progressive vasomotor changes in ischaemic myocardium
TRANSCRIPT

Acta Med Scand (Suppl) 694: 38-44, 1984
Progressive Vasomotor Changes in Ischaemic Myocardium
M.W. Gorman, R.D. Wangler, D.F. DeWitt and H.V. Sparks Jr
From the Departments of Surgery a n d Physiology, Michigan State University, East Lansing. Michigan, U S A
ABSTRACT. Under certain conditions, a progressive increase in vascular resistance occurs within ischaemic myocardium dur- ing the first three hours after coronary ar- tery stenosis. Measurements of vasodilator reserve in the ischaemic region demon- strated that this is at least partly due to an increase in vascular smooth muscle tone. Two hypotheses were suggested as an ex- planation: release of vasoconstrictors with- in the ischaemic area, or a decreasing re- lease of vasodilators. Potential coronary constrictors considered included norepine- phrine, PGF,,, thromboxane A,, and high K'. Each of these substances was elimi- nated as the source of the vasoconstriction by pharmacological studies on ischaemic canine hearts. Measurement of adenosine release from isolated guinea pig hearts pro- vided support for the possibility that the vasoconstriction results from a decreas- ing release of metabolic vasodilatators. Throughout the period of ischaemia, both blood flow and myocardial function were far below the levels that could have been achieved in the presence of the stenosis. We conclude that during moderate ischaemia, myocardial function and blood flow are linked in a positive feedback cycle which promotes reduced ventricular function and coronary blood flow.
Coronary artery stenosis severe enough to produce ischaemia results in an initial dila- tion of resistance vessels within the affected area. Under certain conditions, however, this initial vasodilation is not maintained if the ischaemia continues for more than an hour (1-3). This increase in vascular resistance may be partly due to swelling of myocardial or vascular smooth muscle cells in the is- chaemic region, which can reduce vascular calibre (1, 4, 5, 6). Our own studies indicate that increasing vascular smooth muscle tone also contributes to the increase in resistance (3). Two potential mechanisms for this vaso- constriction are illustrated in Figure 1. First, it is possible that vasoconstrictor substances are released within the ischaemic area. Sec- ond, the production of vasodilator substances
4 R E S I S T A N C E
$.CONTRACTILE F O R C E
I / +METABOLIC ~ A S O D I L A T O R
R E L E A S E
Key words: ischaemic heart disease, coronary cir- culation, radioactive microspheres, alpha-adren- ergic receptors, adenosine.
Fig. I . Two potential positive feedback loops which could produce progressive vasoconstriction within ischaemic myocardium.

Progressive Vasomotor Changes in Ischaemic Myocardium 39
may decline with time, perhaps as a result of decreasing contractile activity. Both of these mechanisms have the potential to become positive feedback loops, which could account for the steadily increasing vascular resist- ance. This paper will report on our attempts to determine the mechanism of this .is- chaemic vasoconstriction..
METHODS
Dog heart preparation
Mongrel dogs were anaesthetized with sodium pentobarbital, intubated, and artificially ventilated with oxygen-enriched room air in order to main- tain arterial blood gases within the physiologi- cal range (7). The heart was exposed through a left lateral thoracotomy, and a segment of the left an- terior descending coronary artery (LAD) was ex- posed 1-3 cm from its origin. A hydraulic occluder was placed around the vessel, and the occluder was connected to a syringe pump to facilitate fine control of the degree of inflation. Just distal to the occluder we inserted a thin catheter (8) with a side hole, which allowed us to measure distal LAD pressure and to make intracoronary infusions directly into the LAD bed (Fig. 2). A catheter for administration of radioactive microspheres was placed in the left atrium, and blood samples for microsphere flow calculations were collected via a cannula in a femoral artery. The hearts were electrically paced at 180 beatslminute. In some cases, venous samples from the ischaemic region were collected via a teflon catheter inserted into the great cardiac vein. Three dogs were instru- mented with Walton-Brodie strain gauge arches within the LAD perfusion territory.
