propranolol, a phosphatidate phosphohydrolase … · 20482 inhibition of protein kinase c by...
TRANSCRIPT

THE JOURNAL OF BIOLOGICAL CHEMISTRY (c. 1992 hy The American Society for Biochemistry and Molecular Biology, Inc
Vol. 267, No. 28, Issue of October 5 , pp. 20481-204@8,1992 Printed in U. S. A .
Propranolol, a Phosphatidate Phosphohydrolase Inhibitor, Also Inhibits Protein Kinase C*
(Received for publication, May 22, 1992)
Silvano SozzaniSO, David E. Agwutl, Charles E. McCallV, Joseph T. O’FlahertyY, Jeffrey D. SchmittS, Jonathan D. Kent$, and Linda C. McPhailStlII From the $Department of Biochemistry and Wection on Infectious Diseases, Department of Medicine, Bowman Gray School of Medicine, Wake Forest University, Winston-Salem, North Carolina 27157
Propranolol, a &adrenergic receptor antagonist, also inhibits phosphatidate phosphohydrolase, the enzyme that converts phosphatidic acid into diacylglycerol. This latter effect has prompted recent use of propran- olol in studies examining the importance of diacylglyc- erol and phosphatidic acid in cellular signalling events. Here, we show that propranolol is also an inhibitor of protein kinase C. At concentrations 1 20 p ~ , propran- olol reduced [3H]phorbol dibutyrate binding (ICs0 = 200 p ~ ) and phorbol myristate acetate-stimulated su- peroxide anion release (IC5o = 130 p ~ ) in human neu- trophils. Scatchard analysis showed that propranolol lowers the number of phorbol diester binding sites without significantly affecting their affinity. In vitro kinetic analysis, performed in a mixed micellar assay with protein kinase C purified from human neutro- phils, suggested a competitive inhibition of propranolol with the cofactor phosphatidylserine. Complex kinetic patterns were observed with respect to diacylglycerol and ATP, approximating competitive and noncompet- itive inhibition, respectively. Taken together, these results suggest that the drug interacts at the level of the regulatory domain of the enzyme. Fifty % inhibi- tion occurred at -150 p M propranolol. Similar levels of inhibition were obtained using exogenous (histone) and endogenous (p47-phox, a NADPH oxidase compo- nent) substrates. Protein kinase C-cu and protein kinase (3-8, two protein kinase C isozymes present in human neutrophils, were inhibited by propranolol in a com- parable manner. In the range of concentrations tested (30-1000 p ~ ) , neither CAMP-dependent protein ki- nase nor neutrophil protein tyrosine kinases were af- fected. The racemic form of propranolol and the (+) and the (-) stereoisomers were equally active, and other @-adrenergic receptor antagonists (pindolol) and agonists (isoproterenol) were inactive. This suggests that the inhibitory action of propranolol on protein kinase C is related to the amphipathic nature of the drug rather than to its &adrenergic receptor blocking ability. Analogs of propranolol were synthesized and found to be more potent protein kinase C inhibitors, with ICs0 values in the 10-20 p~ range. We conclude
* This work was supported in part by National Institutes of Health Grants AI-22564, AI-09169, HL-27799, and HL-26257. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “aduer- tisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
I Supported by a fellowship from L. Berti, Champion Industries, Winston-Salem, NC. Present address: Istituto Ricerche Farmacolo- giche “Mario Negri,” 20157 Milan, Italy.
11 To whom correspondence should be addressed Dept. of Biochem- istry, Bowman Gray School of Medicine, Medical Center Blvd., Winston-Salem, NC 27157. Tel.: 919-716-2621; Fax: 919-716-7671.
that the ability of propranolol to inhibit both protein kinase C and P A phosphohydrolase complicates inter- pretation of results when this drug is used in signal transduction studies. In addition, propranolol may be a useful prototype for the synthesis of new protein kinase C inhibitors.
~~ ~~~ -
There is an increasing amount of data showing that in many cell types, including neutrophils (PMN),’ a phospholi- pase D is activated upon stimulation of specific cell surface receptors (1-4). Phospholipase D activation results in the production of phosphatidic acid (PA), and the PA formed can be dephosphorylated by phosphatidate phosphohydrolase (EC 3.1.3.4) to produce diacylglycerol. Either or both of these lipids may serve second messenger functions, and much in- terest has developed in the use of pharmacologic intervention to investigate their roles in signal transduction.
Propranolol, used clinically as a @-adrenergic receptor an- tagonist (5,6), has recently been found to inhibit the conver- sion of PA to diacylglycerol by PA phosphohydrolase (7, 8). Following this observation, a number of laboratories (9-28) have used propranolol to examine the contribution of diacyl- glycerol derived from PA to cellular signalling processes. In studies by our group and others using this drug in human PMN (19,20, 24,28), we noted that propranolol had opposite effects on the oxidative burst triggered by the chemotactic peptide formyl-Met-Leu-Phe, depending on the presence or absence of cytochalasin B. This prompted us to consider that propranolol may have effects on cellular signalling mecha- nisms other than inhibition of PA phosphohydrolase.
The inhibitory effect of propranolol on PA phosphohydro- lase is common to other cationic amphiphilic drugs, such as chlorpromazine, desmethylimipramine, trifluoperazine, and sphingosine (7, 8, 29-31). Many of these drugs also inhibit protein kinase C (PKC) (32-34). Propranolol shares an am- phipathic nature as well as a charged amino group with other compounds that are potent inhibitors of PKC, such as sphin- gosine (34) and aminoacridines (35). Therefore, we postulated that propranolol might also inhibit PKC activity.
In this paper, we report that propranolol inhibited [3H] phorbol dibutyrate binding and PKC-mediated responses in
The abbreviations and trivial name used are: PMN, human pol- ymorphonuclear leukocytes; 2-ME, 2-mercaptoethanol; C-I, 1-(5-iso- quinolinesulfony1)piperazine; HBSS, Hank’s balanced salt solution; p47-phox, 47-kDa protein component of phagocyte NADPH oxidase; PA, phosphatidic acid PKA, CAMP-dependent protein kinase; PKC, protein kinase C; PMA, phorbol 12-myristate 13-acetate; PMSF, phenylmethylsulfonyl fluoride; PS, phosphatidylserine; TKI, epider- mal growth factor receptor inhibitor 11; Tyr PK, protein tyrosine kinase; diolein, 1,2-dioleyl-racemic-glycerol.
20481

20482 Inhibition of Protein Kinase C by Propranolol
human PMN. Furthermore, propranolol was able to directly inhibit PKC activation by competing with the cofactor phos- phatidylserine and the activator diacylglycerol in a mixed micellar assay. A preliminary series of propranolol analogs was also synthesized, and one analog was at least 10-fold more effective than propranolol for inhibition of PKC. Thus, the ability of propranolol to inhibit PKC should be considered when this drug is used in signal transduction studies. In addition, propranolol may be a useful prototype for the syn- thesis of new PKC inhibitors.
