physicochemical aspects of the contamination of soil and...
TRANSCRIPT

Chemistry for Sustainable Development 11 (2003) 787�801 787
INTRODUCTION
The processes of the interaction of heavymetal (HM) ions with mineral and organic com-ponents of soils and natural waters play animportant part in the genesis of the latter andto a substantial extent determine their prop-erties. The efficiency of these processes variessubstantially depending on the nature of atoxicant and physicochemical parameters ofthe environment in which these processes takeplace [1�3]. Investigation of the kinetics andmechanism of chemical transformation of or-ganic substances in the cultivated soil layerand in surface water taking into account syn-ergistic and antagonistic effects gives the in-
Physicochemical Aspects of the Contaminationof Soil and Hydrosphere with Heavy Metals
VLADISLAV V. GONCHARUK, NELI M. SOBOLEVA and ALEXANDER A. NOSONOVICH
Dumansky Institute of Colloid Chemistry and Chemistry of Water, National Academy of Sciencesof Ukraine, bulvar Vernadskogo 42, Kiev-142, MSP (Ukraine)
E-mail: [email protected]
(Received May 29, 2003)
Abstract
Perspective directions and results of the investigations of physicochemical transformations undergone bymineral and organic components of soil and natural water in the presence of heavy metal ions, which areamong the most widespread pollutants of the biosphere, are considered. The mechanism of the indicatedprocesses is discussed along with the effect of various factors on their kinetics (nature of heavy metals andsubstrate, pH of the medium, action of solar radiation, etc.).
Content
Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 787
Sources of pollution of the environment with heavy metals . . . . . . . . . . . . . . . . . . . . 787
Interaction in the system: soil-forming mineral � heavy metal . . . . . . . . . . . . . . . . . . . 790
Physicochemical processes in the system: humic acids � heavy metal ion . . . . . . . . . . 792
Effect of heavy metals on the transformation of chemical compounds in the biosphere . . . 795
Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 799
formation which is important from the ecolog-ical viewpoint.
SOURCES OF POLLUTION OF THE ENVIRONMENT
WITH HEAVY METALS
Heavy metals enter the environment withdirect discharge of waste water, its leakageand (or) dripping, with river flows, by passingthrough various boundaries between the me-dia: air � water, water � soil, soil � air, etc.The sources of heavy metals in surface watersare also dissolution and destruction (erosion)of rocks and minerals, wastes from metallur-gic, metal-working, oil processing and chemi-cal plants, concentrating mills, mines, etc. With

788 VLADISLAV V. GONCHARUK et al.
the development of engineering, transport andindustry, the relative ecological importance ofeach of these sources changes at all times. Theapplication of waste water and wood wastesin agrotechnics also brings about the threat ofbiospheric pollution since the concentrations ofHM in these wastes are rather high, mg/l: Pb0.30, Zn 0.8, Cr 0.4, Cu 0.35, Cd 0.15 [4].
The level of soil and surface water basincontamination is substantially affected by in-dustry-caused acidification [5]. Acid precipita-tion accelerates desorption of HM ions fromthe surface of soil-forming clay minerals, theirsubstitution by hydrogen ions in humate com-plexes and thus the shift of ion exchange equi-librium toward an increase in the concentra-tion of HM in the contacting aqueous phase,which finally leads to a decrease in the protec-tive properties of soil. An increase in hydro-gen ion content of the soil helps dissolving theoxides of HM present in the soil and promotestheir migration into the surface waters and plantnutrition routes. At a substantially high acidifi-cation degree, also destruction of soil-formingminerals occurs, accompanied by the release ofstructural ions Al and Fe into the contactingmedium [5]. With a high excess of acid, theircomplete destruction is possible, with the for-mation of Al, Fe, Mg salts and silicic acid.
Metal sesquialteral oxides entering the soiland hydrosphere from industry-related sourc-es also take an active part in the formation ofsoil acidity. This process is participated not onlyby the surface groups of oxides but also bycoordination-bound water molecules, the dis-sociation of which results in the formation ofprotons having a substantial effect on the ionexchange processes and on the coordinationinteraction of HM ions with soil components.Additional pollution of natural water with HMis provided through the atmosphere.
Metals in the hydrosphere are concentratedmainly in the surface film, in sediments and inthe biota, while their concentration in watercolumn remains rather small. The levels ofenvironmental pollution with HM and chemi-cal forms in which they are present are essen-tial for ecology of the environment. The HMcontent of surface fresh water varies from sev-eral units to several hundred micrograms perlitre.
The extent of surface water pollution inthe Dnepr region is to a high extent deter-mined by the variations of integral atmosphericpollution index; among HM, prevailing role isplayed by cadmium and manganese. Depend-ing on the properties of media, they are presentin different oxidation degrees as truly solubleand colloid dispersed substances or are presentas the components of mineral and organic sus-pensions thus determining the migration abili-ty of pollutants in the aqueous ecosystem. Withan exception of open sea where impurities arepresent mainly as dissolved substances, thepresence of suspended particles is characteris-tic of the major part of water systems.
The most widespread HM pollutants of soiland natural water are iron, copper, manga-nese, cobalt, which are also biogenic elementsin view of their participation in the synthesisof proteins, enzymes, in photosynthesis, etc.
The oxidation degree of iron is +2 and +3,depending on the oxidation-reduction poten-tial of the medium (Eh). The compounds ofFe(III), which are thermodynamically more sta-ble, occur more frequently that the compoundsof Fe(II). Zinc occupies the second place in sur-face water concentration coming after manga-nese. Its migration occurs mainly in the sus-pended state. Nickel, which is characterized bymedium complex-forming ability, is transportedby river water also in the form of suspensions.Approximately half of Co(II) dissolved in sur-face water is not bound in complexes.
Metals with high oxidation degree occur innatural water mainly in the form of oxygen-containing anions distinguished by poor com-plex-forming ability. Migration in river waterin the form of suspensions is characteristic ofthese HM. Exceptions are chromium and mo-lybdenum, which are mainly (>60 %) trans-ported in dissolved state. Cationic forms of va-nadium and chromium are well absorbed bysuspended particles. Cobalt, molybdenum, va-nadium and chromium are rather stronglybound by the bottom sediments of naturalwater reservoirs, which provides their lowmobility.
The prevailing form of cadmium (II) is metalion (up to 50 %). Due to weak bonding of thistoxicant by the suspended particles, its migra-tion in natural water occurs mainly in the dis-

PHYSICOCHEMICAL ASPECTS OF THE CONTAMINATION OF SOIL AND HYDROSPHERE WITH HEAVY METALS 789
solved state. Mercury (II) migrates in river wa-ter mainly in the form of soluble complexcompounds. The predominant part of Pb(II) ispresent in surface water in suspensions (90�98 %).For the soluble forms of Pb(II), complexingextent is often as high as 98 %.
Among various physicochemical transforma-tions occurring in soil and in natural waterunder the action of industry-related factors(adsorption, ion exchange, chemical and pho-tochemical, oxidation-reduction, catalytic re-actions and so on), migration of HM and theirrelations with the mineral and organic compo-nents of the ecosystem are important. The majorrole in HM migration processes and in theirphysiological activity in natural waters is playedby organic admixtures, the main part of whichis comprised by humic (HA) and fulvic (FA)acids. The latter are dominating in river wa-ter; their content is 20�40 times higher thanthat of HA. In particular, the FA content ofthe water of Kiev water storage pool varieswithin 14�54 mg/dm3, the HA within 0.3�2.0mg/dm3 [8]. The mobility of HM ions is sub-stantially dependent on the amount of organicsubstance in water, its nature, pH redox po-tential of the medium, etc. Fulvic acids andHA possess good complex-forming properties.The complex compounds of HM at definiteconcentrations are toxic for hydrobionts, whiletheir complexes with organic substances do notpossess toxic properties even at high concen-trations, with an exception of some metals.Due to the polydentate character, HM com-plexes with natural organic substances are char-acterized by rather high stability. Humic com-pounds in the systems under consideration formbi-, tri- and even hexadentate coordinationbonds. Not only the presence of complex-form-ing groups in the substrate molecule but alsotheir nature and spatial configuration affectthe strength of complexes. In order to predictthe behaviour of HM during changes in theconditions of the aqueous medium and in an-thropogenic action, important data are pro-vided by the results of investigation of thekinetics and mechanism of complex-formingprocesses under consideration.
