pediatric bone marrow transplantation · pediatric hematology oncology and bone marrow transplant...
TRANSCRIPT

Pediatric Bone MarrowTransplantation
Satya Prakash Yadav, Akshay Sharma, Ravi M. Shah, andPrakash Satwani
ContentsIntroduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3
Indications of BMT . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4Allogeneic BMT . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4Autologous HSCT . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
Matched Sibling Donor BMT . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5Leukemia/Lymphoma . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5Hemoglobinopathy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6Primary Immunodeficiency Disorders (PIDs) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6Inborn Errors of Metabolism . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6Aplastic Anemia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
S. P. Yadav (*)Pediatric Hematology Oncology and Bone MarrowTransplant Unit, Cancer Institute Medanta The MedicityHospital, Gurgaon, Haryana, Indiae-mail: [email protected]; [email protected]
A. SharmaBone Marrow Transplantation & Cellular Therapy St JudeChildren’s Research Hospital, Memphis, TN, USAe-mail: [email protected]
R. M. ShahPediatric Oncology and BMTAlberta Children’s Hospital,Alberta Health Services, Calgary, AB, Canadae-mail: [email protected]
P. SatwaniMYH-Pediatric Bone marrow Transplant Unit MahatmaGandhi Memorial Medical College, Indore, MadhyaPradesh, India
Division of Pediatric Blood and Bone MarrowTransplantation New York-Presbyterian Morgan StanleyChildren’s Hospital, Columbia University, New York, NY,USAe-mail: [email protected]
© Springer Nature Switzerland AG 2020M. Chandy et al. (eds.), Contemporary Bone Marrow Transplantation, Organ and Tissue Transplantation,https://doi.org/10.1007/978-3-319-64938-2_11-1
1

Unrelated Donor BMT . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7History . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7Donor Availability . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8Donor Selection . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8Cell Source Selection . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9Graft Manipulation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11Major Complications Following Unrelated Donor AlloHCT . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11Survival Following Unrelated Donor AlloHCTAmong Children . . . . . . . . . . . . . . . . . . . . . . . 12
T Cell-Replete Haploidentical SCT . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14T Cell-Replete Haploidentical SCT: Beijing (GIAC) Protocol . . . . . . . . . . . . . . . . . . . . . . . . . . 15Haploidentical Stem Cell Transplantation with Posttransplant Cyclophosphamide . . . . 15
Haploidentical Stem Cell Transplantation T Cell Depleted . . . . . . . . . . . . . . . . . . . . . . . . . . 16Principles of T Cell Depletion (Table 3) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17Infection Risk Post-HSCT in TCD Haplo-HSCTApproaches . . . . . . . . . . . . . . . . . . . . . . . . . . 19Evidence in Children . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20
Autologous HSCT . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24Lymphoma . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25Brain Tumors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25Neuroblastoma . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25Other Tumors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25Autoimmune Disorders . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25
Gene Therapy and CAR T Cell Therapy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25Gene Therapy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25CAR T Cells . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
Long-Term Follow-Up of Children Post-BMT . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29Leukemia/Lymphoma/Solid Tumors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29Hemoglobinopathy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30Inherited Bone Marrow Failure Syndromes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30Primary Immunodeficiency Disorders . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30Inborn Error of Metabolism . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30
Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31
Abstract
Blood and marrow transplantation (BMT) forchildren has become a well-established sub-spe-cialty in the last three decades. Some of child-hood disorders like leukemia and severecombined immunodeficiency were the first fewdisorders to be treated successfully by BMT.Great strides have been made to improve out-comes of both autologous and allogeneic stemcell transplant for children for variousmalignantand nonmalignant disorders. Reduced intensityand reduced toxicity conditioning regimes havemade BMT available for children with severemorbidity or poor organ function. With settingup of donor registries, the matched unrelateddonor transplants became routinely available
for Caucasians. But for ethnic minorities andmost people living in the developing world,donors were still not available. Alternate donorBMT have become a reality with improved out-comes. Haploidentical BMT has made donorsavailable for all. Posttransplant cyclophospha-mide has made it feasible to reduce both acuteand chronic graft-versus-host disease in chil-dren. Cellular therapy and gene therapy for chil-dren have taken the field to the next level.Chimeric antigen receptor T cell therapy hasbecome an exciting new treatment option forchildren with B-lymphoblastic leukemia andlymphoma. Gene therapy has become a realityfor immune deficiency disorders and
2 S. P. Yadav et al.

hemoglobinopathies. Long-term follow-up is amust to improve outcomes further.
Keywords
Pediatric · Bone marrow transplant ·Hematopoietic stem cell transplant · Peripheralblood stem cell transplant · Haploidentical ·Conditioning · Reduced intensity ·Myeloablative · Graft-versus-host disease ·TCR alpha-beta depletion · Matched siblingdonor · Matched unrelated donor · Genetherapy · Chimeric antigen receptor
Introduction
Children right from birth and up to 18 years havebeen treated successfully for both malignant andnonmalignant disorders by blood and marrowtransplantation (BMT). Are children differentfrom adults? Yes, of course. Pediatrics is a branchvery different from Internal medicine, so is thesub-specialty of pediatric BMT from adult BMT.Now most developed countries and few develop-ing countries have exclusive pediatric BMT pro-grams and training programs for pediatric fellowsin the field of BMT. This has improved outcomesfor the children undergoing BMT.
Some of the pediatric disorders were the first tobe treated by BMT. Sir Don Thomas was a pioneerwho was awarded Nobel Prize in medicine for hiswork in the field of BMT, and his best-knownwork was on curing children with relapsed acutelymphoblastic leukemia by BMT who werethought to be incurable and were destined to die.Most of his initial work was before the dawn ofthe era of HLA typing. His work in leukemia withallogeneic stem cell transplant was perhaps thefirst suggestion that donor immune system helpscure leukemia. In 1968, Robert Good performedfirst allogeneic BMT in nonmalignant conditionand it was in a child suffering from severe com-bined immunodeficiency (SCID) from his healthysibling. Eleven males had died of the same diseasein his extended family. In 1988, Elaine Gluckman,another pioneer who performed first cord bloodtransplant in the world for a child suffering from
Fanconi anemia from his fully matched sibling’scord cells. This transplant was performed in Parisalthough patient and cord stem cells both camefrom the USA. In early 2000, gene therapy wassuccessfully performed for ADA SCID again in achild and opened up entirely a new field. Latest tobe added to this list of firsts, in 2012, is the pioneerwork of Stephan Grupp and colleagues in 7-year-old Emily Whitehead who was treated success-fully for refractory leukemia by her own geneti-cally modified chimeric antigen receptor T cell(CAR T cell) lymphocytes. This has started anew era of cellular therapy for cancer. BMT forchildren has led the field of BMT for so manydecades now.
Around 30,000 BMT are performed in theUSA annually and another 40,000 in Europe.Sixty percent of these are autologous. BMT forchildren are increasing especially allogeneic. Inchildren, various types of BMT are performed.Autologous is mainly for solid tumors and lym-phomas and rarely autoimmune disorders, whileallogeneic is for both malignant and nonmalignantconditions. Matched sibling donor BMT has tra-ditionally been preferred over other types and hasthe best survival. However, with advent of regis-tries, matched unrelated donor (MUD) BMT hasovertaken MSD, at least for the Caucasians. Fornon-Caucasians initially unrelated cord bloodtransplants became a feasible treatment option.Later one antigen-mismatched donor BMTbecame feasible but had inferior outcomes. How-ever, game changer has been the advent ofhaploidentical related donor BMT which hasmade donor available for all. The most popularhaploidentical related donor BMT platform hasbeen one developed by John Hopkins group, ofin vivo depletion of alloreactive T lymphocytes byposttransplant cyclophosphamide (PTCy)(Luznik et al. 2012). Other platforms are T-replete, Beijing protocol, and TCR alpha-beta Tand B cell-depleted haploidentical BMT. Out-comes with alternative donors like haploidenticaldonors and umbilical cords have improved sub-stantially in the last 15 years as per EBMT surveyof more than 100,000 patients undergoing BMTfor hematological malignancies (Shouval et al.2019). But application of these newer alternate
Pediatric Bone Marrow Transplantation 3

donor BMT to children especially for non-malignant conditions is the most significantadvance in the field.
In children, graft source can be bone marrow(BM), peripheral blood stem cells (PBSC), or cordblood stem cells. Although bone marrow harvestis cumbersome and needs general anesthesia, itcauses less chronic GVHD. Peripheral blood stemcell harvest, on the other hand, is relatively easy toperform except in very young children or infants.The advantage of PBSC is quick engraftment atleast a week before bone marrow, but it causesmore chronic GVHD. Umbilical cord blood stemcells are easily and quickly available from thecord blood banks and tend to cause less GVHD,but problems are slow engraftment at a week laterthan bone marrow and slow immune reconstitu-tion. One cord blood unit is usually sufficient for achild weighing up to 30 kg. Applications of BMTare increasing day by day. To reduce GVHD, TCRalpha-beta T cell depletion has been used success-fully (Locatelli et al. 2017) and CD34-positiveselection has been tried (Keever-Taylor et al.2012). To reduce GVHD, PTCy has been effectiveeven in nonmalignant conditions like sickle cellanemia (Bolanos-Meade et al. 2012). Majorcauses of transplant-related mortality (TRM) inchildren are rejection, infections, and acuteGVHD. These complications also lead to morbid-ity and increased cost. To reduce TRM is a majorgoal specially to improve outcomes with alternatedonors.
Indications of BMT
BMT is performed for children not just for malig-nant but also nonmalignant conditions. It’s eitherallogeneic (stem cells infused from someone else)or autologous (where own stem cells are infused).
Allogeneic BMT
In children it is offered mainly for five categories –leukemia/lymphoma, hemoglobinopathies, bonemarrow failure, primary immunodeficiency, andinborn errors of metabolism.
Leukemia/LymphomaChildhood acute lymphoblastic leukemia (ALL)is highly curable with chemotherapy alone (Puiand Evans 2006). Only a subset of patients in CR1are offered BMT, and they are those who are not inremission after 4–5 weeks of induction, have pres-ence of minimal residual disease after 2–3 monthsof chemotherapy, and have cytogenetic abnormal-ity like MLL gene (Pulsipher et al. 2011). Atrelapse, again patients who relapse very early(within 18 months from diagnosis), have isolatedmarrow relapse in T cell ALL, or have early iso-lated bone marrow relapse in B cell ALL areoffered BMT in CR2 (Oliansky et al. 2012; Peterset al. 2015). All patients in CR3 are taken up forBMT or newer cellular therapies like CAR T celltherapy. For acute myeloid leukemia (AML), allo-geneic BMT is offered for intermediate- or high-risk disease in CR1 (Kolb and Meshinchi 2015).Favorable cytogenetics like t (8,21), t (15,17), andinv16 have good outcomes with chemotherapy. InAPML patients, arsenic oxide as upfront or sal-vage therapy has also reduced need for BMT. Inpatients with juvenile myelomonocytic leukemia(JMML), BMT is the only curative option.Although myelodysplastic syndrome (MDS) inchildren is rare, usually in most children, BMT isits only effective therapy. In lymphomas usuallyautologous stem cell transplant (SCT) is offered.But few patients of lymphoblastic lymphoma orHodgkin lymphoma who have relapsed afterautologous SCT are offered allogeneic SCT(Gross et al. 2010; Satwani et al. 2015a).
HemoglobinopathyGlobally 300,000 children are born with sicklecell disease and 60,000 with thalassemia majorevery year. Both conditions are curable by BMT.It is one of the commonest indications for BMT inIndia for children. Reduced toxicity conditioningto reduce TRM is the new mantra (Satwani et al.2013; Bhatia et al. 2014). BMT in sickle is offeredin those patients with severe disease despitehydroxyurea and/or blood transfusions. In thalas-semia BMT is always a choice as lifelong bloodtransfusions with chelation can be good alterna-tive. Outcomes have improved over the last three
4 S. P. Yadav et al.

decades even with alternate donors for hemoglo-binopathies making BMT a safe option.
Bone Marrow FailureChildren with severe acquired aplastic anemia areoffered BMT from a matched sibling donor as afirst-choice therapy as it has good outcomes>90% cure rates (Barone et al. 2015; Kojima etal. 2000). In those who don’t have a matchedsibling donor, traditionally immune-suppressivetherapy (IST) is offered as therapy (Miano andDufour 2015). But now recent data of upfrontmatched unrelated donor (MUD) has shown sim-ilar outcomes to MSD challenging current algo-rithm (Dufour et al. 2015). For inherited bonemarrow failure syndromes like Fanconi anemiaand dyskeratosis congenita, BMT is alwaysoffered.
Primary Immunodeficiency DisordersThese are rare disorders. Mostly severe combinedimmunodeficiency (SCID) is offered BMT as anemergency. Outcomes are best if BMT isperformed soon after birth. Many a times BMTcan be performed without conditioning in thosewith absent T and NK cells. Other immune defi-ciencies like Wiskott-Aldrich syndrome, chronicgranulomatous disease, leukocyte adhesiondefect, Chediak-Higashi, etc. are offered BMTwith conditioning. If hemophagocytic lymphohis-tiocytosis (HLH) in children is a familial orrelapsed case or central nervous system isinvolved, then BMT is offered.
Inherited Metabolic DisordersAgain, these disorders are very rare and not all arecurable by BMT. Hurler syndrome has maximumdata on success of BMT. Among mucopolysac-charidosis (MPS) Hurler and Hunter syndromesare usually offered BMT before 3 years of age asdevelopmental delay is a contraindication toBMT. X-linked adrenoleukodystrophy is againcurable by BMT provided disease has not severelyaffected the brain.
Autologous HSCT
It is offered for mostly solid tumors and occasion-ally for autoimmune disorders. Solid tumors arerelapsed lymphoma, high-risk/relapsed neuro-blastoma, high-risk/relapsed medulloblastoma,medulloblastoma/ependymoma, relapsed/refrac-tory germ cell tumor, metastatic/relapsed Ewingsarcoma, relapsed Wilms tumor, relapsed retino-blastoma, and relapsed rhabdomyosarcoma.Autoimmune disorders are relapsing/remittingmultiple sclerosis.
Matched Sibling Donor BMT
Traditionally this has had best results in childrenwith TRM <10%. It’s always the first choice as agraft source in children undergoing BMT(Shouval et al. 2019). We will discuss here resultsof MSD BMT in various diseases in children.
Leukemia/Lymphoma
In ALL, outcomes have been excellent for MSDBMT in CR1 and CR2 (D’Souza 2018). Totalbody irradiation (TBI)-based conditioning is stillthe preferred conditioning for ALL BMT (Peterset al. 2015). As TBI has a lot of long-term sideeffects, results of ongoing FORUM trial compar-ing TBI-based conditioning versus thiotepa,busulfan, and fludarabine (TBF) or thiotepa,treosulfan, and fludarabine (TTF) are eagerlyawaited. In AML, myeloablative conditioninghas better outcomes (Selim et al. 2019). But chil-dren with poor general condition going into BMTmay be offered reduced intensity conditioning(RIC) with busulfan-fludarabine (BuFlu) orfludarabine-melphalan (FluMel). Outcomes inMSD are improving. For chronic myeloid leuke-mia (CML), BMT is offered only in blast crisis orTKI resistant. As TKI affect growth of childrenwith CML, idea of stopping TKI is somethingwhich needs to be tried. Otherwise BMT can stillbe an option. For lymphoma, mostly it is autolo-gous SCT, but few cases of lymphoblastic lym-phoma with relapse or high-risk disease and some
Pediatric Bone Marrow Transplantation 5

cases of Hodgkin lymphoma relapse after autolo-gous SCT are offered allogeneic SCT (Gross et al.2010; Satwani et al. 2015a). Outcome data arelimited by lack of numbers and publications.
Hemoglobinopathy
Truly MSD has shown great success in curingthalassemia and sickle cell anemia. In sickle suc-cess of >90%, overall survival is reported withMSD and is indicated in children with severedisease (Bhatia et al. 2014; Shenoy 2007, 2013;Gluckman et al. 2017). Reduced toxicity regimeslike BuFlu have improved outcomes (Bhatia et al.2014). Sickle cell anemia patients with age>16 years had inferior outcome post-BMT(Gluckman et al. 2017). Thalassemia patientsprior to transplant have risk factors (poor chela-tion, hepatomegaly, and liver fibrosis) and can beclassified into Pesaro class I (no risk factor), classII (1–2 risk factors), and class III (all three riskfactors). Outcomes post-BMT are affected as perPesaro classification (Lucarelli et al. 1990) studyshowing overall survival of 90% in class I, 80% inclass II, and 61% in class III. A recent study fromIndia has further added another subgroup calledVellore class III high risk (children with hepato-megaly>5 cm and age>7 years) which have verypoor overall survival of 30% after BuCy condi-tioning (Mathews et al. 2007). Main problems inthalassemia specially in class III are high TRM,rejection, and VOD. To overcome this, thiotepa,treosulfan, and fludarabine (TTF) regimen hasproven to be less toxic and improved survival to89% in class III high-risk patients (Mathews et al.2013). Italians have used another approach calledprotocol 26 to improve outcomes in class IIIpatients by using pretransplant immune suppres-sion (PTIS) with azathioprine, hydroxyurea, G-CSF, and fludarabine prior to BMT followed byBuCy conditioning. This improved survival to93% in class III patients (Sodani et al. 2004).
Primary Immunodeficiency Disorders(PIDs)
Advances in donor selection, graft manipulation,conditioning, and treatment of complicationsmean that survival for many PIDs is now around90%. Next-generation sequencing is identifyingnew immunodeficiencies, many of which aretreatable with BMT (Slatter and Gennery 2018).Matched sibling donor BMT has proven to be aboon for children suffering from PID. In T-NKsubset of SCID, BMT has been performed evenwithout conditioning. But in children with otherSCID or in those without major infections, condi-tioning is highly recommended for B cell engraft-ment and improved outcomes. In children withHLH, RIC is better than myeloablative condition-ing (MAC). Campath as part of conditioning hasimproved outcomes but can lead to mixed chime-rism and need for second BMT (Allen et al. 2018).For WAS, outcomes with MSD are best. Long-term outcomes showed non-GVHD-related auto-immunity in 20% of patients who had mixedchimerism due to persistence of host lymphocytes(Ozsahin et al. 2008). Thus, MAC is generallypreferred for WAS BMT.
Inborn Errors of Metabolism
In MPS, X-ALD early transplant from an unaf-fected MSD can effectively stabilize the disease.Maximum experience has been with Hurler syn-dromewhere>600 BMT have been performed. InHurler syndrome and other MPS, rejection is theproblem so MAC is preferred with full-dosebusulfan targeting levels with pharmacokinetics.BMT should be performed before 3 years of age.Always screen the donor. With well-matcheddonor, survival rates >90% have been reported(Boelens et al. 2014).
Aplastic Anemia
For acquired aplastic anemia, MSD BMT is astandard of care. Success >90% and reducedGVHD with bone marrow as a graft source have
6 S. P. Yadav et al.

