reaction kinetics workshop, part i - iscreiscre.org/iscre24/vlachos_iscre-mkm.pdf · reaction...
TRANSCRIPT

1
Catalysis Center for Energy Innovation
Reaction Kinetics Workshop, Part I
CCEI is an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Basic Sciences
University of DelawareDepartment of Chemical and Biomolecular Engineering Center for Catalytic Science and Technology (CCST)Catalysis Center for Energy Innovation (CCEI),
an Energy Frontier Research Center
June 12, 2016
Catalysis Center for Energy Innovation
Types of catalytic kinetic models and microkinetic modeling
Overview of parameter estimation methods and scales
Accuracy
Lateral interactions
Semi‐empirical methods
Thermodynamic consistency
MKM uses
Reactor design, analysis, catalyst discovery
Kinetic Monte Carlo
Outline

2
Catalysis Center for Energy Innovation
Types of catalytic kinetic models and microkinetic modeling
Overview of parameter estimation methods and scales
Accuracy
Lateral interactions
Semi‐empirical methods
Thermodynamic consistency
MKM uses
Reactor design, analysis, catalyst discovery
Kinetic Monte Carlo
Outline
Catalysis Center for Energy Innovation
Surface Reaction Rate Calculation Paradigm
• Hierarchy of calculation of surface reaction rates
Empirical rate-law; typically fitted to experimental data
Langmuir-Hinshelwood rate-law; developed using rate determining step (RDS) and MF theory
Microkinetic analysis;No assumptions on RDS (Dumesic, 1993)
Complexity/Reliability/Accuracy
3
Catalysis Center for Energy Innovation
Reaction A+B Products
Typical power‐low expression
The parameters have no physical significanceExponents unrelated to stoichiometryApparent activation energies can be negative
Prediction is reliable within the given experimental space
Can describe overall rate and heat effects
Simple interpolation modelsNot good for process optimizationCan typically describe only one set of data
Global reaction rate models
effE / RTa beff A B eff effr=k C C ;k =A e
Catalysis Center for Energy Innovation
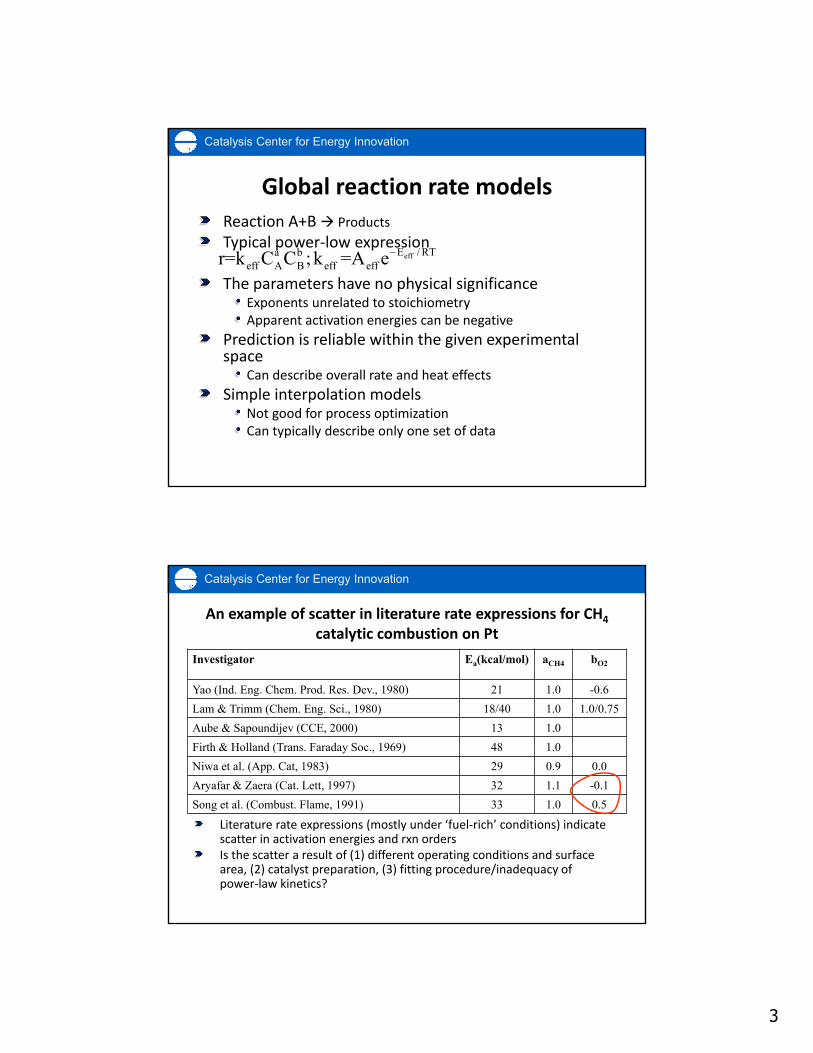
Literature rate expressions (mostly under ‘fuel‐rich’ conditions) indicate scatter in activation energies and rxn ordersIs the scatter a result of (1) different operating conditions and surface area, (2) catalyst preparation, (3) fitting procedure/inadequacy of power‐law kinetics?
An example of scatter in literature rate expressions for CH4
catalytic combustion on Pt
Investigator Ea(kcal/mol) aCH4 bO2
Yao (Ind. Eng. Chem. Prod. Res. Dev., 1980) 21 1.0 -0.6
Lam & Trimm (Chem. Eng. Sci., 1980) 18/40 1.0 1.0/0.75
Aube & Sapoundijev (CCE, 2000) 13 1.0
Firth & Holland (Trans. Faraday Soc., 1969) 48 1.0
Niwa et al. (App. Cat, 1983) 29 0.9 0.0
Aryafar & Zaera (Cat. Lett, 1997) 32 1.1 -0.1
Song et al. (Combust. Flame, 1991) 33 1.0 0.5

4
Catalysis Center for Energy Innovation
LH expressions are based on a priori assumptions and intuition
They are usually fitted using a limited number of data
Multiple rate expressions can describe the same data
Multiple parameters even for the same rate expression may exist
Even if data is well‐fitted, parameters may be physically unreasonable
LH expressions, even if correct, are limited to one regime and cannot describe changes in RDS, MARI, etc. with operating conditions
Summary of LH rate expressions
Catalysis Center for Energy Innovation
CH4 total oxidation (combustion)CH4 + 2O2= CO2 + 2H2O
Steam reforming of CH4
CO2-reforming of CH4
RWGS
LH Rates on Rh
= effectiveness factor

5
Catalysis Center for Energy Innovation
Process is away from equilibrium
Model fits data fairly well
Reactions in series is proposed
Combustion of syngas is important
Comparison of model to data: Conversion
dashed lines=model w/o consecutive combustion of CO and H2; solid lines=model with consecutive CO and H2
combustion
Catalysis Center for Energy Innovation
Models may fit but most often than not include unrealistic parameters
Are the parameters/model reasonable?
Estimated parameters
Hads[kcal/mol]13.939.56.2

6
Catalysis Center for Energy Innovation
Microkinetic Modeling• All relevant elementary reactions
– Written by hand or computer generated1
• No simplifying assumptions
• Rate determining step (RDS), partial equilibrium (PE), quasi‐steady state (QSS), and most abundant reaction intermediate (MARI) are all computed
• Reactor + Catalyst model needed– Use computer software, such as surface
CHEMKIN3, Cantera2, or Matlab
An example1
2( ) 2( ) 2 ( )2
2( )
2( )
2
2 2 ( )
Re
2* 2 *
2* 2 *
* * * *
* * * *
*
g g g
g
g
g
H O H O
Elementary actions
H H
O O
H O HO
HO H H O
H O H O
1Ring: Rangarajan et al., Computers & Chemical Engineering 45, 114 (2012).2Cantera (Matlab Chem Kinetics Package): Goodwin et al., Cantera: An Object-oriented Software Toolkit for Chemical Kinetics, Thermodynamics, and Transport Processes. 2014.3Chemkin (Fortran Chem Kinetics Package): Coltrin; Kee and Rupley, Int. J. Chem. Kinet. 23, 1111 (1991).Coltrin; Kee and Rupley Surface CHEMKIN (Version 4. 0): A Fortran package for analyzing heterogeneous chemical kinetics at a solid-surface---gas-phase interface; SAND-90-8003B; 1991.Reactor Design (commercial kinetics software)
Catalysis Center for Energy Innovation
Types of catalytic kinetic models and microkinetic modeling
Overview of parameter estimation methods and scales
Accuracy
Lateral interactions
Semi‐empirical methods
Thermodynamic consistency
MKM uses
Analysis
Catalyst discovery
Outline

