personalizing oncology with genomics
TRANSCRIPT

White paper
Connectinginsights
Betteroutcomes
Personalizing oncologywith genomicsNext-generation sequencing will transform oncology clinical trials and treatments for cancer.
Philip Breitfeld, M.D., Vice President and Global Head, Therapeutic Centers of Excellence, QuintilesJeff Fitzgerald, Director, Personalized Medicine Integration, Quintiles

2 | www.quintiles.com
Introduction 3
Emerging precision 4
Tools and techniques 5
Exploring genomic features with NGS 7
NGS platform peculiarities 8
NGS in clinical trials 9
Transcriptome analysis – expression profiling 9
Tomorrow’s technology today 10
About the authors 12
Table of contents

3 | www.quintiles.com
By 2025, sophisticated genomics-based tools will have transformed cancer care. Imagine a 60-year-old patient with non-small cell lung cancer (NSCLC). Care will start with a routine tumor biopsy and assessment, based on a standard pathologic examination plus molecular diagnostics, including analysis of single nucleotide variation, translocations, copy-number variations, methylation, transcriptome expression and pathway analysis. Within five days, the oncologist will receive a report of the findings along with treatment recommendations, such as a combination of tyrosine kinase inhibitors and monoclonal antibodies, which will be developed in consideration of publicly available information about treatment outcomes in patients with similar molecular profiles. The report will also include a list of relevant clinical trials. As a result of this three-step process – obtaining molecular data, matching it to publicly available patient-level data sources and translation into a clinical action plan – the patient might live another 35 years. The question is: What will it take to achieve this transformation in oncology?
Before considering this future, we look back at the relationship between cancer agents and potential genomic targets (Figure 1). From 1960–1990, therapies consisted of what oncologists call the sledgehammer approach. The compounds disrupted basic chromosomal mechanisms, such as the replication of DNA. These all-purpose compounds, such as alkylating agents, attacked many tumor types, but they also created adverse side effects because of damage to healthy tissue.
The silver-bullet era emerged during the last decade of the 20th century. For the first time, compounds were aimed at specific molecular targets for particular cancers. For example, trastuzumab targets HER2 receptors in breast cancer, and rituximab targets CD20 proteins in B-cell malignancy. Targeting specific tumor types limits the range of application of these treatments, but also reduces off-tumor effects, which decreases the side effects in comparison with sledgehammer medications.
A second stage of the silver-bullet era truly leveraged genomic alterations. Imatinib, for instance, attacks chronic myelogenous leukemia driven by the BCR-ABL translocation, and gefitinib battles NSCLC sparked by mutations in the epidermal growth factor receptor (EGFR) gene.
In addition, crizotinib provided impressive outcomes in NSCLC patients with the EML4-ALK fusion, which created an opportunity to develop this agent for a sub-population of patients. This provided a direct and fast route to regulatory approval. Despite the initial excitement over the efficacy of these silver-bullet treatments, in some cases resistance has emerged and patients have relapsed, even those who had quickly experienced complete responses.
Sophisticated genomics-based tools will transform cancer care in the coming decade. This paper investigates what will drive this transformation in oncology.
Figure 1 Evolution of cancer drug development paradigm
> alkylating agents> anthracyclines> taxanes
> trastuzumab> rituximab
> imatinib> gefitinib
> RAF and MEK inhibition (melanoma)> c-MET and EGFR inhibition (NSCLC)
Sledgehammer
Targeting oftumor markers
Targeting ofdriver genomic
alterations
Mitigatingdrug resistance
Silver Bullet Emerging Precision
Cytotoxicchemotherapy

4 | www.quintiles.com
These agents take out one target, and that will not necessarily cure a population of patients, due to resistance mechanisms offered by the biologic complexity of cancer (Figure 2). As tumors are heterogeneous in nature, one silver-bullet agent will not destroy all tumor cells. Moreover, a single cancer cell makes use of multiple molecular pathways to sustain replication, which can sometimes circumvent the impact of a single treatment. In addition, one patient might have multiple tumors that vary in the dominant pathobiology. Beyond those challenges, tumors are inherently genetically unstable, making them prone to replication errors and mutations. This leads to resistant clones and an evolution of the disease. These issues of complexity make it difficult to predict the outcome of inhibiting just one part of a cancer ecosystem.
Emerging precision
In 2013, Nature published the Cancer Genome Atlas. This information provides drug developers with clues for aiming a combination of agents at more than one target or pathway in one tumor. It also helps oncologists select the patients most likely to respond to a specific treatment. These advances pave the way for an era of emerging precision. The overall goal is to develop a medication that attacks the right target, or targets, in the right disease, in the right patient, at the right dose to obtain the desired outcome. That requires an understanding of cancer’s molecular complexity, which can be gained through genomic techniques.
