problems 2 answers
TRANSCRIPT

Asymmetric synthesis: problems 2
reagent controlled crotylation
substrate control carbonyl addition
substrate controlled aldol reaction
catalyst controlled Diels-Alder reaction
an enantioselective synthesis of kainic acid
1
BO
OiPrO2C
iPrO2C O
OBn 70%88:12 dr
OH
OBn
Question 1
An easy start. Rationalise the observed diastereoselectivity.
For bonus marks is this an example of matched or mismatched substrate/reagent stereoselectivity (sometimes called double asymmetric induction). Or in other words, learn what matched/mismatched means!
2

BO
OiPrO2C
iPrO2C O
OBn 70%88:12 dr
OH
OBn
Answer
This is taken from the synthesis of callipeltoside A and is an example of the Roush asymmetric crotylation. The Roush reagents are more user friendly than the Brown crotylation reagents, being less moisture sensitive and can be stored for longer periods of time. On the downside they often give slightly lower enantioselectivities than the Brown reagents.
Initially it looks like the selectivity may be harder to rationalise than the Brown reagents …
Angew. Chem. Int. Ed. 2012, 51, 9366
O
OH3COH
OOH
O O
NHO
Cl
O
H
H3CO
3
HO
BH
H
O
O
O
OiPr
OOiPr
BO O
O
O
OiPr
OOiPr
HH
OBnH
BnO
disfavoured lone pair repulsion
vs.
anti-Felkin-Anhdisfavoured
OH
HH
H
BnO
≡
BnOH
H OH
H
attractiveinteraction
Felkin-Anh
… with the stereochemical information being further from the reactive centre.
It turns out that the key interaction is an electronic effect and not a steric effect.
The lone pair of electrons on the aldehyde have a disfavoured interaction with the lone pair of electrons on the lower ester group of the tartrate derivative. This means the aldehyde will prefer to be further from this lower ester.
There is also thought to be an attractive interaction … a formyl hydrogen bond as shown in the scheme.
4

OH
OBn
i. OsO4, NMOii. NaIO4
iii. Zn...............
72% (3 steps)85:15 dr
Br OH
OBn
OH
Question 2
This time an example of substrate control (taken from the same synthesis as Q1). Is the reaction under Felkin-Anh control or chelation control? Justify your answer with the appropriate sketch.
5
OH
OBn
i. OsO4, NMOii. NaIO4
iii. Zn...............
72% (3 steps)85:15 dr
Br OH
OBn
OH
Answer
The first step converts the alkene into an aldehyde. The OsO4 causes dihydroxylation of the alkene. The presence of NMO (N-methylmorpholine N-oxide) allows the use of a sub-stoichiometric quantity of osmium (which is expensive and very toxic).
The 1,2-diol is then oxidatively cleaved by treatment with NaIO4. Basically is a practically simpler version of ozonolysis.
6

H
O
O
ZnLn
OH
OBn
O
OLnZnH
H
OLnZnHH
R
H
H
R
H
Hdisfavoured
OLnZn
OO
LnZn O H
R
H
H
ZnLn
H
HR
or
≡
OH
R
OH
H
HOH
OH
HR≡
The second step involves formation of a propargylic zinc reagent. This is the nucleophile. The zinc also coordinates to both the alcohol and the aldehyde. The two possible half-chair conformations are shown on the right. The one with the large substituent in the psuedo-axial position is disfavoured.
Once we have the probably conformation of the chelated substrate the nucleophile can either add from the top or bottom (Si) face. The approach that gives the chair intermediate and not the twist-boat is favoured.
7
BzOO
i. c-hex2BCl, Et3N
ii. EtCHO
94%> 20:1 dr
i. TBSOTfii. xs CH2=CHCH2MgBr
iii. NaIO483%
O OTBS
i. .......
BF3•OEt2ii. HCl
82%10:1 dr
OTMS
OEt
O
OH
O
A C15H20O4
Question 3
Give the structure of intermediate A. Pay special attention to the diastereoselectivity.
Rationalise the observed high selectivity.Lets face it, I give
you the
diastereoselectivity
(as long as you
understand the
second set of
reactions). So this
question is all about
determining why we
observe this
stereochemistry
because this example
does not obey the
normal steric
arguments!
8