7
Catalysis Center for Energy Innovation
Parameter Estimation of Microkinetic Models
Parameters fitted to data Inability to simultaneously predict multiple experimental sets
Parameters estimated with empirical methods (Bond-Order Conservation) Efficient, reasonable accuracy (2-4 kcal/mol) Limited to small adsorbates
Density functional theory (DFT)-based semi-empirical methods Group additivity, Brønsted-Evans-Polanyi (BEP) relations
Parameters obtained from DFT Fairly accurate (<5 kcal/mol)
Hierarchical methods Empirical or semi-empirical to screen; DFT to refine (zero coverage limit) Include coverage effects
Catalysis Center for Energy Innovation
Hierarchy Enables Rapid Screening ofChemistry, Fuels, and Catalysts
Quantum:ab initio, DFT, TST,
CPMD, QM/MM MD
Continuum: MF-ODEs
Discrete: KMC
Ideal: PFR, CSTR, etc.
Computational Fluid Dynamics
(CFD)
Mesoscopic:PDEs
Discrete:CG-KMC
Pseudo-homogeneous:Transport correlations
Quantum-based correlations:
BEPs, GA, LSRs
Catalyst scale:Reaction rate
Reactor scale:Performance
Electronic scale:Parameter estimation
Accuracy, cost

8
Catalysis Center for Energy Innovation
Types of catalytic kinetic models and microkinetic modeling
Overview of parameter estimation methods and scales
Accuracy
Lateral interactions
Semi‐empirical methods
Thermodynamic consistency
MKM uses
Reactor design, analysis, catalyst discovery
Kinetic Monte Carlo
Outline
Catalysis Center for Energy Innovation
Accuracy of MKM
• Myth: A microkinetic (detailed) model [even with DFT input] can quantitatively describe experimental data
9
Catalysis Center for Energy Innovation
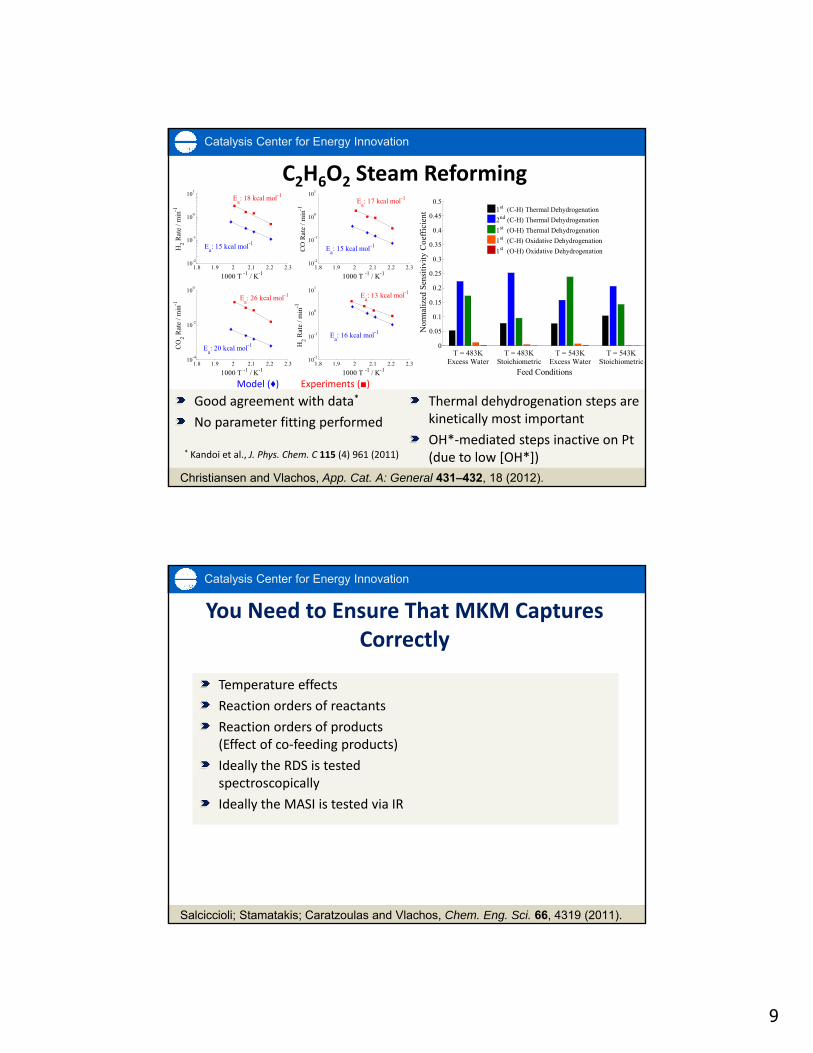
C2H6O2 Steam Reforming
Good agreement with data*
No parameter fitting performed
Thermal dehydrogenation steps are kinetically most important
OH*‐mediated steps inactive on Pt (due to low [OH*])
Christiansen and Vlachos, App. Cat. A: General 431–432, 18 (2012).
1.8 1.9 2 2.1 2.2 2.310
-2
10-1
100
101
Ea: 18 kcal mol-1
Ea: 15 kcal mol-1
1000 T -1 / K-1
H2 R
ate
/ min
-1
1.8 1.9 2 2.1 2.2 2.310
-2
10-1
100
101
Ea: 17 kcal mol-1
Ea: 15 kcal mol-1
1000 T -1 / K-1
CO
Rat
e / m
in-1
1.8 1.9 2 2.1 2.2 2.310
-4
10-2
100
Ea: 26 kcal mol-1
Ea: 20 kcal mol-1
1000 T -1 / K-1
CO
2 Rat
e / m
in-1
1.8 1.9 2 2.1 2.2 2.310
-2
10-1
100
101
Ea: 13 kcal mol-1
Ea: 16 kcal mol-1
1000 T -1 / K-1
H2 R
ate
/ min
-1
Model (♦) Experiments (■)
* Kandoi et al., J. Phys. Chem. C 115 (4) 961 (2011)
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
0.4
0.45
0.5
Nor
mal
ized
Sen
siti
vity
Coe
ffic
ient
T = 483KExcess Water
T = 483KStoichiometric
T = 543KExcess Water
T = 543KStoichiometric
Feed Conditions
1st (C-H) Thermal Dehydrogenation
2nd (C-H) Thermal Dehydrogenation
1st (O-H) Thermal Dehydrogenation
1st (C-H) Oxidative Dehydrogenation
1st (O-H) Oxidative Dehydrogenation
Catalysis Center for Energy Innovation
You Need to Ensure That MKM Captures Correctly
Temperature effects
Reaction orders of reactants
Reaction orders of products (Effect of co‐feeding products)
Ideally the RDS is tested spectroscopically
Ideally the MASI is tested via IR
Salciccioli; Stamatakis; Caratzoulas and Vlachos, Chem. Eng. Sci. 66, 4319 (2011).
10
Catalysis Center for Energy Innovation
• Types of catalytic kinetic models and microkinetic modeling
• Overview of parameter estimation methods and scales
• Accuracy
• Lateral interactions
• Semi‐empirical methods
• Thermodynamic consistency
• MKM uses
– Reactor design, analysis, catalyst discovery
• Kinetic Monte Carlo
Outline
Catalysis Center for Energy Innovation
Lateral Interactions:
Estimation via Hierarchical Estimation Methodology
Myth: After parameterization of a microkinetic model via DFT (or semi‐empirical methods), the model is correct and no further refinement is needed