In a 2011 article in Cell, Doug Hannahan of the Swiss Institute for Experimental Cancer Research and Bob Weinberg of MIT stated that cancer arises from six processes: “sustaining proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, and activating invasion and metastasis.” They also pointed out the fundamental cause of cancer: genome instability. A complex molecular circuit drives each of these processes, and the circuits can interact. This suggests that targeting one step in this complex ecosystem of cancer might be naive. It also reveals the difficulty in predicting what will happen when even one step is inhibited.
As previously noted, activating EGFR mutations can drive NSCLC in some patients. In those patients, inhibiting EGFR leads to good initial clinical outcomes, but the disease often recurs. At least one mechanism for the acquired resistance comes from amplifying the expression of the c-MET gene. This provides the rationale to create a cocktail therapy that inhibits EGFR and c-MET.
As a second example, RAF mutations can be observed in many cases of melanoma. In 2013, a team of scientists from the Royal Marsden National Health Service Foundation Trust and the Institute of Cancer Research in the UK reported in the Journal of Clinical Oncology that therapies blocking RAF can activate a downstream portion of the pathway, which involves the MEK gene. Consequently, a combination that inhibits RAF and MEK might be effective in some patients.
The overall goal in the era of emerging precision is to develop a cancer medication that attacks the right target, or targets, in the right disease, in the right patient, at the right dose to obtain the desired outcome.
Figure 2 The complexity of circuits, individual tumors and populations
Z
Y
Z-coordinate
X-coordinate
Y-coordinate
X Multiple individuals
Multiple circuits/networks in a single cancer cell
Multiple cancer cells in a single tumor in an individual
5 | www.quintiles.com
These examples show that the molecular complexity of cancer demands an understanding of the underlying genomics. These challenges in cancer and drug development require new technologies to guide the efficient and economical development of safe and effective new medicines. Next-generation sequencing (NGS) technologies are already playing a role in providing that broader understanding and helping medical researchers and drug developers address the challenges posed by the molecular complexity of cancer.
Tools and techniques