Microsphere In]ectdo
neumatic Culf
Coronary Calhete
F i g . 2. Dog heart preparation.
Relative ischaemia was created by inflating the LAD occluder until distal coronary pressure fell to approximately 50 mmHg. This pressure was then maintained constant throughout the experiment by making periodic adjustments with the occluder. Therefore, changes in LAD bed flow represent changes in vascular resistance. Microsphere blood flow measurements were made after 30 minutes and 180 minutes of ischaemia in every preparation. Following the 180-minute measurement, a pharma- cological intervention was usually made and flow was measured again. In some series, a control flow measurement was made before beginning isch- aemia. In hearts that were pretreated with phenoxybenzamine, distal LAD pressure had to be lowered to 30 mmHg in order to produce a degree of ischaemia equivalent to untreated hearts at 50 mmHg.
We arbitrarily defined ischaemia as a transmural LAD bed flow less than 75 % of the circumflex bed flow at the 30-minute point. Hearts not meeting this criterion were rejected from the study. In addition, 15 % of the ischaemic hearts did not show a vasoconstriction in the ischaemic area between 30 and 180 minutes. These hearts were also rejected from the study. The reason(s) for the variable oc- currence of ischaemic vasoconstriction is (are) not clear, but may be due to variations in initial myocardial oxygen consumption or temperature
Various drugs were administered either before ischaemia (phenoxybenzamine) or after three hours of ischaemia (adenosine, norepinephrine, indomethacin, phentolamine, propranalol). From the effects of these drugs on vascular resistance in the ischaemic region, we made inferences about the role of several vasoactive substances in the development of ischaemic vasoconstriction.
(i, 2).
Isolated guinea pig heart preparation
The hearts of pentobarbital anaesthetized guinea pigs (male, 200-250 g) were rapidly excised and immersed in ice-cold physiological salt solution. Retrograde aortic perfusion was begun at 37°C. The perfusate was a modified Krebs-Henseleit solution containing glucose and pyruvate, which was oxygenated with 95 70 0, - 5 % CO,, filtered, and not recirculated. Perfusion pressure was main- tained at 60 cmH,O. The left ventricle was vented and the hearts were paced at 300 beatslmin. The pulmonary artery was cannulated for collection of the coronary venous effluent. Venous samples (3 ml) were collected for analysis of adenosine concentration. Samples were evaporated to dry-

40 M.W. Gorman et al.
ness, resuspended in 400 pl water, and analysed by reverse-phase HPLC. Because arterial adeno- sine concentration was zero, adenosine release was calculated as venous effluent concentration times flow.
After an initial 30-minute equilibration period, moderate ischaemia was produced by reducing coronary perfusion pressure from 60 cm to 30 cmH,O. Samples for adenosine analysis were col- lected at the onset of ischaemia and after 10,20,30, 60, 90, and 180 minutes. Perfusion pressure was then further reduced to 15 cmH20 and an addition- al venous sample was collected.
Table 1. Effects of various interventions on vascular resistance in the ischaemic LAD bed. Adenosine, indomethacin, norepine- phrine, and phentolamine were given after 3 hours of ischaemia when ischaemic vaso- constriction had already developed. Phen- oxybenzamine was given before ischaernia was begun, and propranolol was given in phenoxybenzamine-treated hearts after 3 hours of ischaemia. Indomethacin and phen- tolamine were given intravenously; all other drugs via intracoronary infusion. All changes a re statistically significant a t the 0.05 level using the paired t-test.