BzOO
BzOO
BCl
Cy Cy
HH
:NEt3 BzOO
BCy2
Answer
The first step is formation of the boron enolate. This can give either the E- or Z-boron enolate. It is believed that the bulky chlorodicyclohexyborane exclusively gives the E-enolate as shown.
The boron coordinates the carbonyl trans to the bulky stereocentre or cis to the ethyl group to minimise steric interactions. The cyclohexyl substituents then force the methyl group downwards as shown on the right. Deprotonation then gives the E-enolate.
O
R
BCy2Cl
H C
Hvs.
O
R
BCy2Cl
H C
H
9
OO
B
H
H
Et
L
L
HBzO
HOO
H
H
Et
HBzO
≡ BzOO
Et
OH
incorrect
minimisation of A1,3 strain
Directed delivery of
the aldehyde …
Initially, we may think that the stereocentre on the enolate substrate will control the diastereoselectivity as shown above. Here we determine the conformation of the enaolte by minimising A1,3 strain by having the smalles substituent parallel to the enolate double bond.
The aldehyde then approaches the enolate anti to the bulky benzoate ester as shown above. Unfortunately, this gives the wrong diastereoisomer.
Instead it is thought that there is a formyl hydrogen bond between the ester and aldehyde that directs the approach as shown on the right.
C O BL2CH H
O
O PhO
Et H
10

OO
B
H
H
Et
L
L
H O
OPh
OO
B
H
H
Et
L
L
HBzO
OHO
H
H
Et
H OBz
≡BzOO
Et
OH
If we assume hydrogen bonding directs the delivery of the aldehyde then the Zimmerman-Traxler transition state must look like the diagram on the right.
This predicts the correct diastereomer.
11
BzOO OTBS
MgBr
O OTBSO
MgBrPh
O
MgBr
OH OTBSHO
Ph
OHIOO
O O
O OTBSOI
HOHO OO
O OTBS
The next bit is not part of the question but is how the ketone required for the question 4 is formed.
First the Grignard reagent attacks the ketone and removes the ester ‘protecting group’ on the alcohol. This furnishes a diol. The idol can be cleaved with NaIO4 to gibe the necessary ketone.
12

BzOO
i. c-hex2BCl, Et3N
ii. EtCHO
94%> 20:1 dr
i. TBSOTfii. xs CH2=CHCH2MgBr
iii. NaIO483%
O OTBS
i. .......
BF3•OEt2ii. HCl
82%10:1 dr
OTMS
OEt
O
OH
O
A C15H20O4
Question 4
Using the curly arrow notation and appropriate Newman projections, explain how the ketone on the bottom row is transformed into the lactone.
13
O OTBS
OTMS
OEt
BF3•OEt2OTBSO
EtO
OH
R
H
O
≡≡
R
H
O
OTMS
OEt
R
H
HO
OEt
O
Answer
This is simply an example of substrate control so we need to use the Felkin-Anh model to predict/rationalise the diastereochemical outcome.
First draw the Newman projection. Then rotate the C–C bond so that the substituent of the closest stereocentre is perpendicular to the carbonyl group. The silyl ketene acetal then attacks along the Bürgi-Dunitz angle.
14

