O2S�.
Control experiments indicated that under the conditions of
this study the reactions which involved the formation of
H2O2 via O2S� were critical in determining the net rate of
oxidation of Fe(II). The presence of Ah42 always resulted in
higher concentrations of Fe(II) than respective peptide-free
controls (i.e., [Fe2+]T > [Fe2+]C), which suggested a role in
the redox cycling of iron. To identify this role it is revealing
to consider how Fe(II) was generated in preparations in
which Fe(III) was substituted for Fe(II). In the absence of
peptide ca 0.1–0.2 AM Fe(II) was measured following the
addition of 5.0 AM Fe(III). The origin of this Fe(II) was
either as a contaminant of the Fe(III) stock solution or the
reduction of Fe(III) by a constituent of control preparations.
For the latter the rate of reduction of added Fe(III) would be
limited by its rapid precipitation as inert and extremely
insoluble Fe(OH)3(s). This might explain why, unlike Fe(II) +
Al(III), the additional presence of Al(III) in Fe(III) prepara-
tions did not result in higher [Fe2+]C. Contrary to control
preparations, in the presence of Ah42, Fe(III) was rapidly
reduced to the extent that in preparations incubated for 48 h over
50% of added Fe(III) was converted to Fe(II). The rate of
reduction of added Fe(III) increased with increased period of

A. Khan et al. / Free Radical Biology & Medicine 40 (2006) 557–569 567
incubation and though there were no statistically significant
differences in ThT fluorescence between these periods this
dependence was almost certainly associated with the aggrega-
tion state of Ah42. This might be understood in terms of
different affinities of age-dependent forms of Ah42 for binding
either Fe(II) or Fe(III). Binding of iron by organic ligands will
influence the redox potential of the Fe(II)/Fe(III) couple though
not necessarily in a predictable manner [18]. Binding of Fe(III)
will prevent its immediate precipitation as hydroxide and is
likely to increase its susceptibility to reduction whereas binding
of Fe(II) could accelerate its oxidation though it may also have
the opposite effect due to changes in the accessibility of O2 to
the iron centre.
A three-step mechanism is proposed to explain the role of
Ah42 in the auto-oxidation of Fe(II), the reduction of Fe(III),
and how these processes are influenced by Al(III), Zn(II), and
Cu(II) (Fig. 9).
Step 1
Fe(II) and Fe(III) are bound by monomeric Ab42 to increase
the likelihood of their oxidation and reduction, respectively.
The net result of these processes which we measured as [Fe2+]Twill depend upon (i) equilibria among complexed, free, and
precipitated iron, (ii) the redox potential of each of the soluble
forms of iron, and (iii) the dilution of each of these forms in the
preparation. There is no direct evidence in this study that
monomeric Ah42 bound either Fe(II) or Fe(III). There is
quantitative evidence in the literature for an Ah42–Fe(II)
complex though its calculated Kd of 36 AM [22] should mean
that it is unlikely to be stable in preparations herein. Evidence
is presented that preparations predicted to include predomi-
nantly monomeric Ah42 had no significant influence on [Fe2+]T
(Table 1). This would support the tendency for Ah42–Fe(II)
not to be formed in these preparations. There are no
quantitative data in the literature to support the formation of
an Ah42–Fe(III) complex. However, evidence is presented that
Ah42 incubated for only 1 h prevented the immediate
precipitation of Fe(III) and concomitantly accelerated its
reduction (Table 2). Whether these effects were mediated
Fig. 9. Schematic describing the role of the aggregation state of Ah42 in the redox c
Fe(II) and Fe(III), respectively.
through the formation of monomeric Ah42–Fe(III) remains to
be proven.
Step 2
Monomers of Ab42 form oligomers which may include
bound iron. Where oligomerisation brings iron atoms into
closer proximity thus countering any dilution effects redox
reaction rates are accelerated and the influence on [Fe2+]T is
accentuated relative to peptide in monomeric conformations.
There is not any direct evidence in the literature either that
oligomers of Ah42 include bound iron (or other metals) or that
oligomers act as catalysts for the oxidation/ reduction of iron.