RESULTS Intervention Effect on resistance in con- stricted ischaemic region
Dog Hearts
Between the flow measurements at 30 min- utes and three hours, vascular resistance in the ischaemic region increased in untreated hearts by an average of 30 7%. The results of several pharmacological interventions made at the three hour point are summarized in Table I. Intracoronary adenosine infusion dramatically reduced vascular resistance by 74 70. This indicates the presence of sub- stantial vasodilator reserve. When we made an identical adenosine infusion after only 10 minutes of ischaemia, resistance decreased only 24 70 (not statistically significant). Indo- methacin given intravenously after three hours of ischaemia produced a further con- striction of 22 %. Intracoronary norepine- phrine infusion reduced resistance by 54 %, and also increased contractile force develop- ment from 38 70 to 108 % of the non-ischaemic control level. Intravenous phentolamine dropped ischaemic bed resistance by 19 %. Although the results above refer to trans- mural resistance changes, subepicardial and subendocardial flow changed in parallel in all cases except during adenosine infusion, when the subepicardial response was greater than in the subendocardium (3).
In several hearts we measured plasma K' concentration in venous blood drawn from
Adenosine 4 74 70 Indomethacin t 22 7c Norepinephrine 4 5 4 % Phentolamine t 19 7 e Phenoxybenzamine Prevents constriction Phenoxybenzamine + Propranolol Constriction returns
the ischaemic region at various times during the three hour ischaemic interval. We found no significant change in K' over this period (3).
In two series of experiments, phenoxybenz- amine was given intracoronary before begin- ning ischaemia. This pretreatment eliminated the usual increase in ischaemic bed resist- ance over the three-hour period. However, when intracoronary propranalol was given at the three-hour point in phenoxybenz- amine-treated hearts, vascular resistance increased substantially (3).
Guinea pig hearts
Under non-ischaemic control conditions, coronary flow in six hearts was 7.5 k 0.4 mli min/g wet weight, and adenosine release (Rado) was 21 f 2 pmollminlg. Ten minutes after the reduction of perfusion pressure
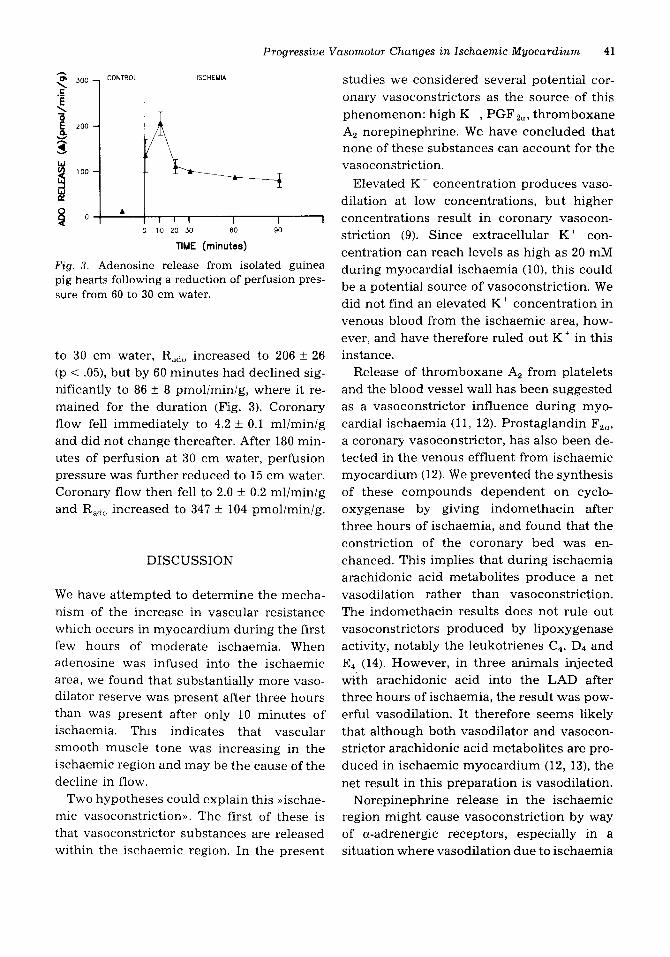
Progressive Vasomotor Changes in lschaemic Myocardium 41
ISCHEMIA
I I 1 0 10 20 30 60 90
TIME (minutes)
F i g . 3. Adenosine release from isolated guinea pig hearts following a reduction of perfusion pres- sure from 60 to 30 cm water.
to 30 cm water, Rado increased to 206 f 26 (p < .05), but by 60 minutes had declined sig- nificantly to 86 k 8 pmoliminig, where it re- mained for the duration (Fig. 3). Coronary flow fell immediately to 4.2 f 0.1 mliminig and did not change thereafter. After 180 min- utes of perfusion at 30 cm water, perfusion pressure was further reduced to 15 cm water. Coronary flow then fell to 2.0 f 0.2 ml/min/g and Rado increased to 347 f 104 pmol/min/g.
