oulu 2012 d 1168 university of oulu p.o.b. 7500 fi...
TRANSCRIPT

ABCDEFG
UNIVERS ITY OF OULU P.O.B . 7500 F I -90014 UNIVERS ITY OF OULU F INLAND
A C T A U N I V E R S I T A T I S O U L U E N S I S
S E R I E S E D I T O R S
SCIENTIAE RERUM NATURALIUM
HUMANIORA
TECHNICA
MEDICA
SCIENTIAE RERUM SOCIALIUM
SCRIPTA ACADEMICA
OECONOMICA
EDITOR IN CHIEF
PUBLICATIONS EDITOR
Senior Assistant Jorma Arhippainen
Lecturer Santeri Palviainen
Professor Hannu Heusala
Professor Olli Vuolteenaho
Senior Researcher Eila Estola
Director Sinikka Eskelinen
Professor Jari Juga
Professor Olli Vuolteenaho
Publications Editor Kirsti Nurkkala
ISBN 978-951-42-9912-4 (Paperback)ISBN 978-951-42-9913-1 (PDF)ISSN 0355-3221 (Print)ISSN 1796-2234 (Online)
U N I V E R S I TAT I S O U L U E N S I S
MEDICA
ACTAD
D 1168
ACTA
Anne-M
ari Moilanen
OULU 2012
D 1168
Anne-Mari Moilanen
IDENTIFICATION OF NOVEL DRUG TARGETS FORTHE TREATMENT OF HEART FAILURE
UNIVERSITY OF OULU GRADUATE SCHOOL;UNIVERSITY OF OULU,FACULTY OF MEDICINE, INSTITUTE OF BIOMEDICINE,DEPARTMENT OF PHARMACOLOGY AND TOXICOLOGY;BIOCENTER OULU


A C T A U N I V E R S I T A T I S O U L U E N S I SD M e d i c a 1 1 6 8
ANNE-MARI MOILANEN
IDENTIFICATION OF NOVEL DRUG TARGETS FOR THE TREATMENTOF HEART FAILURE
Academic dissertation to be presented with the assentof the Doctoral Training Committee of Health andBiosciences of the University of Oulu for public defencein the Auditorium of the Department of Pharmacologyand Toxicology (Aapistie 5 B), on 5 October 2012, at12 noon
UNIVERSITY OF OULU, OULU 2012

Copyright © 2012Acta Univ. Oul. D 1168, 2012
Supervised byProfessor Heikki RuskoahoDocent Jaana Rysä
Reviewed byDocent Ilkka TikkanenDoctor Mikko Turunen
ISBN 978-951-42-9912-4 (Paperback)ISBN 978-951-42-9913-1 (PDF)
ISSN 0355-3221 (Printed)ISSN 1796-2234 (Online)
Cover DesignRaimo Ahonen
JUVENES PRINTTAMPERE 2012

Moilanen, Anne-Mari, Identification of novel drug targets for the treatment ofheart failure. University of Oulu Graduate School; University of Oulu, Faculty of Medicine, Institute ofBiomedicine, Department of Pharmacology and Toxicology; Biocenter Oulu, P.O. Box 5000, FI-90014 University of Oulu, FinlandActa Univ. Oul. D 1168, 2012Oulu, Finland
Abstract
Heart failure (HF) is a complex pathological state, involving simultaneous alterations in severalsignalling pathways and changes in gene programming. In HF, activation of the neurohumoralfactors and renin-angiotensin-aldosterone (RAA) system occurs as a compensatory mechanism tocombat the abnormal ventricular function. Developments in cardiac gene delivery methods haveexerted a significant impact to treat HF and to discover the novel molecular mechanismsassociated with HF and other cardiac diseases.
This study demonstrated that adenovirus–mediated gene delivery of B-type natriuretic peptide(BNP) into the anterior wall of the left ventricle decreased myocardial fibrosis and increasedcapillary density. Post-infarction BNP improved systolic function associated with normalizationof cardiac sarcoplasmic reticulum Ca2+-ATPase (SERCA) 2 expression and phospholambanphosphorylation at Thr17. On the other hand, (Pro)renin receptor ([P]RR) gene delivery resulteddeleterious effects on cardiac function and (P)RR activation induced distinct angiotensin II (AngII)-independent extracellular matrix remodelling and worsening of cardiac function. (P)RR genedelivery resulted in Ang II-independent activation of extracellular-signal regulated (ERK1/2)phosphorylation and increased myocardial fibrosis.
In conclusion, the present study indicates that myocardial BNP gene delivery can achievepleiotropic, context-dependent, favourable effects on cardiac function and that BNP can actlocally as a mechanical load–activated regulator of angiogenesis and fibrosis. These results alsoimplicate that (P)RR blockers may display additional cardiac effects in addition to its ability toevoke effective RAA system blockade. Overall, the findings of this study provide a betterunderstanding of the molecular mechanisms involved in the biological actions of BNP and (P)RR,and identify BNP and (P)RR as potential novel drug targets for the treatment of HF.
Keywords: (pro)renin receptor, B-type natriuretic peptide, cardiac gene transfer, cardiachypertrophy, heart failure, ventricular remodelling


Moilanen, Anne-Mari, Uusien kohdegeenien tunnistaminen sydämenvajaatoiminnan hoitoon. Oulun yliopiston tutkijakoulu; Oulun yliopisto, Lääketieteellinen tiedekunta, Biolääketieteenlaitos, Farmakologia ja toksikologia; Biocenter Oulu, PL 5000, 90014 Oulun yliopistoActa Univ. Oul. D 1168, 2012Oulu
Tiivistelmä
Neuroendokriinisellä aktivaatiolla, jonka seurauksena aiheutuu muun muassa verisuonten supis-tumista ja laajenemista sekä nesteen kertymistä elimistöön, on tärkeä merkitys sydämen vajaa-toiminnan kehittymisessä. Neuroendokriininen aktivaatio kompensoi sydämen vajaatoiminnanseurauksena tapahtuvaa kammioiden poikkeavaa toimintaa. Yksi keskeisimmistä verisuoniasupistavista tekijöistä on reniini-angiotensiini-aldosteroni (RAA) -järjestelmä, ja verisuonia laa-jentaviin tekijöihin kuuluvat muun muassa natriureettiset peptidit, kuten B-tyypin natriureetti-nen peptidi (BNP) ja A-tyypin natriureettinen peptidi. Geeninsiirtomenetelmillä on ollut merkit-täviä vaikutuksia uusien hoitomenetelmien kehittämisessä, sydämen vajaatoiminnan syiden sel-vittämisessä ja uusien kohdegeenien tunnistamisessa sydämen vajaatoiminnan hoitoon.
Väitöskirjan tutkimustulokset osoittivat, että suora adenovirusvälitteinen geeninsiirto rotansydämen vasemman kammion etuseinään on toimiva menetelmä uusien kohdegeenien löytämi-seksi sydämen vajaatoiminnan hoitoon. BNP:n geeninsiirto vähensi merkitsevästi fibroosinmuodostumista ja lisäsi verisuonten uudismuodostumista sydämessä. Sydäninfarktin jälkeenBNP paransi sydämen systolista toimintaa, johon liittyi aktiivisen kalsiumpumpun, SERCA2:nja fosfolambaani-proteiinin fosforylaation normalisoituminen. (Pro)reniini reseptorin ([P]RR)geeninsiirto aiheutti angiotensiini II:sta riippumatonta solunulkoisen matriksin uudelleenmuo-toutumista ja sydämen toiminnan huonontumista sekä lisääntynyttä sydämen fibroosia.
Väitöskirjatutkimus antaa uutta tietoa solunsisäisistä molekulaarisista mekanismeista sydän-soluissa. BNP geeninsiirto aiheutti sydämen tautitilasta riippuvia suotuisia tapahtumia, ja se toi-mi paikallisesti muun muassa fibroosia ehkäisevänä tekijänä. (P)RR geeninsiirtotulosten perus-teella voidaan olettaa, että (P)RR:n salpaus saattaa olla uusi tehokas hoitokeino sydämen vajaa-toiminnan hoitoon.
Asiasanat: (pro)reniini reseptori, B-tyypin natriureettinen peptidi, geeninsiirto,sydämen hypertrofia, sydämen vajaatoiminta


7
Acknowledgements
This work was carried out at the Department of Pharmacology and Toxicology,
Institute of Biomedicine, University of Oulu, during the years 2006–2012. I
hereby wish to sincerely acknowledge all the people who have contributed to this
work and supported me during these years. I am grateful to the former head of the
department, Professor Emeritus Olavi Pelkonen for providing inspiring and
encouraging atmosphere towards my research.
Above all, I am deeply grateful to my supervisor Professsor Heikki
Ruskoaho. Heikki has always been very enthusiastic and supportive supervisor.
He has given me much freedom in my research which I think has enabled me to
become more persistent researcher. I also offer my warmest thanks to Jaana Rysä
and Raisa Serpi. Thank you for sharing your knowledge of science and guiding
me through my studies.
Thanks to the reviewers of my thesis, Docent Ilkka Tikkanen and Ph.D.
Mikko Turunen for their expert and critical comments. They helped me
significantly to clarify my research and thoughts. I also wish to thank Ewen
MacDonald for his excellent revision of the English language of this thesis.
My warmest thanks are due to Professor Olli Vuolteenaho for his scientific
collaboration and valuable comments. I also want to thank Heikki Tokola for his
friendship and quidance along the way. I wish to thank all my other co-authors
Minna Ala-Kopsala, Jani Aro, Hanna Leskinen, Jouko Levijoki, Aki Manninen,
Erja Mustonen, Juha Näpänkangas, Tuula Salo, Zoltán Szabó, Meeri Sutinen and
Olli Tenhunen for contributing their expertise to this work.
I owe my thanks to Marja Arbelius, Kati Lampinen, Kaisa Penttilä, Sirpa
Rutanen, Kirsi Salo, Erja Tomperi and Mirja Vahera for their precise expert
assistance in the laboratory. Sirpa and Kaisa deserve special thanks for guiding
me in the lab and for sharing quite memorable moments with me. Raija Hanni,
Esa Kerttula, Marja Räinä and Pirkko Viitala are warmly acknowledged for their
kind help in all practical matters. I also wish to thank the personnel at the
Laboratory Animal Center.
This Ph.D. journey began in Sikaosasto, where I spent the first four years. My
deepest graditude belongs to our sikaosasto-team Leena Kaikkonen, Sini
Kinnunen, Elina Koivisto, Anna-Maria Kubin, Tuomas Peltonen and Marja Tölli.
We have so many unforgettable memories, in our closed compartment. Tuomas,
thank you for sharing your constructive feedback about science and life.

8
I wish to thank all the former and current members of our research group. I
am grateful for all the good times we have shared during these years. I wish to
express my warmest thanks to my colleagues Alicia Jurado Acosta, Teemu
Karvonen, Annina Kelloniemi, Risto Kerkelä, Johanna Magga, Pauli Ohukainen,
Harri Pennanen, Ábel Perjés, Virva Pohjolainen, Tarja Saiho, Hanna Säkkinen,
Anna-Maria Tolonen, Johanna Ulvila, Laura Vainio and my other colleagues for
their kind and helpful attitude towards my work and for their friendship during
these years. Especially, I wish to thank my dear friend Outi Renko for sharing so
many unforgettable moments around and outside the office and lab. Your
friendship has truly been the icing on the cake. The other staff at the Department
of Pharmacology and Toxicology is most warmly acknowledged for creating a
warm and pleasant place to work during these years.
I owe my heartfelt thanks to my friends who have not forgotten me during
these years. In particular, I want to thank Anne, Leena, Janne, Kaisa and Satu.
You have been a life-long friends and I am so happy you are in my life.
My warmest thanks go to my parents, Ulla and Seppo. They deserve great
admiration and thanks for the support they have given me throughout my life. My
sister Sara and my brother Tuomas deserve my sincere thanks. I also wish to
thank my parents-in-law Sirkka and Esko, and my sister-in-law Arja and her
husband Ari for their help and support in many ways. My beloved furry buddies
Kaapo and Roope deserved to be mentioned too, even though they are not
particularly interested in my research.
Finally, I dedicate this work to my family. My husband Kari, who has always
encouraged and supported me in my work. Thank you for believing in me during
this project. Last but not least, Kalle, being your mom is the most cherished and
important thesis of my life. Thank you for showing me that it is about time to
focus on something far more worthwhile than a doctoral thesis.
This research project has been financially supported by the Academy of
Finland CoE, Emil Aaltonen Foundation, the Finnish Foundation for
Cardiovascular Research, Orion Diagnostica and Sigrid Jusélius Foundation.
Oulu, August 2012 Anne-Mari Moilanen

9
Abbreviations
AAV adeno-associated virus
AC adenylyl cyclase
Ang II angiotensin II
ANP atrial natriuretic peptide
ANOVA analysis of variance
AT1-R Ang II type 1 receptor
AT2-R Ang II type 2 receptor
ATPase adenosine triphosphatase
BCL-2 B-cell lymphoma-2
BNP B-type natriuretic peptide
Ca2+ calcium
CaMKII calcium-calmodulin-dependent protein kinase II
cAMP 3’, 5’-cyclic adenosine monophosphate
cDNA complementary DNA
cGMP 3’, 5’-cyclic guanosine monophosphate
CNP C-type natriuretic peptide
CTGF connective tissue growth factor
CMV cytomegalovirus
DAPI diamidinophenylindole dihydrochloride
DNA deoxyribonucleic acid
DRP deoxyribonuclease resistant particles
ECL enhanced chemiluminescence
eGFP enhanced green fluorescent protein
EIA enzyme immunoassay
ER endoplasmic reticulum
ERK extracellular signal regulated kinase
FGF fibroblast growth factor
G-protein guanine nucleotide binding protein
GαS G-protein α-subunit
GC guanylyl cyclase
GPCR G-protein-coupled receptor
GRK G-protein-coupled protein kinase
hBNP human BNP
HIF-1α hypoxia-inducible factor-1α
HF heart failure

10
HPLC high–pressure liquid chromatography
HSP27 heat shock protein 27
I-1 inhibitor type 1
ifu infectious unit
IGF insulin-like growth factor
i.p. intraperitoneally
ir immunoreactive
JNK c-Jun N-terminal kinase
kb kilobase
LAD left anterior descending coronary artery
LSD least significant difference
LV left ventricle
LVEDD left ventricular end diastolic diameter
LVESD left ventricular end systolic diameter
MAPK mitogen-activated protein kinase
MHC myosin heavy chain
MI myocardial infarction
MMP matrix metalloproteinase
mRNA messenger RNA
NP natriuretic peptide
NPPA natriuretic peptide precursor A
NPPB natriuretic peptide precursor B
NT amino-terminal
NYHA New York Heart Association
PBSF pre-B-cell growth stimulating factor
PI3K phosphatidylinositol-3 kinase
PKA protein kinase A
PLB phospholamban
PLZF promyelocytic zinc finger protein
PP1 protein phosphatase type 1
proBNP profragment of BNP
(P)RR (pro)renin receptor
rAAV recombinant AAV
RAA renin-angiotensin-aldosterone
RIA radioimmunoassay
RNA ribonucleic acid
RSV Rous sarcoma virus

11
RT-qPCR reverse transcriptase quantitative polymerase chain reaction
RyR ryanodine receptor
s.c. subcutaneously
Ser serine
SERCA sarcoplasmic reticulum Ca2+-ATPase
SD Sprague-Dawley
SDF stromal derived growth factor
SEM standard error mean
SR sarcoplasmic reticulum
TBS tris-buffered saline
TGF transforming growth factor
Thr threonine
TNF-α tumor necrosis factor-α
TUNEL terminal deoxynucleotidyl transferase dUTP nick end labeling
V-ATPase vacuolar H+-adenosine trisphosphatase
VEGF vascular endothelial growth factor
VP viral protein
18S ribosomal 18S
β-AR β-adrenergic receptor
βARKct carboxyl-terminus of the β-adrenergic receptor kinase

12

13
List of original papers
The thesis is based on the following articles, which are referred to in the text by
their Roman numerals:
I Moilanen AM, Rysä J, Mustonen E, Serpi R, Aro J, Tokola H, Leskinen H, Manninen A, Levijoki J, Vuolteenaho O & Ruskoaho H (2011) Intramyocardial BNP gene delivery improves cardiac function through distinct context-dependent mechanisms. Circ Heart Fail 4: 483–95.
II Ala-Kopsala M, Moilanen AM, Rysä J, Ruskoaho H & Vuolteenaho O (2010) Characterization of molecular forms of N-terminal B-type natriuretic peptide in vitro. Clin Chem 56: 1822–9.
III Moilanen AM, Rysä J, Serpi R, Mustonen E, Szabó Z, Aro J, Näpänkangas J, Tenhunen O, Sutinen M, Salo T & Ruskoaho H (2012) (Pro)renin receptor triggers distinct angiotensin II-independent extracellular matrix remodelling and deterioration of cardiac function. PLoS ONE 7(7): e41404.
In addition, some unpublished data are presented.

