multiple epiphyseal dysplasia: clinical and radiographic features, differential diagnosis and...
TRANSCRIPT

Best Practice & Research Clinical RheumatologyVol. 22, No. 1, pp. 19–32, 2008
doi:10.1016/j.berh.2007.11.009
available online at http://www.sciencedirect.com2
Multiple epiphyseal dysplasia: clinical
and radiographic features, differential
diagnosis and molecular basis
Sheila UngerStaff Geneticist and ESDN Coordinator
Department of Paediatrics and Institute of Human Genetics, University of Freiburg, Freiburg, Germany
Luisa BonafeHead
Division of Molecular Paediatrics, Centre Hospitalier Universitaire Vaudois, Lausanne, Switzerland
Andrea Superti-Furga*
Director and Chair
Department of Paediatrics, University of Freiburg, Freiburg, Germany
Multiple epiphyseal dysplasia is one of the more common skeletal dysplasias but it can still bedifficult to diagnose. The presenting signs are often rheumatological (‘joint pain’) or neurological(‘myopathy’) in nature, and the cardinal feature of skeletal dysplasia (short stature) may not bepresent. A radiographic skeletal survey is necessary to delineate the pattern of generalized de-layed epiphyseal ossification and changes in epiphyseal contour. Once the diagnosis of multipleepiphyseal dysplasia has been established, careful examination of the radiographs can help todetermine which genes should be analysed. Mutations in at least six different genes can causemultiple epiphyseal dysplasia, and it can be either dominant or recessive. Molecular diagnosisis important for accurate prognosis and genetic counselling.
Key words: multiple epiphyseal dysplasia; COMP; COL9A1; COL9A2; COL9A3; MATN3; DTDST;bone age; delayed epiphyseal ossification.
* Corresponding author. Centre for Paediatrics and Adolescent Medicine, Freiburg University Hospital,
Mathildenstr. 1, D-79106 Freiburg, Germany, Tel.: þ49 761 270 4305; Fax: þ49 761 270 4454.
E-mail address: [email protected] (A. Superti-Furga).
1521-6942/$ - see front matter ª 2007 Published by Elsevier Ltd.

20 S. Unger et al
Multiple epiphyseal dysplasia (MED) is a mild form of skeletal dysplasia. Genetically, it isfairly heterogeneous with mutations in COMP, DTDST (SLC26A2), MATN3, COL9A1,COL9A2 and COL9A3 capable of producing the clinical picture of MED. While full-blown, severe phenotypic expressions of mutations in genes such as COMP, DTDST(SLC26A2) and MATN3 produce quite distinct phenotypes, less severe mutationstend to blend into the relatively mild phenotype of MED. Nonetheless, the distinctgenetic forms of MED can usually be distinguished by virtue of subtle clinical andradiographic signs and by their mode of inheritance. Clinically, the disease manifestswith joint disease and sometimes with reduced growth. Radiographically, there isboth a delay in ossification (‘maturation’) of epiphyses and changes in their shape.Originally, MED was subdivided into ‘Fairbanks’ and ‘Ribbing’ forms but this has notproved useful.
COMMON CLINICAL FEATURES
As a rule, the MED phenotype is not recognizable at birth or during the first 1–2 yearsof life. A newborn destined to develop MED shows normal length and normal bodyproportions. One exception to this rule is the presence of club foot at birth insome cases, with recessive MED caused by mutations in DTDST.1
A common presenting sign is joint pain affecting the hip and knee joints, occur-ring, at least initially, after physical exercise. During childhood, progressive deviationfrom the normal growth curve can occur, resulting in mild to moderate short stat-ure (around or slightly below the third percentile) by the age of 5–6 years. However,there are many examples of adults with MED of normal stature.2 Muscular hypotoniais frequent in young children with MED caused by COMP mutations (sometimes evenfrank myopathy).3 Thus, a typical history would be that of a child who was clinicallynormal at birth, but with some delay in motor development, and who starts to com-plain about joint pain after a long walk, after a ball game or after physical exercise.Paediatric examination may reveal a deviation of the growth curve. At that time, a di-agnosis of MED in the child may be supported by a family history of similar symp-toms in one of his parents or older siblings, or of a history of chronic joint disease,sometimes leading to total hip arthroplasty at a relatively early age, in one of his par-ents or grandparents.
Changes in the femoral heads occurring in school-age children with MED may re-semble those seen in ‘idiopathic’ necrosis of the femoral head (Legg-Calve-Perthes dis-ease). While the majority of cases of Perthes disease are limited to one side and arerarely (as far as is known) the expression of a generalized skeletal anomaly, in caseswhere Perthes disease is familial, bilateral or associated with short stature, a clinicalexamination and a radiographic skeletal survey should be performed to rule out MED.
As the mildest phenotype of skeletal dysplasia, MED affects the epiphyses of tubularbones, including metacarpals, metatarsals and phalanges; the metaphyses and vertebralbodies are only affected slightly or not at all. The effect on the epiphyses manifests on ra-diographs as a maturational delay. The pattern of delay and the changes in shape are helpfulin distinguishing the different genetic forms of MED. The type of MED needs to be deter-mined before providing the patient and their family with precise genetic counselling.