DISCUSSION
We have attempted to determine the mecha- nism of the increase in vascular resistance which occurs in myocardium during the first few hours of moderate ischaemia. When adenosine was infused into the ischaemic area, we found that substantially more vaso- dilator reserve was present after three hours than was present after only 10 minutes of ischaemia. This indicates that vascular smooth muscle tone was increasing in the ischaemic region and may be the cause of the decline in flow.
Two hypotheses could explain this .ischae- mic vasoconstriction)). The first of these is that vasoconstrictor substances are released within the ischaemic region. In the present
studies we considered several potential cor- onary vasoconstrictors as the source of this phenomenon: high K +, PGFz", thromboxane A2 norepinephrine. W e have concluded that none of these substances can account for the vasoconstriction.
Elevated K t concentration produces vaso- dilation at low concentrations, but higher concentrations result in coronary vasocon- striction (9). Since extracellular K + con- centration can reach levels as high as 20 mM during myocardial ischaemia (lo), this could be a potential source of vasoconstriction. We did not find an elevated K + concentration in venous blood from the ischaemic area, how- ever, and have therefore ruled out K + in this instance.
Release of thromboxane A, from platelets and the blood vessel wall has been suggested as a vasoconstrictor influence during myo- cardial ischaemia (11, 12). Prostaglandin Fzu, a coronary vasoconstrictor, has also been de- tected in the venous effluent from ischaemic myocardium (12). We prevented the synthesis of these compounds dependent on cyclo- oxygenase by giving indomethacin after three hours of ischaemia, and found that the constriction of the coronary bed was en- chanced. This implies that during ischaemia arachidonic acid metabolites produce a net vasodilation rather than vasoconstriction. The indomethacin results does not rule out vasoconstrictors produced by lipoxygenase activity, notably the leukotrienes C4, D4 and E4 (14). However, in three animals injected with arachidonic acid into the LAD after three hours of ischaemia, the result was pow- erful vasodilation. It therefore seems likely that although both vasodilator and vasocon- strictor arachidonic acid metabolites are pro- duced in ischaemic myocardium (12, 13), the net result in this preparation is vasodilation.
Norepinephrine release in the ischaemic region might cause vasoconstriction by way of a-adrenergic receptors, especially in a situation where vasodilation due to ischaemia

42 M.W. Gorman et al.
is already present (15). This hypothesis was supported by the results of the phenoxybenz- amine experiments, where a-blockade prior to ischaemia completely eliminated the usual vasoconstriction during the ensuing three hours. In addition, phentolamine given in already constricted preparations produced vasodilation in the ischaemic region. Both phenoxybenzamine and phentolamine, how- ever, can increase the neuronal release of norepinephrine by blockade of presynaptic ti,-receptors (17). If norepinephrine release actually produces a net vasodilation in the ischaemic area (as it does in normal myo- cardium), then these a-receptor antagonists could have prevented ischaemic vasocon- striction by increasing norepinephrine re- lease and producing a metabolic vasodilation. Two series of experiments demonstrated that this was in fact what occurred. When nor- epinephrine was infused into the ischaemic region after three hours, it produced both vasodilation and increased contractile force development. Therefore, norepinephrine does produce net vasodilation in the ischaemic area. Furthermore, when propranalol was ad- ded to phenoxybenzamine-pretreated hearts after three hours of ischaemia, the vasocon- striction prevented by phenoxybenzamine re- turned. This suggests that phenoxybenz- amine was stimulating norepinephrine re- lease. We therefore conclude that norepine- phrine is not the source of ischaemic vaso- constriction.