OTBSO
EtO
OHHCl
OHO
EtO
OHHCl
O
OH
O
Finally, if we treat this molecule with acid we perform two reactions. The first removes the silyl TBS protecting the group. The second is the acid promoted transesterification or lactonisation.
15
PhSOEt
O NCu
N
OO
Ph Ph
22SbF6 cat. 10%,
92%15:1 endo:exo
> 95% ee
SPh
OEtO SPh
O
OEt
endo exo
Question 5
Welcome to the world of asymmetric catalysis. Explain both the enantio- and diastereoselectivity of the Diels-Alder reaction shown above.
16
SPhO
EtOSPh
O
EtO
HOMO
LUMO
EtO
OSPh
HH
SPh
OEtO
Answer
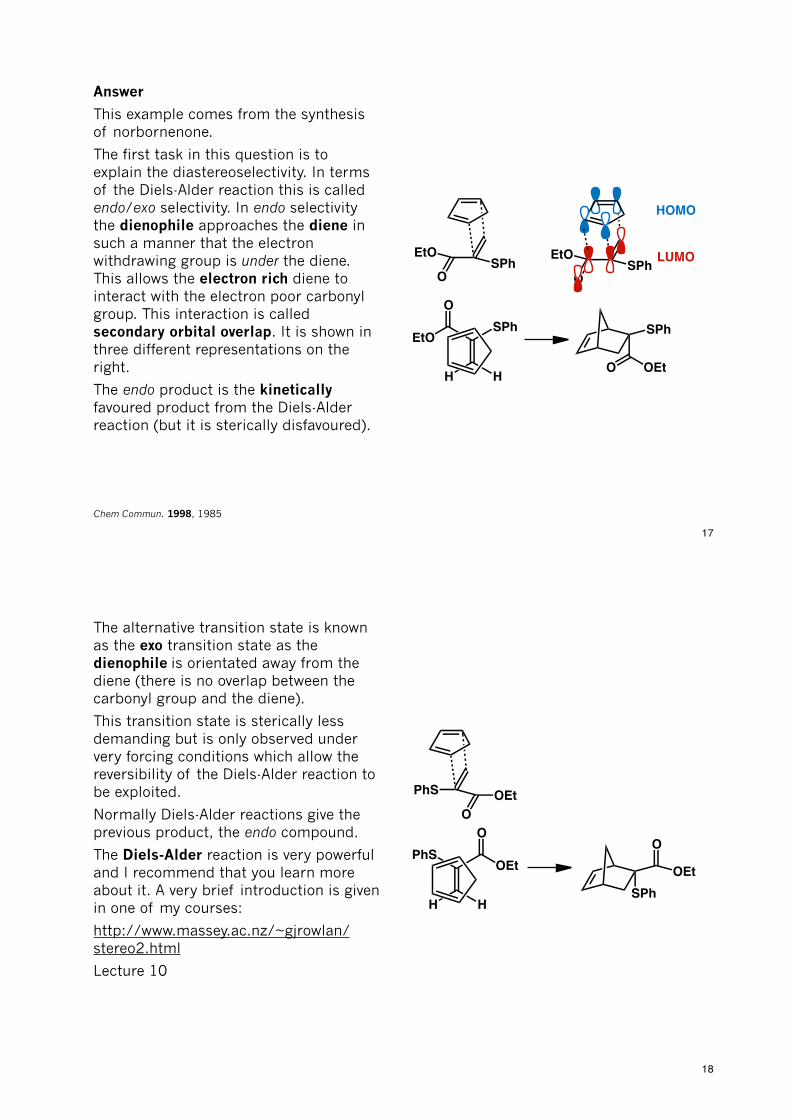
This example comes from the synthesis of norbornenone.
The first task in this question is to explain the diastereoselectivity. In terms of the Diels-Alder reaction this is called endo/exo selectivity. In endo selectivity the dienophile approaches the diene in such a manner that the electron withdrawing group is under the diene. This allows the electron rich diene to interact with the electron poor carbonyl group. This interaction is called secondary orbital overlap. It is shown in three different representations on the right.
The endo product is the kinetically favoured product from the Diels-Alder reaction (but it is sterically disfavoured).
Chem Commun. 1998, 1985
17
PhS
PhS
HHSPh
OOEt
OEt
OO
OEt
The alternative transition state is known as the exo transition state as the dienophile is orientated away from the diene (there is no overlap between the carbonyl group and the diene).
This transition state is sterically less demanding but is only observed under very forcing conditions which allow the reversibility of the Diels-Alder reaction to be exploited.
Normally Diels-Alder reactions give the previous product, the endo compound.
The Diels-Alder reaction is very powerful and I recommend that you learn more about it. A very brief introduction is given in one of my courses:
http://www.massey.ac.nz/~gjrowlan/stereo2.html
Lecture 10
18