11
Catalysis Center for Energy Innovation
Lateral Interactions Are Typically Critical
Adsorbate heterogeneity arises due to coverage effectsCombinatorial problem in a priori estimating kinetic parameters due to coverage effects
x (nm)
y (nm)
Spatial Occupation Probability Map for CO*
0 0.5 1 1.5
0
0.5
1
1.5
0
0.2
0.4
0.6
0.8
1
Review of Catalyst and Kinetic Modeling: Salciccioli et al., Chem. Eng. Sci. 66, 4319 (2011)
Catalysis Center for Energy Innovation
Hierarchical Multiscale Modeling Principle: Dealing with Size (No. of Reactions and Coverage)
Start with a sufficiently simple, physical model at each scaleAutomatic mechanism generation
First‐principles based semi‐empirical parameter estimation
Link all models
Identify important scale and parameter(s)Sensitivity and flux analyses
Use higher level theory for this scale and parameter(s)Kinetically relevant steps, inclusion of coverage effects, internal diffusion, etc.
Iterate
First‐principles accuracy at orders of magnitude reduced cost
12
Catalysis Center for Energy Innovation
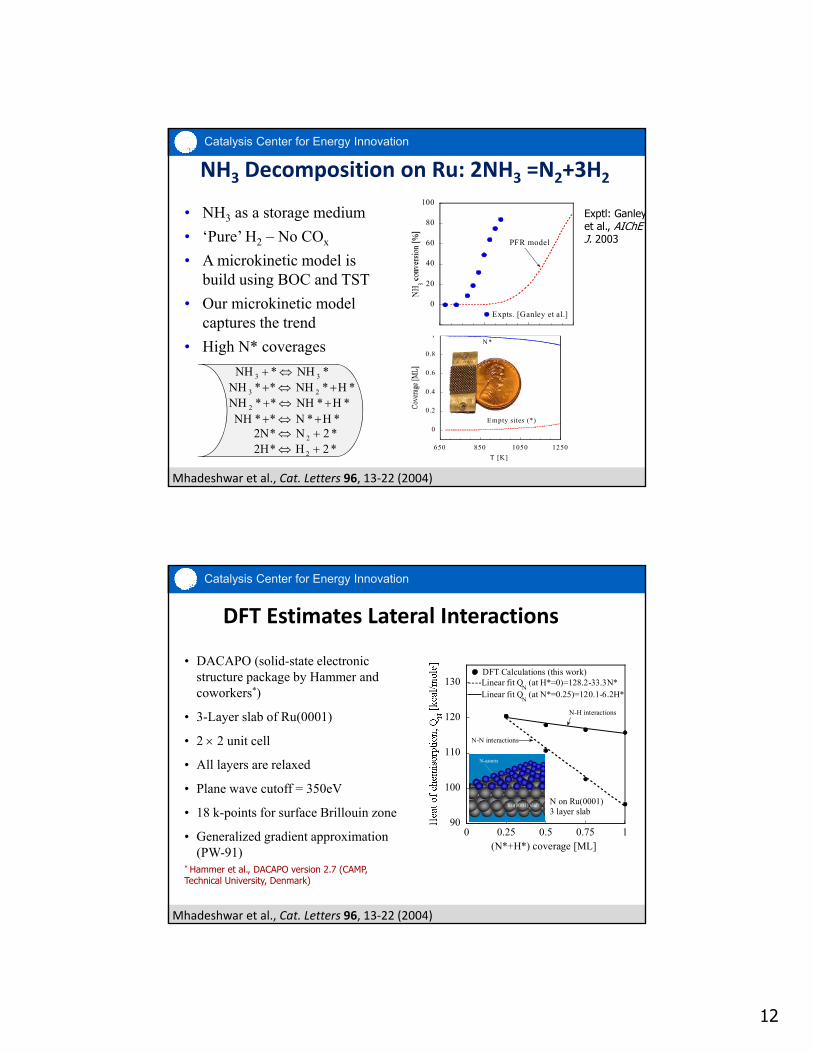
NH3 Decomposition on Ru: 2NH3 =N2+3H2
• NH3 as a storage medium
• ‘Pure’ H2 – No COx
• A microkinetic model is build using BOC and TST
• Our microkinetic model captures the trend
• High N* coverages
196 m x 84 m x 1078 m
Mhadeshwar et al., Cat. Letters 96, 13‐22 (2004)
0
0.2
0.4
0.6
0.8
1
650 850 1050 1250
T [K]
N*
Empty sites (*)
0
20
40
60
80
100
Expts. [Ganley et al.]
PFR model
Exptl: Ganleyet al., AIChE J. 2003
*NH*NH 33 *H*NH**NH 23
*H*NH**NH 2 *H*N**NH *2N*2N 2 *2H*2H 2
Catalysis Center for Energy Innovation
DFT Estimates Lateral Interactions
• DACAPO (solid-state electronic structure package by Hammer and coworkers*)
• 3-Layer slab of Ru(0001)
• 2 2 unit cell
• All layers are relaxed
• Plane wave cutoff = 350eV
• 18 k-points for surface Brillouin zone
• Generalized gradient approximation (PW-91)
* Hammer et al., DACAPO version 2.7 (CAMP, Technical University, Denmark)
90
100
110
120
130
0 0.25 0.5 0.75 1(N*+H*) coverage [ML]
DFT Calculations (this work)Linear fit Q
N (at H*=0)=128.2-33.3N*
N on Ru(0001) 3 layer slab
Linear fit QN (at N*=0.25)=120.1-6.2H*
N-N interactions
N-H interactions
Mhadeshwar et al., Cat. Letters 96, 13‐22 (2004)
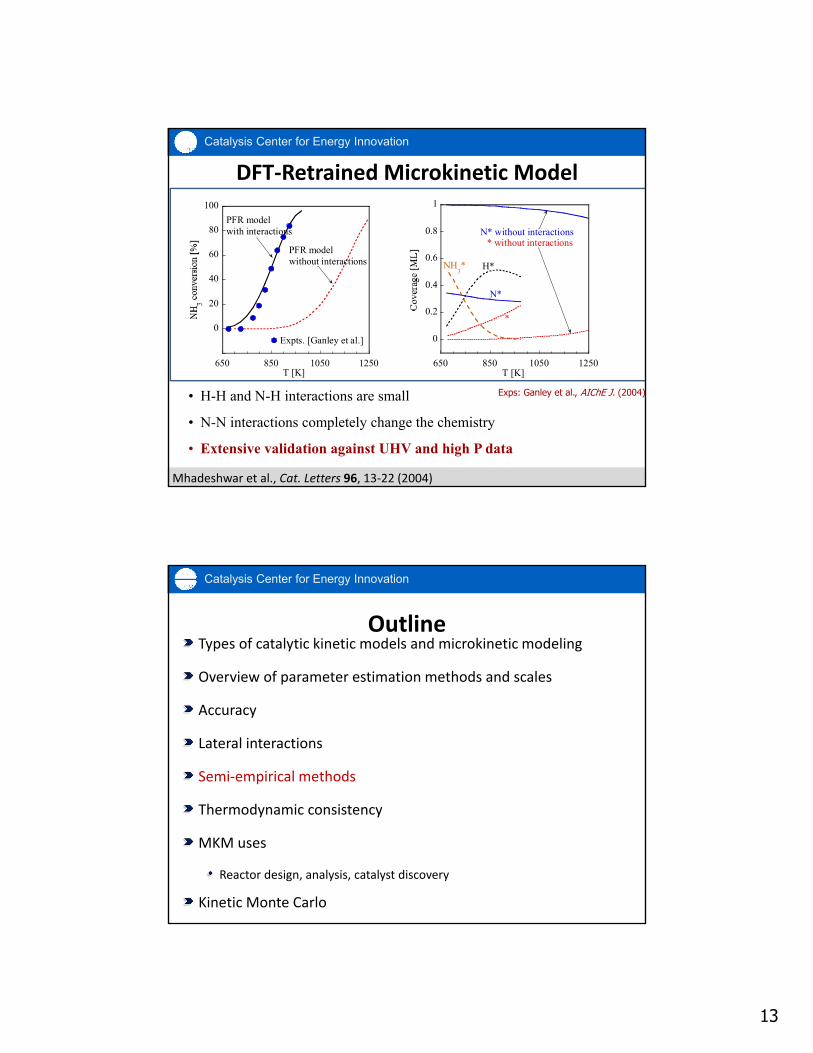
13
Catalysis Center for Energy Innovation
Exps: Ganley et al., AIChE J. (2004)
DFT‐Retrained Microkinetic Model
• H-H and N-H interactions are small
• N-N interactions completely change the chemistry
• Extensive validation against UHV and high P data
0
20
40
60
80
100
650 850 1050 1250
Expts. [Ganley et al.]
PFR modelwithout interactions
T [K]
PFR modelwith interactions
0
0.2
0.4
0.6
0.8
1
650 850 1050 1250T [K]
*
N* without interactions* without interactions
H*
N*
NH3*
Mhadeshwar et al., Cat. Letters 96, 13‐22 (2004)
Catalysis Center for Energy Innovation
Types of catalytic kinetic models and microkinetic modeling
Overview of parameter estimation methods and scales
Accuracy
Lateral interactions
Semi‐empirical methods
Thermodynamic consistency
MKM uses
Reactor design, analysis, catalyst discovery
Kinetic Monte Carlo
Outline

14
Catalysis Center for Energy Innovation
Myth: First‐principles (DFT) MKM can be developed for any mechanism and feedstock
Estimation with first‐principles semi‐empirical methods (FPSEM) is possible
Refinement of important parameters is feasible
Semi‐Empirical Estimation Methods
Catalysis Center for Energy Innovation
Modeling Reactions of Large Molecules is Challenging
Combinatorial explosion in number of calculations for first‐principles (DFT) calculations
Semi‐empirical methods can potentially identify relevant species and reactions instantaneously
Major advances in systematic development of semi‐empirical methods and understanding of errors
10
102
103
104
105
106
IntermediatesReactions
Num
ber
of C
alcu
latio
ns
HO
HOOH
HO
OH
OHHO
OHHO
HO
HO
OH
OHHO
HO
Sugars