As a rule, the high-molecular complexes ofFe(III), Hg(II), Cr(III), Pb(II), Cu(II) accountfor 95�100 % of the total content of dissolved
HM forms; for the case of Zn(II), Cd(II), Co(II),Ni(II), Mn(II), high-molecular complexes ac-count for less than 70 %. For Fe(III) and Pb(II)ions, the formation of complexes with highermolecular mass (>10 thousand) is characteristic.A clearly exhibited trend is observed for theCo(II), Zn(II) and Cu(II) cations to form com-plexes with molecular mass 1 to 10 thousand,for the anions of Ni(II) and Cd(II) <1000. Themetals can be placed in a row according totheir ability to form complexes with FA [7]:Mn(II) < Fe(II) ∼ Cd(II) < Co(II) < Zn(II) <Pb(II) < Ni(II) ~ Fe(III) < Cu(II) < Hg(II).
Within the recent decade, pollution of soilwith HM has got the global character. The ma-jor sources of soil pollution with HM are aero-sol industrial emissions of oxides and sulphidesof elements, automobile emissions, solid in-dustrial wastes, cattle breeding farms, over-flow of rivers polluted with waste water, etc.[4]. The sediments of waste water from the citywaste disposal plants contain elements, mg/kg:Zn 50�8000, Cu 150�4000, Pb 52�600, Cd 3�165, Cr 225�4200, Ni 3�1400 [9].
Under the conditions of anomalously highamount of HM arriving with atmospheric pre-cipitation, an overwhelming part of HM is re-tained mainly in the upper humus layers ofsoil.
A widespread form of HM migration inblack soil is the truly soluble form. In the grayforest soil, metals are transported in suspen-sions. The presence of HM causes changes inthe status of humic substances, structure, pHand biological properties of the soil, whichresults in partial (or complete in some cases)loss of the soil fertility.
As a result of anthropogenic pollution, HMenter soil in the form of oxides and varioussalts, both water-soluble and almost insolubleones (sulphides, sulphates, arsenates, etc.). SomeHM are rather widely spread in nature (iron,titanium, manganese, etc.). The majority ofHM belong to microelements because of theirlow content in the environment (<10 %). Nev-ertheless, the role of many HM is rather highin the soil genesis, especially for its organicconstituent.
Lead, cadmium and mercury are among themost dangerous metal pollutants of soil. Thebackground concentrations of these elements

790 VLADISLAV V. GONCHARUK et al.
in the ground are: 0.5�1.7 mg/kg for Cd andPb, and 0.02�0.50 mg/kg for Hg [4].
The migration of lead in soil occurs in var-ious chelate forms with organic components,and as a result of the action of the groundfauna along the rootage. In this process, hori-zontal migration of lead along the drainagechannels is possible. An insignificant part oflead is transported by the wind with soil parti-cles. The concentration of lead in soil is smallin comparison with its amount in the atmo-sphere [10]. Relative availability of cadmiumfor plants is due to its presence in the soil inion-exchange form. Its migration is strongly af-fected by pH of the medium [11]. Nickel easi-ly migrates into the soil layer; however, therootage of most plants usually creates a barri-er providing safety with respect to this metal.
The stability of complex compounds, whichare formed in the systems HM � surface ofsoil-forming component and HM � metaboliccentre, plays the major role in providing buff-er properties of soil. The HM are placed in thefollowing row according to the degree of inhi-bition of soil respiration: Ag > Hg > Zn > Sn >Sb > Tl > Ni > Pb > Cu > Co > Cd > Bi [12]. Forrather high levels of soil pollution with HM,their inactivation occurs as a result of satura-tion of active centres in metabolites and insoil-forming components.
The content of metal pollutants in largerivers of Ukraine is ten times higher than theMPC; these rivers relate to polluted and heavilypolluted ones according to the internationalstandards. Only in the town near the Dnieperriver, the amount of the accumulated indus-trial wastes with high HM content is, mln t:ash and slag � 28.2, metallurgical slag � 119,wastes of nonferrous metallurgy � 331, wastesof chemical industry � 5.4 [13].
In comparison with the 50-ies of the 20thcentury, the content of manganese in theZaporozhye water reservoir increased by afactor of about 1.7, copper 1.1, zinc 7.8, cobaltnearly 70. The concentration of chromiumreached 100 mg/l, cadmium 0.3 mg/l. Theconcentration of manganese in separate regionsof the water reservoir is 5�370 mg/l, zinc49�157, copper 2�12, lead 40�59 and iron44�75 mg/l.
The bottom sediments of the Zaporozhyewater reservoir turned into a gigantic store ofHM, mg/kg of the dry substance: 131.6 Cr,12.5 Cu, 348.6 Mn, 672.6 Fe. Total content ofthe indicated elements in the sediment layer0�2 cm thick exceeds hundreds tons. In thedeep layers of bottom sediments of some riv-ers the concentration of Zn, Cu and Fe is 2.49,0.077 and 9.36 g/kg of silt, respectively.
As a result of the application of coppercompounds as fertilizers, the level of pollu-tion in the layer 0�20 cm of the black soils ofSouthern Ukraine is 5�7 mg/kg, up to 70 %of this amount being present in the mobileform.
The correlation coefficients characterizing thedependencies of the response of biotic con-stituent of natural ecosystems to the industry-related contamination with HM have ratherlarge values (>70 %) in the region under con-sideration.
INTERACTION IN THE SYSTEM:
SOIL-FORMING MINERAL � HEAVY METAL
Soil is a slow-moving medium, and the pol-lutants migrate in it slower than they do inwater or in air; because of this, the concen-trations of toxicants increase gradually. TheHM are accumulated in soils mainly in the formof insoluble and weakly soluble compoundsand undergo various physicochemical transfor-mations: hydrolysis, sorption, transformationas a result of oxidative-reductive and photo-catalytic reactions, etc. The rate and depth ofthe indicated transformations depend on theproperties of HM and their compounds (sizesof atoms and ions, density, molecular mass,redox potential, concentration of anions, etc.).The interaction of the latter with the HM ionsin some cases helps transferring them into dif-ficultly soluble compounds (phosphates, car-bonates, sulphates) or chelate complexes (HA,organic acids, amino acids, etc.), which affectsthe protective action of the biota. From theecological viewpoint, the distribution of toxi-cants between the liquid and solid phases ofsoil is an important parameter on which theavailability of toxicants for plants and theiraccumulation in the latter depend. In order to

PHYSICOCHEMICAL ASPECTS OF THE CONTAMINATION OF SOIL AND HYDROSPHERE WITH HEAVY METALS 791
TABLE 1
Constants of Langmuir�s equation characterizing adsorption of HM cations on Ca forms of minerals
(numerator: b, ml/µeq, denominator: a, µeq/g)
Mineral Cu2+ Ni2+ Co2+ Mn2+ Cd2+
Kaolinite 0.70/55 0.65/52 0.60/55 0.53/53 0.77/52
Montmorillonite 0.65/10 000 0.60/980 0.50/980 0.45/990 0.42/970
Vermiculite 0.50/1350 0.50/1330 0.45/1360 0.40/1400 0.33/1300
Hydromica 0.65/250 0.65/250 0.55/260 0.53/260 0.50/230
Palygorskite 0.75/150 0.70/145 0.70/150 0.60/145 0.50/140
Fig. 1. EPR spectra of Cu (II) ions adsorbed on the basalsurface of montmorillonite (a) and on the side faces(b): a, b � humidity: 20 and 1.5 %, respectively.
estimate toxicity of HM, one should have thedata on the mechanism of fixation of HM ionsin different components of the soil.
In revealing the regularity of HM distribu-tion in the environment, sorption processes areimportant in the soil composed of the organicand mineral components with relatively largespecific surface. The mineral component is rep-resented mainly by layered silicates and metalhydroxides. According to the ability to enterthe exchange complex of soil-forming miner-als, which is characterized quantitatively bythe value of b constant in Langmuir equation,the investigated cations form the following row:Cu > Ni > Mn > Cd (Table 1) [6].