14

15
Contents
Abstract
Tiivistelmä
Acknowledgements 7 Abbreviations 9 List of original papers 13 Contents 15 1 Introduction 19 2 Review of the literature 21
2.1 Heart failure ............................................................................................ 21 2.2 Cardiac remodelling ................................................................................ 24
2.2.1 Post-infarction left ventricular remodelling .................................. 24 2.2.2 Cardiac remodelling and hypertrophy .......................................... 26
2.3 Gene delivery vectors for cardiovascular gene therapy .......................... 28 2.3.1 Nonviral vectors ........................................................................... 29 2.3.2 Adenoviral vectors ....................................................................... 30 2.3.3 Adeno-associated viral vectors ..................................................... 31 2.3.4 Lentiviral vectors .......................................................................... 33 2.3.5 Other gene transfer vectors ........................................................... 34
2.4 In vivo myocardial gene delivery techniques .......................................... 34 2.4.1 Direct myocardial gene delivery ................................................... 35 2.4.2 Intravascular gene delivery ........................................................... 37
2.5 Gene targets for the treatment of heart failure ........................................ 39 2.5.1 Calcium handling proteins ............................................................ 39 2.5.2 β-adrenergic system ...................................................................... 49 2.5.3 Angiogenic factors ....................................................................... 53 2.5.4 Anti-apoptotic genes ..................................................................... 54 2.5.5 Stem cell homing factors .............................................................. 56 2.5.6 Cardiac natriuretic peptides .......................................................... 58 2.5.7 Renin-angiotensin aldosterone system ......................................... 61 2.5.8 Other potential gene targets for the treatment of heart
failure ........................................................................................... 64 2.6 Clinical trials in gene therapy for heart failure........................................ 67
2.6.1 Sarcoplasmic reticulum Ca2+-ATPase ........................................... 69 2.6.2 Adenylyl cyclase 6 ....................................................................... 71 2.6.3 Stromal derived growth factor-1 ................................................... 71

16
3 Aims of the research 73 4 Materials and methods 75
4.1 Recombinant adenoviral vectors (I, II and III) ........................................ 76 4.2 Animal studies (I, II and III) ................................................................... 77
4.2.1 Intramyocardial gene transfer (I, II and III) .................................. 77 4.2.2 Acute myocardial infarction (I and III) ........................................ 78 4.2.3 Angiotensin II–mediated hypertension (I and III) ........................ 78 4.2.4 Losartan treatments with osmotic minipumps (III) ...................... 78
4.3 Echocardiographic measurements (I and III) .......................................... 79 4.4 Telemetric monitoring (I) ........................................................................ 79 4.5 Protein analysis (I, II and III) .................................................................. 80
4.5.1 Extraction of cytoplasmic protein (I, II and III) ........................... 80 4.5.2 Western blot analyses (I, II and III) .............................................. 80 4.5.3 Immunoprecipitation (III) ............................................................. 82 4.5.4 cGMP assay (I) ............................................................................. 82 4.5.5 Gelatin zymography (III) .............................................................. 83 4.5.6 Immunohistochemistry (I and III) ................................................ 83
4.6 RNA analysis (I and III) .......................................................................... 85 4.6.1 Isolation and analysis of RNA (I and III) ..................................... 85
4.7 Immunoassays (I and II) .......................................................................... 86 4.7.1 Radioimmunoassay and HPLC (I and II) ..................................... 86 4.7.2 Determination of BNP antibodies ................................................. 87
4.8 Histology and image analysis (I and III) ................................................. 87 4.9 Statistical analysis ................................................................................... 89
5 Results 91 5.1 Augmentation of left ventricular gene expression by adenoviral
gene delivery (I, II and III) ...................................................................... 91 5.1.1 Human BNP gene delivery (I and II) ............................................ 91 5.1.2 (Pro)renin receptor gene delivery (III) ......................................... 96 5.1.3 LacZ gene delivery (I and III) ...................................................... 97
5.2 Cardiac effects of intramyocardial BNP gene delivery (I) ...................... 98 5.2.1 BNP as a regulator of cardiac fibrosis .......................................... 99 5.2.2 Coronary angiogenesis after BNP gene transfer ......................... 103 5.2.3 Hemodynamics in rats overexpressing BNP in the heart ............ 103 5.2.4 Activation of cardiac gene expressions and signalling
pathways by BNP gene ............................................................... 105 5.3 Cardiac effects of direct (P)RR gene delivery (III) ............................... 107

17
5.3.1 Cardiac function in rats overexpressing (P)RR in the heart ....... 107 5.3.2 Angiotensin II-independent and dependent effects
triggered by (P)RR ..................................................................... 110 5.3.3 Activation of cardiac hypertrophic marker genes by (P)RR ....... 112 5.3.4 Activation of ERK1/2 and p38 MAPK/HSP27 pathways
by (P)RR ..................................................................................... 113 5.3.5 (P)RR interaction with PLZF ..................................................... 114 5.3.6 Wnt signalling and V-ATPase pathway after (P)RR gene
transfer ........................................................................................ 115 6 Discussion 117
6.1 Characterization of the efficiency of adenoviral–mediated gene
delivery (I and III) ..................................................................................117 6.1.1 Immune reaction and inflammatory response following
adenovirus–mediated gene delivery (I) ...................................... 118 6.2 BNP as a therapeutic target for the treatment of heart failure (I) ...........119
6.2.1 Antifibrotic and angiogenic effects of BNP gene delivery
in normal heart ........................................................................... 122 6.2.2 Functional role of BNP after myocardial infarction ................... 123 6.2.3 Effects of BNP gene delivery in an experimental model of
angiotensin II–mediated hypertension ........................................ 125 6.2.4 Context-dependent effects of BNP in the heart .......................... 125
6.3 (P)RR as a therapeutic target for the treatment of heart failure
(III) ........................................................................................................ 127 6.3.1 Functional effects in normal heart triggered by (P)RR ............... 128 6.3.2 Activation of extracellular matrix remodelling .......................... 128 6.3.3 Angiogenetic and apoptotic responses ....................................... 129 6.3.4 Hypertrophic stimuli activated by (P)RR gene transfer ............. 129 6.3.5 ERK1/2 pathway activation by (P)RR ........................................ 129 6.3.6 PLZF interaction with (P)RR ..................................................... 130 6.3.7 Effects of (P)RR gene delivery to Wnt signalling ...................... 130 6.3.8 Role of (P)RR gene delivery in experimental models of
MI and angiotensin II–mediated hypertension ........................... 130 6.3.9 (P)RR as a multifunctional protein ............................................. 131
7 Summary and conclusions 133 References 135 Original articles 173

18

19
1 Introduction
Heart failure (HF) is one of the most common cardiovascular diseases with high
morbidity and mortality, and its prevalence is rapidly increasing as the mean age
of the population advances (Roger et al. 2012). There are diverse causes of
cardiovascular disease, but coronary artery disease is the most common cause of
systolic HF while diastolic HF is more common in patients with hypertension
(Jessup & Brozena 2003, McMurray & Pfeffer 2005). Worsening of chronic
systolic or diastolic dysfunction is the most common form of acute HF, where
frequent hospitalizations are associated with high mortality and morbidity
(Flaherty et al. 2009). A number of drugs, particularly beta-blockers and drugs
acting on the RAA system have been shown to improve symptoms, lower
hospitalization and death rates in patients who have left ventricular systolic
dysfunction (Jessup & Brozena 2003, McMurray & Pfeffer 2005).
HF is a pathophysiological state in which the heart is unable to supply
sufficient blood flow with the requirements of metabolizing tissues and thus, to
sustain the normal organ function (Francis 2001). In HF, activation of the
neurohumoral factors and RAA system occurs as a compensatory mechanism of
abnormal ventricular function. Remodelling of the left ventricle (LV) and
alterations in the LV geometry represent adaptive mechanisms to improve cardiac
function (Mann & Bristow 2005). As a consequence, prolonged remodelling leads
to the failure of compensatory mechanisms and deterioration of cardiac function.
In myocyte level, abnormal molecular events results in alterations in the
excitation-contraction coupling, hypertrophy, the cardiac myocyte death and
cardiac fibrosis (Jessup & Brozena 2003, Mann & Bristow 2005).
New therapeutic approaches, such as gene therapy, may have a significant
impact on the treatment of HF. This may require the discovery of novel molecular
mechanisms associated with HF. Gene therapy may make it possible to modulate
several molecular targets which are difficult to modulate pharmacologically and
investigate molecular biological approaches on the development of cardiac
diseases (Lips et al. 2003). Definition of potential therapeutic targets, gene vector
development, and in vivo gene delivery techniques have been the most important
steps in the development of gene therapy. Recombinant viral vector systems as
gene delivery vehicles have proved to be the most powerful tool for cardiac gene
transfer. Presently, adenovirus and adeno-associated virus (AAV) vectors are the
vehicles showing the greatest promise in molecular based studies of heart disease
(Davis et al. 2008, Kawase et al. 2011, Vinge et al. 2008).

20
Potential gene-based therapies for the treatment of HF have been validated in
a variety of animal models. Moreover, transgenic and knockout models of cardiac
hypertrophy and heart failure have proved valuable in understanding the effects of
target genes on cardiac function (Ly et al. 2007, Rapti et al. 2011).
HF is a complex pathological state, consisting of the simultaneous alterations
in several signalling pathways. The most extensively studied targets for the
treatment of HF are β-adrenergic receptor (β-AR) signalling and calcium (Ca2+)
handling proteins and stimulation of cardiac angiogenesis (Katz et al. 2011).
Furthermore, gene transfer techniques have been a valuable approach for studying
the effects of signalling pathways on myocyte function and for identifying the
cellular targets within signalling cascades (Davis et al. 2008). The translation of
viral based studies from animal models of cardiac disease to humans is now
progressing in many clinical trials (Gao & Hammond 2011, Hajjar et al. 2008,
Jaski et al. 2009, Kawase et al. 2011).
In the present study, novel drug targets for the treatment of HF were
examined. The direct myocardial effects of BNP and (P)RR on cardiac function
were studied by using adenovirus–mediated gene delivery in normal adult rat
hearts and in hearts during the remodelling process after myocardial infarction
(MI) and in Ang II–induced hypertension. In addition, potential molecular
mechanisms triggering myocardial remodelling effects of cardiac function were
investigated and molecular forms of BNP were studied after gene delivery.

21
2 Review of the literature
2.1 Heart failure
HF is a syndrome which manifests with several symptoms, combined with
objective evidence of cardiac dysfunction (Fig. 1). In general, the symptoms
associated with HF are shortness of breath at rest or during exertion, fatigue, signs
of fluid retention (e.g. pulmonary congestion or ankle swelling) with the objective
evidence being abnormalities of the structure or function of the heart (Dickstein et al. 2008).
In HF, the capability of the heart to pump blood in response to systemic
demands becomes attenuated. HF can be induced by common disease stimuli,
such as, long-standing hypertension, MI or ischemia associated with coronary
artery disease, valvular insufficiency and stenosis. Moreover, myocarditis due to
an infectious agent, congenital malformations, familial hypertrophic and dilated
cardiomyopathies and diabetic cardiomyopathy act as inducers of HF (Berenji et al. 2005, Heineke & Molkentin 2006, Lips et al. 2003).
The aetiological factor conferring the major relative risk for LV systolic
dysfunction is ischemic heart disease. Asymptomatic structural or functional heart
abnormalities account for precursors of symptomatic HF and are combined with a
high death rate (Dickstein et al. 2008, McDonagh et al. 1997, Wang et al. 2003).
One of the most efficient predictors for the development of HF is the
presence of LV hypertrophy (Maron 1997). The New York Heart Association
(NYHA) functional classification has been devised to estimate the impact of HF
of patients. NYHA classifies patients with HF into four categories (I, II, III, IV)
with a higher class indicating more severe symptoms, more limitations in physical
activity and worse health (Bennett et al. 2002, Holland et al. 2010).
In community studies, the 5-year mortality is about 50–60% and the annual
mortality was found to be 10–20% in patients with mild–moderate symptoms
requiring hospital admission and as high as 40–60% in patients with severe HF
(Dargie et al. 1996). McDonah et al. (1997) showed that ischemic heart disease is
present in 83% of patients with LV systolic dysfunction. Nevertheless,
hypertension alone was not more common in patients with than without LV
systolic dysfunction. However, McKee et al. (1971) reported ischemic heart
disease as the precursor of chronic HF in 10% of patients with chronic HF.
Ischemic heart disease was involved in hypertension in 39% of patients. Eriksson

22
et al. (1989) reported that hypertension was the most significant predictor for the
development of chronic HF. Moreover, in the Framingham study McKee et al. (1971) revealed that hypertension accounted for 75% of the patients of chronic
HF. Differences between studies can occur due to the fact that hypertension has
become more easily detectable and treatment is available for these conditions
(McDonagh et al. 1997).
Fig. 1. Pathophysiology of heart failure due to left ventricular dysfunction. Modified
from McMurray & Pfeffer 2005.
Acute heart failure versus chronic heart failure
Acute HF and chronic HF are commonly used to characterize patients with HF.
Worsening HF on a background of chronic HF is the most common form of HF
and it is long-term condition which is associated with the heart undergoing
adaptive responses, such as dilation and hypertrophy (Dickstein et al. 2008).
Acute HF is commonly used to refer to de novo acute HF (the medical emergency
of life-threatening pulmonary oedema) or decompensation of chronic HF
(Dickstein et al. 2008, Swedberg et al. 2005). Acute HF is characterized by the
signs pulmonary congestion (Swedberg et al. 2005).
23
Diastolic heart failure versus systolic heart failure
Distinction of chronic HF is made between diastolic HF and systolic HF. These
two separate syndromes appear to have similar signs, symptoms and prognosis,
although morphological and functional changes are distinctive. In systolic HF, the
LV is dilated and the ejection fraction becomes reduced, whereas in diastolic HF,
the LV is not dilated and the ejection fraction is preserved, which is why diastolic
HF is often diagnosed when symptoms and signs of HF occur in the presence of
normal ejection fraction at rest. Moreover, the neurohumoral abnormalities appear
to be similar in both syndromes (Chatterjee & Massie 2007, Swedberg et al. 2005).
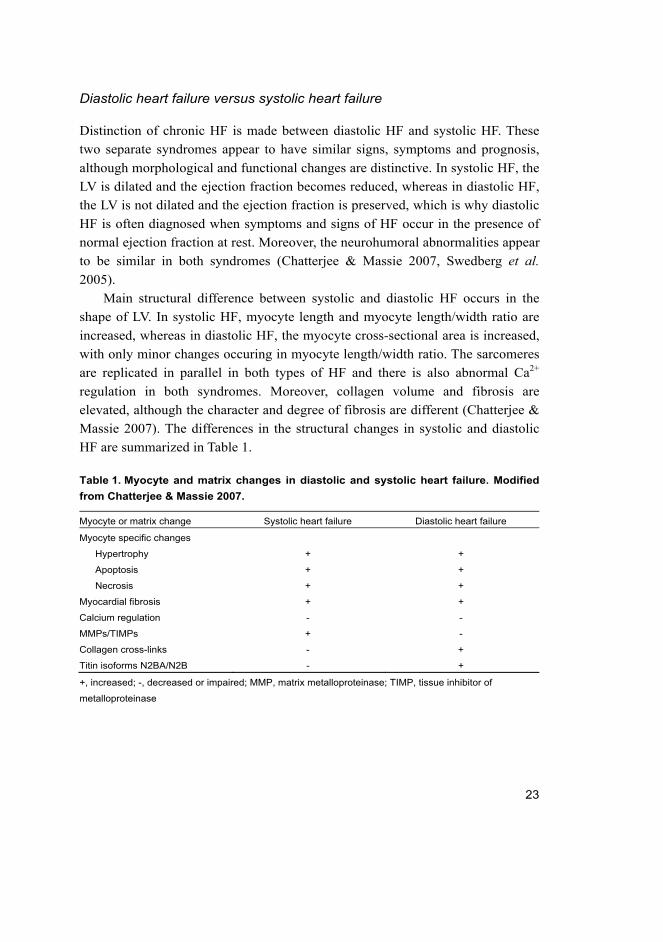
Main structural difference between systolic and diastolic HF occurs in the
shape of LV. In systolic HF, myocyte length and myocyte length/width ratio are
increased, whereas in diastolic HF, the myocyte cross-sectional area is increased,
with only minor changes occuring in myocyte length/width ratio. The sarcomeres
are replicated in parallel in both types of HF and there is also abnormal Ca2+
regulation in both syndromes. Moreover, collagen volume and fibrosis are
elevated, although the character and degree of fibrosis are different (Chatterjee &
Massie 2007). The differences in the structural changes in systolic and diastolic
HF are summarized in Table 1.
Table 1. Myocyte and matrix changes in diastolic and systolic heart failure. Modified
from Chatterjee & Massie 2007.
Myocyte or matrix change Systolic heart failure Diastolic heart failure
Myocyte specific changes
Hypertrophy + +
Apoptosis + +
Necrosis + +
Myocardial fibrosis + +
Calcium regulation - -
MMPs/TIMPs + -
Collagen cross-links - +
Titin isoforms N2BA/N2B - +
+, increased; -, decreased or impaired; MMP, matrix metalloproteinase; TIMP, tissue inhibitor of
metalloproteinase