The diverse population of tumor types and the complex molecular mechanisms involved in cancer drive the need for characterized biomarkers, which can distinguish tumors molecularly. These biomarkers must be reliable and easily accessible. As an example, every cell contains genomic DNA and transcribed RNA, making nucleic acids stable, easy-to-obtain sources from available tissue. NGS can be used to identify and routinely analyze nucleic acid-based biomarkers.
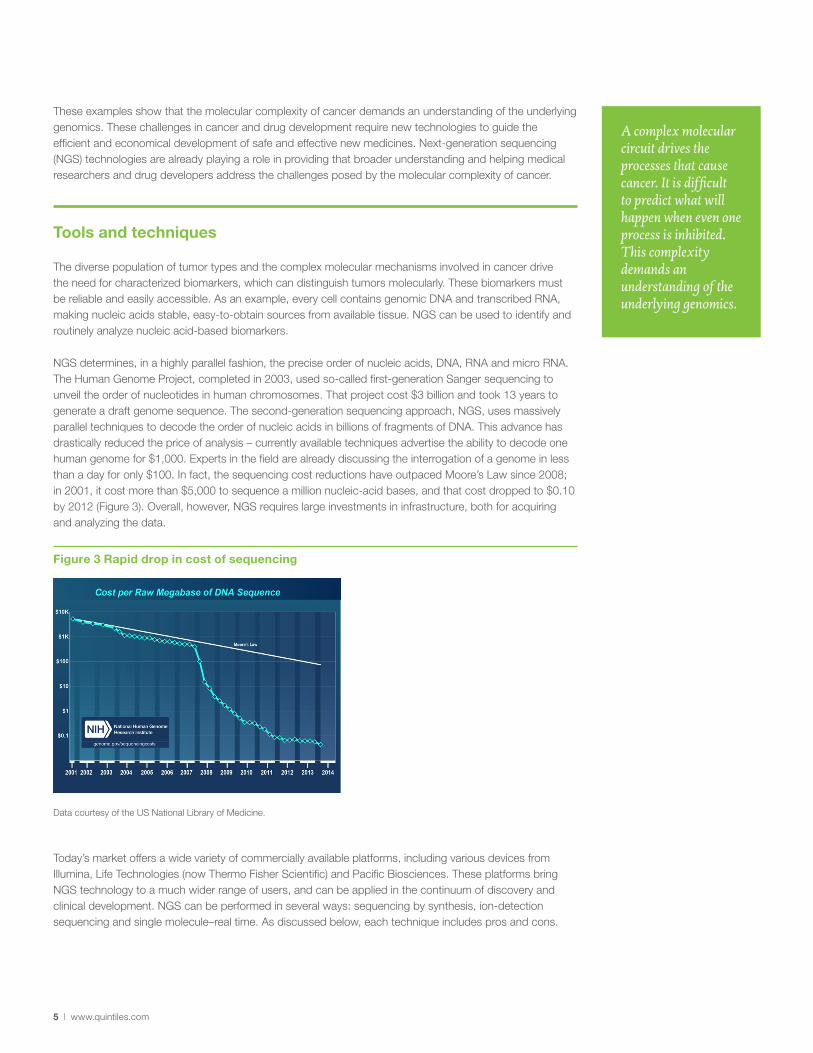
NGS determines, in a highly parallel fashion, the precise order of nucleic acids, DNA, RNA and micro RNA. The Human Genome Project, completed in 2003, used so-called first-generation Sanger sequencing to unveil the order of nucleotides in human chromosomes. That project cost $3 billion and took 13 years to generate a draft genome sequence. The second-generation sequencing approach, NGS, uses massively parallel techniques to decode the order of nucleic acids in billions of fragments of DNA. This advance has drastically reduced the price of analysis – currently available techniques advertise the ability to decode one human genome for $1,000. Experts in the field are already discussing the interrogation of a genome in less than a day for only $100. In fact, the sequencing cost reductions have outpaced Moore’s Law since 2008; in 2001, it cost more than $5,000 to sequence a million nucleic-acid bases, and that cost dropped to $0.10 by 2012 (Figure 3). Overall, however, NGS requires large investments in infrastructure, both for acquiring and analyzing the data.
Today’s market offers a wide variety of commercially available platforms, including various devices from Illumina, Life Technologies (now Thermo Fisher Scientific) and Pacific Biosciences. These platforms bring NGS technology to a much wider range of users, and can be applied in the continuum of discovery and clinical development. NGS can be performed in several ways: sequencing by synthesis, ion-detection sequencing and single molecule–real time. As discussed below, each technique includes pros and cons.
A complex molecular circuit drives the processes that cause cancer. It is difficult to predict what will happen when even one process is inhibited. This complexity demands an understanding of the underlying genomics.
Figure 3 Rapid drop in cost of sequencing
Data courtesy of the US National Library of Medicine.
6 | www.quintiles.com
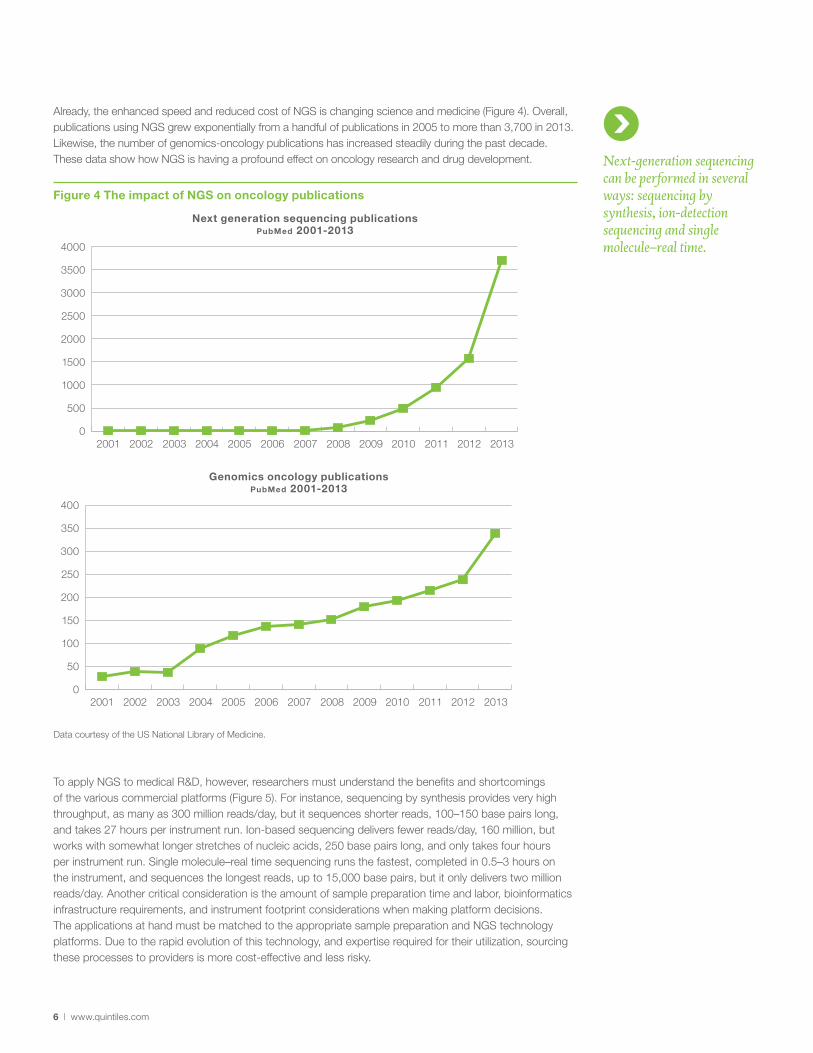
Already, the enhanced speed and reduced cost of NGS is changing science and medicine (Figure 4). Overall, publications using NGS grew exponentially from a handful of publications in 2005 to more than 3,700 in 2013. Likewise, the number of genomics-oncology publications has increased steadily during the past decade. These data show how NGS is having a profound effect on oncology research and drug development.