been reported (Barone et al. 2015; Kojima et al.2000). Regimens initially were ATG + cyclophos-phamide 200 mg/kg, but later to reduce rejectionin multi-transfused aplastic anemia patients,fludarabine and cyclophosphamide (FluCy) andATG has been used with good success similar toadults (George et al. 2007). Campath-based con-ditioning is a newer option.
Among inherited bone marrow failure syn-dromes, Fanconi anemia is the commonest. Allchildren diagnosed with aplastic anemia shouldhave chromosome breakage study and/or muta-tion test to rule out Fanconi anemia, as thesechildren are very sensitive to radiotherapy andchemotherapy especially alkylating agents. MSDBMT has good outcomes, but TBI should beavoided as part of conditioning, and dose of cyclo-phosphamide should not exceed 40 mg/kg maxand serotherapy should be given (64). Alwaysscreen the sibling donor for Fanconi anemia.DKC is a difficult transplant and RIC regimen ispreferred. Post-BMT, patients have to be moni-tored for lung toxicity and liver fibrosis (Alter2017). In pure red cell aplasia, steroid dependenceand no response to steroids are indications forBMT, and it should be with MAC regimen toablate old bone marrow (Alter 2017).
Unrelated Donor BMT
Allogeneic hematopoietic cell transplantation(alloHCT) has the potential to cure various malig-nant and nonmalignant diseases among children(Satwani et al. 2015b). While survival amongchildren undergoing alloHCT has improved overthe past decades, many children still sustain sig-nificant morbidity or mortality. A major factorassociated with suboptimal outcomes followingalloHCT is a lack of an HLA (human leukocyteantigen)-matched sibling (Gragert et al. 2014). Inthe absence of an HLA-matched sibling, childrencan still undergo a successful alloHCT to curemalignant and nonmalignant diseases (Shenoyand Boelens 2015). However, the process can bevery challenging and requires intense resources.In this section, we will discuss the issues associ-ated with unrelated donor alloHCT for children.
History
Early TransplantsIn 1957, E. Donnall Thomas published a paperdescribing the first six patients who received allo-HCTat Cooperstown Hospital (Columbia Univer-sity, NY) (Thomas et al. 1957). All of the patientsdied soon after their transplants due to rejection orcomplications. At that time the HLA wasunknown and the resulting mismatch that likelyoccurred contributed to poor outcome, either dueto host-versus-graft or graft-versus-host interac-tions. A major breakthrough in the field of allo-HCT happened when Jean Dausset, Rose Payne,and Jon van Rood discovered HLA (Dausset1958). Utilizing HLA matching between patientsand donors, Thomas et al. conducted another allo-HCTstudy in 100 patients with leukemia (Thomaset al. 1971). Thirteen percent of patients in thisstudy achieved long-term remission. Encouraged,Thomas conducted yet a third alloHCT study forsubjects in early-stage acute myeloid leukemia(Lucchini et al.; Thomas et al. 1971). Fifty percentof patients achieved a cure. Despite several chal-lenges and failures, Dr. Thomas persevered, as hestrongly believed in the power of the allogeneicimmune system to eradicate leukemia. For hisoutstanding efforts in developing and advancingthe field of hematopoietic cell transplantation, Dr.E. Donnall Thomas received a Nobel Prize in1990.
Introduction of RegistriesChildren paved the way for unrelated donor trans-plantation. In 1973, the first successful unrelateddonor transplantation for nonmalignant diseaseoccurred in a 2-year-old boy, Simon Bostic.Simon was diagnosed with an immunodeficiencyand had no familial matched donor. The success ofSimon’s alloHCT inspired the establishment ofthe Anthony Nolan Bone Marrow Registry in theUK (1974) founded and named by the mother ofan earlier child who was unable to receive a trans-plant as there was no method of connecting amatched individual who was willing to donate toa child in need. Prior to the start of bone marrowdonor registries, the opportunity for alloHCT field
Pediatric Bone Marrow Transplantation 7

was limited to patients who had an HLA-matchedsibling.
Hansen et al. reported the first successfulunrelated donor alloHCT for malignant disease(Hansen et al. 1980). The patient was a 10-yearfemale with acute lymphoblastic leukemia (ALL)in second complete remission (CR). Her siblingswere not HLA-matched. However, her HLA typ-ing revealed that the patient had inherited twocommon HLA haplotypes that were frequentlyfound in North American Caucasians. One of thefive unrelated donors tested by mixed lymphocytereaction was an 8/8 HLA match. The patient wasconditioned with cyclophosphamide (Tiercy2016) (120 mg/kg) and a single dose of totalbody irradiation (TBI) (1000cGY) followed bybone marrow infusion. GVHD prophylaxis wasmanaged with methotrexate. The patient did nothave any major complications post-alloHCTincluding graft-versus-host disease (GVHD).The successful outcome of this case was one ofseveral reasons to establish unrelated donorregistries.
The family of a patient, Laura Graves, a 10-year-old patient with leukemia, started NationalBone Marrow Registry now one of the largestbone marrow registries (now known as theNMDP). This program facilitated the first MUD(matched unrelated donor) alloHCT in 1987. TheNational Marrow Donor Program (NMDP) hasprovided close to 100,000 unrelated donors’hematopoietic cells. Currently, more than 34 mil-lion potential bone marrow donors are registeredworldwide.
Umbilical Cord Blood TransplantsAnother important milestone in the history ofalloHCT was the introduction of umbilical cordblood transplantation (UCBT). The first success-ful UCBT occurred more than 25 years ago in aninternational collaboration for a patient with aFanconi anemia (Auerbach et al. 1990). Cordblood stem cells require less strict HLA matching,but can be associated with slower hematopoieticrecovery. To overcome this challenge, cord bloodexpansion strategies are being developed. Over30,000 UCBTs have been performed and over
600,000 UCB units have been stored for trans-plantation worldwide (Ballen et al. 2013).
Donor Availability
The undisputed best outcomes in alloHCT arewith HLA-matched siblings. This is the gold stan-dard. However, only about 30% of patients whoneed an alloHCT have such a donor available(Gragert et al. 2014). That number is even lowerfor families with multiple siblings who can beaffected with the same nonmalignant (hemoglo-binopathies and immunodeficiency) disorders.Unrelated adult bone marrow registries and publiccord blood banks have tried to close that gap.However, the fact is that volunteer adult donorsare predominantly Caucasians. Subsequently, it isoften challenging to find a fully matched unrelateddonor for patients who belong to racial/ethnicminorities (Gragert et al. 2014). The probabilitiesof finding 8/8 HLA-matched unrelated donor forvarious ethnicities are described in Table 1. TheNMDP and other registries are making a con-certed effort to recruit bone marrow donors fromracial/ethnic minorities. It is expected that theadditional recruitment of racially diverse donors,especially from ethnic minorities, will improvethe probability of identifying an 8/8 HLA-matched unrelated donor by 4–7% (Gragert et al.2014).
Donor Selection
The impact of HLA matching was demonstratedin a study from the NMDP/Center for Interna-tional Blood and Marrow Transplant Research(CIBMTR), which included adults and childrenwith malignant disease (n ¼ 8003): 8/8 HLA-matched (n ¼ 5449), 7/8 HLA-matched(n ¼ 2071), or 6/8 HLA-matched (n ¼ 483)(Pidala et al. 2014). HLA matching wasperformed at HLA-A, HLA-B, HLA-C, andHLA-DRB1. The important finding of this studywas that any single mismatch was associated withsignificantly worse overall survival. There was noevidence that a mismatch (HLA class I vs. II or A
8 S. P. Yadav et al.

vs. DRB1) at any of the individual loci was bettertolerated than others. In 2019, NMDP updated theguidelines for the HLA typing of donors andpatients. HLA typing was extended from eightloci to ten loci with the addition of DPB1. Themost critical recommendation is that HLA typingshould be performed by DNA-based methods athigh resolutions for HLA- HLA-A, HLA-B,HLA-C, HLA-DRB1, and HLA-DPB1 loci(Dehn et al. 2019). Some centers also analyzeHLA-DQB1, HLA-DRB3/4/5, HLA-DQA1, andHLA-DPA1. However, these alleles have not beenshown in isolation to substantially affect overallsurvival. In certain situation when a patient hasmultiple donors who match at HLA-A, HLA-B,HLA-C, HLA-DRB1, and HLA-DPB1 loci, iftime permits centers should attempt to match atHLA-DQB1, HLA-DRB3/4/5, HLA-DQA1, andHLA-DPA1 to further decrease the risk of graftrejection and GVHD.
When patients have multiple 10/10 HLA-matched donors, several other characteristicsmay influence the determination of the best donor:
• The age of the donor (older donors had inferioroverall survival probably due to lower CD34cells and a higher number of memory T cells inthe product)
• Cytomegalovirus serostatus (prefer a donorwith serostatus similar to the patient)
• Sex (avoid female donors for male patients)• ABO compatibility (avoid major mismatch)• Prior pregnancies (nulliparous females poten-
tially have less alloreactive T cells)• Body weight (obese donors are at high risk for
anesthesia-related complications)
AlgorithmIn the current era, with improvement in the out-comes of haploidentical transplants, virtuallyevery eligible patient can undergo alloHCT(Luznik et al. 2012). Patients without an HLA-matched sibling have several options includingunrelated donor (match and mismatched), umbil-ical cord blood (matched and mismatched), andhaploidentical donors (sibling, parents, and otherrelatives). Recently, a large European Bone Mar-row Transplant registry (EBMT) observationalretrospective study validated results of aCIBMTR study (Shouval et al. 2019). In bothstudies, a similar hierarchy of donor selectionwas established. Patients with an HLA-matchedsibling had the best outcomes, followed by HLA-matched unrelated donors, followed by otherdonor sources. Based on the results of these stud-ies, we have developed a stepwise algorithm fordonor selection (Fig. 1).
Cell Source Selection
Three commonly utilized cell sources for alloHCTare bone marrow, peripheral blood stem cells(PBSC), and cord blood. Each source has itsadvantage and disadvantage as detailed in Table2. In a large BMT/CTN randomized study, adultpatients receiving PBSC had faster hematopoieticengraftment, resulting in better early outcomeswhen compared to bone marrow. However, theincidence of chronic GVHD was much loweramong patients who received bone marrow,which not only resulted in better late outcomesbut also a better quality of life (Anasetti et al.2012; Lee et al. 2016). Similarly, in children
Table 1 National Marrow Donor Program model for the probability of finding an 8/8 HLA-matched unrelated donor forvarious ethnic groups
Race Ethnicity Likelihood of finding an 8/8 HLA match donor (%)
White European 75
Middle EasternNorth African
46
Hispanics 34–40
Asian Southeast Asians 27
South Asians 33
Black All ethnicities 16–19
Pediatric Bone Marrow Transplantation 9

utilization of PBSC when compared to bone mar-row had resulted in a higher rate of acute andchronic GVHD, higher incidence of transplant-related mortality, and inferior overall survival(Eapen et al. 2004; Keesler et al. 2018).According to the CIBMTR, bone marrow is the
preferred source of hematopoietic cells for chil-dren. Among patients younger than 18 years old,bone marrow remains the most common graftsource for unrelated donors accounting for 55%,followed by UCB which is 26% and PBSC whichis 19% (D’Souza 2018). However, procuring bone
Fig. 1 Algorithm for donor selection
Table 2 Pros and cons of various unrelated donor hematopoietic cell sources
Bone marrow PBSCCordblood
Donor preparation None G-CSF None
Donor risk Related to prolonged generalanesthesiaPain and anemia post-harvest
Related to apheresis catheterplacement
None
Cell procurement 6–8 weeks 6–8 weeks 1–2 weeks
CD34 cell count in the product High Higher Low
T cell count in the product High Very high Low
Time for neutrophil engraftment Fast Fastest Slow
Time for platelet engraftment Fast Fastest Slow
Acute graft-versus-host disease Low High Low
Chronic graft-versus-host disease Low High Low
Immune recovery in the absence ofGVHD
Fast Fast Slow
10 S. P. Yadav et al.

marrow, or PBSC, from an unrelated donor maytake 6–8 weeks. Cord blood or haploidenticaldonor might be a potential option in more urgentsituations.
Graft Manipulation
CD34 selection, alpha-beta T cell depletion, orPTCy reduce the incidence of GVHD amongpatients eligible for unrelated alloHCT withmatched or mismatched donors or haploidenticalalloHCT (Locatelli et al. 2017; Keever-Taylor etal. 2012; Luznik et al. 2012). Patients receivingCD34-selected graft with little to no number of Tcells have a lower risk of aGVHD, but in turn havea higher risk of viral infections, posttransplantlymphoproliferative disorder, and disease relapse.Alpha-beta T cell depletion is a relatively newertechnology. Currently only a few centers havemastered this technique for haploidentical allo-HCT. It is hypothesized that the removal ofalpha-beta T cells will result in a lower incidenceof GVHD and leftover gamma-delta T cells in thegraft provide protection against disease relapse.However, viral infections continue to be a majorissue. Both CD34 selection and alpha-beta T celldepletion can lead to a higher incidence of graftfailure.
PTCy regimen is another type of graft manip-ulation. Johns Hopkins Medical University popu-larized this methodology in haploidenticaltransplantation. Results of PTCy in terms ofaGVHD, disease relapse, and overall survival arecomparable to unrelated donor alloHCT (Luzniket al. 2012; Bolanos-Meade et al. 2012). Theoutstanding success and relative ease of adminis-tration have resulted in PTCy being utilizedworldwide. It has been used not only forhaploidentical but also for unrelated donor allo-HCT. The way PTCy works is not entirely clear.PTCy most likely eliminates activated allo-reactive T cells in vivo, which may result in alower incidence of GVHD.
Major Complications FollowingUnrelated Donor AlloHCT
Complications following unrelated donor allo-HCT can lead to mortality. CIBMTR reportedthat for the years 2015–2016, unrelated alloHCTmortality related to infection, organ failure, andGVHD accounts for 51% of deaths before100 days and 35% of deaths after 100 days(D’Souza 2018). In a prospective ALL trial atBerlin-Frankfurt-Muenster (BFM), HLA-matched sibling transplants were compared tomatched unrelated donor (MUD) transplants(Pichler et al. 2019). The incidence of bacterial,fungal, and viral infections was higher in theMUD alloHCTs probably due to the intenseimmunosuppression needed to prevent GVHD.Two major sources of complications followingalloHCT are high-dose chemotherapy utilized inconditioning regimens and immune-mediatedhost versus graft reaction or graft-versus-hostreaction. Myeloablative doses of chemotherapyor radiation therapy can result in organ injury,mucositis, sinusoidal obstruction syndrome,bleeding, and bacterial and fungal infectionsrelated to pancytopenia and viral infections dueto immunoablation. In patients undergoingunrelated donor transplant, immune-mediatedreactions can cause significant morbidity andmor-tality. Unrelated donor alloHCT is associated witha higher risk of graft failure. The more the HLAmismatch, the higher the risk for graft failure. Riskof graft failure is higher when cord blood is uti-lized or unrelated grafts are T cell depleted(Cluzeau et al. 2016). As expected, graft failuresare associated with poor outcomes and increasethe cost of alloHCT (Bourgeois et al. 2019).
Acute and chronic graft-versus-host disease isprobably the most challenging complication asso-ciated with unrelated alloHCT. Incidence ofaGVHD can be as high as 60% followingunrelated alloHCT (Socie and Blazar 2009). Var-ious factors are associated with the occurrence ofaGVHD: HLA mismatch (8/10 > 9/10 > 10/10),hematopoietic cell source (PBSC>BM > UCB),and graft manipulation (T cell replete> T celldepletion). For the last four decades, the mainstayof treatment for aGVHD has been corticosteroids,
Pediatric Bone Marrow Transplantation 11

which themselves are associated with serious sideeffects that can result in significant morbidity andmortality. Corticosteroids are only effective intwo-thirds of patients. Salvage therapies of ste-roid-refractory aGVHD result in intense immuno-suppression that ultimately results in a very highincidence of viral, fungal, and bacterial infections.Many patients succumb to these infections (Ricciet al. 2019). Incidence of cGVHD is also higherfor patients undergoing unrelated alloHCT.Chronic GVHD can result in significant morbid-ity, poor quality of life, and as well as substantialresource utilization (Anasetti et al. 2012).
Survival Following Unrelated DonorAlloHCT Among Children
The improvement in HLA typing, molecular mon-itoring for infections, and availability of newerantimicrobial agents and tailoring of conditioningregimen based on the disease and comorbiditieshave resulted in a decrease in transplant-relatedmortality in adults (Gooley et al. 2010). One cansafely assume that transplant-related mortality hasdecreased in children as well (Horan et al. 2011).In the next few paragraphs, the outcomes ofunrelated donor alloHCT among children aredescribed.
Malignant Disease
Acute Lymphoblastic LeukemiaThe majority of children with ALL are cured withmulti-agent, targeted, and risk-adapted chemo-therapy protocols. In the current era, only asmall percentage of patients (10–15%) requiresalloHCT (Pui and Evans 2006). Indications foralloHCT have evolved over time (Pulsipher et al.2011). The majority of the transplants now areperformed for the patients who suffer a relapsewithin 36 months of diagnosis or have a high levelof minimal residual disease (MRD) at the end ofconsolidation (Pulsipher et al. 2011; Oliansky etal. 2012).
In a recent CIBMTR report, among the 1494patients younger than 18 years receiving an HLA-matched sibling transplant for ALL between 2006
and 2016, the 3-year probabilities of survival were74%, 60%, and 45% for patients with early (CR1),intermediate (CR2), and advanced disease (CR3,primary induction failure or relapsed disease),respectively. The corresponding probabilities ofsurvival among the 2827 recipients of anunrelated donor transplant were 68%, 57%, and47% (D’Souza 2018).
In the prospective multinational BFM study,among children with high-risk ALL, alloHCTout-comes were compared between HLA-matchedsibling donors and MUDs. (Peters et al. 2015).Four-year leukemia-free survival was similarbetween matched sibling donor (MSD) (71%)and MUD (67%) alloHCT. However, incidenceof transplant-related mortality was higher forMUD compared to MSD (10 vs. 3%,p ¼ 0.017). This recent study is reflective ofadvances in the current alloHCT practices in thewestern world where transplant-related mortalityfollowing myeloablative total body irradiation-based conditioning regimen utilizing matchedunrelated donor is relatively low.
Acute Myelogenous LeukemiaOver the decades, moderate success has beenaccomplished in the management of AML. Incomparison to ALL, the outcome of AML patientsremains suboptimal (Kolb and Meshinchi 2015).Like ALL, indication for alloHCT in pediatricAML has evolved over time; patients in CR1 areonly eligible if they have high-risk cytogenetics ormolecular mutations and high-level MRD at theend of induction or secondary AML (Pulsipher etal. 2010). The majority of the alloHCT for pedi-atric AML is performed for patients who suffer adisease relapse. AML patients meeting criteria foralloHCT should promptly proceed to alloHCTwith the best available donor. EBMT analyzedretrospective registry data of children whoreceived alloHCT for AML in CR1. In thisstudy, children receiving busulfan/cyclophospha-mide/melphalan had superior leukemia-free andoverall survival compared to other regimens uti-lized. However, patients receiving MUD alloHCThad inferior leukemia-free (64 vs. 59%) and over-all survival (61% vs. 70%) when compared toMSD alloHCT (Lucchini et al. 2017). In a recent
12 S. P. Yadav et al.