The character of binding the HM cationsand the efficiency of their adsorption on thebasal and side faces of minerals were investi-gated by means of EPR. Binding of the adsor-bate with the basal face of a mineral is pref-erably of electrostatic character, while at theside faces of aluminosilicate the decisive roleis played by coordination interaction of HMions with active centres of the adsorbate [14].
The EPR spectrum of Cu(II) ions adsorbedon the basal surface of the mineral wasinterpreted on the basis of the flat-squarecoordination of 2+
2 6Cu(H O) with additionaloxygen atoms of the surface bearing negativecharge (Fig. 1, a). The parameters of the EPRspectra (g|| = 2.083 and g⊥= 2.306) correspond tothe values for Cu(II) complexes in which thecharacter of surroundings and the symmetryof polyhedron for the coordination sphere ofthe central ion are similar [15].
The EPR spectrum of Cu(II) ions adsorbedon the side faces of the mineral is a sum ofthe considered and auxiliary spectra of lower
intensity with the parameters: g|| = 2.05 andg⊥= 2.099 (see Fig. 1, b). The obtained data pro-vide the evidence of two different states ofcopper ions; the symmetry of one of them ishigher than that of the other, which agreeswith the results of sorption studies of the na-ture of montmorillonite surface. Sorption ofCu(II) ions is provided by the excessive nega-tive charge of the apical oxygen atoms in oneof the states of the adsorbate with distortedoctahedral coordination. In another state, oc-tahedral complexes of copper (II) are formed;with a decrease in the humidity of the miner-al, they are transformed into the complexeswith flat square configuration with partial sub-stitution of water by negatively charges oxy-gen atoms. With complete dehydration of thesample, a broad signal is observed in the EPRspectra, which corresponds to tetrahedral sur-rounding of copper ions by apical oxygen at-oms of the side faces of the mineral.
The kinetics of HM migration in soils is to agreat extent dependent on relative content ofthe soil-forming aluminosilicate minerals andtheir ion exchange properties. In the case whenthe predominant role in adsorption processesis played by the basal faces of minerals, thebinding of adsorbate to their active centreshas electrostatic character. This promotes mi-

792 VLADISLAV V. GONCHARUK et al.
Scheme 1.
gration of HM, especially in the case of suffi-cient wetting of the soil. Migration occurs as aresult of the gradient of HM ion concentrationand as a consequence of their ion exchangedisplacement by potassium, magnesium andcalcium ions into the contacting phase, whichcauses accumulation of HM along the routesof plant nutrition and worsening of the eco-logical parameters of soil. In the case of sorp-tion of HM ions via coordination mechanism,which takes place under the conditions of se-lective sorption on the side faces of soil-form-ing minerals, the negative influence decreasessubstantially as a result of a decrease in mi-gration ability of the cations under consider-ation.
PHYSICOCHEMICAL PROCESSES IN THE SYSTEM:
HUMIC ACID � HEAVY METAL ION
In spite of low content of natural organicsubstances in soil in comparison with mineralones, they determine the major physicochem-ical and biological properties of the soil andplay an important part in humification, final-ly affecting the fertility of soil. In the compli-cated composition of the organic substancesof soil humus, the central place from the view-point of importance in soil genesis is occupiedby HA and FA, which are weakly dissociatedpolybasic organic acids.
The processes of sorption and complex for-mation in the HM � HA system are importantfor ecology of soil and hydrosphere, becausethey not only determine migration propertiesof toxicants but also are initial stages of manychemical and photochemical transformationswhich are underwent by mineral and organiccomponents of the indicated objects of thebiosphere.
The interaction of HM cations with theorganic components of soil occurs in morecomplicated manner than with the soil-forming
minerals, mainly due to the features ofchemical structure of n atural organicsubstances. Since the nitrogen content of HA isabout an order of magnitude lower than theoxygen content, the major donor centresproviding HM binding on the surface of thesorbent are carboxylic and phenol groups inthe oxygen-containing fragments of HA(Scheme 1).
The pKa values for carboxylic groups are4�5, and for phenol hydroxyls they are 9�10[5]. For pH 3, the pKa of complexes formed byFe(III) and Al(III) ions with fulvinic acids are0.1 and 3.7, respectively. For Mg(II), Mn(II),Zn(II), Cd(II), Pb(II), Ni(II) and Cu(II), pKa forthe same pH vary within the range 1.9�3.3and increase up to 2.1�4.2 with pH increasedto 5 [16].
The divalent cations can be arranged in thefollowing row according to their ability to formcomplexes with FA: Cu(II) = Pb(II) > Hg(II) >Zn(II) = Ni(II) = Co(II) = Mn(II) = Cd(II).
In addition to carboxylic and hydroxylgroups, complex formation is participated alsoby the fragments of HA containing amino-car-boxyl, imine and other groups [19]. As a resultof the interaction of nitrogen atoms of theindicated groups with HM ions, partial displace-ment of oxygen-containing ligands from theinternal coordination sphere of cation occurs.The influence of nitrogen-containing functionalgroups of HA on the sorption of Cu(II) ionsincreases with a decrease in the metal to HAmass ratio. In the case of high HM content,carboxylic and phenol groups play predomi-nant role in sorption. Amino acid fragmentspossess low sorbing ability in comparison withother nitrogen-containing groups [20].
The interaction of HM with HA was inves-tigated with the help of sorption equilibriummethod, IR and EPR [21�23]. The stability con-stants of HM complexes decrease in phase withthe limiting values of sorption determined from
PHYSICOCHEMICAL ASPECTS OF THE CONTAMINATION OF SOIL AND HYDROSPHERE WITH HEAVY METALS 793
TABLE 2
Stability constants Kstab, limiting sorption aand the degree of saturation of the exchange
capacity ϕ of HA with HM ions
Adsorbate Kstab a, µmol/g ϕ, %
Mn2+ 2.2 ²06 182 7.74
Cd2+ 2.2 ²06 190 8.09
Ni2+ 2.4 ²06 205 8.72
Ñî2+ 2.5 ²06 220 9.36
Pb2+ 4.0 ²08 410 17.45
Cu2+ 4.3 ²08 420 17.87
Fe+3 ².2 ²015 635 40.53
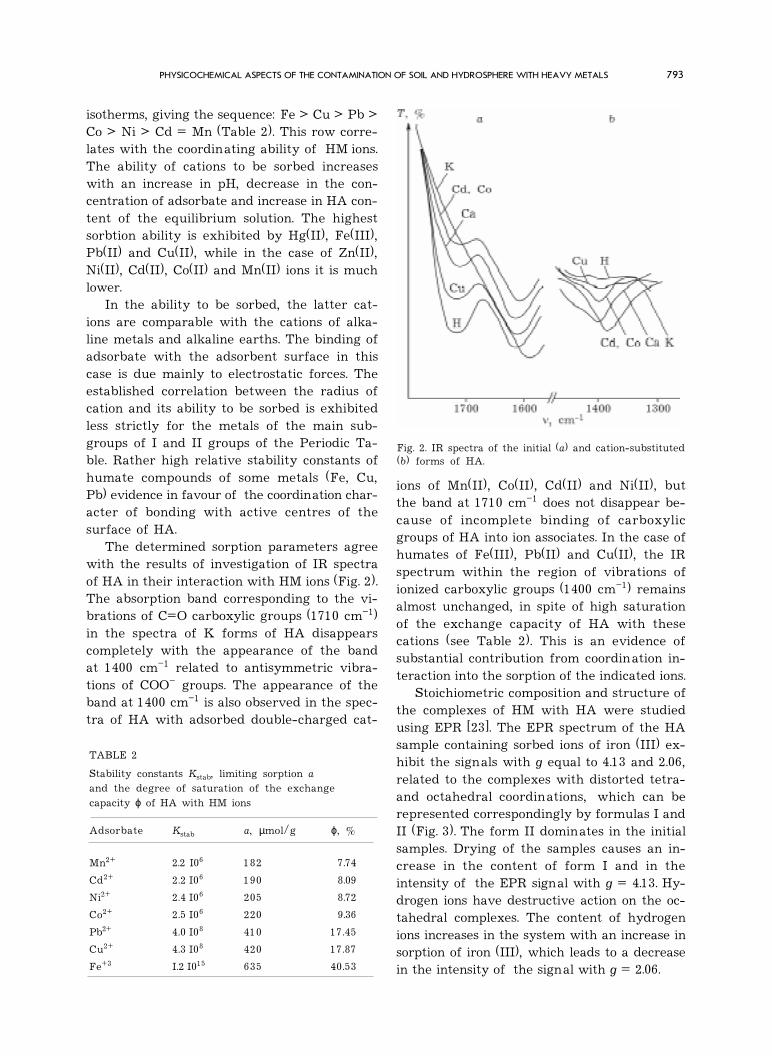
Fig. 2. IR spectra of the initial (a) and cation-substituted(b) forms of HA.