24
2.2 Cardiac remodelling
Cardiac remodelling is the process by which mechanical, neurohumoral and
genetic factors modify ventricular size, shape and function (Jessup & Brozena
2003, Sutton & Sharpe 2000). LV remodelling may be physiological and adaptive
during normal growth or it can be pathological due to clinical conditions, such as
MI, cardiomyopathy, hypertension and valvular heart disease (Sutton & Sharpe
2000). In general, increased interstial fibrosis, loss of myocytes and hypertrophy
are the most commonly encountered features of cardiac remodelling (Jessup &
Brozena 2003, Sutton & Sharpe 2000).
Several factors are involved in the LV remodelling process. The first
initiating stimuli for cardiac hypertrophy have been segregated into
biomechanical and stretch‒sensitive mechanisms or neurohumoral mechanisms
(Heineke & Molkentin 2006). Biomechanical signals are mediated through
internal stretch–sensitive receptors, which converge on intracellular signal-
transduction circuits to mediate the cardiac growth response. However, activation
of endogenous neurohumoral systems plays a major role in cardiac remodelling
and thus, in the progression of HF. Circulating or tissue levels of neurohumoral
factors such as noradrenaline, Ang II, aldosterone, endothelin, vasopressin and
cytokines are increased in patients with HF (Teerlink 1996). Neurohumoral
factors induce sodium retention and peripheral vasoconstriction, thus imposing
greater hemodynamic stresses on the ventricle. Moreover, myocardial fibrosis can
be stimulated by direct toxic effects of neurohumoral factors on cardiac cells.
Direct deleterious effects of neurohumoral activation on the myocytes and
interstium may alter the performance and phenotype of cardiac cells (Hunt et al. 2005).
2.2.1 Post-infarction left ventricular remodelling
During post-infarction ventricular remodelling, the loss of myocardium cells
results in divergent loading conditions in the border zone of the infarction area
and in a remote noninfarcted myocardium. Increased loading conditions induce
dilatation and complex architectural changes involving both the infarcted and
noninfarcted myocardium, including hypertrophic changes and the formation of a
discrete collagen scar (Jessup & Brozena 2003, Sutton & Sharpe 2000).
Post-infarction remodelling has been divided into an early phase (within 72
hours) and a late phase (beyond 72 hours) (Sutton & Sharpe 2000). In early

25
remodelling, there is expansion of the infarct zone, disproportionate thinning and
LV chamber dilatation of the infarct segment and these processes begin within
hours of the acute infarction and result in the elevation of diastolic and systolic
wall stress (Weisman & Healy 1987). Expansion of the infarct zone causes
deformation of the border zone and the remote myocardium, which in
nonischemic segments changes due to the Frank-Starling mechanism and
achieves increased total segment shortening (Lew et al. 1985). The major reasons
for infarct expansion are loss of myocardial extracellular matrix, involving
degradation of the intermyocyte collagen structures by serine (Ser) proteases and
the activation of matrix metalloproteinases (MMPs) released from neutrophils
(Fig. 2) (Cleutjens et al. 1995, Creemers et al. 2001).
The neurohumoral activation has been regarded as a marker of hemodynamic
function (Morita et al. 1993, Yoshitomi et al. 1998). Atrial natriuretic peptide
(ANP) and BNP are cardiac natriuretic peptides (NP) which are activated in the
acute phase of MI and can be assessed as indicators of LV remodelling after MI.
NPs decrease intravascular volume and systemic vascular resistance, normalize
ventricular filling and ameliorate cardiac function. Abnormalities of
hemodynamic function also trigger the sympathetic adrenergic system.
Catecholamine synthesis is stimulated in the adrenals and they are also released
from the sympathetic nerve terminals leading to activation of the RAA system.
Consequently, production of ANP and BNP is stimulated. Increased shortening
and elevated heart rate from sympathetic stimulation results hyperkinesis of the
noninfarcted myocardium and transiently circulatory compensation (Sigurdsson
& Swedberg 1996, Sutton & Sharpe 2000).
The inflammatory response and cytokine elaboration play a crucial role after
MI. After MI, macrophages, monocytes and neutrophils migrate into the infarct
zone, resulting intracellular signalling and neurohumoral activation, thus
localizing the inflammatory response (Sutton & Sharpe 2000). Cytokines, such
tumour necrosis factor-α (TNF-α) and interleukin-6 are released from
myocardium after myocardial ischemic injury and are involved in regulating
myocyte survival, cellular apoptosis and in triggering the cellular inflammatory
response. Moreover, cytokines mediate repair and remodelling through activating
MMPs and collagen formation, integrin regulation, angiogenesis and progenitor
cell mobilization (Nian et al. 2004).
Progressive remodelling of the remote areas of the LV characterizes late LV
remodelling. As a consequence, chamber dilatation, myocardial hypertrophy, re-
expression of a fetal phenotype and progressive deterioration in systolic pump

26
function are observed (Sam et al. 2000, Sutton & Sharpe 2000). Furthermore, the
LV remodelling during late post-MI is associated with increased apoptosis in
myocardium remote from the area of ischemic injury. In the mouse MI model, a
progressive increase in the number of apoptotic myocytes in myocardium remote
from the area of ischemic damage developed from 1 to 6 months (Sam et al. 2000).
Fig. 2. Post-infarction left ventricular remodelling. HF, heart failure; LV, left ventricular;
MI, myocardial infarction. Modified from Abbate et al. 2002, Shamhart & Meszaros
2010.
2.2.2 Cardiac remodelling and hypertrophy
Cardiac hypertrophy can be defined as an increase in the myocardial mass (Frey
& Olson 2003, Lips et al. 2003) and in consequence, ventricular wall stress
becomes reduced by myocyte growth. Several extrinsic and intrinsic stimuli are
involved to trigger this hypertrophic process in response to increased wall stress
and thus, cardiac hypertrophy normalizes the increased wall tension as an attempt
to abrogate the initial stimulus (Frey & Olson 2003).
Three types of cardiac hypertrophic growth have been described: normal
growth, growth induced by physical conditioning and growth induced by
pathological stimuli (Fig. 3) (Dorn & Force 2005). Physiological cardiac
hypertrophy is observed generally during cardiac postnatal growth, pregnancy or
exercise–induced cardiac growth, which is regulated mainly by the growth

27
hormone/insulin-like growth factor (IGF) axis via signalling through the
phosphatidylinositol-3 kinase (PI3K)/Akt pathway (Dorn & Force 2005).
Pathological hypertrophic myocyte growth is defined as the metabolic, structural
and functional remodelling of the heart (Berenji et al. 2005). Pathological
hypertrophy is a result of the cellular response to an increase in biomechanical or
neurohumoral stress, and it is triggered by autocrine and paracrine factors such as
adrenaline, noradrenaline, Ang II and aldosterone (Berenji et al. 2005, Dorn &
Force 2005, Heineke & Molkentin 2006, Lips et al. 2003). Pathological
hypertrophic growth of myocytes develops in patients with hypertension, obesity,
valvular heart disease, or prior MI or as a result of gene mutation encoding of
contractile protein (Berenji et al. 2005).
Ultimately, pathological cardiac hypertrophy predisposes individuals to HF,
arrhythmia and sudden death (Berenji et al. 2005, Heineke & Molkentin 2006).
Nowadays, hypertrophic growth is established as a marker for increased risk of
developing chronic HF and thus, hypertrophy is considered to be a maladaptive
process (Berenji et al. 2005). Well-known intracellular mediators of hypertrophy
are protein kinases and phosphatases such as mitogen activated protein kinase
(MAPK), Janus kinases, cyclin-dependent kinase-9, Ca2+/calmodulin-dependent
protein kinase II (CaMKII) and calmodulin-dependent phosphatases (Molkentin
2004, Zhang & Brown 2004).

28
Fig. 3. Cardiac hypertrophy can be classified as physiological, pathological or
developmental hypertrophy. LV, left ventricle; RV, right ventricle. Modified from
Heineke & Molkentin 2006.
2.3 Gene delivery vectors for cardiovascular gene therapy
Several gene delivery vectors for cardiovascular gene therapy are currently being
investigated to provide either a transient or a permanent transgene expression.
Gene delivery vectors can be divided roughly into two categories: nonviral and
recombinant viral vectors. Nonviral vectors and adenoviral, AAV and lentiviral
vectors have been most commonly investigated cardiac gene delivery methods.
Adenovirus and AAV vectors have a high transduction efficacy in blood vessel
walls, skeletal muscle, heart and liver. Furthermore, lentivirus vectors have been
shown to be applicable for gene delivery in cardiovascular gene therapy, but
further vector modification will be needed to improve their efficacy (Table 2)
(Davis et al. 2008, Kawase et al. 2011, Korpisalo & Yla-Herttuala 2010, Ly et al. 2007, Rissanen & Yla-Herttuala 2007, Vinge et al. 2008).

29
Table 2. Viral vectors for cardiac gene delivery. Modified from Kawase et al. 2011.
Viral Vector Adenovirus AAV Lentivrus
Genome dsDNA ssDNA ssRNA
Duration of expression Transient (7–14 days) Long-term Long-term
Insert capacity 7–30kb 4.8kb 7–10kb
Advantages Highly efficient entry Low immune response;
high in vivo efficiency
Low immune response;
high efficiency in some
cases
Disadvantages Cytotoxic and
immunogenic effects
Limited insert size;
complex to prepare
Biosafety of parental
virus
Clinical trial approved Yes Yes No
AAV, adeno associated virus; dsDNA, double stranded DNA; ssDNA, single stranded DNA; ssRNA, single
stranded RNA
2.3.1 Nonviral vectors
The first reported direct myocardial injection of naked plasmid deoxyribonucleic
acid (DNA) was performed in 1990 (Lin et al. 1990). The β-galactosidase
reporter gene was administered to the apex of the rat heart and LacZ positive
cardiomyocytes were observed 3 days after administration. The results of this
study revealed that β-galactosidase was detected histochemically for at least 4
weeks after gene transfer (Lin et al. 1990). Subsequently, Buttrick et al. (1992)
reported that expression of the transferred gene was restricted to the vicinity of
the injection site.
Nonviral vectors have many advantages in disease models of gene therapy.
For instance, practically there is no size limitation, which allows the utilization of
large transgenes. Furthermore, nonviral vectors can be produced in large amounts
using standard techniques, they are cheap, very stable and can be stored for
prolonged periods. Two other major benefits are their low immunogenic potential
and low toxicity (Muller et al. 2007, Rapti et al. 2011, Rissanen & Yla-Herttuala
2007).
Lipids and polymers have been developed to form complexes with DNA to
improve transfection efficiency. Plasmid-liposome complexes delivered in mice
by tail vein injection resulted plasmid transfer in the heart and lung at 9–11 days
post-injection, but no plasmid transfer was detected in liver and kidney (Stewart
et al. 1992). Furthermore, tail vein injection of a plasmid-lipid complex resulted

30
in gene expression in myocardium, and also in several other tissues such as
skeletal muscle, lung, spleen and liver (Hofland et al. 1997).
Thus far, direct plasmid injection for heart disease has proved too
challenging, because of the low transfection efficiency and restricted expression
of the transferred gene. Nevertheless, direct myocardial injection has been a
useful tool for basic cardiac research because of its simplicity and the low costs of
production (Ly et al. 2007, Rapti et al. 2011).
2.3.2 Adenoviral vectors
Adenoviruses were first isolated from adenoid tissue (Rowe et al. 1953) and they
are the most significant cause of upper respiratory infections (Douglas 2007). The
wild type adenovirus genome is approximately 35 kilobase (kb) in length, of
which up to 30 kb can be replaced with foreign DNA (Verma & Somia 1997).
Adenoviruses are non-enveloped viruses containing a linear double stranded DNA
genome, which remains episomal after infection.
Recombinant human adenoviral vectors are the most extensively used viral
vectors in experimental gene therapy models. Adenovirus serotype 2 and serotype
5 are the most commonly used viral vectors for clinical gene therapy for
cardiovascular diseases, mainly because of their transduction efficiencies
(Douglas 2007, Nabel 1995). Cytomegalovirus (CMV) and Rous sarcoma virus
(RSV) are the most frequently used viral promoters and they are able to express
genes with a large distribution into tissues (Griscelli et al. 1997). The genes
essential for viral replication are deleted and the vector cannot replicate, thus
adenovirus–mediated gene transfer reaches its maximal effect within the first 2–5
days and the vector is limited by short-term gene expression (about 2 weeks post-
injection) (O'Donnell 2012, Rysa et al. 2010, Tenhunen et al. 2006b).
Adenoviral vectors have been studied for cardiac gene therapy in rodents,
large animals and in humans. Originally, Guzman et al. (1993) and Kass-Eisler et al. (1993) reported that direct injection of replication-deficient recombinant
adenovirus vectors provides a simple and effective but short-term method of
myocardial gene transfer in rats. The myocardium has been targeted with
adenovirus vectors also in mice (Stratford-Perricaudet et al. 1992), rabbits (Barr
et al. 1994) and pigs (French et al. 1994). Subsequently, Giordano et al. (1996)
reported increased myocardial vascularization after intracoronary gene transfer of
fibroblast growth factor (FGF) -5, and Hajjar et al. (1998) demonstrated
depressed contractility after the adenoviral gene transfer of phospholamban (PLB)