Table 1 gives an overview of the distinctive features of the various forms of MED. Inthe following sections, some of the features that are useful in the differential diagnosisbetween the various forms of MED will be presented. The MED forms are discussed inorder of decreasing incidence.
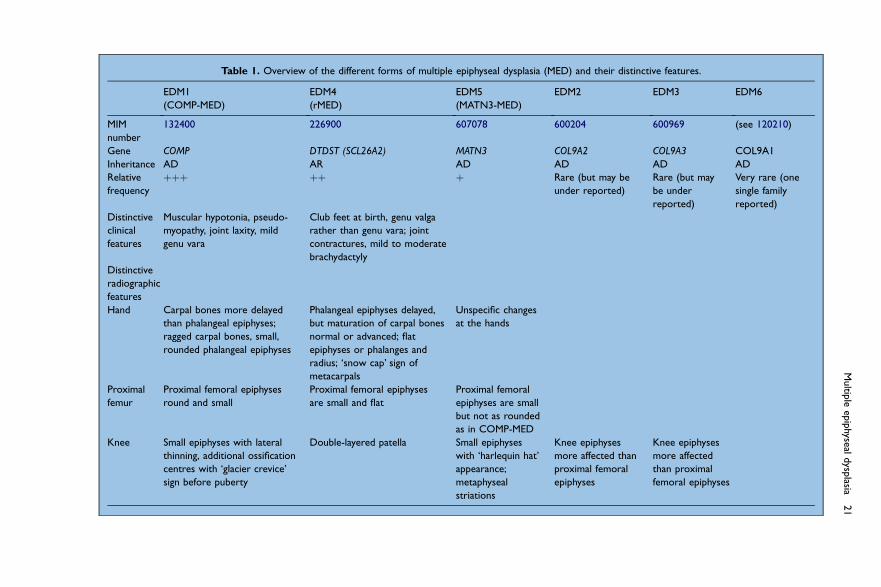
Table 1. Overview of the different forms of multiple epiphyseal dysplasia (MED) and their distinctive features.
EDM1
(COMP-MED)
EDM4
(rMED)
EDM5
(MATN3-MED)
EDM2 EDM3 EDM6
MIM
number
132400 226900 607078 600204 600969 (see 120210)
Gene COMP DTDST (SCL26A2) MATN3 COL9A2 COL9A3 COL9A1
Inheritance AD AR AD AD AD AD
Relative
frequency
þþþ þþ þ Rare (but may be
under reported)
Rare (but may
be under
reported)
Very rare (one
single family
reported)
Distinctive
clinical
features
Muscular hypotonia, pseudo-
myopathy, joint laxity, mild
genu vara
Club feet at birth, genu valga
rather than genu vara; joint
contractures, mild to moderate
brachydactyly
Distinctive
radiographic
features
Hand Carpal bones more delayed
than phalangeal epiphyses;
ragged carpal bones, small,
rounded phalangeal epiphyses
Phalangeal epiphyses delayed,
but maturation of carpal bones
normal or advanced; flat
epiphyses or phalanges and
radius; ‘snow cap’ sign of
metacarpals
Unspecific changes
at the hands
Proximal
femur
Proximal femoral epiphyses
round and small
Proximal femoral epiphyses
are small and flat
Proximal femoral
epiphyses are small
but not as rounded
as in COMP-MED
Knee Small epiphyses with lateral
thinning, additional ossification
centres with ‘glacier crevice’
sign before puberty
Double-layered patella Small epiphyses
with ‘harlequin hat’
appearance;
metaphyseal
striations
Knee epiphyses
more affected than
proximal femoral
epiphyses
Knee epiphyses
more affected
than proximal
femoral epiphyses
Multip
leep
iphyseal
dysp
lasia21

22 S. Unger et al
FREQUENCY OF MED
There are no precise estimates regarding the collective incidence of MED. Based onthe number of cases seen in growth clinics, rheumatology or genetics clinics, and com-pared with conditions whose incidences are known more precisely such as achondro-plasia and osteogenesis imperfecta, it seems reasonable to give a prevalence ofapproximately one in 20 000 for MED. This figure is almost certainly an underestima-tion, as making the diagnosis of MED in an index case often reveals a significant numberof affected relatives who had not been diagnosed previously.
More is known about the relative incidence of the various types of MED. It is cer-tain that MED caused by the COMP (cartilage oligomeric matrix protein) gene (EDM1;COMP-MED) is the most common form, accounting for at least half of cases. The sec-ond most common form, at least in Europe, is the recessive form of MED caused bymutations in the DTDST (SLC26A2) (diastrophic dysplasia sulphate transporter) gene(EDM4; rMED for recessive MED). This form accounts for approximately one-quarterof MED cases. The remaining 25% of MED cases are split between the genes MATN3,COL9A1, COL9A2 and COL9A3. Matrilin-3 mutations seem to be significantly morecommon than mutations in any of the three collagen 9 genes, but the number of casesis still relatively small and it is possible that collagen 9 cases may be under reportedbecause of the lower availability of mutation analysis for those genes.