isotherms, giving the sequence: Fe > Cu > Pb >Co > Ni > Cd = Mn (Table 2). This row corre-lates with the coordinating ability of HM ions.The ability of cations to be sorbed increaseswith an increase in pH, decrease in the con-centration of adsorbate and increase in HA con-tent of the equilibrium solution. The highestsorbtion ability is exhibited by Hg(II), Fe(III),Pb(II) and Cu(II), while in the case of Zn(II),Ni(II), Cd(II), Co(II) and Mn(II) ions it is muchlower.
In the ability to be sorbed, the latter cat-ions are comparable with the cations of alka-line metals and alkaline earths. The binding ofadsorbate with the adsorbent surface in thiscase is due mainly to electrostatic forces. Theestablished correlation between the radius ofcation and its ability to be sorbed is exhibitedless strictly for the metals of the main sub-groups of I and II groups of the Periodic Ta-ble. Rather high relative stability constants ofhumate compounds of some metals (Fe, Cu,Pb) evidence in favour of the coordination char-acter of bonding with active centres of thesurface of HA.
The determined sorption parameters agreewith the results of investigation of IR spectraof HA in their interaction with HM ions (Fig. 2).The absorption band corresponding to the vi-brations of C=O carboxylic groups (1710 cm�1)in the spectra of K forms of HA disappearscompletely with the appearance of the bandat 1400 cm�1 related to antisymmetric vibra-tions of COO� groups. The appearance of theband at 1400 cm�1 is also observed in the spec-tra of HA with adsorbed double-charged cat-
ions of Mn(II), Co(II), Cd(II) and Ni(II), butthe band at 1710 cm�1 does not disappear be-cause of incomplete binding of carboxylicgroups of HA into ion associates. In the case ofhumates of Fe(III), Pb(II) and Cu(II), the IRspectrum within the region of vibrations ofionized carboxylic groups (1400 cm�1) remainsalmost unchanged, in spite of high saturationof the exchange capacity of HA with thesecations (see Table 2). This is an evidence ofsubstantial contribution from coordination in-teraction into the sorption of the indicated ions.
Stoichiometric composition and structure ofthe complexes of HM with HA were studiedusing EPR [23]. The EPR spectrum of the HAsample containing sorbed ions of iron (III) ex-hibit the signals with g equal to 4.13 and 2.06,related to the complexes with distorted tetra-and octahedral coordinations, which can berepresented correspondingly by formulas I andII (Fig. 3). The form II dominates in the initialsamples. Drying of the samples causes an in-crease in the content of form I and in theintensity of the EPR signal with g = 4.13. Hy-drogen ions have destructive action on the oc-tahedral complexes. The content of hydrogenions increases in the system with an increase insorption of iron (III), which leads to a decreasein the intensity of the signal with g = 2.06.

794 VLADISLAV V. GONCHARUK et al.
Fig. 3. EPR spectra of HA with the sorbed Fe(III) ions:a � initial HA, b � HA sample containing 90 µmol/gFe(III) and exposed to vacuum at 375 K for 3 h; c � air-dry sample containing 90 µmol/g Fe(III), 5 % H2O.
Fig. 4. EPR spectra of HA with the sorbed Cu(II) ions:a, b � Cu-humate containing 150 and 420 µmol/gof copper, respectively.
The EPR spectra of HA samples with smallcontent of sorbed copper (II) (<20 % of theamount of �COOH groups) contain a signaldescribed by spin Hamiltonian for the axial sym-metry (g|| = 2.332; g⊥= 2.049; A|| = 11.6 kA/m,A⊥= 1.5 kA/m) which corresponds to the octa-hedral complex with two carboxylic groups ofHA and four water molecules in the coordina-tion sphere (Fig. 4, III). When the content ofsorbed copper (II) exceeds ion exchange capac-ity of HA, the EPR spectrum becomes morecomplicated due to superposition of the linecorresponding to form IV. The ions of copper(II) are easily displaced from form IV into thecontacting solution, even by alkaline metals oralkaline earths, while form III is destroyed onlyunder strong acidification (pH < 1). The dataobtained are in accordance with the results ofinvestigations carried out using the sorptionequilibrium method [21].
Unlike the ions of iron (III) and copper (II),manganese (II) cations are weaker sorbed onHA. Independently of the amount of sorbedcations, only one line is observed in the EPRspectrum; it has hyperfine structure withg = 2.06 and a0 = 7.164 kA/m and correspondsto octahedral aqua complexes of manganese(II) interacting with the carboxylic groups ofHA via the electrostatic mechanism. Dehydra-tion of manganese humate is accompanied bya decrease in a0 to 6.2 kA/m, which indicateschanges in the composition of the coordina-tion sphere of Mn(II) ion due to partial substi-tution of water molecules by carboxylic groups.After wetting the sample its EPR parametersrecover.
The coordination interaction of HM ions withthe sorption centres of HA leads to a decreasein the concentration of free radicals and bi-radicals � products of humification of organicsubstances in soil, unlike the cations of thefirst and second groups which have only slighteffect on paramagnetic properties of HA. Withan increase in the content of many HM cations,the intensity of the main signal (g = 2.003)decreases linearly and almost disappears whenits exchange capacity is saturated completely.The coordination interaction of HM ions withparamagnetic centres (PMC) of HA leads todeactivation of free radicals in them and tothe inhibition of redox processes playing thepredominant role in the soil genesis.
A special group of chemical reactions pro-ceeding in soil includes the interaction of clayishminerals and humic substances with HM oxidesand sulphides resulting in the destruction ofsoil components and in an increase in the levelof pollution of biospheric objects with HM ions.
In the investigations of the kinetics andmechanism of dissolution of copper (II) oxideand sulphide in contact with the soil-formingcomponents, we used natural montmorillonite,its H and Ca forms, and HA isolated frombrown coal. Similarly to the interaction of thesoil-forming components with Cu(II) ions in thedissolved state, in the case under considerationwe also observed the formation of complexeswith octahedral and tetrahedral configurations;their relative content in the system varied with
PHYSICOCHEMICAL ASPECTS OF THE CONTAMINATION OF SOIL AND HYDROSPHERE WITH HEAVY METALS 795
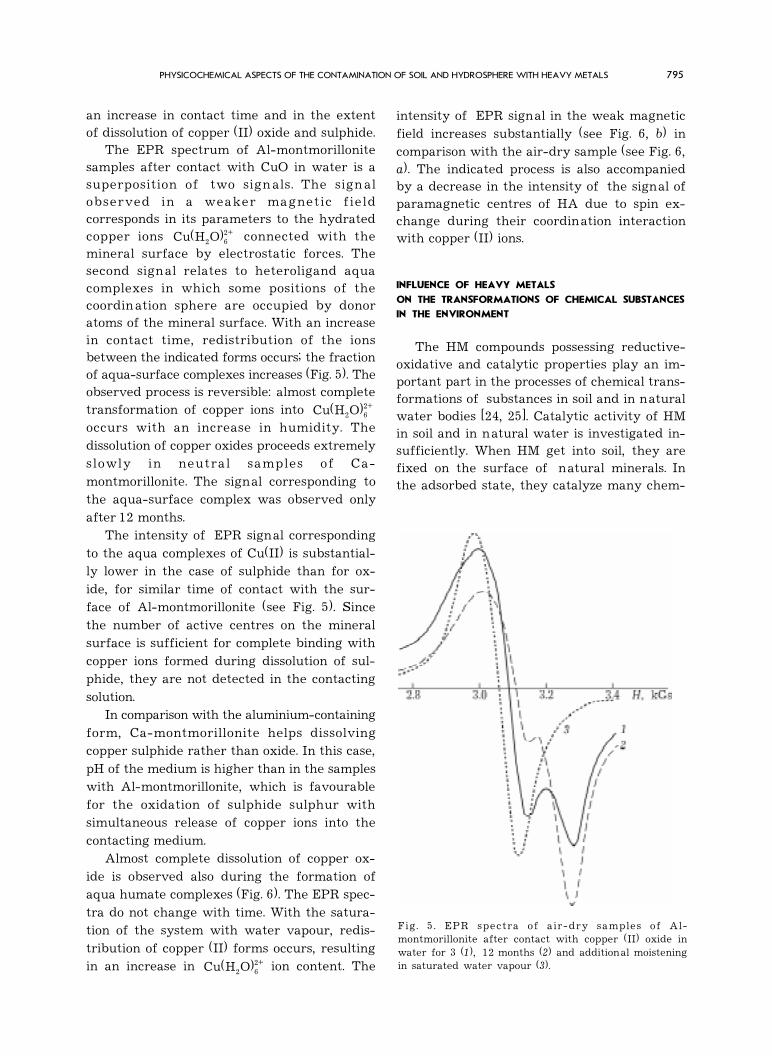
Fig. 5 . EPR spectra of air-dry samples of Al-montmorillonite after contact with copper (II) oxide inwater for 3 (1), 12 months (2) and additional moisteningin saturated water vapour (3).