Evidence is presented for significant increases in [Fe2+]T in
preparations of Ah42 in which oligomers were predicted to
have formed and in which further ageing of peptide did not
result in higher ThT fluorescence (Table 1). However,
increased [Fe2+]T were not recorded in preparations of Ah42
in which oligomers but not h-pleated fibrils were identified
(Table 2; Fig. 8). Because of the rapidity with which 3.52 AMAh42 aggregated under the conditions of this study it is not
possible to identify unequivocally if either monomeric or
oligomeric forms of peptide bound either form of iron.
Therefore it cannot be determined from the data if these forms
of bound iron were involved in the effects of Ah42 on the
Fe(II)/Fe(III) couple.
Step 3
Oligomers assemble into immature and mature b-pleatedfibrils. Iron may be retained by or titrated from newly formed
fibrils and free iron may be bound by newly formed fibrils. This
step is probably most representative of aggregation states in the
majority of peptide preparations investigated in this study. The
most significant effects upon [Fe2+]T were measured in
preparations incubated for 24 or 48 h and implicated either a
particular form of peptide or a specific change in peptide form
in the observed increases in [Fe2+]T. There is evidence in the
literature that fibrillar Ah42 binds Fe(II) much more tightly
(Kd = 0.2 AM) than monomeric Ah42 [22] though similar data
ycling of added Fe(II). Open and filled circles are qualitative representations of

A. Khan et al. / Free Radical Biology & Medicine 40 (2006) 557–569568
are not available for Fe(III). Evidence is presented that some
form of Ah42 bound Fe(III) and accelerated its reduction.
Fe(II), either added or originating from the reduction of
Fe(III), could have been bound by fibrillar peptide, thereby
stabilising it against oxidation to the extent that it accumu-
lated in preparations in this form. Fe(II) would remain
available for binding by 1,10-phenanthroline and would,
therefore, be measured as [Fe2+]T. The slower oxidation and
subsequent titration of peptide-bound Fe(II) would result in its
precipitation as Fe(OH)3(s) and concomitantly the deposition
of Ah42 as mature double-stranded fibrils. The delayed
precipitation of Fe(III) in many of the preparations was
observed by TEM and may have reflected the kinetically
preferred formation of Fe(OH)3(s) relative to its complexation
by fibrillar forms of Ah42.
This three-step mechanism predicts that Ah42 influenced the
redox cycling of iron by delaying its precipitation as highly
insoluble and consequently redox-inactive Fe(OH)3(s). Ah42 is
not redox active per se, a conclusion which is supported by a
consensus in the literature that Ah42 does not spontaneously
form free radicals in the absence of either added or contaminant
metals [23,24]. [Fe2+]T was measured as both Fe(II) in solution
and Fe(II) bound to particular forms of Ah42 with the latter
being important in the mechanism by which high concentra-
tions of Fe(II) were generated from the addition of Fe(III). The
reduction of Fe(III) was fueled by the presence in all
preparations of contaminating concentrations of Fe(II) which
through its auto-oxidation and the formation of O2S� facilitated
the reduction of peptide-bound Fe(III) and the subsequent
titration of Fe(II) into solution. Auto-oxidation of Fe(II)
catalysed the further reduction of Fe(III) with higher [Fe2+]Tin Ah42 preparations aged for 24 and 48 h reflecting a role for
aggregated Ah42 in either binding of a higher proportion of
added Fe(III) or binding of Fe(II) such that its oxidation was
delayed. A combination of these competitive equilibria,
including the propensity for 1,10-phenanthroline to titrate
peptide-bound Fe(II), was responsible for [Fe2+]T in each
preparation and any such combination was further influenced
by the presence of Al(III), Cu(II), and Zn(II). For example, the
action of Al(III) in increasing [Fe2+]T in all preparations is
explained by both its unusual redox activity, the formation of
the putative AlO2S2+ inhibiting the formation of H2O2 while at
the same time accelerating the reduction of Fe(III), and its
acceleration of the formation of the fibrillar form of Ah42. The
latter was clearly evident in preparations which included both
Cu(II) and Al(III) in which in the absence of peptide Al(III)
had little influence upon [Fe2+]C due to the rapid dismutation of
O2S� by Cu(II). In the presence of peptide Al(III) reversed the
effects of Cu(II) on [Fe2+]T by promoting the oligomerisation
and fibrillisation of Ah42 and, since a proportion of the added
Cu(II) was bound by Ah42 and was no longer available for the
dismutation of O2S�, by competing more effectively to bind
O2S� thereby inhibiting the formation of H2O2. Cu(II) in the
absence of additional Al(III) effectively inhibited the redox
cycling of iron by both accelerating the formation of H2O2 [7]
and, importantly, precipitating Ah42 in a nonfibrillar and
critically non-Fe(II)-binding form [10]. The latter was con-
firmed by TEM though a limited number of oligomeric forms
of peptide were observed in these preparations. Zn(II)
influenced peptide effects on [Fe2+]T primarily through its
binding of O2S�. The formation of a putative Zn(II)–O2
S�complex acted to increase [Fe2+]T through the inhibition of the
formation of H2O2 [25] though this effect was not influenced
significantly by the presence of Ah42. Thus there was little
evidence that the oligomeric and fibrillar forms of Ah42 which
were present in Zn(II) preparations contributed to the redox
cycling of iron. Similarly there was no evidence that the
putative Zn(II)–O2S� complex was effective in reducing
Fe(III) only in competing with Al(III) for binding by O2S�.