NCu
N
OO
Ph PhO
EtO
SPh
H
H
NCu
N
OO
Ph PhO
EtO
SPh
H
H
vs.
disfavoured favoured
O
EtO
SPh
H
H
SPh
OEtO
≡
Once we have determined that the reaction proceeds through the endo transition state we have to consider the influence of the chiral Lewis acid as this will control the enantioselectivity.
A copper(II) catalyst is most likely to be square planar. As a result of the sulfide substituent the substrate (the dienophile) is bidentate and forms a rigid chelate. The orientation of coordination is unimportant as the box ligand is C2 symmetric.
When the substrate coordinates to the copper ...
19
NCu
N
OO
Ph PhO
EtO
SPh
H
H
NCu
N
OO
Ph PhO
EtO
SPh
H
H
vs.
disfavoured favoured
O
EtO
SPh
H
H
SPh
OEtO
≡
... the phenyl substituent of the sulfide is forced away from the ligand as shown in the diagram. This minimises non-bonding interactions between the phenyl group and the oxazoline ring and/or the phenyl on the ligand.
The diene (in this case cyclopentadiene, a very common test substrate) then approaches anti to the phenyl of the sulfide. This avoids the interaction between the diene and the phenyl. In the drawings on the right this means it approaches from the bottom face and gives the molecule shown.
20

PMBOO
OBnN
RuH2N
PhPh
Ts
Cl HCO2H:Et3N
83%95% ee
PMBO
OBn
OHH
Question 6
Here we are going to go through the synthesis of (–)-α-kainic acid, a neuroexcitatory amio acid used in research into the CNS.
First, investigate the Noyori transfer hydrogenation and see if you can rationalise the stereoselectivity.
NH
CO2H
CO2H
21
N
TsN
PhH
HPhRuH
N
TsN
PhH
HPhRu
OH
H
O
H
N
TsN
PhH
HPhRu
OH
H
O
HN
TsN
PhH
HPhRu
HH
H
Answer
The following questions concern the synthesis of kainic acid.
This first step is an example of a Noyori transfer hydrogenation.
This reaction employs formic acid as a source of hydrogen as this is more practical than the use of hydrogen gas. The mechanism on the right shows how this occurs. Hydrogen bonding between acid and amine positions the formic acid next to the reactive centre. Reductive elimination gives a metal-hydride and carbon dioxide.
J. Org. Chem. 2013, 78, 3355
22

N
TsN
PhH
HPhRu
HH
H
O
R
R
N
TsN
PhH
HPhRu
HH
H
OR
R
HH
OR
R
≡
OHH
OBn
OPMB
vs.
One of the advantages of the Noyori diamine catalysts is that they do not require additional functionality within the substrate to coordinate the catalyst and substrate together. This can be achieved by a simple hydrogen bond between the carbonyl and the ammonium group.
There are two possible orientations for the substrate. Calculations (and the selectivity of the reaction) suggest that substrates with unsaturated functionality (alkynes, alkenes & aromatics) form an attractive interaction ...
23
N
TsN
PhH
HPhRu
HH
H
O
R
R
N
TsN
PhH
HPhRu
HH
H
OR
R
HH
OR
R
≡
OHH
OBn
OPMB
vs.
... between the π-system and a hydrogen atom of the aromatic ring of the catalyst.
This is a simplification of the mechanism, which undoubtedly involves the solvent molecules as well as the substrate and catalyst but it is a useful predictive model.
24

PMBO
OBn
OHHRed-Al®
86% PMBOOHH
OBn
Red-Al® = sodium bis(2-
methoxyethoxy)aluminiu
m hydride
Question 7
This to is a stereoselective reaction (although many people forget that alkenes are stereocentres as soon as they move onto asymmetric synthesis). How does reduction with Red-Al® result in formation of the E (trans) alkene exclusively?
As a clue I will just say that the alcohol is important and that the structure of Red-Al® is given on the right.
OAl
H
O HO
ONa
25
PMBO
R2
OHH
OAl
H
O HO
ONa
PMBO
R2
OH Al(OR)2
OAl
H
O HO
O
PMBOAlO
R2
OROR
H
PMBOOHH
OBn
H
H
H
The Red-Al serves two functions in this reaction; it activates the alkyne and acts as the hydride source. It is thought that the alcohol reacts with Red-Al first, the aluminium Lewis acid is then tethered to the substrate and this is essential for stereoselectivity. The electron deficient aluminium activates the electron rich alkyne. A second equivalent of Red-Al attacks the activated alkyne, delivering a hydride and forcing the alkyne to attack the aluminium to give a five-membered ring. The formation of the ring forces the hydride and the aluminium onto opposite faces of the alkene. The C–Al bond can be protonated (or halogenated) with retention of stereochemistry.
26