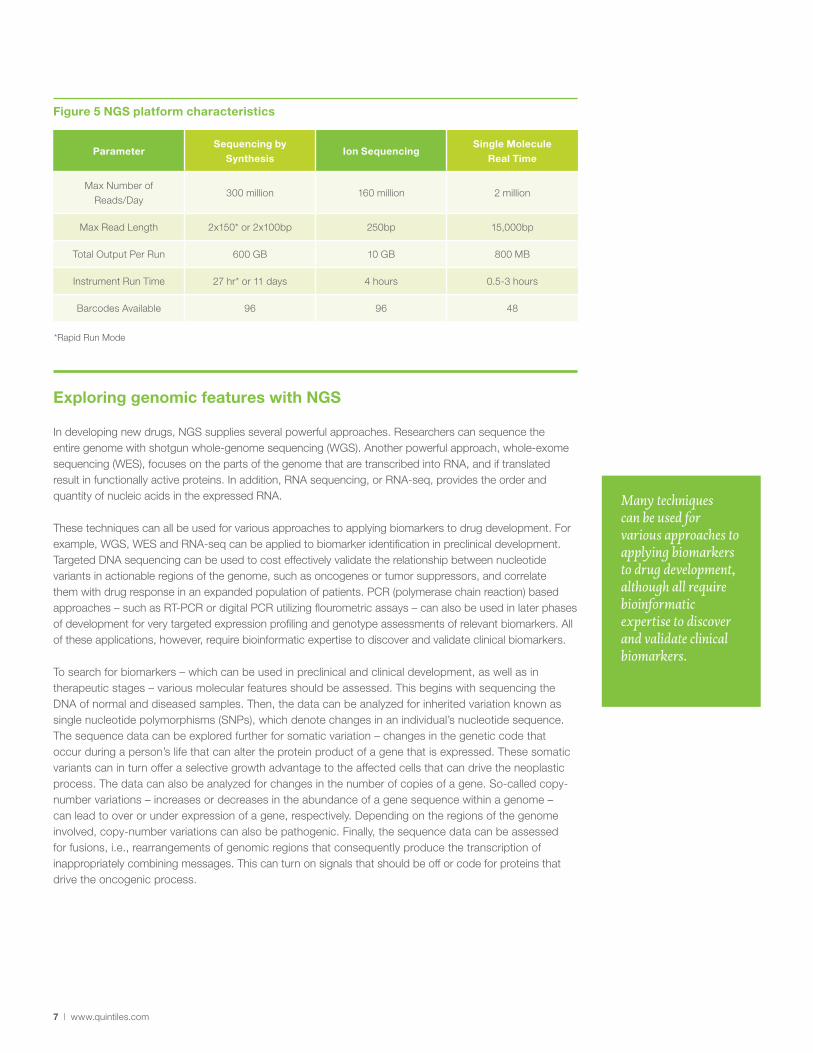
To apply NGS to medical R&D, however, researchers must understand the benefits and shortcomings of the various commercial platforms (Figure 5). For instance, sequencing by synthesis provides very high throughput, as many as 300 million reads/day, but it sequences shorter reads, 100–150 base pairs long, and takes 27 hours per instrument run. Ion-based sequencing delivers fewer reads/day, 160 million, but works with somewhat longer stretches of nucleic acids, 250 base pairs long, and only takes four hours per instrument run. Single molecule–real time sequencing runs the fastest, completed in 0.5–3 hours on the instrument, and sequences the longest reads, up to 15,000 base pairs, but it only delivers two million reads/day. Another critical consideration is the amount of sample preparation time and labor, bioinformatics infrastructure requirements, and instrument footprint considerations when making platform decisions. The applications at hand must be matched to the appropriate sample preparation and NGS technology platforms. Due to the rapid evolution of this technology, and expertise required for their utilization, sourcing these processes to providers is more cost-effective and less risky.
Next-generation sequencing can be performed in several ways: sequencing by synthesis, ion-detection sequencing and single molecule–real time.