an increase in contact time and in the extentof dissolution of copper (II) oxide and sulphide.
The EPR spectrum of Al-montmorillonitesamples after contact with CuO in water is asuperposition of two signals. The sign alobserved in a weaker magnetic fieldcorresponds in its parameters to the hydratedcopper ions 2+
2 6Cu(H O) connected with themineral surface by electrostatic forces. Thesecond signal relates to heteroligand aquacomplexes in which some positions of thecoordination sphere are occupied by donoratoms of the mineral surface. With an increasein contact time, redistribution of the ionsbetween the indicated forms occurs; the fractionof aqua-surface complexes increases (Fig. 5). Theobserved process is reversible: almost completetransformation of copper ions into 2+
2 6Cu(H O)occurs with an increase in humidity. Thedissolution of copper oxides proceeds extremelyslowly in neutral samples of Ca-montmorillonite. The signal corresponding tothe aqua-surface complex was observed onlyafter 12 months.
The intensity of EPR signal correspondingto the aqua complexes of Cu(II) is substantial-ly lower in the case of sulphide than for ox-ide, for similar time of contact with the sur-face of Al-montmorillonite (see Fig. 5). Sincethe number of active centres on the mineralsurface is sufficient for complete binding withcopper ions formed during dissolution of sul-phide, they are not detected in the contactingsolution.
In comparison with the aluminium-containingform, Ca-montmorillonite helps dissolvingcopper sulphide rather than oxide. In this case,pH of the medium is higher than in the sampleswith Al-montmorillonite, which is favourablefor the oxidation of sulphide sulphur withsimultaneous release of copper ions into thecontacting medium.
Almost complete dissolution of copper ox-ide is observed also during the formation ofaqua humate complexes (Fig. 6). The EPR spec-tra do not change with time. With the satura-tion of the system with water vapour, redis-tribution of copper (II) forms occurs, resultingin an increase in 2+
2 6Cu(H O) ion content. The
intensity of EPR signal in the weak magneticfield increases substantially (see Fig. 6, b) incomparison with the air-dry sample (see Fig. 6,a). The indicated process is also accompaniedby a decrease in the intensity of the signal ofparamagnetic centres of HA due to spin ex-change during their coordination interactionwith copper (II) ions.
INFLUENCE OF HEAVY METALS
ON THE TRANSFORMATIONS OF CHEMICAL SUBSTANCES
IN THE ENVIRONMENT
The HM compounds possessing reductive-oxidative and catalytic properties play an im-portant part in the processes of chemical trans-formations of substances in soil and in naturalwater bodies [24, 25]. Catalytic activity of HMin soil and in natural water is investigated in-sufficiently. When HM get into soil, they arefixed on the surface of natural minerals. Inthe adsorbed state, they catalyze many chem-

796 VLADISLAV V. GONCHARUK et al.
Fig. 6. EPR spectrum of air-dry HA sample after contactwith copper (II) oxide in water for 12 months (a) and afteradditional moistening in saturated water vapour (b).
ical reactions in soil or in soil solution, in par-ticular destruction processes [26, 27].
The exchange interaction of HM ions withnatural minerals causes substantial enhance-ment of their catalytic activity toward the ox-idation of sulphide sulphur in water. The rateof the catalytic reaction on natural alumino-silicates (kaolinite, palygorskite, clinoptilolite,etc.) the exchange complex of which containsions: Ni(II), Co(II), Fe(III), Mn(II) and Cr(III),is 1�3 orders of magnitude higher than therate of non-catalytic oxidation [28, 29].
Specific catalytic activity of a catalyst de-pends on the nature of the exchange cation. Inthe case of kaolinite and meta-kaolinite, Niform is the most active one, while the activityof other forms is 1�2 orders of magnitudelower. The investigated exchange cations on thesurface of the indicated catalysts can be placedin the following row according to their catalyt-ic activity: Ni(II) > Fe(III) > Co(II) > Mn(II). Forcrystalline kaolinite, a large difference is ob-served between the activity of Ni form andother cation forms, while for meta-kaolinitethis difference is insignificant. This is explainedby changes in the structure of meta-kaoliniteunder thermal treatment and by stronger fix-ation of the exchange ions Fe(III), Co(II), Mn(II)on meta-kaolinite matrix in comparison withNi(II). In the case of clinoptilolite, the activityof the exchange centres decreases in the row:Ni(II) > Cu(II) > Fe(III) > Cr(III) > Co(II) >Mn(II) > Zn(II). Catalytic activity of one andthe same type of the exchange complex onthe matrices of different nature decreases in
the row: crystalline kaolinite > palygorskite >meta-kaolinite > clinoptilolite.
Differences in the process rate for the crys-talline kaolinite and palygorskite in compari-son with meta-kaolinite are explained by small-er exchange capacity of the latter. With largedifference in the exchange capacity betweenclinoptilolite and meta-kaolinite (about 2 or-ders of magnitude), specific reaction rate val-ues are comparable for the indicated minerals.This is explained by the fact that not all theNi(II) ions in the catalyst are catalytically ac-tive. The values of specific catalytic activityfor meta-kaolinite, crystalline kaolinite andpalygorskite are of the same order of magni-tude; however, the value for the crystallinesample is about two times higher. The latter isexplained by changes in the chemical proper-ties of the surface during the formation ofmeta-kaolinite from crystalline kaolinite. Theexchange ions in meta-kaolinite go irrevers-ibly deeper into the pseudo-hexagonal wellsof the basal surface and become non-exchange-able. Exchangeable ions are only those at theside face, while in the crystalline kaolinite boththe ions on the basal surface and the ions ofthe side space are exchangeable. Less rigidlyfixed ions on crystalline kaolinite can pass intothe contacting medium; as a result, they par-ticipate to some extent also in the homogenouscatalytic oxidation of the substrate.
Changes in the catalytic activity are sub-stantially affected by the non-equilibrium en-ergy state of ions fixed on side faces and thebasal surface of aluminosilicates. The catalyticactivity of clinoptilolite is 2 orders of magni-tude lower than that of kaolinite and palygor-skite, while the exchange capacity of theformer is 1�2 orders of magnitude higher.
The kinetics of the oxidation of S2� ionson the indicated aluminosilicates is describedby the equation
w = k[HS�][O2]0.2[Cat]n (1)
where n depends on the nature of the catalyst(Cat).