EXPERIMENTAL PROCEDURES
Materials
[y3'P]ATP (2-10 Ci/mmol) and [3H]phorbol dibutyrate ([3H] PDBu) (19 Ci/mmol) were obtained from Du Pont-New England Nuclear Products (Boston, MA); Hank's balanced salt solution (HBSS) was from GIBCO; and Type HA 0.45-pm filters were from Millipore. The materials used for SDS-polyacrylamide gels were from Bio-Rad. The following were obtained from Sigma: 1,2-dioleyl-ra- cemic-glycerol (diolein), histone Type 111-S, histone Type 11-A, poly(Glu,Tyr) 4:1, bovine brain-purified protein kinase A (P-3891), 2-mercaptoethanol (2-ME), ATP, phorbol 12-myristate 13-acetate (PMA), PDBu, phenylmethylsulfonyl fluoride (PMSF), leupeptin, cytochrome c (Type 111), Triton X-100, diisopropylfluorophosphate, Me2S0, and EGTA. The kinase inhibitor l-(5-isoquinolinesul- fony1)piperazine (C-I) was synthesized by Dr. M. Thomas (Bowman Gray School of Medicine) as described (36). The epidermal growth factor receptor tyrosine kinase inhibitor number 11 (TKI) (37) was a kind gift of Dr. C.-K. Huang (University of Connecticut). Brain- purified phosphatidylserine (PS) was from Avanti Polar Lipids Inc. (Pelham, AL).
Drugs
(+)Propranolol was from Ayerst Laboratories Inc. (New York, NY); (+)propranolol and (-)propranolol were from Aldricb; (-)ti- molol, (+)pindolol, and (-)isoproterenol were from Sigma. All the drugs were freshly dissolved in water with the exception of pindolol, which was dissolved in Me2S0.
Synthesis of Propranolol Analogs
All chemicals for organic synthesis were purchased from Aldrich and Lancaster Synthesis. As depicted in Fig. 1, hexadecyl allyl ether (B) was prepared by the alkylation of hexadecanol (A) with allyl bromide using potassium t-butoxide and t-butyl ammonium iodide (go%+ yield). The epoxidation of ( B ) was accomplished using m- chloroperoxybenzoic acid. Epoxide ring opening of (C), leading to JS- X-112, was effected using isopropylamine. JS-XI40 was synthesized by the epoxide ring opening of glycidyl-1-naphthyl ether (D) using cyclohexylamine. Analogs were purified by column chromatography. All compounds have proton-NMR and mass spectra (ammonia chem- ical ionization) consistent with their defined structures.
PMN Isolation
PMN were isolated from heparinized venous blood by dextran sedimentation and Isolymph centrifugation as previously described
D JS-XI-60
FIG. 1. Structures and synthesis of propranolol analogs. See "Experimental Procedures" for details. PTC, phase transfer catalyst.
(38). After hypotonic lysis of the contaminating erythrocytes, PMN (>95% purity) were suspended in modified HBSS (HBSS containing 4.2 mM sodium bicarbonate and 10 mM Hepes, pH 7.4) at the appropriate concentration and used in subsequent assays.
rH]PZ)Bu Binding
PMN (5 X 106/ml) were layered over 400 p1 of silicone oil (Versilube F-50), preincubated with propranolol at 37 "C for 5 min, and treated with 125 pmol of [3H]PDBu (-5,000 dpm) for 4 additional min as previously described (39). Tubes were centrifuged (12,000 X g; 1 min) to free cells from supernatant fluids. Both fractions were counted for radioactivity. Data for Scatchard analyses were obtained in the pres- ence of either buffer or 200 PM propranolol, using 125 pmol of [3H] PDBu and increasing amounts of unlabeled PDBu. In all cases, nonspecific binding was evaluated in the presence of 10 p~ cold PDBu. Scatchard analyses used LIGAND SCAPRE/SCAFIT pro- grams.
PMN Superoxide Release
The release of superoxide anion (0;) from PMN was measured as the reduction of extracellular cytochrome c essentially as previously described (36). Briefly, 1 X lo6 PMN in modified HBSS were prein- cubated for 5 min at 37 "C, in the presence of M cytochalasin B and 80 FM cytochrome c, with various concentrations of propranolol and in a final volume of 1.5 ml. PMA (100 ng/ml) or Me2S0 was added and the reaction was stopped after 5 min by immersion in a melting ice bath. Reaction mixtures were cleared by centrifugation (200 X g, 10 min), and the amount of reduced cytochrome c in supernatants was determined by measuring the absorbance at 550 nm in a Cary 2390 dual-beam spectrophotometer (Varian Instru- ments, Sunnyvale, CA) against a blank sample not exposed to PMN. The amount of 0; released was calculated using an extinction coef- ficient for reduced cytochrome c of 21 nM" cm" (401, and results were expressed as nanomoles of 0,/5 min/l X lo6 PMN.
Partial Purification of Protein Kinase C from PMN
PMN (5 X lO'/ml) in modified HBSS were incubated with 1 mM diisopropylfluorophosphate for 5 min at 4 "C, washed twice with modified HBSS, and disrupted by sonication in extraction buffer (50 mM Tris-HC1, pH 7.5, 50 mM 2-ME, 1 mM PMSF, 2 mM EGTA, 10 pg/ml leupeptin). The supernatant obtained after a 150,000 x g centrifugation was loaded onto a DEAE-Sephacel column equilibrated with Buffer A (50 mM Tris-HC1, pH 7.5,50 mM 2-ME, 2 mM EGTA, and 0.1% Triton X-100). The enzyme was eluted with Buffer A containing 150 mM NaCl and had phosphotransferase activity of 4 nmol/min/mg protein at optimal activation conditions (12 mol% PS, 2.5 mol% diolein, and 200 p~ Ca*+). PKC-a and PKC-6 isozymes were isolated by hydroxylapatite chromatography essentially as de- scribed (41). Briefly, the enzyme obtained after DEAE-Sephacel chromatography was loaded onto a hydroxylapatite column equili- brated with Buffer B (20 mM KxPO,, 0.5 mM EDTA, 0.5 mM EGTA, 0.067% (v/v) 2-ME, 10% glycerin, pH 7.5) . The enzyme was eluted with a 240-ml linear (20-280 mM) K,PO, gradient and resolved in two peaks of activity; peak I (PKC-P) and peak I1 (PKC-a). The identity of the two peaks was assessed by the use of isozyme-specific anti-peptide antisera' (42).
Protein Kinase Assays
Mixed Micellar Assay for PKC-PKC activity was assayed as the transfer of 3zP from [y-32P]ATP to histone H1 (Type 111-S) using Triton X-lOO/lipid mixed micelles (43). The required amounts of PS and diolein were dried under a stream of nitrogen in glass tubes and solubilized in 3% Triton X-100 by vortexing and incubation at 30 "C for 10 min. The reaction mixture, in a volume of 100 111, consisted of 35 mM Tris-HC1, pH 7.5, 0.4 mM EGTA, 10 mM MgC12, 10 mM 2- ME, 0.6 mM CaC12 (0.2 mM final free [Ca"] (44)), 12.5 p1 of mixed micelles, 640 pg/ml histone Type 111-S, and 50 pM ATP (containing 1 X lo6 cpm 32P). Mixtures were prewarmed for 5 min at 30 "C, and the reaction was started by adding 25 pl of the enzyme. The incubation was carried out for 10 min, and terminated with the addition of 1 ml of 25% trichloroacetic acid and 0.5 mg of bovine brain albumin. Precipitates were filtered on Millipore filters. The filters were counted in a Beckman scintillation counter.
' S. Sozzani, J. D. Kent, M. Ellenburg, M. Wooten, J. Wilson, D. Qualliotine-Mann, and L. C. McPhail, manuscript in preparation.

Inhibition of Protein Kinase C by Propranolol 20483
Conventional Assay for PKC-PKC activity was measured as pre- viously described (36, 45). Assay conditions were similar to those described in the mixed micellar assay except that sonicated lipids (2.5 pg/ml PS and 2 pg/ml diolein) were substituted for the mixed micelles, and the final assay volume was 250 pl.