PMBOOHH
OBn
H
H
HO
O
DCC, DMAP
80% PMBOOBn
O
O
i. LiHMDS, TMSCl, –78°C to rtii. CH2N2
79%
O
O
OBnPMBO
DCC =
dicyclohexylcarbodiimide
DMAP =
dimethylaminopyridine
LiHMDS = lithium
hexamethyldisilazide
[lithium
bis(trimethylsilyl)amide]
Question 8
If you completed last weeks problems then this sequence should offer no challenge (if you haven’t looked at last weeks questions then you can either cheat and look at the answers (thus spoiling the enjoyment) or have a go now).
Reagents are given on the right.
NC
N
N
N
27
PMBOOBn
O
O
PMBOOBn
O OTMS
Answer
The first step is esterification and I will not go through the mechanism of this reaction (if you don’t know it or can’t work it out go through your undergraduate notes, it will in be in there somewhere).
The second step involves stereoselective enolate/silyl ketene acetal formation. It is essential that we control the geometry of the ‘enolate’ as this effects the relative stereochemistry of the two new stereocentres formed in the rearrangement.
Under standard conditions such as this esters invariably give the (OSi)-E-enolate. One simplistic interpretation of the Ireland model says this is due to the fact the ester substituent can rotate out of the way so is smaller than the substituents of the base and hence the enolate substituent will be better accommodated on the same face as the alkoxy substituent.
28

O
HHPMBOOTMS
HOBn
O
HHPMBOOTMS
HOBn
≡
H H
O
O
OBnPMBO
H
H
Once the silyl ketene acetal has formed the molecule is set-up for the Ireland-Claisen rearrangement. This proceeds through the standard Zimmerman-Traxler-like 6-membered transition state. The existing stereocentre controls the conformation of the transition state with the larger substituent adopting the pseudo-equatorial position. All other substituents positions are fixed as they are on alkenes.
The representations on the right show the reaction and the stereochemical outcome.
29
O
O
OBnPMBO
i. DIBALii. Ac2O,
DMAP, Et3Niii. DDQ
73% (3 steps)O
OBnHO
O
DDQ = 2,3-dichloro-5,6-
dicyano-1,4-
benzoquinone
DDQ is a mild oxidant
Simple functional group transformations. I have only included it so that you know how we get to each of the key intermediates (and so that you can practise your synthesis since that is the point of the course!).
O
ONC
NC Cl
Cl
30

O
O
OBnPMBO
i. DIBALii. Ac2O,
DMAP, Et3Niii. DDQ
73% (3 steps)O
OBnHO
O
DDQ = 2,3-dichloro-5,6-
dicyano-1,4-
benzoquinone
DDQ is a mild oxidant
DIBAL reduces the ester to an alcohol.
Acetic anhydride with dimethylaminopyridine as a catalyst and triethylamine as a base forms a new ester (an acetate).
The DDQ deprotects the PMB ether. It acts as an oxidant, accepting a hydride from the PMB group and effectively, oxidising the PMB ether to an aldehyde and releasing the free alcohol. This only works for the electron rich PMB group and not normal benzyl ethers.
O
ONC
NC Cl
Cl
31
AcO
OBnHO
Ti(iOPr)4, (–)-DET, t-BuOOH
82%100% de
AcO
OBnHO
O
Question 9
This is another example of the Sharpless Asymmetric Epoxidation. See if you can determine the product, obviously paying special attention to the chemoselectivity (or is it regioselectivity?) (and not looking at the next slide).
32

