università degli studi del molise - road.unimol.itroad.unimol.it/bitstream/2192/31/1/tesi dottorato...
TRANSCRIPT

Università degli Studi del Molise
Facoltà di Scienze Matematiche Fisiche e Naturali
dottorato di ricerca in
Biochimica e Chimica Applicate
SVILUPPO DI METODI ANALITICI PER DETERMINAZIONI AFFIDABILI
IN CAMPO AMBIENTALE ED ALIMENTARE Coordinatore Tutor Ch.mo prof. S. Passarella Ch.mo prof. M.V. Russo
Dottorando Dr. Ivan Notardonato
XXII CICLO

I
Sommario Introduzione 2 Tecniche di separazione: principi generali 4 Estrazione in fase solida: SPE 6
Legame ionico 6Legame idrogeno 9Interazione dipolo-dipolo 9Interazione dipolo-dipolo indotto o forze di Wan der Waal 10Legame dipolo istantaneo-dipolo indotto o forze di dispersione di London 11
Vantaggi della Solid Phase Extraction 11
Fasi solide: caratteristica e scelta dell’adsorbente 14
Adsorbenti polari non derivatizzati 14Adsorbenti polari, chimicamente legati, nella cromatografia a fase normale 15Adsorbenti non polari, chimicamente legati, impiegati nella cromatografia a fase inversa 16Adsorbenti carichi, chimicamente legati, impiegati nella cromatografia a scambio ionico 17Adsorbenti polimerici 17Il carbone attivo 18Il Carobograph 18
Scelta della fase adsorbente 20
Gas cromatografia 22
Principi della gas cromatografia 22La colonna cromatografica 23Velocità di flusso del gas di trasporto 26L’effetto della temperatura 27
Rivelatori 29
Il rivelatore a ionizzazione di fiamma (FID) 29Il rivelatore a conducibilità termica (TCD) 30Il rivelatore azoto-fosforo (NPD) 30Il rivelatore a cattura di elettroni (ECD) 30
Le tecniche ifenate 31

II
La spettrometria di massa 33Sistema pompa da vuoto 33Introduzione del campione 33Camera di ionizzazione 33
Sorgenti 35
Impatto elettronico (EI) 35Ionizzazione chimica (CI) 36Ionizzazione a elettrospray (ESI) 37
Analizzatore 37
Analizzatore magnetico 38Analizzatore a doppia focalizzazione 38Analizzatore a quadrupolo 39Analizzatore a trappola ionica 40Analizzatore a tempo di volo (TOF) 41
Rivelatore 42
Lo spettro di massa 42
Bibliografia 44 Abstract 47 Ftalati in matrici idroalcoliche: preconcentrazione su “Carbograph 1” ed analisi mediante GC-FID o GC-MS 48
Introduzione 48
Obiettivo 50
Materiali e metodi 51Materiali 51Metodi 54
Isoterme di adsorbimento 55
Preparazione delle soluzioni 55Estrazione delle soluzioni 55Preparazione dello standard interno 56Costruzione delle isoterme di adsorbimento 57
Volumi di breakthrough 62
Preparazione delle soluzioni e della fase adsorbente 62Estrazione dei soluti e concentrazione delle soluzioni 62Costruzione delle curve di breakthrough 63
Determinazione del solvente di estrazione 67

III
Applicazione del metodo a campioni reali 69 LOD e LOQ ottenuti in GC-FID 71
Errore intraday ed interday in GC-FID 72
Analisi allo spettrometro di massa degli ftalati 73
LOD e LOQ ottenuti in GC-MS 78
Errore intraday ed interday in GC-MS 79
Conclusioni 80
Bibliografia 81 Determinazione dell’acrilammide in prodotti da forno mediante GC-ECD e GC-MS 83
Introduzione 83
Obiettivo 84
Caratteristiche della molecola 85
Metodi analitici in uso per la determinazione dell’acrilammide nei prodotti alimentari 86
Materiali e metodi 87
Materiali 87Metodi 88
Reazione di derivatizzazione 89
LOD e LOQ ottenuti in GC-ECD 91
Errore intraday ed interday in GC-ECD 91
Retta di taratura ottenuta in GC-ECD 92
Analisi del campione alimentare mediante GC-ECD 93
Analsi allo spettrometro di massa dell’acrilammide derivatizzata 94
LOD e LOQ ottenuti in GC-MS 105
Errore intraday ed interday in GC-MS 105

IV
Schema riassuntivo frammentazione 106
Determinazione quantitativa dell’acrilammide in spettrometria di massa 107
Conclusioni 109
Bibliografia 110 Allegato 1: vino bianco 111 Allegato 2: vino rosso 113 Allegato 3: dimetyl phtalate 115 Allegato 4: dietyl phtalate 116 Allegato 5: dibutyl phtalate 117 Allegato 6: isobutyl-cicloesyl phtalate 118 Allegato 7: benzil-butyl phtalate 119 Allegato 8: bis(2-etil-exil) phtalate 120 Allegato 9: MS-MS del di-derivato dell’acrilammide 121 Allegato 10: MS-MS del tri-derivato dell’acrilammide 123

1
p a r t e b i b l i o g r a f i c a

2
INTRODUZIONE
La presenza di sostanze tossiche nel nostro ecosistema è stato sempre un problema
di grande rilevanza e influenza sociale sia per la natura sia per l’uomo.
Nel corso degli anni chimici, biologi e scienziati di varia natura, hanno
evidenziato diverse sostanze tossiche presenti nell’ambiente, derivanti sia da fonti
naturali sia da fonti industriali. Soprattutto da queste ultime, si continuano a
produrre e rilasciare, ogni anno, migliaia di sostanze, la cui tossicità, impatto
sanitario ed ambientale resta in alcuni casi ancora da definire [1].
Alcune di queste sostanze si presentano molto pericolose per la natura e per la
salute pubblica; infatti, sono caratterizzate da elevata o probabile tossicità,
persistenza, bioaccumulabilità e bioconcentrazione.
La tossicità è spesso legata alla struttura molecolare di questi composti. Infatti, il
grado di tossicità dipende dal modo in cui queste molecole possono interferire e/o
interagire con i meccanismi del corpo umano o della vita.
La persistenza, come la tossicità, di queste sostanze è legata alla loro struttura
molecolare [2, 3, 4].
La bioaccumulabilità è legata alla maggiore affinità che queste sostanze
presentano per i sistemi apolari o poco polari di quanto non lo siano per l’acqua.
In altre parole, diffondono nei tessuti grassi dove sono adsorbiti e concentrati [5,
6, 7, 8, 9].
La bioconcentrazione, invece, è legata all’aumento, in modo consistente, della
concentrazione media di molte sostanze procedendo lungo la catena alimentare.
Analizzando la catena alimentare, o meglio il complesso delle interazioni delle

3
catene alimentari all’interno di un ecosistema, notiamo che un essere vivente
tende a nutrirsi con quantità di cibo che attinge da livelli inferiori. Purtroppo, in
questa serie di passaggi, si tende a trattenere, piuttosto che ad eliminare, la
maggior parte delle sostanze tossiche assunte con il cibo [10, 11, 12]. Una
sostanza chimica la cui concentrazione aumenta lungo la catena alimentare, è
definita sostanza biomagnificata e il fenomeno è chiamato biomagnificazione o
amplificazione biologica [1].
Per la maggior parte, le sostanze tossiche o potenzialmente pericolose presenti
nell’ambiente o negli alimenti sono state studiate ed analizzate, per cercare di
caratterizzarne il comportamento e/o ridurne la concentrazione a livello
ambientale.
È da tener presente che, in una determinazione quantitativa, il problema di queste
sostanze è la loro bassissima concentrazione, infatti si parla molto spesso di ppm,
ppb o addirittura ppt; concentrazioni molto piccole anche per strumenti attuali
molto costosi. Nasce, quindi, l’esigenza di preconcentrare queste sostanze in
modo da renderle determinabili anche con strumenti poco costosi o presenti nella
maggior parte dei laboratori.
In questi anni mi sono occupato di mettere a punto e validare metodi analitici per
diverse sostanze di interesse ambientale ed alimentare.
Nella presente tesi viene affrontato il problema degli ftalati nel vino e
l’acrilammide in prodotti da forno.

4
TECNICHE DI SEPARAZIONE: PRINCIPI GENERALI
L’esigenza di separare e/o determinare singoli componenti di miscele complesse,
ha portato allo sviluppo di numerose tecniche di separazione. Ne sono esempio la
distillazione frazionata, la cristallizzazione, la filtrazione, la centrifugazione,
l’elettroforesi capillare, l’estrazione con solvente, e così via.
Tali tecniche sfruttano ed amplificano differenze, anche minime, fra le diverse
specie chimiche in modo da consentire una separazione efficiente.
Tra le tecniche sopra menzionate, quella più utilizzata nella preparazione del
campione analitico, specialmente da soluzioni acquose, è l’estrazione liquido-
liquido. Tale tecnica si esegue portando a contatto con la soluzione in cui si trova
un certo soluto, un solvente che sia praticamente immiscibile con il primo, in
modo che il soluto possa distribuirsi tra i due solventi in ragione della sua e della
loro natura o delle condizioni sperimentali.
La legge con cui avviene la distribuzione di un soluto tra due solventi è la legge di
Nerst , che può così enunciarsi: “un soluto si distribuisce fra due solventi
immiscibili tra loro, quando non ci sono interazioni soluto-solvente, in modo che
il rapporto tra le sue concentrazioni è costante”. Il parametro che meglio esprime
l’enunciato di Nerst è il coefficiente di distribuzione o di ripartizione (KD). Al fine
di chiarire il significato del coefficiente di ripartizione si consideri il sistema
costituito dal soluto A e da due solventi immiscibili tra loro: solvente polare (S1);
solvente apolare (S2). Se la sostanza A si trova inizialmente nella soluzione S1
quando quest’ultima viene dibattuta con il solvente S2, si ha il seguente equilibrio:
AS1 ↔ AS2

5
La frazione del composto A che solubilizza nella fase S2 è tale che è sempre
costante il rapporto
1
2
S
SD A
AK =
in cui AS1 ed AS2 rappresentano le concentrazioni del soluto nei due solventi e KD è
il coefficiente di distribuzione della specie in considerazione. Esso è costante a
temperatura costante. La legge della distribuzione così espressa non è però
termodinamicamente rigorosa, poiché nell’espressione sopra riportata si è fatto
uso delle concentrazioni invece delle attività; ma questa approssimazione è del
tutto lecita ai fini pratici.
Nonostante l’estrazione liquido-liquido sia una delle tecniche separative più
utilizzate, essa comporta notevoli problemi; ad esempio: impiego di grossi
quantitativi di solventi spesso tossici; potenziale contaminazione per l’uso di
molta vetreria; elevato tempo di analisi necessario per l’estrazione; concentrazione
di volumi elevati di solvente con relativa esaltazione delle impurità in esso
contenute; elevato numero di travasi con possibilità di errori da parte
dell’operatore; probabili perdite per evaporazione; costi di trasporto, considerando
che tale operazione è eseguibile solo in laboratorio.
Per questi, ed altri motivi, nasce l’esigenza di studiare nuove tecniche per il
campionamento, in modo da minimizzare i problemi d’analisi sopracitati ed
ottenere risultati altrettanto affidabili.
Una tecnica attualmente in via di grande sviluppo, utilizzata in questo lavoro, è la
Solid Phase Extraction (SPE).

6
ESTRAZIONE IN FASE SOLIDA: SPE
Un processo molto comune a cui di solito un campione è sottoposto quando si
devono analizzare componenti presenti in tracce è il processo di arricchimento o
preconcentrazione.
Molte delle sostanze inquinanti oggetto di analisi chimiche, infatti, sono in genere
disperse in matrici complesse e sono presenti a livello di concentrazioni molto
basse, si tratta quasi sempre di mg/L (ppm) e µg/L (ppb) o addirittura valori
inferiori.
Queste basse concentrazioni non permettono di analizzare direttamente il
campione, poiché sono molto vicine ai limiti di rilevabilità dello strumento. Per
cui nasce l’esigenza di trattare il sistema per disporre di soluzioni a
concentrazione adeguate alla sensibilità strumentale.
La manipolazione del campione, quindi, diventa una necessità inderogabile che
però ha il difetto di modificare il sistema, per questo si rischia di avere una non
perfetta rispondenza dei valori misurati ai valori reali.
Per mantenere la fase di preconcentrazione abbastanza attendibile, in primo luogo
vanno ridotte al minimo le perdite degli analiti nei passaggi del processo. La
condizione ideale si otterrebbe qualora la quantità di sostanza recuperata fosse
uguale alla quantità di partenza. In tutti gli altri casi si commettono errori, anche
rilevanti, nella determinazione quantitativa. In secondo luogo, bisogna assicurarsi
un’adeguata efficienza di concentrazione. Un sistema di arricchimento valido
deve portare a incrementi della concentrazione adeguati alle metodologie
strumentali impiegate per le successive analisi. Infatti, per composti presenti in

7
ppb o quantità inferiori, bisogna avere incrementi di concentrazione di un fattore
maggiore di 1000. Infine, particolare cura, va posta nel non introdurre impurità ed
artefatti nelle soluzioni finali.
Le tecniche preparative di estrazione in fase solida si stanno sempre più
diffondendo in campo ambientale ed alimentare, in risposta alla crescente
domanda di metodi pratici, riproducibili, veloci e altamente selettivi. La
tecnologia di estrazione in fase solida è una tecnica ampiamente utilizzata per la
preparazione di campioni di diversa natura, come i pesticidi clorurati, PCBs,
diossine ecc. ottenuti da campioni ambientali. La tradizionale SPE utilizza
colonnine in polipropilene contenenti un materiale adsorbente (fase solida)
impaccato, a cui si lega in modo specifico il soluto che si vuole purificare e/o
determinare. Con uno o più lavaggi si eliminano dalla fase solida adsorbente i
composti interferenti e con l'aggiunta di un solvente adatto si riesce a recuperare il
soluto o i soluti di interesse.
Un momento importante dello studio è rappresentato dalla scelta dell’ adsorbente,
poiché permette di minimizzare le interferenze senza influenzare la sensibilità del
metodo. Sono apprezzabili anche il condizionamento della cartuccia, il lavaggio
della cartuccia e l’eluizione dell’analita. Il condizionamento prepara la fase
stazionaria in modo da garantire una ritenzione ottimale dell’analita. Il solvente di
lavaggio deve essere scelto in modo da eliminare le interferenze permettendo
all’analita di rimanere adsorbito sulla fase stazionaria. Una buona soluzione è
spesso rappresentata dall’utilizzo di un solvente in cui l’analita è insolubile o poco
solubile. Nella riestrazione dell’analita dalla fase stazionaria, il solvente stabilisce
un legame più forte con il soluto rispetto al precedente e quindi lo eluisce. Il

8
solvente utilizzato nella riestrazione dei soluti deve avere una forte affinità per
gli analiti, in modo da stabilire con essi un legame più forte rispetto al precedente.
Qui di seguito viene schematizzata una colonna usata per la S.P.E.
Nel meccanismo di una SPE la fase solida viene in contatto diretto con la fase
liquida e con i soluti in essa contenuti. Affinché si verifichi la separazione
dell’analita dalla fase liquida, occorre che l’analita si leghi alla fase solida, o
meglio al sito attivo della fase solida, occorre quindi che la forza di legame fra
analita (A) e sito dell’adsorbente (F) sia più elevato di quello esistente fra analita
(A) e fase liquida (S).
Si
O
Si
O
Si
O
Si
O
Si
F
F A
A
F
F
AF
A
A
A
A
A
A
A
A
A
S
S
S
S
S
S S
S
S
S
S
S
S
S
setti porosifase adsorbente
eluizione

9
Le forze che si possono istaurare sono legami ionici, legami idrogeno, interazione
dipolo-dipolo, interazione dipolo-dipolo indotto (o forze di Van Der Waals) e
forze di dipolo-dipolo istantaneo ( o di dispersione di London). Di seguito, darò
una breve descrizione dei legami che caratterizzano la SPE:
Legame ionico
Alcuni atomi raggiungono l'ottetto stabile acquistando o cedendo degli elettroni.
In questo modo l'atomo acquista una carica elettrica positiva o negativa, a seconda
che ceda o acquisti un elettrone. Un atomo che cede uno o più elettroni verrà ad
avere un numero di protoni maggiore di quello degli elettroni; non risulterà quindi
neutro ma elettricamente positivo. Si dice che è diventato uno ione positivo. Un
atomo che acquista uno o più elettroni verrà ad avere un numero di elettroni
maggiore di quello dei protoni; non è più neutro, ma elettricamente negativo. Si
dice che è diventato uno ione negativo. Il legame che si stabilisce tra due atomi di
questo tipo si dice legame ionico ed è dato dalle forze elettriche opposte che si
attraggono. Il legame ionico consiste proprio nell'attrazione elettrostatica tra ioni
di segno opposto.
Legame idrogeno
E’ un tipo di legame che si instaura quando un atomo di idrogeno è legato
chimicamente ad un atomo molto elettronegativo. In queste condizioni, si
sviluppa una frazione di carica positiva sull'idrogeno che si polarizza (δ+) ed una
frazione negativa sull'altro atomo (δ-). Inoltre è necessario che sul secondo atomo
sia presente almeno una coppia di elettroni di non legame. Quando una seconda
molecola si avvicina, si orienta in modo da esporre la propria coppia di elettroni
liberi verso l'idrogeno ed in questo modo si genera una grande forza di attrazione

10
elettrostatica. Tipici atomi molto elettronegativi sono Ossigeno, Azoto, ecc. Il
legame idrogeno è la più forte delle interazioni tra molecole in termini di energia.
L 'intensità della forza di legame dipende dall'atomo legato all'idrogeno e dalla
coppia di elettroni libera. Il legame idrogeno può risultare una forza di legame
importante e discriminante rispetto ad altre forze di legame che si verificano sulle
superfici dei solidi adsorbenti.
Interazione dipolo-dipolo
Quando due atomi generici differenti (X,Y) sono legati chimicamente, a causa
della loro differente capacità di attrarre gli elettroni, si instaura tra loro una
differenza di posizione tra il baricentro delle cariche positive (+) e negative (-)
generando un dipolo elettrico e la molecola si dice polare. Due dipoli elettrici
vicini tendono ad orientarsi in modo che il baricentro del primo dipolo (+) sia
vicino a quello (-) del secondo dipolo e ad attrarsi elettrostaticamente. L'effetto è
simile al precedente, difatti il legame idrogeno non è che un tipo particolare di
interazione dipolo-dipolo, ma in questo caso l'attrazione non è potenziata dalla
presenza di elettroni liberi. Dopo il legame idrogeno, le interazioni dipolo sono le
forze di attrazione intermolecolare maggiori. La loro intensità dipende
dall'intensità del dipolo elettrico.
Interazione dipolo-dipolo indotto o forze di Van Der Waals.
Quando una molecola polare si avvicina ad una non polare induce in quest'ultima
un dipolo elettrico di minore intensità che perdura fintanto che le due molecole
restano vicine. Si genera così attrazione come per il dipolo-dipolo. L'intensità è
proporzionale al dipolo che induce polarizzazione e dalla polarizzabilità della

11
seconda molecola, grandezza che a sua volta cresce con la superficie della
molecola.
Interazione dipolo istantaneo-dipolo indotto o forze di dispersione di London.
Gli elettroni che si muovono continuamente attorno ad un nucleo creano
piccolissimi dipoli istantanei, che inducono a loro volta dipoli istantanei su
molecole vicine. Queste forze sono debolissime, ma la loro somma genera una
risultante che tiene assieme molecole non polari. Sono proporzionali alla
superficie delle molecole interagenti.
VANTAGGI DELLA SOLID PHASE EXTRACTION
La tradizionale tecnica preparativa di estrazione con solvente costituisce, in molte
metodiche, una tappa di congestione e rallentamento; in molti casi, questa tecnica
non porta ad un campione da analizzare adeguato, per recupero e compatibilità
analitica, alle sempre più sofisticate strumentazioni analitiche. Inoltre, l’uso di
grosse quantità di solventi, tossici e volatili, costituisce un serio pericolo per gli
operatori e per l’ambiente.
L’estrazione in fase solida, al contrario, condotta su colonnine preimpaccate, è in
grado di unire al concetto di selettività quello di una certa e sostanziosa
diminuzione del solvente estraente. Altre caratteristiche favorevoli, nella scelta
della tecnica SPE, sono i tempi rapidi d’analisi; la buona selettività, dovuta alla
varietà dei solidi adsorbenti; l’assenza o quasi di evaporazioni del solvente in
quanto gli analiti sono riestratti con piccole quantità di solventi; la scarsa necessità
di trasporto in laboratorio, poiché il campione può essere preparato già sul posto

12
di prelievo; l’elevata automazione in grado di trattare lo stesso campione con
diverse colonnine e con diverse combinazioni di solventi.
Le principali funzioni dell’estrazione in fase solida sono riconducibili
all’arricchimento di tracce, al clean up ed alla conservazione del campione. Nel
primo caso, per una specie con forte affinità (attrazioni relative dei componenti in
soluzione, sia per il solvente sia per la superficie dell’adsorbente) l’adsorbimento
avverrà in una piccola superficie del letto adsorbente. Nel secondo caso, le
interazioni più o meno selettive che si verificano tra i diversi costituenti di un
campione e l’adsorbente, permette, in alcuni casi, la separazione di un certo
gruppo di sostanze da altre durante lo step dell’arricchimento. Nel terzo caso,
grazie al carattere relativamente inerte dei diversi materiali adsorbenti, un analita
adsorbito può rimanere inalterato per un periodo di tempo molto lungo se è tenuto
nelle condizioni opportune. Questo aspetto è fondamentale perché riduce
notevolmente la quantità di campione da trasportare in laboratorio e dà la
possibilità di effettuare campionamenti in situ, specialmente in luoghi dove non è
facile il trasporto di notevoli quantità di campioni.
Per queste ed altre ragioni, in questi ultimi anni, l’estrazione in fase solida si
identifica nella tecnica di separazione più ampiamente usata, che ha segnato una
svolta nei metodi di preconcentrazione e arricchimento.
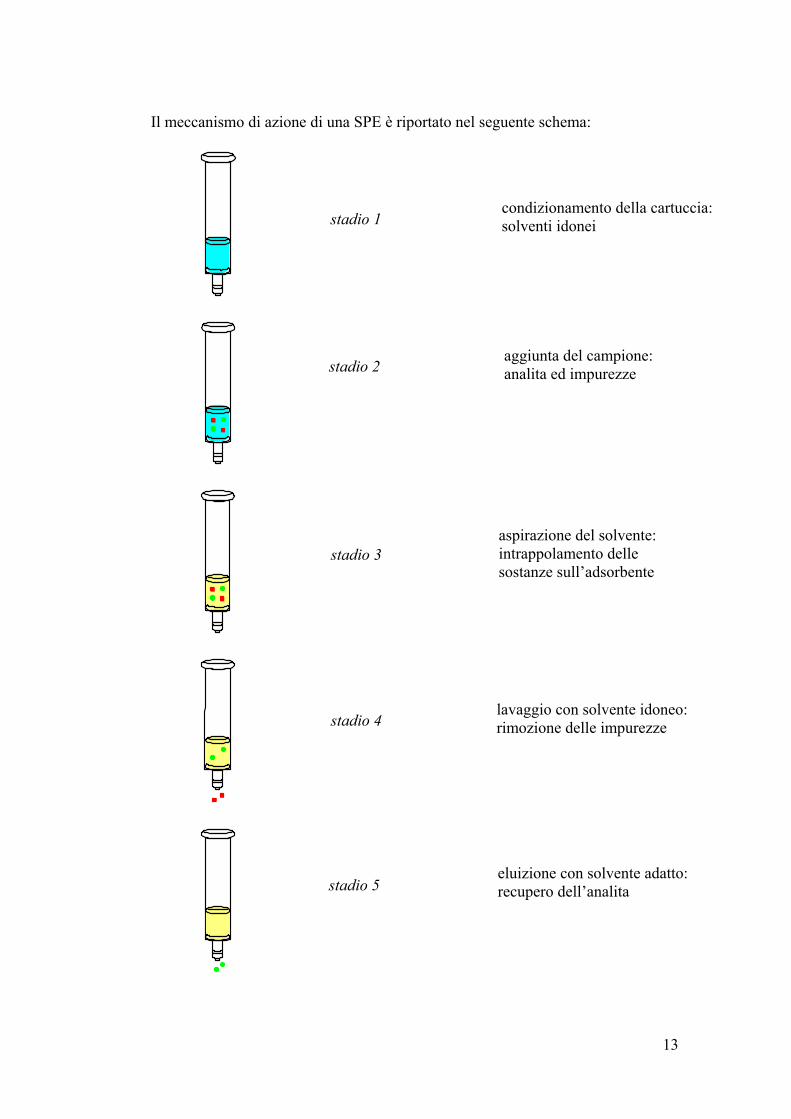
13
Il meccanismo di azione di una SPE è riportato nel seguente schema:
stadio 1
stadio 2
stadio 3
lavaggio con solvente idoneo: rimozione delle impurezze
stadio 5
condizionamento della cartuccia: solventi idonei
aggiunta del campione: analita ed impurezze
aspirazione del solvente: intrappolamento delle sostanze sull’adsorbente
stadio 4
eluizione con solvente adatto: recupero dell’analita

14
FASI SOLIDE: CARATTERISTICHE E SCELTA DELL’ADSORBENTE
Adsorbenti polari non derivatizzati
Con tale termine s’intende una sostanza solida, senza modifiche di sorta, che sia
in grado di interagire con le molecole di soluto presenti in soluzione, che viene a
contatto con la fase solida stessa. Le fasi solide non modificate più utilizzate sono
la Silice, l’Allumina e il Florosil. Esse sono particolarmente adatte a separare
composti polari (aldeidi, alcoli, alogenuri organici) da solventi non polari.
La silice non modificata può essere strutturalmente schematizzata:
I gruppi silanolici possono dar luogo a legami idrogeno con molecole opportune
come, ad esempio, la benzilammina:
Se la benzilammina è solubilizzata nella miscela binaria esano/dietiletere (3:1),
l’interazione benzilammina-gel di silice risulta più forte dell’interazione solvente-
SiHO
SiHO
HSi
OH OH OH
SiHO
SiHO
HSi
OH OH O
CH2NH
H
H

15
gel di silice. Tale fenomeno spiega come l’ammina si trattiene sulla fase
stazionaria ed il solvente fluisce attraverso il gel di silice. Per questa ragione,
l’eluizione dei soluti trattenuti è effettuata con solventi che hanno la capacità di
instaurare interazioni più forti con la fase fissa rispetto ai soluti stessi. In generale,
si può affermare che i composti basici sono trattenuti con più efficacia dalla silice,
la quale risulta leggermente acida, mentre i composti acidi sono fortemente
adsorbiti da fasi solide basiche, come l’allumina. Nel caso dell’esempio sopra
riportato, l’eluizione del soluto sarà effettuata con metanolo poiché esso formerà
legami idrogeno più forti con il gruppo silanolico rispetto a quelli stabiliti dagli
stessi con la benzilammina:
Anche l’acqua è in grado di interagire con i gruppi silanolici per formare legami
idrogeni perciò l’attività dell’adsorbente è funzione del grado della sua umidità.
Adsorbenti polari, chimicamente legati, nella cromatografia a fase normale
Derivatizzando i gruppi silanolici con mono-, di- e trialogenosililderivati, o con
mono-, di-, e trialcossisililderivati si ottengono i silossani:
SiH
O
SiH
O
SiH
OH
O
OH
Si
CH3
CH3
(CH2)xR
SiHO
SiHO
HSi
OH OH OCH2H2N
H
H O CH3

16
Nel caso poi, che la fase solida venga trattata con un derivato trifunzionale
(triclorosililderivato) il risultato è il seguente:
Come si può notare, in entrambi i derivati è possibile notare la presenza di gruppi
silanolici (-SiOH) liberi. Questi ultimi possono essere derivatizzati con
trimetilclorosilano. La fase solida adsorbente ottenuta, non presenta nessun
gruppo silanolico ed il responsabile dell’adsorbimento è a carico del gruppo R. La
fase solida di silice modificata offre una diversa forza di ritenzione nei confronti
degli analiti e consente una maggiore duttilità. I carboidrati, ad esempio, si legano
troppo fortemente alla silice tal quale e perciò non ne sarebbe possibile la
successiva eluizione.
La cromatografia a fase normale con adsorbenti polari legati si ha quando il
gruppo R è costituito da un gruppo ciano (-CN), da un gruppo ammino (-NH2) o
da un gruppo diolo (OH OH).
Adsorbenti non polari, chimicamente legati, impiegati nella cromatografia a fase
inversa
Sono adsorbenti costituiti da silice modificata con gruppi R1 (radicali alchilici),
che risultano meno polari del solvente in cui sono solubilizzati gli analiti. La
dinamica dell’adsorbimento è la seguente: il soluto con parte meno polare,
solubilizzato in un solvente polare, si lega ad un gruppo R1 non polare, sulla fase
solida. Le interazioni fra gruppi non polari sono le interazioni di Van Der Waals. I
SiHO
SiHO
HSi
OH O O
Si
Rx(H2C) OH

17
gruppi R1 non polari legati alla silice possono essere: l’ottadecile, l’ottile, il
cicloesile, il fenile, il butile ed altri. L’analita viene eluito dalla colonna con
solventi apolari.
Adsorbenti carichi, chimicamente legati, impiegati nella cromatografia a scambio
ionico
Queste fasi di silice modificata, hanno un radicale R2 costituito da un gruppo
funzionale carico, i più utilizzati sono il gruppo –SO3- ed il gruppo -N+(CH3)3.
Tali fasi fisse, in genere, sono impiegate per l’estrazione di acidi e basi da
soluzioni acquose. Il gruppo –SO3- è un forte scambiatore cationico ed è
impiegato per l’estrazione di analiti basici dalle soluzioni mentre il gruppo
-N+(CH3)3 è un forte scambiatore anionico ed è usato per analiti acidi. I parametri
da ottimizzare per condurre un’analisi cromatografia a scambio ionico con silice
modificata sono molto importanti, tra cui ricordiamo il pH, il controione, la forza
ionica ed il flusso.
Adsorbenti polimerici
Oltre ai materiali adsorbenti fino ad ora osservati, sono stati messi a punto, per
specifiche esigenze, altri tipi di adsorbenti aventi struttura polimerica.
Tali adsorbenti, spesso, permettono di ottenere un’alta selettività, difficilmente
ottenibile con i materiali adsorbenti esaminati precedentemente. Witzenbacher ed
altri [13], nella preconcentrazione di metabolici dell’atrazina hanno affrontato tale
questione. In prima istanza hanno utilizzato l’ottadecile legato (cromatografia a
fase inversa). Tale sistema, pur risultando valido per l’analita in questione, si è
dimostrato inefficiente per i suoi metaboliti (composti altamente polari). La

18
difficoltà è stata superata impiegando un adsorbente polimerico: resina di
polistirene-divinilbenzene.
Il carbone attivo
Il carbone ha la proprietà di rimuovere le impurezze da soluzioni e da gas.
Le impurezze sono adsorbite sul carbone, precedentemente attivato. La natura
porosa di questo materiale è, in gran parte, la causa delle sue caratteristiche
adsorbenti. Industrialmente il processo di attivazione implica il riscaldamento di
un materiale carbonioso di partenza, parzialmente ossidato, per rimuovere le
impurezze non carboniose. La fase gassosa ossidante, impiegata nella fase di
attivazione, può contenere ossigeno, anidride carbonica ed acqua [14]. La natura
dei siti del carbone attivo è stata ampiamente studiata e le caratteristiche dei siti,
in genere, riflettono quelle dei composto aromatici ossigenati. Quasi sempre vi
sono gruppi carbossilici, forme enoliche di 3-dichetoni, gruppi fenolici, legami
C-O e C-O-C.
Il Carbograph
L'utilizzo del Carbograph come adsorbente per preconcentrare tracce di sostanze
inquinanti presenti in campioni ambientali, è in continuo aumento. Da diversi anni
si è dimostrato come le sue caratteristiche soddisfano pienamente le esigenze di
laboratori privati, enti di ricerca ed università.
Le diverse aree superficiali delle varie tipologie di Carbograph consentono
l'intrappolamento e il rilascio efficiente di un vasta gamma di composti, dai più
volatili ai medio-alto bollenti essendo possibile l'utilizzo simultaneo di 2 o 3 tipi
di Carbograph nello stesso tubo adsorbente.

19
Tabella: riepilogo fasi adsorbenti commerciali
ADSORBENTE POLARE
NON MODIFICATO
STRUTTURA SITO
DELL’ADOSORBENTE
SILICE
FLORISIL
ALLUMINA
-SiOH
Mg2SiO3
Al2O3
ADSORBENTE MODIFICATO
CON R POLARE:
CROMATOGRAFIA A FASE NORMALE
STRUTTURA SITO
DELL’ADOSORBENTE
SILICE CON GRUPPO CIANO
SILICE CON GRUPPO AMMINO
SILICE CON GRUPPO DIOLO
-CH2-CH2-CH2-CN
-CH2-CH2-CH2-NH2
-(CH2)3-O-CH2-CH-CH2
OH OH
ADSORBENTE MODIFICATO
CON R APOLARE:
CROMATOGRAFIA A FASE INEVRSA
STRUTTURA SITO
DELL’ADOSORBENTE
SILICE CON OTTADECILE
SILICE CON OTTILE
SILICE CON ETILE
SILICE CON CICLOESILE
-C18
-C8
-C2
-CH2-CH2-
ADSORBENTE MODIFICATO
CON R CARICO:
CROMATOGRAFIA A SCAMBIO
IONICO
STRUTTURA SITO
DELL’ADOSORBENTE
Si CON AMMINA QUATERNARIA
Si CON Ac. CARBOSSILICO
Si CON Ac. SOLFONICO
-CH2-N+(CH3)2-C18
-CH2-CH2-COOH
-(CH2)3-SO2H

20
SCELTA DELLA FASE SOLIDA ADSORBENTE
La scelta dell’adsorbente, per l’estrazione di un dato analita, rappresenta lo step
fondamentale negli studi della separazione in fase solida.
I concetti affrontati in precedenza non possono che essere solo orientativi.
Ciò dipende dal fatto che quando si esaminano casi reali, le variabili coinvolte in
tali applicazioni sono molteplici e di importanza non trascurabile. Basti solo
pensare alla complessità delle strutture molecolari delle sostanze organiche che si
analizzano. La maggior parte di queste è caratterizzata da una parte con carattere
polare un’altra con carattere apolare. In questo caso, quale sarà l’adsorbente
adatto? Ci sono molecole non polari, solubili in solventi apolari e viceversa.
Inoltre la quantità di fase adsorbente, la sua granulometria ed il suo
condizionamento influenzano l’adsorbimento. Spesso è necessario valutare le
interazioni “secondarie” dell’analita, che si instaurano attraverso zone della
molecola e non con il sito principale. Bisogna stabilire, con margini molto
ristretti, la velocità di flusso, la possibilità che siano presenti contemporaneamente
altre sostanze ed il solvente, che potrebbe legarsi fortemente con il soluto e quindi
non consentire l’adsorbimento, oppure il solvente potrebbe competere con
l’analita stesso per l’adsorbimento. In sintesi, la complessità del problema riflette
la complessità della struttura chimica stessa della molecola. E’, perciò, molto
difficile indicare con assoluta certezza, a priori, l’adsorbente adatto e le condizioni
d’analisi. Ciò richiede uno studio particolareggiato in cui, sempre considerando i
concetti basilari della separazione in fase solida, il ruolo principale è svolto dalle

21
prove preliminari con sostanze a titolo noto. Solo dopo l’ottimizzazione del
processo si può passare ai campioni reali.
In generale, si può affermare che gli analiti dotati di elevata polarità solubilizzati
in solventi non polari saranno trattenuti da adsorbenti aventi funzioni polari non
derivatizzate e funzioni polari chimicamente legate mentre i soluti apolari
solubilizzati in solventi polari saranno adsorbiti, facilmente, da fasi solide aventi
funzioni apolari chimicamente legate.
In ultimo, gli analiti ionici o ionizzabili preferiscono come fasi solide adsorbenti
quelle che presentano una netta separazione di carica.