Some researchers have been unable to obtain molecular confirmation in a majorityof MED cases, and have thus suggested that one or more ‘major’ genes for MED mayexist.4 Other groups have been far more successful, suggesting that a stringent clinicaland radiographic evaluation is needed in making a diagnosis of MED, in case otheradult-onset and spurious phenotypes are included.5,6 However, it is clear that someMED cases, even familial ones, are caused by genes other than those known todate. Gene mapping and mutation analysis will certainly reveal other MED genes,although they are not likely to be very common.
COMP-MED (EDM1) (MIM 132400)
This is the most common form of MED. It is dominantly inherited and while several de-novo mutations have been reported, a family history can be elicited in more than half ofcases. As in other MED forms, the newborn is clinically healthy and measurements arenormal. Motor development in the first months is normal but onset of walking can bedelayed or progress can be slower than average. This may be caused by laxity of largejoints (knees and hips), muscular hypotonia, and (rarely) mild myopathy with mildly ele-vated creatine kinase and/or evidence of myopathy on muscle biopsy. Joint pain is com-mon. It is not rare for children to present to neuropaediatric clinics for hypotonia ordelayed or ‘uncoordinated’ walking. Growth retardation may be observed after 2 yearsof age, and a slight disproportion with mild shortening of the arms and legs with a normaltrunk can occur. During the clinical course, muscular hypotonia becomes less prominentin late childhood but joint laxity persists, leading to premature osteoarthritis of the hipsand knees. Joint replacement at the hip joints may be required in the third decade or later.
Radiographic changes of COMP-MED
In COMP-MED, there is a delay in appearance of the carpal bones. During ossification,their contour is not smooth but irregular, sometimes jagged. The phalanges may be
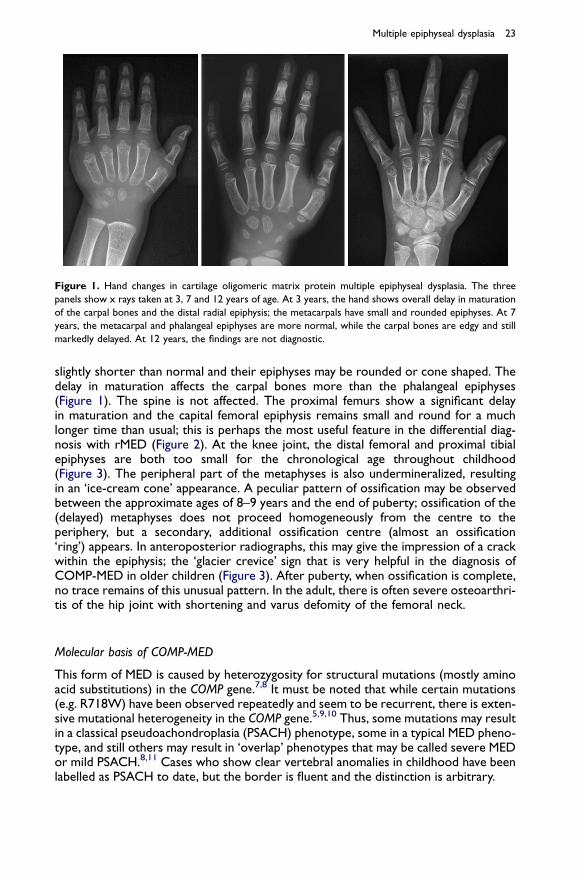
Figure 1. Hand changes in cartilage oligomeric matrix protein multiple epiphyseal dysplasia. The three
panels show x rays taken at 3, 7 and 12 years of age. At 3 years, the hand shows overall delay in maturation
of the carpal bones and the distal radial epiphysis; the metacarpals have small and rounded epiphyses. At 7
years, the metacarpal and phalangeal epiphyses are more normal, while the carpal bones are edgy and still
markedly delayed. At 12 years, the findings are not diagnostic.
Multiple epiphyseal dysplasia 23
slightly shorter than normal and their epiphyses may be rounded or cone shaped. Thedelay in maturation affects the carpal bones more than the phalangeal epiphyses(Figure 1). The spine is not affected. The proximal femurs show a significant delayin maturation and the capital femoral epiphysis remains small and round for a muchlonger time than usual; this is perhaps the most useful feature in the differential diag-nosis with rMED (Figure 2). At the knee joint, the distal femoral and proximal tibialepiphyses are both too small for the chronological age throughout childhood(Figure 3). The peripheral part of the metaphyses is also undermineralized, resultingin an ‘ice-cream cone’ appearance. A peculiar pattern of ossification may be observedbetween the approximate ages of 8–9 years and the end of puberty; ossification of the(delayed) metaphyses does not proceed homogeneously from the centre to theperiphery, but a secondary, additional ossification centre (almost an ossification‘ring’) appears. In anteroposterior radiographs, this may give the impression of a crackwithin the epiphysis; the ‘glacier crevice’ sign that is very helpful in the diagnosis ofCOMP-MED in older children (Figure 3). After puberty, when ossification is complete,no trace remains of this unusual pattern. In the adult, there is often severe osteoarthri-tis of the hip joint with shortening and varus defomity of the femoral neck.