31
by open-chest direct injection into the ventricle. Weig et al. (2000) were the first
to document changes in myocardial function following direct intramyocardial
adenovirus–mediated gene delivery in rats; they observed enhanced cardiac
global contractility with gene transfer of vasopressin V2 receptors.
French et al. (1994) described an effective transduction in the hearts of
domestic swine. Depending on the vector concentrations, transduction efficiencies
were up to 75% in the cardiomyocytes around the needle track after direct
injection of adenoviral vectors into the hearts. In fact, gene expression was very
local and did not spread far from injection site. The expressions of transferred
genes were at their highest at 7 days and decreased thereafter. Furthermore,
leukocytic infiltration was observed near to the transduced cardiomyocytes
(French et al. 1994).
Adenoviral vectors have several limitations with respect to their use in HF
gene therapy and clinical applications have proved very challenging. Dai et al. (1995) revealed that an intense immune reaction is behind the short-term
expression of adenoviral–mediated gene transfers. In general, capsin proteins can
stimulate innate and adaptive immune responses and evoke inflammation. As a
consequence, the appearance of neutralizing antibodies against capsin protein
leads to the elimination of the vector (Jooss & Chirmule 2003). In attempts to
reduce the immunological response of adenovirus vector tropism, the fiber coat
protein has been modified by genetic engineering. Transduction of uninfected
cells e.g. antigen presenting cells can be avoided and the efficiency of gene
transfer is increased (Gwathmey et al. 2011). Long-term adenoviral–mediated
gene expression can be achieved if the recombinant adenoviral vectors are
introduced into nude mice or into mice that are given both the adenoviral vector
and immunosuppressing agents (Dai et al. 1995).
2.3.3 Adeno-associated viral vectors
AAVs are human-specific members of the Parvoviridae family. The wild type
AAV has single-stranded DNA and the size of genome is about 4.8 kb. The AAV
genome contains two major genes; the Rep gene codes Rep proteins (Rep 76, Rep
68, Rep 52 and Rep 40), which are involved in AAV replication and the rescue of
the virus, and the Cap gene encodes for AAV structural proteins including viral
protein (VP) VP-1, VP-2 and VP-3, which forms the icosahedral capsid, within
which the replicated genome is packaged (Ponnazhagan et al. 2001, Srivastava et al. 1983, Wasala et al. 2011). The particle size of AAV vectors is approximately

32
20–25 nm in diameter, which is much smaller than that of the adenovirus particle
(approximately 100 nm in diameter including fibers). Thus, AAV vectors can
bypass the blood vessel pores and extracellular matrix much easier than their
adenovirus counterparts (Li et al. 2003). VP3 is a major capsid protein and AAVs
are classified into different serotypes based on amino acid sequence of the capsid
proteins (Wasala et al. 2011).
AAV is dependent on an adenovirus or some other helper virus, such as
herpes viruses to supply the essential gene products that allow AAV to undergo a
productive infection. Different serotypes of AAV have been identified to contain
variations in the amino acid sequence of capsid protein, which suggests their
potential utility in gene therapy applications (Rutledge et al. 1998).
Svensson et al. (1999) demonstrated recombinant AAV (rAAV) vector–
mediated heart gene transfer for the first time in 1999. After coronary artery
perfusion of rAAV containing the LacZ gene, β-galactosidase expression was
detected in <1% of cardiomyocytes at 2 weeks after perfusion and in up to 50% of
cardiomyocytes at 4 to 8 weeks after perfusion. Furthermore, direct
intramyocardial injection of rAAV containing LacZ did not cause any myocardial
inflammation or myocyte necrosis (Svensson et al. 1999). Subsequently, Li et al. (2003) delivered rAAV2 vector containing LacZ gene into the heart of healthy
hamsters. Effective gene transfer was achieved in up to 90% of the
cardiomyocytes and LacZ gene expression was sustained for more than 1 year (Li
et al. 2003).
Several AAV serotypes have been tested for cardiac gene delivery (Wang et al. 2011). rAAV6–mediated gene transfer injected via the tail vein to mice
induced extensive gene transfer in the heart and skeletal muscles (Gregorevic et al. 2004). Furthermore, rAAV8 and rAAV9 vectors appear to be promising for
systemic gene transfer to the heart and muscle. Wang et al. (2005) compared the
efficiency of rAAV1, rAAV2, rAAV5, rAAV6, rAAV7 and rAAV8 in mice and
hamsters after a single injection via intraperitoneal or intravenous routes. They
revealed that rAAV8 was able to cross the vascular barrier effectively and both
skeletal and cardiac muscles were transduced with rAAV8. Moreover, rAAV8 and
rAAV9 transduced tissues more ubiquitously than other serotypes (Zincarelli et al. 2008) and rAAV9 has been revealed to be the most cardiotropic serotype in the
mouse and rat (Bish et al. 2008a). Nevertheless, rAAV8 and rAAV9 also display
tropism for other organs such as liver, skeletal muscle and pancreas by peripheral
vein injection (Inagaki et al. 2006).

33
Even if the immune response against rAAVs is low and transient when
compared to that evoked by adenoviral vectors, several studies have reported
significant seropositivity for rAAV2, rAAV1, rAAV5 and rAAV6 in humans
(Boutin et al. 2010, Calcedo et al. 2009, Halbert et al. 2006). It is notable that
immune systems between murine models and in humans display notable
differences (Zaiss & Muruve 2008) and the immune response to rAAV vectors
appears to be a major obstacle to clinical trials (Rapti et al. 2011, Rissanen & Yla-
Herttuala 2007).
2.3.4 Lentiviral vectors
Lentiviruses belong to the retrovirus family. These viruses have enveloped
capsids and a stranded ribonucleic acid (RNA) genome and they are able to infect
both proliferating and non-proliferating cells. They can package an approximately
8–10 kb genome. The viral genome is reverse transcribed into double-stranded
DNA, which gradually integrates into the host genome, thus enabling the ability
to achieve long-term stable transgene expression. Lentiviral–mediated gene
delivery enables sustained expression for over 9 months in rodent brains
(Makinen et al. 2006) and for over 6 months in liver, muscle, eye or pancreatic-
islet cells (Blomer et al. 1997, Miyoshi et al. 1997, Verma & Somia 1997). The
first developed and most commonly used lentiviral vectors are based on the
human immunodeficiency virus type 1, which has been disabled and developed as
a vector for gene delivery (Trono 2000).
Lentiviral vectors efficiently transduce adult rat cardiomyocytes. Lentiviral–
mediated gene expression peaked at day 3 and declined by about 4–fold at day 14.
Thereafter gene expression remained stable up to week 10 (Fleury et al. 2003).
Furthermore, Zhao et al. (2002) demonstrated lentiviral gene transfer into
neonatal and adult cardiac myocytes in vitro. The achieved transduction efficiency
was 70% in adult cardiomyocytes and 100% in neonatal cardiomyocytes. Niwano
et al. (2008) demonstrated in the ischemic cardiomyopathy model that lentiviral–
mediated SERCA2 gene transfer effectively improved cardiac function, achieved
favourable molecular remodelling, prevented LV remodelling after MI and
improved the survival rate.
Potential applications of lentiviral vectors for human gene therapy have not
been extensively explored. The major limitation of lentiviral–mediated gene
transfer is related to biosafety. The pathogenicity of the parental virus is a major

34
concern even though the safety features of lentiviral vectors have been improved
and non-essential regulatory genes removed (Kawase et al. 2011, Trono 2000).
2.3.5 Other gene transfer vectors
In hybrid vectors, nonviral vectors are combined with viral vectors. Hybrid
vectors have been widely studied in oligonucleotide-based gene therapy models
(Kaneda 1999, Yasufumi 2000). In general, virosomes are hybrid vectors based on
liposomes. The most studied virosome of with potential cardiovascular gene
therapy applications is hemagglutinating virus of Japan-liposome (Aoki et al. 1997, Kaneda 1999). Virosomes introduce DNA directly into the cytoplasm, and
they contain DNA and DNA-binding nuclear protein in order to enhance
expression of the gene (Ly et al. 2007).
There are some other uncommon viral based vectors such as Epstein-Barr
virus (Tomiyasu et al. 2000), foamy virus (Mergia & Heinkelein 2003) and
simian virus 40 (Strayer et al. 2006) which have been reported to have possible
application in cardiac gene therapy.
2.4 In vivo myocardial gene delivery techniques
In vivo gene deliveries for cardiovascular diseases have focused on the
development of methods to deliver genes in vascular cells and cardiac myocytes
(Nabel 1995, Rissanen & Yla-Herttuala 2007). The gene delivery method should
be technically efficient and simple, feasible, inexpensive, safe and well tolerated,
and only transduce specific regions of the targeted tissue (Davis et al. 2008). The
main gene delivery techniques used in vivo are direct injection into the
myocardium (French et al. 1994, Tenhunen et al. 2006, Rysa et al. 2010) and
intravascular systemic delivery (Bridges et al. 2005, O'Donnell & Lewandowski
2005) (Fig. 4). Furthermore, several specific methods have been developed for
vector delivery within these main categories (Davis et al. 2008).
35
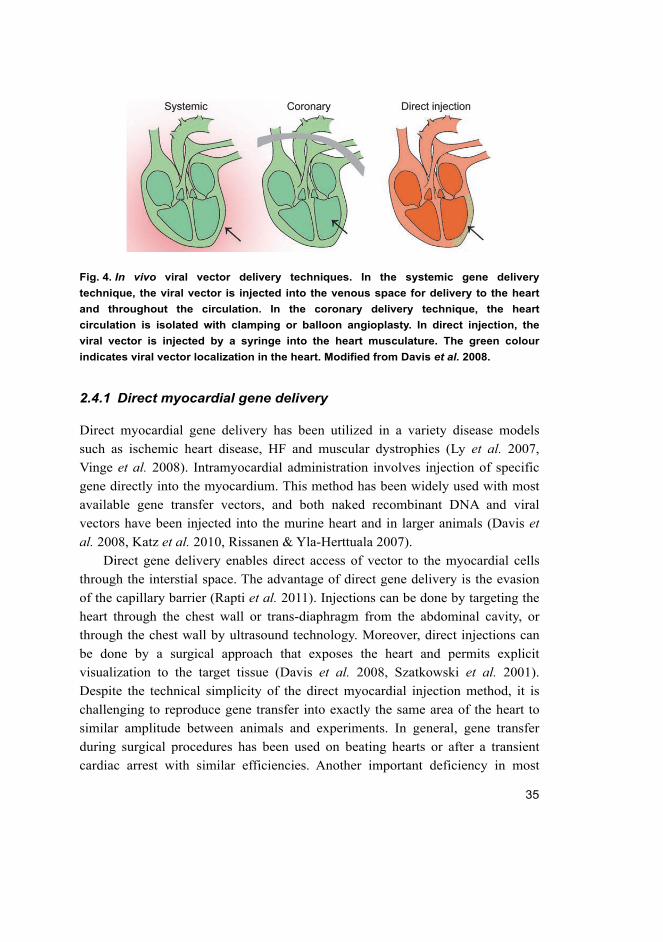
Fig. 4. In vivo viral vector delivery techniques. In the systemic gene delivery
technique, the viral vector is injected into the venous space for delivery to the heart
and throughout the circulation. In the coronary delivery technique, the heart
circulation is isolated with clamping or balloon angioplasty. In direct injection, the
viral vector is injected by a syringe into the heart musculature. The green colour
indicates viral vector localization in the heart. Modified from Davis et al. 2008.
2.4.1 Direct myocardial gene delivery
Direct myocardial gene delivery has been utilized in a variety disease models
such as ischemic heart disease, HF and muscular dystrophies (Ly et al. 2007,
Vinge et al. 2008). Intramyocardial administration involves injection of specific
gene directly into the myocardium. This method has been widely used with most
available gene transfer vectors, and both naked recombinant DNA and viral
vectors have been injected into the murine heart and in larger animals (Davis et al. 2008, Katz et al. 2010, Rissanen & Yla-Herttuala 2007).
Direct gene delivery enables direct access of vector to the myocardial cells
through the interstial space. The advantage of direct gene delivery is the evasion
of the capillary barrier (Rapti et al. 2011). Injections can be done by targeting the
heart through the chest wall or trans-diaphragm from the abdominal cavity, or
through the chest wall by ultrasound technology. Moreover, direct injections can
be done by a surgical approach that exposes the heart and permits explicit
visualization to the target tissue (Davis et al. 2008, Szatkowski et al. 2001).
Despite the technical simplicity of the direct myocardial injection method, it is
challenging to reproduce gene transfer into exactly the same area of the heart to
similar amplitude between animals and experiments. In general, gene transfer
during surgical procedures has been used on beating hearts or after a transient
cardiac arrest with similar efficiencies. Another important deficiency in most

36
rodent studies is that the sites of injection to nonseptal walls of the ventricle are
limited, for example, direct injections to atrial tissue is unfeasible. In addition,
some damage to the tissue will occur along the needle track. Gene delivery to the
septal wall, trabeculae and papillary muscles is also limited, even if an ultrasound
guidance method has been used in attempts to improve gene transfer to the septal
wall (Davis et al. 2008).
Moreover, vector and vector dosage can influence transduction deficiency.
Adenovirus- and rAAV–mediated gene transfer with direct injection has resulted
in a strong response of the ventricular tissue (Bish et al. 2008b, French et al. 1994, Szatkowski et al. 2001). However, injection with retrovirus-based vectors
has induced much lower gene expression and a reduced area of transduced cardiac
tissue (Bonci et al. 2003, Zhao et al. 2002).
Lin et al. (1990) demonstrated that the delivery of naked recombinant DNA,
encoding the LacZ gene, into the LV free wall via a left thoracotomy resulted in
patchy gene expression and was observed within a few millimeters of the
injection site. In large animal models, multiple injections to achieve gene
expression over a clinically pertinent area of myocardium were performed e.g. by
using a grid to define the sites of injection and to avoid coronary vessels (French
et al.1994).
Grossman et al. (2002) revealed that the injection volume is crucial issue.
When endocardial injection volume of neutron-activated microspheres was 10 μl,
almost all of the injected microspheres were retained in the myocardium, whereas
at injection volumes of 100 μl, only 20% were retained in the myocardium, and
via epicardial administration, a mere 10% were retained in the myocardium
(Grossman et al. 2002).
One application of the direct gene transfer method is to inject the vector into
the pericardial space. Adenoviral injection of LacZ into the pericardial sac of rat
hearts resulted in attenuated gene expression (Fromes et al. 1999). Injection of a
mixture including adenovirus and proteolytic enzymes, such as collagenase and
hyaluronidase led to a large diffusion of the transgene activity, reaching up to
40% of the myocardium. The pattern of expression was located mainly in the
neighboring pericardial area (Fromes et al. 1999).
In “gene gun” technology, the vector consists of gold particles complexed
with plasmid DNA. In this method, the heart is surgically exposed and cardiac
musculature is bombarded with the gold particles under gas pressure (Matsuno et al. 2003, Nishizaki et al. 2000). The method has several limitations, such as high
death rate, gene expression levels are not robust and the transduced cells are

37
limited to a superficial layer of the myocardium and even there expression is
weak (Matsuno et al. 2003, Nishizaki et al. 2000, Umeda et al. 2002).
Some of the studies have reported the effects of addition of adjuvants such as
proteases or hyaluronidase within the injectants, to improve the poor penetration
levels. The addition of enzymes can facilitate vector diffusion through the tissue
by degrading the extracellular matrix, thus yielding efficient transduction (Davis
et al. 2008, Fromes et al. 1999, Kuriyama et al. 2000, Pitard et al. 2002).
However, the effects of adjuvants to the cardiac musculature are unclear and need
accurate control (Davis et al. 2008).
2.4.2 Intravascular gene delivery
In animal models, intracoronary gene delivery can be performed in either a
retrograde manner into the coronary sinus or through antegrade coronary artery
delivery (Logeart et al. 2006, Raake et al. 2004). These techniques are catheter
based and thus, enable a remote invasive percutaneous method (Logeart et al. 2006). Catheters are used to deliver plasmid DNA, adenovirus and rAAV vectors
into the right atrium and to the root of the aorta above the sinuses for access to the
coronary arteries, e.g. in rodents (O'Donnell & Lewandowski 2005), dogs
(Bridges et al. 2005) and sheep (Humpl et al. 2005). Ultrasound and fluoroscopy
are generally used as the guiding modality (Davis et al. 2008).
Catheter-based percutaneous antegrade coronary myocardial gene transfer is
the most widely used application in human gene therapy (Hajjar et al. 2008, Katz
et al. 2010, Rapti et al. 2011). The percutaneous antegrade epicardial coronary
artery infusion cardiac gene delivery method has been used in one human clinical
trial (Jaski et al. 2009).
Raake et al. (2004) applied adenoviral vectors via surgical intramyocardial
delivery and selective pressure-regulated retroinfusion. Percutaneous selective
pressure-regulated retroinfusion of the coronary veins in pigs resulted in increased
and homogenous reporter gene expression in the left anterior descending coronary
artery (LAD) when compared with surgical gene transfer (Raake et al. 2004).
Kaspar et al. (2005) used indirect intracoronary delivery for rats. rAAV2–
mediated enhanced green fluorescent protein (eGFP) gene transfer resulted
transgene expression lasting up to 12 months, with a gradient expression across
the LV wall. The transgene was expressed in the epicardium much more than in
the endocardium (Kaspar et al. 2005).