22
GAS CROMATOGRAFIA
PRINCIPI DELLA GAS CROMATOGRAFIA [15-16-17]
La gas cromatografia (GC) è una tecnica cromatografica di grande importanza per
la separazione e l’analisi di miscele gassose o gassificabili.
Solitamente tale tecnica cromatografica si divide in due sottogruppi e cioè la
cromatografia gas-solido e la cromatografia di gas-liquido.
Queste due tecniche sono simili nei loro aspetti generali ma differiscono nelle
caratteristiche della fase stazionaria. Nella cromatografia gas-solido la fase
stazionaria è costituita da un solido adsorbente, mentre nella cromatografia di
ripartizione gas-liquido la fase stazionaria è costituita da un supporto inerte sul
quale è stato depositato un film di un liquido di ripartizione. La fase mobile è
costituita in ambedue i casi da un gas inerte.
La gas cromatografia è in grado di analizzare e separare non solo campioni
gassosi, ma anche liquidi o solidi purché siano sufficientemente volatilizzabili e
stabili in stato vapore. La fase stazionaria assorbe parzialmente o adsorbe il
campione mentre la fase gassosa lo trasporta lungo la colonna impaccata o
capillare. La separazione dei componenti, di una miscela iniettata, procede
attraverso una ripartizione tra la fase gassosa e la fase liquida adsorbita sul
supporto solido inerte. Le differenti caratteristiche delle fasi stazionarie e le
molteplici interazioni che si instaurano fra queste ultime e gli analiti, costituenti
le miscele, sono i responsabili della separazione. I componenti che vengono

23
maggiormente trattenuti (elevata interazione chimico-fisica dei soluti con la fase
stazionaria) dalla fase stazionaria si muovono più lentamente con il flusso della
fase mobile. Al contrario, i componenti che vengono debolmente trattenuti dalla
fase stazionaria, si muovono più rapidamente. In conseguenza di queste differenze
di mobilità i componenti del campione vengono separati.
Il gas inerte o la fase mobile passa attraverso un rivelatore. Il segnale del detector
è continuamente monitorato attraverso un registratore in cui i picchi dei singoli
componenti sono relazionati al tempo di eluizione degli stessi. L’area dei rispettivi
picchi è proporzionale alla quantità relativa di ciascun analita costituente il
campione in esame. Tuttavia, il tempo di ritenzione, il tempo che intercorre tra
l’iniezione del campione e il centro di uno specifico picco, può essere usato come
conferma per la identificazione di un determinato analita utilizzando un campione
noto con le stesse condizioni sperimentali ed osservando il medesimo tempo di
ritenzione. Tale raffronto potrebbe essere ripetuto utilizzando una colonna a
differente polarità al fine di convalidare il precedente accordo dei tempi di
ritenzione. Il tempo di ritenzione è influenzato da diverse variabili come il flusso
del gas di trasporto, la temperatura della colonna, la lunghezza e la composizione
della colonna.
LA COLONNA CROMATOGRAFICA
Il maggior impiego della gas cromatografia rispetto a tutte le altre tecniche di
separazione è dovuto alla sua alta selettività e capacità di separare componenti
volatili in una miscela, anche complessa. Un elevato numero di separazioni

24
possono essere ottenute con appropriate condizioni di esperimento. Probabilmente
il parametro critico, nella conduzione di un’analisi gas cromatografica (gas-
liquido), è la scelta del liquido della fase stazionaria della colonna.
La colonna cromatografica rappresenta la parte essenziale dell’apparecchio: il suo
dimensionamento, la scelta appropriata del materiale di riempimento e le modalità
usate per riempirla, sono determinanti ai fini dell’efficienza che se ne potrà
ottenere. Compito della colonna è di contenere la fase stazionaria ed al tempo
stesso essere permeabile al gas vettore.
A seconda del modo in cui la fase fissa è disposta all’interno della colonna
possiamo distinguere:
- colonne a riempimento (o impaccate), in cui la fase stazionaria si presenta come
un mezzo poroso al flusso del gas vettore. Nella cromatografia gas-liquido, il
liquido di ripartizione è disperso su di un supporto inerte, mentre nella gas-solido,
l’adsorbente è un solido granulare o una polvere fine supportata.
- colonne capillari (open tubular columns) in cui la fase stazionaria è disposta
sulle pareti come un film sottile o polvere.
La trattazione che segue riguarda le colonne di ripartizione gas-liquido.
La funzione del solvente (fase liquida) è di determinare la ripartizione
differenziale dei componenti la miscela in esame (analiti); la sua scelta viene
quindi fatta sulla base dell’intervallo di ebollizione dei soluti e della loro natura
chimica. Una via di classificazione della fase liquida, quindi della fase stazionaria,
è quella relativa alla diversa polarità. La scelta della fase liquida segue alcune
regole generali. Una colonna non selettiva o non polare riesce
approssimativamente a separare soluti simili, in accordo con il loro punto di

25
ebollizione mentre una colonna polare separerà componenti in base alla loro
polarità, piuttosto che al loro punto di ebollizione.
In generale le colonne polari riescono a trattenere composti polari più fortemente
rispetto a sostanze meno polari o a sostanze non polari.
La grandezza della costante di distribuzione del campione tra la fase liquida
stazionaria e la fase gassosa è molto più importante della volatilità o punto di
ebollizione dei componenti, nel determinare l’ordine di eluizione. Tuttavia, i
campioni fluiscono in ordine crescente del punto di ebollizione se, naturalmente,
tutti appartengono alla stessa serie omologa.
Il fattore principale che determina il tempo di ritenzione per un dato componente
nella colonna (a temperatura costante) è il grado di interazione che ha luogo tra la
molecola del campione e la fase liquida.
I vari meccanismi di attrazione intermolecolare determinano il ritardo del
cammino di una data molecola trasportata lungo la colonna dalla fase mobile
gassosa. In conclusione, è opportuno che la struttura del supporto sia
macroporosa, per essere permeabile al gas di trasporto e al tempo stesso trattenere
una considerevole quantità di liquido. L’area superficiale del supporto deve essere
sufficientemente grande; tuttavia pori troppo fini sarebbero occlusi dal solvente,
per cui l’area esposta allo scambio risulterebbe diminuita. Ettre [18], a seguito di
uno studio sull’effetto dell’area superficiale, afferma che il suo valore non
dovrebbe superare 1 m2/g.
La superficie del supporto solido deve essere chimicamente inerte poiché non
deve intervenire nel processo di separazione gas cromatografico.

26
I supporti di più vasto impiego derivano dagli scheletri di diatomee, poiché questi
presentano caratteristiche di struttura e superficie che si avvicinano all’idealità.
Commercialmente si annoverano i seguenti supporti [19]: Chromosorb W;
Chromosorb P; Embacel; Gas-Chrom.
Velocità di flusso del gas di trasporto
L’efficienza di una colonna cromatografica, nella separazione dei componenti di
una miscela, dipende dal numero di equilibri che si instaurano tra la fase liquida
stazionaria e la fase mobile gassosa.
L’efficienza, generalmente, è citata in termini di altezza equivalente di un piatto
teorico (Height Equivalent Theoretical Plate – HETP), che può essere inteso come
la lunghezza minima necessaria affinché si stabilisca l’equilibrio di ripartizione
del campione tra le due fasi: liquida stazionaria e mobile gassosa inerte. In altri
termini, si può pensare che la colonna cromatografica consista di un numero di
zone adiacenti in ognuna della quali c’è spazio sufficiente affinché un soluto si
equilibri completamente tra le due fasi (piatto teorico).
Il numero, N, di questi piatti dipende dalla lunghezza della colonna e dalla
velocità di flusso del gas di trasporto. Maggiore è la lunghezza della colonna più
alto è il numero dei piatti teorici. Tuttavia, più corta è la lunghezza di HETP, più
piatti sono presenti nell’unità di lunghezza della colonna, di conseguenza
maggiore è l’efficienza di quest’ultima. Il numero N è calcolato prendendo in
considerazione il tempo di ritenzione osservato e l’ampiezza del picco
cromatografico secondo la seguente equazione:
2
16 ⎟⎠⎞
⎜⎝⎛=
Wt
N R
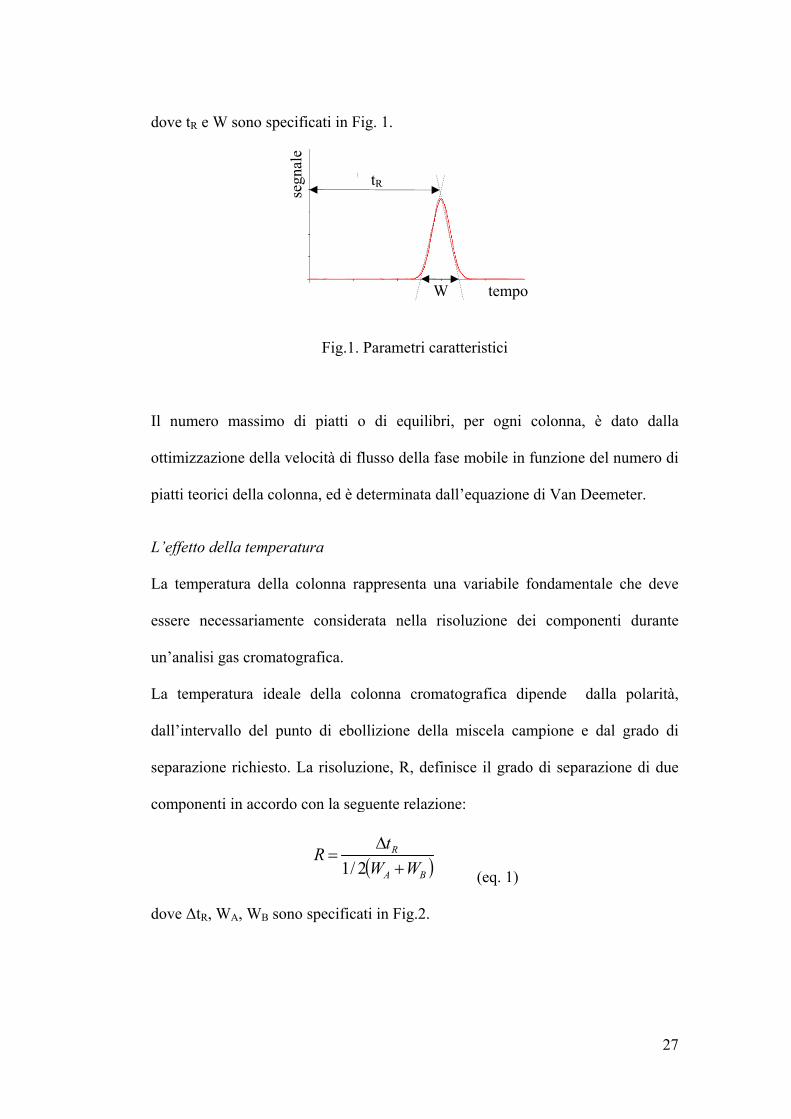
dove tR e W
Il numero
ottimizzaz
piatti teori
L’effetto d
La temper
essere ne
un’analisi
La tempe
dall’interv
separazion
componen
dove ΔtR,
W sono spe
o massimo
zione della v
ici della col
della temper
ratura della
cessariamen
gas cromat
eratura idea
vallo del pu
ne richiesto
nti in accord
WA, WB so
ecificati in F
Fig.1.
di piatti
velocità di f
lonna, ed è d
ratura
a colonna r
nte conside
tografica.
ale della c
unto di ebo
o. La risoluz
do con la seg
R =/1
ono specifica
segn
ale
Fig. 1.
. Parametri
o di equili
flusso della
determinata
rappresenta
erata nella
olonna cro
ollizione de
zione, R, de
guente relaz
( )BA
R
WWt+
Δ2
ati in Fig.2.
tR
caratteristic
ibri, per o
a fase mobil
a dall’equaz
una variab
risoluzion
omatografica
ella miscela
efinisce il g
zione:
) (eq.
temW
ci
ogni colonn
le in funzion
zione di Van
bile fondam
e dei com
a dipende
a campione
grado di sep
1)
mpo
na, è dato
ne del nume
n Deemeter
mentale che
mponenti du
dalla pol
e e dal gra
parazione d
27
dalla
ero di
.
deve
urante
larità,
do di
di due

Fig.2. P
E’ evident
parametro
La miglio
temperatu
in un aum
Generalme
ebollizion
tempo di r
Per analit
convenien
discontinu
denominat
iniziale, la
mantenuti
composti b
gli alto
dell’increm
Per calcolar
te dall’eq.
o R è un num
ore risoluzio
ura, tuttavia,
mento del tem
ente, una t
ne di una mi
ritenzione.
ti caratteriz
nte increme
uo a second
ta “program
a velocità d
sotto contr
basso bolle
bollenti so
mento progr
lre la risoluzi
seguenti
1 che i picc
mero grande
one si ottien
, si traduce
mpo d’anali
temperatura
iscela di so
zzati da un
entare la te
da l’efficie
mma di te
dell’aument
rollo. Come
nti sono ben
ono eluiti
rammato de
se
gnal
e
W
ione di un ci parametri:
chi cromato
e.
ne a bassa
in un incre
isi.
a leggermen
oluti ha com
n ampio in
emperatura
nza della s
emperatura”
to della tem
e conseguen
n separati d
dalla colo
ella tempera
ΔtR
WA
cromatogram: ΔtR, WA, W
ografici son
temperatura
emento del t
nte superior
me effetto un
ntervallo d
della colo
separazione
”. In tale
mperatura e
nza di quan
dalla bassa t
onna in un
atura.
temWB
mma devonWB.
no perfettam
a. La scelta
tempo di el
re alla med
na discreta
del punto d
onna, in m
e, attraverso
procedura,
la tempera
nto espresso
temperatura
n tempo m
mpo
o essere not
mente risolti
a della più
luizione e q
dia del pun
diminuzion
di ebollizio
modo contin
o una proc
la temper
atura finale
o si desume
a iniziale, m
minore a
28
ti i
i se il
bassa
quindi
nto di
ne del
one è
nuo o
edura
ratura
sono
che i
mentre
causa

29
RIVELATORI
In gas cromatografia la fase mobile non prende parte al processo di separazione,
ma agisce esclusivamente quale gas vettore; può quindi essere scelto un qualsiasi
gas permanente indifferentemente per ogni tipo di separazione. Pertanto la misura
di una proprietà fisica o chimica in cui le sostanze da rilevare si differenziano dal
gas di trasporto, consente di visualizzare l’eluizione dei componenti: questa viene
tradotta per mezzo di un “detector” in un segnale, generalmente elettrico, e quindi
registrabile con un opportuno strumento. Il rivelatore ideale per la gas
cromatografia deve avere le seguenti caratteristiche: sensibilità adeguata; buona
stabilità e riproducibilità; una risposta lineare alla quantità d’analita che si estenda
per diversi ordini di grandezza; una temperatura d’esercizio che vada da
temperatura ambiente fino ad almeno 400°C; un tempo di risposta breve che
risulti indipendente dal flusso; elevata affidabilità e facilità d’uso; fattori di
risposta più o meno uniformi nei confronti di tutti gli analiti, oppure al contrario
una sensibilità specifica e prevedibile verso una o più classi di composti; metodo
di rilevazione non distruttivo.
Il rilevatore a ionizzazione di fiamma (FID)
È uno dei rivelatori più diffusi e di uso più generale in gas cromatografia. La
maggior parte dei composti organici, quando viene pirolizzata alla temperatura di
una fiamma idrogeno/aria, genera ioni ed elettroni che possono condurre elettricità
attraverso una fiamma. La corrente generata viene inviata ad un amplificatore.

30
Il rivelatore FID presenta un ampio intervallo di risposta lineare e un basso
rumore di fondo; ma, essendo un rivelatore universale, non presenta una elevata
sensibilità.
Il rivelatore a conducibilità termica (TCD)
Si basa sulla variazione della conducibilità termica della corrente di gas di
trasporto prodotta dalla presenza di molecole di analita. La sensibilità di tale
rivelatore è poco inferiore a quella del FID.
Il rilevatore azoto- fosforo (NPD)
È un rivelatore selettivo nei confronti di composti organici contenenti fosforo e
azoto. Se lo si confronta con il FID risulta 500 volte più sensibile per molecole
che contengono fosforo e azoto.
Il rivelatore a cattura di elettroni (ECD)
Opera in modo molto simile a quello di un contatore proporzionale per la misura
di raggi X. In questo strumento la fase mobile effluente dalla colonna passa sopra
un emettitore di particelle β. Gli elettroni emessi dall’emettitore provocano la
ionizzazione del gas vettore e la produzione di un flusso di elettroni. Tale
rivelatore è molto sensibile, specialmente nei confronti di quelle molecole che
posseggono un gruppo elettronaccettore come un alogeno, un perossido, un
chinone ed un nitrogruppo.

31
LE TECNICHE IFENATE
La gascromatografia o la cromatografia liquida è spesso accoppiata con alcune
tecniche spettroscopiche ed elettrochimiche. Il risultato sono i cosiddetti metodi
accoppiati o tecniche ifenate. Tali tecniche rappresentano mezzi molto validi per
indagare ed identificare i componenti di una miscela complessa. Se con la tecnica
gascromatografica si riesce ad avere una opportuna separazione dei composti
presenti nel campione in esame, la spettrometria di massa è capace di dare
informazioni qualitative su ognuno dei componenti costituenti il campione.
Le metodiche ifenate di più largo utilizzo sono:
- gas cromatografia-spettrometria di massa (GC-MS);
- gas cromatografia-spettrometria infrarossa (GC-FTIR);
- gas cromatografia-emissione atomica (GC-AES);
- cromatografia liquida-spettrometria di massa (LC-MS).
La tecnica GC-MS rappresenta, attualmente, la metodica accoppiata che offre
maggiore garanzia. Molte ditte produttrici di apparecchiature scientifiche offrono
strumentazioni per gascromatografia che sono direttamente accoppiate a
spettrometri di massa di vario tipo.
La spettrometria di massa e’ una tecnica analitica di delucidazione strutturale
basata sulla ionizzazione di una molecola e sulla sua successiva frammentazione
in ioni di diverso rapporto massa/carica (M/z).
Il principio su cui si basa è il seguente: una molecola è ionizzata per espulsione di
un elettrone; il catione radicalico che si forma (ione molecolare) in parte si
frammenta dando molecole e/o radicali neutri, non rilevabili dallo strumento, in

32
parte generando cationi e/o radicali cationi (ioni frammento), rilevabili dallo
strumento. Lo ione molecolare e i vari ioni che si originano per frammentazione
vengono discriminati sulla base del loro rapporto massa/carica e rivelati da un
detector.
L’esperimento di spettrometria di massa consiste dunque nella ionizzazione di
molecole in fase gassosa, nella separazione dei diversi ioni prodotti e nella loro
rivelazione. Il risultato dell’esperimento è lo spettro di massa, che rappresenta
l’abbondanza relativa degli ioni in funzione del loro rapporto massa/carica.
Questa tecnica consente di misurare le masse molecolari e di ottenere dei profili di
frammentazione che sono specifici per ciascun composto; ogni composto ha
quindi una propria impronta digitale registrata in un database. In questo modo, si
può individuare la formula di struttura di composti sconosciuti, e dal confronto
con la banca dati, avere informazioni qualitative inequivocabili sul composto.

33
LA SPETTROMETRIA DI MASSA
Lo spettrometro di massa può essere schematizzato nel seguente modo:
Sistema pompa da vuoto
Il vuoto, che si aggira intorno ai 10-6 – 10-7 torr, è necessario per due motivi:
innanzi tutto serve ad impedire la presenza dei gas atmosferici all’interno del
sistema, che, urtando con le molecole ionizzate, ne causerebbero una perdita di
ionizzazione; inoltre è di fondamentale importanza perché elimina dal sistema di
ionizzazione tutte le molecole neutre o indesiderate che lo strumento non rileva.
Introduzione del campione
L’introduzione del campione nella camera di ionizzazione può essere fatta sia allo
stato solido, usando una sonda, che allo stato liquido o gassoso, usando un sistema
di valvole che permettono di accedere alla camera di ionizzazione senza che
questa venga a contatto con l’esterno. Attualmente, lo spettrometro di massa è
largamente utilizzato come rilevatore di sistemi cromatografici, gassosi o solidi
che sia.
Camera di ionizzazione
Se una molecola è investita in fase vapore da un fascio di elettroni di notevole
energia cinetica si può avere per urto la sua ionizzazione a ione positivo o
negativo. In genere gli strumenti sono regolati per lavorare unicamente con ioni

34
positivi, i quali possono spontaneamente o per urto decomporsi in una serie di
frammenti di massa inferiore e questi a loro volta in altri.
Ogni molecola avrà quindi una sua frammentazione caratteristica e specifica che
dipenderà sia dalla natura delle molecole sia dalle condizioni operative di
ionizzazione.
Il campione viene ionizzato in un’apposita camera di ionizzazione, in cui il fascio
di elettroni viene prodotto da una sorgente ionica che varia a seconda della tecnica
utilizzata.
In genere gli elettroni sono emessi da un filamento caldo di tungsteno o renio, e
passano attraverso un condotto, che crea il raggio, nella parte centrale della
camera che contiene il campione gassoso.
La frazione di elettroni che non urta contro le molecole è raccolta da una trappola
per gli elettroni, le molecole che non sono ionizzate sono allontanate dalla pompa
da vuoto, mentre quelle ionizzate sono accelerate e convogliate verso
l’analizzatore attraverso un campo elettrico (cono di Skimmer).
Il sistema di ionizzazione svolge un ruolo essenziale nella spettrometria di massa,
perché da esso dipende anche il numero, la natura e l’abbondanza dei frammenti
molecolari che compaiono nello spettro di massa. Per questo motivo le tecniche
utilizzate sono numerose e alcune di esse danno origine a particolari varianti nella
spettrometria di massa.
Tra i vari dispositivi alcuni consentono di analizzare solo frammenti positivi, altri
invece, permettono la rivelazione anche di ioni negativi. Inoltre alcune tecniche di
ionizzazione sono decisamente potenti, operano cioè ad alta energia e portano ad

35
una frammentazione spinta (Tecniche HARD), altre invece operano a bassa
energia producendo un numero inferiore di ioni (Tecniche SOFT).
SORGENTI
Di seguito riporto le tre tecniche di ionizzazione più comuni, con una breve
spiegazione di ognuna. In base al tipo di sorgente utilizzata la ionizzazione
primaria del campione viene realizzata in diverso modo; le tecniche più utilizzate
sono:
1) impatto elettronico (E.I.)
2) ionizzazione chimica (C.I.)
3) electrospray (E.S.I.)
Impatto elettronico (EI)
La ionizzazione per impatto elettronico è la tecnica più comune. Un filamento di
tungsteno incandescente emette un fascio di elettroni che, accelerati verso un
anodo posto dalla parte opposta al filamento, acquistano un’elevata energia (ca. 70
eV). Quando questi elettroni vengono a contatto con la sfera elettronica di una
molecola le trasferiscono la loro energia, provocando l’espulsione di un elettrone
con formazione di un radical-catione (ione molecolare) M+*.
Tutti gli ioni positivi sono respinti da una piastra, tenuta ad un potenziale positivo,
verso una serie di piastre forate, tenute a potenziale positivo crescente, dette
piastre acceleratrici. Nel loro tragitto gli ioni subiscono un’accelerazione

36
proporzionale al potenziale delle piastre acceleratrici e vengono espulsi, attraverso
una fenditura di uscita.
Questo tipo di ionizzazione è hard. Gli ioni vengono generati ad un livello
energetico molto alto e si possono avere frammentazioni estese che lasciano poco
o nulla dello ione molecolare. Per risolvere questo problema sono state messe a
punto altre tecniche di ionizzazione, dette tecniche soft (e sono le seguenti).
Ionizzazione Chimica (CI)
La ionizzazione chimica viene utilizzata quando gli ioni molecolari prodotti con il
metodo dell’impatto elettronico sono troppo poco stabili e si frammentano
completamente facendo spesso scomparire il picco molecolare.
Questa e’ una tecnica di ionizzazione più “mild”, che si basa sull’interazione del
campione vaporizzato con un reagente ionizzato, che di solito e’ un acido di
Bronsted gassoso.
I reagenti più usati sono quelli che derivano dalla ionizzazione ad impatto
elettronico del metano
CH4 + e- CH4
+˙ + 2e-
CH4+˙ + CH4 CH5
+ + CH3˙
Il CH5+ funge da acido di Bronsted, quindi se la molecola M ha un’affinità per il
protone più alta di quella del metano si avrà la formazione dello ione [M+H]+.
CH5+ + M [M+H]+ + CH4
Gli ioni [M+H]+ (detti quasimolecolari) non possiedono una energia così elevata e
quindi subiscono una minore frammentazione. In genere la ionizzazione chimica
dà dei frammenti molecolari più significativi di quanto non faccia l’impatto
elettronico.

37
La CI e’ particolarmente adatta a molecole come idrocarburi, alcoli, esteri,
ammine, amminoacidi, piccoli peptidi che in condizioni di EI darebbero una
frammentazione eccessiva.
La particolarità è che nello spettro vedremo lo ione molecolare + 1.
Ionizzazione elettrospay (ESI)
Il campione, sciolto in un solvente polare, è nebulizzato a pressione atmosferica
dentro la camera di ionizzazione attraverso un ago tenuto ad un alto potenziale
elettrico. Le goccioline di spray, che si sono caricate positivamente per azione del
campo elettrico, vengono attratte verso una "lente di estrazione di ioni", che
grossolanamente è costituito da un capillare mantenuto sotto vuoto e a un
potenziale negativo; in tal modo il sovente evapora e gli ioni carichi sono
accelerati verso l'analizzatore. Questa tecnica di ionizzazione è largamente usata
negli strumenti HPLC-MS.
ANALIZZATORE
L’analizzatore consente di differenziare gli ioni generati in base al loro rapporto
massa/carica.
I più comuni sono:
- l’analizzatore magnetico
- l’analizzatore a doppia focalizzazione
- l’analizzatore a quadrupolo
- l’analizzatore a trappola ionica
- l’analizzatore a tempo di volo

38
L’analizzatore magnetico
E' l'analizzatore più usato, perché consente di ottenere le risoluzioni migliori. E’
costituito da un tubo lungo circa 1 metro, piegato con un raggio di curvatura r' ed
immerso in un campo magnetico H. Gli ioni che escono dalla camera di
ionizzazione entrano nel tubo analizzatore e, per effetto del campo magnetico,
subiscono una deviazione dalla loro traiettoria rettilinea (deflessione). La nuova
traiettoria curvilinea ha un raggio di curvatura r che è direttamente proporzionale
alla quantità di moto dello ione e inversamente proporzionale al campo
magnetico.
Analizzatore a doppia focalizzazione
Aggiungendo dopo l'analizzatore magnetico un filtro elettrostatico il percorso
degli ioni positivi viene focalizzato ulteriormente in direzione dal campo elettrico
statico

39
Nel settore elettrostatico gli ioni non vengono separati in funzione del rapporto
massa/carica, ma solo focalizzati in base alla loro energia traslazionale; questo
perché altrimenti nel settore successivo, quello magnetico, ioni con ugual rapporto
m/z ma differente energia traslazionale seguirebbero traiettorie diverse,
diminuendo la risoluzione dello strumento. Così la risoluzione può raggiungere
100˙000 e oltre. Ciò permette di misurare la massa esatta fino alla quarta cifra
decimale. Gli spettrometri ad alta risoluzione di questo genere sono
apparecchiature complicate e costose, che solitamente non si trovano nei
laboratori di analisi
Analizzatore a quadrupolo
E’ costituito da quattro barre cilindriche metalliche, lunghe circa 20 cm., che
delimitano il "cammino" percorso dagli ioni provenienti dalla camera di
ionizzazione e diretti al detector. Le barre sono mantenute ad un potenziale
elettromagnetico oscillante, in modo che quando le due sbarre verticali hanno
potenziale positivo quelle orizzontali l’hanno negativo, e viceversa.
Gli elettroni, accelerati dalle piastre acceleratrici, entrano nel tunnel delimitato
dalle barre e vengono respinti dai poli positivi ed attratti dai negativi.

Tuttavia,
traiettoria
che, per u
tale per cu
nel sistem
Operando
far uscire
quadrupol
e un mino
Analizzato
Può esser
anziché p
ionica trat
a causa d
a zig zag e
una certo va
ui il moto d
ma di rivelaz
quindi una
ioni a mas
lo ha una ris
r costo.
ore a trappo
re considera
ermettere a
ttiene tutti g
dell’oscillaz
e finiscono
alore di freq
diventa sinu
ione (fotom
a scansione
ssa molecol
soluzione pi
ola ionica
ato una var
agli ioni di
gli ioni al su
zione del
con lo scar
quenza di o
usoidale e ri
moltiplicator
di frequenz
lare crescen
iù bassa (<
riante dell'a
attraversar
uo interno.
quadrupolo
ricarsi su un
oscillazione
iescono ad
re).
za di oscilla
nte. Rispetto
1000), ma t
analizzatore
re il campo
o gli ioni
na delle bar
e, hanno un
uscire dal t
zione del ca
o all’analiz
tempi di sca
e a quadrup
o quadrupo
assumono
rre, tranne q
’energia cin
tunnel ed en
ampo è pos
zzatore a tu
ansione più
polo; qui in
olare, la trap
40
o una
quelli
netica
ntrare
ssibile
ubo, il
bassi
nfatti,
ppola

41
Questa variante dell'analizzatore a quadrupolo usa tre elettrodi, un elettrodo
anulare posto fra due elettrodi semisferici di entrata e uscita, per intrappolare ed
accumulare gli ioni in una cavità di volume ristretto, la cosiddetta trappola ionica
(Ion Trap), allo scopo di ottenere una elevata sensibilità. I due elettrodi laterali
hanno un piccolo foro al centro attraverso il quale passano gli ioni. Gli ioni sono
tenuti in orbita all’interno della trappola ionica secondo il campo di stabilita della
stessa, che può essere regolato agendo sui potenziali degli elettrodi. Infatti, lo
spettro di massa è generato variando il potenziale elettrico in modo da espellere in
sequenza dalla trappola verso il rivelatore gli ioni secondo un valore m/z
crescente.
Analizzatore a tempo di volo (TOF)
Il principio su cui si basa questo analizzatore e’ che ioni di differente valore
massa/carica hanno uguale energia, ma differente velocità dopo l’accelerazione
subita nella camera di ionizzazione.
Ne deriva che il tempo che ciascuno mette per attraversare l’analizzatore è
differente e quindi arrivano con tempi diversi al rilevatore.

42
RIVELATORE
Come collettore e rivelatore degli ioni si usa comunemente un moltiplicatore
elettronico, costituito da una serie di elettrodi in cascata. Quando uno ione arriva
sul primo elettrodo questo emette un fascio di elettroni che vanno a colpire il
secondo elettrodo, il quale a sua volta emette una quantità maggiore di elettroni e
così via.
Il risultato è una forte amplificazione del segnale che viene poi digitalizzato ed
elaborato infine dal calcolatore dello spettrometro per la presentazione dello
spettro di massa.
LO SPETTRO DI MASSA
Lo spettro di massa si presenta quindi come un insieme di linee verticali (picchi)
di intensità diversa, ciascuna corrispondente al valore di massa di uno ione
frammento.
Il picco a valore di massa più elevato è quello relativo allo ione molecolare. In
genere, la corrente ionica è normalizzata a 100, ossia il picco più alto (picco base)
ha valore 100, indipendentemente dal fatto che esso sia il picco molecolare o
meno e tutti gli altri picchi sono ad esso normalizzati.
Dallo spettro di massa si può risalire dunque alla struttura di un composto
incognito, attribuendo ai singoli ioni una composizione elementare e ricostruendo
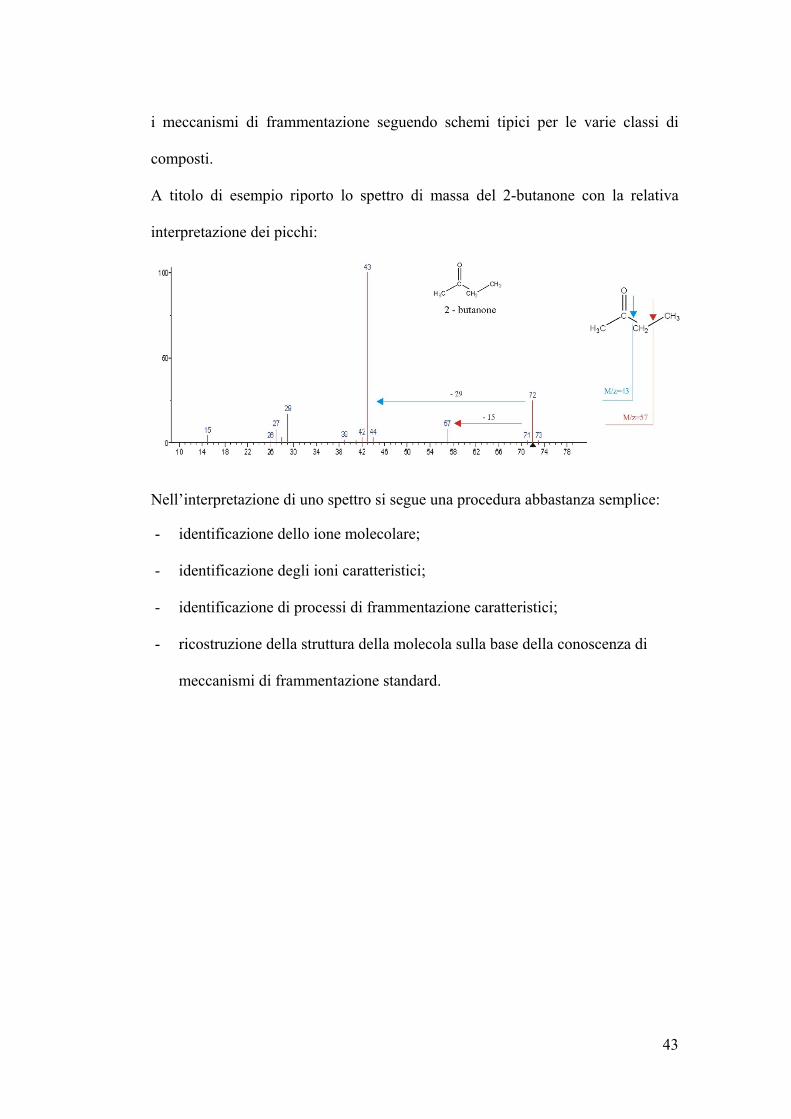
43
i meccanismi di frammentazione seguendo schemi tipici per le varie classi di
composti.
A titolo di esempio riporto lo spettro di massa del 2-butanone con la relativa
interpretazione dei picchi:
Nell’interpretazione di uno spettro si segue una procedura abbastanza semplice:
- identificazione dello ione molecolare;
- identificazione degli ioni caratteristici;
- identificazione di processi di frammentazione caratteristici;
- ricostruzione della struttura della molecola sulla base della conoscenza di
meccanismi di frammentazione standard.

44
BIBLIOGRAFIA
1 C. Baird, M. Cann; Chimica Ambientale, Zanichelli, cap. 7, pag 211-242. (2006) 2 A.C. Hogenboom, J.A. Van Leerdam, P. de Voogt; Accurate mass screening and
identification of emerging contaminants in environmental samples by liquid chromatography-hybrid linear ion trap Orbitrap mass spectrometry; Journal Of Crhomatogrphy A, 1216, 3, 510-519. (2009)
3 J.L.M. Vidal, P. Plaza-Bolanos, R. Romero-Gonzalez; Determination of pesticide transformation products: A review of extraction and detection methods; Journal Of Crhomatogrphy A, 1216, 40, 6767-6788. (2009)
4 N. Borghesi, S. Corsolini, S. Focardi; Levels of polybrominated diphenyl ethers (PBDEs) and organochlorine pollutants in two species of Antarctic fish (Chionodraco hamatus and Trematomus bernacchii); Chemosphere, 73, 2, 155-160. (2008)
5 F. Suja, B.K. Pramanik, SM. Zain; Contamination, bioaccumulation and toxic effects of perfluorinated chemicals (PFCs)in the water environment: a review paper; Water Science and Technology, 60, 6, 1533-1544. (2009)
6 M.G.H. Berntssen, C.N. Glover, DHF- Robb; Accumulation and elimination kinetics of dietary endosulfan in Atlantic salmon; Aquatic Toxicology, 86, 1, 104-111. (2008)
7 H.B. Ozkoc, G. Bakan, S. Ariman; Distribution and bioaccumulation of organochlorine pesticides along the Black Sea coast; Environmental Geochemistry and Health, 29, 1, 59-68. (2007)
8 B.J. Konwick, A.W. Garrison, JT. Avants; Bioaccumulation and biotransformation of chiral triazole fungicides in rainbow trout; Aquatic Toxicology, 80, 4, 372-381. (2006)
9 B.H.T. Poon, C.K.M. Leung, C.K.C. Wong; Polychlorinated biphenyls and organochlorine pesticides in human adipose tissue and breast milk collected in Hong Kong; Archives Of Environmental Contamination And Toxicology, 49, 2, 274-282. (2005)
10 Y. Wang, Y. Li, J. Ding; Estimation of bioconcentration factors using molecular electro-topological state and flexibility; Sar And Qsar In Environmental Research ,19, 3-4, 375-395. (2008)
11 J.E. Weinstein, T.R. Garner; Piperonyl butoxide enhances the bioconcentration and photoinduced toxicity of fluoranthene and benzo[a]pyrene to larvae of the grass shrimp (Palaemonetes pugio); Aquatic Toxicology, 87, 1, 28-36. (2008)
12 L.R. Bordajandi, G. Gomez, M.A. Fernandez; Study on PCBs, PCDD/Fs, organochlorine pesticides, heavy metals and arsenic content in freshwater fish species from the River Turia (Spain); Chemosphere, 53, 2; 163-171. (2003)

45
[13] Buchheit, Witzenbaker, Wotschokowsky; Junker; International Laboratory news.
(1988) [14] E.D. Goldeberg; Black carbon in the environment; p.17 John Wiley & sons Inc., New
York (1985) [15] A.D. Skoog; J.L. Leary; Chimica Analitica Strumentale; EdiSES srl, Napoli. (1995) [16] A.D. Skoog, D.M. West, F.J. Holler; Fundamentals of Analytical Chemistry;
Saunders College Publ; Int. Ed., New York. (1988) [17] M. Taramasso; Gas Cromatografia; Franco Angeli Editore. (1966) [18] L.S. Ettre; Journal of Chromatography, 4, 166-169. (1960) [19] D.M. Ottenstein; Journal of Chromatography, 1, 11. (1963)

46
p a r t e s p e r i m e n t a l e

47
Abstract
The presence of toxic substances in food is a current problem of great concern and
of social influence, both for nature and human beings. Over the years many new
substances present in the environment, derived from industrial processes and from
natural sources, have been studied, characterized and the majority of them are
listed as toxic compounds. Some of them are considered particularly dangerous,
hence the need of monitoring them with the aim of reducing their concentration in
the environment. In this work, two compounds classified as potential carcinogens
have been analysed: phthalates in the wines and acrylamide in the cooked food.
The study on phthalates has been developed and validated using a method of
enrichment and preconcentration, with solid-phase extraction (SPE) system and
using like adsorbent phase “carbobraph 1”. Analyses of phthalates were
conducted using chromatographic techniques like Gas Chromatography-Flame
Ionization Detector (GC-FID) systems and compounds confirmed by Gas
Chromatography-Ion Trap Mass Spectrometry (IT-GC/MS) method. The
determination of acrylamide was carried out by technique of derivatization with
halogenated agent like trifluoro-acetic anhydride (TFAA). The presence of
halogens makes the derivative of acrylamide identifiable with ECD detector. Then
the derivate of acrylamide was studied and characterized with IT-GC/MS system.
It was necessary the use of IT-GC/MS system to increase the sensitivity of the
method and to confirm the compounds. Furthermore both methods are simple,
reliable, reproducible and not expensive.