15
Catalysis Center for Energy Innovation
DFT‐Based Estimation Methods
Surface thermochemistry via group additivity
Brønsted‐Evans Polanyi relations for reaction barriers of homologous series
Transfer thermo from one material to another (linear scaling relations)
Perform high‐throughput microkinetic modeling (MKM) for materials prediction
Identify key steps and refine them via higher level theory
Linear Scaling Relations
• Libraries of atomic binding energies
• Binding energies of AHx species vs. heteroatom A valency
• Metal transferability
Group Additivity
• Single metal thermochemistry
• Thermodynamic consistency
• Screen adsorbates
Brønsted Evans Polanyi (BEP)
• Activation energies• Reaction rate constants
• Screen reactions
Microkinetic Model
•Compute rates, conversion, selectivity, abundant species
• Identify key adsorbates and reactions
•Refine thermochemistry and barriers via DFT
• Include adsorbate-adsorbate interactions
Review: Salciccioli et al., Chem. Eng. Sci. 66, 4319 (2011);Salciccioli et al., J. Phys. Chem. C 114, 20155 (2010); J. Phys. Chem. C 116, 1873 (2012); Sutton and Vlachos, ACS Catal. 2, 1624 (2012); J. Catal. 297, 202 (2013)
Catalysis Center for Energy Innovation
• Binding for oxygenates traced to (CHyOx) groups
• Use alcohols and dehydrogenated alcohol intermediates to develop groups
• Include contributions for ΔHf,298, S(T) and Cp(T)
GroupΔHf,298 Value
[kcal/mol][C(C,H2)-O(M,H)] -50.2[C(C,H,M)-O(H)] -46.8[C(C,M2)-O(H)] -39.3[C(C,H2)-O(M)] -26.1[C(C,H,M)-O(M)] -26.5[C(C,M,M)-O(M)] -33.4[C(C,M)=O] -33.4[C(C2,H)-O(M,H)] -51.1[C(C2,M)-O(H)] -42.9[C(C2,H)-O(M)] -25.1[C(C2,M)-O(M)] -23.0
C
HH
OH
Pt Pt Pt
C
H
H
H
Group Additivity for Adsorbed Species
Salciccioli, Y. Chen, and Vlachos, J. Phys. Chem. C 114(47), 20155 (2010).
O
C CH2
A BO
HC CO

16
Catalysis Center for Energy Innovation
Group Additivity for Adsorbed SpeciesSecond‐order effects included:
4‐member ring strainWeak interactionsHydrogen bonding
Calculations of ΔHf,298 of C2HxO2 and C3HxO3 species show good quantitative agreement
This method can be used for initial screening of larger hydrocarbons and oxygenates
-160
-120
-80
-40
-160-120-80-40
C2H
xO
2
C3H
xO
3
Gro
up a
ddit
ivit
y [k
cal/
mol
]
DFT calculated [kcal/mol]
Values are taken with respect to the most highly hydrogenated species in the gas phase (C2H6O2, C3H8O2) and H adsorbed on a separate slab
Salciccioli, Y. Chen, and Vlachos, J. Phys. Chem. C 114(47), 20155 (2010).Salciccioli et al., J. Phys. Chem. C 114, 20155 (2010); J. Phys. Chem. C 116, 1873 (2012)
Catalysis Center for Energy Innovation
Molecular binding energy is linearly dependent on atomic binding energy1
These correlations relate molecular binding energy to atomic binding energy on different surfacesMultidentate species can be accounted for by summing the contributions of each bond to the surface2
Transferability Between Metals
[1] Abild‐Pedersen et al. PRL 99, (2007)[2] Salciccioli, Y. Chen, and Vlachos, J. Phys. Chem. C 114(47), 20155 (2010)
M Pt A,i,M A,i,Pt S ROHA,i
Q Q Q Q x E E
C
HH OH C
HH OH
C
HOHC
HOH≈
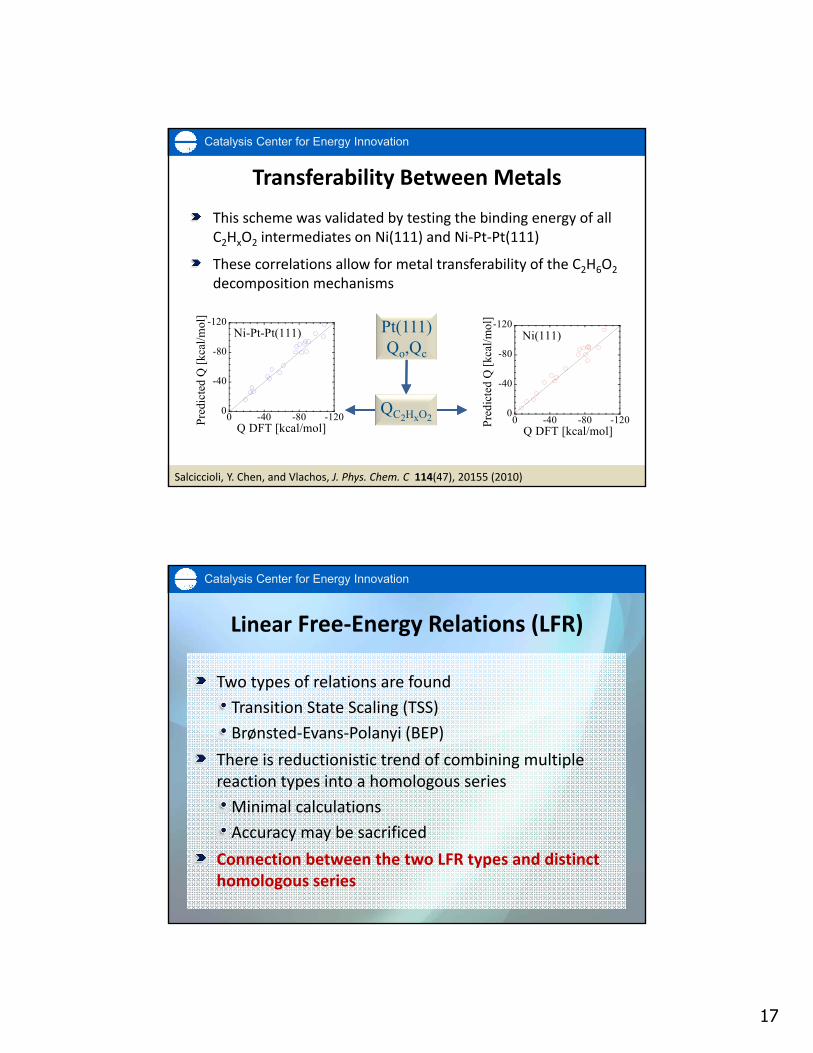
17
Catalysis Center for Energy Innovation
This scheme was validated by testing the binding energy of all C2HxO2 intermediates on Ni(111) and Ni‐Pt‐Pt(111)
These correlations allow for metal transferability of the C2H6O2
decomposition mechanisms
-120
-80
-40
0-120-80-400
Pre
dict
ed Q
[kc
al/m
ol]
Q DFT [kcal/mol]
Ni(111)-120
-80
-40
0-120-80-400P
redi
cted
Q [
kcal
/mol
]
Q DFT [kcal/mol]
Ni-Pt-Pt(111) Pt(111)Qo,Qc
QC2HxO2
Salciccioli, Y. Chen, and Vlachos, J. Phys. Chem. C 114(47), 20155 (2010)
Transferability Between Metals
Catalysis Center for Energy Innovation
Linear Free‐Energy Relations (LFR)
Two types of relations are found
Transition State Scaling (TSS)
Brønsted‐Evans‐Polanyi (BEP)
There is reductionistic trend of combining multiple reaction types into a homologous series
Minimal calculations
Accuracy may be sacrificed
Connection between the two LFR types and distinct homologous series

18
Catalysis Center for Energy Innovation
Distinct Homologous Series Developed
Performed DFT calculations for methane, methanol, ethane, and ethanol
45 stable intermediates and 124 transition states
Considered C‐H (a and b positions), O‐H, C‐C, C‐O, and C‐OH reactions
Statistical tests are used
-1
0
1
2
3
4
5
-3 -2 -1 0 1 2 3
Combined C-HO-HCombined C-CC-OHC-O
Ac
tiv
ati
on
En
erg
y (
eV
)
Heat of Reaction (eV)
Sutton and Vlachos, ACS Catal. 2 1624 (2012); J. Catal. 297, 202 (2013)
Catalysis Center for Energy Innovation
Profound computational savings(Important) information content remains the same
1
102
104
106
CP
U h
rs
DFT
Scre
en
conf
orm
ers
via
GA
DFT
GA
/BE
P->
DFT
GA/BEP
Glycerol thermal decomposition
Chen et al., J. Phys. Chem. 115(38), 18707 (2011)
Computational Savings from Hierarchy