Figure 4 The impact of NGS on oncology publications
0
500
1000
1500
2000
2500
3000
3500
4000
2001 2002 2003 2004 2005 2006 2007 2008 2009 2010 2011 2012 2013
Next generation sequencing publicationsPubMed 2001-2013
0
50
100
150
200
250
300
350
400
2001 2002 2003 2004 2005 2006 2007 2008 2009 2010 2011 2012 2013
Genomics oncology publicationsPubMed 2001-2013
Data courtesy of the US National Library of Medicine.
7 | www.quintiles.com
Exploring genomic features with NGS
In developing new drugs, NGS supplies several powerful approaches. Researchers can sequence the entire genome with shotgun whole-genome sequencing (WGS). Another powerful approach, whole-exome sequencing (WES), focuses on the parts of the genome that are transcribed into RNA, and if translated result in functionally active proteins. In addition, RNA sequencing, or RNA-seq, provides the order and quantity of nucleic acids in the expressed RNA.
These techniques can all be used for various approaches to applying biomarkers to drug development. For example, WGS, WES and RNA-seq can be applied to biomarker identification in preclinical development. Targeted DNA sequencing can be used to cost effectively validate the relationship between nucleotide variants in actionable regions of the genome, such as oncogenes or tumor suppressors, and correlate them with drug response in an expanded population of patients. PCR (polymerase chain reaction) based approaches – such as RT-PCR or digital PCR utilizing flourometric assays – can also be used in later phases of development for very targeted expression profiling and genotype assessments of relevant biomarkers. All of these applications, however, require bioinformatic expertise to discover and validate clinical biomarkers.
To search for biomarkers – which can be used in preclinical and clinical development, as well as in therapeutic stages – various molecular features should be assessed. This begins with sequencing the DNA of normal and diseased samples. Then, the data can be analyzed for inherited variation known as single nucleotide polymorphisms (SNPs), which denote changes in an individual’s nucleotide sequence. The sequence data can be explored further for somatic variation – changes in the genetic code that occur during a person’s life that can alter the protein product of a gene that is expressed. These somatic variants can in turn offer a selective growth advantage to the affected cells that can drive the neoplastic process. The data can also be analyzed for changes in the number of copies of a gene. So-called copy-number variations – increases or decreases in the abundance of a gene sequence within a genome – can lead to over or under expression of a gene, respectively. Depending on the regions of the genome involved, copy-number variations can also be pathogenic. Finally, the sequence data can be assessed for fusions, i.e., rearrangements of genomic regions that consequently produce the transcription of inappropriately combining messages. This can turn on signals that should be off or code for proteins that drive the oncogenic process.
Many techniques can be used for various approaches to applying biomarkers to drug development, although all require bioinformatic expertise to discover and validate clinical biomarkers.
Figure 5 NGS platform characteristics
ParameterSequencing by
SynthesisIon Sequencing
Single Molecule Real Time
Max Number of Reads/Day
300 million 160 million 2 million
Max Read Length 2x150* or 2x100bp 250bp 15,000bp
Total Output Per Run 600 GB 10 GB 800 MB
Instrument Run Time 27 hr* or 11 days 4 hours 0.5-3 hours
Barcodes Available 96 96 48
*Rapid Run Mode

8 | www.quintiles.com
As mentioned previously, heterogeneity in a tumor can cause resistance to therapy. Often, a single tumor consists of subpopulations with different sequence variants, which can be determined with NGS. In a so-called polyclonal tumor, one subclone might include common somatic mutations, meaning that they exist in high frequency, and another subclone could include those mutations plus subclone-specific ones, which appear at a low frequency. With NGS and a reference “normal” sequence, the sequence of the common and subclone-specific mutations that can contribute to therapeutic resistance can be simultaneously discovered. Importantly, NGS can pick out rare somatic events because of the extraordinary number of measurements that can be derived from this technology’s output.
NGS platform peculiarities
When dealing with rare somatic events, however, errors must also be considered. All sequencing platforms and sample preparation procedures involved can lead to measurement errors. This can make it difficult to distinguish between a rare subclone-specific mutation and a sequence error. Consequently, changes in sequence data should be validated before moving ahead. For example, an orthogonal sequencing technique can be used to determine if the subclone-specific mutations still appear in the data. Similarly, bioinformatic methods have been developed to assess specific base call changes and can be used to distinguish between an error and a rare mutation. To minimize false positives, researchers should work with providers that are very familiar with the various NGS platforms and their error profiles, and have tools and capabilities available to perform validation experiments.
WGS, for example, is capable of supplying the sequence of the entire genome but the so-called depth – amount of redundant measurements of an individual sample’s nucleotide base composition – is relatively low compared with targeted methods. That provides fewer reads to detect rare events (or heterogeneity) in a tumor, thereby potentially leading to a higher risk that rare variation within a sample may go undetected.