Cyclic AMP-dependent Protein Kinase (PKA) Activity-PKA ac- tivity was measured as previously described (36). The reaction mix- ture (35 mM Tris-HC1, pH 7.5, 10 mM MgC12, 10 p M CAMP, 50 p M ATP (containing 2 X lo6 cpm 32P), and 160 wg/ml histone Type 11-A, in a final volume of 250 pl) was preincubated for 5 min, and the reaction was started with 50 pl of crude cytosol or 4 pg of the purified enzyme. The incubation was terminated after 5 min with the addition of 1 ml of 25% trichloroacetic acid. The phosphotransferase activity obtained in the presence of cAMP minus the activity in the absence of cAMP was considered as PKA activity.
Protein Tyrosine Kinase (Tyr PK) Activity-Tyr PK activity was measured as previously described in PMN (46). The reaction mixture (20 mM Hepes, 10 mM MgC12, 10 mM MnC12, 100 p M Na3V04, 7 mg/ mlp-nitrophenylphosphate, 0.5 mg/ml poly(Glu,Tyr) 4:1, and 10 p M ATP (containing 2 X lo6 cpm 32P), in a final volume of 250 pl) was prewarmed for 5 min at 30 "C, and the reaction was started with 20 pg of cytosol or 10 pg of membrane fraction (prepared as described below). The incubation was terminated after 5 min with the addition of 2 ml of 20% trichloroacetic acid containing 10 mM sodium pyro- phosphate. Tyr PK activity was considered as the difference between the phosphotransferase activity obtained in the presence and in the absence of substrate. All protein kinase assays were performed under conditions of linearity for the time of incubation and enzyme concen- tration.
Subcellular Fractionation of PMN Cytosolic and membrane fractions were separated on a discontin-
ous sucrose density gradient as previously described (47). Briefly, purified PMN were resuspended at 1 X 108/ml in sonication buffer (11% (w/v) sucrose, 130 mM NaC1, 1 mM EGTA, 0.5 mM PMSF, pH 7.0) and disrupted by sonication. Sonicates were centrifuged at 800 x g for 10 min and the supernatants (in 11% sucrose sonication buffer) were layered over a discontinuous sucrose gradient to achieve a final ratio of 2:l:l (v/v) of 11, 15, 40% (w/v), respectively. After centrifugation at 150,000 X g for 30 min, the top layer (cytosol) was collected and further centrifuged at 150,000 X g for 1 h to pellet any contaminating particulate material. The membrane fraction was col- lected at the 15%/40% interface.
In Vitro PKC-dependent Phosphorylation of p47-phx Cytosolic protein from resting PMN and membrane protein from
PMA-stimulated PMN (100 ng/ml PMA for 30 s at 37 "C, as described in Ref. 38) were phosphorylated in vitro (48) using pooled PKC-a and PKC-/3 isolated by hydroxylapatite chromatography. The reac- tion mixture (30 mM Tris-HC1, pH 7.5, 2 mM EGTA, 7.5 mM MgC12, 21 pg/ml PS, 80 ng/ml PMA, 10 pl pooled PKC, 100 pg/ml subcellular fraction, in a final volume of 114 pl) was preincubated for 5 min at 30 "C, and the reaction was started with the addition of 6 pl of [y- 3ZP]ATP. After 2 min, the reaction was terminated with the addition of 30 pl of 5X Laemmli sample buffer (49). The samples were boiled for 5 min, and the protein (4 pg/lane) separated on 8-15% gradient polyacrylamide slab gels (1.5 X 140 X 320 mm) as described (47, 49). After electrophoresis, the gels were silver-stained (50), dried, and exposed to film (Kodak X-Omat) at -70 "C. The autoradiograms were scanned with an LKB Ultroscan XL laser densitometer equipped with GelScan XL software (LKB-Produkter AB, Bromma, Sweden).
Protein Determination
Protein concentrations were determined by either the method of Bradford (51) or the bicinchoninic method (52) using the BCA Protein Assay Kit (Pierce).
RESULTS
Inhibition by Propranolol of 0; Release from Human PMN-In a first series of experiments it was found that propranolol was able to inhibit the release of 0; from PMN stimulated with PMA, a direct PKC activator. Cells were preincubated with the racemic form of the drug for 5 min and then stimulated with an optimal concentration of PMA (160 nM) for 5 additional min. (?)Propranolol caused a concentra-
tion-dependent inhibition beginning at 220 p ~ , reaching half- maximal at -125 FM, and achieving complete inhibition at 300 p~ (Fig. 2 A ) . Similar results were obtained with the two stereoisomers of propranolol or in the absence of cytochalasin B (data not shown). Under these assay conditions, propranolol did not affect cell viability assessed by trypan blue dye exclu- sion (290%).
Inhibition by Propranolol of PHIPDBu Binding to Human PMN-Since PMA binds to and activates PKC, the observed inhibition of 0; release by propranolol suggests the possibility of a direct effect of the drug on this kinase. To further explore this possibility, we evaluated the ability of propranolol to inhibit binding of [3H]PDBu to PMN. As shown in Fig. 2B, concentrations of (f)propranololz 20 p~ reduced binding of [3H]PDBu to PMN. Half-maximal inhibition occurred at approximately 150 p ~ , a concentration similar to the half- maximal inhibition value obtained in the 02 release assay (Fig. 2 A ) . Scatchard analysis (Fig. 2B, insert) showed that propranolol lowered the number of [3H]PDBu binding sites (420,000 and 264,000 receptors/cell in buffer and 200 PM (+)propranolol-treated PMN, respectively), without signifi- cantly affecting their affinity (Kd = 19.2 and 24.7 nM in control and 200 pM (+)propranolol-treated, respectively).
I n Vitro Inhibition of PKC Activity by Propranolol-The ability of propranolol to directly affect PKC activity was investigated in a Triton X-100 mixed-micellar assay contain- ing 8 mol% PS, 2.5 mol% diolein and in the presence of 200 p~ Ca2+. As shown in Fig. 3, the racemic form, as well as the
I A I
8 25 \ I 100 I ow
[propranolol] (pM) 0.15 ,
0 10 1W 1 ow [propranolol] (pM)
FIG. 2. Effect of propranolol on superoxide release and [3H] PDBu binding in PMN. A, inhibition of superoxide release by propranolol. 1 x lo6 PMN were preincubated for 5 min at 37 "C with buffer or the indicated concentration of propranolol and then exposed for 5 additional min to 100 ng/ml PMA. Results are the mean * S.D. for three experiments. The 0; release induced by PMA in the absence of propranolol (29 rt 4 nmol/min/106 PMN) was used as 100% in each experiment. B, inhibition of [3H]PDBu binding to PMN by propranolol. 5 X lo6 PMN were preincubated at 37 "C for 5 min with the indicated concentrations of propranolol and exposed for 4 addi- tional min to 125 pmol of [3H]PDBu. Nonspecific binding was deter- mined in the presence of 10 p~ unlabeled PDBu. Results are shown as the mean * S.E. of eight experiments. Inset, Scatchard analysis of [3H]PDBu binding in the presence of buffer (0) or 200 p~ propranolol (0). Receptor number (Re) and dissociation constant (K,,) were deter- mined with the LIGAND program and were: R, = 420,000 * 67,200/ cell and 264,000 2 74,00O/cell for buffer and propranolol, respectively; Kd = 19.2 and 24.7 nM for buffer and propranolol, respectively. The Scatchard plots best fit a single receptor model. Each point is the mean of 10 experiments.