38
In animal models, gene delivery into the coronary circulation has been shown
to increase the vector dwelling time in the coronary vessels if one conducts cross-
clamping of the aorta and pulmonary artery i.e. an increased transduction
efficiency (Hajjar et al. 1998, Hayase et al. 2005, Ikeda et al. 2002). The catheter-
based technique allows the adenovirus containing solution to circulate down into
the coronary arteries and perfuse the heart without requiring direct manipulation
of these vessels. The transgene expression was relatively homogenous and diffuse
throughout the myocardium (Hajjar et al. 1998). In the cross-clamping technique,
the heart circulation is isolated with clamping or balloon angioplasty. In general,
the animal is placed on a heart-lung bypass with induced cardioplegia and the
blood is washed out and replaced with the permeabilize buffer. The viral vector is
delivered under pressure and allowed to have a dwell time (Bridges et al. 2005,
Davis et al. 2008). Moreover, balloon catheters are used to occlude the vascular
outflow of the heart. Hayase et al. (2005) occluded anterior interventricular vein
during left anterior descending artery delivery, and the great cardiac vein at the
entrance of the middle cardiac was occluded during left circumflex artery delivery
in an attempt to increase the dwell time and the pressure of the delivered
adenovirus.
In larger animals, myocardial gene delivery conducted with the
cardiopulmonary bypass technique have achieved prolonged exposure of the
vasculature to the vector and allow crossing to take place. Cardiopulmonary
bypass provides increased dwelling time, removal blood cells from the
circulation, the possibility to apply lower temperatures via cold crystalloid
cardioplegia, and the possibility to the recirculate gene delivery solution through
the coronary system (Bridges et al. 2005, Davidson et al. 2001, Jones et al. 2002).
Tail vein injections are a commonly used rAAV delivery method. One major
limitation of tail vein injection is the possibility of co-infection also in noncardiac
tissues, such as skeletal muscle and liver (Zincarelli et al. 2008). In general, rAAV
gene delivery via tail vein injections is well-tolerated and does not result in
immune responses or significant toxicity (Davis et al. 2008, Gregorevic et al. 2004, Muller et al. 2006).
There are several limitations to intravascular gene delivery. For instance, it is
possible that blood contains neutralizing antibodies to the vector. The blood also
contains proteins, such as albumin and platelets, which may absorb the delivery
vector (Davis et al. 2008). The presence of the blood complement system may
also attenuate response of gene transfer vectors. Furthermore, the vector must also
cross physical barriers, such as endocardial cells and the capillary endothelium.

39
Several rAAV serotypes are able to penetrate the endothelial barrier to reach
cardiomyocyte with different efficiencies (Di Pasquale & Chiorini 2006). rAAV8
is the most efficient vector for crossing the blood vessel barrier in order to
achieve systemic gene transfer in cardiac muscle in vivo, whereas rAAV1 and
rAAV6 were less effective in crossing the blood vessel barrier (Wang et al. 2005).
Furthermore, there are limitations to animal studies in their ability to predict
human responses. Intravascular viral gene delivery involves a large amount of
viral solution and this can lead to organ toxicity and an allergic reaction (Raper et al. 2003).
Chemical substances, e.g. vasodilatory and permeabilizing agents, have been
used to facilitate transfer of gene delivery vector from vascular lumen to the
myocardium. The gene delivery vector has to overcome the capillary wall barrier
so that it can reach the interstial space (Nagata et al. 2001). Several agents have
been used to increase gene transfer efficiency in preclinical studies, including
nitroprusside, nitroglycerin, serotonin, bradykinin, histamine, substance P,
sildenafil, adenosine, heparin and vascular endothelial growth factor (VEGF)
(Hillegass et al. 2001, Kawase et al. 2011, Nagata et al. 2001, Rapti et al. 2011),
although VEGF has been the most commonly used microvascular permeabilizing
agent (Rapti et al. 2011).
2.5 Gene targets for the treatment of heart failure
HF is a complex pathological state and characterizing of the mechanisms at the
molecular, neurohumoral and hemodynamic levels has identified several
promising gene targets for the treatment of HF. So far, the most detailed studied
gene therapy targets of HF have attempted enhancement of contractility via β-AR
pathways and Ca2+ handling proteins as well as anti-apoptotic and angiogenesis
associated proteins (Chaanine et al. 2010, Katz et al. 2010, Katz et al. 2011).
2.5.1 Calcium handling proteins
Ca2+ signalling during excitation-contraction coupling is an essential feature of
the cardiomyocyte (Berridge et al. 2003, Bers 2002). Defects in Ca2+ handling
protein with impaired sarcoplasmic reticulum (SR) Ca2+ uptake and release have
been a focus of molecular HF research because Ca2+ homeostasis is disturbed in
failing myocytes (del Monte et al. 2001). Impaired intracellular Ca2+ homeostasis
and alterations of Ca2+ handling proteins have been revealed in both experimental

40
studies (Bing et al. 1991, Hasenfuss 1998) and human HF (Go et al. 1995,
Gwathmey et al. 1987, Hasenfuss 1998).
Excitation-contraction coupling initiates stimulation of Ca2+–induced Ca2+
release in SR. Depolarization activates voltage-gated L-type Ca2+-channels of the
T-tubule to allow Ca2+ appearance into the cardiomyocyte (Vinge et al. 2008).
The influx of Ca2+ triggers the ryanodine receptor (RyR) to open the Ca2+ release
channel of SR, releasing Ca2+ from the SR into the cytosol. RyRs are a family of
Ca2+ release channels, and they form a linkage between the T tubules in the
cardiomyocytes and the SR (Kawase et al. 2011). The concentration of Ca2+ in the
cytosol is increased, triggering cardiomyocyte contraction through Ca2+ binding
to troponin C (Bers 2002). Relaxation of the sarcomere is initiated when Ca2+ is
removed from the cytosol. Ca2+ detaches from troponin C and returns to the SR
via the action of SERCA2a or its extrusion from the cardiac cell via the
sarcolemmal Na+/Ca2+ exchange (Bers & Despa 2006).
SERCA2a activity is regulated by PLB, a SR transmembrane protein. In its
unphosphorylated form, PLB inhibits SERCA2a function, whereas the
phosphorylation of PLB relieves this inhibitory effect resulting in increased
SERCA2a activity with improved SR Ca2+ reuptake and release (Kawase et al. 2011, Ly et al. 2007). Serine/threonine-protein phosphatase type 1 (PP1), the
major SR phosphatase dephosphorylates specifically PLB in the heart (Nicolaou
et al. 2009, Pathak et al. 2005). This mechanism by which stimulation of the β-
adrenergic axis induces phosphorylation of a PP1 inhibition with 3’,5’-cyclic
adenosine monophosphate (cAMP)-dependent protein kinase A (PKA) activation
and PLB phosphorylation results in enhancement of cardiac contractility
(Chaanine et al. 2010, Kawase et al. 2011, Ly et al. 2007, Vinge et al. 2008).
Taken together, HF is characterized by several defects in the Ca2+-handling
proteins and reversal of those effects by gene therapy techniques has shown
promising results in the treatment of HF. Excitation-contraction coupling in
cardiac myocytes is summarized in Fig. 5.
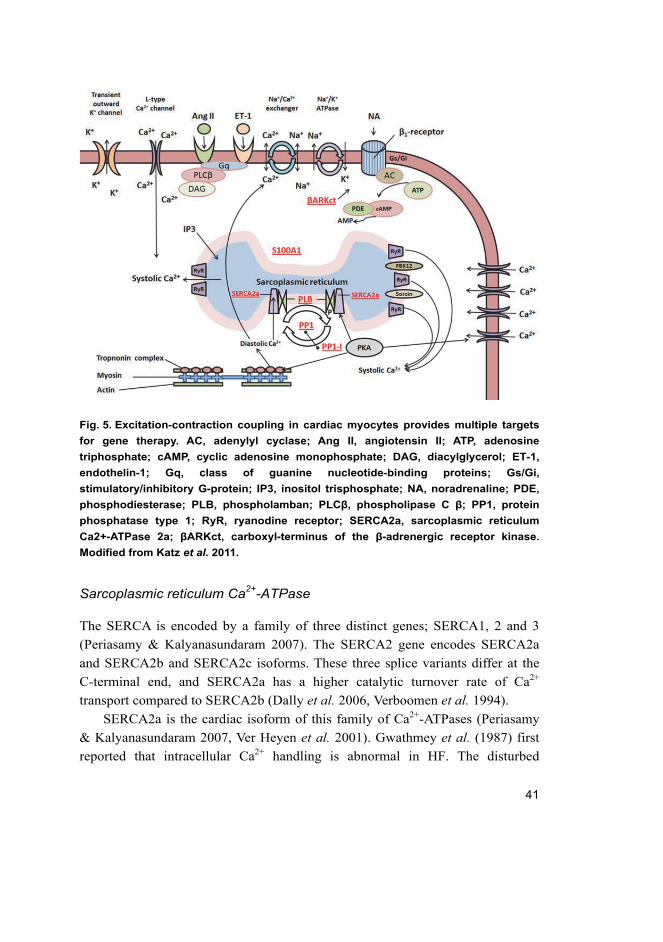
41
Fig. 5. Excitation-contraction coupling in cardiac myocytes provides multiple targets
for gene therapy. AC, adenylyl cyclase; Ang II, angiotensin II; ATP, adenosine
triphosphate; cAMP, cyclic adenosine monophosphate; DAG, diacylglycerol; ET-1,
endothelin-1; Gq, class of guanine nucleotide-binding proteins; Gs/Gi,
stimulatory/inhibitory G-protein; IP3, inositol trisphosphate; NA, noradrenaline; PDE,
phosphodiesterase; PLB, phospholamban; PLCβ, phospholipase C β; PP1, protein
phosphatase type 1; RyR, ryanodine receptor; SERCA2a, sarcoplasmic reticulum
Ca2+-ATPase 2a; βARKct, carboxyl-terminus of the β-adrenergic receptor kinase.
Modified from Katz et al. 2011.
Sarcoplasmic reticulum Ca2+-ATPase
The SERCA is encoded by a family of three distinct genes; SERCA1, 2 and 3
(Periasamy & Kalyanasundaram 2007). The SERCA2 gene encodes SERCA2a
and SERCA2b and SERCA2c isoforms. These three splice variants differ at the
C-terminal end, and SERCA2a has a higher catalytic turnover rate of Ca2+
transport compared to SERCA2b (Dally et al. 2006, Verboomen et al. 1994).
SERCA2a is the cardiac isoform of this family of Ca2+-ATPases (Periasamy
& Kalyanasundaram 2007, Ver Heyen et al. 2001). Gwathmey et al. (1987) first
reported that intracellular Ca2+ handling is abnormal in HF. The disturbed

42
function of SR was suggested in early studies showing that the SR Ca2+ pump
messenger RNA (mRNA) levels were reduced in the failing heart (Arai et al. 1992, Arai et al. 1993, Mercadier et al. 1990 Takahashi et al. 1992). Furthermore,
protein levels of SERCA2a were found to be decreased in relation (Hasenfuss et al. 1994, Meyer et al. 1995) to total protein, to calsequestrin, to the RyR and to
PLB in human HF (Meyer et al. 1995). On the other hand, in some studies, there
were no differences in the expression of SERCA2 between nonfailing and failing
myocardium (Movsesian et al. 1994, Munch et al. 1998). Moreover, the
heterogeneity of SERCA expression levels in failing hearts has been suggested to
depend on age, gender, drug treatment, severity of disease and methodological
differences (Periasamy & Kalyanasundaram 2007).
Early in vitro studies revealed that overexpression of SERCA2a in failing
human ventricular cardiomyocytes enhanced contractility and relaxation velocity
and normalization of Ca2+ handling (del Monte et al. 1999). In a rat model of
pressure overload hypertrophy in the transition to HF, SERCA2a gene transfer
restored SERCA2a expression and adenosine triphosphatase (ATPase) activity to
nonfailing levels. Furthermore, SERCA2a overexpression normalized LV systolic
and diastolic function. The size of LV was reduced and the slope of the end
systolic pressure/dimension relationship was restored (Miyamoto et al. 2000). In a
rat model of HF, SERCA2a gene transfer via of a catheter-based technique
normalized LV volumes (del Monte et al. 2001). Furthermore, overexpression of
SERCA2a in failing heart restored the levels of phosphocreatine and ATP
enabling normalized Ca2+ transport and in improving cardiac energetics (del
Monte et al. 2001).
SERCA2-knockout mice studies have demonstrated that homozygous null
SERCA2-/- mice die early in development (Periasamy et al. 1999). In
heterozygous SERCA2+/- mice, the SERCA2a mRNA in heart was reduced by
45% and SERCA2 protein levels and maximal velocity of Ca2+ uptake into the SR
by 35%. 12–16-week-old SERCA2+/- mice appeared to be healthy and no cardiac
pathology was exhibited (Periasamy et al. 1999). When SERCA+/- mice hearts
were stressed in physiological demands via pressure overload, a decrease in
SERCA pump level led to cardiac dysfunction and HF much more quickly than in
the wild type control mice. Furthermore, SERCA2+/- mice exhibited attenuated
intracellular Ca2+ homeostasis and decreased myocardial contractility (Schultz et al. 2004). SERCA2+/- mice showed an exaggerated response to transverse aortic
constriction (Schultz et al. 2004), whereas the transgenic mouse model
overexpressing SERCA2a showed better survival and preserved contractile in

43
early HF in mice with chronic pressure overload (Ito et al. 2001). In addition,
work done in a transgenic rat model confirmed the role of SERCA2 in Ca2+
homeostasis and contractile function (Muller et al. 2003). Overexpression of rat
SERCA2 in the hearts of transgenic mice rescued disturbed Ca2+ cycling and
enhanced myocardial contractility and relaxation (He et al. 1997).
Interestingly, overexpression of SERCA2a has been reported to increase
glucose oxidation, which accompanies the improved contractile phenotype of the
heart (Belke et al. 2007). Cardiac-restricted transgenic mice expressing the
mutant SERCA2a, which disrupted the functional association of PLB was
associated with an increase in Ca2+ affinity of SERCA2a in microsomes isolated
from hearts, suggesting that overexpressed SERCA2a had been integrated into SR
membranes in the heart of transgenic mice. Overexpression of the high Ca2+
affinity mutant SERCA2a induced attenuation of pressure overload–induced
cardiac hypertrophy and enhancement of cardiac contractility (Nakayama et al. 2003). Interestingly SERCA2a-transgenic rats suffered increased mortality 24h
after MI compared to wild type rats. A higher mortality rate was associated with a
higher frequency of ventricular arrhythmias. In that study, no beneficial effects
were attributable to SERCA2a overexpression at the end of 6 months (Chen et al. 2004).
During β-adrenergic stimulation, the expected enhancement of failing canine
myocyte contraction was abrogated by SERCA2a (Hirsch et al. 2004). Despite
these disadvantages, in a large-animal, volume-overload model of HF, long-term
overexpression of SERCA2a by rAAV1–mediated gene transfer via intracoronary
gene delivery preserved systolic function, prevented diastolic dysfunction and
improved ventricular remodelling in pigs (Kawase et al. 2008). At present, from
the point of view of gene therapy, increasing the amount of SERCA2 protein has
appeared to be the most promising gene therapy target in HF (see also 2.6.1).
Phospholamban
SERCA2a activity is regulated by its interaction with a 52 amino acid
phosphoprotein, PLB (James et al. 1989, Tada et al. 1975). Interactions between
PLB and SERCA2a control the Ca2+ content of the SR and thus, cardiac
contractility. In vitro experiments have shown that PLB can be phosphorylated at
Ser 16 by cAMP- and 3’,5’-cyclic guanosine monophosphate (cGMP)-dependent
protein kinases (PKA and protein kinase G, respectively) and at threonine (Thr)
17 by CaMKII and at Ser10 by protein kinase C (Drago & Colyer 1994,