In addition, different forms of NGS work better in different stages of drug development. WES isolates regions of the genome that get transcribed and eventually translated into proteins. Variants found in these regions are more likely to be pathogenic, which makes this a useful tool for zeroing in on potential drug targets. Further into the development cycle, researchers might choose PCR enrichment to very selectively amplify regions of interest, because this technology provides a high level of focused sequencing depth of coverage. This redundancy in measurement yields the capability to detect more rare events in a heterogeneous population of cells.
As a project focuses more closely on a specific region of the genome, researchers need equally focused tools. This can lead to switching from using WGS to WES and, ultimately, to studying targeted genomic loci with PCR panels. Likewise, increasing sample sizes contribute to the need to progress from one technique to another. For example, many more samples are likely to be used at the stage of analyzing targeted gene loci than when looking at the entire genome.
Each of these technologies, however, comes with advantages and disadvantages. On the upside, WGS is comprehensive and unbiased, provides genome structure, and can be used in discovery without requiring a panel; on the downside, WGS has limited sensitivity to detect rare variants within a sample, can expose many variants of unknown significance and can provide an overload of data, resulting in high costs for data storage and analysis.
WES is also comprehensive, provides some genome structure and panels are available off the shelf, but it too is limited in its sensitivity to detect rare events and can be biased in coverage (some regions of the target sequence are well measured while others are underrepresented), which leaves gaps in the data. When using PCR panels, the results are very specific for particular regions of interest and the results are very actionable; however, this technology includes shortcomings, such as its limited biological breadth, limited ability to detect rearrangements and the fact that primers can fail on some samples due to heterogeneity of sequence in the regions in which PCR is primed.
Next-generation sequencing can pick out rare somatic events because of the extraordinary number of measurements that can be derived from this technology’s output.
As a project focuses more closely on a specific region of the genome, researchers need equally focused tools.
9 | www.quintiles.com
NGS in clinical trials
As an added benefit, genes for ADME – absorption, distribution, metabolism and excretion – can be screened in parallel with predictive biomarkers. In doing so, however, researchers must obtain informed consent for genetic testing. Such a study should also collect samples from everyone; then, if a safety signal arises, the data for every patient can be analyzed retrospectively without sampling bias.
To use NGS in a clinical trial, other factors must also be considered. In preparation for a study, researchers should determine the format of samples. For instance, formalin-fixed, paraffin-embedded (FFPE) samples pose additional technical challenges (base modifications due to formalin fixation) versus fresh-frozen tissues, which present logistical sample-handling issues. For FFPE samples, the NGS technology must provide a high depth of sequencing coverage, keeping in mind that the specificity of the results is proportional to the error rate of the NGS method. The study preparation should also estimate the turnaround time needed for prospective and retrospective studies. Prospective trials, for instance, need results quickly to plan the next steps in a patient’s treatment in the context of a clinical trial. To validate the results, orthogonal methods are also recommended. A collection of other concerns must be addressed. Will enrichment methods be needed for regions of interest, such as oncogenes or tumor suppressors? The technology and team for computing and bioinformatics plus data interpretation and reporting must be assembled. The biomarkers selected must be assessed for analytical and clinical validity. If one is considering an FDA-approved diagnostic test, the process must also provide clinical utility. Regulatory standards must be followed, and steps toward development of an in vitro diagnostic (IVD) need to be considered. Many of these steps require collaboration with experts in these fields.