48
FTALATI IN MATRICI IDROALCOLICHE:
PRECONCENTRAZIONE SU “CARBOGRAPH 1” ED
ANALISI MEDIANTE GC-FID E GC-MS
INTRODUZIONE
Durante il mio dottorato di ricerca ho realizzato e messo a punto uno studio sulla
determinazione qualitativa e quantitativa di ftalati presenti in soluzioni
idroalcoliche mediante una tecnica di preconcentrazione in fase solida (SPE). Ho
scelto questa classe di composti perché loro sono molto utilizzati nella
fabbricazione delle plastiche, soprattutto nel polietilene tereftalato (PET), e sono
stati dichiarati probabili cancerogeni se presi in quantità elevate [1-2-3]. In
letteratura sono presenti degli elaborati che trattano gli ftalati in altre matrici
alimentari e non, ad esempio nell’acqua [4-5], nel latte [6], nei giocattoli per
l’infanzia [7], eccetera.
Sono state scelte matrici idroalcoliche, in particolar modo vini, perché la
letteratura è carente nell’argomento, infatti allo stato attuale esistono pochissimi
lavori a riguardo, uno di questi tratta il problema utilizzando la tecnica SPME [8].
Ho pensato che queste sostanze in qualche modo potessero diffondere all’interno
della soluzione che contengono, causando probabili intossicazioni.
La necessità di preconcentrare tali sostanze nasce da un bisogno strumentale,
infatti gli strumenti attuali non sono in grado di determinare concentrazioni così
basse quali quelle degli ftalati o di altri microinquinanti presenti in natura. Si parla
infatti ppm, ppb o concentrazioni inferiori.

49
Innanzi tutto, ho studiato le interazioni che si instaurano fra gli analiti scelti e la
fase adsorbente in condizioni statiche, con la relativa costruzione delle isoterme di
adsorbimento, ed in condizioni dinamiche, con la relativa costruzione della curva
di breakthrough. Una volta studiata la ripartizione, il KD, fra gli analiti e la fase
adsorbente ed il volume di rottura sono andato alla ricerca del solvente di
estrazione più idoneo, il solvente quindi che con la minima quantità recupera, per
desorbimento, quantitativamente gli analiti intrappolati.
Infine ho applicato il metodo a campioni reali di vino bianco e di vino rosso. Ho
fatto questo ultimo step per vedere innanzi tutto un applicazione del metodo
studiato su campioni reali, ed inoltre per vedere se la matrice reale, una matrice
complessa, in qualche modo va ad influenzare i risultati ottenuti in condizioni
standard.
Ho fatto uno studio preliminare utilizzando la tecnica GC-FID, cioè una tecnica
che abbina la gas cromatografia ad un rilevatore universale. Per migliorare la
robustezza e la sensibilità del metodo ho concluso lo studio utilizzando la tecnica
GC-MS, cioè una gas cromatografia abbinata alla spettrometria di massa. Diventa
necessario utilizzare uno spettrometro di massa per una questione di sensibilità e
precisione, infatti utilizzando questa tecnica si abbassa la sensibilità rispetto al
FID anche di ordini di grandezza di circa 105-106 volte. L’analisi allo spettrometro
di massa mi ha permesso di ricavare e generalizzare sperimentalmente tutte le
frammentazioni degli ftalati presi in esame. Inoltre, utilizzando questa tecnica, ho
sensibilmente abbassato i limiti di rilevabilità strumentali (LOD), effettuando
analisi in Full Scan, in SIM (Selected Ion Monitoring) ed in MS/MS.

50
OBIETTIVO
Scopo di questo lavoro è lo studio di un adsorbente apolare, il “carbograph 1”, un
carbone semi-grafitato, per la preconcentrazione di ftalati da soluzioni
idroalcoliche. La scelta è caduta su questo adsorbente perché nel nostro
laboratorio da tempo sono studiati adsorbenti polari (-CN, -diol, -NH2) e non
polari (-C8, -C18, XAD-2) per analizzare PCB, pesticidi clorurati, diossine,
furani, composti aromatici solforati e composti ritenuti tossici o potenzialmente
tossici [9-10-11-12-13-14-15-16-17-18].
Per ottimizzare le condizioni sperimentali per l’ottenimento del massimo
rendimento sono state studiate le interazioni tra l’adsorbente e i soluti esaminati in
soluzione idroalcolica (10% v/v di etanolo), allo scopo di simulare l’ambiente del
vino. Come anticipato nella parte introduttiva, a tale scopo sono state costruite
sperimentalmente le Isoterme di Adsorbimento e le Curve di Breakthrough in
condizioni controllate di flusso, temperatura, pH e salinità. Successivamente sono
stati esaminati diversi solventi per ottenere il desorbimento degli analiti nel minor
volume possibile.
Il metodo analitico studiato e messo a punto è stato applicato per l’analisi di
alcuni campioni di vino rosso e di vino bianco.

51
MATERIALI E METODI
Materiali
La fase adsorbente scelta per questo studio è un carbone semi-grafitato, il
“Carbograph 1” (80-100 Mesh, area superficiale 70, 80 m2/g).
Tutte le analisi sono state effettuate con l’aggiunta di uno standard interno:
antracene 2,4 mg/mL disciolto in acetone.
Sono state analizzate sostanze appartenenti alla famiglia degli ftalati, esteri
dell’acido ftalico, peculiari per il loro largo utilizzo come agenti plastificanti.
Dovendo scegliere fra una vasta gamma di ftalati, ho scelto alcuni standard
rappresentativi l’intera classe. Ho cercato quindi, in base alle caratteristiche
strutturali e alle limitazioni strumentali, di scegliere nel modo migliore i miei
standard in base ai gruppi sostituenti la molecola di base.
Si ricorda la struttura generale di uno ftalato.
O
O
O
O
R1
R2
La denominazione e le rispettive proprietà chimico fisiche dipendono dal tipo di
sostituente che prende il posto di R1 e di R2. In base a queste considerazioni ed in
base alle disponibilità di standard presenti in laboratorio, ho deciso di operare con
i seguenti ftalati:

52
Dimetyl phthalate, DMP
Dietyl phthalate, DEP
Dibutyl phthalate, DBP
Isobutyl-cicloesil phthalate, iBcEP
Benzil-butyl phthalate, BBP
Bis-(2etyl-exil) phthalate, DEHP
In questo modo sono riuscito a prendere tre (DMP, DEP, DBP) ftalati con
sostituenti alifatici a catena crescente, due (iBcEP, BBP) ftalati con sostituenti
differenti ed uno (DEHP) con sostituenti ramificati.
Le caratteristiche strutturali e chimiche degli analiti studiati sono riportate nella
tabella successiva.
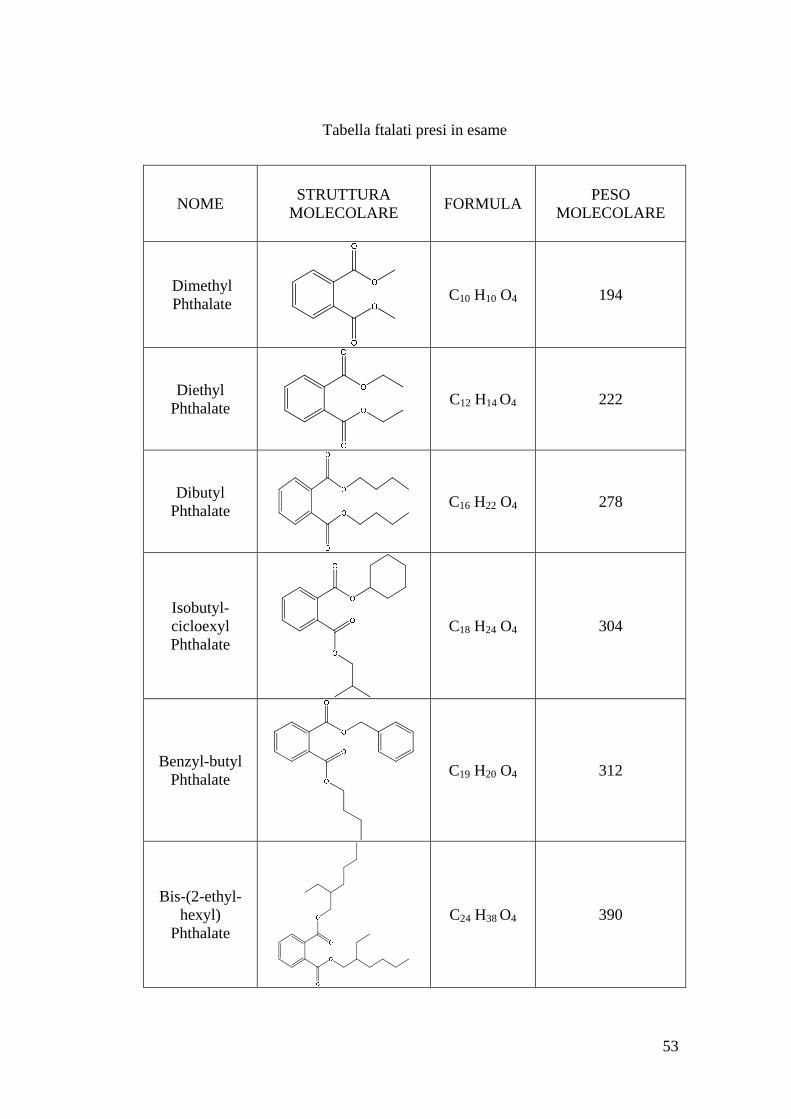
53
Tabella ftalati presi in esame
NOME STRUTTURA MOLECOLARE FORMULA PESO
MOLECOLARE
Dimethyl Phthalate C10 H10 O4 194
Diethyl Phthalate C12 H14 O4 222
Dibutyl Phthalate C16 H22 O4 278
Isobutyl-cicloexyl Phthalate
C18 H24 O4 304
Benzyl-butyl Phthalate C19 H20 O4 312
Bis-(2-ethyl-hexyl)
Phthalate C24 H38 O4 390

54
Metodi
Per le analisi è stato impiegato un gascromatografo della DANI, modello 86.10
HT, equipaggiato con un iniettore PTV (Programmed Temperature Vaporizer),
una colonna capillare ed un rivelatore a ionizzazione di fiamma (FID).
Le programmate del PTV e del forno sono schematizzate a fondo pagina.
Il PTV e regolato in modo da aprire lo spillo dopo 1.30 min. La temperatura di
esercizio del rilevatore è fissata a 300 °C. La colonna è in silice fusa con fase
chimicamente legata (SE 54 - 5% fenile, 95 % dimetilsilossano), con L=30 m, I.D.
(diametro interno) 250 µm e df (spessore del film) di 0.25 µm, fornita dalla
Teknokroma.
La fase mobile è l’idrogeno (prodotto con un generatore di idrogeno della Packard
modello 9400) con una velocità di flusso circa 1 mL/min (rilevato
sperimentalmente). Lo strumento è interfacciato a un PC con il software di
elaborazione specifico per gas cromatografia “Clarity”.
Inoltre, tutti gli esperimenti sono stati svolti a salinità controllata, utilizzando il
cloruro di sodio all’interno delle soluzioni. Questo sale è di fondamentale
importanza, infatti essendo un sale completamente solubile in acqua, modifica la
forza ionica della soluzione, diminuendo la solubilità degli ftalati (Salting-Out
Effect) [19]
condizioni operative PTV condizioni operative del forno
800 °C/min
60°C
280°C 1’
30’’ 1’
10 °C/min
50°C
280°C4’

55
ISOTERME DI ADSORBIMENTO
Preparazione delle soluzioni
Come prima cosa è stata preparata una soluzione contenente i 6 ftalati ad una
concentrazione di 100 µg/mL. Ho usato tale soluzione come punto di partenza per
tutte le prove effettuate.
Per la costruzione delle isoterme di adsorbimento sono state preparate differenti
soluzioni a concentrazione nota e crescente dei composti. Queste concentrazioni
variano in un range compreso tra 10 e 200 ng/mL.
Per ogni concentrazione da analizzare, sono stati preparati 250 mL di soluzione, di
cui 100 mL costituiscono il riferimento (testa), altri 100 mL sono impiegati per gli
esperimenti (coda).
Tutte le soluzioni coda a diversa concentrazione sono state analizzate in beute da
100 mL aggiungendo in ognuna 100 mg di fase adsorbente. In questo modo le
soluzioni contenenti gli analiti entrano in contatto con la fase adsorbente.
L’adsorbimento delle sostanze analizzate è avvenuto in condizioni statiche,
lasciando le soluzioni in contatto con la fase adsorbente per 24 ore, a temperatura
ambiente (25 °C), a pH e salinità controllati. Una precauzione è stata quella di
tenere le beute al buio per evitare che all’interno potessero avvenire reazioni foto-
degradative.
Estrazione delle soluzioni
Dopo aver filtrato le soluzioni in modo da dividere la fase solida dalla fase
liquida, ho estratto gli analiti con esano, un solvente organico apolare immiscibile
con l’acqua. La fase d’estrazione è una delle più delicate dal punto di vista

56
operativo, poiché comporta evaporazione del solvente e a volte perdita
dell’analita. Per l’estrazione si è usato un imbuto separatore da 250 mL. In questo
imbuto sono stati versati i 100 mL di soluzione precedentemente ottenuti dalla
filtrazione. L’estrazione dei composti è stata effettuata utilizzando aliquote di 4
mL di esano, ripetendo l’operazione per 3 volte (3x4mL). Il volume raccolto di
solvente è stato concentrato sotto flusso costante di azoto fino ad un volume di
circa 100 µL.
Per avere un riscontro pratico sulla quantità di analiti adsorbiti dal carbograph 1,
oltre a preparare queste soluzioni, sono state analizzate anche le soluzioni testa.
Le soluzioni testa rispecchiano le rispettive code sotto il profilo della
concentrazione. La differenza sta solo nel fatto che la testa non subisce alcuna
manipolazione ed è quindi usata come riferimento. Le teste sono infatti preparate
prelevando 100 mL da ognuna delle soluzioni di partenza, ed estratte direttamente
utilizzando la stessa metodica descritta prima, in altre parole non subiscono
l’influenza della fase adsorbente.
Le soluzioni di riferimento sono molto importanti; infatti, dall’analisi al GC della
testa e della coda possiamo risalire per differenza alla quantità di analiti adsorbiti
dalla fase adsorbente.
Preparazione dello standard interno
Lo standard interno (SI) è un composto aggiunto in quantità nota e costante a
tutte le soluzioni da analizzare. Lo SI solitamente viene aggiunto alla soluzione
prima di effettuare l’iniezione al GC, quando il campione non subisce ulteriori
trattamenti. La funzione dello SI all’interno della soluzione è quella di dare un

57
riferimento costante, dando così la possibilità di normalizzare tutti i dati. In questo
caso lo SI è stato aggiunto dopo aver concentrato le soluzioni a circa 100 µL.
La condizione necessaria per l’utilizzo dello standard interno è che il suo picco
deve essere risolto e paragonabile con quello degli analiti presenti in soluzione.
Prima di mettere, quindi, lo S.I. all’interno delle soluzioni sono state fatte delle
prove sperimentali in modo tale da ottimizzare la sua concentrazione.
Costruzione delle isoterme di adsorbimento
Una volta concentrate le soluzioni a un volume di circa 100 µL sono state eseguite
le analisi gas cromatografiche. Delle varie concentrazioni prese in esame, è stata
analizzata sia la testa sia la coda. Tutte le analisi sono state eseguite rapportando
le aree dei picchi degli analiti con l’area del picco dello standard interno, relativo
alla stessa soluzione.
Per calcolare le quantità di ftalati presenti nelle code si è usata la semplice
proporzione:
AreaTesta : QunatitàTesta = AreaCoda : QuantitàCoda
Per cercare la quantità presente nella coda, quindi, si va a calcolare:
QuantitàCoda = (AreaCoda / AreaTesta) x QuantitàTesta
I parametri presenti a destra dell’equazione sono tutti noti: le due aree sono quelle
calcolate, l’altro parametro rappresenta la quantità di analita presente in 100 mL di
soluzione.
Una volta calcolate le quantità degli analiti presenti nelle code, conoscendo quelle
presenti nelle teste, per differenza è calcolata la quantità di analiti trattenuti dalla
fase adsorbente.
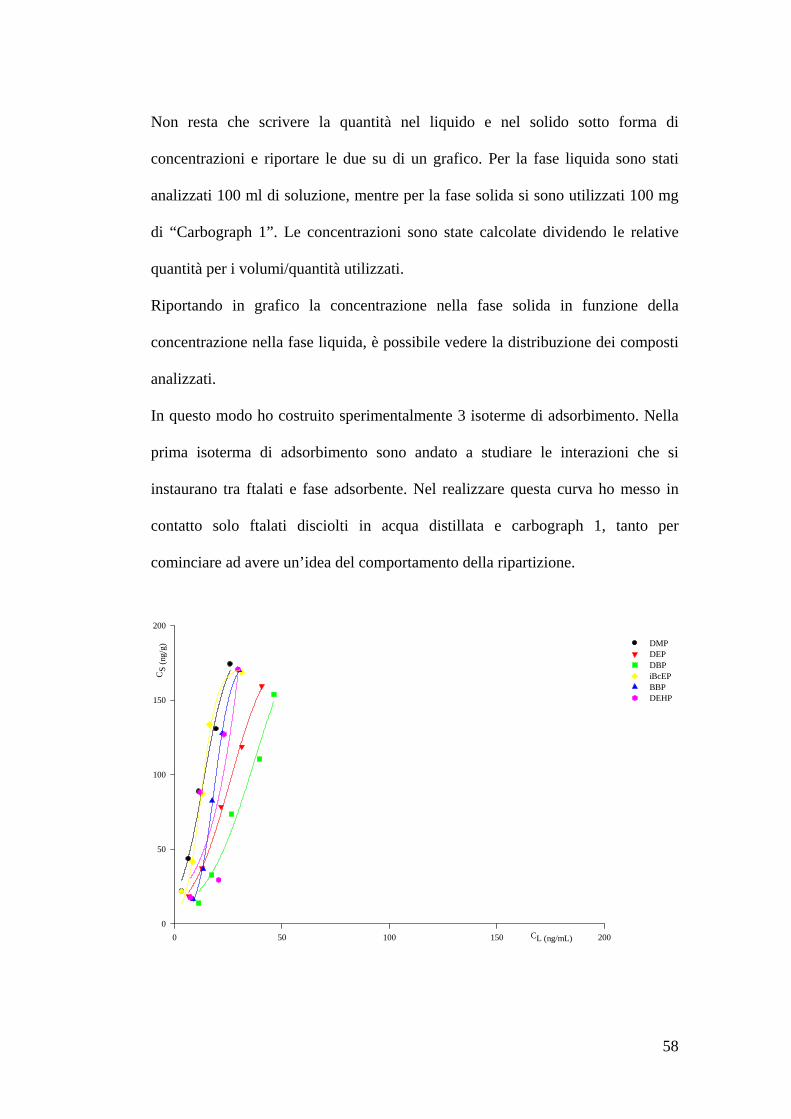
58
Non resta che scrivere la quantità nel liquido e nel solido sotto forma di
concentrazioni e riportare le due su di un grafico. Per la fase liquida sono stati
analizzati 100 ml di soluzione, mentre per la fase solida si sono utilizzati 100 mg
di “Carbograph 1”. Le concentrazioni sono state calcolate dividendo le relative
quantità per i volumi/quantità utilizzati.
Riportando in grafico la concentrazione nella fase solida in funzione della
concentrazione nella fase liquida, è possibile vedere la distribuzione dei composti
analizzati.
In questo modo ho costruito sperimentalmente 3 isoterme di adsorbimento. Nella
prima isoterma di adsorbimento sono andato a studiare le interazioni che si
instaurano tra ftalati e fase adsorbente. Nel realizzare questa curva ho messo in
contatto solo ftalati disciolti in acqua distillata e carbograph 1, tanto per
cominciare ad avere un’idea del comportamento della ripartizione.
CL (ng/mL)0 50 100 150 200
CS
(ng/
g)
0
50
100
150
200
DMPDEPDBPiBcEPBBPDEHP
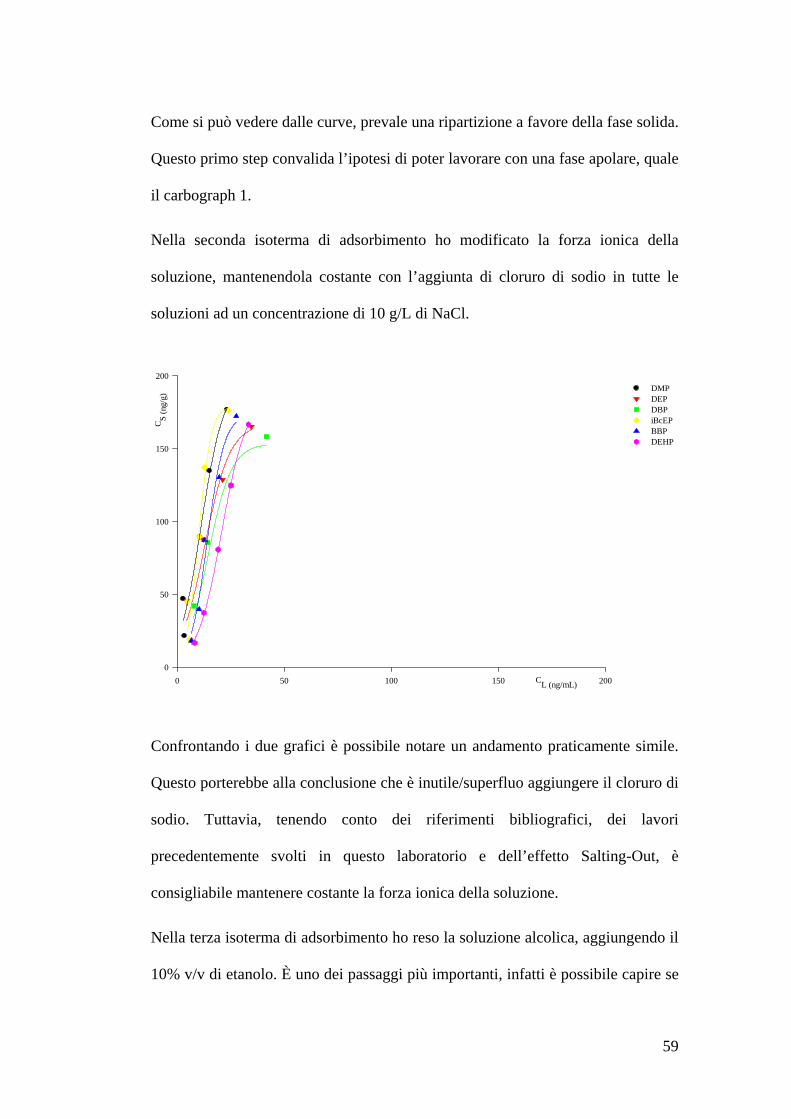
59
Come si può vedere dalle curve, prevale una ripartizione a favore della fase solida.
Questo primo step convalida l’ipotesi di poter lavorare con una fase apolare, quale
il carbograph 1.
Nella seconda isoterma di adsorbimento ho modificato la forza ionica della
soluzione, mantenendola costante con l’aggiunta di cloruro di sodio in tutte le
soluzioni ad un concentrazione di 10 g/L di NaCl.
CL (ng/mL)0 50 100 150 200
C S (n
g/g)
0
50
100
150
200DMPDEPDBPiBcEPBBPDEHP
Confrontando i due grafici è possibile notare un andamento praticamente simile.
Questo porterebbe alla conclusione che è inutile/superfluo aggiungere il cloruro di
sodio. Tuttavia, tenendo conto dei riferimenti bibliografici, dei lavori
precedentemente svolti in questo laboratorio e dell’effetto Salting-Out, è
consigliabile mantenere costante la forza ionica della soluzione.
Nella terza isoterma di adsorbimento ho reso la soluzione alcolica, aggiungendo il
10% v/v di etanolo. È uno dei passaggi più importanti, infatti è possibile capire se
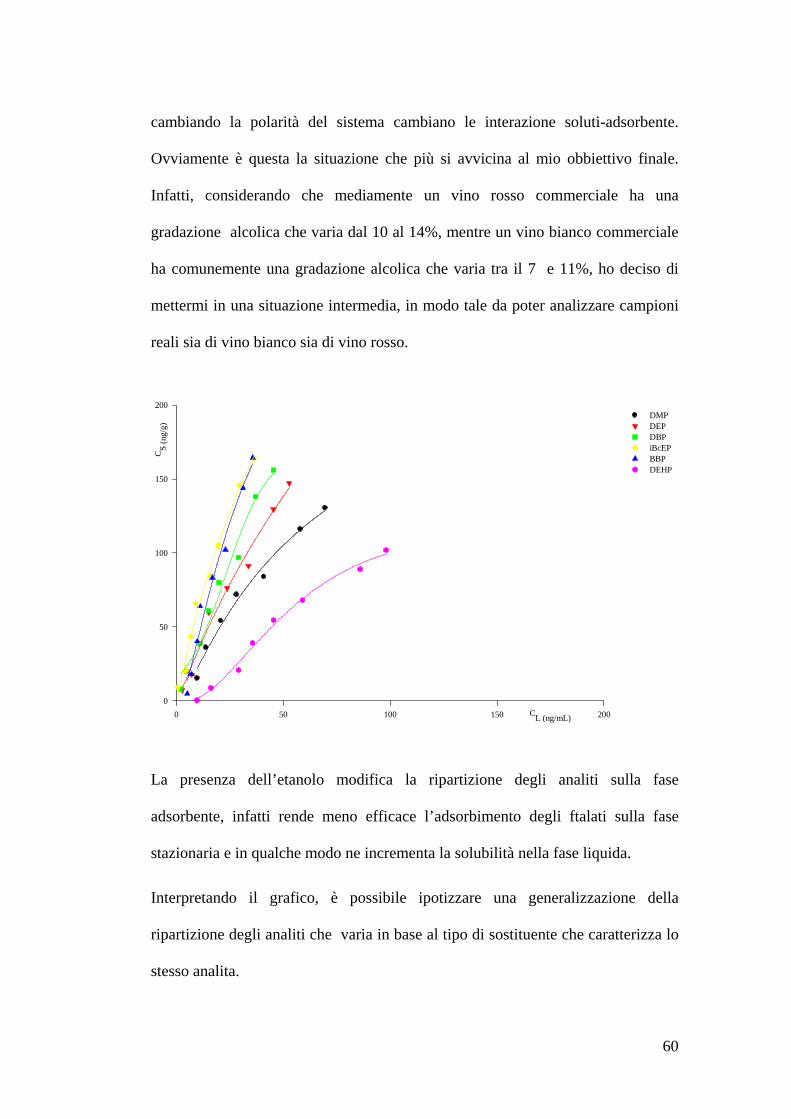
60
cambiando la polarità del sistema cambiano le interazione soluti-adsorbente.
Ovviamente è questa la situazione che più si avvicina al mio obbiettivo finale.
Infatti, considerando che mediamente un vino rosso commerciale ha una
gradazione alcolica che varia dal 10 al 14%, mentre un vino bianco commerciale
ha comunemente una gradazione alcolica che varia tra il 7 e 11%, ho deciso di
mettermi in una situazione intermedia, in modo tale da poter analizzare campioni
reali sia di vino bianco sia di vino rosso.
CL (ng/mL)0 50 100 150 200
C S (n
g/g)
0
50
100
150
200DMPDEPDBPiBcEPBBPDEHP
La presenza dell’etanolo modifica la ripartizione degli analiti sulla fase
adsorbente, infatti rende meno efficace l’adsorbimento degli ftalati sulla fase
stazionaria e in qualche modo ne incrementa la solubilità nella fase liquida.
Interpretando il grafico, è possibile ipotizzare una generalizzazione della
ripartizione degli analiti che varia in base al tipo di sostituente che caratterizza lo
stesso analita.

61
Infatti si può notare che il DBP presenta una ripartizione più spostata verso la fase
solida rispetto al DEP, che a sua volta presenta una ripartizione più spostata verso
la fase solida del DMP. Questa prima osservazione ha permesso di arrivare alla
conclusione che la ripartizione dei composti verso il carbograph 1 che hanno in
sostituzione catene alifatiche aumenta all’aumentare del numero di atomi di
carbonio che costituiscono la catena alifatica.
È possibile notare, inoltre, che i composti con gruppi differenti, quali iBcEP e il
BBP, presentano una ripartizione prevalentemente favorevole alla fase adsorbente.
Un comportamento anomalo è seguito dal DEHP, che presenta una ripartizione
quasi al 50% fra la fase adsorbente e la fase liquida.
Lo studio delle isoterme di adsorbimento evidenzia che i soluti esaminati, nelle
condizioni sperimentali adottate, hanno una particolare affinità verso la fase
stazionaria.
Questo risultato pone il “Carbograph 1” come un buon adsorbente per
l’arricchimento degli ftalati da soluzioni idroalcoliche.

62
VOLUMI DI BREAKTHROUGH
Preparazione delle soluzioni e della fase adsorbente
La fase adsorbente è stata impaccata in una cartuccia in polipropilene, utilizzando
setti porosi specifici di bloccaggio. Tutte le cartucce sono state costruite
manualmente, avendo disponibile la fase adsorbente sotto forma di granuli
finemente macinati. Quindi, prima di ogni prova analitica, ogni cartuccia è stata
lavata, impaccata, attivata e condizionata utilizzando opportuni solventi.
Ho preparato una soluzione idroalcolica al 10% v/v di etanolo, 10 g/L di NaCl,
contenente tutti gli ftalati, ognuno ad una concentrazione di 50 ng/mL.
Ho fatto passare questa soluzione attraverso la cartuccia raccogliendo
consecutivamente frazioni di 100 mL (code), in condizioni dinamiche controllate,
ad un flusso di 6-8 mL/mim.
Estrazione dei soluti e concentrazione delle soluzioni
Come fatto per la costruzione delle curve di isoterme di adsorbimento, tutte le
soluzioni coda di ftalati sono statte estratte e concentrate (sotto flusso costante di
azoto) a 100 µl con n-esano.
Anche in questo caso per avere un riscontro sulla quantità di ftalati adsorbiti dalla
fase adsorbente, è stata preparata la soluzione testa, soluzione di riferimento. Le
teste sono state preparate direttamente dalle soluzioni iniziali preparate per le
curve di breakthrough.

63
Anche in questo caso la testa è molto importante. Infatti, dall’analisi al GC della
testa e della coda possiamo risalire al volume di soluzione che può passare
attraverso la cartuccia affinché tutti gli analiti siano quantitativamente adsorbiti.
Costruzione delle curve di breakthrough
Sono state analizzate sia la testa sia le code per via gas cromatografica. Tutti i
calcoli sono stati eseguiti rapportando le aree dei picchi degli analiti con l’area del
picco dello standard interno, relativo alla stessa soluzione.
In seguito sono state calcolate le concentrazioni degli analiti presenti in ogni coda
(Ci) e la concentrazione degli analiti presenti nella testa (C0).
Infine, riportando su un grafico la concentrazione riscontrata in percentuale nelle
code in funzione dei millilitri di soluzione passati attraverso la cartuccia,
costruiamo le curve di breakthrough dei composti analizzati.
Ho costruito 2 curve di breakthrough, in condizione dinamiche, lavorando a
temperatura ambiente (circa 25 °C), a flusso costante (controllato con una pompa
da vuoto), a forza ionica e a polarità costante, facendo passare e raccogliendo in
continuo frazioni da 100 mL di campione (code). La prima curva di breakthrough
è stata costruita impaccando una cartuccia di polipropilene con 100 mg di fase
adsorbente.
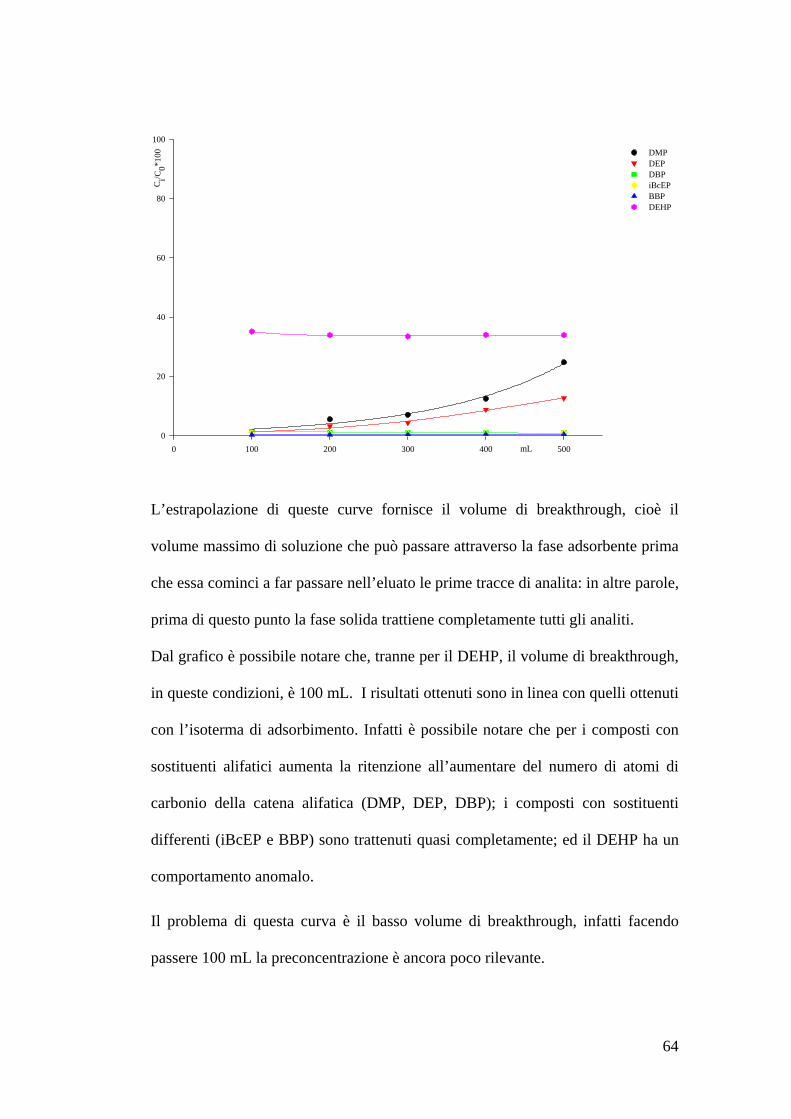
64
mL0 100 200 300 400 500
Ci/C
0*100
0
20
40
60
80
100DMP DEP DBP iBcEP BBP DEHP
L’estrapolazione di queste curve fornisce il volume di breakthrough, cioè il
volume massimo di soluzione che può passare attraverso la fase adsorbente prima
che essa cominci a far passare nell’eluato le prime tracce di analita: in altre parole,
prima di questo punto la fase solida trattiene completamente tutti gli analiti.
Dal grafico è possibile notare che, tranne per il DEHP, il volume di breakthrough,
in queste condizioni, è 100 mL. I risultati ottenuti sono in linea con quelli ottenuti
con l’isoterma di adsorbimento. Infatti è possibile notare che per i composti con
sostituenti alifatici aumenta la ritenzione all’aumentare del numero di atomi di
carbonio della catena alifatica (DMP, DEP, DBP); i composti con sostituenti
differenti (iBcEP e BBP) sono trattenuti quasi completamente; ed il DEHP ha un
comportamento anomalo.
Il problema di questa curva è il basso volume di breakthrough, infatti facendo
passere 100 mL la preconcentrazione è ancora poco rilevante.
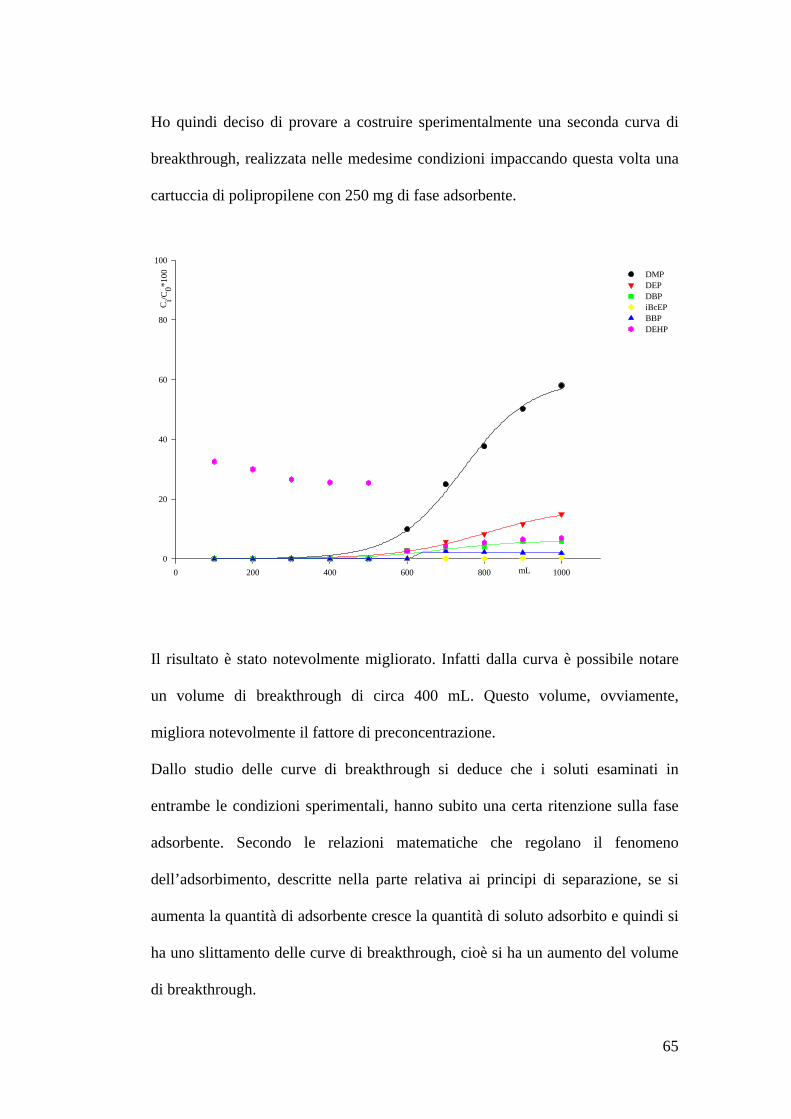
65
Ho quindi deciso di provare a costruire sperimentalmente una seconda curva di
breakthrough, realizzata nelle medesime condizioni impaccando questa volta una
cartuccia di polipropilene con 250 mg di fase adsorbente.
mL0 200 400 600 800 1000
Ci/C
0*100
0
20
40
60
80
100DMP DEP DBP iBcEP BBP DEHP
Il risultato è stato notevolmente migliorato. Infatti dalla curva è possibile notare
un volume di breakthrough di circa 400 mL. Questo volume, ovviamente,
migliora notevolmente il fattore di preconcentrazione.
Dallo studio delle curve di breakthrough si deduce che i soluti esaminati in
entrambe le condizioni sperimentali, hanno subito una certa ritenzione sulla fase
adsorbente. Secondo le relazioni matematiche che regolano il fenomeno
dell’adsorbimento, descritte nella parte relativa ai principi di separazione, se si
aumenta la quantità di adsorbente cresce la quantità di soluto adsorbito e quindi si
ha uno slittamento delle curve di breakthrough, cioè si ha un aumento del volume
di breakthrough.