AcO
OBnHO
Ti(iOPr)4, (–)-DET, t-BuOOH
82%100% de
AcO
OBnHO
O
Answer
The chemoselectivity is easy to understand. For the Sharpless Asymmetric Epoxidation to proceed it must coordinate to the substrate. It does this through an alcohol group and as a result the SAE invariably occurs at allylic alcohols (although I realise there is an example in my notes that is not allylic). So the allylic alcohol reacts considerably faster than the isolated alkene.
33
OH
"O" D-(–)-DET unnatural isomer
R
You are not expected to draw the postulated transition state. All you need to say is that the Sharpless Asymmetric Epoxidation is predictable and that the (–)-enantiomer of the tartrate ligand would be expected to cause epoxidation from the top face.
The mnemonic that shows the selectivity of each ligand is given on the right. Remember, the alcohol goes in the bottom right corner and the alkene goes backwards into the plane of the page.
34

AcO
OBnHO
O
TBSCl, imidazole
94%AcO
OBnTBSO
O
i. K2CO3, MeOHii. DIAD, Ph3P, (PhO)2P(O)N3
45% (2 steps)N3
OBnTBSO
O
DIAD is the ispropyl
version of DEAD. Due
to its increased
steric bulk it is less
prone to side
reactions that result
in the formation of
hydrazides (attack at
the carbonyl group).
More functional group interconversions. See if you can work out a mechanism for step ii.
As a clue this is an example of the Mitsunobu reaction that was covered in the third year.
35
O N
ON O
OPh3P
OiPr N
ON OiPr
OPh3P
PO
N3PhOPhO
OiPr N
ON OiPr
OPh3P
P(O)(OPh)2
R OH
R O
N3
N3PPh3
R N3
Ph3P=O
The order of events is protection of the primary alcohol with the silyl group.
The acetate is then removed by base-mediated hydrolysis.
The primary alcohol is substituted with an azide possibly by the mechanism given on this page.
First the triphenylphosphine is activated by reaction with DIAD. This creates an anion that can displace the azide anion form DPPA. The alcohol is then activated by reaction with the cationic phosphine. Finally, SN2 displacement gives the product.
36

N3
OBnTBSO
O
i. TBAFii. PPh3, H2O
iii. Boc2O, Et3N
61% (3 steps)N H
Ot-BuO
OH
OBn
OH
Question 10
This is a straight forward, SN2-like, substitution reaction. Suggest a reason why the reaction gives the pyrrolidine (5-membered) ring preferentially over formation of the piperidine ring (6-membered).
Make sure you are happy that this is the only diastereomeric product.
37
R NN
N:PPh3
R NNN
Ph3PR N
N NPPh3
R NPPh3
HO
H
N2
R NH
PPh3
OH
R NH
PPh3
OH
R NH2
P3P=O
Answers
The use of triphenylphosphine and water to reduce an azide to an amine is a classic reaction. For those of you that are interested the mechanism is given above. As is frequently the case the driving force for this reaction is the formation of triphenylphosphine oxide and, of course, nitrogen.
38

HNHBnO
H HOH
H
O
HNHBnO
H HOH
H
HO
The transition state of the cyclisation can be approximated as the diagram above. This is based on a chair-like transition state with two of the three substituents in the pseudo-equatorial position. It is impossible to get all of them equatorial; this is one of the reasons the synthesis of kainic acid is so challenging.
In the cyclisation the amine has to approach the C–O σ* antibonding orbital of the epoxide. This is 180° to the C–O bond. Hopefully the drawing above shows that this should be possible for the formation of the pyrrolidine (5-membered ring) but would be more challenging for the 6-ring.
This transition state also explains the diastereoselectivity.
39
N HOt-BuO
OH
OBn
OH
i. NaIO4ii. NaBH4
iii. Li•naphthalene
71%N H
Ot-BuO
OH
OH
i. Jones reagentii. TFA
62%NH
CO2HH
CO2H
No more stereochemistry to worry about. These are just the ‘end-game’ transformations to get to the final product.
The diol is cleaved with the periodate and the resulting aldehyde is reduced to an alcohol with sodium borohydride.
The lithium naphthalenide is a reductant (source of electrons) and is used to cleave the benzyl ether.
The two primary alcohols are then oxidised to carboxylic acids with Jones reagent. Surprisingly, this does not cleave the Boc group so deprotection is achieved with TFA (trifluoroacetic acid).
40