Transcriptome analysis – expression profiling
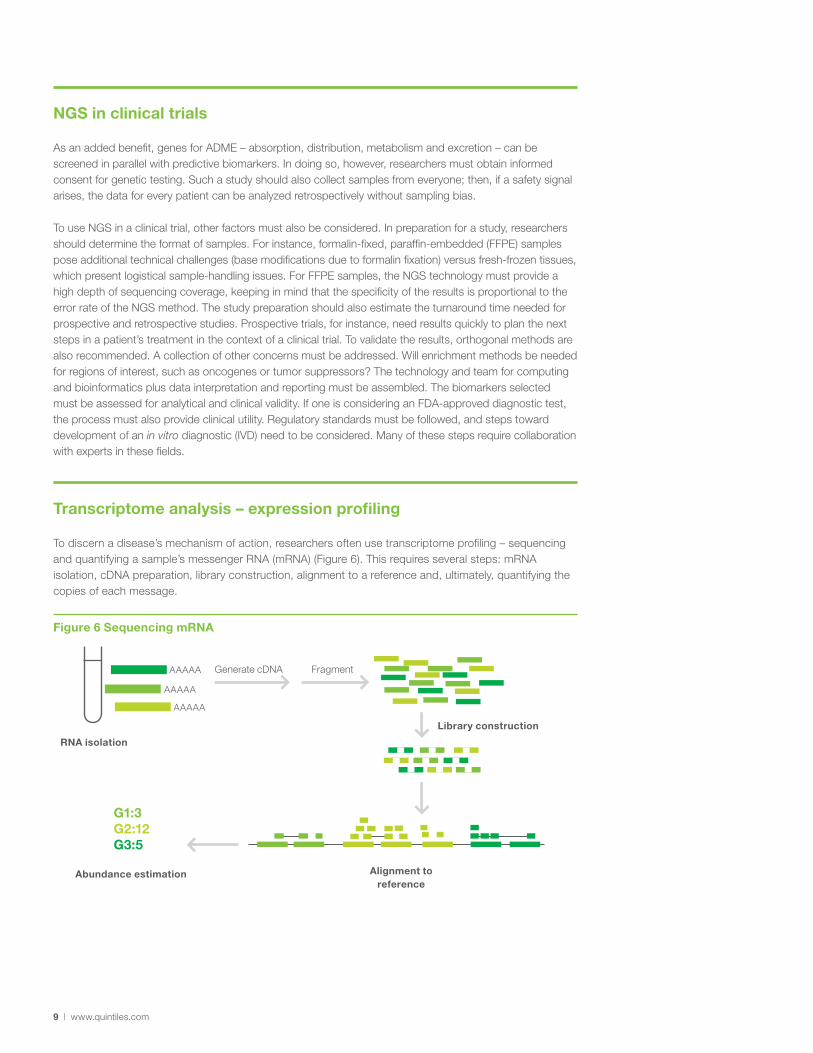
To discern a disease’s mechanism of action, researchers often use transcriptome profiling – sequencing and quantifying a sample’s messenger RNA (mRNA) (Figure 6). This requires several steps: mRNA isolation, cDNA preparation, library construction, alignment to a reference and, ultimately, quantifying the copies of each message.
Figure 6 Sequencing mRNA
AAAAA
AAAAA
AAAAA
Generate cDNA Fragment
Library construction
Alignment to reference
RNA isolation
Abundance estimation
G1:3G2:12G3:5

10 | www.quintiles.com
RNA-seq can also determine the role of alternate splicing in a disease. Humans produce more than 100,000 proteins from only about 21,000 genes, and various splicing arrangements account for the ability to make far more proteins than there are genes. This accounts for the complexity of biology seen at the protein level, and it can also drive the complexity of disease. Understanding the role of splicing in a disease, however, reveals new opportunities to cure or constrain it.
Expression profiling can also be used to divide a disease into subgroups and provide insights into prognosis. Diffuse large B-cell lymphoma, for example, exists in two forms: germinal center B-cell-like and activated B-cell-like. As George Wright of the National Cancer Institute and his colleagues reported in the October 19, 2003 Proceedings of the National Academy of Sciences, expression profiling can distinguish samples based on subgroups, because the disease types vary in the genes that get over- or under-expressed. By knowing the disease-driven changes in gene expression, a therapy can be assessed by its ability to revert expression to the patterns found in healthy tissue.
RNA-seq can also reveal gene fusions. Briefly, if two sequences of nucleotides are typically located far apart in physical reference genome space but keep turning up closer together in individual RNA sequencing reads, gene fusion may have taken place. This can also be confirmed with fluorescence in situ hybridization (FISH), but that requires an expert in cytogenetics to read the results, and gene fusions can only be examined one by one. By contrast, RNA-seq can simultaneously find multiple gene fusions, but it is more difficult to set up the data analysis. Once an expert-developed system of analysis is created, however, the results of RNA-seq for gene fusions are more objective and easier to interpret. Overall, NGS offers a cost-effective means to screen multiple biomarkers in a single experiment. This requires less tissue than serial testing, such as the multiple singleplex reactions used in FISH and immunohistochemistry. This does not imply that NGS replaces confirmatory methods at the protein level, but it provides a very effective first pass where positive, validated biomarkers exist.
Tomorrow’s technology today
To see if it is feasible to take today’s genomic technology to improve matching of patients to clinical trials, Quintiles partnered with the US Oncology Network. In the typical approach to matching a NSCLC patient with a trial, for example, the patient would be tested for an activating EGFR mutation. If that returned a negative result, the patient could be tested for the EML4-ALK fusion. Alternatively, a patient could be screened for multiple genomic alterations and the resulting information could be used to select the best protocol from a large menu of trial options.
The Quintiles team studied patients with metastatic colorectal cancer. Using a 50-gene profile developed with an Ion Torrent PGM NGS platform, we searched for potential drug-targetable genomic alterations. The results show that the cycle from tumor-sample acquisition to reporting to the clinician can be completed in about two weeks (Figure 7). We believe that this general method of genomic screening for multiple genomic alterations will become frequently used for selecting patients for clinical trials, as well as in oncology practices to help inform treatment decisions.