66
Il DHEP nei primi 500 mL di eluato viene perso costantemente per circa il 30%,
per poi essere trattenuto in quantità apprezzabili nel prosieguo dell’eluizione
(fenomeno dell’auto-adsorbimento). La spiegazione del fenomeno è che tale
composto si leghi ai siti attivi della fase adsorbente, formando uno strato liquido
monostadio (mono-layer) sul quale si solubilizza e quindi aumenta l’adsorbimento
del DHEP. In altre parole esso diventa, con il passare della soluzione attraverso il
carbone semi-grafitato, fase stazionaria di se stesso, di conseguenza aumenta la
ritenzione in modo significativo.
Anche in questo caso, come spiegato precedentemente nelle curve di breakthrough
ottenute con 100 mg di fase stazionaria, si vede come l’affinità dei soluti alla fase
stazionaria è in completo accordo con le deduzione fatte per le isoterme di
adsorbimento.
Il volume di breakthrough estrapolato per i composti analizzati è di circa 400 mL
(fatta eccezione per il DEHP) se si usano 250 mg di adsorbente.
Il volume di breakthrough così calcolato è un risultato teorico, infatti
nell’applicazione del metodo solitamente si lavora ad un volume di circa il
60-70% del volume calcolato. Questa precauzione serve a dare una certa
tranquillità nel trattenere quantitativamente tutti gli analiti presi in esame, senza
perdere nulla. Per questo motivo, per fare l’arricchimento degli ftaltati su una
cartuccia in polipropilene impaccata con 250 mg di carbograph 1 ho deciso di
utilizzare un volume di soluzione pari a 250 mL.
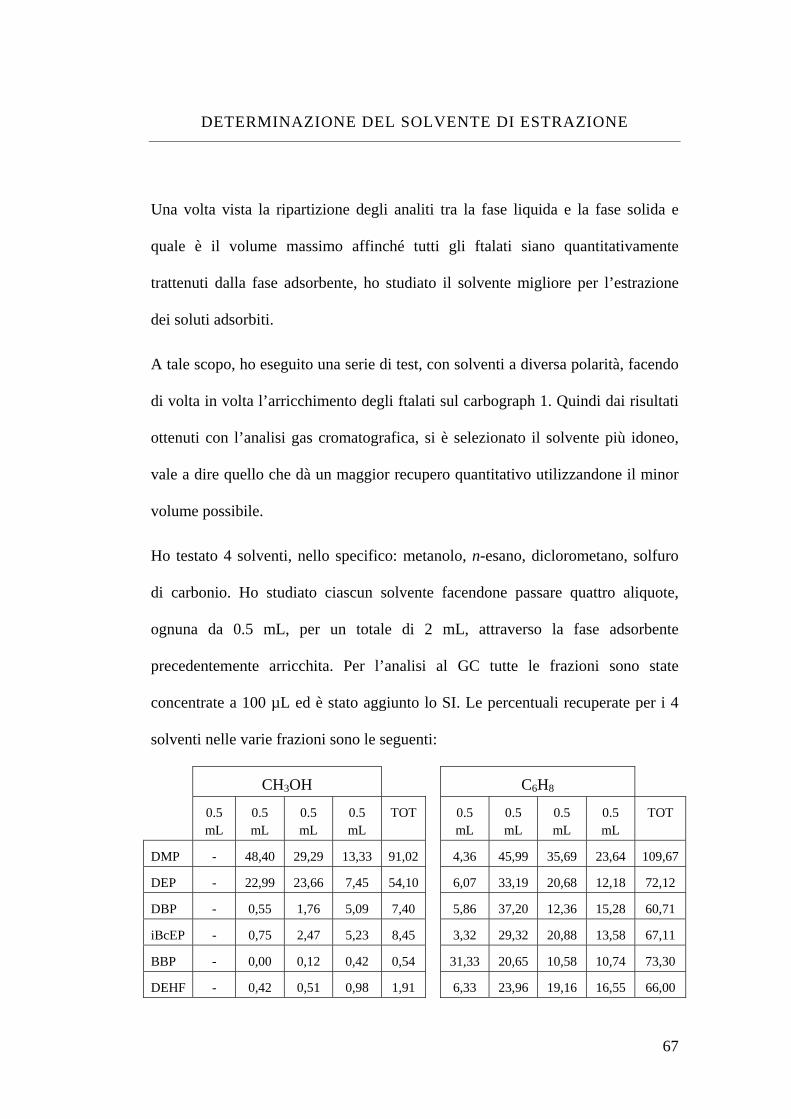
67
DETERMINAZIONE DEL SOLVENTE DI ESTRAZIONE
Una volta vista la ripartizione degli analiti tra la fase liquida e la fase solida e
quale è il volume massimo affinché tutti gli ftalati siano quantitativamente
trattenuti dalla fase adsorbente, ho studiato il solvente migliore per l’estrazione
dei soluti adsorbiti.
A tale scopo, ho eseguito una serie di test, con solventi a diversa polarità, facendo
di volta in volta l’arricchimento degli ftalati sul carbograph 1. Quindi dai risultati
ottenuti con l’analisi gas cromatografica, si è selezionato il solvente più idoneo,
vale a dire quello che dà un maggior recupero quantitativo utilizzandone il minor
volume possibile.
Ho testato 4 solventi, nello specifico: metanolo, n-esano, diclorometano, solfuro
di carbonio. Ho studiato ciascun solvente facendone passare quattro aliquote,
ognuna da 0.5 mL, per un totale di 2 mL, attraverso la fase adsorbente
precedentemente arricchita. Per l’analisi al GC tutte le frazioni sono state
concentrate a 100 µL ed è stato aggiunto lo SI. Le percentuali recuperate per i 4
solventi nelle varie frazioni sono le seguenti:
CH3OH C6H8
0.5 mL
0.5 mL
0.5 mL
0.5 mL
TOT 0.5 mL
0.5 mL
0.5 mL
0.5 mL
TOT
DMP - 48,40 29,29 13,33 91,02 4,36 45,99 35,69 23,64 109,67
DEP - 22,99 23,66 7,45 54,10 6,07 33,19 20,68 12,18 72,12
DBP - 0,55 1,76 5,09 7,40 5,86 37,20 12,36 15,28 60,71
iBcEP - 0,75 2,47 5,23 8,45 3,32 29,32 20,88 13,58 67,11
BBP - 0,00 0,12 0,42 0,54 31,33 20,65 10,58 10,74 73,30
DEHF - 0,42 0,51 0,98 1,91 6,33 23,96 19,16 16,55 66,00
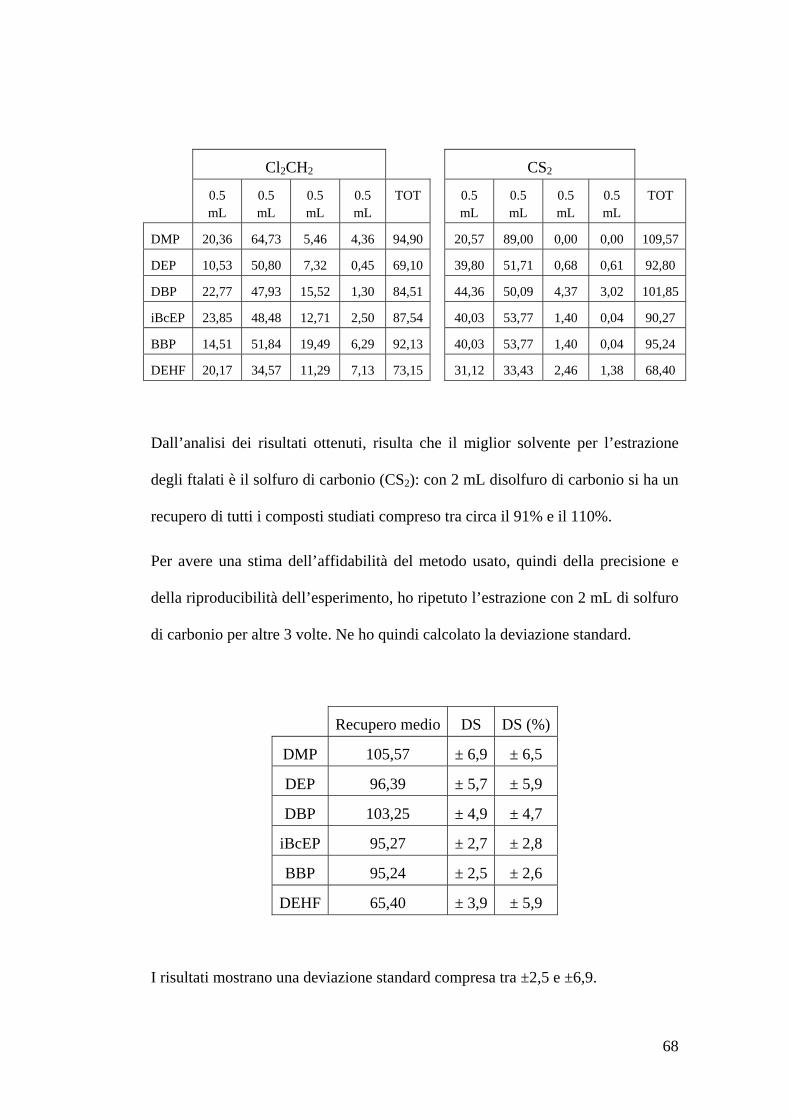
68
Cl2CH2 CS2
0.5 mL
0.5 mL
0.5 mL
0.5 mL
TOT 0.5 mL
0.5 mL
0.5 mL
0.5 mL
TOT
DMP 20,36 64,73 5,46 4,36 94,90 20,57 89,00 0,00 0,00 109,57
DEP 10,53 50,80 7,32 0,45 69,10 39,80 51,71 0,68 0,61 92,80
DBP 22,77 47,93 15,52 1,30 84,51 44,36 50,09 4,37 3,02 101,85
iBcEP 23,85 48,48 12,71 2,50 87,54 40,03 53,77 1,40 0,04 90,27
BBP 14,51 51,84 19,49 6,29 92,13 40,03 53,77 1,40 0,04 95,24
DEHF 20,17 34,57 11,29 7,13 73,15 31,12 33,43 2,46 1,38 68,40
Dall’analisi dei risultati ottenuti, risulta che il miglior solvente per l’estrazione
degli ftalati è il solfuro di carbonio (CS2): con 2 mL disolfuro di carbonio si ha un
recupero di tutti i composti studiati compreso tra circa il 91% e il 110%.
Per avere una stima dell’affidabilità del metodo usato, quindi della precisione e
della riproducibilità dell’esperimento, ho ripetuto l’estrazione con 2 mL di solfuro
di carbonio per altre 3 volte. Ne ho quindi calcolato la deviazione standard.
Recupero medio DS DS (%)
DMP 105,57 ± 6,9 ± 6,5
DEP 96,39 ± 5,7 ± 5,9
DBP 103,25 ± 4,9 ± 4,7
iBcEP 95,27 ± 2,7 ± 2,8
BBP 95,24 ± 2,5 ± 2,6
DEHF 65,40 ± 3,9 ± 5,9
I risultati mostrano una deviazione standard compresa tra ±2,5 e ±6,9.
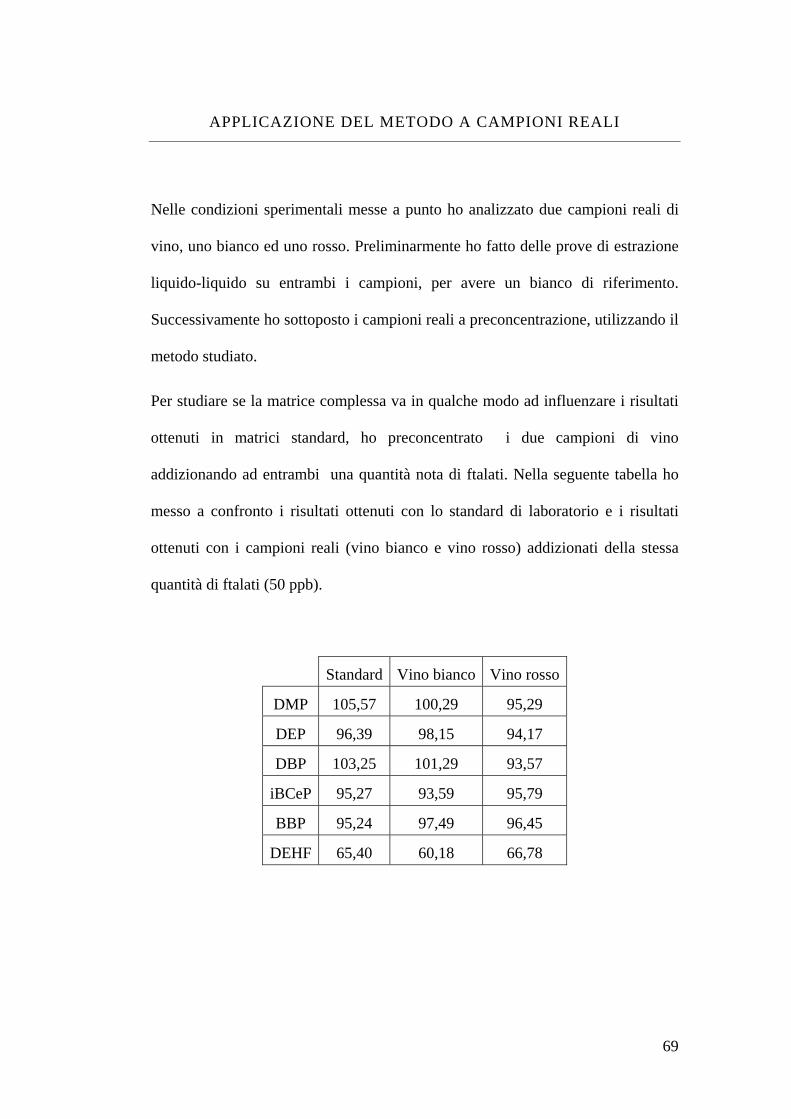
69
APPLICAZIONE DEL METODO A CAMPIONI REALI
Nelle condizioni sperimentali messe a punto ho analizzato due campioni reali di
vino, uno bianco ed uno rosso. Preliminarmente ho fatto delle prove di estrazione
liquido-liquido su entrambi i campioni, per avere un bianco di riferimento.
Successivamente ho sottoposto i campioni reali a preconcentrazione, utilizzando il
metodo studiato.
Per studiare se la matrice complessa va in qualche modo ad influenzare i risultati
ottenuti in matrici standard, ho preconcentrato i due campioni di vino
addizionando ad entrambi una quantità nota di ftalati. Nella seguente tabella ho
messo a confronto i risultati ottenuti con lo standard di laboratorio e i risultati
ottenuti con i campioni reali (vino bianco e vino rosso) addizionati della stessa
quantità di ftalati (50 ppb).
Standard Vino bianco Vino rosso
DMP 105,57 100,29 95,29
DEP 96,39 98,15 94,17
DBP 103,25 101,29 93,57
iBCeP 95,27 93,59 95,79
BBP 95,24 97,49 96,45
DEHF 65,40 60,18 66,78

70
Una lettura della tabella ci dice che lo standard di laboratorio può essere
considerato come il campione reale, infatti se si considera l’errore (riportato
successivamente) i dati possono essere considerati statisticamente uguali.
Analizzando invece i campioni reali tal quali, sono stati trovati i seguenti
composti:
Concentrazione calcolata (ppb) ± DS
Vino bianco Vino rosso
DBP 40 ± 2,5 68 ± 4,3
BBP 17 ± 1,0 -
DEHP 9 ± 0,5 3 ± 0,8
I composti rinvenuti purtroppo sono quelli catalogati come i più tossici tra tutti gli
ftalati, ma questi inducono danni evidenti soltanto se somministrati in quantità
elevate. Per cui i risultati ottenuti in questa analisi non rappresentano un rischio
significativo per la salute.
I cromatogrammi relativi alle analisi svolte sul vino bianco sono nell’allegato 1,
mentre quelli relativi al vino rosso sono nell’allegato 2.

71
LOD E LOQ OTTENUTI IN GC-FID
Per dimostrare la sensibilità del metodo ho calcolato sperimentalmente il limite di
rilevabilità (calcolato considerando un picco con altezza almeno 3 volte la
deviazione standard del rumore di fondo) e il limite di quantificazione (calcolato
considerando un picco con altezza almeno 7 volte la deviazione standard del
rumore di fondo) per ogni composto analizzato.
Riporto i valori ottenti nella seguente tabella.
LOD* (3 volte il noise) LOQ* (7 volte il noise)
quantità
(ng) DS
DS ( % )
quantità
(ng) DS
DS ( % )
DMP 66 ± 4.2 ± 6.40 DMP 110 ± 3.0 ± 2.74 DEP 31 ± 1.8 ± 5.71 DEP 59 ± 1.9 ± 3.14 DBP 26 ± 1.2 ± 4.52 DBP 48 ± 1.0 ± 2.24
iBcEP 30 ± 1.5 ± 4.84 iBcEP 55 ± 2.1 ± 3.76 BBP 62 ± 4.9 ± 7.09 BBP 95 ± 2.2 ± 2.29
DEHP 23 ± 1.2 ± 5.14 DEHP 45 ± 1.6 ± 3.45
* valore ottenuto sulla media di tre determinazioni
Sia i LOD sia i LOQ si dimostrano essere quantità accettabili per un rivelatore
universale quale il FID. Per scendere ulteriormente con le concentrazioni
occorrono tecniche di spettrometria di massa, come descritto successivamente.
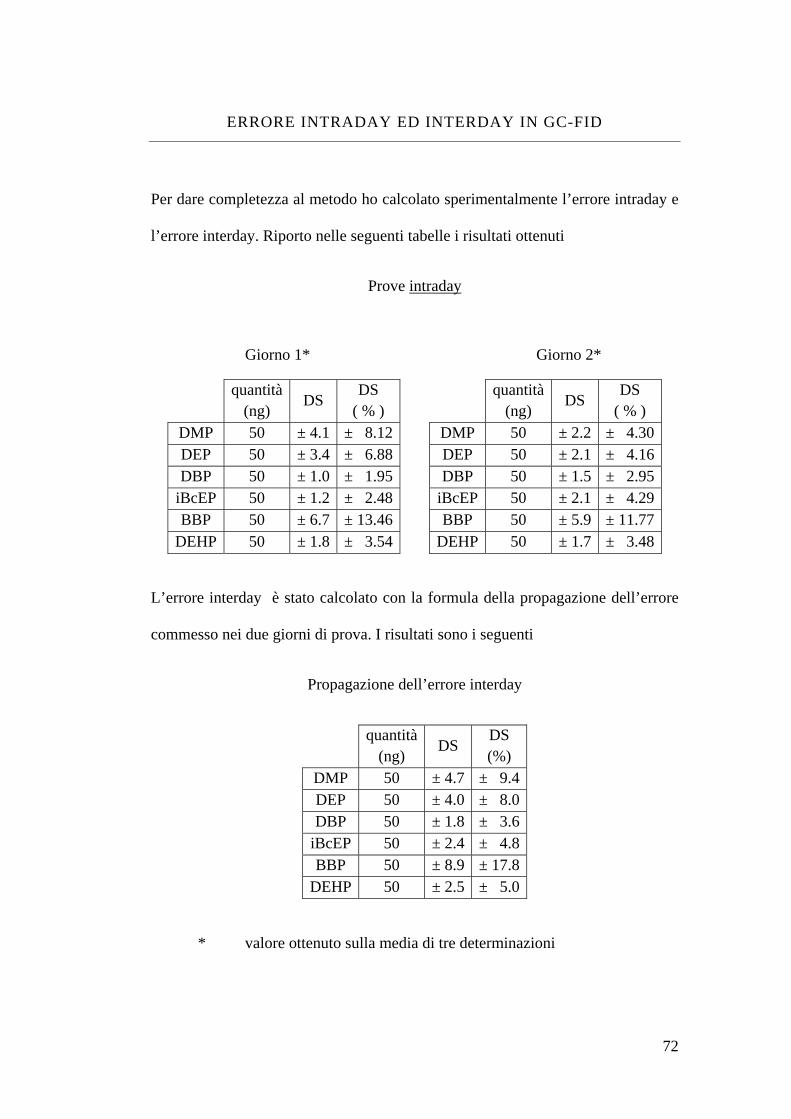
72
ERRORE INTRADAY ED INTERDAY IN GC-FID
Per dare completezza al metodo ho calcolato sperimentalmente l’errore intraday e
l’errore interday. Riporto nelle seguenti tabelle i risultati ottenuti
Prove intraday
Giorno 1* Giorno 2*
quantità
(ng) DS
DS ( % )
quantità
(ng) DS
DS ( % )
DMP 50 ± 4.1 ± 8.12 DMP 50 ± 2.2 ± 4.30DEP 50 ± 3.4 ± 6.88 DEP 50 ± 2.1 ± 4.16DBP 50 ± 1.0 ± 1.95 DBP 50 ± 1.5 ± 2.95
iBcEP 50 ± 1.2 ± 2.48 iBcEP 50 ± 2.1 ± 4.29BBP 50 ± 6.7 ± 13.46 BBP 50 ± 5.9 ± 11.77
DEHP 50 ± 1.8 ± 3.54 DEHP 50 ± 1.7 ± 3.48
L’errore interday è stato calcolato con la formula della propagazione dell’errore
commesso nei due giorni di prova. I risultati sono i seguenti
Propagazione dell’errore interday
quantità
(ng) DS
DS (%)
DMP 50 ± 4.7 ± 9.4DEP 50 ± 4.0 ± 8.0DBP 50 ± 1.8 ± 3.6
iBcEP 50 ± 2.4 ± 4.8BBP 50 ± 8.9 ± 17.8
DEHP 50 ± 2.5 ± 5.0
* valore ottenuto sulla media di tre determinazioni

73
ANALISI ALLO SPETTROMETRO DI MASSA DEGLI FTALATI
Lo studio degli ftalati è proseguito con un’analisi con lo spettrometro di massa.
Questa tecnica ha permesso di confermare i risultati ottenuti con il rilevatore
universale FID. Inoltre la sensibilità e la precisione dello strumento hanno
permesso di abbassare notevolmente i limiti di rilevabilità della metodica
proposta.
Lo strumento utilizzato è un gas cromatografo con un rivelatore a trappola ionica,
prodotto dalla Thermofisher. L’intero strumento è interfacciato con un PC e il
software di analisi è l’Xcalibur, specifico per la spettrometria di massa.
Di seguito riporto tutte le condizioni operative dello strumento dall’iniettore, al
forno, al rivelatore. È da tener presente che i dati descritti sono frutto di uno
studio preliminare, fatto in modo tale da ottimizzare e/o massimizzare il risultato
cromatografico. Le condizioni riportate, dunque, sono quelle che hanno dato le
risposte migliori.
L’iniettore PTV è regolato in modalità splitless; la temperatura iniziale è di 60 °C;
la velocità di splittaggio è di 30 mL/min; l’apertura dello spillo è stata fissata a
1:30 min e la pulizia del setto è mantenuta sotto flusso cotante di elio ad
1 mL/min. L’iniettore è programmato secondo il seguente schema:
Pressure (kPa)
Rate (°C/min)
Temp. (°C)
Time (min.)
Flow (mL/min)
Injection 70 0.05 50 Evap. 140 14.5 200 1:00 Transfer 210 14.5 280 2:00 Cleaning 14.5 310 10:00 70

74
Come per le analisi svolte con il FID, il forno è equipaggiato con una colonna
capillare in silice fusa con fase chimicamente legata (SE 54 - 5% fenile, 95 %
dimetilsilossano), con L=30 m, I.D. (diametro interno) 250 µm e df (spessore del
film)=0.25 µm. A differenza dell’analisi al FID, dove il carrier gas era l’idrogeno,
nella spettrometria di massa la fase mobile è l’elio ultrapuro, con una velocità di
flusso di circa 1 mL/min (rilevato sperimentalmente).
Le condizioni operative del forno sono riportate nella seguente tabella:
rate (°C/min) Temp (°C) Hold time (min) Initial 50 1:00 Ramp 1 10 300 4:00
Poiché il rivelatore di massa è una parte distinta e separata dal forno, particolare
attenzione va data alla Transfer Line (TL), unità che funge da collegamento tra il
forno e la trappola ionica. Solitamente la TL viene regolata ad una temperatura di
circa 20-30 °C in meno rispetto alla temperatura di esercizio massima del forno.
Questa piccola differenza fa in modo che nell’eventualità ci fosse un problema di
condensazione dell’analita, esso andrebbe a condensare sulla TL, senza creare
otturazioni nella colonna. Per questo motivo la temperatura della TF è stata
impostata a 270 °C.
Infine la temperatura di esercizio della trappola ionica è regolata a 250 °C. La
temperatura di esercizio della trappola è inferiore a quella della TF, e questo
porterebbe ad una contraddizione, in quanto si potrebbe avere una condensazione
sulla trappola e quindi una perdita di sensibilità. Ovviamente questo non è
possibile poiché la trappola è sottoposta ad un vuoto molto spinto, in questo modo
tutte le molecole che condensano vengono risucchiate dal vuoto.

75
Lo strumento è dotato di due sistemi di ionizzazione, l’impatto elettronico (EI) e
la ionizzazione chimica (CI). In questo caso è stato utilizzando come sorgente di
ionizzazione l’impatto elettronico (EI) positivo, caratterizzato da un potenziale
standard di 70 eV. Inoltre il damping gas, un gas ausiliario (elio) all’interno della
trappola ionica, è stato fissato a 0.3 mL/min.
Per la determinazione degli ftalati, sono state eseguite analisi in Full Scan e in
SIM. In entrambi i casi, l’acquisizione del cromatogramma è stata fatta partire da
10:00 min. Questa è una accortezza che solitamente è utilizzata in spettrometria di
massa, infatti il ritardo dell’accensione del filamento, quindi della registrazione
del cromatogramma, impedisce a spurghi di colonna ed a bande del fronte del
solvente di sovraccaricare la camera di ionizzazione, evitando possibili lesioni.
Tutte le analisi in Full Scan (FS) sono state eseguite in un range che va da 50 a
450 amu. È ovviamente inutile sovraccaricare la trappola con frammenti ancora
più pesanti considerando che il composto più grande ha una massa di 390 amu.
Oltre ad eseguire analisi in FS sono state eseguite analisi in SIM (Selected Ion
Monitoring). Lo strumento permette di fare l’analisi in SIM sia impostando il
frammento prescelto in acquisizione direttamente sulla trappola ionica, in questo
caso è il rivelatore che seleziona la corrente ionica che genera quel frammento;
oppure può essere fatto utilizzando il software, in questo caso è il software che va
a selezionare fra tutti i frammenti acquisiti quello prescelto dall’operatore. I due
modi di operare presentano vantaggi e svantaggi a seconda della situazione in cui
ci si trova. Lo studio delle frammentazioni dei vari ftalati ottenuti in FS ha
permesso di generalizzare il comportamento a cui questi composti sono soggetti
all’interno della trappola ionica, che può essere schematizzato come segue:
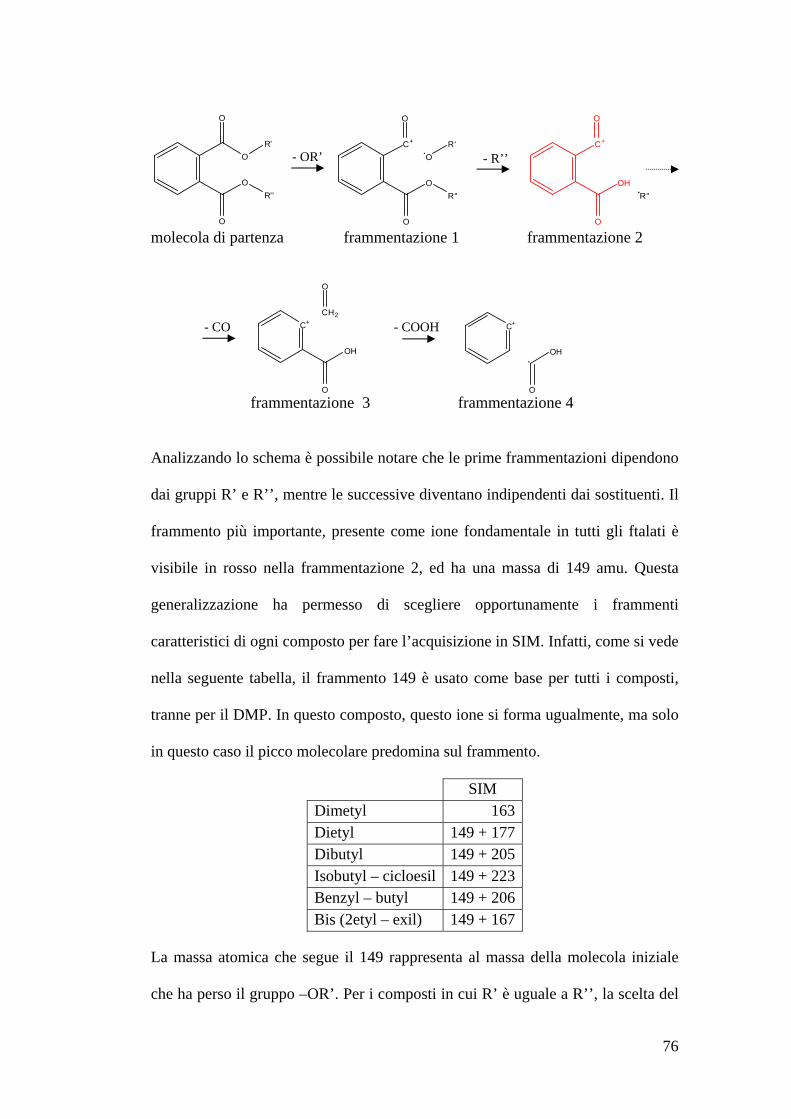
76
molecola di partenza frammentazione 1 frammentazione 2
frammentazione 3 frammentazione 4
Analizzando lo schema è possibile notare che le prime frammentazioni dipendono
dai gruppi R’ e R’’, mentre le successive diventano indipendenti dai sostituenti. Il
frammento più importante, presente come ione fondamentale in tutti gli ftalati è
visibile in rosso nella frammentazione 2, ed ha una massa di 149 amu. Questa
generalizzazione ha permesso di scegliere opportunamente i frammenti
caratteristici di ogni composto per fare l’acquisizione in SIM. Infatti, come si vede
nella seguente tabella, il frammento 149 è usato come base per tutti i composti,
tranne per il DMP. In questo composto, questo ione si forma ugualmente, ma solo
in questo caso il picco molecolare predomina sul frammento.
SIM Dimetyl 163Dietyl 149 + 177Dibutyl 149 + 205Isobutyl – cicloesil 149 + 223Benzyl – butyl 149 + 206Bis (2etyl – exil) 149 + 167
La massa atomica che segue il 149 rappresenta al massa della molecola iniziale
che ha perso il gruppo –OR’. Per i composti in cui R’ è uguale a R’’, la scelta del
OR'
OR''
O
O
C+
OR'
OR''
O
O
C+
OHR''
O
O
C+CH2
OH
O
O
C+
OH
O
- OR’ - R’’
- CO - COOH