Molecular basis of COMP-MED
This form of MED is caused by heterozygosity for structural mutations (mostly aminoacid substitutions) in the COMP gene.7,8 It must be noted that while certain mutations(e.g. R718W) have been observed repeatedly and seem to be recurrent, there is exten-sive mutational heterogeneity in the COMP gene.5,9,10 Thus, some mutations may resultin a classical pseudoachondroplasia (PSACH) phenotype, some in a typical MED pheno-type, and still others may result in ‘overlap’ phenotypes that may be called severe MEDor mild PSACH.8,11 Cases who show clear vertebral anomalies in childhood have beenlabelled as PSACH to date, but the border is fluent and the distinction is arbitrary.
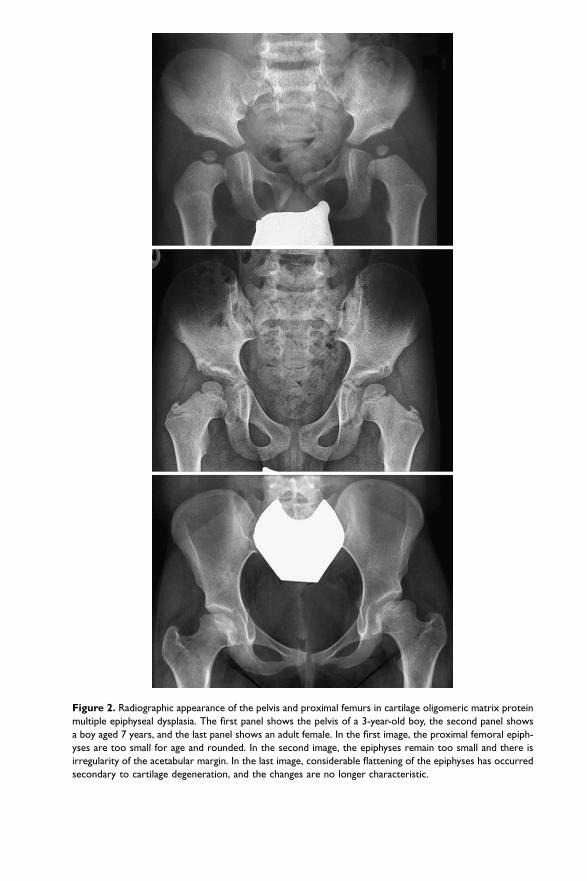
Figure 2. Radiographic appearance of the pelvis and proximal femurs in cartilage oligomeric matrix protein
multiple epiphyseal dysplasia. The first panel shows the pelvis of a 3-year-old boy, the second panel shows
a boy aged 7 years, and the last panel shows an adult female. In the first image, the proximal femoral epiph-
yses are too small for age and rounded. In the second image, the epiphyses remain too small and there is
irregularity of the acetabular margin. In the last image, considerable flattening of the epiphyses has occurred
secondary to cartilage degeneration, and the changes are no longer characteristic.

Figure 3. The three panels show knee x rays of siblings taken at 7 and 11 years of age as well as of their
mother (cartilage oligomeric matrix protein R718W mutation). At 7 years, the epiphyses at the knee are
small and their lateral border, in particular, is thin. At 11 years, there is an accessory ‘ring’ of cartilage os-
sification that gives the impression of fragmentation (the ‘glacier crevice’ sign). In the adult, the changes are
no longer discernible; the only remaining feature is the small size of the epiphyses.
Multiple epiphyseal dysplasia 25
Recessive MED (rMED; EDM4; DTDST-MED) (MIM 226900)
Clinical features
In approximately one-third of individuals with rMED, there is a history of congenitalclub feet; otherwise, nothing suggests the diagnosis at birth. Growth is often normal,and although most individuals with rMED are slightly shorter than normal, there ismuch variability with some individuals having heights above the mean, even as adults.12
Unlike in COMP-MED, there is no joint laxity but rather joint limitation, and the pre-senting sign can be restriction of movement at the hip joint, knee joint or even inter-phalangeal joints. A more common presenting symptom is joint pain. The mostcommon clinical pattern is that of a child of normal height complaining of chronic jointpain and with some degree of diffuse joint limitation. Malposition or dislocation of theproximal radius may be present, resulting in inability to extend the elbow fully. A milddegree of genu valgum is common. In older children and adults, there is concomitantulnar incurvation of the index finger and radial incurvation of the fifth finger ina ‘bracket’ pattern. The great toe may be more prominent than the other toes, anda large sandal gap may be present. Palatal cleft or cystic ear transformation, commonin diastrophic dysplasia, are not observed.1
Radiographic changes in rMED
Whereas COMP-MED is characterized by small, roundish epiphyses, rMED is charac-terized by flat epiphyses (Figure 4). Carpal ossification delay is not a feature of rMED;instead, carpal age may be slightly advanced. Conversely, phalangeal epiphyses areboth delayed in appearance and too thin or flat, and sometimes thinned on oneside only. The distal epiphyses of radius and ulna are also flattened. The distal

Figure 4. Radiographic features of the hand in recessive multiple epiphyseal dysplasia caused by DTDST mu-
tations at 1, 6 and 12 years of age. In this condition, carpal ossification is not delayed but sometimes slightly
advanced, while the metacarpal and phalangeal epiphyses are flat and delayed. Flattening of the distal meta-
carpal epiphyses gives a ‘snow cap’ appearance. Overall, the phalanges and metacarpal are somewhat short
and plump.