20484 Inhibition of Protein Kinase C by Propranolol
120 , I
100 1 0 0 0
(PM)
FIG. 3. Effect of &adrenergic compounds on human PMN PKC activity. PKC activity was measured in a Triton X-100 mixed- micellar assay in the presence of 8 mol% PS, 2.5 mol% diolein, and 200 PM Ca2+, as described under “Experimental Procedures.” The activity in the absence of the drugs (830 f 75 pmol/min/mg; n = 4) was defined as 100%. Drugs were freshly dissolved in water. Results shown with propranolol are the mean f S.E. of three experiments. Data with timolol and isoproterenol are the means of two experi- ments. 0, (f)propranolol; 0, (-)propranolol; A, (+)propranolol; 0, (-)timolol; I, isoproterenol.
two optically active stereoisomers, of propranolol caused a marked inhibition of PKC activity. The inhibition was dose- dependent, and the three forms showed a comparable inhibi- tory activity with ICbo values of 135 f 7,155 + 13, and 188 k 32 pM (n = 3) for (+)propranolol, (-)propranolol, and (+)pro- pranolol, respectively. Under the assay conditions, the bulk concentration of PS was 390 p~ and that of diolein was 134 p ~ . Thus, on a molar basis, 50% inhibition of PKC activity was obtained at 30-50% the concentration of PS and 100- 150% the concentration of diolein in the assay.
Two other compounds, an antagonist (timolol) and an agonist (isoproterenol) of the /?-adrenergic receptor (Z), were also studied. Neither of these compounds was an effective inhibitor of PKC activity, and only high concentrations of timolol had a significant effect (40% inhibition at 1 mM; Fig. 3). In addition, pindolol, another &adrenergic receptor antag- onist, failed to inhibit PKC activity using a conventional PKC assay (19) (data not shown). In the same type of assay, the ICbo for (+)propranolol was 250 p M (n = 2).
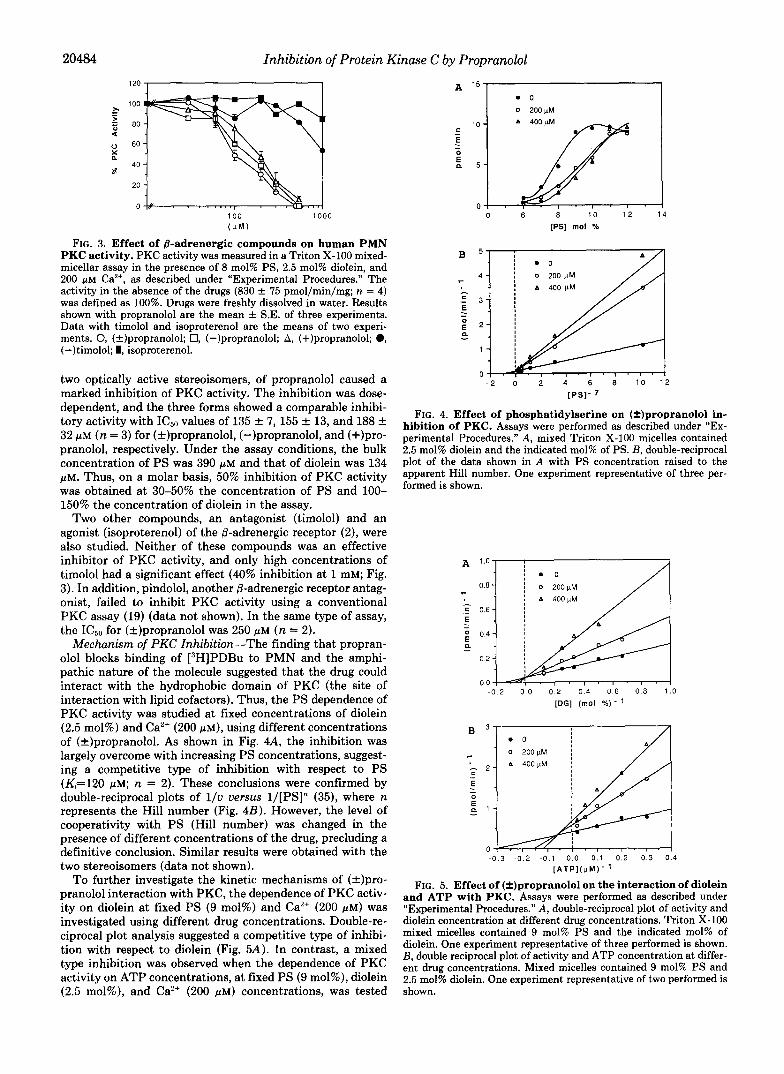
Mechanism of PKC Inhibition-The finding that propran- olol blocks binding of [3H]PDBu to PMN and the amphi- pathic nature of the molecule suggested that the drug could interact with the hydrophobic domain of PKC (the site of interaction with lipid cofactors). Thus, the PS dependence of PKC activity was studied at fixed concentrations of diolein (2.5 mol%) and Ca2+ (200 p ~ ) , using different concentrations of (?)propranolol. As shown in Fig. 4A, the inhibition was largely overcome with increasing PS concentrations, suggest- ing a competitive type of inhibition with respect to PS (K,=120 p ~ ; n = 2). These conclusions were confirmed by double-reciprocal plots of l / v versus l/[PS]” (35), where n represents the Hill number (Fig. 4B). However, the level of cooperativity with PS (Hill number) was changed in the presence of different concentrations of the drug, precluding a definitive conclusion. Similar results were obtained with the two stereoisomers (data not shown).
To further investigate the kinetic mechanisms of (+)pro- pranolol interaction with PKC, the dependence of PKC activ- ity on diolein at fixed PS (9 mol%) and Ca2+ (200 pM) was investigated using different drug concentrations. Double-re- ciprocal plot analysis suggested a competitive type of inhibi- tion with respect to diolein (Fig. 5A). In contrast, a mixed type inhibition was observed when the dependence of PKC activity on ATP concentrations, at fixed PS (9 mol%), diolein (2.5 mol%), and Ca2+ (200 p ~ ) concentrations, was tested
A 1 5 ~
I m o
0 200pM 1 - . 0 6 8 i o 12 1 4
[PSI mol 36
“2 0 2 4 6 8 1 0 12 [PSI - 7
FIG. 4. Effect of phosphatidylserine on (*)propranolol in- hibition of PKC. Assays were performed as described under “Ex- perimental Procedures.” A , mixed Triton X-100 micelles contained 2.5 mol% diolein and the indicated mol% of PS. B, double-reciprocal plot of the data shown in A with PS concentration raised to the apparent Hill number. One experiment representative of three per- formed is shown.
-0 2 0 0 0.2 0.4 0 6 0 .8 1.0
[DG] (mol % ) -
[ A T P I ( P M ) -
FIG. 5. Effect of (*)propranolol on the interaction of diolein and ATP with PKC. Assays were performed as described under “Experimental Procedures.” A , double-reciprocal plot of activity and dioletn concentration at different drug concentrations. Triton X-100 mixed micelles contained 9 mol% PS and the indicated mol% of diolein. One experiment representative of three performed is shown. B, double reciprocal plot of activity and ATP concentration at differ- ent drug concentrations. Mixed micelles contained 9 mol% PS and 2.5 mol% diolein. One experiment representative of two performed is shown.

Inhibition of Protein Kinase C by Propranolol 20485
(Fig. 5B). However, a slight curvature of the points for each line could be observed in both panels, suggesting complex kinetic patterns. Finally, (+)propranolol was found to be a noncompetitive inhibitor of PKC activity with respect to the protein substrate (e.g. histone) (data not shown).