77
secondo valore è obbligata, mentre per i composti in cui R’ è diverso da R’’, è
stato scelto come secondo valore il frammento con picco più alto tra i due.
Tuttavia lo schema così presentato porta ad una generalizzazione grossolana della
frammentazione degli ftalati, infatti studiando i frammentogrammi mi sono
accorto che alcuni ioni, in determinati step di frammentazione, tendono ad
assorbire per poi rilasciare una molecola di acqua. Questo fenomeno avviene
soprattutto sul frammento 2 e a volte sul frammento 1.
Tutti i frammenti ipotizzati per ogni composto sono stati confermato in
GC/MS/MS. A titolo di esempio riporto la frammentazione del DEP. Un quadro
completo dei composti analizzati è riportato negli allegati dal 3 al 9.
frammentogramma del di-etyl ftalato
I picchi caratteristici sono spiegati con le seguenti frammentazioni
prova_FS_dopo_estate #529 RT: 14.40 AV: 1 SB: 93 14.59-14.98 , 13.91-14.30 NL: 4.65E5T: + c Full ms [ 50.00-450.00]
60 80 100 120 140 160 180 200 220m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Relative Abundance
149.1
177.0
176.0
150.2121.1
105.165.0 178.0104.1 122.176.1 93.0 106.1 148.274.1 221.0133.250.9 151.194.1 195.0175.0 179.179.0 107.1 202.3
C+
O
O
O
C+
OH
O
O
C+
OH
O
- CH2CH3
+ H+
- COC
O
O
O
O
- OCH2CH3
Exact Mass: 222,09 Exact Mass: 177,05 Exact Mass: 149,02 Exact Mass: 121,03
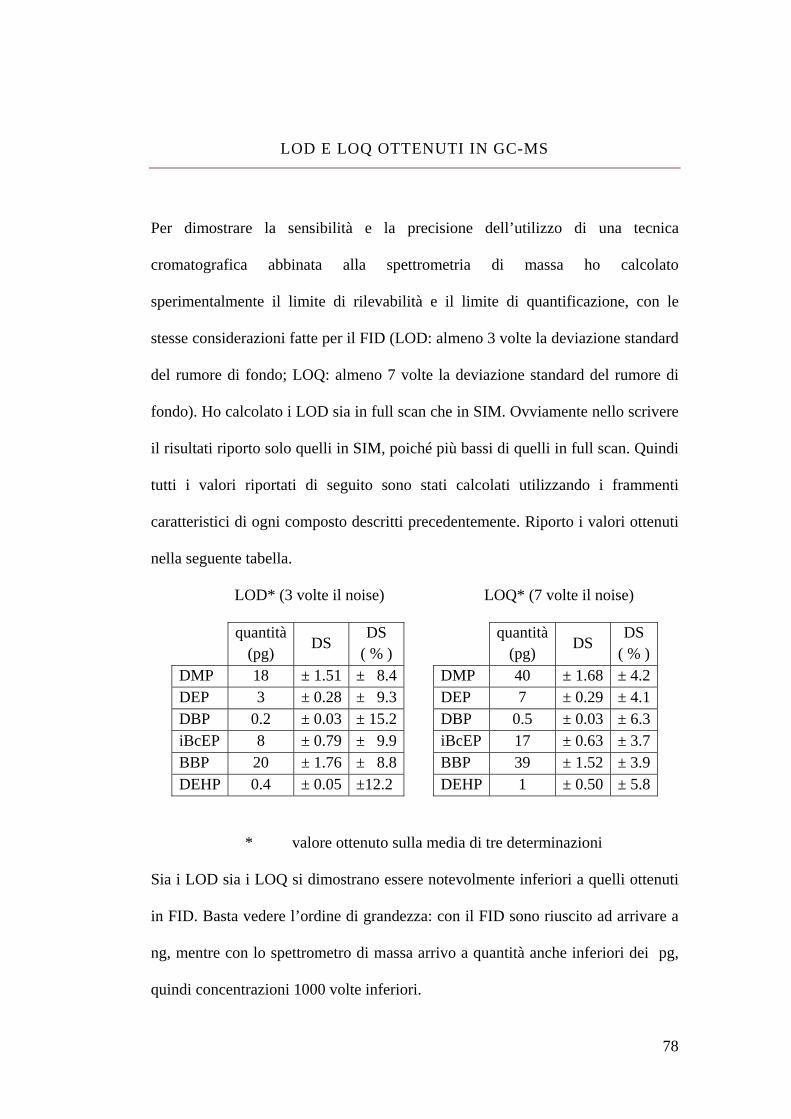
78
LOD E LOQ OTTENUTI IN GC-MS
Per dimostrare la sensibilità e la precisione dell’utilizzo di una tecnica
cromatografica abbinata alla spettrometria di massa ho calcolato
sperimentalmente il limite di rilevabilità e il limite di quantificazione, con le
stesse considerazioni fatte per il FID (LOD: almeno 3 volte la deviazione standard
del rumore di fondo; LOQ: almeno 7 volte la deviazione standard del rumore di
fondo). Ho calcolato i LOD sia in full scan che in SIM. Ovviamente nello scrivere
il risultati riporto solo quelli in SIM, poiché più bassi di quelli in full scan. Quindi
tutti i valori riportati di seguito sono stati calcolati utilizzando i frammenti
caratteristici di ogni composto descritti precedentemente. Riporto i valori ottenuti
nella seguente tabella.
LOD* (3 volte il noise) LOQ* (7 volte il noise)
quantità (pg)
DS DS
( % ) quantità
(pg) DS
DS ( % )
DMP 18 ± 1.51 ± 8.4 DMP 40 ± 1.68 ± 4.2 DEP 3 ± 0.28 ± 9.3 DEP 7 ± 0.29 ± 4.1 DBP 0.2 ± 0.03 ± 15.2 DBP 0.5 ± 0.03 ± 6.3 iBcEP 8 ± 0.79 ± 9.9 iBcEP 17 ± 0.63 ± 3.7 BBP 20 ± 1.76 ± 8.8 BBP 39 ± 1.52 ± 3.9 DEHP 0.4 ± 0.05 ±12.2 DEHP 1 ± 0.50 ± 5.8
* valore ottenuto sulla media di tre determinazioni
Sia i LOD sia i LOQ si dimostrano essere notevolmente inferiori a quelli ottenuti
in FID. Basta vedere l’ordine di grandezza: con il FID sono riuscito ad arrivare a
ng, mentre con lo spettrometro di massa arrivo a quantità anche inferiori dei pg,
quindi concentrazioni 1000 volte inferiori.
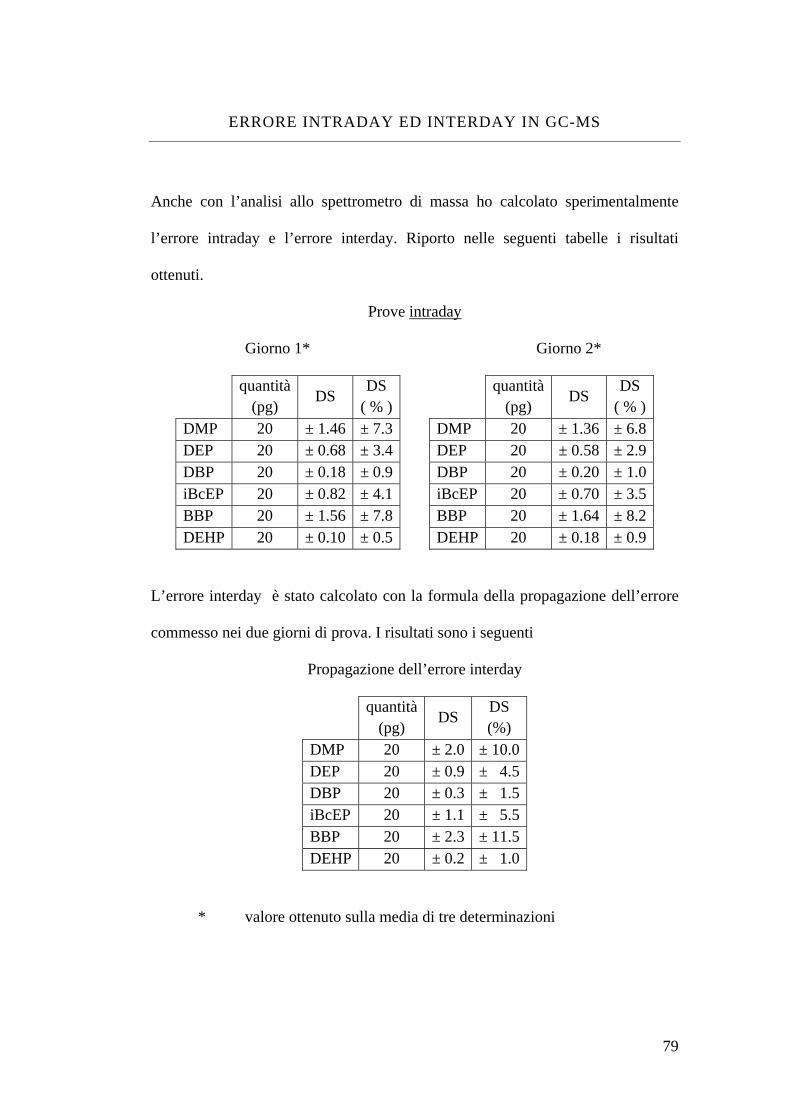
79
ERRORE INTRADAY ED INTERDAY IN GC-MS
Anche con l’analisi allo spettrometro di massa ho calcolato sperimentalmente
l’errore intraday e l’errore interday. Riporto nelle seguenti tabelle i risultati
ottenuti.
Prove intraday
Giorno 1* Giorno 2*
quantità (pg)
DS DS
( % ) quantità
(pg) DS
DS ( % )
DMP 20 ± 1.46 ± 7.3 DMP 20 ± 1.36 ± 6.8 DEP 20 ± 0.68 ± 3.4 DEP 20 ± 0.58 ± 2.9 DBP 20 ± 0.18 ± 0.9 DBP 20 ± 0.20 ± 1.0 iBcEP 20 ± 0.82 ± 4.1 iBcEP 20 ± 0.70 ± 3.5 BBP 20 ± 1.56 ± 7.8 BBP 20 ± 1.64 ± 8.2 DEHP 20 ± 0.10 ± 0.5 DEHP 20 ± 0.18 ± 0.9
L’errore interday è stato calcolato con la formula della propagazione dell’errore
commesso nei due giorni di prova. I risultati sono i seguenti
Propagazione dell’errore interday
quantità (pg)
DS DS (%)
DMP 20 ± 2.0 ± 10.0DEP 20 ± 0.9 ± 4.5DBP 20 ± 0.3 ± 1.5iBcEP 20 ± 1.1 ± 5.5BBP 20 ± 2.3 ± 11.5DEHP 20 ± 0.2 ± 1.0
* valore ottenuto sulla media di tre determinazioni

80
CONCLUSIONI
Nel corso del lavoro sono emersi molti dati sperimentali che hanno dimostrato il
funzionamento del metodo proposto e della fase adsorbente presa in esame.
Il metodo proposto si adatta molto bene alla determinazione qualitativa e
quantitativa di ftalati presenti in matrici idroalcoliche. Infatti l’applicazione a
campioni reali del metodo proposto ha permesso di dimostrare che esso è
applicabile anche a matrici complesse con risultati sufficientemente in linea con
quelli ottenuti da soluzioni standard.
Inoltre, dallo studio della ripartizione tra gli analiti e la fase adsorbente, sembra
sia possibile allargare il campo di analisi ad altri campioni alimentari.
L’utilizzo di apparecchiature poco costose e presenti nella maggior parte dei
laboratori (il gas cromatografo), la praticità e la semplicità nell’operare, la poca
manualità richiesta all’operatore, l’utilizzo di pochi solventi e l’utilizzo di
cartucce SPE reperibili in commercio a basso costo, rendono il metodo molto
economico.
Infine, il limite di rilevabilità, il limite di quantificazione, gli errori intraday ed
interday ottenuti sia con il GC-FID e confermati in GC-MS, rendono il metodo
sensibile, affidabile e riproducibile.

81
BIBLIOGRAFIA
[1] K. Romanowicz, T. Misztal, B. Barcikowski, Genistein; A phytoetstrogen, effectively
modulates luteinizing homone and prolactin secretion in ovariectomized ewes during seasonal anestru;. Neuroendocrinology, 79 (2), 73-81. (2004)
[2] S. Kanno, S. Hirano, F.Kayama; Effects of phytoestrogens in MC3T3-E1 cells. Toxicology, 1961 (1-2), 137-145. (2004)
[3] R.M. Harris, D.M. Wood, L. Bottomley, S. Blagg, K. Owen, P.J. Hughes, R.H. Waring, C.J. Kirk; Phytoestrogens are potent inhibitors of estrogen sulfation: implications for breast cancer risk and treatment; J Clin Endocrinol Metab, 89 (4), 1779-1787. (2004)
[4] Cao, Xu-Liang; Determination of phthalates and adipate in bottled water by headspace solid-phase microextraction and gas chromatography/mass spectrometry; Food Researche Division, Bureau of Chemical Safety, Food Directorate, Health. Canada
[5] S.Jara, C.Lysebo, E.Lundanes; Determination of phthalates in water samples
using polystyrene solid-phase extraction and liquid chromatography quantification; Departement of Chemistry, University of Oslo, Norway (2000).
[6] P.T. Anh, T.X.T. Nguyen, T.V. Hue; Solid phase extraction and capillary GC analysis
of phtalate esters in milk powder; Abstract of Papers of the American Chemical Society, 213, 139-anyl, (1997)
[7] S.Ratogi; Gas chromatography analysis of phtalate esters in plastic toys; National
Environmental Research Institute, Department of Environmental Chemistry. Denmark.
[8] J.D. Carrillo, M.P. Martinez, M.T. Tena; Determination of phthalates in wine by head space solid-phase microextraction followed by gas chromatography-mass spectrometry: use of deuterated phthalates as internal standards; Department of Chemistry, University of La Rioja, Spain.
[9] M.V. Russo, G. Goretti, A. Liberti; Rapid determination of chlorinated pesticides
using CN-bonded cartridge followed by GC-ECD; Chromatographia, 35, 290-294. (1993) [10] M.V. Russo, G. Goretti, T. Nevigato; Separation of Polychlorinated Biphenylis from
Chlorinated Pesticides using Aminopropyl Bonded-Phase Cartridge and Determination by GC-ECD; Chromatographia, 48, 293-298. (1998)
[11] M.V. Russo, G. Goretti, T. Nevigato; Sequential solid-phase extract in with
Cyanopropil Bonded-Phase Cartridge for trace enrichment of PCB’s Chlorinated Pesticides for water samples; Chromatographia, 50 (7/8), 446-452. (1999)
[12] M.V. Russo; Fast Solid Phase Extraction Polychlorobiphenylis and Chlorinated
Pesticides Residues from Mussels Using Sep-Pack Cartridges; Chromatographia, 51 (1/2), 71-76. (2000)
[13] M.V. Russo; Diol Sep-Pack Cartridges for polychlorobiphenyls and Chlorinated
Pesticides Enrichment from water samples; determination by GC-ECD; Chromatographia, 53, 93-98. (2000)

82
[14] M.V. Russo, P. Avino; Determination of 1,2,4- and 1,3,5-Trichlorobenzenes in water
samples by solid-phase extraction and gas-chromatografy coupled to electron capture; Analytical Letters, 34 (6), 1003-1013. (2001)
[15] M.V. Russo, P. Avino; SPE with cyanopropyl bonded-phase cartridge for trace
enrichment of dioxins and chlorinated pesticides from water samples; The Second European Bioremediation Conference, 597-600 Chania, Crete, Greece, June 30 – July 4 (2003)
[16] M.V. Russo, V. Di Lorenzo, G. Cinelli, I. Notardonato, P. Avino; SPE with a mixture of aminopropyl- and C18 bonded porous silica for enrichment of Aromatic Sulphur Compounds as “markers” of crude oil sea water pollution: determination by GC-FID and GC-MS; Cemepe/Secotox Conference 24-28 giugno, Skianthos Island, Grecia. (2007)
[17] M.V. Russo, P. Avino, I. Notardonato, G. Cinelli; Cyanopropyl bonded-phase cartridges for trace enrichment of dioxins and chlorinated pesticides from water sample; CHROMATOGRAPHIA, 69, 709-717. (2009)
[18] P. Avino, I. Notardonato, G. Cinelli and M.V. Russo; Aromatic sulphur compounds enrichment from seawater in crude oil contamination by solid phase extraction; Current Analytical Chemistry, 5 (4), 339-346. (2009)
[19] S. Jara, C. Lysebo, E. Lundanes; Determination of phthalates in water samples using polystyrene solid-phase extraction and liquid chromatography quantification. Departement of Chemistry, University of Oslo, Norway. (2000)

83
DETERMINAZIONE DELL’ACRILAMMIDE IN PRODOTTI
DA FORNO MEDIANTE GC-ECD E GC-MS.
INTRODUZIONE
Alcuni anni fa, un lavoro sulla presenza dell’acrilammide in alcuni prodotti
alimentari cotti, soprattutto nelle patate fritte, ha lanciato l’allarme per la salute
dei consumatori [1-2]. L’assenza di questa molecola nei cibi crudi o bolliti ha
fatto avanzare l’ipotesi della sua formazione durante i processi di trattamento
termico ad alte temperature.
La via di sintesi dell’acrilammide non è ancora del tutto chiara, ma l’ipotesi più
attendibile pare sia la stretta relazione con la reazione di Maillard, detta anche di
imbrunimento non enzimatico. In modo particolare la sintesi di acrilammide
sembra dipendere dalla presenza nella matrice alimentare di un amminoacido,
l’asparagina [3-4-5-6]. La formazione di questa molecola, inoltre, sembra essere
legata anche alla presenza di zuccheri riducenti, quali glucosio e fruttosio.
In altre parole, la quantità di acrilammide che si può formare nei prodotti
alimentari è strettamente legata alla presenza di precursori nella matrice e ad
alcuni parametri fisici, quali ad esempio la temperatura, il ph, eccetera [7-8].
L’Agenzia Internazionale di Ricerca sul Cancro (IARC) ha classificato
l’acrilammide come un probabile agente cancerogeno per l’uomo. Quindi, è
necessario conoscere la presenza di questo composto nei vari prodotti alimentari e
limitarne il quantitativo quanto più possibile.

84
OBIETTIVO
Scopo di questo lavoro è lo studio di un metodo quali/quantitativo, per la
determinazione dell’acrilammide in cibi sottoposti a cottura. In particolare,
l’obiettivo del lavoro è lo studio di un metodo alternativo ai metodi presenti in
letteratura, che sfrutta come tecnica di analisi la derivatizzazione con agente
alogenato abbinata alla successiva analisi gas cromatografica.
A seguito di uno studio dei più comuni agenti derivatizzanti presenti sul mercato,
allo scopo di rendere la molecola rilevabile con un rilevatore a cattura di elettroni,
ho deciso di derivatizzare l’acrilammide con l’anidride trifluoroacetica (TFAA).
L’acrilammide derivatizzata è stata rivelata in primis con l’ECD, successivamente
si è passati alla conferma ed alla caratterizzazione del derivato tramite GC-MS.
L’uso di un tale sistema presenta diversi vantaggi, quali ad esempio la semplicità
e la praticità strumentale, basata sull’utilizzo di uno strumento (il
gascromatografo) economico e presente nella maggior parte dei laboratori.
Nella messa a punto del metodo, sono andato alla ricerca e all’ottimizzazione
delle quantità e delle concentrazioni dei reagenti da impiegare.
Infine il metodo è stato applicato su alcuni prodotti alimentari con particolare
attenzione ai prodotti per la prima colazione e a patatine croccanti in busta,
largamente consumati, soprattutto dai ragazzi.

85
CARATTERISTICHE DELLA MOLECOLA
Commercialmente è prodotta come precursore nella sintesi di poliacrilammide,
sostanza usata prevalentemente come agente flocculante nella chiarificazione
delle acque potabili e nel trattamento delle acque di scarico urbane ed industriali
[9]. Altri usi comuni di questa molecola sono quelli nell’industria tessile, della
carta, della plastica e dei cosmetici [10]. L’acrilammide, inoltre, è utilizzata nella
preparazione di gel di poliacrilammide in uso nei laboratori scientifici per tecniche
elettroforetiche. L’acrilammide è anche una componente del fumo di sigarette, e
questo conferma che deriva dal riscaldamento di materiale organico.
Le caratteristiche chimiche dell’acrilammide sono riportate di seguito:
formula chimica
struttura chimica
peso molecolare DL50 (ratti)
CH2CHCONH2 71,08 g/mol 107-203 mg/Kg
Si presenta in forma di piccoli cristalli di colore bianco. È altamente solubile in
acqua e in altri solventi a polarità inferiore, come metanolo, etanolo, acetone,
acetonitrile, acetato di etile, benzene. Può polimerizzare se raggiunge il punto di
ebollizione e se esposta ai raggi UV [11].
In forma solida è stabile a temperatura ambiente, mentre le soluzioni sono stabili
fino ad un anno se conservate a basse temperature.
NH2
O

86
METODI ANALITICI IN USO PER LA DETERMINAZIONE
DELL’ACRILAMMIDE NEI PRODOTTI ALIMENTARI
Per l’analisi dell’acrilammide sono in uso diversi metodi analitici, tutti basati su
tecniche cromatografiche. Le due tecniche cromatografiche di largo impiego sono
la gascromatografia (GC) e la cromatografia liquida (LC), quasi sempre abbinate
alla spettrometria di massa.
Per l’analisi gas cromatografica dell’acrilammide il punto fondamentale è la
derivatizzazione della molecola. I metodi di derivatizzazione dell’acrilammide
riportati in letteratura si basano sul metodo EPA 8032A e, cioè, sulla
bromurazione dell’acrilammide per formare il derivato 2,3-
dibromopropionammide, che può essere analizzato attraverso GC-ECD o GC-MS
per basse concentrazioni.
Un altro metodo di derivatizzazione è quello che prevede la silanizzazione
dell’acrilammide in seguito a reazione con Bis-(trimetil-silil)trifluoroacetammide
(BSTFA) [12].
Non molto tempo fa, è stato messo a punto anche un metodo GC-MS senza
preventiva derivatizzazione dell’acrilammide [13].
A differenza della cromatografia solida, la cromatografia liquida ha il vantaggio di
poter analizzare l’acrilammide direttamente: è possibile iniettare nello strumento
l’estratto acquoso degli alimenti. Anche in questo caso si abbina la cromatografia
liquida alla spettrometria di massa [14].
Tuttavia, esistono studi che dimostrano la possibilità di analizzare l’acrilammide
anche attraverso HPLC-UV, usando una colonna specifica della Dionex. [15].

87
L’estrazione dell’acrilammide dalla matrice alimentare avviene utilizzando acqua
come solvente, vista l’elevata solubilità di questa molecola in fase acquosa. Per la
purificazione si può ricorrere a centrifugazione ed estrazione in fase solida (SPE).
Per la successiva analisi in GC, però, è necessario recuperare l’analita in un
solvente organico. L’estrazione dell’acrilammide può essere effettuata anche
tramite estrazione con solvente (Soxhlet o ASE).
MATERIALI E METODI
Materiali
Le soluzioni oggetto di analisi sono state preparate utilizzando polvere di
acrilammide, prodotto della Sigma-Aldrich, con purezza ≥ 99%.
Allo scopo di ottenere un derivato rilevabile con l’ECD, come agente
derivatizzante è stata scelta l’anidride trifluoroacetica (TFAA), fornita dalla
Supelco in confezioni da 10 fiale ognuna da 1 mL.
L’acetonitrile (CH3CN), la piridina, l’esano, il solfato di sodio anidro (Na2SO4)
sono stati acquistati da Sigma-Aldrich e da Carlo Erba Reagenti.
Anche in questo studio le analisi sono state effettuate utilizzando uno standard
interno: 1,2,3,4,5,6-cloro-cicloesano (lindane), fornito dalla Carlo Erba.
Inoltre sono stati utilizzati un Rotavapor, estrattori Soxhlet, vetreria e materiale
vario di laboratorio.

88
Metodi
Per le analisi è stato impiegato un gascromatografo della DANI, modello 86.10
HT, equipaggiato con un iniettore PTV (programmed temperature vaporizer), una
colonna capillare ed un rivelatore a cattura di elettroni (ECD).
Le programmate del PTV e del forno sono schematizzate a fondo pagina.
L’apertura dello spillo del PTV è fissato dopo 1:30 minuti. La pressione
dell’idrogeno usato come carrier gas (rilevata sperimentalmente) è pari a 1
mL/min.
Il forno è equipaggiato con una colonna in silice fusa con fase chimicamente
legata (SE 54 - 5% fenile, 95 % dimetilsilossano), con L=30 m, I.D. (diametro
interno) 250 µm e df (spessore del film)=0.25 µm, fornita dalla Supelco.
La temperatura di esercizio del rilevatore è fissata a 280°C, con un flusso costante
di azoto pari a 1,4 mL/min. Il volume iniettato è 1 μL, opportunamente prelevato.
Lo strumento è interfacciato a un PC con il software di elaborazione specifico dei
dati sperimentali “Clarity”.
Inoltre, tutti gli esperimenti sono stati svolti aggiungendo solfato anidro di sodio
alle soluzioni da iniettare. Essendo il solfato di sodio un sale che tende fortemente
ad idratarsi, cattura l’eventuale presenza di acqua e fa in modo che la stessa non
va a rompere il derivato formato.
Programmata PTV Programmata forno
800 °C/min
60°C
280°C 1’
30’’
30’’
8 °C/min
1’
5 °C/min
70°C
280°C4’
130°C

89
REAZIONE DI DERIVATIZZAZIONE
Il protocollo di analisi per la derivatizzazione della molecola, ottenuto in vial
ambrate da 2 mL, è riportato sinteticamente nello schema seguente:
• 50 µL della soluzione di acrilammide disciolta in acetonitrile;
• 3 µL di piridina pura, necessaria a prevenire l’acidificazione della
soluzione;
• 10 µL di anidride trifluoro-acetica (calcolata stechiometricamente in forte
eccesso);
• reazione a 65 °C per 30 minuti a bagnomaria;
• 50 µL di una soluzione di MeOH/NaOH (170 mg/L), per neutralizzare
l’eccesso di TFAA;
• essiccamento del campione sotto flusso di N2
• recupero con 100 µL di toluene ed aggiunta dello standard interno, 10µL
(lindane, 0.47 ppm)
• Analisi in GC-ECD o GC-MS (1 μL).
Tutte le quantità che ho descritto nel protocollo per la determinazione
dell’acrilammide sono state vagliate e calcolate in base a prove sperimentali, al
fine di ottimizzare il risultato cromatografico. A tale scopo, ho calcolato
sperimentalmente l’effetto quantitativo del volume iniziale, la concentrazione e il
volume della piridina, il volume di TFAA, il tempo e la temperatura di reazione,
la soluzione per meglio ottenere la neutralizzazione, quale e quanto solvente
utilizzare per il recupero del composto derivatizzato e la concentrazione ottimale
dello S.I.
Oltre alle prove di ottimizzazione ho fatto anche prove di cinetica di formazione e
di degradazione.
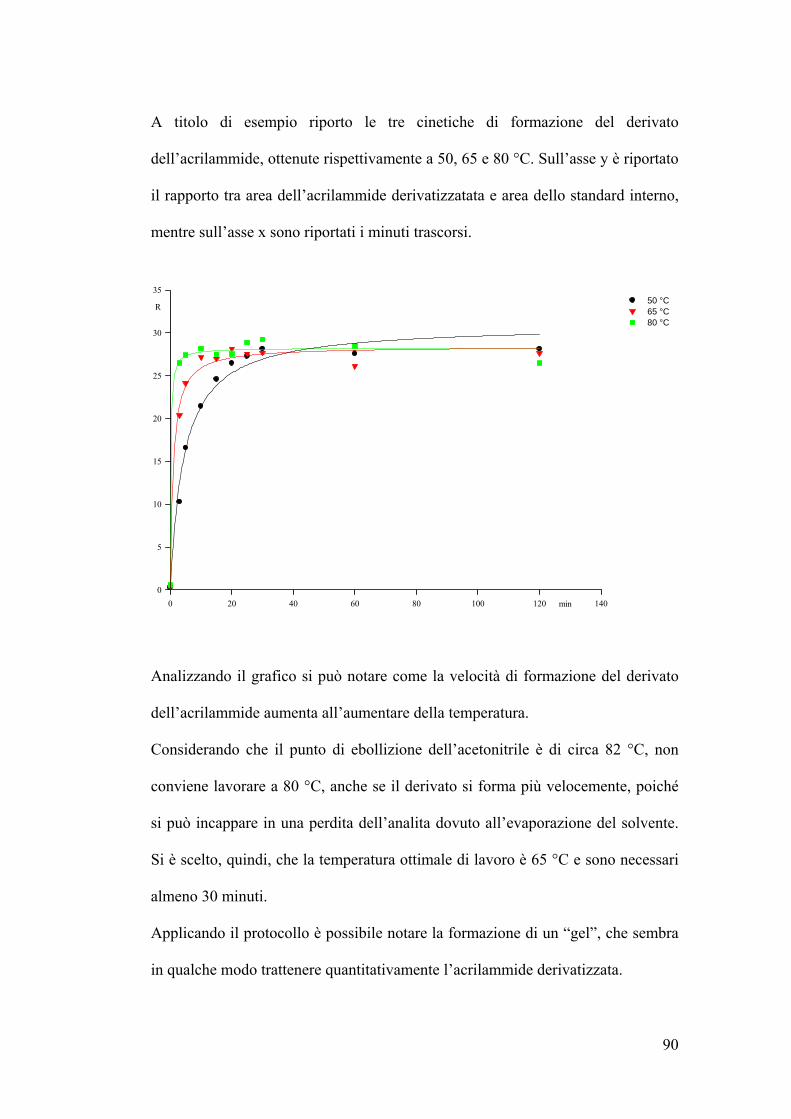
90
A titolo di esempio riporto le tre cinetiche di formazione del derivato
dell’acrilammide, ottenute rispettivamente a 50, 65 e 80 °C. Sull’asse y è riportato
il rapporto tra area dell’acrilammide derivatizzatata e area dello standard interno,
mentre sull’asse x sono riportati i minuti trascorsi.
min0 20 40 60 80 100 120 140
R
0
5
10
15
20
25
30
3550 °C65 °C80 °C
Analizzando il grafico si può notare come la velocità di formazione del derivato
dell’acrilammide aumenta all’aumentare della temperatura.
Considerando che il punto di ebollizione dell’acetonitrile è di circa 82 °C, non
conviene lavorare a 80 °C, anche se il derivato si forma più velocemente, poiché
si può incappare in una perdita dell’analita dovuto all’evaporazione del solvente.
Si è scelto, quindi, che la temperatura ottimale di lavoro è 65 °C e sono necessari
almeno 30 minuti.
Applicando il protocollo è possibile notare la formazione di un “gel”, che sembra
in qualche modo trattenere quantitativamente l’acrilammide derivatizzata.

91
LOD E LOQ OTTENUTI IN GC-ECD
Nelle condizioni sviluppate e messe a punto, sono andato alla ricerca del LOD e
del LOQ utilizzando come rivelatore l’ECD. I dati ottenuti sono il linea con le
risposte di un rivelatore ECD, infatti il limite di rilevabilità è di 10 pg in quantità
assoluta e il limite di quantificazione è 78 pg in quantità assoluta. Nel seguente
schema riporto il LOD e il LOQ con la relativa deviazione standard calcolata su
tre determinazioni.
LOD LOQ
quantità
(pg) DS*
DS* (%)
quantità (pg)
DS* DS* (%)
acrilammide derivatizzata
10 ±
1.68 ±
16.8 78
± 3.25
± 4.16
ERRORE INTRADAY ED INTERDAY AL GC-ECD
Considerando che il limite di quantificazione è di 78 pg/µL, quindi 78 ppb, ho
calcolato l’errore intraday ed interday considerando una concentrazione di 100
ppb. Ho ripetuto tre analisi in due giorni differenti. Riporto le stime degli errori
nelle seguenti tabelle, calcolate sulla media di tre determinazioni giornaliere
giorno 1 giorno 2 interday
quantità
(pg) DS*
DS* (%)
DS* DS* (%)
DS DS (%)
acrilammide derivatizzata
100 ±2.13 ±2.13 ±2.94 ±2.94 ±3.63 ±3.63
*DS calcolata su tre determinazioni
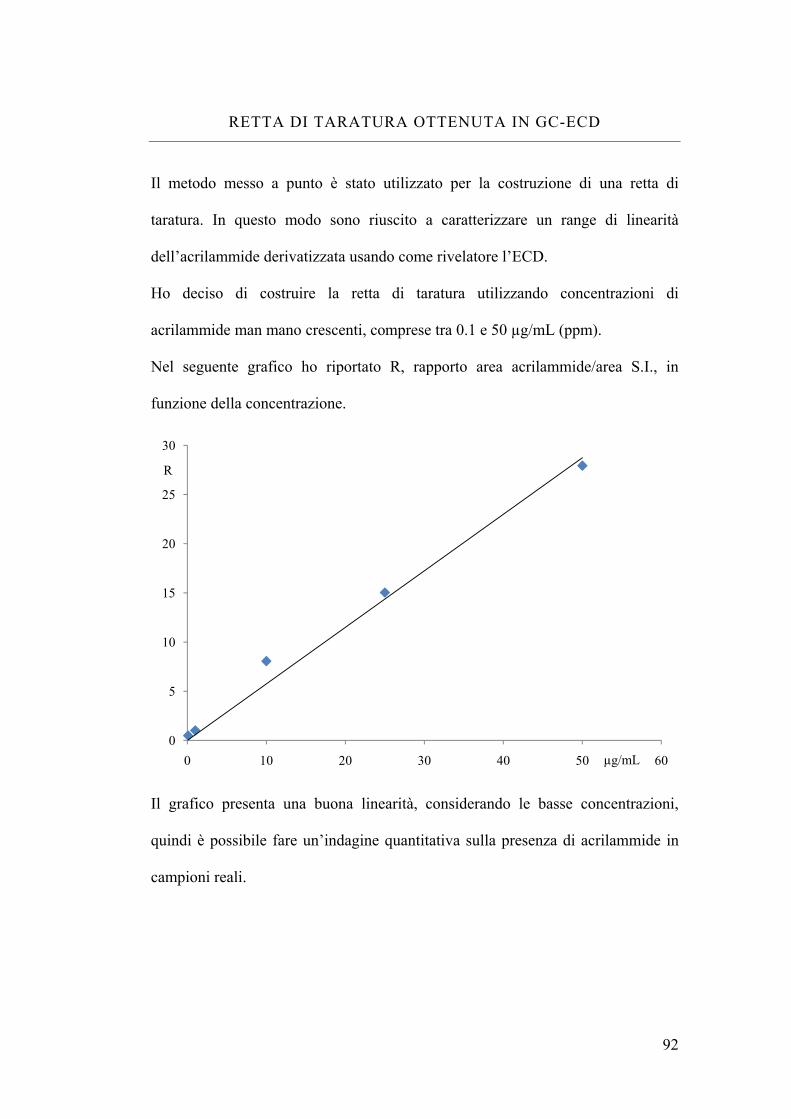
92
RETTA DI TARATURA OTTENUTA IN GC-ECD
Il metodo messo a punto è stato utilizzato per la costruzione di una retta di
taratura. In questo modo sono riuscito a caratterizzare un range di linearità
dell’acrilammide derivatizzata usando come rivelatore l’ECD.
Ho deciso di costruire la retta di taratura utilizzando concentrazioni di
acrilammide man mano crescenti, comprese tra 0.1 e 50 µg/mL (ppm).
Nel seguente grafico ho riportato R, rapporto area acrilammide/area S.I., in
funzione della concentrazione.
Il grafico presenta una buona linearità, considerando le basse concentrazioni,
quindi è possibile fare un’indagine quantitativa sulla presenza di acrilammide in
campioni reali.
0
5
10
15
20
25
30
0 10 20 30 40 50 60
R
µg/mL