By knowing the disease-driven changes in gene expression, a therapy can be assessed by its ability to revert expression to the patterns found in healthy tissue.
Figure 7 Cycle of genomics sequencing to match patients with clinical trials
Patient presents – IC patient enrollment
sample collection
Quintiles Labs – Sample processing
– DNA isolation
EA – Genomic testing
EA – Bioinfomatic
analysis
Clinical annotation
and reporting

11 | www.quintiles.com
To make such techniques widely applicable for research or clinical practice, scientists and oncologists need noninvasive biomarkers, especially for longitudinal screening for chronic clinical management. These biomarkers could come from circulating tumor cells (CTCs) or cell-free/plasma DNA. Using cell-sorting techniques and proteomics markers, CTCs can be distinguished from healthy cells in the blood. Monitoring the presence or absence of CTCs might be correlated with the progression of disease. Also, CTCs and NGS might be combined to analyze the development of resistance to treatment in individual cells or interacting cell populations. Likewise, cell-free/plasma DNA might be sequenced with NGS to use as a surrogate to indicate tumor recurrence. With today’s technology, those DNA molecules can be counted even when they exist as only one molecule in 250,000.
Thinking more generally, genomics will change oncology in at least four ways. First, intervention trials will test combinations of rationally selected agents targeted for specific niche populations of patients with a specific array of genomic alterations in a particular tumor context. Second, real-world observational trials and registries are expected to be able to collect patient-level clinical and genomic data on well-characterized cohorts of patients. Third, the public domain will include readily available, patient-level data linked to genomic data. Fourth, data aggregators and analytic tools will efficiently and rapidly sort through these patient-level data sets to improve the design of new agent development programs and clinical decision-making.
Using genomics-based techniques like these, our 2025 NSCLC patient of the future, and patients with a wide range of other cancer types, will receive personalized treatment. More importantly, this treatment will be more effective, safer and less likely to be overturned by resistance-driven recurrence of the cancer. In short, genomics-based techniques will change the entire oncology ecosystem from the very beginning of drug discovery through treatment.
Genomics-based treatment will be more effective, safer and less likely to be overturned by resistance-driven recurrence of the cancer.

About the authors
12 | www.quintiles.com
Philip Breitfeld, M.D.Vice President and Global Head, Therapeutic Centers of Excellence, Quintiles Philip Breitfeld, M.D., is vice president and Global Head of Quintiles’ Therapeutic Centers of Excellence (COE). In this role, Dr. Breitfeld has overall responsibility for Quintiles’ Therapeutic COEs – cross-functional teams of therapeutically aligned experts, focused on optimizing customers’ product development efforts.
Dr. Breitfeld has more than 25 years of experience in oncology, including 20 years of experience in academic medical institutions in the U.S., and seven years of experience in the biopharmaceutical industry focused on oncology drug development and execution of clinical programs. Prior to joining Quintiles, he held senior oncology clinical development positions at BioCryst and Merck Serono. Dr. Breitfeld has global experience in most all malignant hematology and oncology indications, with biologics, small molecules, cancer vaccines and hematopoietic stem cell transplantation.
Dr. Breitfeld received his undergraduate degree from Princeton, his medical degree from the University of Rochester, and was a clinical and research fellow (Pediatric Hematology-Oncology) at the Dana-Farber Cancer Institute at Harvard. He completed a second fellowship in Medical Informatics sponsored by the National Library of Medicine and was a visiting scientist at the Whitehead Institute at MIT.
Jeff FitzgeraldDirector, Personalized Medicine Integration, QuintilesJeff possesses more than a decade of successful sales and support experience in the genomics tools sector. In his current position, he supports the integration of genomic technologies to address biomarker analysis in sponsors’ clinical trials.
Previously while at RainDance Technologies, he cultivated new business opportunities in research and clinical markets through the use of its sequence enrichment solutions in conjunction with various next generation sequencing platforms. In previous sales and support roles at Affymetrix, he supported key genome and academic research centers in the use of expression, resequencing, and genotyping microarrays. Prior to his commercial experience, Jeff sought to identify molecular markers responsible for tumor progression while performing research in the Department of Pathology at Brigham and Women’s Hospital and Harvard Medical School.
Jeff holds a Bachelor of Science from the University of Vermont in Microbiology and Molecular Genetics.

Cop
yrig
ht ©
201
4 Q
uint
iles.
All
right
s re
serv
ed.
15.0
012-
1-10
.14
Contact usToll free: 1 866 267 4479 Direct: +1 973 850 7571Website: www.quintiles.com/consulting Email: [email protected]