26 S. Unger et al
metacarpal epiphyses are not round as normal but slightly flat and broadened, andtend to embrace the metacarpal bone itself, like ‘snow melting from a roof’. The ra-diographic appearance of the hand in rMED is very different from that in COMP-MED, and while hands are usually clinically unaffected, hand x rays should be partof the diagnostic work-up of possible MED cases. The spine is not affected in child-hood, but a moderate degree of scoliosis and some degree of irregularities of thevertebral bodies can be seen in older children and adults. As in the genetically relateddisorder diastrophic dysplasia, there may be a reduction of the vertebral interpedic-ular distance from L1 to L5. At the pelvis, the most distinctive sign is progressive flat-tening of the proximal femoral epiphysis (Figure 5). The epiphysis may be normal orjust mildly flattened until 4 or 5 years of age, but then progressively decreases inheight, and the femoral neck becomes broader than normal, giving the epiphysisa ‘moon sickle’ appearance. Unlike in COMP-MED, the femoral neck does not de-form to a varus position, but rather to a valgus position.
A particular ossification variant of the patella seems to be a distinctive and diagnos-tically helpful finding.1,12 The so-called double-layer patella (Figure 5) is most oftenseen in rMED but has been reported in one case of COL9-MED.13 The two layersare actually formed from two different ossification centres, residing in two differenttendons, which fail to fuse.
Molecular basis of rMED
Unlike the other forms of MED, this form is inherited as a recessive trait. The phe-notype of rMED was delineated originally in a series of 18 individuals homozygous forthe R279W mutation in DTDST.1 This remains the genotype most often found, al-though other ‘mild’ mutations, such a C653S, result in the same phenotype when ho-mozygous, and compound heterozygosity for R279W or C653S and other mutations
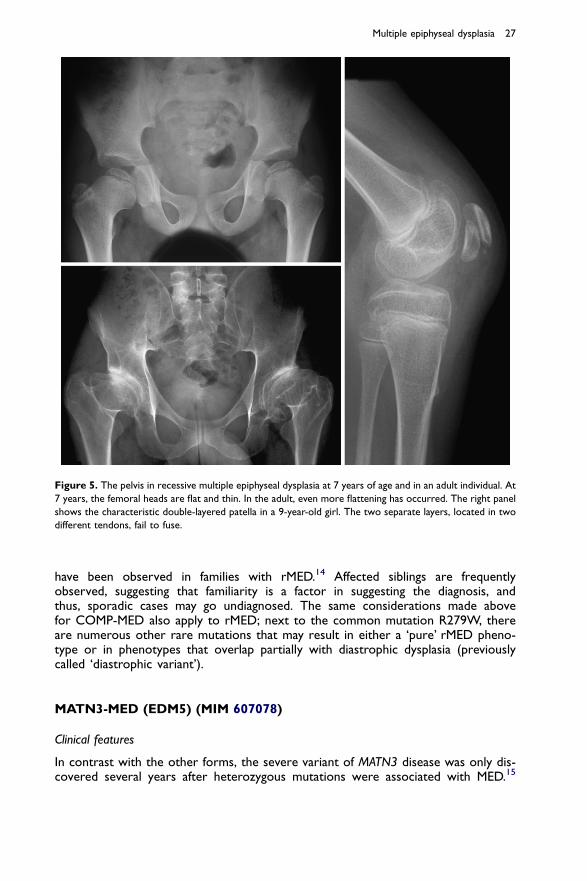
Figure 5. The pelvis in recessive multiple epiphyseal dysplasia at 7 years of age and in an adult individual. At
7 years, the femoral heads are flat and thin. In the adult, even more flattening has occurred. The right panel
shows the characteristic double-layered patella in a 9-year-old girl. The two separate layers, located in two
different tendons, fail to fuse.
Multiple epiphyseal dysplasia 27
have been observed in families with rMED.14 Affected siblings are frequentlyobserved, suggesting that familiarity is a factor in suggesting the diagnosis, andthus, sporadic cases may go undiagnosed. The same considerations made abovefor COMP-MED also apply to rMED; next to the common mutation R279W, thereare numerous other rare mutations that may result in either a ‘pure’ rMED pheno-type or in phenotypes that overlap partially with diastrophic dysplasia (previouslycalled ‘diastrophic variant’).