Despite the observations by both ThT fluorescence and TEM
that preparations including Zn(II) T Al(III) included both
oligomeric and fibrillar Ah42 the highest [Fe2+]T for these
preparations were recorded for peptide preparations aged for
only 1 h and this tended to exclude a major role for the
aggregation state of the peptide in the redox cycling of iron in
the presence of Zn(II). The possible explanation for this is that
the free radical which was most closely associated with the
peptide_s role in these processes, O2S�, was effectively
mopped up by Zn(II). Support for this notion is that Al(III)
increased both [Fe2+]C and [Fe2+]T in the presence of Zn(II) and
this was most probably due to the competitive binding of
Al(III) and Zn(II) by O2S�.
One further major observation appeared to support the
notion that a fibrillar form of Ah42 bound and stabilised Fe(II).
In every preparation in which fibrillar material was recorded by
ThT fluorescence or TEM the addition of Fe(II) was followed
immediately by a statistically significant reduction in ThT
fluorescence (Table 3). These effects upon ThT fluorescence
were not observed in those preparations in which Fe(III) was
substituted for Fe(II) even though Fe(II) was subsequently
generated from the reduction of the added Fe(III). We contend
that the changes in ThT fluorescence which followed the
addition of Fe(II) were not brought about by redox events per
se, for example, the oxidation of amino acid residues, such as
methionine-35 [26], altering the propensity of peptide to form
fibrils, but by the Fe(II)-induced precipitation of fibrillar
peptide such that fewer sites were subsequently available for
binding of ThT [10,15]. The bound Fe(II) might still be
available to 1,10-phenathroline if, as is predicted, binding of
Fe(II) stabilised it against auto-oxidation.
In conclusion, the major role of Ah42 in the redox cycling of
iron was probably in binding Fe(III) and thereby delaying its
precipitation as redox-inactive Fe(OH)3(s). Further aggregation
state-specific binding of both Fe(II) and Fe(III) determined
critical equilibria involved in the formation of H2O2 via O2S� in
favour of maintaining Fe(II) in solution. The additional presence
of Al(III), Cu(II), and Zn(II) influenced both the aggregation
state of Ah42, and therefore its binding of Fe(II) and Fe(III), and
the redox chemistry, most specifically through direct interactions
with O2S�. It is the latter free radical anion which is now heavily
implicated in ROS-mediated neurotoxicity [27–30] and it is
demonstrated herein that in the presence of Ah42 any predicted
O2S�-induced toxicity would be exacerbated by a pathophysio-
logically significant concentration of Al(III) while both Cu(II)

A. Khan et al. / Free Radical Biology & Medicine 40 (2006) 557–569 569
and Zn(II), providing that Al(III) was absent, would mitigate
against any oxidative damage.
Acknowledgments
A.K. is funded by NIH Grant R01AG02030-01A1. Dr. O.
Exley is thanked for her help with the figures.