44
Movsesian et al. 1984, Simmerman et al. 1986). Phosphorylations of PLB at
Ser16 by the cAMP-dependent PKA pathway and at Thr17 by the CaMKII
pathway are the crucial determinants of the positive inotropic and cardiac
relaxation effects evoked by β-adrenergic stimulation (Bartel et al. 2000,
Lindemann et al. 1983, Lindemann & Watanabe 1985, Mattiazzi et al. 2005,
Wegener et al. 1989). Phosphorylation of PLB at Ser16 by protein kinase G has
positive inotropic (contractility) and lusitropic (relaxation rate) effects in the heart
(Pierkes et al. 2002) and it has been associated with the regulation of smooth
muscle relaxation (Mundina-Weilenmann et al. 2000). Phosphorylation of PLB at
Ser10 by protein kinase C has not been observed in the intact heart (Edes &
Kranias 1990). In its dephosphorylated form, PLB binds to SERCA2a and inhibits
Ca2+ activity, whereas phosphorylation of PLB reverses Ca2+-pump inhibition and
increases SERCA2a activity and Ca2+ uptake into the SR (Chu & Kranias 2002).
Several studies have revealed that increased activity of PLB can contribute to
reduced SR function and cardiac contractility in failing human hearts (Meyer et al. 1995, Schwinger et al. 1999).
Overexpression of wild type PLB in the heart of transgenic mice resulted in
an inhibition of Ca2+ transport by the SR, reduced Ca2+ kinetics and basal left
ventricular systolic function. Similarly, contractile parameters in ventricular
myocytes were decreased compared to control myocytes (Dash et al. 2001,
Kadambi et al. 1996). In addition, stimulation with the β-agonist, isoproterenol,
prevented inhibitory effects by stimulating phosphorylation of PLB, thus relieving
its inhibitory effects on the SERCA2a affinity for Ca2+ (Dash et al. 2001). In PLB
transgenic mice, cardiac contraction rate and prolongation of relaxation phase
were significantly depressed by the β-receptor antagonist, propranolol, indicative
of improved sympathetic tone (Dash et al. 2001).
Transgenic mice overexpressing superinhibitory PLB mutants
(Aspargine27Alanine, Leucine37Alanine, Isoleucine40Alanine and
Valine49Glycine) was associated with increased inhibition of the affinity of
cardiac SERCA2a for Ca2+, which resulted in disturbed Ca2+ handling and
decreased fractional shortening of cardiac myocytes (Haghighi et al. 2001,
Schmidt et al. 2002, Zhai et al. 2000, Zvaritch et al. 2000).
The loss of PLB in various animal models of HF has been studied. For
instance, PLB-knockout mice studies have shown that ablation of PLB alters
cardiac function by enhancing myocardial performance without changing heart
rate and attenuated contractile responses to β-adrenergic stimulation (Luo et al. 1994). Subsequently, crossbreeding PLB-knockout mice into mouse models of

45
genetic cardiomyopathy resulted in rescue of cardiac function (Minamisawa et al. 1999, Sato et al. 2001). Calsequestrin overexpressing mice have been cross-bred
with PLB-knockout mice which led to a reversal of the depressed cardiac
contractile parameters (Sato et al. 2001). Furthermore, PLB ablation prevented
systolic dysfunction and improved exercise performance in a mouse model of
hypertrophic cardiomyopathy (Freeman et al. 2001). In hyperdynamic PLB-
knockout mice, cardiac compensation against a chronic aortic stenosis was similar
to that seen in wild type counterparts (Kiriazis et al. 2002).
From the point of view of gene therapy, a dominant negative mutant of PLB
(phosphomimetic mutation at Ser16, named S16E) has been generated. rAAV2–
mediated overexpression of PLB-S16E prevented cardiac deterioration in a
hamster cardiomyopathy model (Hoshijima et al. 2002) and in rats after acute MI
(Iwanaga et al. 2004). Silencing of PLB expression in a sheep HF model after
intracoronary delivery of adenovirus expressing PLB-S16E led to increased
SERCA2a activity and improved systolic and diastolic LV function (Kaye et al. 2007).
RNA interferency therapy has also been used to downregulate PLB
expression in rats with HF. rAAV9-RNA interference vector expressing a small
hairpin RNA targeting PLB (rAAV9-shPLB) was delivered into the aortic roots in
rats with HF (Suckau et al. 2009). Cardiac PLB expression was suppressed to
25% and SERCA2a expression was increased in HF rats. Moreover, systolic and
diastolic cardiac functions were restored and cardiac dilatation became
normalized, cardiac hypertrophy, cardiomyocyte diameter and cardiac fibrosis
were reduced rAAV9-shPLB treated hearts in HF (Suckau et al. 2009).
Furthermore, in cultured rat neonatal cardiomyocytes, adenovirus based anti-
sense RNA of PLB resulted in an increased Ca2+ affinity of SR calcium pump and
improved Ca2+ uptake (Eizema et al. 2000, He et al. 1999). In failing human
cardiomyocytes, decreasing PLB levels by applying small interfering RNA
knockdown method has been shown to improve single-cell contractility, Ca2+
handling and the frequency response was restored to normal in the failing
cardiomyocytes (del Monte et al. 2002).
Role of PLB in pathophysiology in human HF remains unclear. Several
studies have indicated that the levels of PLB protein remain unchanged with end-
stage ischemic or dilated cardiomyopathy (Bohm et al. 1994, Flesch et al. 1996,
Linck et al. 1996, Movsesian et al. 1994, Schwinger et al. 1995). Some studies
noted a decrease in PLB mRNA levels in human HF, but this decrease was not
associated with a decrease in PLB protein (Feldman et al. 1991, Linck et al.

46
1996) or its levels decreased only slightly with dilated cardiomyopathy (Meyer et al. 1995). In failing human myocardium, the phosphorylation of PLB at Ser16
and Thr17 has been reported to be decreased, evidence for an increased inhibitory
function by PLB (Dash et al. 2001, Schwinger et al. 1999). Hence, although PLB
ablation in animal models confers several benefits on cardiac function in HF,
there are difficulties and controversies to extrapolate findings from animal models
to humans and thus, studies targeting PLB need confirmation.
Protein phosphatase type 1 and Inhibitor type-1
Cardiac contractile performance is regulated by the PP type 1 and type 2, which
are members of the Ser/Thr phosphatases. PP1 is localized in the SR membranes
and it is a negative regulator of β-adrenergic signalling (Steenaart et al. 1992).
Although SERCA2a activity in myocytes is controlled by PLB, inhibition of PP1
activity by two endogenous inhibitors type 1 (I-1) and type 2 is needed for Ca2+
homeostasis and cardiac contractility (Raake et al. 2011). It is known that β-
adrenergic signalling activates I-1 on phosphorylation of Thr35 by PKA and
thereby promotes inhibition of PP1 activity. Thus, enhanced PKA–mediated
protein phosphorylation resulted in amplification of the β-agonist response in
cardiac muscle (Carr et al. 2002).
In a transgenic mouse model, the levels of the catalytic subunit of PP1 were
increased in hearts, resulting in impaired cardiac contractility and dilated
cardiomyopathy (Carr et al. 2002). Furthermore, deletion of I-1 resulted in a
moderate increase in PP1 activity and decreased β-adrenergic–mediated Ca2+
signalling. Although no alterations in the PLB, SERCA or calsequestrin
expression levels were observed, phosphorylation of PKA phosphorylation of
PLB at the CaMKII site, Thr17, was significantly reduced in I-1 knockout hearts.
The same study also reported that in isolated failing human cardiomyocytes,
inhibition of PP1 by a constitutively active I-1 via intracoronary adenovirus–
mediated expression could improve the contractile response to a β-agonist (Carr
et al. 2002). In failing human hearts with dilated cardiomyopathy, there were no
changes in total I-1 protein levels, but phosphorylation of I-1 was reduced and
contractile function was enhanced in the presence or absence of β-agonist.
Furthermore, SERCA levels were decreased and the degree of phosphorylation of
PLB on both Ser16 and Thr17 was decreased in failing human hearts (Carr et al. 2002).

47
Double transgenic mouse model with cardiac specific expression of a
constitutively active and truncated form of I-1 resulted in enhanced basal
contractility associated with increased PLB phosphorylation at Ser16 and Thr17,
and enhanced Ca2+ uptake into the SR. Active I-1 expression improved contractile
function and recovery, and reduced infarct size after ischemia/reperfusion–
induced injury (Nicolaou et al. 2009). Moreover, adenoviral gene delivery of
active I-1 resulted in restoration of myocardial contractility, partially reversed
remodelling and overactivated p38 MAPK, without any alteration in ERK1/2 or
c-jun N-terminal kinase (JNK) activation in rats with pressure overload–induced
HF (Pathak et al. 2005). Transgenic mice with cardiac-specific overexpression of
the rat full-length I-1 complementary DNA (cDNA) developed cardiac
hypertrophy and mild dysfunction (El-Armouche et al. 2008). The same study
demonstrated that I-1-knockout hearts displayed unchanged maximal contractile
responses to β-adrenergic stimulation and I-1-knockout hearts were partially
protected against lethal catecholamine–induced arrhythmias and from
hypertrophy and dilatation induced by isoprenaline. This protection was
associated with a reduction in the degree of phosphorylation of PLB and RyR (El-
Armouche et al. 2008).
In conclusion, targeting PP1 and its inhibitor protein to increase SERCA2a
activity may prove be a potential gene therapy target in human HF, although there
is an obvious need for further studies to clarify the mechanisms in failing human
heart and also in larger experimental animal models.
S100A1
S100A1 belongs to the S100 protein family and it is the most abundant S100
protein isoform in cardiomyocytes. In ventricular cardiomyocytes, S100A1 is
found at the junctional and longitudinal SR, at the sarcomere, and within the
mitochondria (Kato & Kimura 1985, Most et al. 2007). S100A1 is a low
molecular weight Ca2+ cycling protein (Most et al. 2001) and its protein and
mRNA levels become reduced in human ischemic and dilated cardiomyopathies
(Remppis et al. 1996).
S100A1 overexpression by adenoviral gene transfer has indicated that the
enhanced Ca2+ cycling in rat neonatal ventricular cardiomyocytes mainly involves
SR Ca2+ fluxes, whereas endocytosed S100A1 alters intracellular Ca2+ -turnover
through sarcolemmal modulation (Most et al. 2005). In a post-infarction rat HF
model, adenoviral S100A1 gene transfer normalized S100A1 expression and

48
restored myocardial contractility of the failing myocardium. The overexpression
of S100A1 induced an interaction with RyR2 in failing myocardium and an
interaction with SERCA2a in vitro. Moreover, S100A gene transfer normalized
reduced intracellular Ca2+ transients and SR Ca2+ load due to increased Ca2+
uptake and decreased the RyR–mediated Ca2+ leak from the SR (Most et al. 2004). Moreover, in failing human myocytes, adenoviral S100A1 overexpression
reversed the pathophysiological features which are typically encountered in
human failing myocardium (Brinks et al. 2011).
Targeted deletion of the S100A1 gene caused an impaired cardiac
contractility response to hemodynamic stress (Du et al. 2002). Heterozygous and
homozygous S100A1 knockout mice exhibited a relatively normal cardiac
function and heart rate basally, but displayed reduced contractile function
responses to β-adrenergic stimulation and enhanced transsarcolemmal Ca2+ influx
(Du et al. 2002). Infarcted S100A1-knockout mice hearts responded with acute
contractile decompensation and accelerated transition to HF, a rapid onset of
cardiac remodelling with augmented apoptosis and elevated mortality as
compared to their wild type counterparts (Most et al. 2006). Consequently,
overexpression of the S100A1 gene in the heart of transgenic mice showed a
stabilized LV function after MI and maintained inotropic reserve (Most et al. 2006). Myocardial targeted transgenic overexpression of S100A1 has led to an
improvement of cardiac function in mice under baseline conditions and in
response to β-adrenergic stimulation. S100A1 overexpression enhanced
cardiomyocyte SR Ca2+ cycling, which was associated with an elevated SR Ca2+
content and enhanced SR Ca2+–induced Ca2+ release (Most et al. 2003).
In a rat model of HF, S100A1-rAAV6 was delivered via intracoronary
injection (Pleger et al. 2007). S100A1 gene transfer performed in rats 10 weeks
after MI resulted in improved SR Ca2+ handling, contractile function and LV
remodelling compared to control HF rats. Furthermore, HF rescue was seen in
those individual ventricular myocytes where restoration of intracellular Ca2+
transients were enhanced and single myocyte contractility had been normalized
(Pleger et al. 2007). In a study conducted in a post-ischemic pig HF model,
rAAV9–mediated S100A1 gene transfer exhibited a long-term therapeutic
efficiency in a preclinical setting. Two weeks after MI S100A1-rAAV9 was
administered by retrograde coronary venous delivery. At 14 weeks S100A-treated
pigs showed an improvement in cardiac function, reversed cardiac remodelling
and normalized cardiomyocyte Ca2+ cycling, SR Ca2+ handling and energy
homeostasis (Pleger et al. 2011).

49
2.5.2 β-adrenergic system
β-AR signalling regulates the rate and force of cardiac contraction in response to
catecholamines. The heterodimeric guanine nucleotide binding protein (G-
protein) second messenger system is activated by binding of agonist
(noradrenaline, adrenaline). In myocytes, activation results in stimulatory G-
protein α-subunit (GαS) dissociation which then stimulates adenylyl cyclase (AC)
to increase the intracellular level of cAMP, which subsequently activates PKA.
The physiological effects of β-AR stimulation are mediated via PKA-dependent
phosphorylation of downstream targets such as troponin-I, PLB and L-type
calcium channels (Ly et al. 2007, Rapti et al. 2011, Vinge et al. 2008).
The β-adrenergic signalling becomes deranged in HF. Abnormalities of β-
adrenergic signalling induce changes which lead to β-adrenergic receptor
downregulation and decreased responsiveness to β-agonists in failing human
myocardium (Bristow et al. 1982, Brodde et al. 2006). Gene-based studies have
suggested that genetic manipulation of the myocardial β-AR system can enhance
cardiac function in HF.
β-Adrenergic receptors
Adrenergic receptors belong to the G-protein-coupled receptor (GPCR)
subfamily. β1-AR and β2-AR are predominant forms in cardiomyocytes. In the
human heart, β1-AR predominates: the ratio β1-AR:β2-AR is about 70%:30% in
the atria and 80%:20% in the ventricles (Brodde et al. 1991, Brodde et al. 2006).
Both β-AR subtypes act through the AC-cAMP-PKA-pathway and evoke positive
inotropic and chronotropic effects. However, β2-AR possesses less efficacy in the
ventricles and only stimulation of β1-AR causes maximal increases in force of
contraction (Kaumann et al. 1989, Motomura et al. 1990). In atria, β2-AR acts
less efficiently than in ventricles. Stimulation of β1-AR can also promote
apoptosis of cardiomyocytes (Shizukuda & Buttrick 2002, Zaugg et al. 2000).
Furthermore, β2-AR have been shown to couple also to the Gi-protein in rat and
murine heart, and in that way to induce antiapoptosis (Steinberg 1999, Xiao et al. 1995, Zhu et al. 2001).
In disease conditions, the desensitization of β1-AR and β2 -AR can occur and
there is an increase in the ratio β1-AR:β2-AR (Rapti et al. 2011). Transgenic
cardiac overexpression (30–fold) of human β1-AR in mice was associated with
significant cardiomyopathy (Engelhardt et al. 2001, Engelhardt et al. 2004).