Fractionation of human PMN extracts by successive DEAE-Sephacel and hydroxylapatite chromatography yields two major peaks of PKC activity, corresponding to isoforms a and @ of PKC2 (53). These isoforms were isolated and tested in the presence of (&)propranolol. Both PKC forms were inhibited by the drug to a similar extent, with Ki = 66 and 128 p~ for PKC-CY and PKC-p, respectively, when PS concen- trations were varied (data not shown, n = 2).
Effect of Propranolol on the Phosphorylation of p47-phox Phosphoprotein-Histone H1 was used as an exogenous sub- strate protein for all the experiments described above. Thus, it was of interest to confirm the inhibitory properties of (&)propranolol using an endogenous substrate. A component of the NADPH oxidase enzyme in PMN is a phosphoprotein of 47 kDa (p47-phox) (54). The p47-phox protein is localized in the cytosolic fraction of resting cells and upon cellular stimulation translocates to the membrane (55, 56). This pro- tein is phosphorylated during activation of the oxidase in intact cells (54) and is a substrate for PKC in vitro (48). The effect of propranolol on the in vitro phosphorylation of p47- phox was examined in cytosolic fractions from resting PMN and in membrane fractions from PMA-stimulated PMN. The identity of p47-phox was confirmed in separate experiments by comparison with samples missing this protein from pa- tients with chronic granulomatous disease (Ref. 54; data not shown).
As shown in Fig. 6A, several cytosolic proteins became phosphorylated during a 2-min incubation with PKC in the presence of PKC activators (e.g. PS and PMA, lane 2), but not in their absence (lane I). In the presence of propranolol (500 p ~ ) , an overall decrease in the level of phosphorylation was observed in proteins a t the different molecular weights (Fig. 6A, lane 3). The phosphorylation of p47-phox was also strongly reduced (83% inhibition, Fig. 6B).
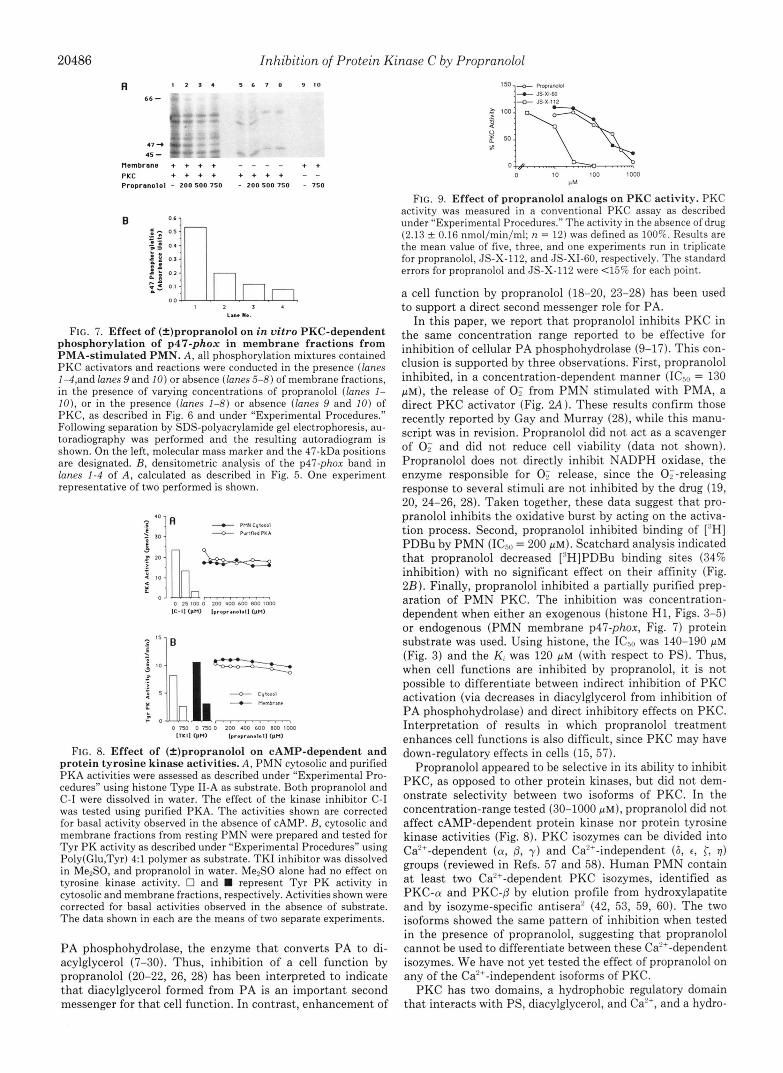
In the membrane fraction, propranolol inhibited, in a con- centration-dependent manner (0-750 p ~ ) , the phosphoryla- tion of p47-phox (Fig. 7A, lanes 1-4, and Fig. 7B). In this subcellular fraction, p47-phox was one of the proteins most sensitive to the effect of the drug (Fig. 7A). As shown in lanes 5-8, no 47-kDa proteins were present when the membrane fraction was omitted from the incubation mixture. The phos- phorylation observed in these experiments appeared to be strictly dependent on the presence of the exogenous PKC preparation (Fig. 7A, lanes 9 and 10). The same levels of inhibition were observed with both cytosolic and membrane fractions in a second set of experiments (data not shown).
Effect of Propranolol on Other Kinases-The specificity of the inhibitory effect of (+)propranolol for PKC was investi- gated. As shown in Fig. 8A, propranolol concentrations up to 1 mM failed to inhibit PKA when both crude PMN cytosol and purified PKA were used. On the other hand, in the same assay, the serine-threonine kinase inhibitor C-I inhibited in a dose-dependent manner the activity of the purified enzyme preparation. Similarly, (&)propranolol did not inhibit Tyr PK activity present in PMN cytosolic and membrane fractions (Fig. 8B) . In the same assay, the Tyr PK inhibitor TKI was able to strongly suppress the kinase activity in both PMN fractions.
Effect of Propranolol Analogs on PKC Activity-A prelimi- nary series of chemical modifications were introduced at the amino and the naphthyl moieties of the propranolol structure
A I 2 3 ~- **
116-
92 -
66 -
45 - 47 -+
31 -
Activators - + + Propranolol - - 500
B
La.. no,
FIG. 6. Effect of (*)propranolol on in vitro PKC-dependent phosphorylation of p47-phox in cytosolic fractions from rest- ing PMN. A , cytosolic proteins were phosphorylated in vitro in the presence or absence of propranolol and/or PKC activators (PS and PMA) as described under “Experimental Procedures.” Proteins were subsequently solubilized and separated on an 8-15% SDS-polyacryl- amide gel. The dried gel was exposed to Kodak X-Omat film, and the resulting autoradiogram is shown. On the left are shown the positions of molecular mass markers and the 47-kDa position. B, absorbance levels of the p47-phox band in lanes I , 2, and 3 of A, determined by densitometric analysis. The values represent the area under the curves expressed as units per mm?. The experiment shown is representative of two performed.
(see Fig. 1 for chemical structures and synthetic scheme). In Fig. 9, the effects of propranolol and the two analogs, JS-XI- 60 and JS-X-112, on PKC activity are shown. The introduc- tion of a bulky cyclohexane ring at the amino group of propranolol (JS-XI-60) did not change PKC inhibition rela- tive to the parent molecule (IC5” = 300 p ~ ) . In contrast, the substitution of the naphthyl group with a long carbon chain (JS-X-112) greatly improved the ability to inhibit PKC by approximately 10-20 fold (ICso = 17 & 1 p ~ , n = 3). Studies are underway to synthesize additional analogs in order to further investigate the structure/function properties of pro- pranolol for PKC inhibition.