For the cation-substituted Ni forms of meta-kaolinite and clinoptilolite n = 0.5; for Ni-pa-lygorskite n = 0.9, which is connected with dif-ferent dispersed state of the catalysts. The mech-anism of oxidation of sulphide sulphur in thepresence of dispersed aluminosilicates with a

PHYSICOCHEMICAL ASPECTS OF THE CONTAMINATION OF SOIL AND HYDROSPHERE WITH HEAVY METALS 797
transition metal ion in the exchange complexincludes the formation of a sulphide of transi-tion metal, active complex of oxygen with thelatter, and adsorption of sulphide sulphur onthe active centre of the mineral surface:
S + H2O + MexSyM pH ≈ 7
2�2 3S O + H2O + MexSyM
pH ≥ 8
where Me is the transition metal ion, M is thecatalyst matrix.
In case of definite levels of pollution, HMplay the role of initiators of many photochem-ical reactions proceeding in the biosphere un-der the action of sunlight [30�33]. Among awide range of physicochemical transformationsproceeding in soil and natural water pollutedwith HM, of high ecological importance arethe processes of photooxidative decompositionof organic compounds both those of industry-related origin (pesticides, surfactants, indus-trial and household wastes, etc.) and humiccompounds, which are important componentsof the fertile layer of the ground [34�41]. Themost important functions of soil include redis-tribution of the energy of absorbed sunlightbetween separate components, which providessubstantial influence on the rate and depth ofmany reductive-oxidative, destructive, isomer-ization reactions, etc. As a result of the latter,the soil undergoes substantial physicochemical,morphological and biological changes. The di-rection and efficiency of these processes de-pend on chemical composition and concentra-tions of HM compounds present in the soil,since they possess high photochemical activity.The importance of these processes increasesgradually because of annual growth of ozoneholes in the Earth�s atmosphere and consequentincrease in the intensity of the UV componentof solar radiation reaching the Earth�s surface.
The molecules of organic substances, HMions and complex compounds which can beformed in their interactions can participate asphotoactive components in the processes underconsideration. In the case of sufficiently highcontent of organic matter in the system, whenthe organic matter accounts for the main
→
→
fraction of the absorbed light, the primarystage of the photochemical transformation isdirect photolysis. The interaction of thus formedradicals with oxygen and HM ions causes deeperdestruction of the organic substance.
In the second case, the primary stages ofphotochemical reaction result in the formationof highly active hydroxyl radicals via thephotoreduction of hydrated HM cation.Hydroxyl radicals generated in this processinteract with the substrate, which leads to theaccumulation of various organic radicals in thesolution. Under the action of atmosphericoxygen, the initial form of HM is recovered,with simultaneous formation of superoxideions, organic peroxide radicals, along with theoxidation of radical fragments, which are theproducts of substrate destruction.
The primary photochemical process can alsooccur with the complex compounds of HM withHA and other organics. Phototransfer of elec-tron occurs in this case between the metal ionand the functional group oh HA included inthe coordination sphere of the cation. The re-duced form of metal, organic radicals and ionradicals formed at the first stages participatein further thermal reactions [44�46].
General and specific features of these pro-cesses can be described considering the mecha-nism of photooxidative destruction of HA un-der the action of molecular oxygen in the pres-ence of iron ions (Fig. 7). Depending on thenature of photoactive component, three mainroutes of photoreaction progress in this systemcan be distinguished. The primary stage of thephotoreaction can be direct photolysis of HAunder the action of the shorter-wavelengthcomponent of sunlight, resulting in the for-mation of radical fragments. The latter inter-act with iron ions generating organic cationsand anions, or with molecular oxygen trans-forming into the corresponding peroxide com-pounds. Intermediates formed in the systemparticipate in various thermal reactions withthe components of the ecosystem giving riseto stable products of the substrate transfor-mation. The efficiency of these processes, whichcompete with the recombination of the pri-mary radicals, is determined by the concen-trations of iron ions and oxygen, as well as byphysicochemical parameters of the medium, pH,
MexSyO2HS�M
(2)
(3)

798 VLADISLAV V. GONCHARUK et al.
Fig. 7. Scheme of the mechanism of HA photooxidationin aqueous media containing Fe(II) ions.
reductive-oxidative potential, nature and con-centration of the background electrolyte, etc.
Initiation of the photochemical process canoccur also as a result of the generation ofhydroxyl radicals, which are the products ofphotooxidation of water. Radical fragmentsformed in the interaction of hydroxyl radicalswith HA molecules participate in the above-considered reductive-oxidative processes withoxygen and iron ions.
Along with the indicated routes of photo-oxidative degradation of HA, the system alsoprovides photochemical transformation of thesubstrate in complex compounds which thesubstrate forms with iron ions. In this case,photoreaction is initiated due to the intra-com-plex electron transfer between the coordinat-ing ion and a functional group of the HA ligand.
Iron ions form complexes with many organ-ic compounds of natural and industry-relatedorigin [46]. As a result of the formation ofFe(II) complexes with a number of natural or-ganic substances (tannic acid, gallic acid, py-rogallol, etc.), the stability of the latter to-ward the action of atmospheric oxygen increases.Complex formation and increase in the stabil-ity of the substrate are promoted by an in-crease in pH of the medium and in the organ-ic substance to Fe(II) ratio. The stability of fer-rous ions to oxidation increases, too.
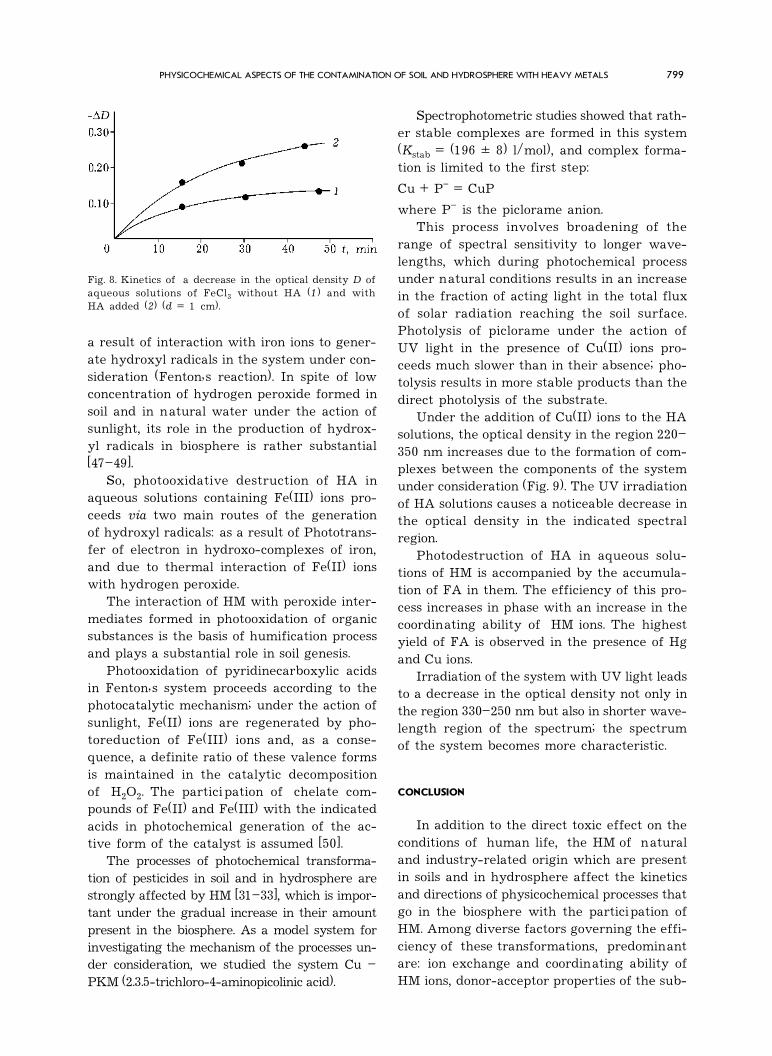
The rate of photoreduction of Fe(III) ionsunder the action of light (>300 nm) in the
inert atmosphere increases noticeably with theintroduction of HA into the solution (Fig. 8).With the mass ratios Fe : HA (1 : 1.5) used inthe experiments, the active light was absorbedalmost completely by humate complexes andfree hydroxo complexes of iron (III).
The efficiency of photooxidation of the sub-strate in the form of complex is substantiallyhigher than in the free state.