study of pediatric AML, investigators from Aus-tralia and New Zealand reported that 5-year prob-ability of survival for patients in CR2 wassignificantly improved during 2005–2013 com-pared to 1998–2004, 44% versus 71%, respec-tively (Selim et al. 2019). The 71% survival inrecent cohort is encouraging, especially when75% of patients received unrelated donor allo-HCT in this study. Another important observationin this study was that patients who received twocycles of re-induction had better survival com-pared to those who received one or three cycles.
LymphomaLeukemia is the most common indication for allo-HCT in children with malignant diseases. How-ever, a small subset of patients with lymphoma arealso considered to be the good candidates foralloHCT due to potential graft versus lymphomaeffect; these diseases include relapsed T cell lym-phoblastic lymphoma, relapsed anaplastic largecell lymphoma, and Hodgkin lymphoma (failedautologous HCT) (Gross et al. 2010; Satwani et al.2015a).
Nonmalignant DiseasesWhile indications for alloHCT for malignant dis-orders in children are decreasing, improved safetyand efficacy of alloHCT procedures may beincreasing the number of patients undergoingalloHCT for nonmalignant disorders (Shenoyand Boelens 2015). In addition, the improvementin genome sequencing resulting in molecularcharacterization of previously undiagnosedimmunological disorders is increasing indicationsfor alloHCT in nonmalignant diseases. The graftversus malignancy effect is not needed in non-malignant disease. In the nonmalignant diseasesetting, every effort should be employed todecrease the occurrence of GVHD, especiallychronic GVHD among patients undergoing elec-tive alloHCT (Shenoy and Boelens 2015).Chronic GVHD can often lead to a significantdecrease in the quality of life for patients whosometimes feel like they are trading one chronicdisease for another. Other toxicities associatedwith alloHCT, i.e., gonadal failure, growth failure,
poor bone health, and secondary malignancies,must be reduced (Shenoy and Boelens 2015).
One potential strategy to reduce late toxicitiesis to decrease the dose of chemotherapy in condi-tioning regimens. However, reduction in chemo-therapy could result in mixed chimerism, which isstill curative in some nonmalignant diseases(Satwani et al. 2013). However, mixed chimerismcould progress into graft failure, which isunpredictable. Optimal reduced intensity condi-tioning regimens that will result in successfulengraftment, low incidence of acute and chronicGVHD, and minimal long-term toxicities remainselusive. For patients with nonmalignant disorders,organ injury due to disease process prior to trans-plant is a key determinant of success. AlloHCT isunlikely to reverse organ damage sustained priorto transplant. However, in some cases diseasestabilization can be achieved. Delay in the diag-nosis among patients with inborn errors of metab-olism can result in brain injury and potentiallymakes them ineligible for transplant. Similarly,among children with primary immunodeficiency,systemic infections can result in poor outcomesfollowing alloHCT.
HemoglobinopathiesMajority of alloHCT for nonmalignant disease areindicated for transfusion-dependent thalassemia(TDT) and sickle cell disease (SCD). In both ofthese diseases, MSD alloHCT is associated with>90% cure rate (Bhatia et al. 2014). The majorityof TDT patients are concentrated in SoutheastAsian and Middle Eastern countries. The bonemarrow transplantation field is rapidly evolvingespecially in India (Weatherall and Clegg 1996;Weatherall 2018). In India, there are not enoughcenters to meet the demand. Lack of awareness ofthe benefits of transplants and financial affordabil-ity remain major challenges.
In a recent collaboration between CIBMTR,centers in India and China published the largeststudy on outcomes of TDT (Li et al. 2019). In thisstudy, data from 1110 patients were analyzed.Thalassemia-free survival was significantly betterfor patients younger than 6 years old (86%) com-pared to 7–15 years (80%) versus older than15–25 years old (63%). The other important
Pediatric Bone Marrow Transplantation 13

information gleaned from this study was that thal-assemia-free survival was similar between HLA-matched related and HLA-matched unrelated allo-HCT (87% vs. 82%). Since age is a major deter-minant, families with a child with thalassemiashould be educated and encouraged to considerHLA typing of the siblings at an early age andproceed to alloHCT promptly if MSD is available.The ideal age for alloHCT for TDT is probablybetween 2 and 6 years old.
Sickle cell disease can progressively result inmulti-organ dysfunction resulting in a shorterlifespan. MSD alloHCT has been associated witha >90% of cure rate. However, it is crucial toperform alloHCT before significant end-organdamage occurs, as transplant is unable to reverseit. AlloHCT should be offered to every patientwith HLA-matched sibling (Bhatia et al. 2014;Shenoy 2007). However, many patients do nothave HLA-matched sibling. In those situations,patients with symptomatic SCD (stroke, multipleVOC, multiple acute chest syndrome events)should be considered for MUD alloHCT,haploidentical alloHCT, or gene therapy (Shenoy2013).
In the study of 1000 patients, the 5-year SCD-free survival was 91.4% following HLA-matchedsibling transplant, similar to a thalassemia studytransplant in which age was a critical factor indetermining an outcome (Gluckman et al. 2017;Li et al. 2019). Patients <16 years old had signif-icantly better SCD-free survival than >16 yearsold. Interestingly, for every 1-year increment inage, there was a 10% increased risk of death. Moreimportantly, the incidence of cGVHD was 14%following MSD alloHCT. However, in a prospec-tive study conducted by BMT CTN, among chil-dren undergoing MUD alloHCT, 1-year incidencerate of chronic GVHD was 62%. High incidenceof chronic GVHD is worrisome (Shenoy et al.2016). Better strategies must be designed toimprove the SCD-free survival and chronicGVHD incidence to increase the acceptability ofMUD alloHCT.
Severe Aplastic AnemiaIdiopathic severe aplastic anemia (Arnold et al.2017) is rare but a life-threatening disorder in
children. SAA patients with HLA-matched sib-ling should promptly proceed to alloHCT. In thecurrent era, >90% patients are cured with MSDalloHCT (D’Souza 2018). The guidelines to man-age patients without HLA-matched siblings wereestablished at a time when MUD transplants werenot very efficacious, and it was recommended thatpatients without a MSD should receive immuno-suppressive therapy (IST) (Barone et al. 2015;Kojima et al. 2000). However, immunosuppres-sive therapy is associated with suboptimal hema-tological response and increases risk of infection(Miano and Dufour 2015). Over the last fewdecades, safety and efficacy of MUD alloHCThas improved significantly. Now many centersconduct upfront MUD or haploidentical alloHCTfor patients with SAA. In a study by Dufour et al.,the survival of MUD was similar to MSD allo-HCT (91% vs. 96%), and event-free survival wassuperior in MUD alloHCT when compared to anIST historical control (Dufour et al. 2015).Upfront MUD versus IST issue remains contro-versial. To get a definitive answer, the PediatricBlood and Marrow Transplant Consortium isconducting “TransIT” trial in North America. Inthis prospective randomized study, clinical out-comes will be compared between children receiv-ing IST versus unrelated donor transplants.
Other Nonmalignant DiseasesSeveral other nonmalignant diseases includingcongenital bone marrow failures, primary immu-nodeficiency disorders, primary hemophagocyticlymphohistiocytosis, inborn error of metabolism,osteopetrosis, etc. could be successfully treatedwith MUD alloHCT (Shenoy and Boelens 2015).
T Cell-Replete Haploidentical SCT
We all inherit one haplotype from each parent orshare a haplotype with relatives in the family orpass on one haplotype to our children. So, withsuccess of haploidentical (half-matched) relateddonor SCT, we all have donors available withinour family. Haploidentical SCT withunmanipulated graft doesn’t need expensive lab-oratory and machines to deplete T cells. This has
14 S. P. Yadav et al.

made this technique spread around the globequickly. As donor registries are lacking in thedeveloping world and ethnic minorities havepoor chance of finding a MUD, T-repletehaploidentical technique has made donor avail-able for almost everyone who needs to undergoBMT. This has given a huge push to the number ofBMT around the world, and haploidentical donorhas already overtaken umbilical cord as a graftsource and is soon going to cross MSD (D’Souza2018). There are two platforms for performing T-replete haploidentical HSCT. The first one is Bei-jing protocol and the other is PTCy (Hopkinsplatform).We will briefly discuss Beijing protocolfirst followed by PTCy in more detail as that hasbeen more commonly used across the globe withgood success.
T Cell-Replete Haploidentical SCT:Beijing (GIAC) Protocol
One-child norm in China made MSD unavailablefor most patients needing BMT. As unrelateddonor registries were lacking in China, this forcedphysicians to find a way of doing BMT by usinghaploidentical donors (parent to child, child toparent, or half matched relatives for others).Thus, Beijing haploidentical protocol was devel-oped. This proved to be a turning point in historyof BMT. The success made available donors forthe largest population in the world. Essentiallyhere there is no T cell depletion, and after givingconditioning, an unmanipulated haploidenticalrelated donor graft is infused. A combined graftfrom the same donor is collected peripheral stemcells and G-mobilized bone marrow and infusedin the recipient after giving myeloablative condi-tioning. Posttransplant for GVHD prophylaxis,intense immune suppression is given (GIAC pro-tocol after G-CSF, intensified immunologic sup-pression, anti-thymocyte globulin, andcombination of PB and BM grafts). Incidence ofGVHD in 250 acute leukemia patients werehigher than those seen with PTCy (46% gradeII � IV aGVHD and 54% cGVHD), whereasalmost all patients had successful engraftment(Huang et al. 2009).
Haploidentical Stem CellTransplantation with PosttransplantCyclophosphamide
PTCy is thought to prevent GVHD by eliminatingrapidly dividing donor T cells induced by themajor HLA mismatch early after thehaploidentical graft infusion. Furthermore, quies-cent progenitor cells and memory T cells in thegraft are less susceptible to cyclophosphamidedue to their high levels of aldehyde dehydroge-nase (Luznik et al. 2012; Jones et al. 1995). In thedeveloping world, haploidentical SCTwith PTCytechnique has become as popular in children as inadults. It has global acceptance and is more pop-ular in the developing world as there is a lack ofdonor registries for MUD BMT. Low cost, easilyavailable, and good success have brought thisBMT option almost at par with MUD especiallyin patients with leukemia (Luznik et al. 2012).Acute GVHD rates are 25–30% and severe grade4 GVHD<5%. Chronic GVHD rates are 15–25%in various reports but much lower than MUD(Luznik et al. 2008). Bone marrow as a graftsource is likely to have less chronic GVHD andless cytokine release syndrome (CRS). CRSincreases risk of TRM. TRM ranges between10% and 20%. Initially, mostly non-myeloablative (NMA) conditioning was usedwhich had high relapse rate of 45%, but nowMAC option has proven to be safe especially forleukemia. Raiola et al. reported grade II–III acuteGVHD incidence of 12% and disease-free sur-vival of 68% after a median follow-up of333 days in a cohort of 50 patients with high-risk hematological malignancies who underwenthaploidentical SCT with PTCy and busulfan ortotal body irradiation (TBI)-based myeloablativeconditioning (Raiola et al. 2013). PBSC as a graftsource leads to higher rate of chronic GVHD.Choice of donor selection is another area ofdebate. Higher HLA mismatch and HLA DR mis-match are preferred in selecting a haploidenticaldonor for leukemia patients for improved out-comes (Solomon et al. 2018).
Pediatric Bone Marrow Transplantation 15

LeukemiaData in pediatric leukemia with PTCy approach isnot as big as adults. Published data is promising inboth ALL and AML (Jaiswal et al. 2016a; Hong etal. 2018). Conditioning NMAversus MAC debateis open. But most centers are moving towardMAC. For AML, it’s TBF, and for ALL, TBI-based regimes are preferred. But superiority ofone regimen over other is yet to be established.Early removal of immune suppression is one strat-egy to prevent relapse. Targeted therapies post-BMT are options in children as well. Role ofdonor lymphocyte infusion (DLI) is yet notestablished. However, DLI is safe post-haploidentical PTCy, and not a huge risk of severeGVHD has been shown (Jaiswal et al. 2016a).
Thalassemia and Sickle Cell AnemiaClearly science has moved ahead! Sickle and thal-assemia burden is huge globally. As matched sib-ling donors are available to only about 30% ofpatients, remaining patients need an option. MUDis not available for most children with sickle andthalassemia who are non-Caucasians. Initially,success of haploidentical PTCy was reported insickle with NMA from Hopkins group. Although100% survival, the high rejection rate of 43% wasa problem (Bolanos-Meade et al. 2012). Later on,Vanderbilt global learning collaborative improvedconditioning by adding thiotepa to Hopkins pro-tocol. It showed 100% survival and disease-freesurvival of 93% (de la Fuente et al. 2019a). NowBMT CTN 1507 trial has opened forhaploidentical SCTwith PTCy for sickle cell ane-mia across the USAwith the same conditioning.
Success of haploidentical PTCy BMT in sickleleads to this approach being used in thalassemiamajor as well. Thiotepa-based conditioning wastried in 12 children in London with overall sur-vival of 89%, whereas US group just increaseddose of TBI to 4 Gy from 2 Gy in 11 children andshowed 100% success (de la Fuente et al. 2019).So essentially it was RIC versus NMA in thisstudy, and both options were good and graftsource in both was bone marrow. Very low rateof GVHDwas impressive. Another approach withPTIS followed by MAC from Thailand showedOS 95% and EFS 94% in cohort of 31 children.
Here PTIS was given with two cycles of FluDex1 month apart starting 60 days before BMT. Con-ditioning was BuFlu followed by PTCy. Heregraft was PBSC. Acute GVHD was seen in 29%patients, and chronic GVHD rate was 16%(Anurathapan et al. 2016).
Primary Immunodeficiency and InbornError of MetabolismData with PTCy in PID is not a lot but is highlypromising. One study reported survival of 75%with reduced toxicity conditioning and PTCyapproach (Rastogi et al. 2018). Another studyfrom India of 16 children who underwenthaploidentical SCT for PID reported OS of 62%.Only 75% children could engraft. Acute GVHDwas seen in 50% and CMV reactivation in 44%(Uppuluri et al. 2019). Another study from Franceof 27 children (22 PID and 5 osteopetrosis)showed OS of 77%. Acute GVHD was seen in44%, chronic GVHD in 24%, and autoimmunityin 29% (Neven et al. 2019). Haploidentical PTCydata on inborn error of metabolism is mostlylacking.
Aplastic AnemiaAgain there is no data on upfront haploidenticalPTCy in severe aplastic anemia. However, Hop-kins group has reported OS and EFS of 100% in aseries of 16 patients of refractory aplastic anemiawho underwent BMT with PTCy approach (13haploidentical donor and 3 MUD). It’s a veryimpressive strategy with low rates of GVHD(DeZern et al. 2017).
Haploidentical Stem CellTransplantation T Cell Depleted
A significant breakthrough in GVHD preventionin HLA-mismatched transplantation was thedepletion of donor T cells by physical or immu-nological methods. This included soybean lectinagglutination, rosette depletion, and monoclonalantibody-mediated methods (Frame et al. 1989;Waldmann et al. 1984; Friedrich et al. 1984), andimmunomagnetic technique refined the processfurther (Booth et al. 2013). The first ex vivo T
16 S. P. Yadav et al.

cell-depleted BMT, using soybean agglutinin androsette formation with sheep red blood cells, wereperformed in children with immunodeficiencysyndromes (Friedrich et al. 1984). Subsequently,Henslee-Downey et al. (Mehta et al. 2004) usedtotal body irradiation (TBI)-based myeloablativeconditioning (MAC) with partial in vitro TCD ofhaploidentical BM using anti-CD3 antibodieswith reasonable success. The introduction of theone-step, semiautomated MACS® device(CliniMACS, Miltenyi Biotec, BergischGladbach, Germany) brought further improve-ment, including the achievement of a medianCD34-cell purity of 97% and an extensive deple-tion of Tand B cells. Pioneering clinical trials withthe Miltenyi semiautomated device wereperformed in children and showed a high engraft-ment rate with a low incidence of acute andchronic GVHD. T cell depletion (TCD), in vivo(e.g., ATG, alemtuzumab) or in vitro (TCD orCD34 selection), has been traditionally used toreduce the risk of GVHD in haploidentical trans-plant (haplo-HSCT) setting, but the outcomesremain poor due to higher risk of host-mediatedgraft rejection, delayed immune recovery, andinfections.
Modern haplo-HSCT platforms (e.g., post-transplant cyclophosphamide) and graft processingtechnologies (e.g., TCRαβ T and B cell depletion)
have tried to address the problems related to haplo-HSCT done with traditional TCD. The outcome ofhaplo-HSCT has now reached “non-inferiority”status in comparison to HLA-matched HSCT formost indications using these modern approaches.The depletion of CD19+ B cells is commonly donetogether with CD3 or TCRαβ depletion to preventthe occurrence of EBV-associated PTLD sinceTCD increases the risk of EBV infection consider-ably. Although the threshold dose of contaminatingB cells is still not defined, no cases of PTLD wereobserved in two multicenter trials with children(Lang et al. 2014) and adults (Federmann et al.2012) after infusion of median numbers of 28 and7 � 103 CD20+ cells/kg BW, respectively.Approaches of mismatched donor ex vivo graftprocessing in current use or under developmentare (1) pan T cell depletion, (2) TCRαβ T and Bcell depletion, and (3) naïve TCD.
Principles of T Cell Depletion (Table 3)
Pan T Cell DepletionTCD can be performed by positive (CD34 enrich-ment) or negative selection (using board panel ofmAbs against T cells). In haplo-HSCT, residual Tcells in the donor graft should not exceed50–100 � 103/kg. The CliniMACS CD3/CD19
Table 3 Principles of various ex vivo TCD platforms for haplo-HSCT
CD3 T celldepletion CD34+ selection or enrichment
TCRαβ T celldepletion
Naïve T celldepletion
Efficacy ofTCD
3.5–4 log 4–5 log 4.5–5 log 4.5 log
CD34 recovery +++ +++ +++ ++
Graft failurerisk
++ +++ +/� +/�
Acute GVHDrisk
+++ +(<10%) + ++ (steroidresponsive)
ChronicGVHD risk
+ Nil Rare Rare
Infection risk +++ ++++ +++ ++
Early GvLeffect
NK cells Poor NK and γδ T cells TM cells
Immunerecovery
Delayed Very delayed Early Early
Conditioning Immune-myeloablative
Myeloablative + intensiveimmune ablation
Myeloablative Myeloablative
Pediatric Bone Marrow Transplantation 17