DISCUSSION
The importance of various lipid second messengers in the modulation of cell function has received much attention over the past few years. The recent demonstration that many extracellular agonists induce the activation of a phospholipase D (1-3) has raised the issue of whether phosphatidic acid has a direct second messenger function or whether the role of PA in cell function is to serve as a precursor for diacylglycerol. To this end, a number of researchers have utilized the p- adrenergic receptor antagonist propranolol as an inhibitor of
20486 Inhibition of Protein Kinase C by Propranolol
A 1 2 x 4 5 6 7 8 9 1 0
66 -
45 - 47 -t
Membrane + + + + - - - - + + PKC Propranolol - 200 500 750 - 200 500 750 - 750
+ + + + + + + + ”
LUl. PI. FIG. 7. Effect of (2)propranolol on in vitro PKC-dependent
phosphorylation of p47-phox in membrane fractions from PMA-stimulated PMN. A, all phosphorylation mixtures contained PKC activators and reactions were conducted in the presence (lanes 1-4,and lanes 9 and 10) or absence (lanes 5-8) of membrane fractions, in the presence of varying concentrations of propranolol (lanes 1- IO), or in the presence (lanes 1-8) or absence (lanes 9 and 10) of PKC, as described in Fig. 6 and under “Experimental Procedures.” Following separation by SDS-polyacrylamide gel electrophoresis, au- toradiography was performed and the resulting autoradiogram is shown. On the left, molecular mass marker and the 47-kDa positions are designated. B, densitometric analysis of the p47-phox band in lanes 1-4 of A, calculated as described in Fig. 5. One experiment representative of two performed is shown.
FIG. 8. Effect of (=)propranolol on CAMP-dependent and protein tyrosine kinase activities. A , PMN cytosolic and purified PKA activities were assessed as described under “Experimental Pro- cedures” using histone Type 11-A as substrate. Both propranolol and C-I were dissolved in water. The effect of the kinase inhibitor C-I was tested using purified PKA. The activities shown are corrected for basal activity observed in the absence of CAMP. B, cytosolic and membrane fractions from resting PMN were prepared and tested for Tyr PK activity as described under “Experimental Procedures” using Poly(Glu,Tyr) 4:l polymer as substrate. TKI inhibitor was dissolved in Me,SO, and propranolol in water. Me2S0 alone had no effect on tyrosine kinase activity. 0 and represent Tyr PK activity in cytosolic and membrane fractions, respectively. Activities shown were corrected for basal activities observed in the absence of substrate. The data shown in each are the means of two separate experiments.
PA phosphohydrolase, the enzyme that converts PA to di- acylglycerol (7-30). Thus, inhibition of a cell function by propranolol (20-22, 26, 28) has been interpreted to indicate that diacylglycerol formed from PA is an important second messenger for that cell function. In contrast, enhancement of
UM
FIG. 9. Effect of propranolol analogs on PKC activity. PKC activity was measured in a conventional PKC assay as described under “Experimental Procedures.” The activity in the absence of drug (2.13 0.16 nmol/min/ml; n = 12) was defined as 100%. Results are the mean value of five, three, and one experiments run in triplicate for propranolol, JS-X-112, and JS-XIBO, respectively. The standard errors for propranolol and JS-X-112 were 4 5 % for each point.
a cell function by propranolol (18-20, 23-28) has been used to support a direct second messenger role for PA.
In this paper, we report that propranolol inhibits PKC in the same concentration range reported to be effective for inhibition of cellular PA phosphohydrolase (9-17). Th’ 1s con- clusion is supported by three observations. First, propranolol inhibited, in a concentration-dependent manner (ICso = 130 p ~ ) , the release of 0; from PMN stimulated with PMA, a direct PKC activator (Fig. 2.4) . These results confirm those recently reported by Gay and Murray (28), while this manu- script was in revision. Propranolol did not act as a scavenger of 0; and did not reduce cell viability (data not shown). Propranolol does not directly inhibit NADPH oxidase, the enzyme responsible for 0; release, since the 0;-releasing response to several stimuli are not inhibited by the drug (19, 20, 24-26, 28). Taken together, these data suggest that pro- pranolol inhibits the oxidative burst by acting on the activa- tion process. Second, propranolol inhibited binding of [‘HI PDBu by PMN (ICso = 200 p ~ ) . Scatchard analysis indicated that propranolol decreased [“HIPDBu binding sites (34% inhibition) with no significant effect on their affinity (Fig. 2B). Finally, propranolol inhibited a partially purified prep- aration of PMN PKC. The inhibition was concentration- dependent when either an exogenous (histone H1, Figs. 3-5) or endogenous (PMN membrane p47-phox, Fig. 7) protein substrate was used. Using histone, the ICso was 140-190 pM (Fig. 3) and the Ki was 120 p~ (with respect to PS). Thus, when cell functions are inhibited by propranolol, it is not possible to differentiate between indirect inhibition of PKC activation (via decreases in diacylglycerol from inhibition of PA phosphohydrolase) and direct inhibitory effects on PKC. Interpretation of results in which propranolol treatment enhances cell functions is also difficult, since PKC may have down-regulatory effects in cells (15, 57).
Propranolol appeared to be selective in its ability to inhibit PKC, as opposed to other protein kinases, but did not dem- onstrate selectivity between two isoforms of PKC. In the concentration-range tested (30-1000 p ~ ) , propranolol did not affect CAMP-dependent protein kinase nor protein tyrosine kinase activities (Fig. 8). PKC isozymes can be divided into Ca”-dependent (CY, @, y) and Ca”-independent ( 6 , c, 3; 1) groups (reviewed in Refs. 57 and 58). Human PMN contain a t least two Ca2+-dependent PKC isozymes, identified as PKC-CY and PKC-fl by elution profile from hydroxylapatite and by isozyme-specific antisera2 (42, 53, 59, 60). The two isoforms showed the same pattern of inhibition when tested in the presence of propranolol, suggesting that propranolol cannot be used to differentiate between these Ca”-dependent isozymes. We have not yet tested the effect of propranolol on any of the Ca2+-independent isoforms of PKC.
PKC has two domains, a hydrophobic regulatory domain that interacts with PS, diacylglycerol, and Ca2+, and a hydro-

Inhibition of Protein Kinase C by Propranolol 20481
philic catalytic domain that interacts with Mg2'-ATP and substrate proteins (61,62). In the past years, several inhibitors of PKC have been described. The inhibition can result from interaction of the inhibitor either with the phospholipid and diacylglycerol binding domain (34, 63) or with the catalytic domain (36,64,65). A number of compounds effective on both regulatory and catalytic domains have also been described (35, 65, 66). In vitro kinetic studies (Figs. 4 and 5) suggested that the interaction of propranolol with PKC is at the level of the regulatory domain. Kinetic analysis was consistent with propranolol inhibition being competitive, with respect to PS (Fig. 4) and diolein (Fig. 5A), and noncompetitive, with respect to ATP (Fig. 5B) and histone (data not shown). However, the complex kinetic patterns observed preclude reaching definitive conclusions concerning the type of inhi- bition.