Independently of which of the indicatediron complexes play the role of photoactivecomponent of the system, an increase in HAcontent of the solution under irradiation shouldlead to an increase in the quantum effect ofphotoreaction. Really, not only the degree ofcomplex-binding of ferric ions increases, butso does also the rate of deactivation of hy-droxyl radicals formed at the initial stage ofthe photochemical process, when the role ofphotoactive component is played by hydroxo-complex Fe(III) ions [44].
It follows from the comparison between theobtained kinetic data that the rate of photo-oxidative process in solutions containing the ionsof Fe(III) and HA exceeds the sum of the ratesof iron ion photoreduction and substratephotodecomposition for the process carried outin the corresponding single-component systems.This is explained not only by the participationof Fe(III) ions in the reactions with radical productsof the direct and auto-oxidative photolysis ofHA, but also by the interaction of the substratewith the primary hydroxyl radicals.
Due to the effect of atmospheric oxygen,the efficiency of photooxidation of HA byFe(III) ions in soil and in the upper layers ofthe surface waters is rather high. The rate ofoxygen absorption is proportion al to theconcentration of humic substance and non-linearly depends on the intensity of light andon pH. Preliminary esterification of carboxylicgroups decreases the consumption of oxygenas a result of photoreaction by 50 %, whichindicates that the oxidation of HA proceedsthrough the primary stage of charge transferfrom carboxylic groups on ferric ions boundwith them through coordination [47].
In the presence of oxygen, also the accu-mulation of hydrogen peroxide occurs throughthe intermediate formation of superoxide an-ion radicals. Hydrogen peroxide decomposes as
PHYSICOCHEMICAL ASPECTS OF THE CONTAMINATION OF SOIL AND HYDROSPHERE WITH HEAVY METALS 799
Fig. 8. Kinetics of a decrease in the optical density D ofaqueous solutions of FeCl3 without HA (1) and withHA added (2) (d = 1 cm).
a result of interaction with iron ions to gener-ate hydroxyl radicals in the system under con-sideration (Fenton�s reaction). In spite of lowconcentration of hydrogen peroxide formed insoil and in natural water under the action ofsunlight, its role in the production of hydrox-yl radicals in biosphere is rather substantial[47�49].
So, photooxidative destruction of HA inaqueous solutions containing Fe(III) ions pro-ceeds via two main routes of the generationof hydroxyl radicals: as a result of Phototrans-fer of electron in hydroxo-complexes of iron,and due to thermal interaction of Fe(II) ionswith hydrogen peroxide.
The interaction of HM with peroxide inter-mediates formed in photooxidation of organicsubstances is the basis of humification processand plays a substantial role in soil genesis.
Photooxidation of pyridinecarboxylic acidsin Fenton�s system proceeds according to thephotocatalytic mechanism; under the action ofsunlight, Fe(II) ions are regenerated by pho-toreduction of Fe(III) ions and, as a conse-quence, a definite ratio of these valence formsis maintained in the catalytic decompositionof H2O2. The participation of chelate com-pounds of Fe(II) and Fe(III) with the indicatedacids in photochemical generation of the ac-tive form of the catalyst is assumed [50].
The processes of photochemical transforma-tion of pesticides in soil and in hydrosphere arestrongly affected by HM [31�33], which is impor-tant under the gradual increase in their amountpresent in the biosphere. As a model system forinvestigating the mechanism of the processes un-der consideration, we studied the system Cu �PKM (2.3.5-trichloro-4-aminopicolinic acid).
Spectrophotometric studies showed that rath-er stable complexes are formed in this system(Kstab = (196 ± 8) l/mol), and complex forma-tion is limited to the first step:
Cu + P� = CuP
where P� is the piclorame anion.This process involves broadening of the
range of spectral sensitivity to longer wave-lengths, which during photochemical processunder natural conditions results in an increasein the fraction of acting light in the total fluxof solar radiation reaching the soil surface.Photolysis of piclorame under the action ofUV light in the presence of Cu(II) ions pro-ceeds much slower than in their absence; pho-tolysis results in more stable products than thedirect photolysis of the substrate.
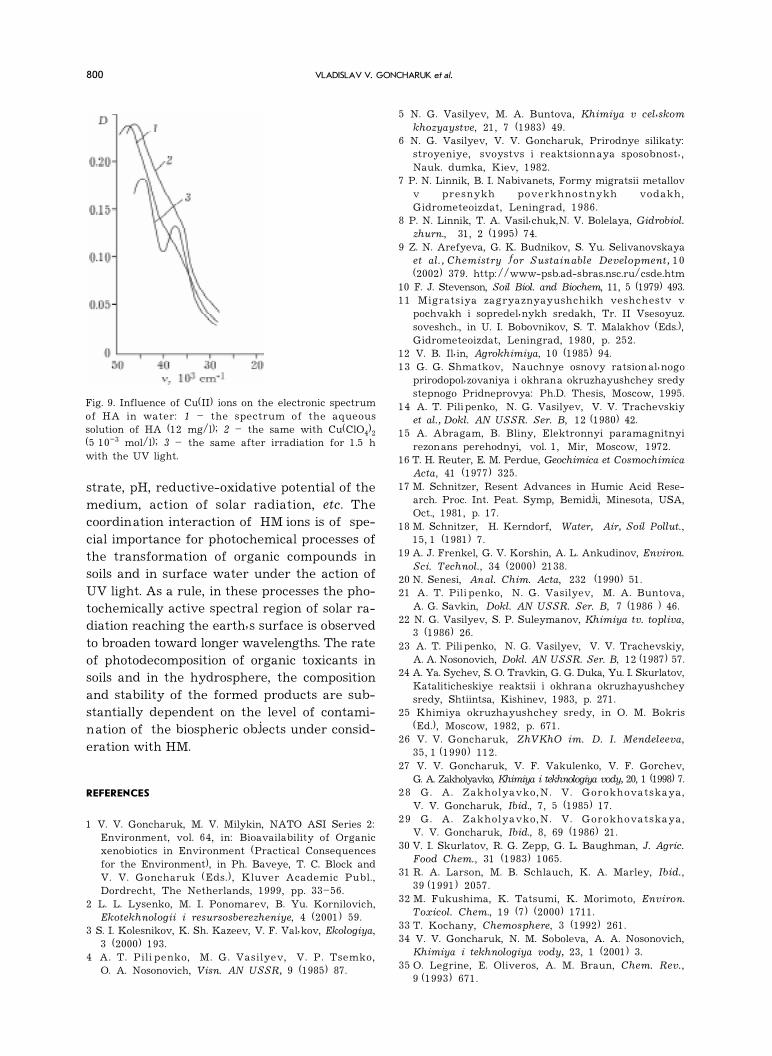
Under the addition of Cu(II) ions to the HAsolutions, the optical density in the region 220�350 nm increases due to the formation of com-plexes between the components of the systemunder consideration (Fig. 9). The UV irradiationof HA solutions causes a noticeable decrease inthe optical density in the indicated spectralregion.
Photodestruction of HA in aqueous solu-tions of HM is accompanied by the accumula-tion of FA in them. The efficiency of this pro-cess increases in phase with an increase in thecoordinating ability of HM ions. The highestyield of FA is observed in the presence of Hgand Cu ions.
Irradiation of the system with UV light leadsto a decrease in the optical density not only inthe region 330�250 nm but also in shorter wave-length region of the spectrum; the spectrumof the system becomes more characteristic.
CONCLUSION
In addition to the direct toxic effect on theconditions of human life, the HM of naturaland industry-related origin which are presentin soils and in hydrosphere affect the kineticsand directions of physicochemical processes thatgo in the biosphere with the participation ofHM. Among diverse factors governing the effi-ciency of these transformations, predominantare: ion exchange and coordinating ability ofHM ions, donor-acceptor properties of the sub-
800 VLADISLAV V. GONCHARUK et al.
strate, pH, reductive-oxidative potential of themedium, action of solar radiation, etc. Thecoordination interaction of HM ions is of spe-cial importance for photochemical processes ofthe transformation of organic compounds insoils and in surface water under the action ofUV light. As a rule, in these processes the pho-tochemically active spectral region of solar ra-diation reaching the earth�s surface is observedto broaden toward longer wavelengths. The rateof photodecomposition of organic toxicants insoils and in the hydrosphere, the compositionand stability of the formed products are sub-stantially dependent on the level of contami-nation of the biospheric objects under consid-eration with HM.