MATN3-MED (EDM5) (MIM 607078)
Clinical features
In contrast with the other forms, the severe variant of MATN3 disease was only dis-covered several years after heterozygous mutations were associated with MED.15

28 S. Unger et al
MATN3 as a cause of MED was found by using linkage in a large four-generation familyfollowed by a candidate gene approach.16 Since then, several cases have been identifiedbut it remains a relatively rare form of MED. A single family has been reported in whicha matrilin mutation was inherited as a recessive trait; the phenotype was that of spon-dylo-epiphyseal dysplasia in homozygous children, and heterozygotes were reportedas phenotypically normal.15
Most patients present in early childhood with knee pain or knee and hip pain.17 Oc-casionally, genu valgum or varum is the presenting feature. Stature is often slightlyreduced compared with unaffected family members, but within the lower portion ofthe normal growth curve.10 The hip changes are progressive and many people requirehip replacements by middle age. This form of MED is associated with a high degree ofintrafamilial variability.17,18
Radiographic changes of MATN3-MED
In many respects, the radiographic features of MATN3-MED resemble those ofCOMP-MED. The epiphyses of the knees and hips are small, delayed in appearance,flattened and sometimes fragmented (Figure 6); they are not as rounded as inCOMP-MED.10 The proximal femoral epiphyses are almost always radiographically af-fected, and are usually smaller than normal (Figure 6). The distal femoral epiphyses thinout at the sides, and when they cone in the femoral metaphysis, they may resemblea ‘harlequin hat’. Longitudinal metaphyseal striations are not uncommon in MATN3-MED. Unlike in COMP-MED, the carpal bones of patients with a MATN3 presentationare delayed in appearance but smooth in contour (Figure 6). Again, one must be cau-tious before generalizing as many patients have not had complete radiographic exam-inations but only hip and knee x rays. If there are no prepubertal hand x rays, it isimpossible to assess maturational delay. Despite the relatively normal stature, severalpatients have been noted to have minor spinal radiographic abnormalities, usually de-scribed as endplate irregularity.10
Figure 6. Features of matrilin-3-associated multiple epiphyseal dysplasia (MATN3-MED) in an 8-year-old girl.
The proximal femoral epiphyses are small and already somewhat flattened. The acetabular margin is better
preserved than in cartilage oligomeric matrix protein (COMP)-MED. The epiphyses at the knee are small and
resemble those of COMP-MED in having thin edges; the shape of the distal femoral epiphyses in MATN3-
MED with the central depression is reminiscent of a ‘harlequin hat’. Metaphyseal striations may be more
common in MATN3-MED than in other forms of MED. The hand shows a general delay of maturation affect-
ing the carpal bones more than the phalangeal epiphyses. There is some phalangeal shortening in this case but
this is not a universal finding in MATN3-MED.

Multiple epiphyseal dysplasia 29
Molecular basis of MATN3-MED
Matrilin-3 is an important component of the cartilage extracellular membrane.19 Likethe other matrilin family members, MATN3 contains a von Willebrand factor A domain,and most of the mutations (but not all) are clustered there.10,16 Most mutations aremissense but at least one single base pair deletion causing a premature stop codonhas been reported.8
COL9-MED (EDM2, EDM3, EDM6; MIM 600204 AND 600969)
Clinical features
COMP was the first gene associated with MED.7 However, it was recognized thatCOMP mutations accounted for only a percentage of MED cases. By using linkageanalysis, a second MED (EDM2) locus was identified on chromosome 1 in the re-gion of COL9A2.20 Indeed, a splice site mutation was subsequently identified in thisgene.24 Later, analogous mutations were identified in the two other genes that en-code the remaining components of type IX collagen (COL9A1 and COL9A3).21,22
However, COL9-MED appears to be quite rare in North America and Europewith only a few families reported. COL9-MED is a relatively benign form of the dis-order, presenting late in the first decade of life with knee pain and stiffness. As withCOMP-MED, a clinical picture mimicking a myopathy can be the presentingfeature.23
Radiographic changes of COL9-MED