References
[1] Tsai, J.; Grutzendler, J.; Duff, K.; Gan, W.-B. Fibrillar amyloid
deposition leads to local synaptic abnormalities and breakage of neuronal
branches. Nat. Neurol. 7:1181–1183; 2004.
[2] Good, P. F.; Werner, P.; Hsu, A.; Olanow, C.; Perl, D. P. Evidence for
neuronal oxidative damage in Alzheimer_s disease. Am. J. Pathol.
149:21–27; 1996.
[3] Dong, J.; Atwood, C. S.; Anderson, V. E.; Siedlak, S. L.; Smith, M. A.;
Perry, G.; Carey, P. R. Metal binding and oxidation of amyloid-h within
isolated senile plaque cores: Raman microscopic evidence. Biochemistry
42:2768–2773; 2003.
[4] Smith, M. A.; Harris, P. L. R.; Sayre, L. M.; Perry, G. Iron accumulation
in Alzheimer_s disease is a source of redox-generated free radicals. Proc.
Natl. Acad. Sci. USA 94:9866–9868; 1997.
[5] Beauchemin, D.; Kisilevsky, R. A method based on ICP-MS for the
analysis of Alzheimer_s amyloid plaques. Anal. Chem. 70:1026–1029;
1998.
[6] Sayre, L. M.; Perry, G.; Harris, P. L. R.; Liu, Y.; Schubert, K. A.; Smith,
M. A. In situ oxidative catalysis by neurofibrillary tangles and senile
plaques in Alzheimer_s disease: a central role for bound transition
metals. J. Neurochem. 74:270–279; 2000.
[7] Huang, X.; Atwood, C. S.; Hartshorn, M. A.; Multhaup, G.; Goldstein,
L. E.; Scarpa, R. C.; Cuajungco, M. P.; Gray, D. N.; Lim, J.; Moir, R. D.;
Tanzi, R. E.; Bush, A. I. The Ah peptide of Alzheimer_s disease directly
produces hydrogen peroxide through metal ion reduction. Biochemistry
38:7609–7616; 1999.
[8] Rottkamp, C. A.; Raina, A. K.; Zhu, X.; Gaier, E.; Bush, A. I.;
Atwood, C. S.; Chevion, M.; Perry, G.; Smith, M. A. Redox-active
iron mediates amyloid-h toxicity. Free Radic. Biol. Med. 30:447–450;
2001.
[9] Monji, A.; Utsumi, H.; Ueda, T.; Imoto, T.; Yoshida, I.; Hashioka, S.;
Tashiro, K.-I.; Tashiro, N. The relationship between the aggregational
state of the amyloid-h peptides and free radical generation by the
peptides. J. Neurochem. 77:1425–1432; 2001.
[10] House, E.; Collingwood, J.; Khan, A.; Korchazhkina, O.; Berthon, G.;
Exley, C. Aluminum, iron, zinc and copper influence the in vitro
formation of amyloid fibrils of Ah42 in a manner which may have
consequences for metal chelation therapy in Alzheimer_s disease. J. Alzh.Dis. 6:291–301; 2004.
[11] Bondy, S. C.; Guo-Ross, S. X.; Pien, J. Mechanisms underlying the
aluminum-induced potentiation of the pro-oxidant properties of transi-
tion metals. Neurotoxicology 19:65–72; 1998.
[12] Bondy, S. C.; Guo-Ross, S. X.; Truong, A. T. Promotion of transition
metal-induced reactive oxygen species formation by h-amyloid. Brain
Res. 799:91–96; 1998.
[13] Yang, E. Y.; Guo-Ross, S. X.; Bondy, S. C. The stabilisation of ferrous
iron by a toxic h-amyloid fragment and by an aluminum salt. Brain Res.
839:221–226; 1999.
[14] Yegorov, D. Y.; Kozlov, A. V.; Azizova, O. A.; Vladimorov, Y. A.
Simultaneous determination of Fe(III) and Fe(II) in water solutions and
tissue homogenates using desferal and 1,10-phenanthroline. Free Radic.
Biol. Med. 15:565–574; 1993.
[15] Levine, H. Thioflavine-T interaction with synthetic Alzheimer_s-disease
beta amyloid peptides—Detection of amyloid aggregation in solution.
Protein Sci. 2:404–410; 1993.