REFERENCES
1 V. V. Goncharuk, M. V. Milykin, NATO ASI Series 2:Environment, vol. 64, in: Bioavailability of Organicxenobiotics in Environment (Practical Consequencesfor the Environment), in Ph. Baveye, T. C. Block andV. V. Goncharuk (Eds.), Kluver Academic Publ.,Dordrecht, The Netherlands, 1999, pp. 33�56.
2 L. L. Lysenko, M. I. Ponomarev, B. Yu. Kornilovich,Ekotekhnologii i resursosberezheniye, 4 (2001) 59.
3 S. I. Kolesnikov, K. Sh. Kazeev, V. F. Val�kov, Ekologiya,3 (2000) 193.
4 A. T. Pili penko, M. G. Vasilyev, V. P. Tsemko,O. A. Nosonovich, Visn. AN USSR, 9 (1985) 87.
5 N. G. Vasilyev, M. A. Buntova, Khimiya v cel�skomkhozyaystve, 21, 7 (1983) 49.
6 N. G. Vasilyev, V. V. Goncharuk, Prirodnye silikaty:stroyeniye, svoystvs i reaktsionnaya sposobnost�,Nauk. dumka, Kiev, 1982.
7 P. N. Linnik, B. I. Nabivanets, Formy migratsii metallovv presnykh poverkhnostnykh vodakh,Gidrometeoizdat, Leningrad, 1986.
8 P. N. Linnik, T. A. Vasil�chuk,N. V. Bolelaya, Gidrobiol.zhurn., 31, 2 (1995) 74.
9 Z. N. Arefyeva, G. K. Budnikov, S. Yu. Selivanovskayaet al., Chemistry for Sustainable Development, 10(2002) 379. http://www-psb.ad-sbras.nsc.ru/csde.htm
10 F. J. Stevenson, Soil Biol. and Biochem, 11, 5 (1979) 493.11 Migratsiya zagryaznyayushchikh veshchestv v
pochvakh i sopredel�nykh sredakh, Tr. II Vsesoyuz.soveshch., in U. I. Bobovnikov, S. T. Malakhov (Eds.),Gidrometeoizdat, Leningrad, 1980, p. 252.
12 V. B. Il�in, Agrokhimiya, 10 (1985) 94.13 G. G. Shmatkov, Nauchnye osnovy ratsional�nogo
prirodopol�zovaniya i okhrana okruzhayushchey sredystepnogo Pridneprovya: Ph.D. Thesis, Moscow, 1995.
14 A. T. Pili penko, N. G. Vasilyev, V. V. Trachevskiyet al., Dokl. AN USSR. Ser. B, 12 (1980) 42.
15 A. Abragam, B. Bliny, Elektronnyi paramagnitnyirezonans perehodnyi, vol. 1, Mir, Moscow, 1972.
16 T. H. Reuter, E. M. Perdue, Geochimica et CosmochimicaActa, 41 (1977) 325.
17 M. Schnitzer, Resent Àdvances in Humic Acid Rese-arch. Proc. Int. Peat. Symp, Bemidji, Minesota, USA,Oñt., 1981, ð. 17.
18 M. Schnitzer, H. Kerndorf, Water, Air, Soil Pollut.,15, 1 (1981) 7.
19 A. J. Frenkel, G. V. Korshin, A. L. Ankudinov, Environ.Sci. Technol., 34 (2000) 2138.
20 N. Senesi, Anal. Chim. Acta, 232 (1990) 51.21 A. T. Pili penko, N. G. Vasilyev, M. A. Buntova,
A. G. Savkin, Dokl. AN USSR. Ser. B, 7 (1986 ) 46.22 N. G. Vasilyev, S. P. Suleymanov, Khimiya tv. topliva,
3 (1986) 26.23 A. T. Pili penko, N. G. Vasilyev, V. V. Trachevskiy,
A. A. Nosonovich, Dokl. AN USSR. Ser. B, 12 (1987) 57.24 A. Ya. Sychev, S. O. Travkin, G. G. Duka, Yu. I. Skurlatov,
Kataliticheskiye reaktsii i okhrana okruzhayushcheysredy, Shtiintsa, Kishinev, 1983, p. 271.
25 Khimiya okruzhayushchey sredy, in O. M. Bokris(Ed.), Moscow, 1982, p. 671.
26 V. V. Goncharuk, ZhVKhO im. D. I. Mendeleeva,35, 1 (1990) 112.
27 V. V. Goncharuk, V. F. Vakulenko, V. F. Gorchev,G. A. Zakholyavko, Khimiya i tekhnologiya vody, 20, 1 (1998) 7.
28 G. A. Zakholyavko,N. V. Gorokhovatskaya,V. V. Goncharuk, Ibid., 7, 5 (1985) 17.
29 G. A. Zakholyavko,N. V. Gorokhovatskaya,V. V. Goncharuk, Ibid., 8, 69 (1986) 21.
30 V. I. Skurlatov, R. G. Zepp, G. L. Baughman, J. Agric.Food Chem., 31 (1983) 1065.
31 R. A. Larson, M. B. Schlauch, K. A. Marley, Ibid.,39 (1991) 2057.
32 M. Fukushima, K. Tatsumi, K. Morimoto, Environ.Toxicol. Chem., 19 (7) (2000) 1711.
33 T. Kochany, Chemosphere, 3 (1992) 261.34 V. V. Goncharuk, N. M. Soboleva, A. A. Nosonovich,
Khimiya i tekhnologiya vody, 23, 1 (2001) 3.35 O. Legrine, E. Oliveros, A. M. Braun, Chem. Rev.,
9 (1993) 671.
Fig. 9. Influence of Cu(II) ions on the electronic spectrumof HA in water: 1 � the spectrum of the aqueoussolution of HA (12 mg/l); 2 � the same with Cu(ClO4)2(5 10�3 mol/l); 3 � the same after irradiation for 1.5 hwith the UV light.

PHYSICOCHEMICAL ASPECTS OF THE CONTAMINATION OF SOIL AND HYDROSPHERE WITH HEAVY METALS 801
36 O. C. Zafiriou, T. Tousot-Dubien, R. G. Zepp, R. G. Zi-ka, Environ. Sci. Technol., 18, 12 (1984) 358A.
37 D. Peterson, D. Watson, W. Wihterlin, Bull. Environ.Contam. Toxicol., 44 (1990) 744.
38 J. Kochany, R. T. Maquire, J. Agric. Food Chem.,42 (1994) 42.
39 L. Cox et al., Chemosphere, 33, 10 (1996) 2057.40 P. Backlund, Ibid., 25, 12 (1992) 1869.41 P. Schmitt-Kopplin, Environ. Sci. Technol . ,
32, 17 (1998) 2531.42 V. V. Goncharuk, N. M. Soboleva, A. A. Nosonovich,
Khimiya i tekhnologiya vody, 20, 6 (1998) 608.43 R. G. Zepp, Hum. Subst. and Role Environ. Rept Dahlem
Worshop Workshof. Berlin, March 29 � April 3, 1987,Chichester etc., 1988, pð. 193�214.
44 A. I. Kryukov, V. P. Sherstuyk, I. I. Dilung, Fotoperenoselektrona i yego prikladnye aspekty, Nauk. dumka,Kiev, 1982.
45 T. L. Theis, P. C. Singer, Environ. Sci. Technol.,8, 6 (1974) 569.
46 C. T. Miles, P. L. Brezonik, Ibid., 15, 9 (1981) 1089.47 W. T. Cooper, D. R. S. Lean, Ibid., 23, 11 (1989) 1425.48 W. T. Cooper, R. G. Zika, R. G. Petasne and J. M. C. Plane,
Ibid., 22, 10 (1988) 1156.49 C.-H. Liao, M. D. Gurol, Ibid., 25, 12 (1995) 3007.50 E. G. Solozhenko, N. M. Soboleva, V. V. Goncharuk,
Ukr. khim. zhurn., 56, 4 (1990) 439.51 A. A. Nosonovich, N. M. Soboleva, N. G. Vasilyev,
V. V. Goncharuk, Khimiya i tekhnologiya vody,9, 4 (1987) 320.