Due to the rarity of the condition and the symptoms being mainly limited to theknees, there is a paucity of radiographs available for molecularly proven COL9-MED cases; therefore, one must be cautious about making generalizations. Theknees are the anatomical site most consistently radiographed, and often have signif-icant abnormalities such as flattened and irregular epiphyses.24,25 There is a generaltrend towards more severe involvement of the knees than the hips compared withCOMP-MED.25,26 COL9-MED seems to be more common in Japanese populations,and several families have been documented. These families demonstrate that despitea lack of symptoms, many epiphyses are radiographically abnormal, especially at thewrists and ankles.5,25 Few patients have had spinal x rays but those are reported asnormal.23,27
Molecular basis of COL9-MED
Type IX collagen is a structural component of the extracellular matrix and is a het-erotrimer composed from one chain each of COL9A1, COL9A2 and COL9A3. It isa fibril-associated collagen with interrupted triple helices. Each peptide chain hasthree collagenous domains separated by four non-collagenous domains.28 Interest-ingly, all mutations found to date have been of the exon skipping variety predictedto cause a loss of amino acids in the third collagenous domain.21,22,24,29 Despitecomplete sequencing of the gene in several MED patients, no other mutationshave been found.5
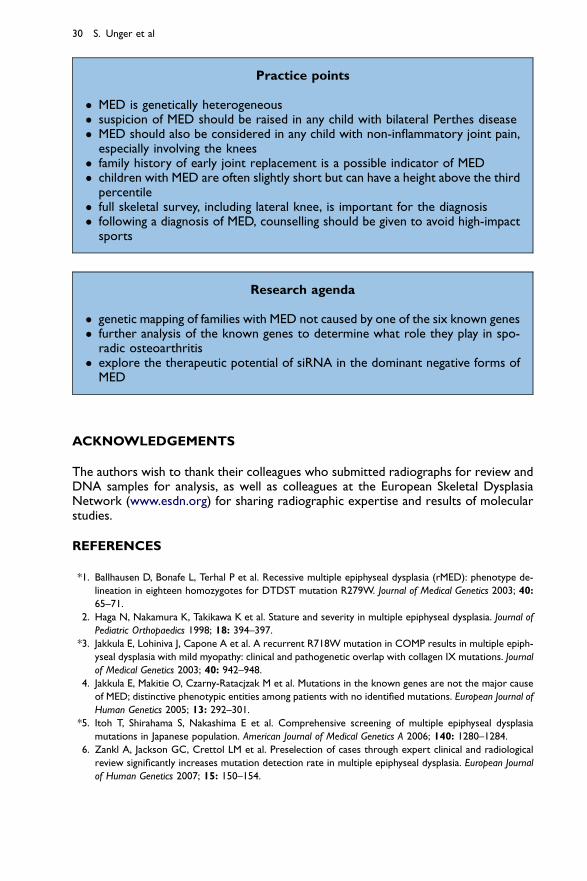
Practice points
� MED is genetically heterogeneous� suspicion of MED should be raised in any child with bilateral Perthes disease� MED should also be considered in any child with non-inflammatory joint pain,
especially involving the knees� family history of early joint replacement is a possible indicator of MED� children with MED are often slightly short but can have a height above the third
percentile� full skeletal survey, including lateral knee, is important for the diagnosis� following a diagnosis of MED, counselling should be given to avoid high-impact
sports
Research agenda
� genetic mapping of families with MED not caused by one of the six known genes� further analysis of the known genes to determine what role they play in spo-
radic osteoarthritis� explore the therapeutic potential of siRNA in the dominant negative forms of
MED
30 S. Unger et al
ACKNOWLEDGEMENTS
The authors wish to thank their colleagues who submitted radiographs for review andDNA samples for analysis, as well as colleagues at the European Skeletal DysplasiaNetwork (www.esdn.org) for sharing radiographic expertise and results of molecularstudies.
REFERENCES
*1. Ballhausen D, Bonafe L, Terhal P et al. Recessive multiple epiphyseal dysplasia (rMED): phenotype de-
lineation in eighteen homozygotes for DTDST mutation R279W. Journal of Medical Genetics 2003; 40:
65–71.
2. Haga N, Nakamura K, Takikawa K et al. Stature and severity in multiple epiphyseal dysplasia. Journal of
Pediatric Orthopaedics 1998; 18: 394–397.
*3. Jakkula E, Lohiniva J, Capone A et al. A recurrent R718W mutation in COMP results in multiple epiph-
yseal dysplasia with mild myopathy: clinical and pathogenetic overlap with collagen IX mutations. Journal
of Medical Genetics 2003; 40: 942–948.
4. Jakkula E, Makitie O, Czarny-Ratacjzak M et al. Mutations in the known genes are not the major cause
of MED; distinctive phenotypic entities among patients with no identified mutations. European Journal of
Human Genetics 2005; 13: 292–301.
*5. Itoh T, Shirahama S, Nakashima E et al. Comprehensive screening of multiple epiphyseal dysplasia
mutations in Japanese population. American Journal of Medical Genetics A 2006; 140: 1280–1284.
6. Zankl A, Jackson GC, Crettol LM et al. Preselection of cases through expert clinical and radiological
review significantly increases mutation detection rate in multiple epiphyseal dysplasia. European Journal
of Human Genetics 2007; 15: 150–154.

Multiple epiphyseal dysplasia 31
7. Briggs MD, Hoffman SM, King LM et al. Pseudoachondroplasia and multiple epiphyseal dysplasia due to
mutations in the cartilage oligomeric matrix protein gene. Nature Genetics 1995; 10: 330–336.
*8. Briggs MD & Chapman KL. Pseudoachondroplasia and multiple epiphyseal dysplasia: mutation re-
view, molecular interactions, and genotype to phenotype correlations. Human Mutation 2002; 19:
465–478.
9. Briggs MD, Mortier GR, Cole WG et al. Diverse mutations in the gene for cartilage oligomeric matrix
protein in the pseudoachondroplasia-multiple epiphyseal dysplasia disease spectrum. American Journal of
Human Genetics 1998; 62: 311–319.