93
ANALISI DEL CAMPIONE ALIMENTARE MEDIANTE GC-ECD
Parallelamente all’ottimizzazione del nuovo metodo proposto per la
determinazione dell’acrilammide, ho condotto un’indagine sul contenuto
dell’acrilammide in alcuni prodotti alimentari, in particolare su tre tipi di biscotti
secchi (A,B,C). Per precauzione, preferisco omettere i nomi dei biscotti utilizzati.
Per l’estrazione dell’acrilammide da questi biscotti è stato utilizzando come
solvente l’acetonitrile e come estrattore un Soxhlet. La quantità di biscotti pesata,
circa 1 grammo per ognuno, è stata finemente macinata ed omogeneizzata
utilizzando un mortaio. Una precauzione è stata quella di aggiungere una piccola
quantità di Na2SO4 anidro per assorbire l’eventuale umidità. La quantità di
campione macinata è stata posta nel Soxhlet ed estratta per circa 8 ore con
acetonitrile. L’estratto così ottenuto è concentrato al Rotavapor fino ad un volume
di circa 2 mL. 50 µL di questa soluzione vengono derivatizzati secondo il metodo
messo a punto.
Per la determinazione quantitativa dell’acrilammide è stata utilizzata la retta di
taratura precedentemente calcolata. Riporto nella seguente tabella i valori ottenuti
espressi in µg/Kg, unità imposta dalla letteratura:
campione peso (g)
acrilammide(µg/Kg)*
± DS* ± DS (%)
A 1.007 413 4.7 1.1 B 1.017 350 12.0 3.4 C 1.075 426 9.1 2.1
* valore ottenuto sulla media di 3 determinazioni
94
I valori ottenuti per questi campioni rientrano nei range riportati in letteratura,
quindi danno conferma sulla validità ed affidabilità del metodo sviluppato.
ANALISI ALLO SPETTROMETRO DI MASSA
DELL’ACRILAMMIDE DERIVATIZZATA
Una volta confermata la presenza di un derivato dell’acrilammide, ho cercato di
determinare qualitativamente questa molecola usando lo spettrometro di massa.
Questa tecnica ha permesso di confermare i risultati ottenuti con l’ECD e mi ha
permesso di studiare e caratterizzare il derivato che si forma.
Lo studio sulla formazione del derivato dell’acrilammide è stato effettuato con
uno spettrometro di massa, con sistema a trappola ionica (Polaris Q), collegato ad
un gas cromatografo (Trace GC Ultra), il tutto interfacciato ad un PC dotato di un
software d’elaborazione specifico (Xcalibur).
Di seguito riporto tutte le condizioni operative dello strumento dall’iniettore, al
forno, al rivelatore. L’iniettore PTV è regolato in modalità splitless; la
temperatura iniziale è di 50 °C; la velocità di splittaggio è di 30 mL/min;
l’apertura dello spillo è stata fissata a 1:30 min e la pulizia del setto è mantenuta
sotto flusso costante di elio ad 1 mL/min. L’iniettore è programmato secondo il
seguente schema:
Pressure
(kPa) Rate
(°C/min) Temp. (°C)
Time (min.)
Flow (mL/min)
Injection 70 50 0.05 50 Evap. 140 14.5 280 1:00
Transfer 210 14.5 280 2:00 Cleaning 14.5 310 10:00 70
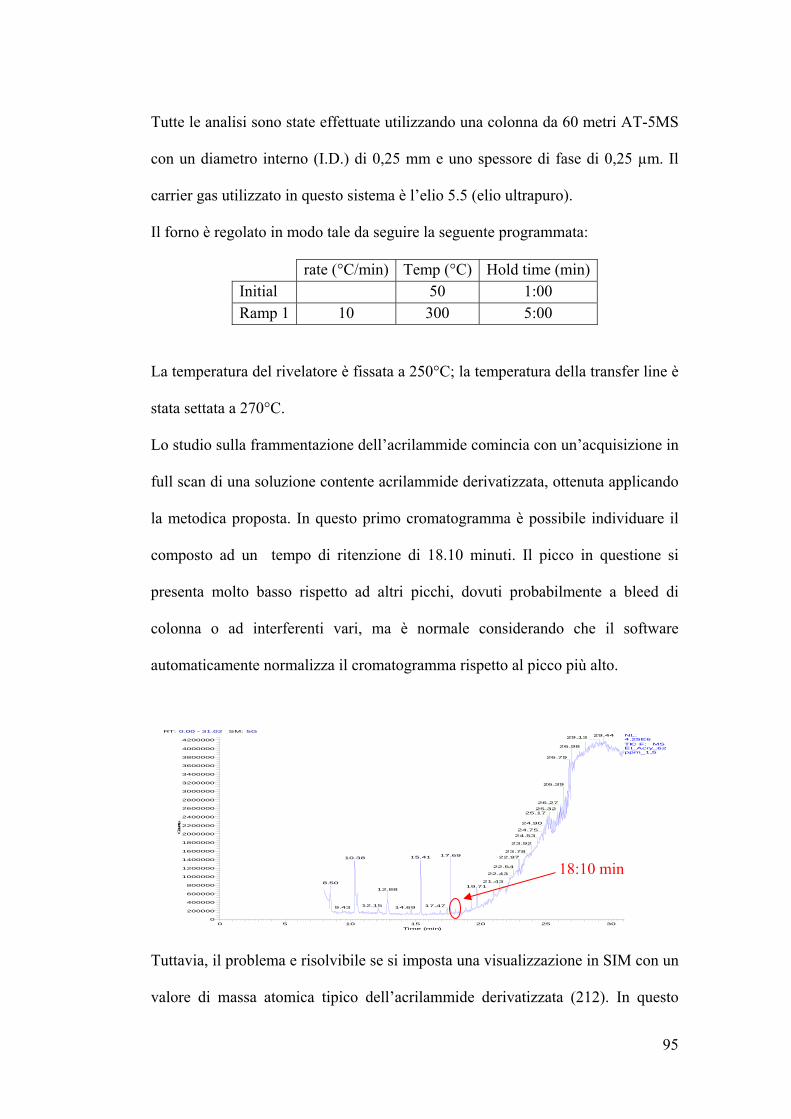
95
Tutte le analisi sono state effettuate utilizzando una colonna da 60 metri AT-5MS
con un diametro interno (I.D.) di 0,25 mm e uno spessore di fase di 0,25 µm. Il
carrier gas utilizzato in questo sistema è l’elio 5.5 (elio ultrapuro).
Il forno è regolato in modo tale da seguire la seguente programmata:
rate (°C/min) Temp (°C) Hold time (min) Initial 50 1:00 Ramp 1 10 300 5:00
La temperatura del rivelatore è fissata a 250°C; la temperatura della transfer line è
stata settata a 270°C.
Lo studio sulla frammentazione dell’acrilammide comincia con un’acquisizione in
full scan di una soluzione contente acrilammide derivatizzata, ottenuta applicando
la metodica proposta. In questo primo cromatogramma è possibile individuare il
composto ad un tempo di ritenzione di 18.10 minuti. Il picco in questione si
presenta molto basso rispetto ad altri picchi, dovuti probabilmente a bleed di
colonna o ad interferenti vari, ma è normale considerando che il software
automaticamente normalizza il cromatogramma rispetto al picco più alto.
Tuttavia, il problema e risolvibile se si imposta una visualizzazione in SIM con un
valore di massa atomica tipico dell’acrilammide derivatizzata (212). In questo
RT: 0.00 - 31.02 SM: 5G
0 5 10 15 20 25 30Time (min)
0
200000
400000
600000
800000
1000000
1200000
1400000
1600000
1800000
2000000
2200000
2400000
2600000
2800000
3000000
3200000
3400000
3600000
3800000
4000000
4200000
Counts
29.4429.13
26.98
26.79
26.39
26.27
25.3225.17
24.90
24.7524.53
23.92
23.7817.69 22.9715.4110.38
22.54
22.43
21.438.5019.71
12.88
12.15 17.4714.699.43
NL:4.25E6TIC F: MS EI_Acry_62ppm_1,5
18:10 min
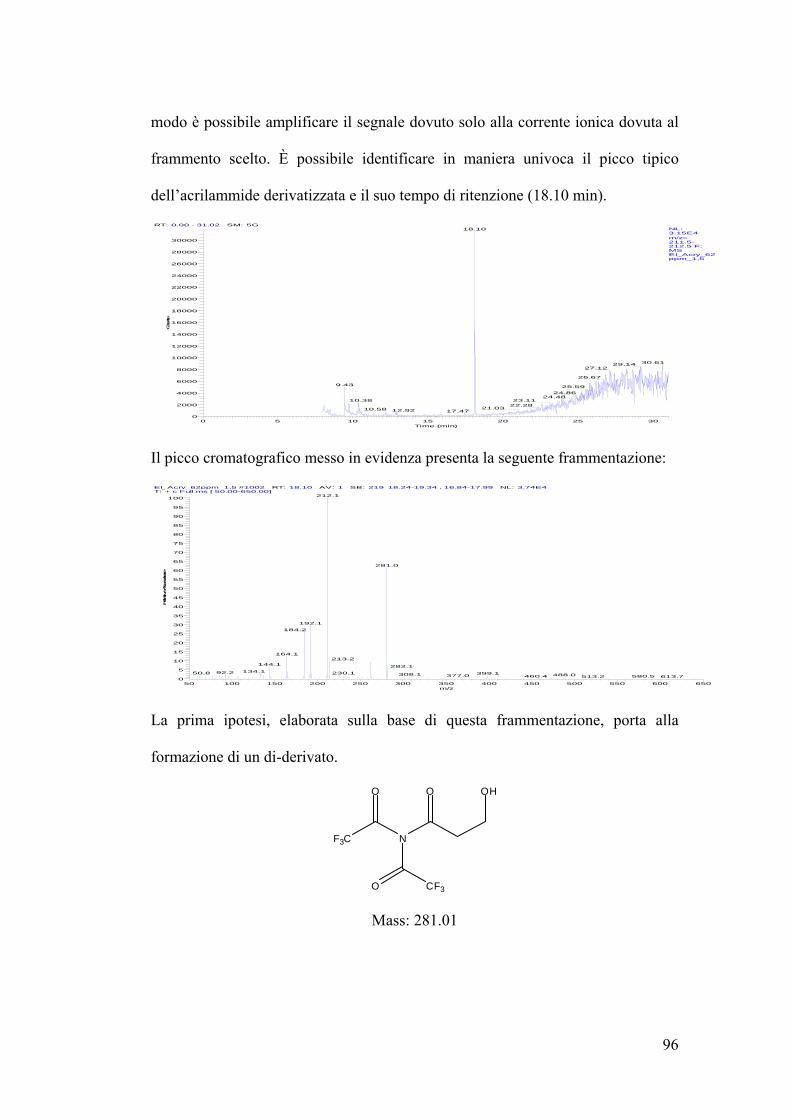
96
modo è possibile amplificare il segnale dovuto solo alla corrente ionica dovuta al
frammento scelto. È possibile identificare in maniera univoca il picco tipico
dell’acrilammide derivatizzata e il suo tempo di ritenzione (18.10 min).
Il picco cromatografico messo in evidenza presenta la seguente frammentazione:
La prima ipotesi, elaborata sulla base di questa frammentazione, porta alla
formazione di un di-derivato.
Mass: 281.01
RT: 0.00 - 31.02 SM: 5G
0 5 10 15 20 25 30Time (min)
0
2000
4000
6000
8000
10000
12000
14000
16000
18000
20000
22000
24000
26000
28000
30000
Counts
18.10
30.6129.1427.12
26.67
9.43 25.5924.86
24.4823.1110.38
22.2821.0310.58 12.92 17.47
NL:3.15E4m/z= 211.5-212.5 F: MS EI_Acry_62ppm_1,5
EI_Acry_62ppm_1,5 #1002 RT: 18.10 AV: 1 SB: 219 18.24-19.34 , 16.84-17.99 NL: 3.74E4T: + c Full ms [ 50.00-650.00]
50 100 150 200 250 300 350 400 450 500 550 600 650m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Relative Abundance
212.1
281.0
192.1
184.2
164.1213.2
144.1 282.1134.192.250.8 230.1 399.1308.1 488.0377.0 460.4 580.5513.2 613.7
CF3O
F3C
O
N
O OH

97
I picchi più significativi che possono essere notati sul frammentogramma hanno
un segnale m/z relativamente di 281, 262, 212, 192, 184 e 164. Tali frammenti
sono stati spiegati con la seguente frammentazione:
• 281, ione molecolare; ottenuto con l’inserimento di 2 gruppi –COCF3, con
l’apertura del doppio legame e l’inserimento di un gruppo –OH
• 262, ottenuto con la perdita di un fluoro dal 281
• 212, ottenuto con la perdita di un gruppo –CF3 dal 281
• 192, ottenuto con la perdita di un HF dal 212 o dalla perdita di un –CF3 e
di un HF dal 281
• 184, ottenuto con la perdita di un CO dal 212 o dalla perdita di un –CF3 ed
un CO dal 281
• 164, ottenuto con la perdita di un HF dal 184 o con la perdita di un CO dal
192
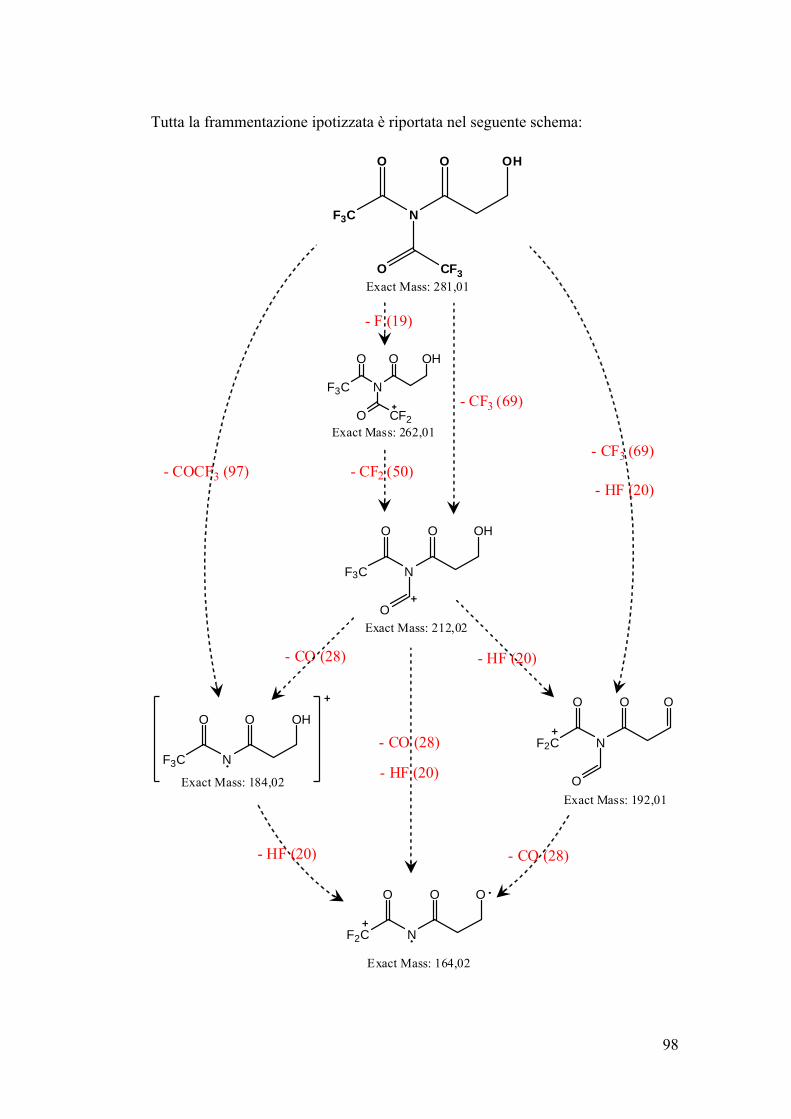
98
Tutta la frammentazione ipotizzata è riportata nel seguente schema:
CF2O
F3C
O
N
O OH
O
F3C
O
N
O OH
F3C
O
N
O OH
O
F2C
O
N
O O
F2C
O
N
O O
- COCF3 (97)
- F (19)
- CF3 (69)
- CF2 (50)
- CO (28) - HF (20)
- HF (20) - CO (28)
- CO (28)
- HF (20)
CF3O
F3C
O
N
O OH
- CF3 (69)
- HF (20)
Exact Mass: 281,01
Exact Mass: 262,01
Exact Mass: 212,02
Exact Mass: 184,02Exact Mass: 192,01
Exact Mass: 164,02

99
Per ottenere la conferma che i frammenti risultanti dal primo frammentogramma
sono caratteristici della molecola di-derivata dell’acrilammide e per avere
conferma della frammentazione ipotizzata, sono state acquisite prove in MSMS su
ogni frammento caratteristico. I risultati ottenuti sono riportati nell’allegato 9.
Nell’allegato è possibile vedere la corrispondenza tra il tempo di ritenzione
dell’acrilammide derivatizzata (18.10 minuti) e i rispettivi frammentogrammi
ottenuti in MSMS con ione precursore specifico. Tutti i cromatogrammi sono stati
acquisiti in SIM 212 (frammento più abbondante). Inoltre l’acquisizione dei vari
cromatogrammi parte da 15 minuti e finisce a 20 minuti per una questione di
comodità.
I frammentogrammi riportati sono quelli che meglio rendono l’idea di
frammentazione in MSMS (ione precursore circa un decimo dei vari ioni figli).
Per ottenere questi risultati ogni frammentogramma riportato è stato provato con
vari filtri (funzione disponibile sul software di elaborazione Xcalibur). Purtroppo
non è possibile generalizzare il valore del filtro applicato, poiché ogni frammento
preso in considerazione risponde in maniera differente, quindi ad ogni frammento
va associato un valore che deve essere determinato sperimentalmente di volta in
volta. La determinazione del valore di questo filtro è rapida e semplice e può
essere fatta con un’unica iniezione del composto e va chiaramente ripetuta per
ogni frammento considerato.
Tutti i frammentogrammi in MSMS dei vari frammenti analizzati concordano
sulla frammentazione ipotizzata precedentemente, quindi in una prima analisi
sembra essere confermata la formazione del di–derivato dell'acrilammide. Ma . . .
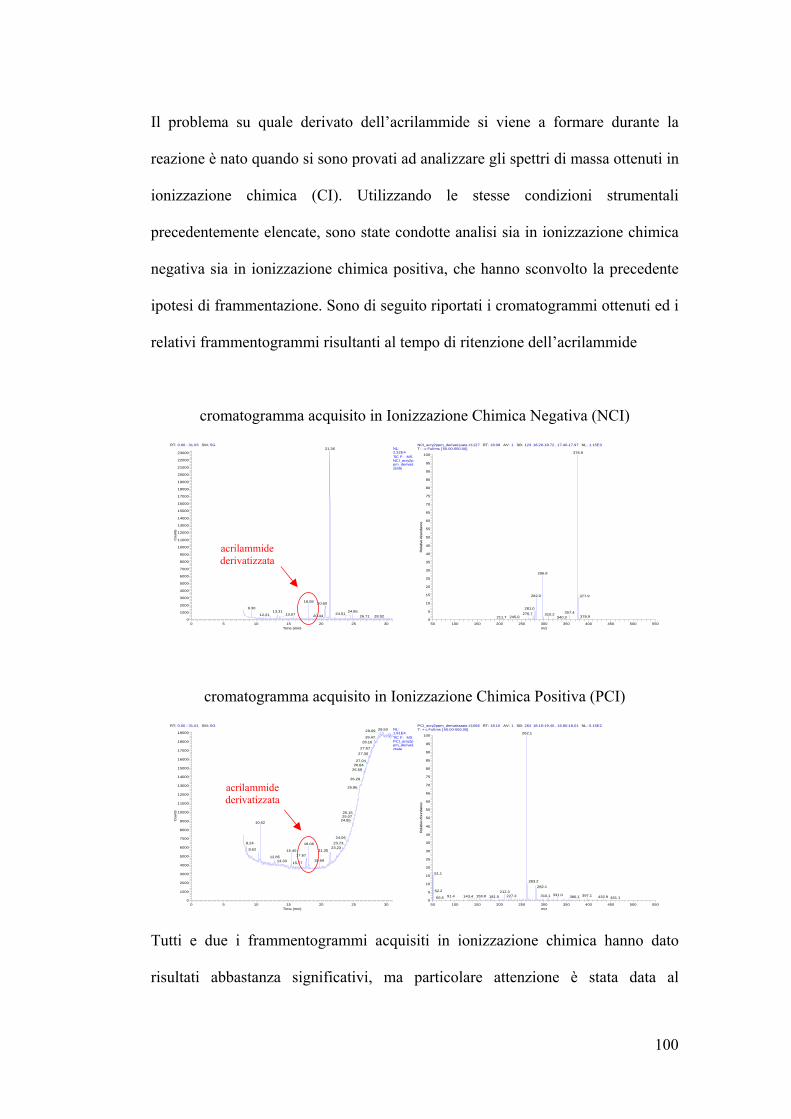
100
Il problema su quale derivato dell’acrilammide si viene a formare durante la
reazione è nato quando si sono provati ad analizzare gli spettri di massa ottenuti in
ionizzazione chimica (CI). Utilizzando le stesse condizioni strumentali
precedentemente elencate, sono state condotte analisi sia in ionizzazione chimica
negativa sia in ionizzazione chimica positiva, che hanno sconvolto la precedente
ipotesi di frammentazione. Sono di seguito riportati i cromatogrammi ottenuti ed i
relativi frammentogrammi risultanti al tempo di ritenzione dell’acrilammide
cromatogramma acquisito in Ionizzazione Chimica Negativa (NCI)
cromatogramma acquisito in Ionizzazione Chimica Positiva (PCI)
Tutti e due i frammentogrammi acquisiti in ionizzazione chimica hanno dato
risultati abbastanza significativi, ma particolare attenzione è stata data al
RT: 0.00 - 31.03 SM: 5G
0 5 10 15 20 25 30Time (min)
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
10000
11000
12000
13000
14000
15000
16000
17000
18000
19000
20000
21000
22000
23000
Cou
nts
21.36
18.08 20.609.30
13.31 24.8524.5113.5712.21 20.44 26.71 28.92
NL:2.32E4TIC F: MS NCI_acry2ppm_derivatizzata
NCI_acry2ppm_derivatizzata #1127 RT: 18.08 AV: 1 SB: 123 18.20-18.72 , 17.40-17.97 NL: 1.15E3T: - c Full ms [ 50.00-550.00]
50 100 150 200 250 300 350 400 450 500 550m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ativ
e A
bund
ance
376.9
296.8
282.0 377.9
281.0357.4276.7 310.2 378.9245.0211.7 340.3
RT: 0.00 - 31.01 SM: 5G
0 5 10 15 20 25 30Time (min)
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
10000
11000
12000
13000
14000
15000
16000
17000
18000
19000
Cou
nts
29.5029.09
28.4728.16
27.6727.36
27.0426.84
26.59
26.29
25.86
25.1525.07
24.8510.62
24.068.24 23.7318.08
23.238.62 21.3515.4017.6712.85
19.6913.33 15.77
NL:1.91E4TIC F: MS PCI_acry2ppm_derivatizzata
PCI_acry2ppm_derivatizzata #1066 RT: 18.10 AV: 1 SB: 264 18.15-19.40 , 16.80-18.01 NL: 5.15E2T: + c Full ms [ 50.00-550.00]
50 100 150 200 250 300 350 400 450 500 550m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ativ
e A
bund
ance
262.1
51.1
263.2282.1
52.2 212.3331.0 397.1310.1227.3158.891.4 143.4 181.5 380.1 432.966.6 451.1
acrilammide derivatizzata
acrilammide derivatizzata

101
cromatogramma acquisito in ionizzazione chimica negativa. Infatti analizzando
questo cromatogramma in corrispondenza del tempo di ritenzione tipico
dell’acrilammide derivatizzata (18.10 minuti), è possibile notare un frammento
molto alto rispetto a tutti gli altri, con un rapporto m/z di 377. Questo frammento
ha dato l’input ad ipotizzare la formazione di un tri-derivato dell’acrilammide.
L’analisi dei due cromatogrammi ha portato, quindi, all’ipotesi di una seconda
frammentazione, dovuta alla formazione del tri-derivato dell’acrilammide:
mass: 376.99
La spiegazione della seconda ipotesi, vale a dire della formazione del tri-derivato
dell’acrilammide, è stata ricercata partendo da due cromatogrammi
Cromatogramma A Cromatogramma B
CF3O
O
N
O O CF3
O
F3C
RT: 0.00 - 31.02 SM: 5G
0 5 10 15 20 25 30Time (min)
0
5000
10000
15000
20000
25000
30000
35000
40000
45000
50000
55000
60000
Cou
nts
18.09
28.92
28.0626.86
25.509.44 25.38
24.1822.99
10.26 21.1015.7212.14
NL:6.17E4m/z= 211.5-212.5 F: MS Acry2h
RT: 0.00 - 31.02 SM: 5G
0 5 10 15 20 25 30Time (min)
0
5000
10000
15000
20000
25000
30000
35000
40000
45000
50000
55000
60000
65000
70000
75000
80000
85000
90000
95000
100000
105000
110000
115000
120000
125000
130000
135000
Cou
nts
18.09
28.5127.3526.389.44 25.53
24.9523.6610.26 22.0012.15 15.73
NL:1.38E5m/z= 211.5-212.5 F: MS Ei_Acry3minuti_fullscan
Acry2h #1360 RT: 18.09 AV: 1 SB: 103 18.19-18.53 , 17.53-17.94 NL: 6.89E4T: + c Full ms [ 40.00-400.00]
50 100 150 200 250 300 350 400m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Relative Abundance
212.1
281.0
192.1258.0
308.0184.1
164.1213.2
262.1377.0
144.1 230.0 282.0 309.1134.178.0 106.2 358.051.1 253.1 378.0307.1 332.0

102
Il cromatogramma A preso in considerazione è relativo al campione acrilammide
derivatizzata, dove la reazione si è prolungata a 2 ore, ma è possibile fare le stesse
considerazioni anche su campioni dove il tempo di reazione è stato inferiore (3
minuti, cromatogramma B). L’unico picco visibile in modalità SIM a 212, che
rappresenta il frammento comune sia alla acrilammide di-derivatizzata sia tri-
derivatizzata, è quello a RT di 18.09. Tutti gli altri picchi sembrano essere bleed
di colonna. Relativamente a questo picco, oltre ai frammenti indagati e spiegati
(frammentazione del 281) è possibile notare anche altri frammenti, in particolare
il 377, 358, 308, 258, 230 e il 202. Nel frammentogramma è possibile notare la
presenza del frammento 378 con altezza del 10% rispetto al frammento 377, tale
rapporto è compatibile con l’abbondanza isotopica complessiva della molecola.
I frammenti più abbondanti per questa seconda ipotesi di frammentazione sono
stati spiegati nel seguente modo:
• 377, ione molecolare, ottenuto con inserimento di 3 molecole di –COCF3
ed una molecola d’acqua
• 358, ottenuto con la perdita di –F dal 377
• 308, ottenuto con la perdita di un –CF3 dal 377
• 280, ottenuto con la perdita di –CO dal 308 o dalla perdita di un –CF3 ed
un CO dal 377
• 258, ottenuto con la perdita di un –CF2 dal 308 o dalla perdita di un –CF3
ed un –CF2 dal 377
• 230, ottenuto dalla perdita di un –CO dal 258
• 202, ottenuto dalla perdita di un –CO dal 230

103
È da notare che tutte queste frammentazioni non danno il frammento 281. Il 281 e
tutte le sue frammentazioni sono spiegabili se il frammento 280 lega un idrogeno.
Per rendere valida anche la prima ipotesi di frammentazione occorre che il tri-
derivato perde complessivamente un gruppo trifluoroacetato (-COCF3) e lega un
H+, formando il di-derivato.
Considerando però che il tempo di ritenzione dei due composti è praticamente lo
stesso, è un controsenso, in cromatografia, affermare che si tratta di due composti
differenti che vengono rilevati con lo stesso tempo di ritenzione.
Tuttavia l’ipotesi sulla formazione del di-derivato dell’acrilammide è spiegabile e
plausibile se lo si intende come prodotto secondario del tri-derivato per reazioni
non controllabili che avvengono all’interno della trappola ionica stessa.
Anche in questo caso, per dare senso e conferma, alle frammentazioni ipotizziate
nella formazione del tri-derivato, sono stati condotti esperimenti MSMS. Tutti i
risultati ottenuti sono riportati nell’allegato 10.
L’analisi MSMS ha dato conferma della frammentazione ipotizzata quindi della
formazione del tri-derivato dell’acrilammide.
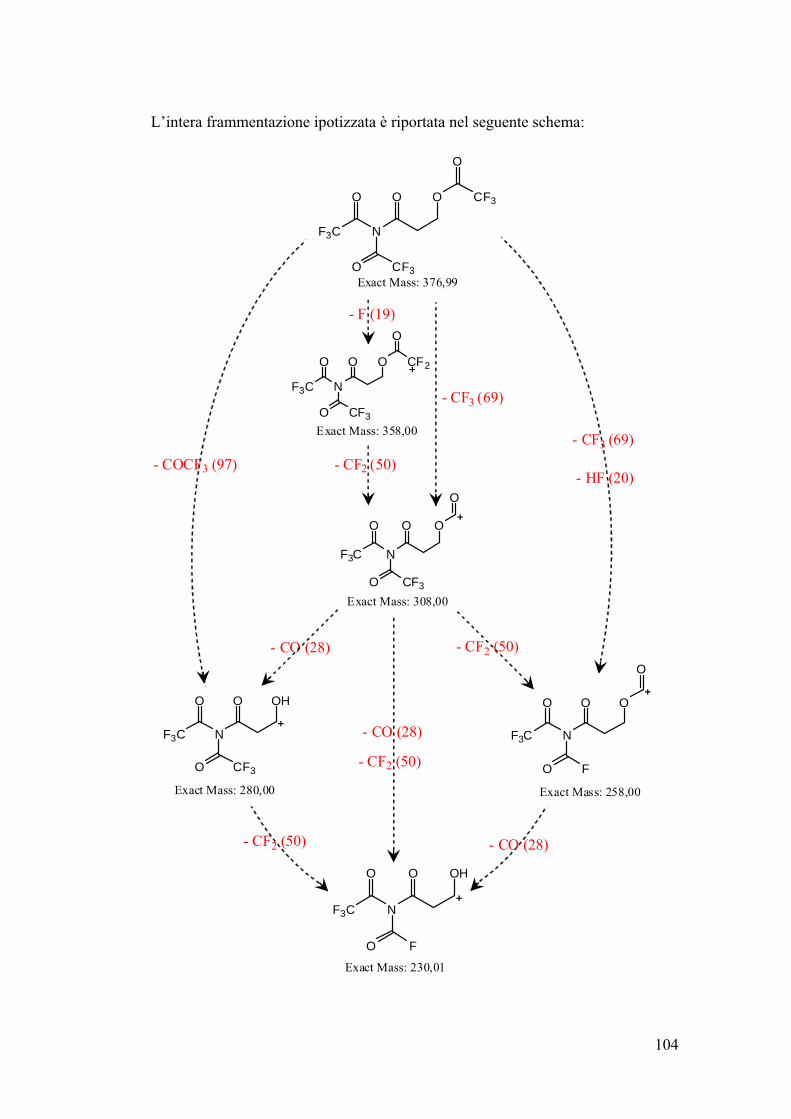
104
L’intera frammentazione ipotizzata è riportata nel seguente schema:
- COCF3 (97)
- F (19)
- CF3 (69)
- CF2 (50)
- CO (28)
- CF2 (50) - CO (28)
- CO (28)
- CF3 (69)
- HF (20)
CF3O
O
N
O O CF3
O
F3C
CF3O
O
N
O O CF2
O
F3C
CF3O
O
N
O O
O
F3C
CF3O
O
N
O OH
F3C
Exact Mass: 280,00
Exact Mass: 308,00
Exact Mass: 358,00
Exact Mass: 376,99
FO
O
N
O O
O
F3C
Exact Mass: 258,00
FO
O
N
O OH
F3C
Exact Mass: 230,01
- CF2 (50)
- CF2 (50)

105
LOD E LOQ OTTENUTI IN GC-MS
Nelle condizioni sviluppate e messe a punto, sono andato alla ricerca del LOD e
del LOQ utilizzando come rivelatore lo spettrometro di massa. Tutte le
acquisizioni sono state fatte in modalità SIM con un frammento base di 212. Nel
seguente schema riporto il LOD e il LOQ con la relativa deviazione standard
calcolata su tre determinazioni.
LOD LOQ
quantità
(pg) DS*
DS* (%)
quantità (pg)
DS* DS* (%)
acrilammide derivatizzata 6 ± 1.1 ± 18.8 52 ± 3.25 ± 6.25
ERRORE INTRADAY ED INTERDAY AL GC-MS
Considerando che il limite di quantificazione è di 52 pg/µL, quindi 52 ppb, ho
calcolato l’errore intraday ed interday considerando una concentrazione di 100
ppb. Ho ripetuto tre analisi in due giorni differenti. Riporto le stime degli errori
nelle seguenti tabelle, calcolate sulla media di tre determinazioni giornaliere
giorno 1 giorno 2 interday
quantità
(pg) DS*
DS* (%)
DS* DS* (%)
DS* DS* (%)
acrilammide derivatizzata
100 ±4.80 ±4.80 ±3.94 ±3.94 ±6.21 ±6.21
*DS calcolata su tre determinazioni
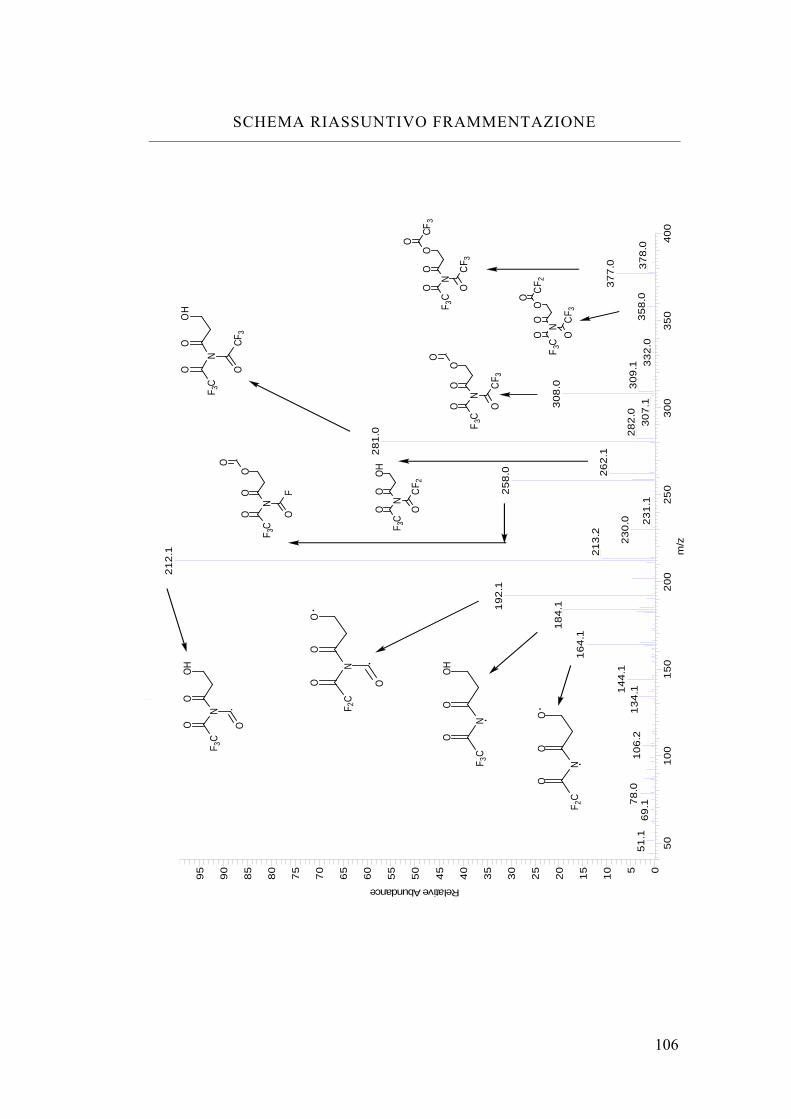
106
SCHEMA RIASSUNTIVO FRAMMENTAZIONE
Acr
y2h
#136
0R
T:18
.09
AV
:1
SB
:83
618
.41-
21.3
9 , 1
4.46
-17.
70N
L:6.
87E
4T:
+ c
Ful
l ms
[ 40.
00-4
00.0
0]
5010
015
020
025
030
035
040
0m
/z
05101520253035404550556065707580859095100
Relative Abundance
212.
1
281.
0
192.
125
8.0
308.
018
4.1
164.
121
3.2
262.
137
7.0
144.
123
0.0
282.
030
9.1
78.0
134.
110
6.2
51.1
358.
037
8.0
69.1
307.
123
1.1
332.
0
CF3
OO
N
OO
CF3
O
F 3C
CF3
OONO
OCF
2
O
F 3C
CF3
OO
N
OO
O
F 3C
CF3
O
F 3C
O
N
OO
H
FOO
N
OO
O
F 3C
CF2
O
F 3C
O
N
OOH
O
F 3C
O
N
OOH
O
F 2C
O
N
OO
F 3C
O
N
OOH
F 2C
O
N
OO

107
DETERMINAZIONE QUANTITATIVA DELL’ACRILAMMIDE
IN SPETTROMETRIA DI MASSA
Come prima cosa ho costruito una retta di taratura per avere una corrispondenza e
una riproducibilità dei dati ottenuti con l’analisi in ECD, avendo conservato gli
estratti di biscotto. I risultati ottenuti sono riportati nel seguente schema:
campioneGC-ECD
(µg/Kg ± DS)GC-MS
(µg/Kg ± DS)A 413 ± 4.7 381 ± 19.2 B 350 ± 12.0 226 ± 24.6 C 426 ± 9.1 395 ± 18.9
E’ possibile notare i valori di acrilammide determinati con i due sistemi possono
essere considerati dello stesso ordine di grandezza. Con questa approssimazione
ho deciso di effettuare una determinazione con un altro campione alimentare.
La determinazione dell’acrilammide con lo spettrometro di massa è stata fatta su
patatine fritte in busta. L’acrilammide è stata estratta dal campione reale
utilizzando l’estrattore Soxhlet, utilizzando i tempi e le modalità previste dalla
metodica.
È stata costruita una retta di taratura utilizzando soluzioni standard di acrilammide
derivatizzata a concentrazione crescente comprese tra 0.1 e 10 ppm.
Inoltre tutti i cromatogrammi sono stati acquisiti in modalità SIM, settando
opportunamente lo strumento in base ai tempi di ritenzione e ai frammenti
caratteristi sia dell’acrilammide sia dello standard interno.
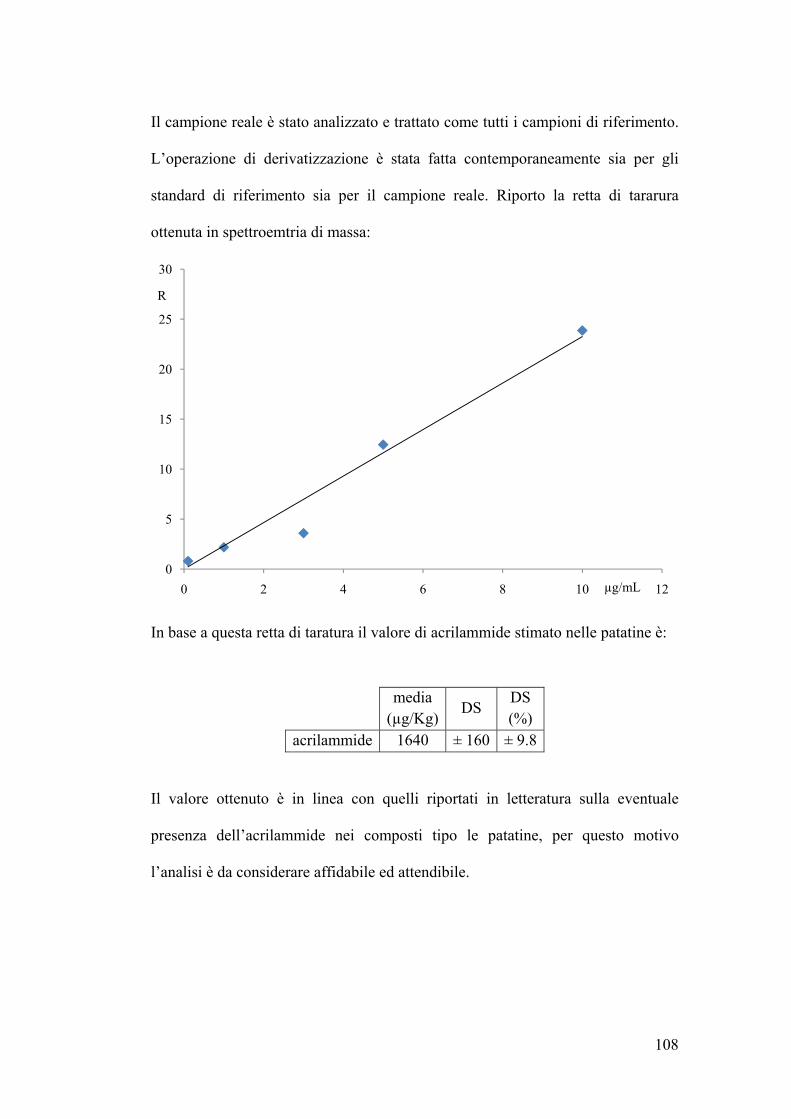
108
Il campione reale è stato analizzato e trattato come tutti i campioni di riferimento.
L’operazione di derivatizzazione è stata fatta contemporaneamente sia per gli
standard di riferimento sia per il campione reale. Riporto la retta di tararura
ottenuta in spettroemtria di massa:
In base a questa retta di taratura il valore di acrilammide stimato nelle patatine è:
media
(µg/Kg)DS
DS (%)
acrilammide 1640 ± 160 ± 9.8
Il valore ottenuto è in linea con quelli riportati in letteratura sulla eventuale
presenza dell’acrilammide nei composti tipo le patatine, per questo motivo
l’analisi è da considerare affidabile ed attendibile.
0
5
10
15
20
25
30
0 2 4 6 8 10 12
R
µg/mL

109
CONCLUSIONI
Il metodo proposto permette di determinare in modo attendibile l’acrilammide nei
prodotti alimentari.
La derivatizzazione della molecola con composti alogenati aumenta la sensibilità
del metodo ed è particolarmente adatta per essere applicata a matrici complesse
quali quelle alimentari.
Il metodo, applicato sia su soluzioni standard che su campioni reali, ha mostrato
una elevata precisione e accuratezza analitica.
Il metodo innovativo standardizzato ha consentito l’eliminazione della fase di
clean-up nella preparazione del campione con una significativa riduzione del
tempo di analisi e del costo analitico legato all’impiego delle cartucce per
l’estrazione in fase solida.
Inoltre, sono sufficienti per l’analisi pochi grammi di campione e piccoli volumi
di solventi con riduzione di costi per l’acquisto dei reagenti e per lo smaltimento
dei residui.
Il metodo sviluppato può essere applicato per le analisi routinarie
dell’acrilammide per la valutazione della qualità e della sicurezza d’uso degli
alimenti vista l’elevata sensibilità strumentale e la praticità di applicazione.