The inhibitory effect of propranolol on the regulatory do- main of PKC may be due to its membrane-interactive prop- erties. Comparable levels of inhibition were obtained with (f)propranolol and with the (-) and (+) stereoisomers (Fig. 3). Other @-adrenergic receptor antagonists (e.g. (-)timolol and (+)pindolol) or agonists ((-)isoproterenol) did not show the same ability as propranolol in inhibiting PKC activity (Fig. 3 and data not shown). In addition, PKC inhibition was observed at higher concentrations (-100-fold) than those needed to antagonize the p-adrenergic receptor (6). (-)Pro- pranolol preferentially interacts with the p-adrenergic recep- tor, while both stereoisomers share equally in local anesthetic and membrane-stabilizing properties (67-69). These obser- vations suggest that the effect of propranolol on PKC is not related to interaction with the p-adrenergic receptor, but rather to the anesthetic and membrane-stabilizing properties of the molecule.
The anesthetic effect of propranolol and some other adren- oreceptor antagonists is related to the lipid-solubility/mem- brane interaction ability of these drugs (69). Propranolol and chlorpromazine can penetrate and specifically bind to PS and phosphatidylinositol present on the inner face of platelet membrane (69). This property may be related to their ability to inhibit PKC, since chlorpromazine (32, 33) and other phospholipid-interacting drugs, such as imipramine, dibu- caine, phentolamine, tetracaine, verapamil (32), aminoacri- dines (35), adriamycin (66), palmitoylcarnitine (70), trifluo- perazine, fluphenazine, haloperidol, and chlorprothixene (71), have all been shown to be inhibitors of PKC.
To further explore the features of propranolol responsible for PKC inhibition, we have begun synthesis of propranolol analogs, testing the requirement for particular portions of the molecule. Propranolol, as an in vivo drug, can be administered at relatively high concentrations (e.g. 1.3 g/day (Ref. 72)) because of its low toxicity. Since PKC inhibitors are generally rather toxic, we also wished to create new molecules that could maintain the low toxicity properties and more effec- tively inhibit PKC activity. Results with two analogs are reported in this paper (Fig. 9) and suggest that an increase in hydrophobicity and membrane solubility by replacement of the naphthyloxy moiety with a hexadecyloxy group (JS-X- 112) increases potency for PKC inhibition. In contrast, an unhindered hydroxyethylamine moiety is not necessary for PKC inhibition, since replacement of an isopropylamine group with a cyclohexylamine group (JS-XI-60) did not affect inhibitory potency. Studies are currently underway to synthe- size and characterize additional analogs and to better inves- tigate their in vitro and in vivo effects.
In conclusion, our results demonstrate that propranolol is a PKC inhibitor and is not specific for inhibition of PA
phosphohydrolase. This increases the level of complexity for the use of propranolol in signal transduction studies and indicates that interpretation of previously published results (18-28) and future studies using propranolol should consider the lack of specificity of this drug. In addition, our results suggest that the structure of propranolol can be modified to yield analogs of increased potency for inhibition of PKC. Therefore, it may be possible to create new, more effective compounds for the study of cellular signalling mechanisms.
Acknowledgments-At Wake Forest University we thank Sue Cou- sart and Mary Ellenburg for performing the 0; release assays, Diane Qualliotine-Mann for development and assistance with the tyrosine kinase assay, Mary Ellenburg and Dr. Peter Leone for development of the in vitro phosphorylation assay of PMN proteins, Bill Small for help with initial kinetic studies, and Dr. Mike Thomas for synthesis of the protein kinase inhibitor C-I. In addition, we thank Drs. Rick Woodman and John Curnutte (Research Institute at Scripps Clinic) for providing us with blood from two patients with the p47-phox- deficient form of chronic granulomatous disease, Drs. Marie Wooten (Auburn University) and Mary Makowske (Memorial Sloan-Ketter- ing Cancer Center) for their kind gifts of anti-peptide antisera specific for protein kinase C isoforms (a, 0, y ) , and Dr. C.-K. Huang (Uni- versity of Connecticut) for the erbstatin analog inhibitor of epidermal growth factor receptor tyrosine kinase activity.
2. 1.
3.
4.
5.
6.
7. 8.
9.
10.
11. 12.
13.
14.
15.
16. 17.
18. 19.
20.
21.
22.
23. 24.
25.
26.
27.
28. 29.
31. 30.
32.
33.
34.
35.
REFERENCES Exton, J. H. (1990) J . Biol. Chem. 266 , 1-4 Billah, M. M., and Anthes, J. C. (1989) Biochem. J. 269,281-291 Thompson, N. T., Bonser, R. W., and Garland, L. G. (1991) Trends
Agwu, D. E., McPhail, L. C., Chabot, M. C., Daniel, L. W., Wykle, R. L.,
Beta-blocker Heart Attack Trial Research Group (1982) J. Am. Med. Assoc.
Stiles, G. L., Caron, M. G., and Lefkowitz, R. J. (1984) Physiol. Reu. 6 4 ,
Koul, O., and Hauser, G. (1987) Arch. Biochem. Biophys. 253,453-461 Jamal, A., Martin, A,, Gomez-Munoz, A., and Brindley, D. N. (1991) J.
Cabot, M. C., Welsh, C. J., Cao, H.-T., and Chabbott, H. (1988) FEBS Lett.
Billah, M. M., Eckel, S., Mullmann, T. J., Egan, R. W., and Siegel, M. I.
Qian, A., and Drewes, L. R. (1990) J. Biol. Chtm. 266,3607-3610 Martinson, E. A,, Trilivas, I., and Brown, J. H. (1990) J. Biol. Chem. 265 ,
Lopez-Barahona, M., Kaplan, P. L., Cornet, M. E., Diaz-Meco, M. T., 22282-22287
Larrodera, P., Diaz-Laviada, I., Municio, A. M., and Moscat, J. (1990) J.
Mullmann. T. J.. Sieeel. M. I.. Eean. R. W.. and Billah. M. M. (1990) J. Biol. Chem. 266,9022-9026
Pharmacol. Sci. 12,404-408
and McCall, C. E. (1989) J. Bzol. Chem. 264,1405-1413
247,1707-1714
661-743
Biol. Chem. 266,2988-2996
2 3 3 , 153-157
(1989) J. Biol. Chem. 2 6 4 , 17069-17077
Immunol. 144'. 19&i908 ' ' , ~ ~~~ ~~~~
~~, ~ ~ . . ~ ~ . . ~, ~
Bishop, W. R., August, J., Petrin, J. M., and Pai, J.-K. (1990) Biochem. J. 9.aQ AG5-A7?!
English, D., Taylor, G., and Garcia, J. G. N. (1991) Blood 77, 2746-2756 Song, J., Pfeffer, L. M., and Foster, D. A. (1991) Mol. Cell. Biol. 11 , 4903-
Liscovitch, M., and Amsterdam, A. (1989) J. Biol. Chem. 264,11762-11767 Rossi, F., Grzeskowiak, M., Della Bianca, V., Calzetti, F., and Gandini, G.
English, D., and Taylor, G. S. (1991) Biochem. Biophys. Res. Commun.
Lin, P., Wiggan, G. A., and Gilfillan, A. M. (1991) J. Immunol. 146,1609-
Lin, P., Wiggan, G. A., Welton, A. F., and Gilfillan, A. M. (1991) Biochem.
Metz, S. A., and Dunlop, M. (1991) Biochem. Pharmacol. 4 1 , Rl-R4 Agwu, D. E., McPhail, L. C., Sozzani, S., Bass, D. A,, and McCall, C. E.
Della Bianca, V., Grzeskowiak, M., Lissandrini, D., and Rossi, F. (1991)
Hardy, S. J., Robinson, B. S., Poulos, A., Harvey, D. P., Ferrante, A,, and
Kanaho, Y., Kanoh, H., Saitoh, K., and Nozawa, Y. (1991) J. Immunol.