The results mentioned so far have elimi- nated all the potential coronary vasoconstric- tors that seem likely to be present in is- chaemic myocardium. There is always the possibility that an unknown .substance X,) causes the vasoconstriction, but ischaemic vasoconstriction might also be caused by a decreasing release of vasodilators (Fig. 1, outer loop). The adenosine release measure- ments from isolated guinea pig hearts pro- vide support for this hypothesis. Adenosine release rose by an order of ten after 10 min-
utes of hypoperfusion, but declined to a steady state value of roughly four times the control level by 60 minutes. The decline in Rado is not due to substrate depletion, because a further drop in perfusion pressure after 180 minutes dramatically increases Rado. These isolated guinea pig hearts did not ex- hibit ischaemic vasoconstriction. The situa- tion in the dog heart might be different, how- ever, since the humoral and neural influences present in the blood-perfused heart may pro- mote more vascular smooth muscle tone than in the isolated guinea pig heart. In summary, evidence for the .decreasing vasodilator re- leasen hypothesis is still incomplete, but this seems to be a promising area for further re- search.
A recent study by Sunnergren and Rovetto (18) demonstrated that ischaemic vasocon- stiiction in the isolated rat heart could be reversed by treatment with hyaluronidase. This was attributed to the prevention of oedema formation. It seems unlikely that oedema formation could increase vascular resistance by extravascular compression in the dog heart, because some of the vasodila- tor reserve should be recruited to compen- sate for this. This may not be the case in the rat heart, however. Another possible inter- pretation of their results is that by reducing oedema, hyaluronidase increases substrate availability and prevents a fall in metabolic activity during the ischaemic period. This would fit the hypothesis diagrammed in the outer loop of Figure 1. Hyaluronidase reduces myocardial necrosis and increases collateral blood flow after coronary artery occlusion in dogs (19, 20). Its effect on blood flow after coronary artery stenosis, however, is un- known.
If the outer loop in Figure 1 is indeed the mechanism of ischaemic vasoconstriction, this challenges some of the current thinking about blood flow regulation in ischaemic myocardium. The traditional view is that myocardial performance in ischaemic areas

Progressive Vasomotor Changes i n Ischaemic Myocardium 43
is limited by the flow. Our results suggest that what frequently happens in that flow is limited by performance. Of course in severely ischaemic regions where substrate availabili- ty is too low to maintain cell viability, flow certainly limits performance. This would be the case in most of the ischaemic region, for example, following coronary occlusion. Our results demonstrate, however, that with se- vere coronary artery stenoses most of the myocardium in the affected area, if stimu- lated, is capable of both increased perform- ance and increased flow. The norepinephrine infusion experiments demonstrate this most clearly. When subjected to moderate is- chaemia, the myocardium apparently reaches a new steady state where oxygen supply (flow) and demand (performance) are once again in balance, but at reduced levels. This happens even though both flow and perform- ance could be maintained at much higher 1 evels.
Why does the myocardium behave this way? It would seem at first glance that main- tained myocardial performance might have important survival value. Perhaps the answer lies in the cost of maintained performance during moderate ischaemia. Suppose, for example, that adenosine release is responsi- ble for the matching of myocardial blood flow to performance (21). During ischaemia, this would necessitate an increased release of adenosine, which could deplete the ade- nine nucleotide pool if it continued long enough. Reducing regional myocardial per- formance may represent the best long-term strategy for survival.