110
BIBLIOGRAFIA [1] F. Pedreschi, K. Kaack, K. Granby; Reduction of acrylamide formation in potato
slices during frying; Lebensm.–Wiss. u. –Technol., 37, 679–685, (2004)
[2] K. Svensson, L. Abramsso, W. Becker, A. Glynn, K.E. Hellenäs, Y. Lind, J. Rosén; Dietary intake of acrylamide in Sweden; Food and Chemical Toxicology, 41, 1581-1586. (2003)
[3] A. Becalski, B. P.Y. Lau, D. Lewis, S.W. Seaman, S. Hayward, M. Sahagian,
M. Ramesh, Y. Leclerc; Acrylamide in French Fries: Influence of Free Amino Acids and Sugars; Journal of Agricultural and Food Chemistry, 52, 3801-3806. (2004)
[4] D.V. Zyzak, R.A. Sanders, M. Stojanovic, D.H. Tallmadge, B.L. Eberhart,
D.K Ewald, D.C. Gruber, T.R. Morsch, M.A. Strothers, G.P. Rizzi, M.D Villagran; Acrylamide Formation Mechanism in Heated Foods; Journal of Agricultural and Food Chemistry, 51, 4782-4787. (2003)
[5] T.M. Amrein, B. Schönbächler, F. Escher, R. Amadò; Acrylamide in Gingerbread:
Critical Factors for Formation and Possible Ways for Reduction; Journal of Agricultural and Food Chemistry, 52, 4282-4288. (2004)
[6] M. Friedman; Chemistry, Biochemistry and Safety of Acrylamide. A Review; Journal of
Agricultural and Food Chemistry, 51, 4504-4526. (2003) [7] J. S. E. Williams; Influence of variety and processing condition on acrylamide levels in
fried potato crisps; Food Chemistry, 90, 857-881. (2005) [8] D. Taubert, S. Harlfinger, L. Henkes, R. Berkels, E. Schömig; Influence of
Processing Parameters on Acrylamide Formation during Frying of Potatoes; Journal of Agricultural and Food Chemistry, 52, 2735-2739. (2004)
[9] Summary - Acrylamide in Heat-Processed Foods. 2002. www.slv.se
[10] Opinion of the Scientific Committee on Food on new findings regarding the presence of
acrylamide in food. July 3, 2002. www.europa.eu.int [11] Working Group 1: Mechanisms of formation of Acrylamide in Food. Summary Report.
Chicago, 28-30 Ottobre 2002 [12] A. F. Lagalante, M. Felter; Silylation of Acrylamide for Analysis by Solid-Phase
Microextraction/Gas Chomatography/Ion-Trap Mass Spectrometry; Journal of Agricultural and Food Chemistry, 52, 3744-3748. (2004)
[13] F. Tateo, M. Bononi; A GC/MS method for the routine determination of acrylamide in Food; Italian Journal Food Science, 15, 149-151. (2003)
[14] K; Hoenicke, R; Gatermann, W. Harder; Analysis of acrylamide in different foodstuffs using liquid chromatography-tandem mass spectrometry and gas chromatography-tandem mass spectrometry; Analytica Chimica Acta; 520 (1-2), 207-215. (2002)
[15] Dionex, Application Note 409. Fast determination of Acrylamide in Food Samples Using Accelerated Solvent Extraction (ASE) Followed by Ion Chromatography with UV or MS Detection
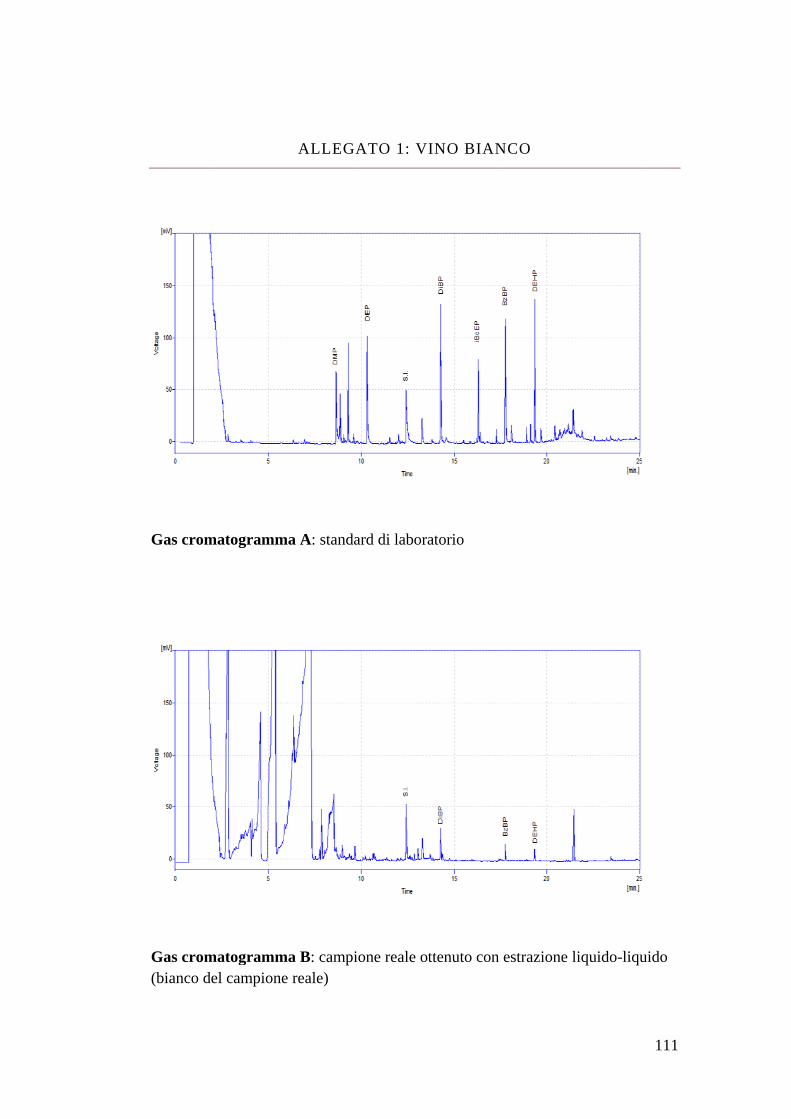
Gas crom
Gas crom(bianco de
matogramm
matogrammel campione
ALLEGA
ma A: standa
ma B: campie reale)
ATO 1: VI
ard di labora
one reale ot
INO BIAN
atorio
ttenuto con
NCO
estrazione liquido-liqu
111
uido
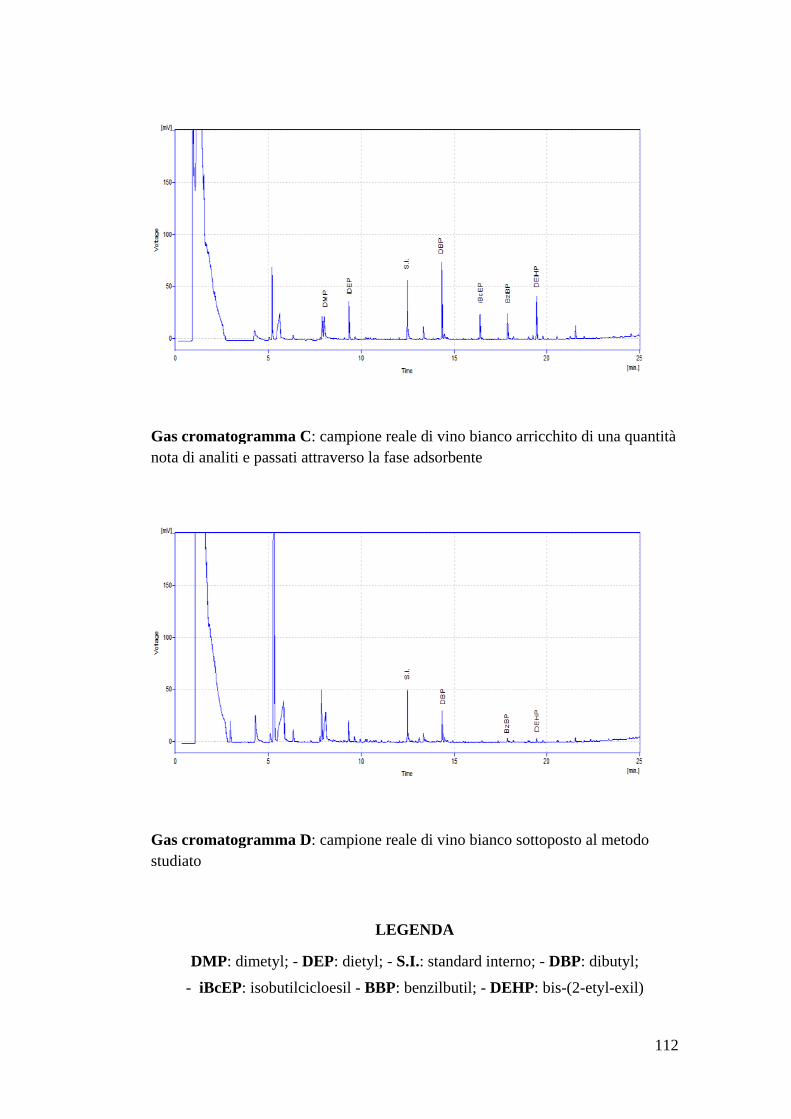
Gas cromnota di ana
Gas cromstudiato
DM
- iB
matogrammaliti e passa
matogramm
MP: dimetyl;
cEP: isobut
ma C: campiati attraverso
ma D: campi
; - DEP: die
tilcicloesil -
ione reale do la fase ad
ione reale d
LEGEN
etyl; - S.I.: s
- BBP: benz
i vino biancsorbente
i vino bianc
NDA
standard int
zilbutil; - D
co arricchito
co sottopost
terno; - DBP
DEHP: bis-(
o di una qua
to al metodo
P: dibutyl;
2-etyl-exil)
112
antità
o
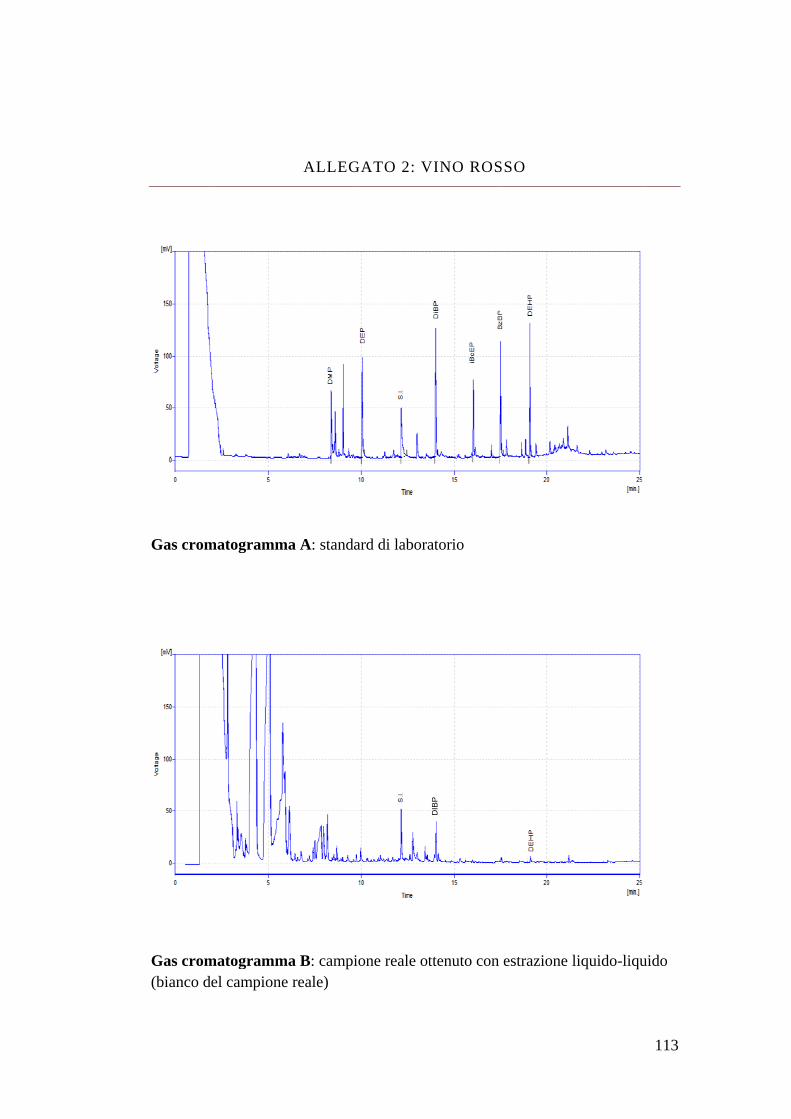
Gas crom
Gas crom(bianco de
matogramm
matogrammel campione
ALLEG
ma A: standa
ma B: campie reale)
GATO 2: V
ard di labora
one reale ot
VINO ROS
atorio
ttenuto con
DIB
P
SSO
estrazione liquido-liqu
113
uido
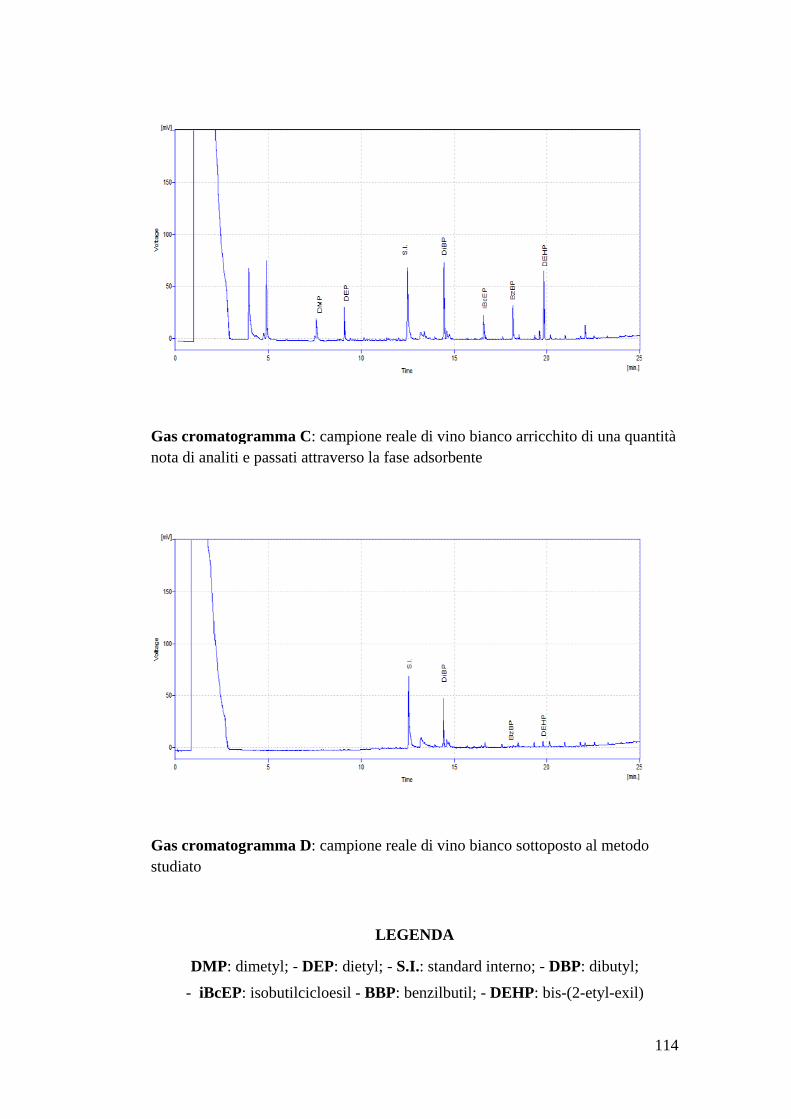
Gas cromnota di ana
Gas cromstudiato
DM
- iB
matogrammaliti e passa
matogramm
MP: dimetyl;
cEP: isobut
ma C: campiati attraverso
ma D: campi
; - DEP: die
tilcicloesil -
ione reale do la fase ad
ione reale d
LEGEN
etyl; - S.I.: s
- BBP: benz
i vino biancsorbente
i vino bianc
NDA
standard int
zilbutil; - D
co arricchito
co sottopost
terno; - DBP
DEHP: bis-(
o di una qua
to al metodo
P: dibutyl;
2-etyl-exil)
114
antità
o
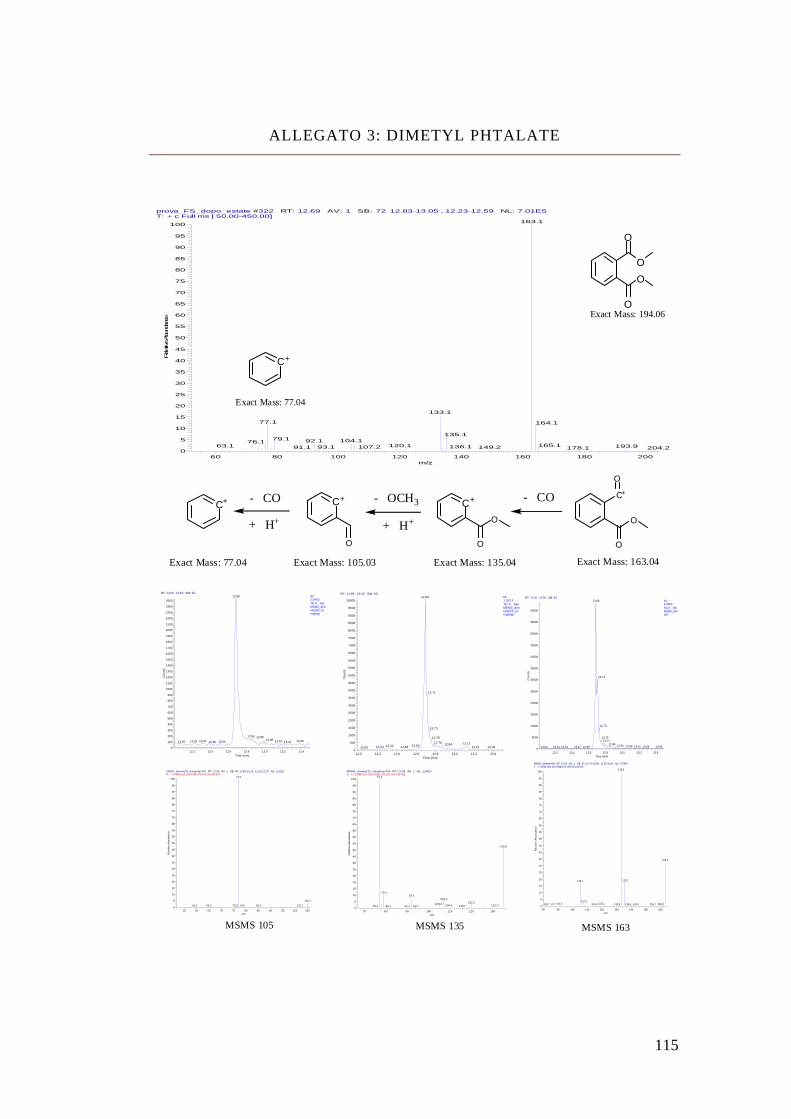
115
ALLEGATO 3: DIMETYL PHTALATE
prova_FS_dopo_estate #322 RT: 12.69 AV: 1 SB: 72 12.83-13.05 , 12.23-12.59 NL: 7.01E5T: + c Full ms [ 50.00-450.00]
60 80 100 120 140 160 180 200m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Relative Abu
ndan
ce
163.1
133.1
77.1 164.1
135.179.1 104.192.176.1
165.1120.163.1 193.9136.1107.293.1 149.291.1 178.1 204.2
O
O
O
OExact Mass: 194.06
C+
Exact Mass: 77.04
MSMS_dimetyl #94 RT: 12.69 AV: 1 SB: 29 12.74-12.89 , 12.58-12.64 NL: 6.70E4T: + c SRM ms2 [email protected] [ 80.00-165.00]
80 90 100 110 120 130 140 150 160m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ativ
e A
bund
ance
133.2
163.2
135.2105.1
107.2120.191.2 162.4132.6 136.481.0 87.7 114.8 155.7141.5
MSMS_dimetyl105_energihigt #97 RT: 12.68 AV: 1 SB: 49 12.80-13.18 , 12.28-12.57 NL: 3.01E3F: + c SRM ms2 [email protected] [ 52.00-106.00]
55 60 65 70 75 80 85 90 95 100 105m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ativ
e A
bund
ance
77.0
105.1
76.2 85.2 102.178.065.059.2
- OCH3
+ H+
- COC+ - CO
+ H+
MSMS 163MSMS 135MSMS 105
O
C+
O
O
Exact Mass: 163.04
C+
O
O
Exact Mass: 135.04
C+
O
MSMS_dimetyl135_energihigt #95 RT: 12.68 AV: 1 NL: 1.89E3F: + c SRM ms2 [email protected] [ 67.00-136.00]
70 80 90 100 110 120 130m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ativ
e A
bund
ance
77.0
135.2
79.192.1
107.1120.1105.0 131.0109.0 115.775.4 92.780.1 91.1
Exact Mass: 105.03
RT: 12.01 - 13.50 SM: 5G
12.2 12.4 12.6 12.8 13.0 13.2 13.4Time (min)
0
5000
10000
15000
20000
25000
30000
35000
40000
45000
50000
55000
60000
Cou
nts
12.69
12.71
12.73
12.7512.77
12.84 12.90 12.99 13.15 13.28 13.4412.31 12.4712.24 12.5512.04
NL:6.33E4TIC F: MS MSMS_dimetyl
RT: 11.99 - 13.50 SM: 5G
12.0 12.2 12.4 12.6 12.8 13.0 13.2 13.4Time (min)
0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
5500
6000
6500
7000
7500
8000
8500
9000
9500
10000
Cou
nts
12.69
12.71
12.73
12.7612.78 13.1212.8412.6312.3212.24 12.48 13.3813.2112.05
NL:1.01E4TIC F: MS MSMS_dimetyl133_energihigt
RT: 12.00 - 13.50 SM: 5G
12.2 12.4 12.6 12.8 13.0 13.2 13.4Time (min)
0
100
200
300
400
500
600
700
800
900
1000
1100
1200
1300
1400
1500
1600
1700
1800
1900
2000
2100
2200
2300
2400
2500
Cou
nts
12.68
12.83 12.8612.98 13.3912.32 13.1312.24 12.5312.05 12.38 13.21
NL:2.54E3TIC F: MS MSMS_dimetyl105_energihigt
Exact Mass: 77.04
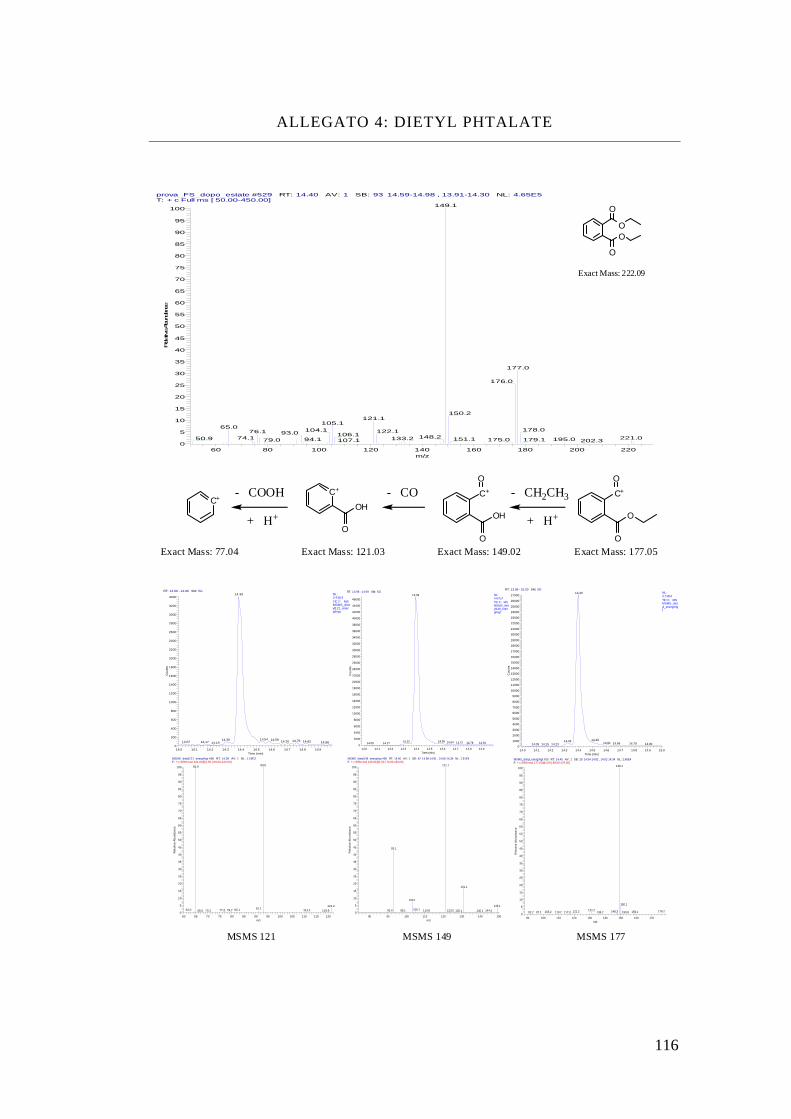
116
ALLEGATO 4: DIETYL PHTALATE
MSMS_dietyl149_energihigt #56 RT: 14.40 AV: 1 SB: 42 14.58-14.91 , 14.06-14.34 NL: 1.91E4F: + c SRM ms2 [email protected] [ 74.00-150.00]
80 90 100 110 120 130 140 150m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ativ
e A
bund
ance
121.1
93.1
131.1
103.1
149.1105.191.4 123.0110.8 144.598.1 126.1 140.1
MSMS_dietyl121_energihigt #56 RT: 14.39 AV: 1 NL: 1.19E3F: + c SRM ms2 [email protected] [ 60.00-122.00]
60 65 70 75 80 85 90 95 100 105 110 115 120m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ativ
e A
bund
ance
93.065.0
121.291.182.162.0 111.179.777.2 119.670.165.6
MSMS_dietyl_energihigt #55 RT: 14.40 AV: 1 SB: 28 14.54-14.82 , 14.02-14.34 NL: 2.86E4F: + c SRM ms2 [email protected] [ 88.00-179.00]
90 100 110 120 130 140 150 160 170m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ativ
e A
bund
ance
149.1
150.1
131.2 176.2121.2 148.3103.2 159.192.2 150.8117.3110.2 136.297.2
prova_FS_dopo_estate #529 RT: 14.40 AV: 1 SB: 93 14.59-14.98 , 13.91-14.30 NL: 4.65E5T: + c Full ms [ 50.00-450.00]
60 80 100 120 140 160 180 200 220m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Relative Abu
ndan
ce
149.1
177.0
176.0
150.2121.1
105.165.0 178.0104.1 122.176.1 93.0 106.1 148.274.1 221.0133.250.9 151.194.1 195.0175.0 179.179.0 107.1 202.3
OO
O
O
Exact Mass: 222.09
C+
O
O
OExact Mass: 177.05
C+
OH
O
OExact Mass: 149.02
C+
OH
O
Exact Mass: 121.03
C+
Exact Mass: 77.04
- CH2CH3
+ H+
- CO- COOH
+ H+
MSMS 121 MSMS 149 MSMS 177
RT: 13.99 - 15.00 SM: 5G
14.0 14.1 14.2 14.3 14.4 14.5 14.6 14.7 14.8 14.9 15.0Time (min)
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
10000
11000
12000
13000
14000
15000
16000
17000
18000
19000
20000
21000
22000
23000
24000
25000
26000
27000
Cou
nts
14.40
14.4914.32 14.61 14.66 14.79 14.9014.2314.09 14.15
NL:2.71E4TIC F: MS MSMS_dietyl_energihigt
RT: 13.98 - 14.99 SM: 5G
14.0 14.1 14.2 14.3 14.4 14.5 14.6 14.7 14.8 14.9Time (min)
0
2000
4000
6000
8000
10000
12000
14000
16000
18000
20000
22000
24000
26000
28000
30000
32000
34000
36000
38000
40000
42000
44000
46000
Cou
nts
14.39
14.5614.32 14.64 14.72 14.78 14.9114.05 14.17
NL:4.67E4TIC F: MS MSMS_dietyl149_energihigt
RT: 13.99 - 14.99 SM: 5G
14.0 14.1 14.2 14.3 14.4 14.5 14.6 14.7 14.8 14.9Time (min)
0
200
400
600
800
1000
1200
1400
1600
1800
2000
2200
2400
2600
2800
3000
3200
3400
Cou
nts
14.39
14.5414.30 14.59 14.7614.70 14.8214.02 14.17 14.9614.19
NL:3.41E3TIC F: MS MSMS_dietyl121_energihigt
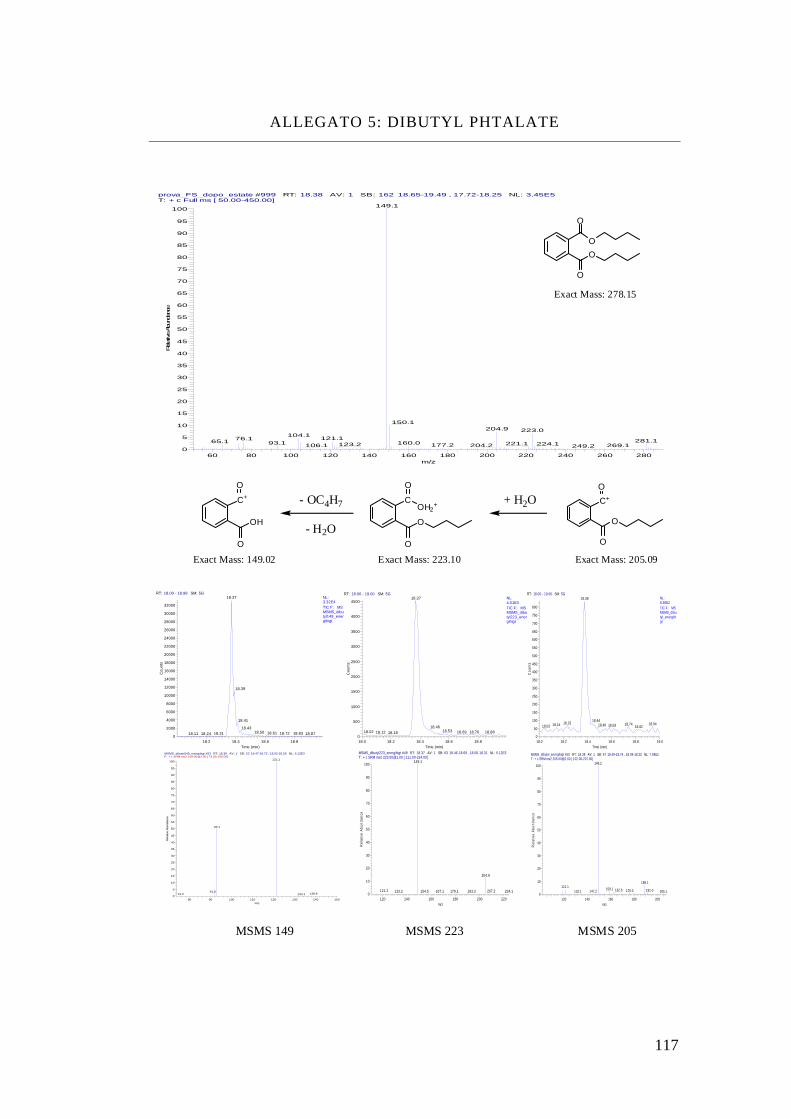
117
ALLEGATO 5: DIBUTYL PHTALATE
prova_FS_dopo_estate #999 RT: 18.38 AV: 1 SB: 162 18.65-19.49 , 17.72-18.25 NL: 3.45E5T: + c Full ms [ 50.00-450.00]
60 80 100 120 140 160 180 200 220 240 260 280m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Relative Abu
ndan
ce
149.1
150.1204.9 223.0
104.1 121.176.1 281.165.1 93.1 160.0 221.1 224.1123.2 177.2 269.1106.1 204.2 249.2
OO
O
O
Exact Mass: 278.15
MSMS_dibutyl_energihigt #50 RT: 18.38 AV: 1 SB: 67 18.49-18.76 , 18.09-18.32 NL: 7.48E2T: + c SRM ms2 [email protected] [ 102.00-207.00]
120 140 160 180 200m/z
0
10
20
30
40
50
60
70
80
90
100
Rel
ativ
e Ab
unda
nce
149.2
189.1121.1 159.1 162.6 191.0176.3147.2132.1 205.1
C+
O
O
O
Exact Mass: 205.09
C
O
O
O
OH2+ + H2OC+
OH
O
O
Exact Mass: 223.10Exact Mass: 149.02
- H2O
- OC4H7
MSMS_dibutyl223_energihigt #49 RT: 18.37 AV: 1 SB: 63 18.46-18.69 , 18.06-18.31 NL: 5.12E3T: + c SRM ms2 [email protected] [ 111.00-224.00]
120 140 160 180 200 220m/z
0
10
20
30
40
50
60
70
80
90
100
Rel
ativ
e Ab
unda
nce
149.1
204.9
121.2 207.2133.2 179.1 224.1193.3167.1154.5
MSMS 205MSMS 223MSMS 149
MSMS_dibutyl149_energihigt #53 RT: 18.38 AV: 1 SB: 23 18.47-18.72 , 18.02-18.28 NL: 6.12E3F: + c SRM ms2 [email protected] [ 74.00-150.00]
80 90 100 110 120 130 140 150m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ativ
e A
bund
ance
121.1
93.1
91.8138.975.3 133.1
RT: 18.00 - 19.00 SM: 5G
18.0 18.2 18.4 18.6 18.8 19.0Time (min)
0
50
100
150
200
250
300
350
400
450
500
550
600
650
700
750
800
Cou
nts
18.38
18.4418.23 18.9418.7418.14 18.48 18.5818.03 18.82
NL:8.38E2TIC F: MS MSMS_dibutyl_energihigt
RT: 18.00 - 19.00 SM: 5G
18.0 18.2 18.4 18.6 18.8Time (min)
0
500
1000
1500
2000
2500
3000
3500
4000
4500
Cou
nts
18.37
18.4618.5318.02 18.69 18.8818.7618.12 18.18
NL:4.51E3TIC F: MS MSMS_dibutyl223_energihigt
RT: 18.00 - 18.99 SM: 5G
18.2 18.4 18.6 18.8Time (min)
0
2000
4000
6000
8000
10000
12000
14000
16000
18000
20000
22000
24000
26000
28000
30000
32000
Cou
nts
18.37
18.39
18.41
18.4318.50 18.61 18.72 18.8318.31 18.8718.11 18.24
NL:3.32E4TIC F: MS MSMS_dibutyl149_energihigt
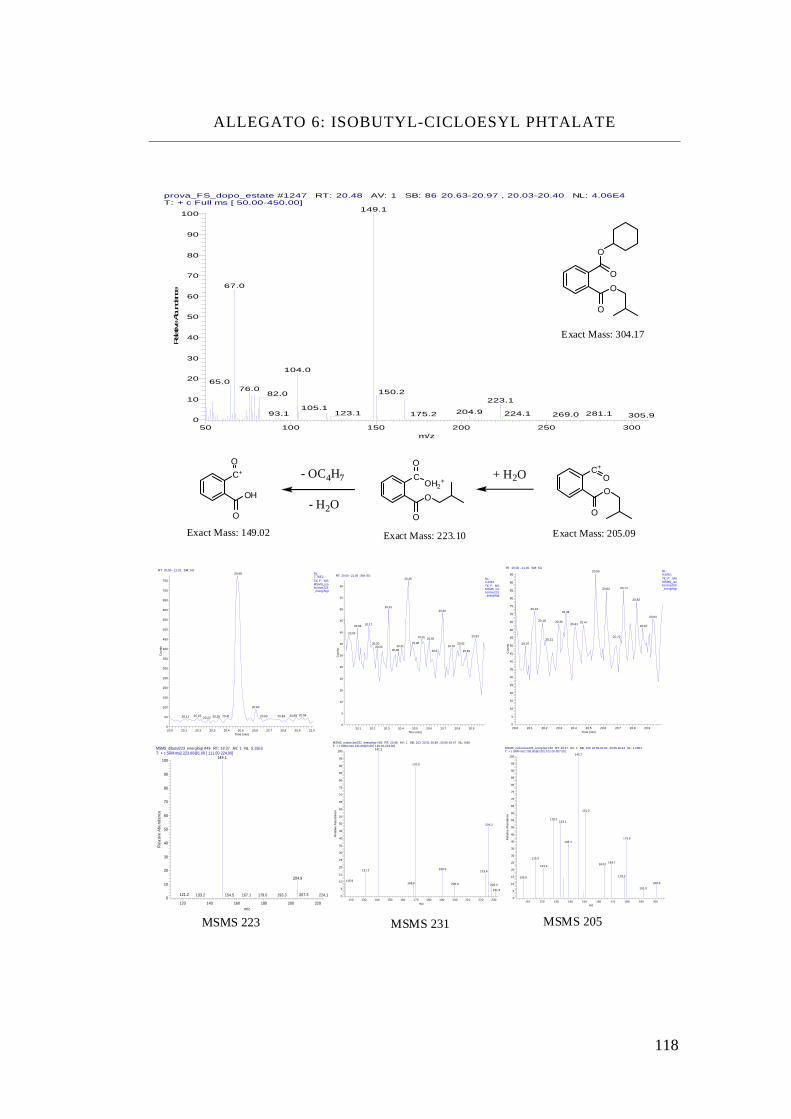
118
ALLEGATO 6: ISOBUTYL-CICLOESYL PHTALATE
prova_FS_dopo_estate #1247 RT: 20.48 AV: 1 SB: 86 20.63-20.97 , 20.03-20.40 NL: 4.06E4T: + c Full ms [ 50.00-450.00]
50 100 150 200 250 300m/z
0
10
20
30
40
50
60
70
80
90
100
Relative Ab
unda
nce
149.1
67.0
104.0
65.076.0 150.282.0
223.1105.1 204.9123.193.1 281.1224.1175.2 269.0 305.9
O
OO
O
Exact Mass: 304.17
+ H2O
- H2O
- OC4H7C+
OO
O
Exact Mass: 205.09
CO
O
O
OH2+
C+O
OH
O
Exact Mass: 223.10Exact Mass: 149.02
MSMS_dibutyl223_energihigt #49 RT: 18.37 AV: 1 NL: 5.15E3T: + c SRM ms2 [email protected] [ 111.00-224.00]
120 140 160 180 200 220m/z
0
10
20
30
40
50
60
70
80
90
100
Rel
ativ
e Ab
unda
nce
149.1
204.9
121.2 207.3179.0133.2 224.1193.3167.1154.5
MSMS 223 MSMS 205
RT: 20.00 - 21.00 SM: 5G
20.0 20.1 20.2 20.3 20.4 20.5 20.6 20.7 20.8 20.9Time (min)
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
Cou
nts
20.55
20.7420.62
20.82
20.1320.34
20.9420.18 20.30 20.4720.41 20.87
20.7220.21
20.07
NL:9.58E1TIC F: MS MSMS_isobcicloe205_energihigt
MSMS_isobcicloe205_energihigt #62 RT: 20.47 AV: 1 SB: 108 20.50-20.92 , 20.05-20.44 NL: 1.23E1T: + c SRM ms2 [email protected] [ 102.00-207.00]
110 120 130 140 150 160 170 180 190 200m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ativ
e A
bund
ance
145.7
151.0
128.2133.1
179.9138.4
115.3169.2164.2121.3
178.3106.9
200.9
191.3
RT: 20.00 - 21.01 SM: 5G
20.0 20.1 20.2 20.3 20.4 20.5 20.6 20.7 20.8 20.9 21.0Time (min)
0
50
100
150
200
250
300
350
400
450
500
550
600
650
700
750
Cou
nts
20.48
20.60
20.9420.19 20.8920.63 20.8420.4120.3520.11 20.22
NL:7.76E2TIC F: MS MSMS_isobcicloe223_energihigt
RT: 20.00 - 21.00 SM: 5G
20.1 20.2 20.3 20.4 20.5 20.6 20.7 20.8 20.9Time (min)
0
5
10
15
20
25
30
35
40
45
50
55
60
Cou
nts
20.45
20.3120.69
20.1720.09
20.0320.9320.55 20.60
20.4820.20 20.8220.7620.4120.23
20.38 20.67 20.86
NL:6.23E1TIC F: MS MSMS_isobcicloe231_energihigt
MSMS_isobcicloe231_energihigt #63 RT: 20.48 AV: 1 SB: 103 20.51-20.89 , 20.06-20.47 NL: 9.88T: + c SRM ms2 [email protected] [ 115.00-233.00]
120 130 140 150 160 170 180 190 200 210 220 230m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ativ
e A
bund
ance
141.2
170.0
226.2
190.5131.2 223.4
115.9168.0 200.0 228.3
231.5
MSMS 231
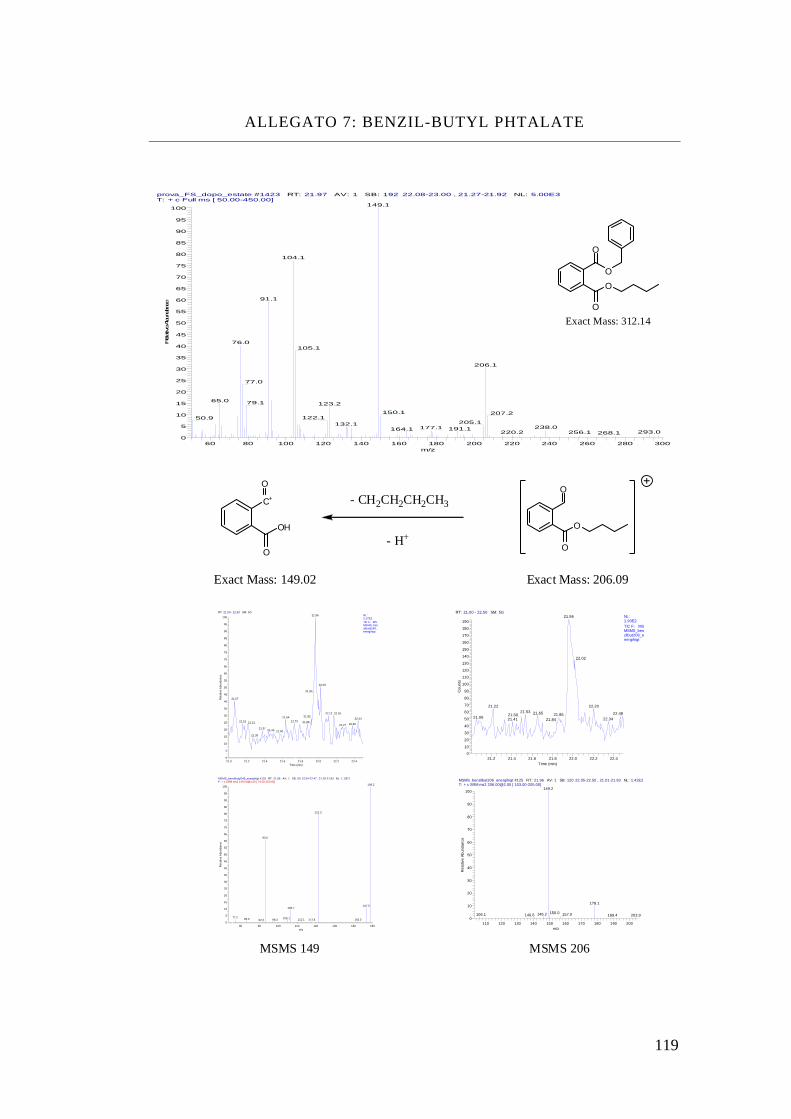
119
ALLEGATO 7: BENZIL-BUTYL PHTALATE
prova_FS_dopo_estate #1423 RT: 21.97 AV: 1 SB: 192 22.08-23.00 , 21.27-21.92 NL: 5.00E3T: + c Full ms [ 50.00-450.00]
60 80 100 120 140 160 180 200 220 240 260 280 300m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Relative Abu
ndan
ce
149.1
104.1
91.1
76.0105.1
206.1
77.0
65.0 79.1 123.2
150.1 207.2122.150.9
205.1132.1238.0177.1 191.1164.1 293.0256.1220.2 268.1
OO
O
OExact Mass: 312.14
C+
OH
O
O
Exact Mass: 149.02
- CH2CH2CH2CH3
RT: 21.00 - 22.50 SM: 5G
21.2 21.4 21.6 21.8 22.0 22.2 22.4Time (min)
0
10
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
Cou
nts
21.96
22.02
21.22 22.2021.53 21.65 22.4821.8621.5021.06 22.3421.41 21.84
NL:1.93E2TIC F: MS MSMS_benzilbut206_energihigt
MSMS_benzilbut206_energihigt #125 RT: 21.96 AV: 1 SB: 120 22.05-22.50 , 21.01-21.93 NL: 1.42E2T: + c SRM ms2 [email protected] [ 103.00-205.00]
110 120 130 140 150 160 170 180 190 200m/z
0
10
20
30
40
50
60
70
80
90
100
Rel
ativ
e Ab
unda
nce
149.2
178.1
150.0146.2 157.0106.1 140.6 203.9189.4
O
O
O
Exact Mass: 206.09
- H+
MSMS 206
RT: 21.00 - 22.50 SM: 5G
21.0 21.2 21.4 21.6 21.8 22.0 22.2 22.4Time (min)
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ativ
e A
bund
ance
21.96
22.03
21.94
21.07
22.12 22.1821.9221.64 22.44
21.7321.15 21.8821.21 22.3822.2721.37 21.45 21.50
21.26
NL:2.17E2TIC F: MS MSMS_benzilbutyl149_energihigt
MSMS_benzilbutyl149_energihigt #133 RT: 21.96 AV: 1 SB: 50 22.04-22.47 , 21.29-21.93 NL: 1.15E2F: + c SRM ms2 [email protected] [ 74.00-150.00]
80 90 100 110 120 130 140 150m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ativ
e A
bund
ance
149.2
121.2
93.0
147.0106.7
77.2 105.183.3 142.3112.198.3 117.892.2
MSMS 149
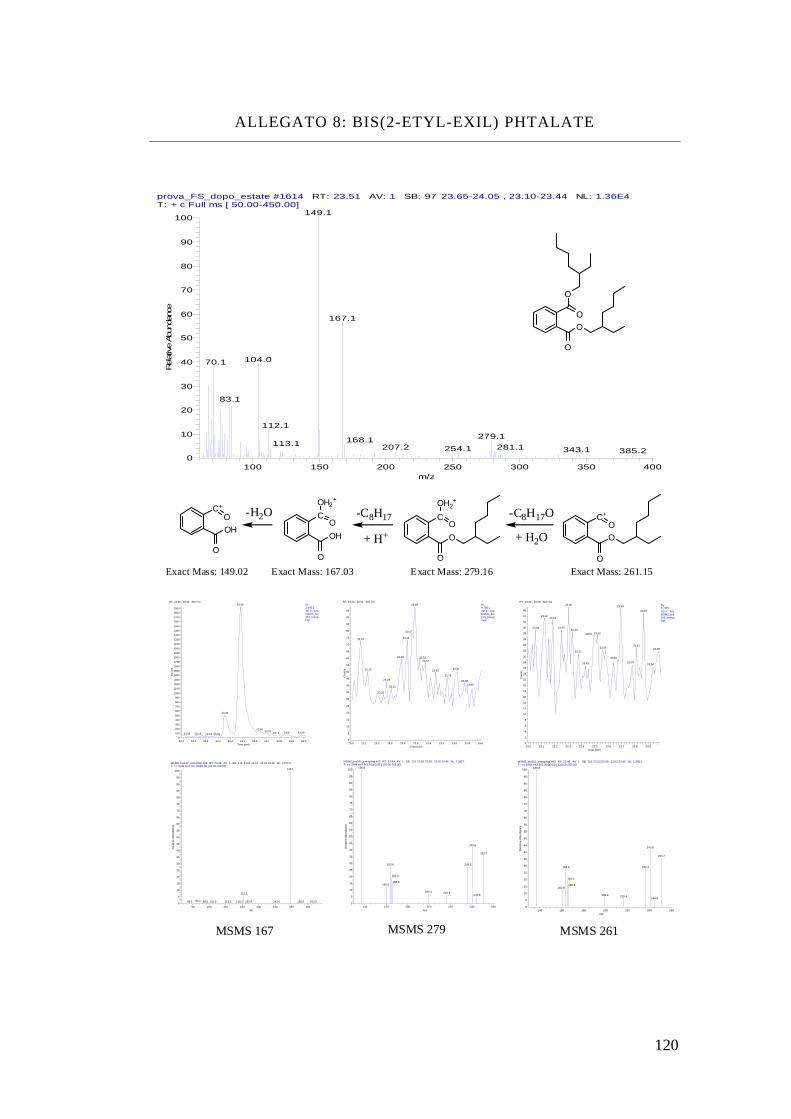
120
ALLEGATO 8: BIS(2-ETYL-EXIL) PHTALATE
prova_FS_dopo_estate #1614 RT: 23.51 AV: 1 SB: 97 23.65-24.05 , 23.10-23.44 NL: 1.36E4T: + c Full ms [ 50.00-450.00]
100 150 200 250 300 350 400m/z
0
10
20
30
40
50
60
70
80
90
100
Relat
ive
Abun
danc
e
149.1
167.1
104.070.1
83.1
112.1279.1168.1113.1 207.2 281.1254.1 343.1 385.2
C+
OO
O
O
OO
O
C
OO
O
OH2+
C
OHO
O
OH2+
C+
OHO
O
Exact Mass: 261.15Exact Mass: 279.16Exact Mass: 167.03Exact Mass: 149.02
-C8H17O
+ H2O
-C8H17
+ H+
-H2O
RT: 23.00 - 23.99 SM: 5G
23.0 23.1 23.2 23.3 23.4 23.5 23.6 23.7 23.8 23.9Time (min)
0
2
4
6
8
10
12
14
16
18
20
22
24
26
28
30
32
34
36
38
40
42
44
46
Cou
nts
23.30 23.69
23.87
23.1223.18
23.2523.0623.33
23.5223.50
23.8123.56 23.98
23.37
23.6723.7823.43 23.94
NL:4.73E1TIC F: MS MSMS_bis261_energihigt
MSMS_bis261_energihigt #63 RT: 23.49 AV: 1 SB: 116 23.53-23.99 , 23.02-23.46 NL: 1.36E1T: + c SRM ms2 [email protected] [ 130.00-262.00]
140 160 180 200 220 240 260m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ativ
e A
bund
ance
136.8
240.5
250.7
236.2163.6
165.0
165.8160.6
199.4 216.4244.6
MSMS 261
MSMS_bis261_energihigt #63 RT: 23.49 AV: 1 SB: 116 23.53-23.99 , 23.02-23.46 NL: 1.36E1T: + c SRM ms2 [email protected] [ 130.00-262.00]
140 160 180 200 220 240 260m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ativ
e A
bund
ance
136.8
240.5
250.7
236.2163.6
165.0
165.8160.6
199.4 216.4244.6
RT: 23.00 - 24.01 SM: 5G
23.0 23.1 23.2 23.3 23.4 23.5 23.6 23.7 23.8 23.9 24.0Time (min)
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
Cou
nts
23.49
23.47
23.4423.07
23.39 23.5323.57
23.8123.13 23.65
23.7523.28 23.88
23.9023.32
23.25
NL:9.78E1TIC F: MS MSMS_bis279_energimed
MSMS 279
RT: 23.00 - 24.01 SM: 5G
23.0 23.1 23.2 23.3 23.4 23.5 23.6 23.7 23.8 23.9 24.0Time (min)
0
100
200
300
400
500
600
700
800
900
1000
1100
1200
1300
1400
1500
1600
1700
1800
1900
2000
2100
2200
2300
2400
2500
2600
2700
2800
2900
Cou
nts
23.48
23.36
23.6323.70 23.87 23.9823.7423.1323.05 23.3123.16
NL:2.94E3TIC F: MS MSMS_bis167_energihigt
MSMS_bis167_energihigt #66 RT: 23.48 AV: 1 SB: 114 23.61-24.01 , 23.02-23.45 NL: 2.87E3T: + c SRM ms2 [email protected] [ 83.00-168.00]
90 100 110 120 130 140 150 160m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ativ
e A
bund
ance
149.1
121.1
93.1 121.8119.1111.388.1 96.8 101.3 155.6141.0 163.3
MSMS 167
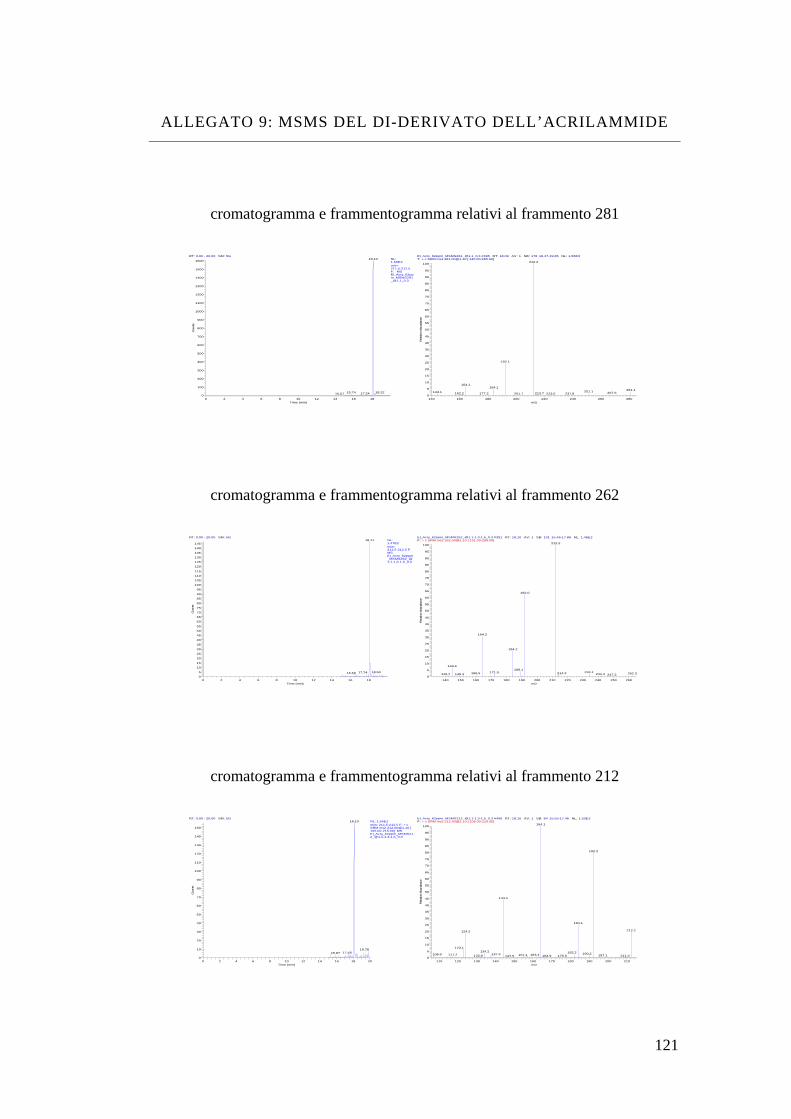
121
ALLEGATO 9: MSMS DEL DI-DERIVATO DELL’ACRILAMMIDE
cromatogramma e frammentogramma relativi al frammento 281
cromatogramma e frammentogramma relativi al frammento 262
cromatogramma e frammentogramma relativi al frammento 212
RT: 0.00 - 20.00 SM: 5G
0 2 4 6 8 10 12 14 16 18Time (min)
0
100
200
300
400
500
600
700
800
900
1000
1100
1200
1300
1400
1500
1600
Cou
nts
18.10
18.2215.74 17.2415.57
NL:1.60E3m/z= 211.5-212.5 F: MS EI_Acry_62ppm_MSMS281_@1,1_0,3
EI_Acry_62ppm_MSMS281_@1,1_0,3 #395 RT: 18.09 AV: 1 SB: 178 18.47-19.85 NL: 1.56E3T: + c SRM ms2 [email protected] [ 140.00-285.00]
140 160 180 200 220 240 260 280m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ative
Abu
ndan
ce
212.2
192.1
164.1184.1
281.1251.1144.1 267.6213.7162.2 177.2 201.7 223.0 237.8
RT: 0.00 - 20.00 SM: 5G
0 2 4 6 8 10 12 14 16 18Time (min)
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
105
110
115
120
125
130
135
140
145
Cou
nts
18.11
18.5017.7416.59
NL:1.47E2m/z= 211.5-212.5 F: MS EI_Acry_62ppm_MSMS262_@1,1-1,3-1,5_0,3
EI_Acry_62ppm_MSMS262_@1,1-1,3-1,5_0,3 #391 RT: 18.10 AV: 1 SB: 101 15.46-17.86 NL: 1.46E2F: + c SRM ms2 [email protected] [ 131.00-265.00]
140 150 160 170 180 190 200 210 220 230 240 250 260m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ative
Abu
ndan
ce
212.2
192.0
164.2
184.2
144.4189.1
234.1171.9160.5 214.3 262.3142.7 241.4149.3 247.2
RT: 0.00 - 20.00 SM: 5G
0 2 4 6 8 10 12 14 16 18 20Time (min)
0
10
20
30
40
50
60
70
80
90
100
110
120
130
140
150
Cou
nts
18.10
19.7817.4815.87
NL: 1.55E2m/z= 211.5-212.5 F: + c SRM ms2 [email protected] [ 106.00-215.00] MS EI_Acry_62ppm_MSMS212_@1,1-1,3-1,5_0,3
EI_Acry_62ppm_MSMS212_@1,1-1,3-1,5_0,3 #406 RT: 18.10 AV: 1 SB: 84 15.55-17.46 NL: 1.53E3F: + c SRM ms2 [email protected] [ 106.00-215.00]
110 120 130 140 150 160 170 180 190 200 210m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ative
Abu
ndan
ce
164.1
192.2
144.1
184.1
212.2124.2
123.1134.2 182.2 190.2137.3106.9 117.2 163.4157.1 197.1132.8 211.4175.5164.9147.5