19
Catalysis Center for Energy Innovation
Hierarchical Multiscale Modeling Principle: Dealing with Size (No. of Reactions and Coverage)
Start with a sufficiently simple, physical model at each scaleAutomatic mechanism generation
First‐principles based semi‐empirical parameter estimation
Link all models
Identify important scale and parameter(s)Sensitivity and flux analyses
Use higher level theory for this scale and parameter(s)Kinetically relevant steps, inclusion of coverage effects, internal diffusion, etc.
Iterate
First‐principles accuracy at orders of magnitude reduced cost
Catalysis Center for Energy Innovation
Speed of Convergence of FPSEM Models
0
0.2
0.4
0.6
0.8
1
300 320 340 360 380 400
DFT02345
Fra
cti
on
al C
on
vers
ion
Temperature (°C)
It takes only a few iterations to converge the semi‐empirical model to be nearly indistinguishable with the DFT‐based MKM
Interaction parameters in the first two iterations are most critical for qualitative agreement
3 2 4 2CH CH OH CH + CO + H
3 2 3 2CH CH OH CH CHO + H
3 2 4 22CH CH OH 3CH + CO
Ethanol MKM/Pt
160 reversible reactions
67 gas and surface species
0
0.1
0.2
0.3
0.4
0.5
0.6
300 320 340 360 380 400
Fra
cti
on
al C
Se
lect
ivit
y, C
H4
Temperature (°C)
Sutton and Vlachos, Chem. Eng. Sci., In press

20
Catalysis Center for Energy Innovation
Types of catalytic kinetic models and microkinetic modeling
Overview of parameter estimation methods and scales
Accuracy
Lateral interactions
Semi‐empirical methods
Thermodynamic consistency
MKM uses
Reactor design, analysis, catalyst discovery
Kinetic Monte Carlo
Outline
Catalysis Center for Energy Innovation
Myth: Your model is thermodynamically consistent
A mechanism based solely on a single DFT mechanism is thermo consistent
Challenges
DFT is not accurate for gas‐phase thermo overall thermo is not accurate; thus, we often mix high level ab initio data or NIST thermo data
Kinetic parameters are adjusted to describe data
Either adjustment leads to thermo incosistencies
Thermodynamic Consistency of MKM
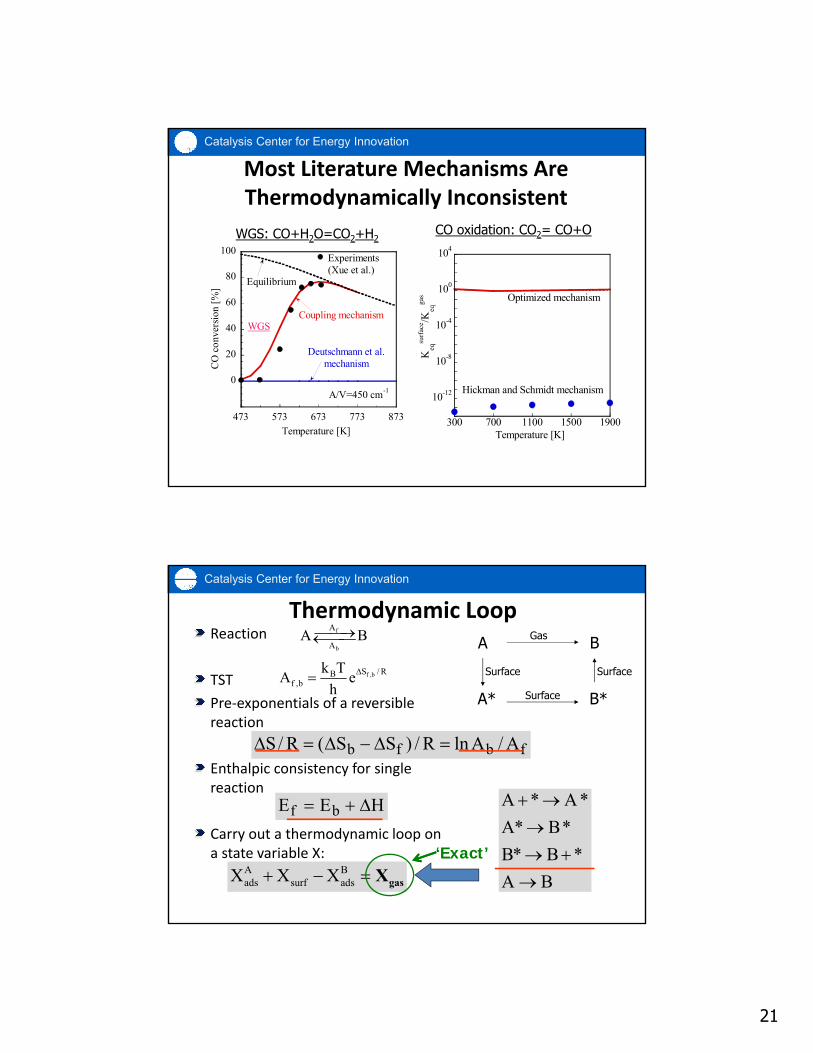
21
Catalysis Center for Energy Innovation
Most Literature Mechanisms Are Thermodynamically Inconsistent
0
20
40
60
80
100
473 573 673 773 873
CO
con
vers
ion
[%]
Temperature [K]
Coupling mechanismWGS
Equilibrium
Experiments(Xue et al.)
A/V=450 cm-1
Deutschmann et al.mechanism
WGS: CO+H2O=CO2+H2
10-12
10-8
10-4
100
104
300 700 1100 1500 1900K
eq
surf
ace /K
eq
gas
Temperature [K]
Hickman and Schmidt mechanism
Optimized mechanism
CO oxidation: CO2= CO+O
Catalysis Center for Energy Innovation
Reaction
TST
Pre‐exponentials of a reversible reaction
Enthalpic consistency for single reaction
Carry out a thermodynamic loop on a state variable X:
Thermodynamic Loop
f ,bS / RBf ,b
k TA e
h
fbfb A/AlnR/)SS(R/S
HEE bf
f
b
A
AA B A B
A* B*
Gas
SurfaceSurface
Surface
A Bads surf adsX X X gasX
A * A*
A* B*
B* B *
A B
‘Exact’

22
Catalysis Center for Energy Innovation
For every surface reaction, one can write a corresponding gas reaction and a thermo loop
The number of surface species usually determines the number of independent variables
Cannot simply change barriers or pre‐exponentials to fit data without paying attention to thermo consistency
Linear Independence and Parameter Tuning
• Mhadeshwar et al., Thermodynamic consistency in microkinetic development of surface reaction mechanisms, Journal of Physical Chemistry B 107, 12721-12733 (2003).
• Salciccioli et al., A review of multiscale modeling of metal-catalyzed reactions: Mechanism development for complexity and emergent behavior, Chem. Eng. Sci. 66, 4319–4355 (2011).
Catalysis Center for Energy Innovation
Types of catalytic kinetic models and microkinetic modeling
Overview of parameter estimation methods and scales
Accuracy
Lateral interactions
Semi‐empirical methods
Thermodynamic consistency
MKM uses
Reactor design, analysis, catalyst discovery
Kinetic Monte Carlo
Outline

23
Catalysis Center for Energy Innovation
Reconcile apparently contradictory experimental data at different conditions (TPD, steady state, various operating conditions)
Mechanistic understanding
Perform reactor optimization
Model‐based design of experiments to assess model
Rational catalyst designComposition
Size
Shape
What Can We Use MKMs for?Tr
aditi
onal
Mod
ern
Catalysis Center for Energy Innovation
Process
Reaction mechanism
Reactordesign
Catalystdiscovery
Catalysis and reaction engineering (experimental and computational)
Chemical kinetics
Microchemical systems for portable and distributed energy
In silico catalyst discovery
MKM at WorkJP8 (cat combustion)NH3
CH4; small HCs (POX, SR, hydrogenolysis, dehydrogenation)Alcohols (SR)WGS, PROX
2NH3=N2+3H2Hansgen et al., Nature Chem. 2, 484 (2010)