Product Line was developed for the simultaneousdepletion of unwanted T and B cells in combina-tion with the CliniMACS System. This approachkeeps stem and progenitor cells untouched andleaves immune effector cells, such as NK cellsand dendritic cells, in the cellular product. There-fore, posttransplant application of a moderatepharmacologic GVHD prophylaxis is necessaryin most patients. Pan TCD has largely been aban-doned in many settings due to high graft failurerates (especially for HLA-mismatched grafts)(Seidel et al. 2005). GVH reaction from donor Tcells in graft reduces the residual population capa-ble of alloreactivity, thus decreasing risk of graftrejection. Utilizing this principle, one can infuselimited number of donor Tcells to recipient (T celladd-back) along with CD34-enriched product.This will reduce the risk of GVHD and helpfight infections in post-HSCT period. Head-to-head comparison between pan TCD approach toHSCT and TCRαβ-depleted HSCT or naïve TCDapproach is not available. However, based onevidence, it is clear that the graft failure incidencesare much higher in delayed immune recovery isfamiliar (Buckley et al. 1999; Pai et al. 2014).
TCRab and B Cell DepletionEx vivo processing of the graft using animmunomagnetic method (CliniMACS, MiltenyiBiotec, Bergisch Gladbach, Germany) results indepletion of GVHD-causing TCRαβ T cells andretention of CD34+ with committed progenitorcells, NK and TCRγδ T cells. A high proportionof NK and TCRγδ T cells promote engraftment(Locatelli et al. 2013) and contribute to rapidimmune reconstitution. In addition, they kill can-cer cells in an MHC-independent manner(Ruggeri et al. 2007) and are involved in anti-CMV responses (Couzi et al. 2015). They do notmediate GVHD and are cytotoxic to mesenchy-mal stromal cells. Optimal graft composition iscrucial for good outcomes, and the following cellthresholds of cells are recommended (per Kg ofrecipient body weight) in graft post-processing:αβ T cells<1� 105, B cells<1� 105, and CD34cells 5–10� 106/kg. Although various condition-ing regimens have been studied with TCRαβhaplo, a MAC with the use of low-dose anti-T
lymphocyte globulin (ATLG, Grafalon) (15 mg/kg total) and rituximab (200 mg/m2) on D-1 is themost common in use. Omitting ATG results in anincreased risk of severe GVHD (Shelikhova et al.2019). Two alkylators are commonly used in con-ditioning for patients with hemoglobinopathies,leukemias, previous GF, and some non-SCIDPIDs. Posttransplant GVHD prophylaxis is notneeded, but some groups use it for a brief duration(2–3 months) with lower CI targets (Shah et al.2018) for nonmalignant indications. In compari-son with other techniques of T cell depletion,including positive selection of CD34+ cells andCD3/CD19 depletion, TCRαβ depletion revealeda comparable or better performance in terms ofCD34 enrichment and CD3/CD19 depletion, withmore constant results and lower coefficients ofvariation (Schumm et al. 2013). Bellicum trial(NCT02065869) is a modification where depletedαβ T cells are genetically modified (BPX-501/rivogenlecleucel) by incorporating CaspaCIDe®
safety switch based on the fusion of humancaspase 9 to human FK506-binding protein andgiven back to recipient around 2 weeks after D-0.These cells are hypothesized to reduce incidenceof viral infections and improve immune reconsti-tution. In case GVHD occurs, the switch can beactivated by rimiducid (AP1903), and alloreactiveT cells get eliminated (Di Stasi et al. 2011). Thisapproach has shown promising results in initialtrials involving children.
The major drawbacks are a requirement ofregulatory approvals for cellular processingwhich is costly (~$15,000 USD/depletion), train-ing of lab personnel, and the need of laboratoryinfrastructure (cell washing platforms,CliniMACS device, flow cytometry support,etc.). Most published studies included theCliniMACS Plus system (Miltenyi Biotec, Ger-many) for graft processing which involves multi-ple manual interventions. Now, an automated cellprocessing closed system is available(CliniMACS Prodigy®) and is simpler to imple-ment and use. Clinical studies using Prodigy® arelacking, and there are concerns regarding losingthe graft if automation is defective during pro-cessing. With T cell depletion, viral infectionrisk increases considerably in early post-HSCT
18 S. P. Yadav et al.

period and needs vigorous monitoring and pre-emptive treatment. However, evidence suggestthe risk may be comparable to MUD transplants(Bertaina et al. 2018).
While low-grade (grade I/II) skin aGVHD iscommonly reported with the TCRαβ haplo, it israrely (<5% incidence) associated with visceral,severe skin, or chronic GVHD even with the useof PBSC in both nonmalignant (Shah et al. 2018)and malignant (Bertaina et al. 2018) conditions.The presence of TCRαβ cells in the graft is a causefor mild aGVHD (Shah et al. 2018), but its lowercontent and a high level of γδ T cells in the graftguarantee near absence of severe aGVHD orcGVHD. This is also true for those with a veryhigh risk of GVHD development as shown byShah et al. (2018) in patients with pre-existingrefractory GVHD resulting from first HSCT(underlying pro-inflammatory milieu) and thenundergoing second HSCT with TCRαβ-depletedgraft. Maschan et al. (2016) reported higher occur-rence of cGVHD (30% at 2 years) with the use ofTCRαβ-depleted graft in children undergoingHSCT for AML, but it was related to the use ofdonor lymphocyte infusion (DLI) post-HSCT. ARussian study (Shelikhova et al. 2016) showed ahigher incidence of acute and chronic GVHDwithequine ATG in conditioning compared to rATGwith TCRαβ-depleted transplant.
Naïve T Cell Depletionαβ T cells exist in the blood, secondary lymphoidorgans, and tissues as distinct naive (TN), effector(TE), and memory (TM) subsets. The latterincludes effector memory (TEM) and central mem-ory (TCM) T cells. The TN [CD45RA+CD62L+]subset is antigen inexperienced and has a morediverse TCR repertoire than TM. Preclinical datashowed TN cells to be a significant contributor toGVHD occurrence (Chen et al. 2007; Anderson etal. 2003), while TM cells give anti-infective andGvL activities. CD45RA-depleted apheresisproducts also contain CD4+ and CD8+ T cellswith intact in vitro action against CMV, adenovi-rus, EBV, and Aspergillus and Candida speciesantigens. Bleakley et al. developed novel two-stepgraft engineering strategy (Bleakley et al. 2014) todeplete CD45RA cells from the graft and retain
pathogen memory-specific T cells. CD34+ HSCs(which also express CD45RA) are initiallyselected from G-CSF-mobilized apheresis prod-ucts, followed by depletion of CD45RA+ cellsfrom CD34-depleted fraction using murine anti-CD45RA monoclonal antibodies directly conju-gated to iron dextran beads. Terminally differen-tiated effector memory re-expressing CD45RA(TEMRA) (CCR7-CD45RA+) cells and B cellsare also removed (Appay et al. 2008). The CD34-CD45RA- fraction from the second selection stepis infused into the patient, along with the CD34+fraction. Post-CD45RA depletion, the targeted Tcell content is 1 � 107 cells/Kg in the graft whichis co-infused with CD34+ selected fraction topatient. The resulting naïve T cell content in theprocessed graft is<7.5� 104/Kg recipient weight(>99.9% depletion). In addition to a 4.5–5.0-logdepletion of naïve T cells, CD45RA-depletedproducts contain a lower number of Treg, Bcells, γδ Tcells, and NK cells, all of which expressCD45RA. CD45RA depletion of the apheresisproduct has been successfully used as post-HSCT DLI strategy (Muller et al. 2018). Theconcept was tested by the same group in 35 adultswith hematological malignancies with promisingoutcomes (Bleakley et al. 2015). Importantly, thefrequency and pattern of aGVHD were similar tothe T-replete graft transplants, but GVHD wasalways responsive to steroids. The estimatedprobability of chronic GVHD was 9% at 2 yearscompared with 50% rates in a contemporarycohort of patients receiving T cell-replete grafts.
Infection Risk Post-HSCT in TCD Haplo-HSCT Approaches
Immediate anti-infective effects are contributedby γδ T with NK cells in TCRαβ haplo. The Tcell numbers rapidly expand post-HSCT afterTCRαβ T cell-depleted HSCT, but it is not asso-ciated with the reconstitution of protective TCRdiversity (Zvyagin et al. 2017). Incidence ofreported systemic viral reactivation after the useof TCRαβ-depleted HSCT ranges from 40% to70% in children. With TCRαβ haplo, BK hemor-rhagic cystitis is not common (Locatelli et al.
Pediatric Bone Marrow Transplantation 19

2017; Balashov et al. 2015; Shah et al. 2018) evenwith MAC regimens. However, a study in patientswith hemoglobinopathies getting cyclophospha-mide-based conditioning regimen with TCRαβdepletion of donor graft reported a 35% incidence(Gaziev et al. 2018). A study assessing TCRαβdepletion showed that despite high viralreactivations, outcomes of transplant are notaffected (Laberko et al. 2017) which are onlychanged when the patient undergoes HSCT withongoing active systemic viral infection (Shah etal. 2018). The risk of EBV reactivation and EBV-related PTLD appear to be very low in patientsundergoing TCRαβ haplo. In a retrospective studyfrom Italy (Bertaina et al. 2018), a lower risk ofbacterial infections in TCRαβ haplo (8%) groupwas found in comparison with MUD (17%) andthe MMUD-HSCT (34%) groups which is partlyexplained by faster engraftment of neutrophils inTCRαβ group.
CD45RA depletion provides a large number ofdonor memory T cells to the recipients and isassociated with enhanced early T cell recoveryand protection against viremia. A study in chil-dren comparing infection occurrence between panTCD platform and naïve TCD platform confirmedthis (Triplett et al. 2018). In this analysis, apartfrom viral infection frequency, prolonged viremiaand adenoviremia were much less familiar withnaïve TCD platform. However, unusually higherrate of HHV-6 encephalitis was found in a 25-children cohort receiving naïve TCD transplants(Sisinni et al. 2018a). The cumulative incidence ofHHV-6 encephalitis was 31%, with an early post-HSCT onset (median 35 days). The authorshypothesized high number of CD4 cells in thedonor graft (reservoir of the virus) as an underly-ing pathogenesis for this occurrence. The mainadvantage of naïve TCD approach over TCRαβhaplo is faster clearance of systemic viral infec-tions post-HSCT (Touzot et al. 2015); however,head-to-head comparison is lacking to assesswhether this translates into survival benefit. Forpatients who cannot clear viral infections beforeD-0, the TCRαβ haplo outcomes are inferior, andnaïve TCD approach may result in better out-comes. In addition, CD45RA-depleted DLI canalso be used post-haplo-HSCT for clearance of
viral infections without high risk of severeGVHD (Brodszki et al. 2016).
Evidence in Children
TCRab and B Cell Depletion
Malignant Illnesses (Table 4)Evidence shows good outcomes for children get-ting haplo-HSCT for leukemias, which dependson CR status before HSCT. A German groupreported their pediatric results with this approachusing a MAC regimen. With a median follow-upof 19 months, the DFS was 51% and 100% forthose who were transplanted in CR. Engraftmentoccurred in 88%, grade III–IV aGVHD in 15%,cGVHD in 19%, and relapse in 47%. Shelikhovaet al. (2019) reported a 49% EFS in those withrefractory AML. Locatelli et al. (2017) reportedoutcomes of 80 children with leukemia recruitedinto a prospective, single-center phase 2 trial.With median 46 months of follow-up, the 5-yearDFS was 71% comparable toMSD orMUD trans-plants (n¼ 92) done during the same period in thestudy institution. In multivariate analysis, only theuse of TBI remained as a factor affecting DFS.These study findings were replicated in a sizeablemulticentric study (Bertaina et al. 2018) involving343 leukemia patients (98 with αβ haplo). It notonly showed comparable cGVHD/DFS with useof TCR-alpha-beta haplo or MUD transplants butsuperior outcomes with haplo compared toMMUD transplants (61% for αβ haplo, 58% forMUD, and 34% for MMUD) (Bertaina et al.2018). However, this study did not show the roleof TBI as a protective factor for relapse.
Nonmalignant Illnesses (Table 5)Use in PID patients has been described in threewell-done studies from different countries withconsistent results, and the detailed description isavailable elsewhere (Shah et al. 2018). UK groupcompared four mismatched HSCT approaches(cord blood, CD34 selection plus T cell add-back, unmanipulated marrow, and TCRαβ haplo)used in PID patients over 11 years (Elfeky et al.2019). This retrospective analysis showed no
20 S. P. Yadav et al.
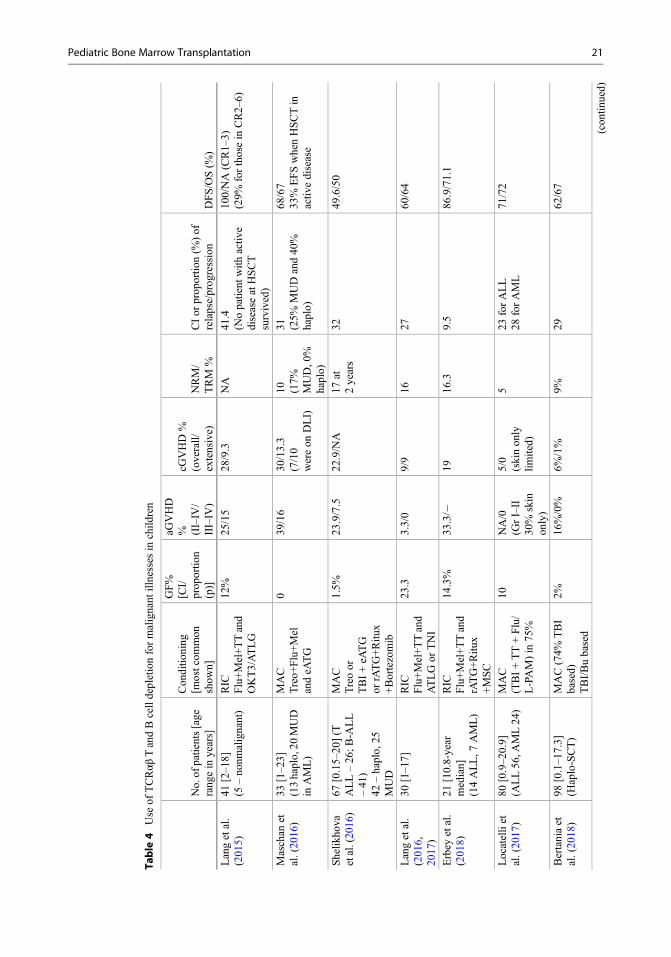
Table
4Use
ofTCRαβ
TandBcelldepletionformalignant
illnesses
inchild
ren
No.of
patients[age
rang
ein
years]
Con
ditio
ning
[mostcom
mon
show
n]
GF%
[CI/
prop
ortio
n(p)]
aGVHD
% (II–IV
/III–IV
)
cGVHD%
(overall/
extensive)
NRM/
TRM
%CIor
prop
ortio
n(%
)of
relapse/prog
ression
DFS/OS(%
)
Langetal.
(201
5)41
[2–1
8](5
–no
nmalignant)
RIC
Flu+Mel+TTand
OKT3/ATLG
12%
25/15
28/9.3
NA
41.4
(Nopatient
with
activ
ediseaseatHSCT
survived)
100/NA(CR1–
3)(29%
forthosein
CR2–
6)
Maschan
etal.(20
16)
33[1–2
3](13haplo,20
MUD
inAML)
MAC
Treo+
Flu+Mel
andeA
TG
039
/16
30/13.3
(7/10
wereon
DLI)
10 (17%
MUD,0
%haplo)
31 (25%
MUDand40
%haplo)
68/67
33%
EFSwhenHSCTin
activ
edisease
Shelik
hova
etal.(20
16)
67[0.15–20
](T
ALL–26
;B-A
LL
–41
)42
–haplo,
25MUD
MAC
Treoor
TBI+eA
TG
orrATG+Ritu
x+Bortezomib
1.5%
23.9/7.5
22.9/NA
17at
2years
3249
.6/50
Langetal.
(201
6,20
17)
30[1–1
7]RIC
Flu+Mel+TTand
ATLGor
TNI
23.3
3.3/0
9/9
1627
60/64
Erbey
etal.
(201
8)21
[10.8-year
median]
(14ALL,7
AML)
RIC
Flu+Mel+TTand
rATG+Ritu
x+MSC
14.3%
33.3/�
1916
.39.5
86.9/71.1
Locatelliet
al.(20
17)
80[0.9–2
0.9]
(ALL56
,AML24
)MAC
(TBI+TT+Flu/
L-PAM)in
75%
10NA/0
(GrI–II
30%
skin
only)
5/0
(skinon
lylim
ited)
523
forALL
28forAML
71/72
Bertaniaet
al.(20
18)
98[0.1–1
7.3]
(Haplo-SCT)
MAC(74%
TBI
based)
TBI/Bubased
2%16
%/0%
6%/1%
9%29
62/67
(con
tinued)
Pediatric Bone Marrow Transplantation 21

Table
4(con
tinue
d)
No.of
patients[age
rang
ein
years]
Con
ditio
ning
[mostcom
mon
show
n]
GF%
[CI/
prop
ortio
n(p)]
aGVHD
% (II–IV
/III–IV
)
cGVHD%
(overall/
extensive)
NRM/
TRM
%CIor
prop
ortio
n(%
)of
relapse/prog
ression
DFS/OS(%
)
+ATLG/rATG
+Ritu
x
Shelik
hova
etal.(20
19)
22[1–1
8]RefractoryAML
MAC
(Treo+
Mel/
TT+Ritu
x/+Bortezomib
Tocilizumab
+Abatacept)
(CD45
RA-
depleted
DLIin
17)
NA
18/(3/4
with
Gr
III–IV
)
23/NA
942
%at2years
49/53
(7patientsreceived
post-
engraftm
entp
roph
ylactic
chem
o)
Abb
reviations:A
ML,acutemyeloid
leuk
emia;A
LL,acutelymph
oblasticleuk
emia;eATG,equ
ineanti-thym
ocyteglob
ulin;rATG,rabbitanti-thym
ocyteglob
ulin;A
TLG,anti-T
lymph
ocyteglob
ulin;Bu,
busulfan;CR,completeremission
;Cy,
cyclop
hosphamide;
Flu,flud
arabine;
GVHD,graft-versus-hostdisease;
MAC,myeloablativ
econd
ition
ing;
MDS,m
yelody
splasticsynd
rome;Mel,m
elph
alan;M
SC,m
esenchym
alstem
cells;N
RM,non
-relapse
mortality;RIC,reduced
intensity
cond
ition
ing;SOT,solid
organtransplant;
TBI,totalb
odyirradiation;
TRM,transplantrelated
mortality;
TT,
thiotepa;V
P16
,etopo
side
22 S. P. Yadav et al.