50
However, human β2-AR cardiac overexpression in mice resulted in increased
basal myocardial AC activity, enhanced atrial contractility and increased LV
function, with no evidence of any adverse cardiac phenotype (Liggett et al. 2000,
Milano et al. 1994) and preserved ventricular contractility after MI (Du et al. 2000). Moreover, a dose-dependent effect of human β2-AR overexpression in
mice has been described; animals with 100–fold overexpression developed a
fibrotic cardiomyopathy and HF, with death occurring at about 41 weeks of age,
whereas 40–fold overexpression led to enhanced basal cardiac function without
any increased mortality (Liggett et al. 2000).
In a rabbit model, adenoviral overexpression of human β2-AR via
intracoronary gene delivery was shown to confer cardioprotection in normal
myocytes and in failing myocytes (Maurice et al. 1999, Shah et al. 2000). In a rat
heterotopic heart transplant model, adenovirus–mediated gene transfer of the
human β2-AR resulted in enhanced cardiac function (Kypson et al. 1999).
Furthermore, administration of the β2-AR-selective agonist, zinterol, improved
basal cardiac performance (Kypson et al. 1999). Another study with adenoviral
overexpression of human β2-AR in a heterotopic transplantation model in the
rabbit revealed that β2-AR overexpression acutely improved LV function in
failing hearts and improved the functional recovery of unloaded failing hearts
(Tevaearai et al. 2002). Although animal studies have suggested that β2-AR
overexpression may be beneficial from the point of view gene therapy, there is a
clear need to clarify the long-term effects of cardiac β2-AR overexpression in HF.
Adenylyl cyclase 6
There are nine known AC isoforms (AC 1 to 9) expressed in mammals, with AC5
and AC6 being the most predominant forms in cardiac myocytes (Ishikawa et al. 1992, Steinberg 1999). There is no published data about AC isoform protein
levels in the adult human heart, but in human LV, AC6 mRNA is predominantly
expressed (Wang & Brown 2004). AC translates the increased catecholaminergic
β-AR–GαS-protein signal into an intracellular cAMP response. In addition, AC6
has been indicated as the rate-limiting step in the β-AR–GαS–AC–mediated
generation of cAMP (Raake et al. 2011, Vinge et al. 2008). In failing left
ventricle, levels of AC5 (Ishikawa et al. 1994) and AC6 (Ishikawa et al. 1994,
Ping et al. 1997) were reduced. Decreased AC activity in the failing heart
correlated with the downregulation and desensitization of β-ARs from
downstream signalling (Rapti et al. 2011).

51
Transgenic mice with cardiac-directed AC6 expression showed no change in
myocardial β-AR number or G-protein content (Gao et al. 1999). The transgenic
mice had structurally normal hearts with normal basal function. However, cardiac
function was increased and myocytes showed increased cAMP production in
response to β-AR stimulation suggesting that AC6 did not alter transmembrane
signalling except when the receptors had been activated. This is in contrast to
receptor/G-protein expression, which induces continuous activation and thus,
detrimental consequences (Gao et al. 1999). Adenoviral AC6 gene transfer in
neonatal cardiac myocytes caused similar results (Gao et al. 1998). It was found
that AC6 overexpressed myocytes respond to agonist stimulation with increasing
in cAMP expression, in parallel to the amount of increased protein. Moreover, the
AC6 content appeared to limit the level of transmembrane β-adrenergic signalling
(Gao et al. 1998). Interestingly, cardiac-directed AC6 overexpression resulted in
increased survival, attenuated adverse LV remodelling and preserved LV
contractile function at 1 week in myocardial ischemia transgenic mice (Takahashi
et al. 2006). Roth et al. (1999) crossbred transgenic mice with cardiac-directed
expression of AC6 with mice with Gq–induced cardiomyopathy. Increased
myocardial AC6 levels in cardiomyopathy restored the cAMP-generating capacity
and increased basal LV function, and also dobutamine–stimulated LV function
was increased significantly. Furthermore, the increased AC6 content prevented
myocardial hypertrophy and improved survival (Roth et al. 1999). Adenoviral–
mediated intracoronary delivery of AC6 resulted in increased contractile
responsiveness in the hearts of normal pigs (Lai et al. 2000). Overexpressed AC6
increased adrenergic responsiveness and thus, increased LV contractile function,
but did not evoke any signs of arrhythmias or increased mean heart rate (Lai et al. 2000). Intracoronary adenovirus–mediated AC6 gene transfer increased LV
function and attenuated deleterious LV remodelling in congestive HF in pigs.
Furthermore, cAMP production was increased and BNP levels were reduced in
LV samples from AC6-treated pigs (Lai et al. 2004). In conclusion, AC6 gene
delivery studies have revealed AC6 as a promising target for use in HF and
clinical trials are now ongoing (see also 2.6.2).
G-protein-coupled protein kinase
G-protein-coupled protein kinases (GRK) regulate the interaction between
activated β-ARs and G-proteins which modulate the receptor activity by
phosphorylation of its carboxyl terminus (Hall & Lefkowitz 2002). The

52
ubiquitously expressed GRK2 is the most highly expressed GRK in the heart.
GRK2 phosphorylates a variety of GPCR. Upon GPCR stimulation, GRK2 binds
to dissociated and membrane-embedded βγ-subunits of G-proteins and
phosphorylates β-ARs, which then attach to an inhibitory protein, β-arrestin
(Aragay et al. 1998, Rengo et al. 2011).
In human HF, increased GRK activity appears to be a major factor
contributing to β-receptor desensitization (Lohse 1995, Rockman et al. 2002).
GRK activity and GRK2 mRNA levels are upregulated both in patients and in
animal models of HF and hypertrophy (Choi et al. 1997, Leineweber et al. 2002,
Ungerer et al. 1993). Inhibition and lowering of GRKs could therefore provide a
therapeutic target for HF (Lohse et al. 2003, Rockman et al. 2002).
There is no suitable pharmacological inhibitor which can achieve GRK2-
targeted inhibition in HF. Carboxyl-terminus of the β-adrenergic receptor kinase
(βARKct) is a miniprotein inhibitor, which inhibits GRK–mediated receptor
phosphorylation thus causing activation of GRK2. βARKct competes with
endogenous GRK for membrane binding and has been shown to inhibit GRK2
activity on several receptors (Inglese et al. 1994).
Transgenic, adenovirus- and rAAV–mediated overexpressions of βARKct
have rescued myocardium of several experimental HF models (Harding et al. 2001, Rengo et al. 2009, Rockman et al. 1998, Rockman et al. 2002).
Intracoronary adenovirus–mediated βARKct overexpression to rabbits 3 weeks
after experimental MI resulted in reversal of ventricular dysfunction (Shah et al. 2001). Moreover, in failing human myocytes, inhibition of GRK1 via adenovirus–
mediated βARKct improved contractile function and β-adrenergic signalling
(Williams et al. 2004). Katz et al. (2012) investigated the effect of molecular
cardiac surgery with recirculating delivery (MCARD)–mediated βARKct gene
transfer on β-AR signalling. Eight weeks after self-complimentary rAAV6–
mediated βARKct gene transfer with the MCARD technique it was found that
there was enhancement of cardiac contractility and relaxation, and preservation of
β-AR signalling. In summary, several studies support the concept that GRK2
inhibition is a potential target for the treatment of HF and thus, large animal
studies are ongoing to clarify the potential of β-ARKct prior to undertaking
clinical trials.

53
2.5.3 Angiogenic factors
Angiogenesis is process of developing new blood vessels from pre-existing
vessels. Furthermore, pre-existing vessels can expand, lengthen or sprout.
Angiogenesis can be physiological or pathological (Rapti et al. 2011, Yla-
Herttuala et al. 2007). Stimulation of cardiac angiogenesis is beneficial to
ischemic and infarcted heart (Bull et al. 2003, Zhao et al. 2010). Vasculogenesis,
in the other hand, is defined as the differentiation of precursor cells into the
endothelial cells and their subsequent formation of a primitive vascular network
(Vailhe et al. 2001). Therapeutic vasculogenesis can be induced by several
angiogenic factors; VEGF, hepatocyte growth factor, FGF, platelet growth factor
and hypoxia inducible factor-1α (HIF-1α); most of the studies have concentrated
on VEGFs (Hoeben et al. 2004, Korpisalo & Yla-Herttuala 2010). In humans,
VEGF gene transfer has been used for the treatment of two diseases, ischemic
peripheral artery disease and coronary artery disease (Gupta et al. 2009, Yla-
Herttuala et al. 2007).
Vascular endothelial growth factor
The mammalian genome encodes five VEGF genes. VEGF-A and VEGF-B play
important roles in angiogenesis in the heart and the transcripts encoding for two
isoforms VEGF-A121 and VEGF-A165, are detected mainly in cells and tissues
which express the VEGF gene (Katz et al. 2010, Yla-Herttuala et al. 2007, Zhao
et al. 2010). The VEGF is glycoprotein that binds to heparin and plays a major
role in the development of the new vasculature in the ischemic myocardium.
Moreover, VEGF is secreted also from intact cells and it appears to be suitable for
gene therapy aimed at improving perfusion into the ischemic myocardium (Symes
2001). In a rat myocardial infarct model, newly formed vessels first appeared at
the border zone between noninfarcted and infarcted myocardium 3 days after MI
and they subsequently extended into the infarcted myocardium (Zhao et al. 2010).
In addition, vascular density in the infarcted myocardium had increased by day 7.
Interestingly, VEGF-A and VEGF-receptors increased significantly at the border
zone on the first day after MI, but not at later stages even during the first week
when angiogenesis was most active (Zhao et al. 2010). Similarly, cardiomyocyte-
specific VEGF knockout mice showed fewer coronary microvessels, thinned
ventricular wall and depressed basal contractile function and displayed an
abnormal response to β-adrenergic stimulation (Giordano et al. 2001).

54
Nonviral lipopolymeric delivery of VEGF-A165 in rabbits with circumflex
artery ligation induced neovascularization and improved fractional shortening and
ejection fraction after MI (Bull et al. 2003). VEGF-A165-treated animals
exhibited an increase in peri-infarct vessel density and a trend towards a smaller
infarct size (Bull et al. 2003). Similarly, Ferrarini et al. (2006) showed in dogs
that myocardial viability with several troponin T-expressing cardiomyocytes was
significantly improved in rAAV–mediated gene transfer of VEGF-A165 during
acute MI. Furthermore, an increased number of α-smooth muscle actin positive
arterioles was observed (Ferrarini et al. 2006). Subsequently, Serpi et al. 2011
reported that adenoviral VEGF-B167A gene delivery prevented the Ang II–
induced diastolic dysfunction associated with the induction of the Akt pathway.
Moreover, VEGF-B167A gene transfer increased the number of c-kit+ cells and
proliferating cardiomyocytes, and also the capillary area of the LV was elevated.
In large animal models, Vera Janavel et al. (2006) injected VEGF-A165
naked DNA intramyocardially into the sheep 1 hour after coronary artery ligation.
Fifteen days later, the infarct size was reduced, arteriogenesis and angiogenesis
increased, peri-infarct fibrosis and myofibroblast proliferation decreased and
mitosis of adult cardiomyocytes enhanced with occasional cytokinesis. Moreover,
Lahteenvuo et al. 2009 reported that intramyocardial adenoviral–mediated gene
delivery of VEGF-B186 increased ejection fraction, collateral artery formation,
blood vessel area and the appearance of apoptosis-resistant cardiomyocytes
around the infarction border zone after acute MI in pigs and rabbits. Furthermore,
cardiomyocyte hyperplasia in the ischemic zone was induced after VEGF-A165
naked plasmid transfection in pigs with chronic myocardial ischemia, resulting in
division of native myocytes, thus supporting the hypothesis that VEGF has
therapeutic effects in diseases characterized by myocardial cell loss (Laguens et al. 2004). In a tachycardia–induced model, adenoviral delivered VEGF-121
(coding 121 amino acid isoform of VEGF) into the LV improved significantly
fractional wall thickening and segmental shortening within 7 days after treatment
(Leotta et al. 2002). In conclusion, because of vasculogenic and angiogenic
properties of VEFG-A, it is considered as one of the most important candidates
for gene therapy.
2.5.4 Anti-apoptotic genes
Several studies have associated cardiomyocyte apoptosis to human HF (Abbate et al. 2003, Kang & Izumo 2000, Narula et al. 1996). Apoptotic myocardial cell

55
death contributes to the pathogenesis of HF, because it results in a decrease the
number of cardiomyocytes and as consequence it evokes a worsening of cardiac
function (Ly et al. 2007). Thus gene targets that promote myocyte survival by
limiting cardiac myocyte loss through inhibition of apoptosis may offer
therapeutic benefits for the treatment of HF. Apoptotic cell death is a strightly-
regulated, energy-dependent process and it is a form of programmed cell death.
Apoptosis becomes activated when the cellular reparative mechanisms fail to
maintain cellular homeostasis and integrity as a result of continuous cellular
insults. Apoptotic signalling cascades are activated in both the endoplasmic
reticulum (ER) and mitochondria in the cardiomyocytes i.e. both of these
organelles can activate apoptotic pathways. However, the molecular mechanisms
of activation of apoptotic proteins are not completely understood (Chaanine et al. 2010, Elmore et al. 2007, Ly et al. 2007).
B-cell lymphoma-2
B-cell lymphoma 2 (BCL-2) is a cytosolic protein localized to the mitochondrial
outer membrane, ER and nuclear envelope, and it has been shown to prevent
cytochrome c release, caspase activation and cell death. BCL-2 has been
associated with the inhibition of apoptosis (Hanada et al. 1995, Hengartner &
Horvitz 1994, Hockenbery et al. 1993).
Transgenic mice overexpressing a human BCL-2 in the heart suffered smaller
infarct sizes, and they displayed improved LV ejection fraction (Brocheriou et al. 2000) and their hearts had fewer terminal deoxynucleodidyl-transferase nick-end
labeling-positive or in situ oligo ligation-positive myocytes after
ischemia/reperfusion (Brocheriou et al. 2000, Chen et al. 2001). In agreement,
adenovirus–mediated gene transfer of BCL-2 could prevent ventricular myocyte
death and apoptosis provoked by p53. These antiapoptotic effects of BCL-2 were
independent of p53 gene expression (Kirshenbaum & de Moissac 1997).
Akt
The Ser/Thr-protein kinase Akt is a mediator of PI3K signalling. Akt is a crucial
mediator of cell size and survival of many cell types (Matsui et al. 2003, Shiojima
& Walsh 2006). In cardiomyocytes, Akt-dependent signalling pathways have been
associated regulation of cardiac growth, contractile function and coronary
angiogenesis (Rota et al. 2005, Shiojima et al. 2005, Shiojima & Walsh 2006). In

56
myocytes, Akt is activated by glycoprotein 130-dependent cytokine stimulation
and it has cardiac hypertrophic (Oh et al. 1998) and an antiapoptotic effects (Zhu
et al. 2001). Moreover, Akt is known to be activated by other growth-promoting
and pro-survival ligand receptor systems such as insulin (Cong et al. 1997), IGF-I
(Kulik et al. 1997) and estrogen (Camper-Kirby et al. 2001).
PI3K and Akt have reduced the rate of cardiomyocyte apoptosis. In rat
neonatal cardiomyocytes, expression of activated forms of either PI3K or
myristoylated Akt1 (membrane targeted Akt1) inhibited hypoxia–induced
apoptosis in vitro (Matsui et al. 1999). Adenoviral–mediated overexpression of
constitutively active Akt protected cardiomyocytes against apoptosis in the
absence of IGF-1 in response to ischemia-reperfusion injury in vivo (Fujio et al. 2000). In cultured cardiomyocytes, adenoviral delivery of dominant-negative
Akt1 (Akt1 Thr308Aspartic acid/Ser473Aspartic acid) prevented IGF-1–mediated
myocyte survival and overexpression of constitutively active Akt promoted
myocyte survival (Fujio et al. 2000), suggesting that activation of this pathway
may well be useful in promoting myocyte survival in the failing heart.
In a rat model of cardiac ischemia/reperfusion injury, direct adenoviral gene
transfer of constitutively active myristoylated Akt1 reduced the infarct size and
the number of apoptotic cells in the region of the injury and restored cardiac
function (Matsui et al. 2001). Cardiac specific transgenic overexpression of
constitutively active Akt increased cardiomyocyte cell size and induced cardiac
hypertrophy (Condorelli et al. 2002, Shioi et al. 2002). Furthermore, cardiac
contractility of Akt-transgenic mice was increased (Condorelli et al. 2002,
Shiraishi et al. 2004). Transgenic mice expressing nuclear targeted Akt–mediated
inhibition of apoptosis without resulting in hypertrophic remodelling, suggesting
that biologically significant targets of Akt activity may be located within the
nucleus. In addition, transgenic mice overexpressing IGF-1 exhibited less
myocyte apoptosis after MI (Li et al. 1997). Furthermore, administration of IGF-1
was able to attenuate myocardial apoptosis and necrosis in response to ischemia-
reperfusion injury (Buerke et al. 1995).
2.5.5 Stem cell homing factors
Factors regulating stem cells are potential targets for gene therapy with aiming to
improve cardiac remodelling and function by inducing reconstitution of
functional myocardium and stimulate the formation of new blood vessels. Stem
cell homing factors are thought to be released from the heart in response to injury,