ACKNOWLEDGMENTS
Supported by American Heart Association Grant 80928 and U.S. Public Health Service Grant HL24232. This work was done during the tenure of Research Fellowships of the Michigan Heart Association awarded to Drs. Gorman and Wangler.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
REFERENCES
Frame LH, Powell WJ. Progressive perfusion impairment during prolonged low flow myo- cardial ischemia in dogs. Circ Res 1976; 39:
Guyton RA, McClenathan JH, Michaelis LL. Evolution of regional ischemia distal to a prox- imal coronary stenosis: Self-propagation of is- chemia. Am J Cardiol 1977; 40: 381--92. Gorman MW, Sparks HV Jr. Progressive cor- onary vasoconstriction during relative ische- mia in canine myocardium. Circ Res 1982; 51:
Willerson JT, Watson JT, Hutton I, Templeton GH, Fixler DE. Reduced myocardial reflow and increased coronary vascular resistance fol- lowing prolonged myocardial ischemia in the dog. Circ Res 1975; 36: 771-81. Powell WJ , DiBona DR, Flores J, Frega N, Leaf A. Effects of hyperosmotic mannitol in reducing ischemic cell swelling and minimiz- ing myocardial necrosis. Circulation 1976; 53
Kloner RA, Ganote CE, Jennings RB. The .no- reflow.> phenomenon after temporary cor- onary occlusion in the dog. J Clin Invest 1974;
Feigl EO, D’Alecy LG. Normal arterial blood pH, oxygen and carbon dioxide tensions in unanesthetized dogs. J Appl Physiol 1972; 32:
Herd JA, Barger AC. Simplified technique for chronic catheterization of blood vessels. J Apll Physiol 1964; 19: 791-2. Bohr DF, Goulet PL. Role of electrolytes in the contractile machinery of vascular smooth muscle. Am J Cardiol 1961; 8: 549-56. Hill JL , Gettes LS. Effect of acute coronary artery occlusion on local myocardial extra- cellular K ‘ activity in swine. Circulation 1980;
Needleman P, Kulkarni PS, Raz A. Coronary tone modulation: formation and actions of prostaglandins, endoperoxides, and throm- boxanes. Science 1977; 195: 409- 12. Lewy RI, Wiener L, Walinsky P, Lefer AM, Silver MJ, Smith JB. Thromboxane release during pacing-induced angina pectoris: pos- sible vasoconstrictor influence on the coronary vasculature. Circulation 1980; 61: 1165-71. Berger HJ , Zaret B, Speroff L, Cohen LS, Wolfson S . Regional cardiac prostaglandin re- lease during myocardial ischemia in anesthe- tized dogs. Circ Res 1976; 38: 566-71.
269- 76.
411-20.
(Suppl I): 45-9.
54: 1496- 1507.
152-3.
61: 768-78.

44 M.W. Gorman et al.
14. Samuelson B. Leukotrienes: mediators of immediate hypersensitivity reactions and inflammation. Science 1983; 220: 568-75.
15. Mudge GH J r , Grossman W, Mills RM Jr , Lesch M, Braunwald E. Reflex increase in coronary vascular resistance in patients with ischemic heart disease. N Eng J Med 1976;
16. Heiisch G , Deussen A. The effects of cardiac sympathetic nerve stimulation on perfusion of stenotic coronary arteries in the dog. Circ Res
17. Langer S Z . Presynaptic receptors and their role in the regulation of transmitter release. Br J Pharmacol 1977; 60: 481-97.
295: 1333-7.
1983; 53: 8- 15.
18. Sunnergren KP, Rovetto MJ. Hyaluronidasr reversal of increased coronary vascular resist- ance in ischemic rat hearts. Am J Physiol 1983;
19. Maroko PR, Libby P, Bloor CM, Sobel BE, Braunwald E. Reduction by hyaluronidase of myocardial necrosis following coronary artery occlusion. Circulation 1972; 46: 430-7.
20. Askenazi J , Hillis LD, Diaz PE, Davis MA, Braunwald E, Maroko PR. The effects of hya- luronidase on coronary blood flow following coronary artery occlusion in the dog. Circ Res
21. Berne RM. The role of adenosine in the regu- lation of coronary blood flow. Circ Res 1980;
245: H183-8.
1977; 40: 566-71.
47: 807- 13.