Table
5Use
ofTCRαβ
TandBcelldepletionin
nonm
alignant
illnesses
inchild
ren
No.
ofpatients
(age)
Con
ditio
ning
GF%
[CI/
prop
ortio
n(p)]
aGVHD(II–IV
/III–IV)
cGVHD
(overall/
extensive)
NRM/
TRM
CI(%
)Infections
EFS/OS
Bertainaetal.
(201
4)23
[0.4–1
2](PID
¼11)
MAC
Treo/Bu+Flu
+TT
ATLG+Ritu
x
16.2
13.1/0
09.3
38forCMVand
adeno
74/91.1@2year
Balasho
vetal.
(201
5)37
[0.2–1
7]10
haplo,
27MUD
Treo
+Flu�M
elrATG
27%
(GFrelatedtoon
lyon
ealky
latoruse)
25.4/2.8
(GrIV
inapatient
who
didno
tgetcond
ition
ing)
5.4/2.7
3.3
46(CMV)
67.7/96.5
@15
mon
ths
Shahetal.2
017
(Shahetal.201
8)26
(0.2–
10.3)
PID
MAC-
Treo+
Flu+TT
in80
.7%
4.2
48.4/4.3
016
.458
.8(CMVand/or
adeno)
80.4/83.9
Deripapaetal.
(201
7)15 NBS
MAC
Bu+Flu+Cy
rATG
200(allHLA-m
atched
dono
rs)
03 deaths
NA(one
patient
died
ofadeno-
hepatitis)
NA/76(vs.30
inthosewith
out
HSCT)
Gazievetal.
(201
8)14
[3–1
5.2]
Thal(11)
SCD(3)
Bu+Cy+TT
+ TBI(2G
y)+HUAzFlu
(D-59to
D-
12)
1428
/�21
/21
CMV64
EBVPTLD23
%(non
ereceived
Ritu
x)
69/84
ImHoetal.
(201
5)21
Flu
+Cy+rATG
+TBI4G
y
0NA
NA
NA
NA
NA/94@
35mon
ths
Abb
reviations:rATG,rabb
itanti-thym
ocyteglob
ulin;Bu,
busulfan;CMV,cytomegalov
irus;Cy,
cyclop
hosphamide;
DOCK
8,dedicatorof
cytokinesis8deficiency;EBV,
Epstein-Barrvirus;Flu,fl
udarabine;MAC,m
yeloablativ
econd
ition
ing;NBS,N
ijmegen
breakage
synd
rome;PID
,primaryim
mun
odeficiency;P
TLD,posttransplantlymph
opro-
liferativedisorder;R
itux,
rituximab;T
reo,
treosulfan;T
T,thiotepa;W
AS,W
isko
tt-Aldrich
synd
rome
Pediatric Bone Marrow Transplantation 23

difference in survival with the use of TCRαβhaplo (76.7% at 2 years) (Elfeky et al. 2019)compared to other approaches. The highlight ofthis comparative analysis was that none of the 30patients undergoing HSCT with TCRαβ haplodeveloped cGVHD and the incidence of aGVHDwas only 11.5%. Gaziev et al. (2018) reported a69% DFS and 84% OS in 14 children with hemo-globinopathies getting TCRαβ haplo after MAC.DFS was higher compared to patients gettingCD34-selected grafts (69% vs. 39%, p ¼ 0.08).In this study, a higher incidence of extensivecGVHD (21%) and CMV reactivation (64%) isreported, and it might be because of higher ATGdose used which might have affected γδ T cells inthe graft. Interestingly, 37% incidence of autoim-munity was reported in patients who did not getrituximab as part of the conditioning regimen.
Immune reconstitution of Tand B cell numbersseems to be robust with TCRαβ haplo and com-parable to HLA-matched transplants (Berger et al.2016). In PID patients getting TCRαβ-depletedhaplo-HSCT, the median time to reachCD4 > 200 was 111 days was 129 days. The Bcell recovery >200 μl happened at median85 days with TCRαβ haplo (Shah et al. 2018) inPID patients. Eighty-two percent of patientsstopped requiring Ig replacement by 1 year post-HSCT after TCRαβ haplo in PID patients (Shah etal. 2018).
Naïve TCDEvidence using naïve TCD in children is sparse,and it is limited to case series or reports. Shook etal. (2015) described use of naïve TCD (single-stepCD45 depletion) haplo-HSCT in eight childrenwith refractory solid tumors. The early outcomeswere promising with no occurrence of GVHD andengraftment with complete donor chimerism in allpatients. A French study tested two-step CD45depletion process in five children with PID andchronic viral infections (Touzot et al. 2015). Fourof five patients successfully engrafted and showedprompt viral clearance. Early T cell responseagainst viral pathogen is detected in two patients.A Spanish group reported outcomes of naïve TCDhaplo-HSCT in 25 children with leukemia (Sisinniet al. 2018b). The cumulative incidence of
aGVHD II–IV and III–IV was 39% and 33%,respectively, and was universally responsive tosteroids except in one patient who required extra-corporeal photopheresis. The cumulative inci-dence of cGVHD at 30 months was 22%. Thecumulative incidence of relapse in this cohortwas 16.6% with 60% survival at 1 year post-HSCT (Sisinni et al. 2018a). St Jude group haspublished feasibility and TRM outcomes of chil-dren with leukemia undergoing CD45RA-depleted HSCT and compared it to children get-ting pan TCD transplants (Triplett et al. 2018).This study showed promising early results andlower infection and TRM risk in this high-riskcohort; the survival outcomes are awaited.
Autologous HSCT
Autologous HSCT is mostly offered for childrenwith lymphoma or solid tumors like brain tumors,neuroblastoma, etc. This procedure is safe and hasvery low TRM<2%. High-dose chemotherapy asconditioning helps in clearing the residual tumorand helps in achieving cure, while autologousstem cells help recover counts after HSCT. Thereis no graft versus tumor effect. Conditioning isalways MAC. Stem cell harvest is usually a chal-lenge as prior chemotherapy and/or radiotherapymakes it difficult to collect the cells and young ageespecially infants with neuroblastoma also makesit difficult to collect cells as it has to be done inintensive care setting with close monitoring of thevitals of the infants and also sedation is needed inyoung children during the procedure. Ideally stemcells should be collected before giving radiother-apy in brain tumors like medulloblastoma or after2–3 cycles of chemotherapy in solid tumors likelymphoma or neuroblastoma to get a good yield.Prior radiotherapy, cisplatin, carboplatin, andother exposure leads to poor stem cell mobiliza-tion. Traditionally G-CSF has been used to mobi-lize stem cells, but now data is emerging thatsingle-dose peg-G-CSF can also be used (Schaubet al. 2013). In poor mobilisers, like in adults,plerixafor can be used for a successful stem cellharvest.
24 S. P. Yadav et al.

Lymphoma
Hodgkin lymphoma with relapse auto-SCT is usu-ally offered autologous SCT. It is highly effectivein those who are chemosensitive before autolo-gous SCT (Garfin et al. 2015). BEAM/LACE arechosen conditioning regimens. In non-Hodgkinlymphoma (NHL), also autologous SCT is offeredespecially for relapsed DLBCL or Burkitt orALCL, etc. (Gross et al. 2010; Satwani et al.2015a).
Brain Tumors
In medulloblastoma, SJMB96 protocol withupfront tandem mini auto-SCT has shown OS of85% in average-risk and 70% in high-risk disease(Gajjar et al. 2006). In infants, baby brain pro-tocols HEAD START I, II, III, and IV have built inautologous SCT to avoid cranial radiotherapy.Radiotherapy free EFS was 29% in HEADSTART III protocol (Davidson et al. 2011).
Neuroblastoma
For neuroblastoma, high-risk auto-SCT is part ofoverall treatment. Matthay et al. reported EFS of40% with chemotherapy, surgery, autologousSCT, radiotherapy, and cis-retinoic acid therapy(Matthay et al. 2009). With addition of anti-GD2antibody to the above regimen, the EFS increasedto 66% (Yu et al. 2010). In a randomized clinicaltrial that included 652 eligible patients with high-risk neuroblastoma, tandem autologous stem celltransplant versus single transplant resulted in 3-year event-free survival of 61.6% versus 48.4%, adifference that was statistically significant (Park etal. 2019). But long-term side effects would remaina concern with tandem transplants.
Other Tumors
In relapsed malignant germ cell tumor, autologousSCT should be offered, and it can lead to cure in40% of cases. Ewing sarcoma auto-SCT has been
offered in metastatic or relapsed cases as lastresort with success of 20% or less. Other solidtumors published data is lacking.
Autoimmune Disorders
In adults with relapsing/remitting multiple sclero-sis, it’s a standard of care and can lead to pro-longed disease stabilization. However, data inchildren is lacking.
Gene Therapy and CAR T Cell Therapy
Gene Therapy
It is a therapeutic modality that involves introduc-ing new genes into the cells or tissues of a personor the manipulation of their pre-existing geneticmaterial to treat or prevent diseases. Althoughthere have been various attempts to develop suc-cessful gene therapies in the past 50 years, thefield has recently seen rapid progress as a resultof the introduction of versatile gene-editing toolsand advances in our understanding of deliveryplatforms. Since 2016, the European and USdrug regulatory agencies have approved six genetherapy products (146), including two chimericantigen receptor (CAR) T cell products and fourproducts targeting monogenic disorders. As of2019, more than 800 cell and gene therapy prod-ucts are in development, including some aimed attreating diseases such as Duchenne muscular dys-trophy, Huntington disease, and Hutchinson-Gilford progeria syndrome – all diseases forwhich no therapeutic interventions have hithertobeen available (146–149).
Monogenic disorders are particularly suited totreatment by gene therapy because they can beaddressed by targeting a single gene or locus,either by supplying a functional copy of the defec-tive gene or by correcting the defective gene itself.The ideal outcome after gene therapy is thesustained long-term expression of the newly intro-duced transgene or the suppression of the defec-tive gene that is producing a deleterious protein.Three main strategies have been used to achieve
Pediatric Bone Marrow Transplantation 25

these goals. The first strategy is to introduce anintegrating vector into a precursor cell thatdelivers the transgene to the cell nucleus(150–159). The transgene then integrates into thehost genome and is passed to every daughter cell.The second strategy is to deliver a non-integratingvector carrying a transgene to a long-lived post-mitotic cell. In this case, the expression of the non-integrating transgene is sustained for the life ofthat cell (160–163). The third strategy involvesediting the host genome with nucleases to accom-plish the desired genetic manipulation (164–169).In this chapter, we will focus on the gene therapyapproaches currently in clinical or advanced pre-clinical development to target hematopoietic stemand progenitor cells (HSPCs) and T cells with thegoal of treating blood and bone marrow disorders.Other relevant applications involving HSPCs andT cells are briefly mentioned, with referencesbeing provided for additional reading.
General Principles of Gene TherapyAlmost all current HSPC and T cell gene therapymethods involve the collection of HSPCs or Tcells, in vitro culture of the cells, transduction ofthe cells with the vector of interest to deliver thetransgene or the genome-editing machinery, and,finally, re-transplantation of the genetically mod-ified cells into patients after appropriate condi-tioning. Key concepts in this process arediscussed below.
Site of Genetic Modification and Collectionof HSPCsMost of the approaches currently used in clinicalpractice for gene therapy of HSPCs involve exvivo (outside the body) transduction of HSPCsharvested from the patient. In this process,HSPCs are extracted from the patient byharvesting bone marrow (150, 158,159) or bycollecting HSPCs (identified by the CD34marker) that have been mobilized from the bonemarrow niche into the peripheral blood(151,152,158). The mode of collection dependson the number of HSPCs needed for gene therapy,as well as on the feasibility of automated collec-tion of mobilized cells from the peripheral bloodvia leukapheresis.
Gene therapy for hemoglobinopathies requiresreplacing a substantial fraction of the patient’shematopoietic progenitors with genetically modi-fied cells; hence, the usual goal is to collect morethan 5–10 � 106/kg CD34+ cells for processing,but it is challenging to obtain such high cell yieldsvia bone marrow harvests. Additionally, recentstudies suggest that the quality of HSPCs obtainedfrom the bone marrow of patients with sickle celldisease may be suboptimal for lentiviral transduc-tion in comparison to peripheral blood-derivedmobilized CD34+ cells (170, 171). Increasedinflammation, cellular aggregation, and contami-nation with early differentiated progenitors withinthe bone marrow niche lead to markedly poorCD34+ HSPC transduction ability and yield inpatients with sickle cell disease (170,171).Hence, for gene therapy in patients with hemoglo-binopathies, mobilized peripheral blood-derivedCD34+ cells are increasingly preferred overHSPCs harvested from the bone marrow.
At the same time, the use of granulocyte col-ony-stimulating factor (G-CSF) – the most com-mon stem cell-mobilizing agent – iscontraindicated in patients with sickle cell diseasebecause of the risk of inducing vaso-occlusivecrises or even death. Fortunately, plerixafor hasbeen demonstrated to be a safe and effectivemobilization agent in patients with sickle celldisease (172,173). Plerixafor reversibly inhibitsthe binding of stromal cell-derived factor 1α(SDF-1α) to its cognate receptor, CXCR4, onHSPCs (174–176). When SDF-1α interacts withCXCR4, it promotes increased homing, quies-cence, and retention of human HSPCs in the mar-row compartment, and its inhibition results in therapid mobilization of CD34+ HSPCs into the cir-culation without inducing hyperleukocytosis(174–176).
Lastly, leukapheresis using automated mononu-clear cell collection/separating devices cannot beperformed in very small children and infants (177),such as those with inherited enzyme defects orimmunodeficiencies. In such situations, a bone mar-row harvest may be the only available means ofcollecting HSPCs for gene therapy (154–156,159).
T cells for CAR T cell development are col-lected from the peripheral blood without cytokine
26 S. P. Yadav et al.

stimulation. The collection is achieved using auto-mated apheresis devices in a manner similar to theperipheral blood-derived HSPC collectiondescribed earlier. Patients with hematologicmalignant neoplasms who need CAR T cell ther-apy usually have a low white blood cell count,making the collection of CD3+ cells challenging.The goal is usually to collect 0.6–2 � 109 CD3+cells (178). Although CAR T cells can bemanufactured with fewer T cells, the transductionefficiency is poor (178). After manufacture, thecells are infused using patient weight-baseddosing.
Platform for Gene TherapyDepending upon whether the genetic modificationis to be performed within the patient’s body or thecells are to be extracted and modified ex vivo, oneor more platforms may be suitable for deliveringthe new genetic material into the cells or tissues ofinterest. Early approaches to ex vivo modificationof HSPCs involved delivering the gene of interestby using a γ-retroviral vector (179–181). Unfor-tunately, many patients treated with these earlyvectors experienced insertional mutagenesis withresultant leukemia (182, 183). These early mis-haps prompted considerable research in this areaand led to the development of much safer vectorsthat are being currently used in clinical applica-tions (150, 184, 185). Lentiviral vectors are thecurrent platform of choice for gene therapybecause they can transduce nondividing HSPCswith high efficiency and have a safer integrationpattern (186, 187). These vectors are being used totreat inherited blood disorders, including hemo-globinopathies, severe combined immunodefi-ciency (SCID), Wiskott-Aldrich syndrome, X-linked adrenoleukodystrophy, and metachromaticleukodystrophy (150–152, 154–158).
Despite their improving safety profile, ex vivogene therapy approaches are prohibitively expen-sive and technically complex, and they can beperformed only in certain facilities. In addition,there are concerns that the extensive manipulationand cytokine stimulation of HSPCs in vitro mightaffect their pluripotency. Myeloablative chemo-therapy is also needed as a conditioning agent fornonmalignant disease before re-transplantation of
the genetically modified cells. This exposure tochemotherapy is associated with nonhematologicearly and late toxicities. To overcome these limita-tions, in vivo gene therapy procedures are beingdeveloped that are highly cost-effective for use inresource-limited settings. Early results from two ofthese approaches, one using a direct IVadministra-tion of foamy virus vectors in a canine model ofSCID-X1 (188) and another involving a helper-dependent adenovirus vector in a murine model ofβ-thalassemia (189), have been encouraging. Sim-ilar approaches with non-integrating hepatotropicadeno-associated virus vectors for treating hemo-philia B have previously been effective, albeit com-plicated by host immune response against thevectors (163).
Conditioning RegimenThe conditioning therapy used before transplanta-tion of genetically modified cells depends on theunderlying disease process. Gene therapy proto-cols that use a lentiviral vector encoding a mutatedadult β-globin with anti-sickling properties(HbAT87Q) in patients with hemoglobinopathieshave employed myeloablative busulfan-basedconditioning before infusion of transducedHSPCs (151, 152). The production of a higherpercentage of red blood cells expressing thisanti-sickling globin from modified HSPCs uponrecovery leads to a greater improvement in theclinical phenotype (151, 152). Similarly, patientsundergoing gene therapy for lysosomal storagediseases such as metachromatic leukodystrophyor enzyme deficiencies such as X-linked adreno-leukodystrophy benefit from genetic correction ofa high percentage of their HSPCs because of theresulting long-term, high-level expression of themissing proteins (155, 156). Thus, myeloablationhas been recommended as a means of replacingmost of the native host HSPCs with geneticallymodified HSPCs. Myeloablation enables the bonemarrow niche to be opened and creates space forthe autologous genetically modified stem cells.Busulfan-based conditioning has been generallywell tolerated, with the expected toxicity profile,although one patient treated on the HGB-206study did develop myelodysplasia syndromeapproximately 3 years after receiving an infusion
Pediatric Bone Marrow Transplantation 27

of modified cells, and that was probably second-ary to busulfan exposure (190). As most of thefocus until recently has been on developing effec-tive gene therapies, this case is a reminder that theimpact of the conditioning agents must also becarefully considered in future trials.
Two recent independent reports of gene ther-apy for infants with X-linked SCID havedescribed the successful use of low-exposure,targeted busulfan conditioning with minimal non-hematologic toxic effects (150, 159). In an ongo-ing trial of gene therapy targeting the FANCA genein patients with Fanconi anemia, the cellular prod-uct is delivered with no conditioning regimenbecause of the increased susceptibility of thesepatients to DNA damage from chemotherapyand their relatively empty marrow niches (169).These approaches to reducing conditioning inten-sity or eliminating it completely are expected toreduce the long-term adverse effects of chemo-therapy exposure in growing children and in thosewho are otherwise at higher risk of complications.Novel non-genotoxic and highly targetedapproaches using CD117 antibody-drug conju-gates are also being tested as a conditioning reg-imen in human trials (191–193). A clinical trial ofa CD117 monoclonal antibody-based condition-ing approach for allogeneic hematopoietic pro-genitor cell transplantation is already underwayin children with SCID (194). Whereas themyeloablative effect of the conditioning chemo-therapy on leukemic cells is beneficial for patientsundergoing transplants to treat malignant hemato-logic disorders, myelotoxicity has no additionalbenefit for patients with nonmalignant blood dis-orders apart from facilitating the replacement ofthe hematopoietic stem cells carrying the defec-tive gene. These highly targeted approaches usingmonoclonal antibodies would be ideal for patientsreceiving autologous transplants of gene-modifiedHSPCs.
Early Clinical Applications
HemoglobinopathiesHemoglobinopathies, including β-thalassemiaand sickle cell disease, have been viewed asideal targets for genetic manipulation since their
molecular basis was identified. However, thecomplex regulation of globin genes had madethe successful clinical application of gene therapychallenging (195). An improved understanding ofthe genetic regulation of globin genes, the identi-fication of several naturally occurring mutationsassociated with high levels of fetal hemoglobin,and the development of new gene-editing tech-niques have ushered in an era of expedited clinicaldevelopment of curative therapies for hemoglo-binopathies. Currently, several gene therapyapproaches for treating β-thalassemia are beingtested in clinical trials or preclinical studies(196). Of these, a phase 3 trial of a vectorencoding a variant adult β-globin (HbAT87Q)showed transfusion independence in patientswith transfusion-dependent β-thalassemia for upto 56 months, resulting in European MedicinesAgency (EMA) approval for Zynteglo (fromBluebird Bio) for patients older than 12 years(197). Other approaches to globin gene transferare currently in early phase clinical trials (198,199). Clinical trials targeting BCL11A, a repres-sor of γ-globin genes, in which short hairpin RNA(200, 201) or CRISPR-Cas9 gene editing is usedto disrupt an erythroid-specific enhancer (202,203), are ongoing, with promising early resultsin patients with β-thalassemia or sickle cell dis-ease. Correction of the mutated β-globin gene insickle cell disease by using site-specific generepair mechanisms is also being evaluated in pre-clinical studies, and human trials are awaited(165, 204–206).
Severe Combined ImmunodeficiencyEarly gene therapy studies of γ-retroviral vectorsin children with adenosine deaminase (ADA)-deficient SCID resulted in the short-lived restora-tion of cellular and humoral immunity (207). Theuse of some of these early vectors resulted ininsertional mutagenesis in trial participants (182,183), leading to the development of much safervectors that are being used today (150, 184, 185).Incorporating low-dose cytoreductive chemother-apy before the infusion of genetically modifiedcells yielded more engraftment and longer sur-vival of gene-corrected cells (159). Recently,low-dose busulfan followed by lentiviral vector
28 S. P. Yadav et al.