*10. Mabuchi A, Haga N, Maeda K et al. Novel and recurrent mutations clustered in the von Willebrand
factor A domain of MATN3 in multiple epiphyseal dysplasia. Human Mutation 2004; 24: 439–440.
11. Rimoin DL, Rasmussen IM, Briggs MD et al. A large family with features of pseudoachondroplasia and
multiple epiphyseal dysplasia: exclusion of seven candidate gene loci that encode proteins of the carti-
lage extracellular matrix. Human Genetics 1994; 93: 236–242.
12. Superti-Furga A, Neumann L, Riebel T et al. Recessively inherited multiple epiphyseal dysplasia with
normal stature, club foot, and double layered patella caused by a DTDST mutation. Journal of Medical
Genetics 1999; 36: 621–624.
13. Nakashima E, Ikegawa S, Ohashi H et al. Double-layered patella in multiple epiphyseal dysplasia is not
exclusive to DTDST mutation. American Journal of Medical Genetics 2005; 133: 106–107.
*14. Rossi A & Superti-Furga A. Mutations in the diastrophic dysplasia sulfate transporter (DTDST) gene
(SLC26A2): 22 novel mutations, mutation review, associated skeletal phenotypes, and diagnostic rele-
vance. Human Mutation 2001; 18: 82.
15. Borochowitz ZU, Scheffer D, Adir V et al. Spondylo-epi-metaphyseal dysplasia (SEMD) matrilin 3 type:
homozygote matrilin 3 mutation in a novel form of SEMD. Journal of Medical Genetics 2004; 41:
366–372.
16. Chapman KL, Mortier GR, Chapman K et al. Mutations in the region encoding the von Willebrand
factor A domain of matrilin-3 are associated with multiple epiphyseal dysplasia. Nature Genetics 2001;
28: 393–396.
*17. Makitie O, Mortier GR, Czarny-Ratajczak M et al. Clinical and radiographic findings in multiple epiph-
yseal dysplasia caused by MATN3 mutations: description of 12 patients. American Journal of Medical Ge-
netics 2004; 125: 278–284.
*18. Mortier GR, Chapman K, Leroy JL & Briggs MD. Clinical and radiographic features of multiple epiphy-
seal dysplasia not linked to the COMP or type IX collagen genes. European Journal of Human Genetics
2001; 9: 606–612.
19. Deak F, Wagener R, Kiss I & Paulsson M. The matrilins: a novel family of oligomeric extracellular matrix
proteins. Matrix Biology 1999; 18: 55–64.
20. Briggs MD, Choi H, Warman ML et al. Genetic mapping of a locus for multiple epiphyseal dysplasia
(EDM2) to a region of chromosome 1 containing a type IX collagen gene. American Journal of Human
Genetics 1994; 55: 678–684.
21. Czarny-Ratajczak M, Lohiniva J, Rogala P et al. A mutation in COL9A1 causes multiple epiphyseal dys-
plasia: further evidence for locus heterogeneity. American Journal of Human Genetics 2001; 69: 969–980.
22. Paassilta P, Lohiniva J, Annunen S et al. COL9A3: a third locus for multiple epiphyseal dysplasia. American
Journal of Human Genetics 1999; 64: 1036–1044.
23. Bonnemann CG, Cox GF, Shapiro F et al. A mutation in the alpha 3 chain of type IX collagen
causes autosomal dominant multiple epiphyseal dysplasia with mild myopathy. Proceedings of the Na-
tional Academy of Sciences of the United States of America 2000; 97: 1212–1217.
24. Muragaki Y, Mariman EC, van Beersum SE et al. A mutation in the gene encoding the alpha 2 chain of
the fibril-associated collagen IX, COL9A2, causes multiple epiphyseal dysplasia (EDM2). Nature Genetics
1996; 12: 103–105.
25. Takahashi M, Matsui Y, Goto T et al. Intrafamilial phenotypic diversity in multiple epiphyseal dysplasia
associated with a COL9A2 mutation (EDM2). Clinical Rheumatology 2006; 25: 591–595.
*26. Unger SL, Briggs MD, Holden P et al. Multiple epiphyseal dysplasia: radiographic abnormalities corre-
lated with genotype. Pediatric Radiology 2001; 31: 10–18.
27. Holden P, Canty EG, Mortier GR et al. Identification of novel pro-alpha2(iX) collagen gene mutations in
two families with distinctive oligoepiphyseal forms of multiple epiphyseal dysplasia. American Journal of
Human Genetics 1999; 65: 31–38.

32 S. Unger et al
28. Pihlajamaa T, Vuoristo MM, Annunen S et al. Human COL9A1 and COL9A2 genes. Two genes of
90 and 15 kb code for similar polypeptides of the same collagen molecule. Matrix Biology 1998;
17: 237–241.
29. Lohiniva J, Paassilta P, Seppanen U et al. Splicing mutations in the COL3 domain of collagen IX cause
multiple epiphyseal dysplasia. American Journal of Medical Genetics 2000; 90: 216–222.