24
Catalysis Center for Energy Innovation
0
0.2
0.4
0.6
0.8
1
200 240 280 320 360
C2H5pi-C2H4sigma-C2H4CCH3Vacancies
Cov
erag
e
Temperature [K]
-1 0 1
H2=50 torr
150665
Normalized Sensitivity Coefficient
j
iij A
RS
ln
ln
Hydrogenation rxns are rate controlling (π-C2H4**→C2H5**, C2H5**→C2H6)
Ethylene Hydrogenation: Analysis
Catalysis Center for Energy Innovation
Ethylene Hydrogenation Reaction
H2 + 2* ↔ 2H*
C2H4 + 2* ↔ π-C2H4**
C2H6 + 3* ↔ C2H5** + H*
π-C2H4** + 2* ↔ σ-C2H4****
π-C2H4** + H* ↔ C2H5** + *
C2H5** + 3* ↔ σ-C2H4**** + H*
σ-C2H4**** ↔ C2H3** + H* + *
C2H2** + H* ↔ C2H3** + *
CHCH3** ↔ CCH3* + H*
CCH3* + 2* ↔ CCH2** + H*
C2H5** + * ↔ CHCH3** + H*
CHCH3** + *↔ C2H3** + H*
C2H3** + * ↔ CCH2** + H*
C2H** + H* ↔ CCH2** + *
10-510-410-310-210-1100101
1 10 100 1000
Reduced modelExperimentalOne step rate expressionC
H4 T
OF
[1/
s]
Ethane Pressure [Torr]
673K
623K
573K
0.01
0.1
1
10
100
100 1000
ExperimentalReduced microkinetic model Reduced rate expression
C2H
6 TO
F [s
-1]
Hydrogen Pressure [Torr]
336 K
298 K
273 K
248 K
223 K
H2 + 2* ↔ 2H*
CH4 + 2* ↔ CH3* + H*
C2H4 + 2* ↔ π-C2H4**
C2H4 + 4* ↔ σ-C2H4****
C2H2 + 2* ↔ C2H2**
C2H6 + 3* ↔ C2H5** + H*
π-C2H4** + 2* ↔ σ-C2H4****
π -C2H4** + H* ↔ C2H5** + *
C2H5** + 3* ↔ σ-C2H4**** + H*
σ-C2H4**** ↔ C2H3** + H* + *
C2H2** + H* ↔C2H3** + *
C2H** + H* ↔ C2H2** +*
CHCH3** ↔ CCH3* + H*
CCH3* + 2* ↔ CCH2** + H*
C2H5** + * ↔ CHCH3** + H*
CHCH3** + *↔ C2H3** + H*
C2H3** + *↔CCH2** + H*
C2H** + H* ↔CCH2** + *
C2H5** ↔ CH3* + CH2*
2CH2* + 2* ↔ σ-C2H4****
C2H3** ↔ CH* + CH2*
C2H2** ↔ 2CH*
C2H** ↔ CH* + C*
CHCH3** ↔ CH3*+ CH*
CH3* + C* ↔ CCH3* + *
CCH2** ↔ CH2* + C*
CHCH3** + 2* ↔ C2H4****
C2H3** ↔ CCH3* + *
C2H2**↔ CCH2**
C* + H* ↔ CH* + *
CH2* + * ↔ CH* + H*
CH3* + * ↔ CH2* + H*
Ethane Hydrogenolysis Reaction
H2 + 2* ↔ 2H*
CH4 + 2* ↔ CH3* + H*
C2H6 + 3* ↔ C2H5** + H*
C2H5** + 3* ↔ σ-C2H4**** + H*
σ-C2H4**** ↔ C2H3** + H* + *
CHCH3** ↔ CCH3* + H*
CCH3* + 2* ↔ CCH2** + H*
C2H5** + * ↔ CHCH3** + H*
CHCH3** + *↔ C2H3** + H*
C2H3** + *↔CCH2** + H*
C2H5** ↔ CH3* + CH2*
CHCH3** ↔ CH3*+ CH*
CH2* + * ↔ CH* + H*CH3* + * ↔ CH2* + H*
Model Reduction to Rate Expressions
Full mechanism
Reduced mechanism
Rate equation
Sensitivity analysisPrincipal component analysis
Elementary rate comparisonRate determining stepPartial equilibriumAbundant adsorbate assumptions
Full mechanism
Reduced mechanism
Rate equation
Full mechanism
Reduced mechanism
Rate equation
Full mechanism
Reduced mechanism
Rate equation
10-510-410-310-210-1100101
Reduced modelExperimental One step rate expressionC
H4 T
OF
[1/
s] 673K623K
573K
PC2H6
= 5 Torr
10-4
10-3
10-2
10-1
100
101
10 100 1000