[16] Exley, C.; Korchazhkina, O. V. Promotion of formation of amyloid fibrils
by aluminum adenosine triphosphate (AlATP). J. Inorg. Biochem.
84:215–224; 2001.
[17] Sengupta, P.; Garai, K.; Sahoo, B.; Shi, Y.; Callaway, D.;
Maiti, S. The amyloid beta peptide (Abeta(1–40)) is thermody-
namically soluble at physiological concentrations. Biochemistry
42:10506–10513; 2003.
[18] Rose, A. L.; Waite, T. D. Kinetic model for Fe(II) oxidation in seawater
in the absence and presence of natural organic matter. Environ. Sci.
Technol. 36:433–444; 2002.
[19] Luo, Q.-H.; Zhang, J.-J.; Hu, X.-L.; Jiang, X.-Q.; Shen, M.-C.; Li, F.-M.
A study on the reaction of copper complex of dioxotetraamine with
superoxide ion by spectrophotometry and pulse radiolysis. Inorg. Chim.
Acta 357:66–74; 2004.
[20] Exley, C. The pro-oxidant activity of aluminium. Free Radic. Biol. Med.
36:380–387; 2004.
[21] Ohtsu, H.; Fukuzumi, S. Coordination of semiquinone and superoxide
radical anions to zinc ion in SOD model complexes that act as the key
step in disproportionation of the radical anions. Chem. - Eur. J.
7:4947–4953; 2001.
[22] Garzon-Rodriguez, W.; Yatsimirsky, A. K.; Glabe, C. G. Binding
of Zn(II), Cu(II) and Fe(II) ions to Alzheimer_s Ah peptide
studied by fluorescence. Bioorg. Med. Chem. Lett. 9:2243–2248;
1999.
[23] Dikalov, S. I.; Vitek, M. P.; Maples, K. R.; Mason, R. P. Amyloid hpeptides do not form peptide-derived free radicals spontaneously, but can
enhance metal-catalysed oxidation of hydroxylamines to nitroxides.
J. Biol. Chem. 274:9392–9399; 1999.
[24] Turnbull, S.; Tabner, B. J.; El-Agnaf, O. M. A.; Twyman, L. J.; Allsop,
D. New evidence that the Alzheimer_s h-amyloid peptide does not
spontaneously form free radicals: an ESR study using a series of spin
traps. Free Radic. Biol. Med. 30:1154–1162; 2001.
[25] Cuajungco, M. P.; Goldstein, L. E.; Nunomura, A.; Smith, M. A.; Lim, J.
T.; Atwood, C. S.; Huang, X.; Farrag, Y. W.; Perry, G.; Bush, A. I.
Evidence that the h-amyloid plaques of Alzheimer_s disease represent
the redox-silencing and entombment of Ah by zinc. J. Biol. Chem.
275:19439–19442; 2000.
[26] Butterfield, D. A.; Boyd-Kimball, D. The critical role of methionine 35
in Alzheimer_s amyloid h-peptide (1–42)-induced oxidative stress and
neurotoxicity. Biochim. Biophys. Acta 1703:149–156; 2005.
[27] Longo, V. D.; Viola, K. L.; Klein, W. L.; Finch, C. E. Reversible
inactivation of superoxide-sensitive aconitase in Ah1-42-treated neuro-
nal cell lines. J. Neurochem. 75:1977–1985; 2000.
[28] Jana, A.; Pahan, K. Fibrillar amyloid-h peptides kill human primary
neurons via NADPH oxidase-mediated activation of neutral sphingo-
myelinase. J. Biol. Chem. 279:51451–51459; 2004.
[29] Markert, C.; Morre, D. M.; Morre, D. J. Human amyloid peptides Ah1-40 and Ah1-42 exhibit NADH oxidase activity with copper-induced
oscillations and a period length of 24 min. BioFactors 20:221–235;
2004.
[30] Park, L.; Anrather, J.; Zhou, P.; Frys, K.; Pitstick, R.; Younkin, S.;
Carlson, G. A.; Iadecola, C. NADPH oxidase-derived reactive oxygen
species mediate the cerebrovascular dysfunction induced by the amyloid
h peptide. J. Neurosci. 16:1769–1777; 2005.