gene therapy in infants with X-linked SCID(SCID-X1) resulted in broad immune reconstitu-tion, with B cell reconstitution being especiallyevident (150). Thus, for the first time, gene ther-apy has restored humoral immunity in patientswith SCID-X1 (150). These developments willundoubtedly change the management of SCID inthe years to come.
CAR T Cells
The approval of novel immunotherapy agents,namely, tisagenlecleucel (from Novartis) andaxicabtagene ciloleucel (from Kite Pharma), bythe FDA in 2017 and by the EMA in 2018 forpatients with refractory acute lymphoblastic leu-kemia (ALL) and non-Hodgkin lymphoma,respectively, has revolutionized the definition oftargeted therapies in oncology. Although thedetails of CAR T cell therapy and the specifics ofdifferent CART cells are beyond the scope of thischapter, let it suffice to say that these therapieshave demonstrated remarkable curative potentialwith respect to previously incurable leukemias.Both of these agents are autologous CD19-directed gene-modified T cells that have a chime-ric T cell receptor with an anti-CD19 single-chainvariable fragment domain coupled to intracellularT cell signalling domains of the T cell receptor(208). CD19 is universally expressed on the sur-face of B cells (including leukemia cells) but is notfound on hematopoietic stem cells; hence, theseCARs redirect cytotoxic T cells toward B cells(and leukemia cells) expressing the CD19 antigen.These CAR T cells not only recognize leukemiacells and mount their effector function againstthem but also expand and proliferate, just likenormal T cells, in response to their target antigenand act like “living” drugs that may persist in theirhosts for several months. Several pediatric studiesusing various CAR T cell products have showngreat early success in treating patients withrelapsed/refractory CD19+ B cell ALL. Majordifferences between these studies include differ-ences in the co-stimulatory domains of the chime-ric receptor, the vector constructs, and the use oflymphodepleting chemotherapy (208–212). The
pivotal multicenter clinical trial of CTL019(ELIANA) showed a 12-month overall survivalof 76% and an event-free survival of 50% inpediatric patients with relapsed or refractory Bcell ALL (213). This trial resulted in the eventualapproval of tisagenlecleucel by the FDA fortreating patients aged 1 to 25 years with refractoryor relapsed (second or subsequent relapse) CD19+ALL. Similar CAR T cell products targeting var-ious other tumor-associated antigens are underinvestigation for treating several other leukemias,lymphomas, and solid tumors, and they may soonbecome frontline therapies replacing conventionalchemotherapy-based regimens.
Long-Term Follow-Up of ChildrenPost-BMT
Around the world, nearly 60,000 BMT areperformed annually and a significant number areperformed for children. Many children are nowlong-term survivors. They have a need to befollowed for late effects of therapy given for can-cer or sequelae of original genetic or immunedisorder and late effects of BMT. Most of thepublished results are with short follow-up of1–3 years post-BMT. Many children die even>2 years post-BMT due to complications otherthan relapse of the original disease and many keepon having issues like chronic GVHD, second can-cer, cataract, cardiac dysfunction, pulmonary tox-icity, infertility, etc. which affects their quality oflife. Normal growth and development of a child orinfant can be affected due to BMT, and they maydevelop psychological and social issues post-BMT especially as they transition into adulthoodfrom childhood (Cupit et al. 2016). Here we willhighlight long-term follow-up needs of certainsubgroups of pediatric BMT (Bhatia et al. 2011).
Leukemia/Lymphoma/Solid Tumors
These children will need to be followed for sideeffects of chemotherapy/radiotherapy as well aslong-term complications of BMT. In late effectclinic, they need to have a good general physical
Pediatric Bone Marrow Transplantation 29

examination, systemic examination, and growthand developmental assessment. Tests for variousorgan dysfunctions need to be performed likecardiac; liver; renal; lungs; central nervous sys-tem; hematological; eyes; ear, nose, and throat;and endocrine. Look for complications or impactof chronic GVHD on various organs like the skin,joints, eyes, liver, lungs, and hematopoietic sys-tem. Monitor for second malignancy. Fertilityneeds to be assessed. Psychological support andsocial support needs for transition into adult carefrom pediatric care.
Hemoglobinopathy
Children with sickle cell anemia would have mor-bidity due to sickle like strokes, lung damage,avascular necrosis of bones, increased risk ofinfections to absent spleen, retinopathy, nephrop-athy, etc. which would need long-term follow-up.They will also have sequelae of BMT of condi-tioning affecting various organs and impact ofchronic GVHD affecting quality of life.
Thalassemia children especially those withPesaro class III prior to BMTwill have iron over-load affecting the liver, heart, skin, and endocrinefunction, puberty, and growth. Children need to bemonitored carefully for these complications.Intensive chelation needs to be restarted afterBMT to reduce iron overload which helpsimprove function of certain vital organs like theheart and liver. Fertility and marriage issueswould need psychosocial support.
Inherited Bone Marrow FailureSyndromes
Children with Fanconi anemia have special needs.At diagnosis itself they may have many organsaffected like congenital heart disease, deafness,short stature, ectopic or single kidney, andepispadias/hypospadias which would need multi-speciality care over a long period of time. Fanconichildren are at risk of developing cancer 300–400times higher than normal population. Risk ofdeveloping myelodysplastic syndrome or acute
myeloid leukemia is very high which reducedsubstantially post-BMT. They have very highrisk of developing head and neck cancers, andthis risk persists post-BMT or increases if theydevelop chronic GVHD post-BMT. Vaccinationagainst human papilloma virus is highlyrecommended post-BMT to reduce risk of cancer.
Dyskeratosis congenita will have high risk ofdeveloping pulmonary and hepatic dysfunctionpost-BMT. Children with Diamond-Blackfan ane-mia may have multiple other birth defects likecleft palate, cleft lip, cataract, cardiac defects,etc. needing multiple surgeries. If heavily trans-fused prior to BMT, then chelation needs to becontinued post-BMT until excess iron is removedfrom the body.
Primary Immunodeficiency Disorders
Children with PID may have associated compli-cations as part of the syndrome (e.g., omen syn-drome affecting skin, developmental delay,autoimmune problems, and increased risk of lym-phoma). Patients of SCID who undergo BMTwithout conditioning may never engraft B lym-phocytes of donor, thus needing intravenousimmunoglobulin replacement therapy for manydecades unless they undergo a second BMT tocorrect this. RIC BMT are commonly performedfor PID, and those with mixed chimerism tend todevelop autoimmunity independent of donor lym-phocytes. Many are very infants when theyundergo BMT for SCID and are at very high riskof developmental delay due to impact of chemo-therapy and steroid on the developing brain.Manymay develop attention deficit hyperactivitydisorder.
Inborn Error of Metabolism
Children with Hurler and Hunter syndrome havemultiple organs affected as part of syndrome. Sothey need different specialist to take care of lungs,eyes, skeleton, heart, and brain complications.Developmental assessment at baseline and thenannually is needed to asses benefit of BMT.
30 S. P. Yadav et al.

Similarly, children with X-linked adrenoleuko-dystrophy need endocrine and neurology teamsto be closely involved in the care even afterBMT. Other rare metabolic disorders may needspecialized care.
Conclusion
Pediatric BMT has come a long way from initialera of doing BMTwithout HLA typing with highmorbidity and mortality. BMT for children hasbecome much safer and less toxic due to betterconditioning regimens, GVHD prophylaxis, andbetter supportive care. Haploidentical donor BMThas expanded donor pool for children. CAR-T celltherapy has opened up the era of cellular therapyto treat cancer in children. Gene therapy is makingfast strides in treating thalassemia and sickle cellanemia.
References
Allen CE, Marsh R, Dawson P, Bollard CM, Shenoy S,Roehrs P et al (2018) Reduced-intensity conditioningfor hematopoietic cell transplant for HLH and primaryimmune deficiencies. Blood 132:1438–1451. https://doi.org/10.1182/blood-2018-01-828277
Alter BP (2017) Inherited bone marrow failure syndromes:considerations pre- and posttransplant. HematologyAm Soc Hematol Educ Prog 2017(1):88–95
Anasetti C, Logan BR, Lee SJ, Waller EK, Weisdorf DJ,Wingard JR, Cutler CS, Westervelt P, Woolfrey A,Couban S, Ehninger G, Johnston L, Maziarz RT,Pulsipher MA, Porter DL, Mineishi S, McCarty JM,Khan SP, Anderlini P, Bensinger WI, Leitman SF,Rowley SD, Bredeson C, Carter SL, Horowitz MM,Confer DL, Blood & Marrow Transplant Clinical Tri-als, N (2012) Peripheral-blood stem cells versus bonemarrow from unrelated donors. N Engl J Med367:1487–1496
Anderson BE, Mcniff J, Yan J, Doyle H, Mamula M,Shlomchik MJ, Shlomchik WD (2003) Memory CD4+T cells do not induce graft-versus-host disease. J ClinInvest 112:101–108
Anurathapan U, Hongeng S, Pakakasama S, SirachainanN, Songdej D, Chuansumrit A, Charoenkwan P,Jetsrisuparb A, Sanpakit K, Rujkijyanont P,Meekaewkunchorn A, Lektrakul Y, Iamsirirak P,Surapolchai P, Satayasai W, Sirireung S, Sruamsiri R,Wahidiyat PA, Ungkanont A, Issaragrisil S, AnderssonBS (2016) Hematopoietic stem cell transplantation forhomozygous β-thalassemia and β-thalassemia/
hemoglobin E patients from haploidentical donors.Bone Marrow Transplant 51:813–818
Appay V, van Lier RA, Sallusto F, Roederer M (2008)Phenotype and function of human T lymphocyte sub-sets: consensus and issues. Cytometry A 73:975–983
Arnold SD, Brazauskas R, He N, Li Y, Aplenc R, Jin Z,Hall M, Atsuta Y, Dalal J, Hahn T, Khera N, Bonfim C,Majhail NS, Diaz MA, Freytes CO, Wood WA, SavaniBN, Kamble RT, Parsons S, Ahmed I, Sullivan K,Beattie S, Dandoy C, Munker R, Marino S, Bitan M,Abdel-Azim H, Aljurf M, Olsson RF, Joshi S,Buchbinder D, Eckrich MJ, Hashmi S, Lazarus H,Marks DI, Steinberg A, Saad A, Gergis U,Krishnamurti L, Abraham A, Rangarajan HG, WaltersM, Lipscomb J, Saber W, Satwani P (2017) Clinicalrisks and healthcare utilization of hematopoietic celltransplantation for sickle cell disease in the USAusing merged databases. Haematologica 102:1823–1832
Auerbach AD, Liu Q, Ghosh R, Pollack MS, Douglas GW,Broxmeyer HE (1990) Prenatal identification of poten-tial donors for umbilical cord blood transplantation forFanconi anemia. Transfusion 30:682–687
Balashov D, Shcherbina A, Maschan M, Trakhtman P,Skvortsova Y, Shelikhova L, Laberko A, Livshits A,Novichkova G, Maschan A (2015) Single-center expe-rience of unrelated and haploidentical stem cell trans-plantation with TCRalphabeta and CD19 depletion inchildren with primary immunodeficiency syndromes.Biol Blood Marrow Transplant 21:1955–1962
Ballen KK, Gluckman E, Broxmeyer HE (2013) Umbilicalcord blood transplantation: the first 25 years andbeyond. Blood 122:491–498
Barone A, Lucarelli A, Onofrillo D, Verzegnassi F,Bonanomi S, Cesaro S, Fioredda F, Iori AP, LadoganaS, Locasciulli A, Longoni D, Lanciotti M, Macaluso A,Mandaglio R, Marra N, Martire B, Maruzzi M, MennaG, Notarangelo LD, Palazzi G, Pillon M, Ramenghi U,Russo G, Svahn J, Timeus F, Tucci F, Cugno C, ZeccaM, Farruggia P, Dufour C, Saracco P, Marrow FailureStudy Group of the Pediatric Haemato-Oncology Ital-ian, A (2015) Diagnosis and management of acquiredaplastic anemia in childhood. Guidelines from the Mar-row Failure Study Group of the Pediatric Haemato-Oncology Italian Association (AIEOP). Blood CellsMol Dis 55:40–47
BergerM, Lanino E, Cesaro S, ZeccaM, Vassallo E, FaraciM, De Bortoli M, Barat V, Prete A, Fagioli F (2016)Feasibility and outcome of haploidentical hematopoi-etic stem cell transplantation with post-transplant high-dose cyclophosphamide for children and adolescentswith hematologic malignancies: an AIEOP-GITMORetrospective Multicenter Study. Biol Blood MarrowTransplant 22:902–909
Bertaina A, Merli P, Rutella S, Pagliara D, Bernardo ME,Masetti R, Pende D, Falco M, Handgretinger R,Moretta F, Lucarelli B, Brescia LP, Li Pira G, Testi M,Cancrini C, Kabbara N, Carsetti R, Finocchi A,MorettaA, Moretta L, Locatelli F (2014) HLA-haploidentical
Pediatric Bone Marrow Transplantation 31

stem cell transplantation after removal of alphabeta+ Tand B cells in children with nonmalignant disorders.Blood 124:822–826
Bertaina A, Zecca M, Buldini B, Sacchi N, Algeri M,Saglio F, Perotti C, Gallina AM, Bertaina V, LaninoE, Prete A, Barberi W, Tumino M, Favre C, Cesaro S,Del Bufalo F, Ripaldi M, Boghen S, Casazza G,RabusinM, Balduzzi A, Fagioli F, Pagliara D, LocatelliF (2018) Unrelated donor vs HLA-haploidenticalalpha/beta T-cell and B-cell depleted HSCT in childrenwith acute leukemia. Blood 132:2594
Bhatia S, Davies SM, Scott Baker K, Pulsipher MA,Hansen J (2011) NHLBI first international consensusconference on late effects after pediatric hematopoieticcell transplantation: etiology and pathogenesis of lateeffects after HCT performed in childhood –methodologic challenges. Biol Blood Marrow Trans-plant 17(10):1428–1435
Bhatia M, Jin Z, Baker C, Geyer MB, Radhakrishnan K,Morris E, Satwani P, George D, Garvin J, Del Toro G,Zuckerman W, Lee MT, Licursi M, Hawks R, SmilowE, Baxter-Lowe LA, Schwartz J, Cairo MS (2014)Reduced toxicity, myeloablative conditioning withBU, fludarabine, alemtuzumab and SCT from siblingdonors in children with sickle cell disease. Bone Mar-row Transplant 49:913–920
Bleakley M, Heimfeld S, Jones LA, Turtle C, Krause D,Riddell SR, Shlomchik W (2014) Engineering humanperipheral blood stem cell grafts that are depleted ofnaive T cells and retain functional pathogen-specificmemory T cells. Biol Blood Marrow Transplant20:705–716
Bleakley M, Heimfeld S, Loeb KR, Jones LA, Chaney C,Seropian S, Gooley TA, Sommermeyer F, Riddell SR,Shlomchik WD (2015) Outcomes of acute leukemiapatients transplanted with naive T cell-depleted stemcell grafts. J Clin Invest 125:2677–2689
Boelens JJ, Orchard PJ, Wynn R (2014) Transplantation ininborn errors of metabolism: current considerations andfuture perspectives. Br J Haematol 167:293–303
Bolanos-Meade J, Fuchs EJ, Luznik L, Lanzkron SM,Gamper CJ, Jones RJ, Brodsky RA (2012) HLA-haploidentical bone marrow transplantation with post-transplant cyclophosphamide expands the donor poolfor patients with sickle cell disease. Blood 120:4285–4291
Booth C, Lawson S, Veys P (2013) The current role of Tcell depletion in paediatric stem cell transplantation. BrJ Haematol 162:177–190
Bourgeois W, Ricci A, Jin Z, Hall M, George D, Bhatia M,Garvin J, Satwani P (2019) Health care utilization andcost among pediatric patients receiving unrelated donorallogeneic hematopoietic cell transplantation. BoneMarrow Transplant 54:691–699
Brodszki N, Turkiewicz D, Toporski J, Truedsson L, DykesJ (2016) Novel treatment of severe combined immuno-deficiency utilizing ex-vivo T-cell depletedhaploidentical hematopoietic stem cell transplantation
and CD45RA+ depleted donor lymphocyte infusions.Orphanet J Rare Dis 11:5
Buckley RH, Schiff SE, Schiff RI, Markert L, WilliamsLW, Roberts JL, Myers LA, Ward FE (1999) Hemato-poietic stem-cell transplantation for the treatment ofsevere combined immunodeficiency. N Engl J Med340:508–516
Chen BJ, Deoliveira D, Cui X, Le NT, Son J, WhitesidesJF, Chao NJ (2007) Inability of memory T cells toinduce graft-versus-host disease is a result of an abor-tive alloresponse. Blood 109:3115–3123
Cluzeau T, Lambert J, Raus N, Dessaux K, Absi L, DelbosF, Devys A, De Matteis M, Dubois V, Filloux M, FortM, Hau F, Jollet I, Labalette M, Masson D, Mercier B,Pedron B, Perrier P, Picard C, Quainon F, Ramounau-Pigot A, Renac V, Van Endert P, Charron D, Peffault DeLa Tour R, Taupin JL, Loiseau P (2016) Risk factorsand outcome of graft failure after HLA matched andmismatched unrelated donor hematopoietic stem celltransplantation: a study on behalf of SFGM-TC andSFHI. Bone Marrow Transplant 51:687–691
Couzi L, Pitard V, Moreau JF, Merville P, Dechanet-Merville J (2015) Direct and indirect effects of cyto-megalovirus-induced gammadelta T cells after kidneytransplantation. Front Immunol 6:3
Cupit MC, Duncan C, Savani BN et al (2016) Childhood toadult transition and long-term follow-up after bloodand marrow transplantation. Bone Marrow Transplant51:176–181
D’Souza A, FC (2018) Current uses and outcomes ofhematopoietic cell transplantation (HCT): CIBMTRsummary slides, 2018
Dausset J (1958) [Iso-leuko-antibodies]. Acta Haematol20:156–166
Davidson TB, Ji L, Haley K et al (2011) Outcome of headstart III multinational protocol for newly diagnosedcentral nervous system (CNS) primitiveneuroectodermal tumors (PNET) of young children[abstract]. J Clin Oncol. 29(15 Suppl); abstr 9519
De La Fuente J, Dhedin N, Koyama T, Bernaudin F,Kuentz M, Karnik L et al (2019a) Haploidenticalbone marrow transplant with post-transplant cyclo-phosphamide plus thiotepa improves donor engraft-ment in patients with sickle cell anemia: results of aninternational learning collaborative. Biol Blood Mar-row Transplant 25(6):1197–1209
De La Fuente J, Hanna R, Gamper C, Symons H et al(2019b) Augmenting non-myeloablative BMT withPTCY using thiotepa or 400 CGY TBI improvesengraftment in patients with transfusion dependentthalassemia: results of a haploidentical transplant con-sortium for hemoglobinopathies [ICHH]. Biol BloodMarrow Transplant 25(3):S310–S311
Dehn J, Spellman S, Hurley CK, Shaw BE, Barker JN,Burns LJ, Confer DL, Eapen M, Fernandez-Vina MA,Hartzman R, Maiers M, Marino SR, Mueller C, PeralesMA, Rajalingam R, Pidala J (2019) Selection ofunrelated donors and cord blood units for
32 S. P. Yadav et al.