Gay, J. C., and Murray, J. J. (1991) Biochim. Biophys. Acta 1096 , 236-242 Holmsen, H., and Dangelmaier, C. A. (1990) Thromb. Hamnostassis 6 4 ,
"-, =."
4908
(1990) Biochem. Biophys. Res. Commun. 168,320-327
176,423-429
1616
Pharmacol. 41 , 1941-1948
(1991) J. Clin. Inuest. 88,531-539
Bwchem. Biophys. Res. Commun. 177,948-955
Murray, A. W. (1991) Eur. J. Biochem. 198,801-806
146,3536-3541
2n7-wl Lavie, Y., Piterman, O., and Liscovitch, M. (1990) FEBS Lett. 277 , 7-10 Mullmann, T. J. Siegel, M. I., Egan, R. W., and Billah. M. M. (1991) J.
Biol. Chem. 266,2013-2016 Mori, T Takai, Y. Minakuchi, R., Yu, B., and Nishizuka, Y. (1980) J.
Biol. ?hem. 266,'8378-8380 Wise, B. C., Glass, D. B., Chou, C.-H. J., Raynor, R. L., Katoh, N.,
Schatzman, R. C., Turner, R. S., Kibler, R. F., and Kuo, J. F. (1982) J. R i d ChPm 257. Ad%-8495
"I Y I I
~ ~ -_ _. _"l _ " _ Hannun, Y. A., Loomis, C. R., Merrill, A. H., Jr., and Bell, R. M. (1986) J.
Hannun, Y. A., and Bell, R. M. (1988) J. Biol. Chem. 263,5124-5131 36. Gerard, C., McPhail, L. C., Marfat, A., Stimler-Gerard, N. P., Bass, D. A,,
Biol. Chem. 261,12604-12609

20488 Inhibition of Protein Ei
37. Yaish, P., Gazit, A,, Gilon, C., and Levitzki, A. (1988) Science 2 4 2 , 933-
38. McPhail, L. C., and Snyderman, R. (1983) J. Clin. Inuest. 7 2 , 192-200 39. O’Flaherty, J. T., Jacobson, D. P., Redman, J. F., and Rossi, A. G. (1990)
40. Massey, V. (1959) Biochim. Biophys. Acta 34,255-256 41. Shearman, M. S., Ogita, K., Kikkawa, U., and Nishizuka, Y. (1989) Methods
42. Makowske, M., Ballester, R., Cayre, Y, and Rosen, 0. M. (1988) J. Biol.
43. Hannun, Y. A,, Loomis, C. R., and Bell, R. M. (1985) J. Biol. Chem. 2 6 0 ,
44. Pires, E. M. V., and Perry, S. V. (1977) Biochem. J . 167 , 137-146 45. Wolfson, M., McPhail, L. C., Nasrallah, V. N., and Snyderman, R. (1985)
46. Berkow. R. L.. Dodson. R. W.. and Kraft. A. S. (1989) Biochim. Bwohvs.
and McCall, C. E. (1986) J. Clin. Inuest. 77, 61-65
935
J. Biol. Chem. 265,9146-9152
Enzymol. 168,347-351
Chem. 263,3402-3410
10039-10043
J. Immunol. 135 , 2057-2062
~ Acta 997,292-300 ’
. . . ” 47. Caldwell, S. E., McCall, C. E., Hendricks, C. L., Leone, P. A., Bass, D. A.,
48. Kramer, I. M., Verhoeven, A. J., van der Bend, R. L., Weening, R. S., and and McPhail, L. C. (1988) J. Clin. Inuest. 8 1 , 1485-1496
Roos. D. (1988) J. Btol. Chem. 263. 2352-2357 49. Laemmi6 U: K. (1970) Nature 227,680-685 50. Wray, W., Boulikas, T., Wray, V. P., and Hancock, R. (1981) Anal. Biochem.
51. Bradford, M. M. (1976) Anal. Biochem. 72,248-254 52. Smith, P. K., Krohn, R. I., Hermanson, G. T., Mallia, A. K., Gartner, F.
H., Provenzano, M. D., Fujimoto, E. K., Goeke, N. M., Olson, B. J., and
53. Pontremoli, S., Melloni, E., Sparatore, B., Mlchetti, M., Salamino, F., and Klenk, D. C. (1985) Anal. Biochem. 1 5 0 , 76-85
54. Clark, R. A. (1990) J. Infect. Dis. 161 , 1140-1147 Horecker, B. L. (1990) J. Biol. Chem. 2 6 5 , 706-712
118 , 197-203
h a s e C by Propranolol 55. Ambruso, D. R., Bolscher, B. G. J. M., Stokman, P. M., Verhoeven, A. J.,
56. Clark, R. A,, Volpp, B. D., Leidal, K. G., and Nauseef, W. M. (1990) J. and Roos, D. (1990) J. Biol. Chem. 265,924-930
Clin. Inuest. 86.-714-721 57. 58. 59.
60. 61. 62.
63.
64. 65.
66.
67.
Nishizuka, Y. (19k) Nature 334,661-665 Bell, R. M., and Burns, D. J. (1991) J. Biol. Chem. 266,4661-4664 Majumdar, S., Rossi, M. W., Fujiki, T., Phillips, W. A., Disa, S., Queen, C.
F., Johnston, R. B., Jr., Rosen, 0. M., Corkey, B. E., and Korchak, H. M. (1991) J. Biol. Chem. 266,9285-9294
Smallwood, J. I., and Malawista, S. E. (1992) J. h u h . Biol. 5 1 , 84-92 Lee, M.-H., and Bell, R. M. (1986) J. Biol. Chem. 26,1,14867-14870 Huang, K. P., and Huang, F. L. (1986) Biochem. Btophys. Res. Commun.
1 ~ ~ 2 3 n - 2 2 6 Turner, R. S., and Kuo, J. F. (1985) in Phospholipids and Cellular Regulation
Loomis, C. R., and Bell, R. M. (1988) J. Biol. Chem. 263 , 1682-1692 II (Kuo, J. F., ed) Vol. 11, pp. 75-110, CRC Press Inc., Boca Raton, FL
Nakadate, T., Jeng, A. Y., and Blumberg, P. M. (1988) Btochem. Pharmocol.
Hannun. Y. A,. Foelesone. R. J.. and Bell. R. M. (1989) J. Biol. Chem. 264.
- - - , - - - - - -
37,1541-1545 ~~
9960-9966 ’ I’ ’
J. Clin. Pharmocol. 5.145-150 Grobecker, H., Lemmer, B., Hellenbrecht, D., and Wiethold, G. (1973) Eur.
68. Lan slet A (1970) Eur. J Pharmacol. 13,6-14 69. Daciary:Pkgent, J., Dufourcq, J., Lussan, C., and Boisseau, M. (1979)
70. Katoh, N., Wrenn, R. W., Wise, B. C., Shoji, M., and Kuo, J. F. (1981) Thromb. Res. 14,15-22
71. Schatzman, R. C., Wise, B. C., and Kuo, J. F. (1981) Biochem. Biophys. Proc. Natl. Acad. Sci. U. S. A. 78,4813-4817
72. Murray, K. T., Reilly, C., Koshakji, R. P., Roden, D. M., Lineberry, M. D., Res. Commun. 98, 669-676
Wood, A. J. J., Siddoway, L. A,, Barbey, J. T., and Woosley, R. L. (1990) J. Clin. Inuest. 8 5 , 836-842