Reduced modelExperimental One step rate expressionC
H4 T
OF
[1/s
]
Hydrogen Pressure [Torr]
673K623K573K
PC2H6
= 25Torr
Ethane Hydrogenolysis
Ethylene Hydrogenation
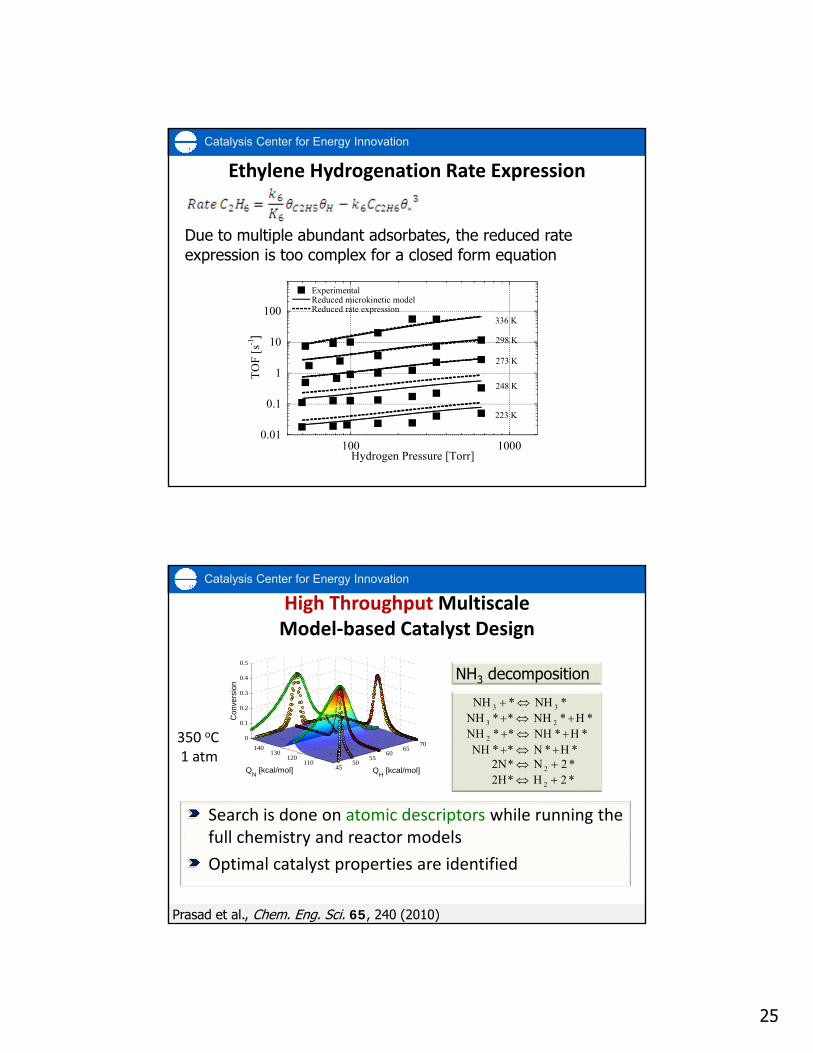
25
Catalysis Center for Energy Innovation
Ethylene Hydrogenation Rate Expression
Due to multiple abundant adsorbates, the reduced rate expression is too complex for a closed form equation
0.01
0.1
1
10
100
100 1000
ExperimentalReduced microkinetic model Reduced rate expression
TO
F [s
-1]
Hydrogen Pressure [Torr]
336 K
298 K
273 K
248 K
223 K
Catalysis Center for Energy Innovation
Search is done on atomic descriptors while running the full chemistry and reactor models
Optimal catalyst properties are identified
High Throughput MultiscaleModel‐based Catalyst Design
350 oC1 atm
Prasad et al., Chem. Eng. Sci. 65, 240 (2010)
4550
5560
6570
110120
130140
0
0.1
0.2
0.3
0.4
0.5
QH
[kcal/mol]QN
[kcal/mol]
Con
vers
ion
NH3 decomposition*NH*NH 33
*H*NH**NH 23 *H*NH**NH 2
*H*N**NH *2N*2N 2 *2H*2H 2
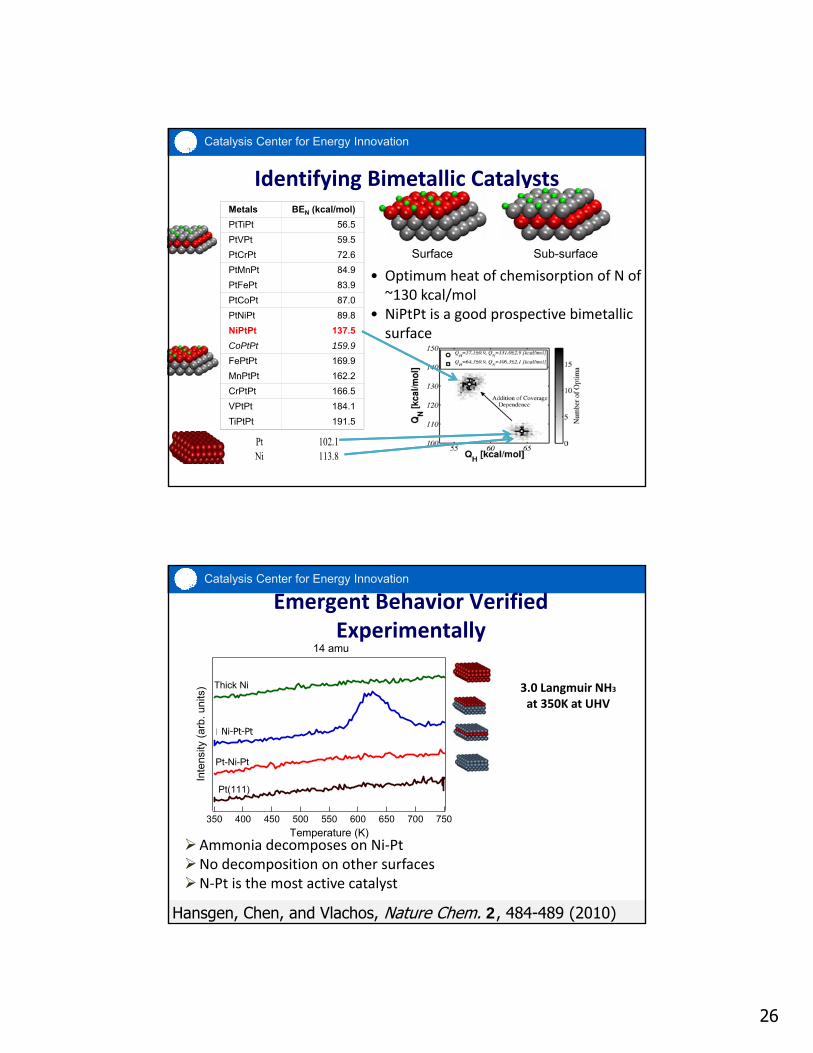
26
Catalysis Center for Energy Innovation
Identifying Bimetallic Catalysts
• Optimum heat of chemisorption of N of ~130 kcal/mol
• NiPtPt is a good prospective bimetallic surface
Surface Sub-surface
Metals BEN (kcal/mol)
PtTiPt 56.5
PtVPt 59.5
PtCrPt 72.6
PtMnPt 84.9
PtFePt 83.9
PtCoPt 87.0
PtNiPt 89.8
NiPtPt 137.5
CoPtPt 159.9
FePtPt 169.9
MnPtPt 162.2
CrPtPt 166.5
VPtPt 184.1
TiPtPt 191.5
Pt 102.1 Ni 113.8
Catalysis Center for Energy Innovation
Emergent Behavior Verified Experimentally
3.0 Langmuir NH3
at 350K at UHV
Ammonia decomposes on Ni‐PtNo decomposition on other surfacesN‐Pt is the most active catalyst
Inte
nsity
(ar
b. u
nits
)
750700650600550500450400350
Temperature (K)
Thick Ni
Ni-Pt
Pt-Ni-Pt
Pt(111)
14 amu
Hansgen, Chen, and Vlachos, Nature Chem. 2, 484-489 (2010)
Ni-Pt-Pt
27
Catalysis Center for Energy Innovation
Types of catalytic kinetic models and microkinetic modeling
Overview of parameter estimation methods and scales
Accuracy
Lateral interactions
Semi‐empirical methods
Thermodynamic consistency
MKM uses
Reactor design, analysis, catalyst discovery
Kinetic Monte Carlo
Outline
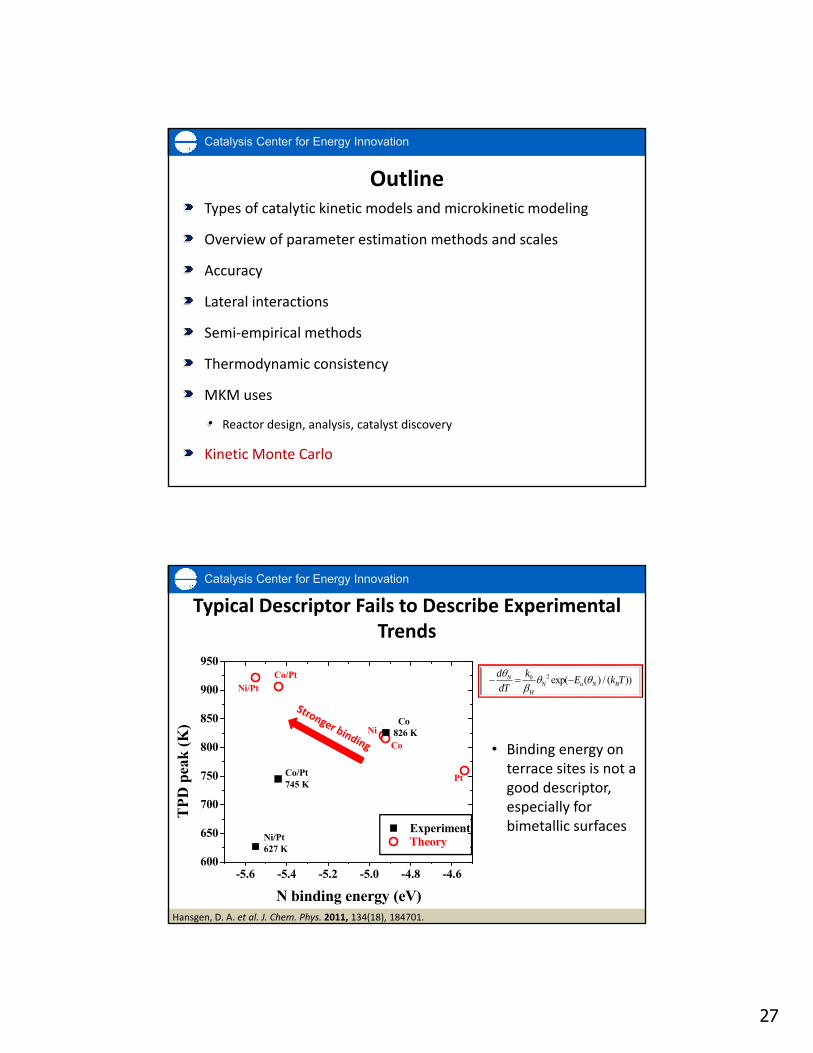
Catalysis Center for Energy Innovation
Co
Ni
Co/Pt
Ni/Pt
Theory
Pt
-5.6 -5.4 -5.2 -5.0 -4.8 -4.6600
650
700
750
800
850
900
950
Co826 K
Ni/Pt627 K
Co/Pt745 K
TP
D p
eak
(K
)
N binding energy (eV)
Experiment
Hansgen, D. A. et al. J. Chem. Phys. 2011, 134(18), 184701.
Typical Descriptor Fails to Describe Experimental Trends
20 exp( ( ) / ( ))NN a N B
H
d kE k T
dT
• Binding energy on terrace sites is not a good descriptor, especially for bimetallic surfaces