hematopoietic cell transplantation: guidelines fromNMDP/CIBMTR. Blood 134:924
Deripapa E, Balashov D, Rodina Y, Laberko A, MyakovaN, Davydova NV, Gordukova MA, Abramov DS, PayGV, Shelikhova L, Prodeus AP, Maschan MA,Maschan AA, Shcherbina A (2017) Prospective studyof a cohort of Russian Nijmegen breakage syndromepatients demonstrating predictive value of low kappa-deleting recombination excision circle (Krec) numbersand beneficial effect of hematopoietic stem cell trans-plantation (HSCT). Front Immunol 8:807
Dezern AE, ZahurakM, Symons H et al (2017) Alternativedonor transplantation with high-dose post-transplanta-tion cyclophosphamide for refractory severe aplasticanemia. Biol Blood Marrow Transplant 23:498–504
Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A,Martinez C, Straathof K, Liu E, Durett AG, Grilley B,Liu H, Cruz CR, Savoldo B, Gee AP, Schindler J,Krance RA, Heslop HE, Spencer DM, Rooney CM,Brenner MK (2011) Inducible apoptosis as a safetyswitch for adoptive cell therapy. N Engl J Med365:1673–1683
Dufour C, Veys P, Carraro E, Bhatnagar N, Pillon M, WynnR, Gibson B, Vora AJ, Steward CG, Ewins AM, HoughRE, De La Fuente J, Velangi M, Amrolia PJ, Skinner R,Bacigalupo A, Risitano AM, Socie G, Peffault DeLatour R, Passweg J, RovoA, Tichelli A, SchrezenmeierH, Hochsmann B, Bader P, Van Biezen A, Aljurf MD,Kulasekararaj A, Marsh JC, Samarasinghe S (2015)Similar outcome of upfront-unrelated and matched sib-ling stem cell transplantation in idiopathic paediatricaplastic anaemia. A study on behalf of the UK paediatricBMT working party, paediatric diseases working partyand severe aplastic anaemia working party of EBMT. BrJ Haematol 171:585–594
EapenM, HorowitzMM, Klein JP, Champlin RE, LoberizaFR Jr, Ringden O, Wagner JE (2004) Higher mortalityafter allogeneic peripheral-blood transplantation com-pared with bone marrow in children and adolescents:the Histocompatibility and Alternate Stem Cell SourceWorking Committee of the International Bone MarrowTransplant Registry. J Clin Oncol 22:4872–4880
Elfeky R, Shah RM, Unni MNM, Ottaviano G, Rao K,Chiesa R, Amrolia P, Worth A, Flood T, Abinun M,Hambleton S, Cant AJ, Gilmour K, Adams S, Ahsan G,Barge D, Gennery AR, Qasim W, Slatter M, Veys P(2019) New graft manipulation strategies improve theoutcome of mismatched stem cell transplantation inchildren with primary immunodeficiencies. J AllergyClin Immunol 144:280
Erbey F, Akcay A, Atay D, Ovali E, Ozturk G (2018)Comparison of outcomes after HLA-matched unrelatedand alphabeta T-cell-depleted haploidentical hemato-poietic stem cell transplantation for children withhigh-risk acute leukemia. Pediatr Transplant 22:e13192
Federmann B, Bornhauser M, Meisner C, Kordelas L,Beelen DW, Stuhler G, Stelljes M, Schwerdtfeger R,Christopeit M, Behre G, Faul C, Vogel W, SchummM,Handgretinger R, Kanz L, Bethge WA (2012)
Haploidentical allogeneic hematopoietic cell transplan-tation in adults using CD3/CD19 depletion and reducedintensity conditioning: a phase II study. Haematologica97:1523–1531
Frame JN, Collins NH, Cartagena T, Waldmann H,O’Reilly RJ, DuPont B, Kernan NA (1989) T celldepletion of human bone marrow. Comparison ofCampath-1 plus complement, anti-T cell ricin A chainimmunotoxin, and soybean agglutinin alone or in com-bination with sheep erythrocytes or immunomagneticbeads. Transplantation 47:984–988
Friedrich W, Goldmann SF, Vetter U, Fliedner TM,Heymer B, Peter HH, Reisner Y, Kleihauer E (1984)Immunoreconstitution in severe combined immunode-ficiency after transplantation of HLA-haploidentical, T-cell-depleted bone marrow. Lancet 1:761–764
Gajjar A, Chintagumpala M, Ashley D, Kellie S, Kun LE,Merchant TE, Woo S, Wheeler G, Ahern V, Krasin MJ,Fouladi M, Broniscer A, Krance R, Hale GA, StewartCF, Dauser R, Sanford RA, Fuller C, Lau C, Boyett JM,Wallace D, Gilbertson RJ (2006) Risk-adaptedcraniospinal radiotherapy followed by high-dose che-motherapy and stem-cell rescue in children with newlydiagnosed medulloblastoma (St Jude Medulloblas-toma-96): long-term results from a prospective, multi-centre trial. Lancet Oncol 7:813–820
Garfin PM, Link MP, Donaldson SS et al (2015) Improvedoutcomes after autologous bone marrow transplanta-tion for children with relapsed or refractory Hodgkinlymphoma: twenty years experience at a single institu-tion. Biol Blood Marrow Transplant 21:326–334
Gaziev J, Isgro A, Sodani P, Paciaroni K, De Angelis G,Marziali M, Ribersani M, Alfieri C, Lanti A, GalluccioT, Adorno G, Andreani M (2018) Haploidentical HSCTfor hemoglobinopathies: improved outcomes withTCRalphabeta(+)/CD19(+)-depleted grafts. BloodAdv 2:263–270
George B, Methews V, Viswabandya A, Kavitha ML,Srivastava A, ChandyM (2007) Fludarabine and cyclo-phosphamide based reduced intensity conditioning(RIC) regimens reduce rejection and improve outcomein Indian patients undergoing allogeneic stem celltransplantation for severe aplastic anemia. Bone Mar-row Transplant 40(1):13–18
Gluckman E, Cappelli B, Bernaudin F, Labopin M, Volt F,Carreras J, Pinto Simoes B, Ferster A, DuPont S, De LaFuente J, Dalle JH, Zecca M, Walters MC, KrishnamurtiL, Bhatia M, Leung K, Yanik G, Kurtzberg J, Dhedin N,Kuentz M, Michel G, Apperley J, Lutz P, Neven B,Bertrand Y, Vannier JP, Ayas M, Cavazzana M,Matthes-Martin S, Rocha V, Elayoubi H, Kenzey C,Bader P, Locatelli F, Ruggeri A, Eapen M,EUROCORD, T. P. W. P. O. T. E. S. F. B., Marrow, T.,The Center for International, B.&Marrow Transplant, R(2017) Sickle cell disease: an international survey ofresults of HLA-identical sibling hematopoietic stemcell transplantation. Blood 129:1548–1556
Gooley TA, Chien JW, Pergam SA, Hingorani S, SorrorML, Boeckh M, Martin PJ, Sandmaier BM, Marr KA,
Pediatric Bone Marrow Transplantation 33

Appelbaum FR, Storb R, McDonald GB (2010)Reduced mortality after allogeneic hematopoietic-celltransplantation. N Engl J Med 363:2091–2101
Gragert L, Eapen M, Williams E, Freeman J, Spellman S,Baitty R, Hartzman R, Rizzo JD, Horowitz M, ConferD, Maiers M (2014) HLA match likelihoods for hema-topoietic stem-cell grafts in the U.S. registry. N Engl JMed 371:339–348
Gross TG, Hale GA, HeW, Camitta BM, Sanders JE, CairoMS, Hayashi RJ, Termuhlen AM, Zhang MJ, DaviesSM, Eapen M (2010) Hematopoietic stem cell trans-plantation for refractory or recurrent non-Hodgkin lym-phoma in children and adolescents. Biol BloodMarrowTransplant 16:223–230
Hansen JA, Clift RA, Thomas ED, Buckner CD, Storb R,Giblett ER (1980) Transplantation of marrow from anunrelated donor to a patient with acute leukemia. NEngl J Med 303:565–567
Hong KT, Kang HJ, Choi JY, Hong CR, Cheon JE, Park JDet al (2018) Favorable outcome of post-transplantationcyclophosphamide haploidentical peripheral bloodstem cell transplantation with targeted busulfan-basedmyeloablative conditioning using intensive pharmaco-kinetic monitoring in pediatric patients. Biol BloodMarrow Transplant 24:2239–2244
Horan JT, Logan BR, Agovi-Johnson MA, Lazarus HM,Bacigalupo AA, Ballen KK, Bredeson CN, CarabasiMH, Gupta V, Hale GA, Khoury HJ, Juckett MB,Litzow MR, Martino R, McCarthy PL, Smith FO,Rizzo JD, Pasquini MC (2011) Reducing the risk fortransplantation-related mortality after allogeneic hema-topoietic cell transplantation: how much progress hasbeen made? J Clin Oncol 29:805–813
Huang X-J, Liu D-H, Liu K-Y et al (2009) Treatment ofacute leukemia with unmanipulated HLA-mismatched/haploidentical blood and bone marrow transplantation.Biol Blood Marrow Transplant 15:257–265
Im HJ, Koh KN, Seo JJ (2015) Haploidentical hematopoi-etic stem cell transplantation in children and adoles-cents with acquired severe aplastic anemia. Korean JPediatr 58:199–205
Jaiswal SR, Chakrabarti A, Chatterjee S et al (2016a)Haploidentical peripheral blood stem cell transplanta-tion with post-transplantation cyclophosphamide inchildren with advanced acute leukemia withfludarabine-, busulfan-, and melphalan-based condi-tioning. Biol Blood Marrow Transplant 22(3):499–504
Jaiswal SR, Zaman S, Chakrabarti A, Sen S, Mukherjee S,Bhargava S et al (2016b) Improved outcome of refrac-tory/relapsed acute myeloid leukemia after post-trans-plantation cyclophosphamide-based haploidenticaltransplantation with myeloablative conditioning andearly prophylactic granulocyte colony-stimulating fac-tor-mobilized donor lymphocyte infusions. Biol BloodMarrow Transplant 22:1867–1873
Jones RJ, Barber JP, Vala MS et al (1995) Assessment ofaldehyde dehydrogenase in viable cells. Blood85:2742–2746
Keesler DA, St Martin A, Bonfim C, Seber A, Zhang MJ,Eapen M (2018) Bone marrow versus peripheral bloodfrom unrelated donors for children and adolescentswith acute leukemia. Biol Blood Marrow Transplant24:2487–2492
Keever-Taylor CA, Devine SM, Soiffer RJ, Mendizabal A,Carter S, Pasquini MC, Hari PN, Stein A, Lazarus HM,Linker C, Goldstein SC, Stadtmauer EA, O’Reilly RJ(2012) Characteristics of clinimacs(R) system CD34-enriched T cell-depleted grafts in a multicenter trial foracute myeloid leukemia-blood and marrow transplantclinical trials network (BMT CTN) protocol 0303. BiolBlood Marrow Transplant 18:690–697
Kojima S, Nakao S, Tomonaga M, Hows J, Marsh J,Gerard S, Bacigalupo A, Mizoguchi H (2000) Consen-sus conference on the treatment of aplastic anemia. Int JHematol 72:118–123
Kolb EA, Meshinchi S (2015) Acute myeloid leukemia inchildren and adolescents: identification of new molec-ular targets brings promise of new therapies. Hematol-ogy Am Soc Hematol Educ Program 2015:507–513
Laberko A, Bogoyavlenskaya A, Shelikhova L,Shekhovtsova Z, Balashov D, Voronin K, KurnikovaE, Boyakova E, Raykina E, Brilliantova V, Pirumova V,Novichkova G, Maschan A, Maschan M (2017) Riskfactors for and the clinical impact of cytomegalovirusand Epstein-Barr virus infections in pediatric recipientsof TCR-alpha/beta- and CD19-depleted grafts. BiolBlood Marrow Transplant 23:483–490
Lang P, Teltschik HM, Feuchtinger T, Muller I, Pfeiffer M,Schumm M, Ebinger M, Schwarze CP, Gruhn B,Schrauder A, Albert MH, Greil J, Urban C,Handgretinger R (2014) Transplantation of CD3/CD19 depleted allografts from haploidentical familydonors in paediatric leukaemia. Br J Haematol165:688–698
Lang P, Feuchtinger T, Teltschik HM, SchwingerW, Schle-gel P, Pfeiffer M, Schumm M, Lang AM, Lang B,Schwarze CP, Ebinger M, Urban C, Handgretinger R(2015) Improved immune recovery after transplanta-tion of TCRalphabeta/CD19-depleted allografts fromhaploidentical donors in pediatric patients. Bone Mar-row Transplant 50(Suppl 2):S6–S10
Lang PJ, Schlegel PG, Meisel R, Schulz AS, Greil J, BaderP, Karitzky S, Holtkamp S, Siewert C, Schumm M,Eyrich MK, Wiesneth M, Bönig H, Handgretinger R(2016) TCR-alpha/beta and CD19 depletedhaploidentical stem cell transplantation followingreduced intensity conditioning in children: first resultsof a prospective multicenter phase I/II clinical trial.Blood 128:389–389
Lang PJ, Schlegel PG, Meisel R, Schulz AS, Greil J, BaderP, Karitzky S, Holtkamp S, Siewert C, Pflitsch S,Schumm M, Eyrich MK, Wiesneth M, Bonig H,Handgretinger R (2017) Safety and efficacy ofTCRalpha/beta and CD19 depleted haploidenticalstem cell transplantation following reduced intensityconditioning in children: results of a prospective mul-ticenter phase I/II clinical trial. Blood 130:214–214
34 S. P. Yadav et al.