122
cromatogramma e frammentogramma relativi al frammento 192
cromatogramma e frammentogramma relativi al frammento 184
cromatogramma e frammentogramma relativi al frammento 164
RT: 0.00 - 20.00 SM: 5G
0 2 4 6 8 10 12 14 16 18Time (min)
0
50
100
150
200
250
300
350
400
450
500
550
600
650
700
750
800
850
900
950
1000
1050
1100
1150
1200
Cou
nts
18.10
18.3315.43
17.70
NL: 1.22E3TIC F: + c SRM ms2 [email protected] [ 96.00-195.00] MS EI_Acry_62ppm_MSMS192_@1,1-1,3-1,5_0,3
EI_Acry_62ppm_MSMS192_@1,1-1,3-1,5_0,3 #412 RT: 18.10 AV: 1 NL: 6.41E2F: + c SRM ms2 [email protected] [ 96.00-195.00]
100 110 120 130 140 150 160 170 180 190m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ative
Abu
ndan
ce
144.1
164.1
124.2
123.1
117.1134.2 192.2163.2 166.0137.2 182.0114.2 179.1145.198.1 162.2133.2102.0 186.5154.2119.2
RT: 0.00 - 20.01 SM: 5G
0 2 4 6 8 10 12 14 16 18 20Time (min)
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ative
Abu
ndan
ce
18.10
16.79 18.35
NL: 8.05E2TIC F: + c SRM ms2 [email protected] [ 92.00-190.00] MS EI_Acry_62ppm_MSMS184_@1,1-1,3-1,5_0,3
EI_Acry_62ppm_MSMS184_@1,1-1,3-1,5_0,3 #413 RT: 18.10 AV: 1 SB: 79 15.60-17.37 NL: 4.05E2F: + c SRM ms2 [email protected] [ 92.00-190.00]
100 110 120 130 140 150 160 170 180 190m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ative
Abu
ndan
ce
124.1
144.1
164.2
123.1
137.2134.2
107.1 117.2 184.2162.298.1
142.2126.8 158.2110.9 182.293.0 167.0149.4
RT: 0.00 - 20.00 SM: 5G
0 2 4 6 8 10 12 14 16 18Time (min)
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ative
Abu
ndan
ce
18.11
15.4715.85 19.60
NL: 3.82E2TIC F: + c SRM ms2 [email protected] [ 82.00-170.00] MS EI_Acry_62ppm_MSMS164_@1,1-1,3-1,5_0,3
EI_Acry_62ppm_MSMS164_@1,1-1,3-1,5_0,3 #421 RT: 18.11 AV: 1 NL: 2.94E2F: + c SRM ms2 [email protected] [ 82.00-170.00]
90 100 110 120 130 140 150 160 170m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ative
Abu
ndan
ce
124.1
144.2
137.1
123.2 124.7114.1 163.3134.193.1 164.1100.1 117.183.1 162.098.1
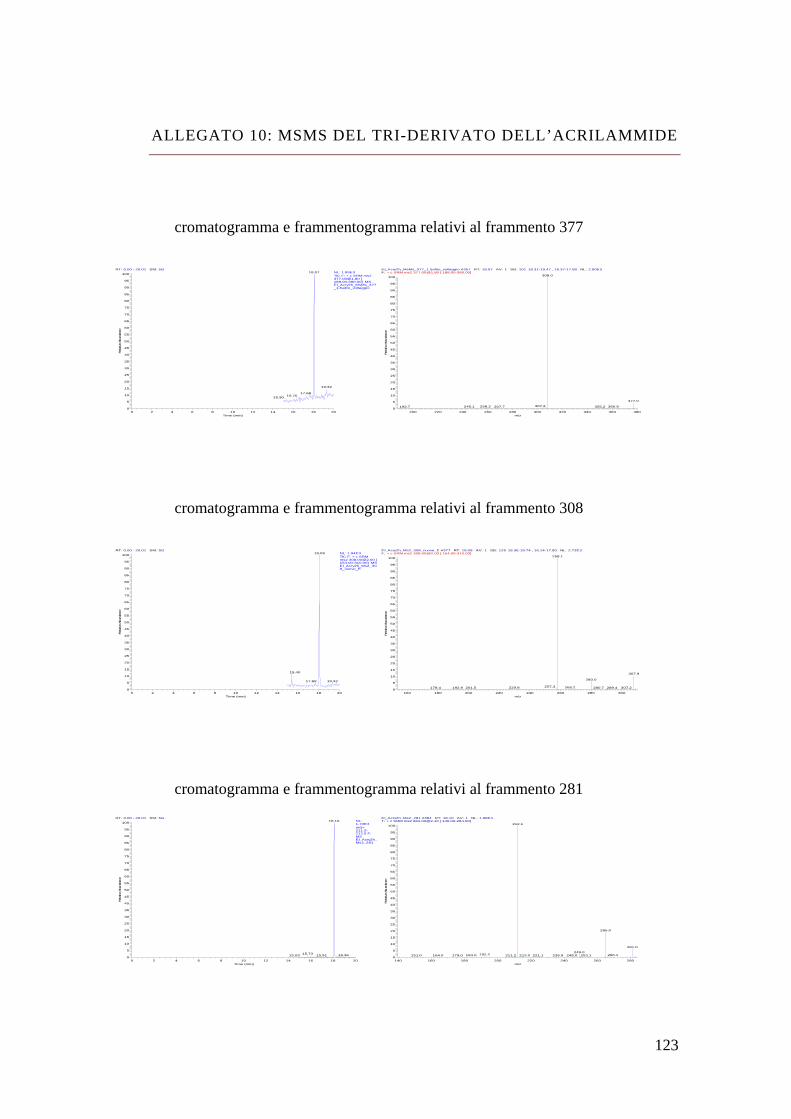
123
ALLEGATO 10: MSMS DEL TRI-DERIVATO DELL’ACRILAMMIDE
cromatogramma e frammentogramma relativi al frammento 377
cromatogramma e frammentogramma relativi al frammento 308
cromatogramma e frammentogramma relativi al frammento 281
RT: 0.00 - 20.01 SM: 5G
0 2 4 6 8 10 12 14 16 18 20Time (min)
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Relative Abu
ndan
ce
18.07
19.32
17.6816.7515.30
NL: 1.99E3TIC F: + c SRM ms2 [email protected] [ 188.00-380.00] MS EI_Acry2h_MsMs_377_1,5altro_voltaggio
EI_Acry2h_MsMs_377_1,5altro_voltaggio #357 RT: 18.07 AV: 1 SB: 101 18.31-19.47 , 16.37-17.80 NL: 2.90E3F: + c SRM ms2 [email protected] [ 188.00-380.00]
200 220 240 260 280 300 320 340 360 380m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Relative Abu
ndan
ce
308.0
377.0
307.2258.2245.1 360.9355.2267.7192.7
RT: 0.00 - 20.01 SM: 5G
0 2 4 6 8 10 12 14 16 18 20Time (min)
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Relative Abu
ndan
ce
18.09
15.40
17.69 19.32
NL: 1.94E3TIC F: + c SRM ms2 [email protected] [ 154.00-310.00] MS EI_Acry2h_Ms2_308_nuove_E
EI_Acry2h_Ms2_308_nuove_E #377 RT: 18.09 AV: 1 SB: 129 18.36-19.74 , 16.14-17.90 NL: 2.73E3F: + c SRM ms2 [email protected] [ 154.00-310.00]
160 180 200 220 240 260 280 300m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Relative Abu
ndan
ce
258.1
307.9
280.0
257.3 266.2229.8178.4 192.9 280.7 289.4201.5 307.2
RT: 0.00 - 20.01 SM: 5G
0 2 4 6 8 10 12 14 16 18 20Time (min)
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Relative Abu
ndan
ce
18.10
15.73 18.3615.9115.03
NL:1.70E4m/z= 211.5-212.5 F: MS EI_Acry2h_Ms2_281
EI_Acry2h_Ms2_281 #384 RT: 18.10 AV: 1 NL: 1.98E4T: + c SRM ms2 [email protected] [ 140.00-284.00]
140 160 180 200 220 240 260 280m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Relative Abu
ndan
ce
212.1
265.0
281.0
249.0192.2 266.1184.0 213.0164.0 248.0 253.1236.9221.1211.2179.0151.0
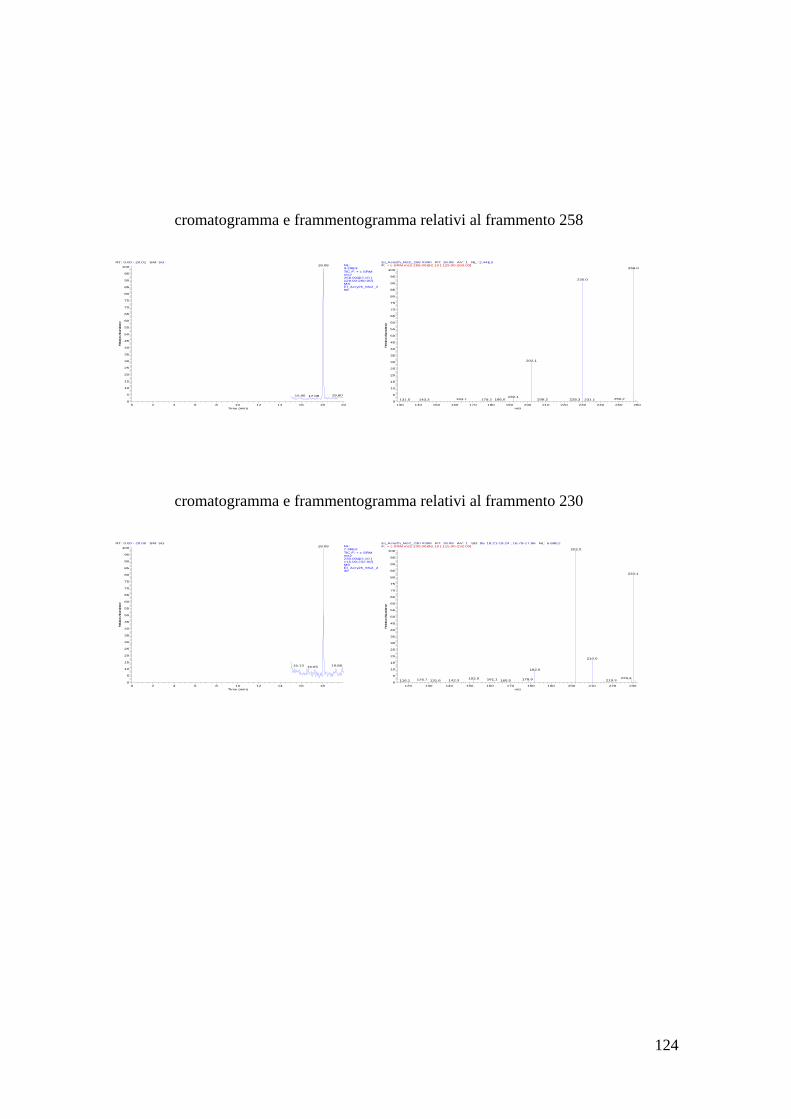
124
cromatogramma e frammentogramma relativi al frammento 258
cromatogramma e frammentogramma relativi al frammento 230
RT: 0.00 - 20.01 SM: 5G
0 2 4 6 8 10 12 14 16 18 20Time (min)
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Relative Abu
ndan
ce
18.08
19.6015.90 17.08
NL:3.29E3TIC F: + c SRM ms2 [email protected] [ 129.00-260.00] MS EI_Acry2h_Ms2_258
EI_Acry2h_Ms2_258 #390 RT: 18.08 AV: 1 NL: 2.44E3F: + c SRM ms2 [email protected] [ 129.00-260.00]
130 140 150 160 170 180 190 200 210 220 230 240 250 260m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Relative Abu
ndan
ce
258.0
230.0
202.1
192.1250.2163.7 228.3186.0 208.2 231.1143.3131.5 179.2
RT: 0.00 - 20.00 SM: 5G
0 2 4 6 8 10 12 14 16 18Time (min)
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Relative Abu
ndan
ce
18.09
19.8615.13 16.65
NL:7.36E2TIC F: + c SRM ms2 [email protected] [ 115.00-232.00] MS EI_Acry2h_Ms2_230
EI_Acry2h_Ms2_230 #399 RT: 18.09 AV: 1 SB: 95 18.21-19.24 , 16.79-17.96 NL: 5.68E2F: + c SRM ms2 [email protected] [ 115.00-232.00]
120 130 140 150 160 170 180 190 200 210 220 230m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Relative Abu
ndan
ce
202.0
230.1
210.0
182.0
229.3152.0 178.9161.1126.7 142.3 219.3131.6 165.5116.1