28
Catalysis Center for Energy Innovation
Tupy et al., ACS Catal. 2, 2290 (2012)
Can be Important
0 2 4 6 8 100
1
2
3x 10
4
Particle Diameter, d (nm)
TOF (s‐1)
Octahedral
Lean Kitchin et al., Surf. Sci. 544, 295, (2003)
Single Metal
0
0.5
1
1.5
2
0 1 2 3 4 5 6 7 8
TO
F, s
-1
Particle size, nm
NH3 on Ru
Bimetallics
CO+O2 Pt/Au Ni/Pt
Stamatakis and Vlachos, in prep.Karim et al., J. Am. Chem. Soc. 131, 12230 (2009)
Tupy et al., ACS Catal. 2, 2290 (2012)
Catalysis Center for Energy Innovation
Instead of simulating dynamics, KMC focuses on rare events
Simulates reactions much faster than Molecular Dynamics
Incorporates spatial information contrary to micro‐kinetic models
The Kinetic Monte Carlo Approach
CO(gas) + OH COOH
reactants
products
Potential Energy Surface
Metal surface
transitionstate
‡
B
E‡k TB
rxnrxn
k T Qk e
h Q
29
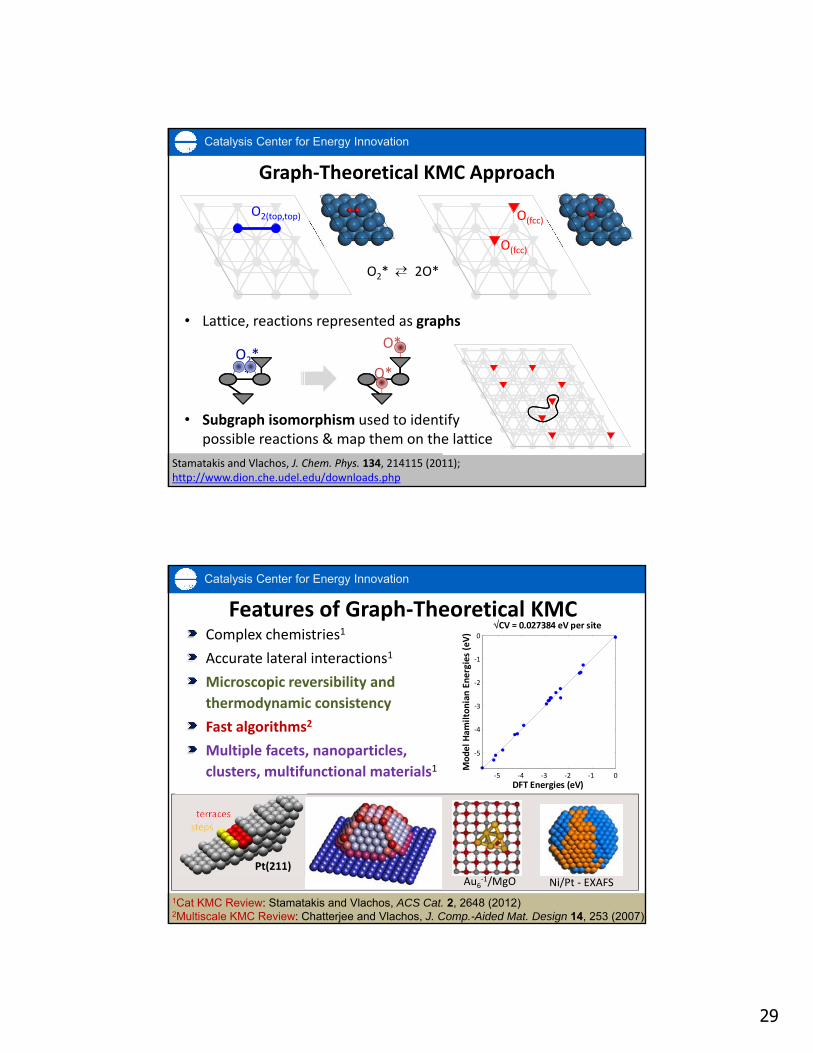
Catalysis Center for Energy Innovation
Graph‐Theoretical KMC Approach
• Lattice, reactions represented as graphs
Stamatakis and Vlachos, J. Chem. Phys. 134, 214115 (2011); http://www.dion.che.udel.edu/downloads.php
O2(top,top)
O(fcc)
O2**
O*
O*
O2* 2O*
O(fcc)
• Subgraph isomorphism used to identify possible reactions & map them on the lattice
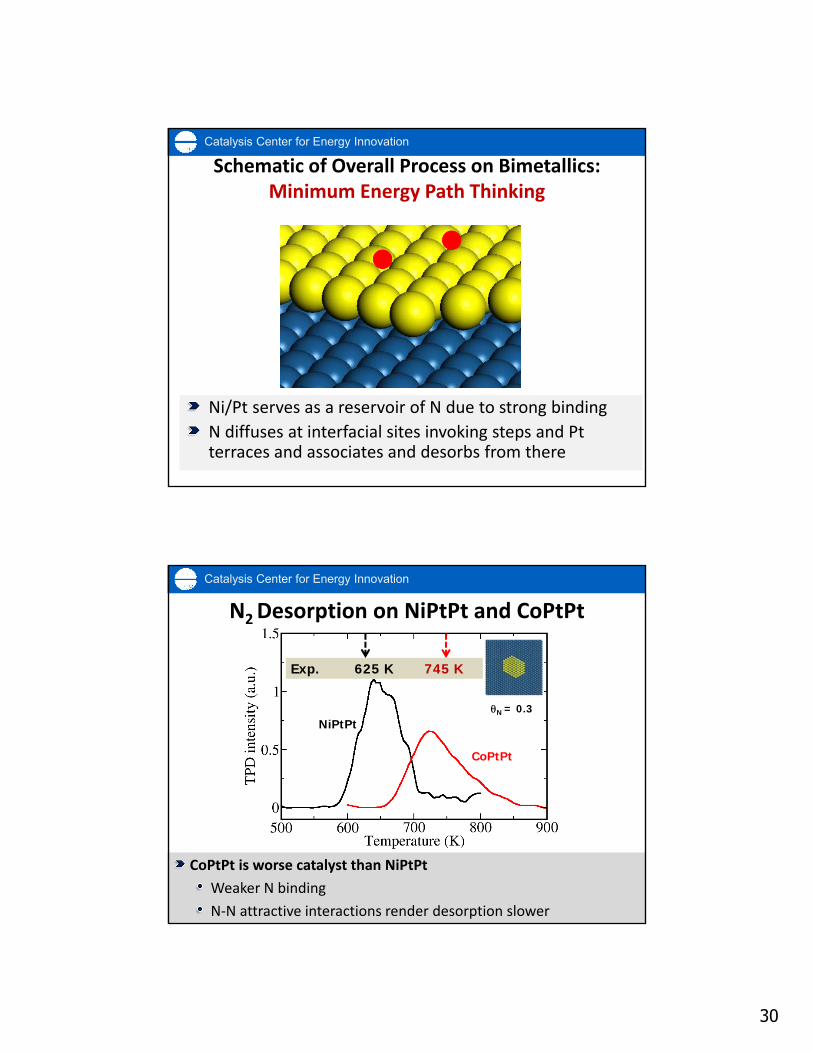
Catalysis Center for Energy Innovation
Features of Graph‐Theoretical KMCComplex chemistries1
Accurate lateral interactions1
Microscopic reversibility and
thermodynamic consistency
Fast algorithms2
Multiple facets, nanoparticles,
clusters, multifunctional materials1
Au6‐1/MgO
stepsterraces
Pt(211)
DFT Energies (eV)
Model H
amiltonian Energies (eV)
CV = 0.027384 eV per site
‐5 ‐4 ‐3 ‐2 ‐1 0
‐5
‐4
‐3
‐2
‐1
0
1Cat KMC Review: Stamatakis and Vlachos, ACS Cat. 2, 2648 (2012)2Multiscale KMC Review: Chatterjee and Vlachos, J. Comp.-Aided Mat. Design 14, 253 (2007)
Ni/Pt ‐ EXAFS
30
Catalysis Center for Energy Innovation
Ni/Pt serves as a reservoir of N due to strong binding
N diffuses at interfacial sites invoking steps and Pt terraces and associates and desorbs from there
Schematic of Overall Process on Bimetallics: Minimum Energy Path Thinking
Catalysis Center for Energy Innovation
NiPtPt
CoPtPt
N = 0.3
Exp. 625 K 745 K
N2 Desorption on NiPtPt and CoPtPt
CoPtPt is worse catalyst than NiPtPt
Weaker N binding
N‐N attractive interactions render desorption slower

31
Catalysis Center for Energy Innovation
First‐Principles KMC Resolves Structure Sensitivity
Co
Ni
Co/Pt
Ni/Pt
Theory
Pt
-5.6 -5.4 -5.2 -5.0 -4.8 -4.6600
650
700
750
800
850
900
950
Co826 K
Ni/Pt627 K
Co/Pt745 K
TP
D p
eak
(K
)
N binding energy (eV)
Experiment
• Flat surfaces: strong N binding results in high desorption temperature, well described by Redhead theory
KMC
Ni-Pt-Pt
• Steps are responsible for the low N2
desorption temperature on Ni/Ptand Co/Pt surfaces
Catalysis Center for Energy Innovation
End