Lee SJ, Logan B,Westervelt P, Cutler C,Woolfrey A, KhanSP, Waller EK, Maziarz RT, Wu J, Shaw BE, Confer D,Horowitz MM, Anasetti C (2016) Comparison ofpatient-reported outcomes in 5-year survivors whoreceived bone marrow vs peripheral blood unrelateddonor transplantation: long-term follow-up of a ran-domized clinical trial. JAMA Oncol 2:1583–1589
Li C, Mathews V, Kim S, George B, Hebert K, Jiang H, LiC, Zhu Y, Keesler DA, Boelens JJ, Dvorak CC,Agarwal R, Auletta JJ, Goyal RK, Hanna R, KasowK, Shenoy S, Smith AR, Walters MC, Eapen M (2019)Related and unrelated donor transplantation for beta-thalassemia major: results of an international survey.Blood Adv 3:2562–2570
Locatelli F, Merli P, Rutella S (2013) At the bedside: innateimmunity as an immunotherapy tool for hematologicalmalignancies. J Leukoc Biol 94:1141–1157
Locatelli F, Merli P, Pagliara D, Li Pira G, Falco M, PendeD, Rondelli R, Lucarelli B, Brescia LP, Masetti R,Milano GM, Bertaina V, Algeri M, Pinto RM,Strocchio L, Meazza R, Grapulin L, Handgretinger R,Moretta A, Bertaina A, Moretta L (2017) Outcome ofchildren with acute leukemia given HLA-haploidentical HSCT after alphabeta T-cell and B-celldepletion. Blood 130:677–685
Lucarelli G, Galimberti M, Polchi P, Angelucci E,Baronciani D et al (1990) Bone marrow transplantationin patients with thalassemia. N Engl JMed 322:417–421
Lucchini G, Labopin M, Beohou E, Dalissier A, Dalle JH,Cornish J, Zecca M, Samarasinghe S, Gibson B,Locatelli F, Bertrand Y, Abdel-Rahman F, Socie G,Sundin M, Lankester A, Sedlacek P, Hamladji RM,Heilmann C, Afanasyev B, Hough R, Peters C, BaderP, Veys P (2017) Impact of conditioning regimen onoutcomes for children with acute myeloid leukemiaundergoing transplantation in first complete remission.An analysis on behalf of the Pediatric Disease WorkingParty of the European Group for Blood and MarrowTransplantation. Biol Blood Marrow Transplant23:467–474
Luznik L, O’Donnell P, Symons H et al (2008) HLA-haploidentical bone marrow transplantation for hemato-logic malignancies using nonmyeloablative condition-ing and high-dose, posttransplantationcyclophosphamide. Biol Blood Marrow Transplant14:641–650
Luznik L, O’Donnell PV, Fuchs EJ (2012) Post-transplan-tation cyclophosphamide for tolerance induction inHLA-haploidentical bone marrow transplantation.Semin Oncol 39:683–693
Maschan M, Shelikhova L, Ilushina M, Kurnikova E,Boyakova E, Balashov D, Persiantseva M, SkvortsovaY, Laberko A, Muzalevskii Y, Kazachenok A,Glushkova S, Bobrynina V, Kalinina V, OlshanskayaY, Baidildina D, Novichkova G, Maschan A (2016)TCR-alpha/beta and CD19 depletion and treosulfan-based conditioning regimen in unrelated andhaploidentical transplantation in children with acute
myeloid leukemia. Bone Marrow Transplant 51:668–674
Mathews V, George B, Deotare U, Lakshmi KM,Viswabandya A et al (2007) A new stratification strat-egy that identifies a subset of class III patients with anadverse prognosis among children with beta thalasse-mia major undergoing a matched related allogeneicstem cell transplantation. Biol Blood Marrow Trans-plant 13:889–894
Mathews V, George B, Viswabandya A, Abraham A,Ahmed R, Ganapule A et al (2013) Improved clinicaloutcomes of high risk beta thalassemia major patientsundergoing a HLAmatched related allogeneic stem celltransplant with a treosulfan based conditioning regimenand peripheral blood stem cell grafts. PLoS One 8(4):e61637
Matthay KK et al (2009) Long-term results for childrenwith high-risk neuroblastoma treated on a randomizedtrial of myeloablative therapy followed by 13-cis-retinoic acid: a children’s oncology group study. JClin Oncol 27:1007–1013
Mehta J, Singhal S, Gee AP, Chiang KY, Godder K, RheeFv F, Derienzo S, O’Neal W, Lamb L, Henslee-Dow-ney PJ (2004) Bone marrow transplantation from par-tially HLA-mismatched family donors for acuteleukemia: single-center experience of 201 patients.Bone Marrow Transplant 33:389–396
Miano M, Dufour C (2015) The diagnosis and treatment ofaplastic anemia: a review. Int J Hematol 101:527–535
Muller N, Landwehr K, Langeveld K, Stenzel J, PouwelsW, van der Hoorn M, Seifried E, Bonig H (2018)Generation of alloreactivity-reduced donor lymphocyteproducts retaining memory function by fully automaticdepletion of CD45RA-positive cells. Cytotherapy20:532–542
Neven B, Diana JS, Castelle M et al (2019) Haploidenticalhematopoietic stem cell transplantation with post-trans-plant cyclophosphamide for primary immunodefi-ciencies and inherited disorders in children. BiolBlood Marrow Transplant 25:1363. https://doi.org/10.1016/j.bbmt.2019.03.009. pii: S1083-8791(19)30184-3
Oliansky DM, Camitta B, Gaynon P, Nieder ML, ParsonsSK, Pulsipher MA, Dillon H, Ratko TA, Wall D,McCarthy PL Jr, Hahn T, American Society for, B. &Marrow, T (2012) Role of cytotoxic therapy with hema-topoietic stem cell transplantation in the treatment ofpediatric acute lymphoblastic leukemia: update of the2005 evidence-based review. Biol Blood MarrowTransplant 18:505–522
Ozsahin H, Cavazzana-Calvo M, Notarangelo LD et al(2008) Long-term outcome following hematopoieticstem-cell transplantation inWiskott-Aldrich syndrome:collaborative study of the European Society for Immu-nodeficiencies and European Group for Blood andMarrow Transplantation. Blood 111(1):439–445
Pai SY, Logan BR, Griffith LM, Buckley RH, Parrott RE,Dvorak CC, Kapoor N, Hanson IC, Filipovich AH,Jyonouchi S, Sullivan KE, Small TN, Burroughs L,
Pediatric Bone Marrow Transplantation 35

Skoda-Smith S, Haight AE, Grizzle A, Pulsipher MA,ChanKW, Fuleihan RL, Haddad E, Loechelt B, AquinoVM, Gillio A, Davis J, Knutsen A, Smith AR, MooreTB, Schroeder ML, Goldman FD, Connelly JA,Porteus MH, Xiang Q, Shearer WT, Fleisher TA,Kohn DB, Puck JM, Notarangelo LD, Cowan MJ,O’Reilly RJ (2014) Transplantation outcomes forsevere combined immunodeficiency, 2000–2009. NEngl J Med 371:434–446
Park JR, Kreissman SG, London WB et al (2019) Effect oftandem autologous stem cell transplant vs single trans-plant on event-free survival in patients with high-riskneuroblastoma: a randomized clinical trial. JAMA 322(8):746–755. https://doi.org/10.1001/jama.2019.11642
Peters C, Schrappe M, Von Stackelberg A, Schrauder A,Bader P, Ebell W, Lang P, Sykora KW, Schrum J,Kremens B, Ehlert K, Albert MH, Meisel R, Matthes-Martin S, Gungor T, Holter W, Strahm B, Gruhn B,Schulz A, Woessmann W, Poetschger U, ZimmermannM, Klingebiel T (2015) Stem-cell transplantation inchildren with acute lymphoblastic leukemia: a prospec-tive international multicenter trial comparing siblingdonors with matched unrelated donors-the ALL-SCT-BFM-2003 trial. J Clin Oncol 33:1265–1274
Pichler H, Lawitschka A, Glogova E, Willasch AM, vonLuettichau I, Lehrnbecher T, Matthes-Martin S, Lang P,Bader P, Sykora KW, Schrum J, Kremens B, Ehlert K,Albert MH, Kuhlen M, Meisel R, Guengoer T, StrahmB, Gruhn B, Schulz A, Woessmann W, Poetschger U,Peters C (2019) Allogeneic hematopoietic stem celltransplantation from unrelated donors is associatedwith higher infection rates in children with acute lym-phoblastic leukemia-a prospective international multi-center trial on behalf of the BFM-SG and the EBMT-PDWP. Am J Hematol 94:880–890
Pidala J, Lee SJ, Ahn KW, Spellman S, Wang HL, AljurfM, Askar M, Dehn J, Fernandez Vina M, Gratwohl A,Gupta V, Hanna R, HorowitzMM,Hurley CK, InamotoY, Kassim AA, Nishihori T, Mueller C, Oudshoorn M,Petersdorf EW, Prasad V, Robinson J, Saber W, SchultzKR, Shaw B, Storek J, Wood WA, Woolfrey AE,Anasetti C (2014) Nonpermissive HLA-DPB1 mis-match increases mortality after myeloablative unrelatedallogeneic hematopoietic cell transplantation. Blood124:2596–2606
Pui CH, EvansWE (2006) Treatment of acute lymphoblas-tic leukemia. N Engl J Med 354:166–178
Pulsipher MA, Hunger SP, Gamis AS,Wall DA, Grupp SA(2010) Allogeneic marrow transplantation in childrenwith acute leukemia: careful comparison with chemo-therapy alternatives required. Leukemia 24:1212–1216
Pulsipher MA, Peters C, Pui CH (2011) High-risk pediatricacute lymphoblastic leukemia: to transplant or not totransplant? Biol Blood Marrow Transplant 17:S137–S148
Raiola AM, Dominietto A, Ghiso A et al (2013)Unmanipulated haploidentical bone marrow transplan-tation and posttransplantation cyclophosphamide for
hematologic malignancies after myeloablative condi-tioning. Biol Blood Marrow Transplant 19:117–122
Rastogi N, Katewa S, Thakkar D, Kohli S, Nivargi S,Yadav SP (2018) Reduced-toxicity alternate-donorstem cell transplantation with posttransplant cyclo-phosphamide for primary immunodeficiency disorders.Pediatr Blood Cancer 65. https://doi.org/10.1002/pbc.26783
Ricci A, Jin Z, Broglie L, Bhatia M, George D, Garvin JH,Hall M, Satwani P (2019) Healthcare utilization andfinancial impact of acute-graft-versus host diseaseamong children undergoing allogeneic hematopoieticcell transplantation. Bone Marrow Transplant 55:384
Ruggeri L, Mancusi A, Capanni M, Urbani E, Carotti A,Aloisi T, Stern M, Pende D, Perruccio K, Burchielli E,Topini F, Bianchi E, Aversa F, Martelli MF, Velardi A(2007) Donor natural killer cell allorecognition of miss-ing self in haploidentical hematopoietic transplantationfor acute myeloid leukemia: challenging its predictivevalue. Blood 110:433–440
Satwani P, Jin Z, Duffy D, Morris E, Bhatia M, Garvin JH,George D, Bradley MB, Harrison L, Petrillo K,Schwartz J, Foley S, Hawks R, Baxter-Lowe LA,Cairo MS (2013) Transplantation-related mortality,graft failure, and survival after reduced-toxicity condi-tioning and allogeneic hematopoietic stem cell trans-plantation in 100 consecutive pediatric recipients. BiolBlood Marrow Transplant 19:552–561
Satwani P, Jin Z, Martin PL, Bhatia M, Garvin JH, GeorgeD, Chaudhury S, Talano J, Morris E, Harrison L, SosnaJ, Peterson M, Militano O, Foley S, Kurtzberg J, CairoMS (2015a) Sequential myeloablative autologous stemcell transplantation and reduced intensity allogeneichematopoietic cell transplantation is safe and feasiblein children, adolescents and young adults with poor-risk refractory or recurrent Hodgkin and non-Hodgkinlymphoma. Leukemia 29:448–455
Satwani P, Kahn J, Jin Z (2015b) Making strides andmeeting challenges in pediatric allogeneic hematopoi-etic cell transplantation clinical trials in the UnitedStates: past, present and future. Contemp Clin Trials45:84–92
Schaub F, Federmann B, Kanz L (2013) Daily G-CSF(filgrastim) and a single dose of pegylated G-CSF(pegfilgrastim) after chemotherapy result in differentkinetics of circulating hematopoetic progenitors: impli-cation for potential stem cell harvesting. Blood 122(21):4513. Retrieved from http://www.bloodjournal.org/content/122/21/4513. Accessed 01 Oct 2019
Schumm M, Lang P, Bethge W, Faul C, Feuchtinger T,Pfeiffer M, Vogel W, Huppert V, Handgretinger R(2013) Depletion of T-cell receptor alpha/beta andCD19 positive cells from apheresis products with theCliniMACS device. Cytotherapy 15:1253–1258
Seidel MG, Fritsch G, Matthes-Martin S, Lawitschka A,Lion T, Potschger U, Rosenmayr A, Fischer G, GadnerH, Peters C (2005) In vitro and in vivo T-cell depletionwith myeloablative or reduced-intensity conditioning
36 S. P. Yadav et al.

in pediatric hematopoietic stem cell transplantation.Haematologica 90:1405–1414
Selim A, Alvaro F, Cole CH, Fraser CJ, Mechinaud F,O’Brien TA, Shaw PJ, Tapp H, Teague L, Nivison-Smith I, Moore AS (2019) Hematopoietic stem celltransplantation for children with acute myeloid leuke-mia in second remission: a report from the AustralasianBone Marrow Transplant Recipient Registry and theAustralian and New Zealand Children's HaematologyOncology Group. Pediatr Blood Cancer 66:e27812
Shah RM, Elfeky R, Nademi Z, Qasim W, Amrolia P,Chiesa R, Rao K, Lucchini G, Silva JMF, Worth A,Barge D, Ryan D, Conn J, Cant AJ, Skinner R, AbdHamid IJ, Flood T, Abinun M, Hambleton S, GenneryAR, Veys P, Slatter M (2018) T-cell receptor alphabeta(+) and CD19(+) cell-depleted haploidentical and mis-matched hematopoietic stem cell transplantation in pri-mary immune deficiency. J Allergy Clin Immunol141:1417–1426.e1
Shelikhova L, Shekhovtsova Z, Balashov D, Boyakova E,Muzalevskyi I, Gutovskaya E, Shipitsina I, ShashelevaD, Khismatullina R, Blagov S,Myakova N, AbrosimovA, Novichkova G, Maschan A, Maschan M (2016)TCRαβ+/CD19+-depletion in hematopoietic stemcells transplantation from matched unrelated andhaploidentical donors following treosulfan or TBI-based conditioning in pediatric acute lymphoblasticleukemia patients. Blood 128:4672–4672
Shelikhova L, Ilushina M, Shekhovtsova Z, Shasheleva D,Khismatullina R, Kurnikova E, Pershin D, Balashov D,Radygina S, Trakhtman P, Kalinina I, Muzalevskii Y,Kazachenok A, Zaharova V, Brilliantova V,Olshanskaya Y, Panferova A, Zerkalenkova E,Baidildina D, Novichkova G, Rumyantsev A, MaschanA, Maschan M (2019) Alphabeta T cell-depletedhaploidentical hematopoietic stem cell transplantationwithout antithymocyte globulin in children withchemorefractory acute myelogenous leukemia. BiolBlood Marrow Transplant 25:e179
Shenoy S (2007) Has stem cell transplantation come of agein the treatment of sickle cell disease? Bone MarrowTransplant 40:813–821
Shenoy S (2013) Hematopoietic stem-cell transplantationfor sickle cell disease: current evidence and opinions.Ther Adv Hematol 4:335–344
Shenoy S, Boelens JJ (2015) Advances in unrelated andalternative donor hematopoietic cell transplantation fornonmalignant disorders. Curr Opin Pediatr 27:9–17
Shenoy S, Eapen M, Panepinto JA, Logan BR, Wu J,Abraham A, Brochstein J, Chaudhury S, Godder K,Haight AE, Kasow KA, Leung K, Andreansky M,Bhatia M, Dalal J, Haines H, Jaroscak J, Lazarus HM,Levine JE, Krishnamurti L, Margolis D, Megason GC,Yu LC, Pulsipher MA, Gersten I, Difronzo N, HorowitzMM, Walters MC, Kamani N (2016) A trial ofunrelated donor marrow transplantation for childrenwith severe sickle cell disease. Blood 128:2561–2567
Shook DR, Triplett BM, Eldridge PW, Kang G, SrinivasanA, Leung W (2015) Haploidentical stem cell
transplantation augmented by CD45ra negative lym-phocytes provides rapid engraftment and excellent tol-erability. Pediatr Blood Cancer 62:666–673
Shouval R, Fein JA, Labopin M, Kroger N, Duarte RF,Bader P, Chabannon C, Kuball J, Basak GW, DuFourC, Galimard JE, Polge E, Lankester A, Montoto S,Snowden JA, Styczynski J, Yakoub-Agha I, MohtyM, Nagler A (2019) Outcomes of allogeneichaematopoietic stem cell transplantation from HLA-matched and alternative donors: a European Societyfor Blood and Marrow Transplantation registry retro-spective analysis. Lancet Haematol 6:e573
Sisinni L, Gasior M, De Paz R, Querol S, Bueno D,Fernandez L, Marsal J, Sastre A, Gimeno R, AlonsoL, Badell I, Lopez-Granados E, Torres J, Medina L,Torrent M, Diaz De Heredia C, Escudero A, Perez-Martinez A (2018a) Unexpected high incidence ofhuman herpesvirus-6 encephalitis after naive T cell-depleted graft of haploidentical stem cell transplanta-tion in pediatric patients. Biol Blood Marrow Trans-plant 24:2316–2323
Sisinni L, Gich I, Torrent M, Badell I (2018b) [Subsequentmalignancies after long-term follow-up of pediatrichematopoietic stem cell transplantation]. An Pediatr(Barc) 90:157
Slatter MA, Gennery AR (2018) Hematopoietic cell trans-plantation in primary immunodeficiency – conven-tional and emerging indications. Expert Rev ClinImmunol 14:103–114. https://doi.org/10.1080/1744666X.2018.1424627
Socie G, Blazar BR (2009) Acute graft-versus-host dis-ease: from the bench to the bedside. Blood114:4327–4336
Sodani P, Gaziev D, Polchi P, Erer B, Giardini C et al(2004) New approach for bone marrow transplantationin patients with class 3 thalassemia aged younger than17 years. Blood 104:1201–1203
Solomon SR, Aubrey MT, Zhang X, Piluso A, Freed BM,Brown S et al (2018) Selecting the best donor forhaploidentical transplant: impact of HLA, killer cellimmunoglobulin-like receptor genotyping, and otherclinical variables. Biol Blood Marrow Transplant24:789–798
Thomas ED, Lochte HL Jr, Lu WC, Ferrebee JW (1957)Intravenous infusion of bone marrow in patients receiv-ing radiation and chemotherapy. N Engl J Med257:491–496
Thomas ED, Buckner CD, Rudolph RH, Fefer A, Storb R,Neiman PE, Bryant JI, Chard RL, Clift RA, Epstein RB,Fialkow PJ, Funk DD, Giblett ER, Lerner KG, ReynoldsFA, Slichter S (1971) Allogeneic marrow grafting forhematologic malignancy using HL-A matched donor-recipient sibling pairs. Blood 38:267–287
Tiercy JM (2016) How to select the best available related orunrelated donor of hematopoietic stem cells?Haematologica 101:680–687
Touzot F, Neven B, Dal-Cortivo L, Gabrion A,Moshous D,Cros G, ChomtonM, Luby JM, Terniaux B, Magalon J,Picard C, Blanche S, Fischer A, Cavazzana M (2015)
Pediatric Bone Marrow Transplantation 37

CD45RA depletion in HLA-mismatched allogeneichematopoietic stem cell transplantation for primarycombined immunodeficiency: a preliminary study. JAllergy Clin Immunol 135:1303–1309.e1-3
Triplett BM, Muller B, Kang G, Li Y, Cross SJ, Moen J,Cunningham L, Janssen W, Mamcarz E, Shook DR,Srinivasan A, Choi J, Hayden RT, Leung W (2018)Selective T-cell depletion targeting CD45RA reducesviremia and enhances early T-cell recovery comparedwith CD3-targeted T-cell depletion. Transpl Infect Dis20(1). https://doi.org/10.1111/tid.12823
Uppuluri R, Sivasankaran M, Patel S et al (2019)Haploidentical stem cell transplantation with post-transplant cyclophosphamide for primary immune defi-ciency disorders in children: challenges and outcomefrom a tertiary care center in South India. J ClinImmunol 39(2):182–187
Waldmann H, Polliak A, Hale G, Or R, Cividalli G, WeissL, Weshler Z, Samuel S, Manor D, Brautbar C et al(1984) Elimination of graft-versus-host disease by in-
vitro depletion of alloreactive lymphocytes with amonoclonal rat anti-human lymphocyte antibody(CAMPATH-1). Lancet 2:483–486
Weatherall DJ (2018) The evolving spectrum of the epide-miology of thalassemia. Hematol Oncol Clin North Am32:165–175
Weatherall DJ, Clegg JB (1996) Thalassemia–a globalpublic health problem. Nat Med 2:847–849
Yu AL et al (2010) Anti-GD2 antibody with GM-CSF,interleukin-2, and isotretinoin for neuroblastoma. NEngl J Med 363:1324–1334
Zvyagin IV, Mamedov IZ, Tatarinova OV, Komech EA,Kurnikova EE, Boyakova EV, Brilliantova V,Shelikhova LN, Balashov DN, Shugay M, SychevaAL, Kasatskaya SA, Lebedev YB, Maschan AA,Maschan MA, Chudakov DM (2017) Tracking T-cellimmune reconstitution after TCRalphabeta/CD19-depleted hematopoietic cells transplantation in chil-dren. Leukemia 31:1145–1153
38 S. P. Yadav et al.