57
to stimulate endogenous tissue repair pathways (Boyle et al. 2011). Expression of
stem cell homing factors have a crucial role in the recruitment of circulating stem
cells. Stem cell mobilization from the bone marrow to the ischemic cardiac area
enables vasculogenesis and aids in the preservation of injured cardiac myocytes.
Stromal derived growth factor (SDF)-1α represents a candidate stem cell homing
factor which can recruit circulating progenitors to sites of ischemic injury (Tang
et al. 2005).
Stromal derived factor-1
SDF-1, also known as CXCL12 or pre-B-cell growth stimulating factor (PBSF) is
a cytokine of the CXC chemokine subfamily, which is transiently expressed for
instance in infarct and peri-infarct regions where it can induce stem cell homing
to the myocardium (Abbott et al. 2004, Askari et al. 2003, Ghadge et al. 2011, Hu
et al. 2007). SDF-1α and SDF-1β are the main isoforms in human and mouse
tissues, which are produced by alternate splicing of the same gene (De La Luz
Sierra 2004). Gene expression of SDF-1α in ischemic injury is regulated by the
transcription factor HIF-1α (Ceradini et al. 2004). The signalling effects of SDF-1
were originally demonstrated to be mediated through G-protein-coupled receptor
CXCR4. SDF-1 binding to CXCR4 receptor stabilizes homodimerization or
heterodimerization of the receptor, enhancing activation of the GPCR, thus
leading to regulation of several signalling cascades (Percherancier et al. 2005).
Binding of SDF-1 to CXCR4 receptor on stem and progenitor cells has been
reported to play a crucial role in the regulation of bone marrow homing and stem
cell implementation into the bloodstream (Ghadge et al. 2011, Lapidot & Petit
2002). Furthermore, SDF-1 binds G-protein-coupled receptor CXCR7 (Sierro et al. 2007) or RDC1 (Balabanian et al. 2005).
Expression of SDF-1α was increased in serum (Leone et al. 2005) and
cardiac tissue samples (Yamani et al. 2005) of patients with MI. Circulating SDF-
1α levels were associated with a number of circulating progenitor cells. Bone
marrow derived stem cells harvestered from mice were released into the
peripheral circulation after MI and these cells were chemoattracted to
homogenates of infarcted myocardium in a SDF-1–dependent manner (Kucia et al. 2004). Abbot et al. (2004) revealed that intracoronary infusion of bone
marrow–derived stem cells after coronary artery ligation induced the recruitment
of stem cells into the mice hearts and adenoviral–mediated gene delivery of SDF-
1 accentuated this effect.

58
Several animal studies have revealed that SDF-1–mediated therapies can be
beneficial after MI (Elmadbouh et al. 2007, Zhang et al. 2007). For instance,
intramyocardial injection of SDF-1α into the peri-infarct zone protected tissue
after an acute ischemic event in mice. The amount of scar formation was
decreased and VEGF–mediated neoangiogenesis was increased with mice
exposed to SDF-1α (Saxena et al. 2008). Local adenovirus–mediated gene
transfer of SDF-1 resulted in increased c-kit+ cell mobilization and improved
cardiac structure and function through angiogenic and antifibrotic actions after
experimental MI in rat hearts (Tang et al. 2010). Overexpression of SDF-1 in
myocardial tissue led to recruitment of endogenous cardiac stem cells to the
infarct border zone (Unzek et al. 2007).
Due to the rapid diffusion and degradation of natural SDF-1, Segers et al. (2007) designed a chemokine called S-SDF-1 (S4V) which was resistant to
MMP-2 and exopeptidase, but retained SDF-1 chemotactic potential.
Intramyocardial delivery of S-SDF-1 (S4V) after MI resulted in increased
capillary density, induction of stem cell recruitment and improved cardiac
function in rat heart.
Some studies have also demonstrated negative effects of SDF-1 therapy in
MI. Pyo et al (2006) reported that adenoviral gene delivery of CXCR4 to cardiac
myocytes accentuated the negative inotropic effects of SDF-1 during Ca2+
stimulation. Furthermore, the β-adrenergic–mediated increase in Ca2+
mobilization and fractional shortening were attenuated (Pyo et al. 2006).
Catheter-based transendocardial injection of SDF-1α into the peri-infarct
myocardium in a myocardial infarct model to pigs resulted in an increase in the
peri-infarct vessel density and a reduction of collagen levels but it failed to
improve LV function (Koch et al. 2006). In conclusion, because of promising
results in animal studies, a trial investigating SDF-1 naked DNA gene delivery by
percutaneous administration to the peri-infarct area is currently recruiting patients
(see also 2.6.3).
2.5.6 Cardiac natriuretic peptides
The NP gene family consists three structurally related, but genetically distinct
peptides. ANP, BNP and C-type natriuretic peptide (CNP) are encoded by
different genes but the mature forms of these natriuretic peptides contain a
distinctive 17 amino acid ring structure with a highly conserved internal sequence
(D'Souza et al. 2004, Ruskoaho 2003).

59
ANP and BNP are cardiac hormones; their secretions are markedly
upregulated during HF (Ruskoaho 2003). Both compounds exert potent diuretic,
natriuretic, vasorelaxant, aldosterone-inhibiting, antifibrotic and antihypertrophic
effects, which are mediated via their common receptor, guanylyl cyclase (GC)-A
(Lee & Burnett 2007, Potter et al. 2009). The human CNP is structurally distinct
from ANP and BNP. The 22 amino acid form of CNP dominates in the systemic
circulation and the 53 amino acid molecule is the major active form of CNP at the
tissue level (Ogawa et al. 1992, Tawaragi et al. 1991).
ANP is secreted mainly by the atria and released in response to wall stretch
(Lang et al. 1985, Ruskoaho et al. 1986). The human gene encoding ANP is
called natriuretic peptide precursor A (NPPA) and it encodes for a 151 amino acid
preprohormone which after proteolytic processing, forms a 126 amino acid
prohormone, proANP, to be stored in atrial granules and cleaved into the 98
amino acid N-terminal fragment and the 28 amino acid active hormone on release
into the circulation (Oikawa et al. 1984, Vuolteenaho et al. 1985, Wu et a. 2002).
The gene encoding human BNP (hBNP), natriuretic peptide precursor B
(NPPB), encodes the 132 amino acid preprohormone which is processed to a 108
amino acid prohormone, profragment of BNP (proBNP). ProBNP is cleaved by
furin to form the 76 amino acid N-terminal BNP and a 32 amino acid active
hormone (Sawada et al. 1997). N-terminal BNP and active BNP are constitutively
produced and released from the atria and ventricles (Mukoyama et al. 1991,
Yoshimura et al. 2001).
B-type natriuretic peptide
BNP is among the earliest cardiac factors induced in response to hemodynamic
load (Ruskoaho 2003) and this mechanical stretch–induced activation of BNP
gene expression is mediated via the GATA transcription factor (Suga et al. 1992).
NPs have emerged as important candidates for the development of therapeutic
agents in cardiovascular disease (Lee & Burnett 2007). In 2001, the human
recombinant form of mature BNP, nesiritide, was approved by the United States
Food and Drug Administration for the treatment of acutely decompensated HF.
Infusion of nesiritide has been used clinically in HF and it possesses a number of
useful properties, such as positive lusitropic activity, vasodilatory features and
positive effects with cardiac reverse remodelling (Burger et al. 2002, Dickstein et al. 2008). Nevertheless, the use of nesiritide has been limited by hypotension and
concerns regarding the worsening of renal function. Intravenous infusion of

60
nesiritide has reduced LV filling pressure, but it has variable effects on cardiac
output, urinary output, sodium excretion and blood pressure. It has been claimed
that adverse renal consequences can be encountered with nesiritide (Sackner-
Bernstein et al. 2005) and careful monitoring of renal function is mandatory
(Dickstein et al. 2008, Roger et al. 2009, Sackner-Bernstein et al. 2005).
However, the effect of nesiritide on mortality is uncertain, and controversy
remains regarding the safety, efficacy, and dosing of BNP in the therapy of acute
HF (Lee & Burnett 2007).
In addition to direct administration of native peptides, several other
approaches alone or in combination with other pharmacologic therapies have been
shown to enhance the function of the NP system: administration of designer NPs,
inhibition of degradation of NPs and their second messenger cGMP, and
stimulation of cGMP generation (Boerrigter et al. 2009). Recently, the feasibility
of an orally delivered, conjugated form of BNP was demonstrated in an
experimental model of Ang II–mediated hypertension (Cataliotti et al. 2008).
In cultured cardiac fibroblasts, BNP has reduced collagen synthesis and
increased MMPs via cGMP-protein kinase G signalling (Tsuruda et al. 2002).
Accordingly, after MI, overexpression of BNP in mice, targeted to the liver, has
been shown to increase MMP-9, but not MMP-2 expression in the infarcted
region. Elevated plasma BNP facilitated neutrophil infiltration into the infarcted
area after MI and the neutrophils were the main source of MMP-9 (Kawakami et al. 2004). Targeted deletion of the GC-A gene resulted in marked cardiac
hypertrophy and fibrosis (Lopez et al. 1995, Oliver et al. 1997) while mice
lacking the BNP gene (Nppb-/-) showed a normal heart size but multifocal fibrotic
lesions in the ventricles. Moreover, no signs of systemic hypertension and
ventricular hypertrophy were noted (Tamura et al. 2000). Thus, genetic studies in
mice collectively support a local role for BNP in the regulation of cardiac fibrosis.
However, transgenic models have exhibited alterations in blood pressure and
cardiac function, (Kawakami et al. 2004, Kishimoto et al. 2009, Lopez et al. 1995, Oliver et al. 1997, Tamura et al. 2000) suggesting that, in part, the cardiac
effects may be mediated indirectly via systemic hemodynamic actions.
Earlier studies have indicated that the NP/GC-A system could be involved
also in the stimulation of angiogenesis. In vitro, BNP/GC-A triggers proliferation
and migration of cultured microvascular endothelial cells of rats by activating
cGMP-dependent protein kinase I (Lin et al. 1995). An increase in circulating
BNP levels resulting from targeted overexpression of the BNP gene in the liver of
mice was able to accelerate vascular regeneration in limb ischemia model.

61
Furthermore, in a pressure overload–induced hypertrophy model, selective
disruption of the endothelial GC-A evoked diminished angiogenesis (Cao &
Gardner 1995, Lin et al. 1995).
2.5.7 Renin-angiotensin aldosterone system
The RAA system plays a crucial role in the homeostatic control of arterial
pressure, tissue perfusion and extracellular volume affecting both blood pressure
and hydroelectrolyte balance. In the RAA system, angiotensinogen serves as a
substrate for renin. Renin catalyses the proteolytic conversion of angiotensinogen
into the decapeptide angiotensin I. Subsequently, angiotensinogen converting
enzyme converts angiotensinogen I to the Ang II, while Ang II type 1 receptor
(AT1-R) is responsible for transducing the cellular effects on Ang II (Griendling et al. 1993). In cardiac tissue, Ang II is mainly produced by local synthesis from
angiotensin I, rather than from any uptake from the systemic circulation (Atlas
2007, Carey & Siragy 2003). (P)RR is the newest member of the renin-
angiotensin system. (P)RR has been suggest to play an important role in the tissue
RAA system via both Ang II-dependent and Ang II-independent pathways
(Nguyen et al. 2002). Moreover, other therapeutic targets for RAA-system have
been suggested. Expression of the Ang II type 2 receptor (AT2-R) is high during
foetal development and there is evidence that despite low levels of expression in
the adult, the AT2-R might inhibit growth and remodelling in the heart and
mediate vasodilation and antiproliferative and apoptotic effects in vascular
smooth muscle. The Ang II type 4 receptors are supposed to mediate the release
of plasminogen activator inhibitor 1 by Ang II and by the N-terminal truncated
peptides: angiotensin III and angiotensin IV. However, the importance of these
receptors remains uncertain. Angiotensin-(1–7) is a heptapeptide fragment of Ang
II and it appears to act via Mas receptor. Angiotensin-(1–7) is presumed to have
protective cardiovascular effects (Atlas 2007, Carey & Siragy 2003). Under physiological conditions, the cardiac RAA system maintains cellular
balance of inhibiting and inducing cell growth and proliferation and mediation of
adaptive responses to myocardial stretch (Atlas 2007, Paul et al. 2006). The
activation of the RAA system and sympathetic hyperactivation play a central role
in pathogenesis of post-infarction LV remodelling and HF (Jessup & Brozena
2003, McMurray & Pfeffer 2005). Ang II promotes peripheral vasoconstriction,
aldosterone and vasopressin secretion, and sodium and water retention, leading to
further deterioration of ventricular function (Holubarsch et al. 1993, Pieruzzi et

62
al. 1995). Moreover, it has been claimed that Ang II may have a major role in
mediating the fibrogenic response after MI and in the development of cardiac
hypertrophy (Holubarsch et al. 1993, Sun et al. 1994). Ang II has also stimulated
heart rate and contractility by facilitating adrenergic neurotransmission in the
heart, resulting in a decrease in coronary flow and the appearance of arrhythmias
(Pieruzzi et al. 1995, Sadoshima & Izumo 1993, Seravalle et al. 1993).
(Pro)renin receptor
(P)RR is a 350 amino acid protein with a single transmembrane domain. High
levels of the (P)RR mRNA are expressed in human brain, heart, placenta and
lower expression levels have been detected in liver, pancreas and kidney and there
is very weak expression in lung and skeletal muscle (Nguyen et al. 2002).
Mahmud et al. (2011) reported high (P)RR mRNA expression in mouse brain and
substantially lower expressions in heart, muscle, liver, intestine, lung and kidney.
Nevertheless, (P)RR protein was highly expressed only in the kidneys of mice. In
mouse heart, liver, intestine and brain, (P)RR protein expression was much lower
and in muscle and lung protein expression was very weak.
Thus far, two (P)RRs have been characterized. The mannose-6-phosphate
receptor is a ubiquitous clearance receptor for renin and its inactive precursor
prorenin (Saris et al. 2001). Specific (P)RR mediates renin and prorenin cellular
effects by activating intracellular signalling pathways. Prorenin is an inactive
precursor of renin and binding of prorenin to (P)RR leads to its activation via a
nonproteolytic mechanism and to the generation of Ang II (Nguyen et al. 2002).
The binding of prorenin or renin to (P)RR triggers extracellular pathways.
The Ang II-dependent pathway is essential for the increase in the prorenin
catalytic activity. The (P)RR by binding to prorenin activates the enzyme activity
of renin resulting in the generation of Ang II at the tissue level (Nguyen et al. 2002). In the Ang II-independent pathway, prorenin binding to the (P)RR initiates
a cascade of signalling events, including activation of MAPK, these being
associated with profibrotic and proliferative action. In most cell types, (P)RR
activation triggers the phosphorylation of ERK1/2 inducing the upregulation of
profibrotic genes. In cardiomyocytes, (P)RR activates the p38 MAPK/heat shock
protein 27 (HSP27) pathway, PI3K p85 and promyelocytic zinc finger protein
(PLZF) (Feldt et al. 2008, Huang et al. 2006, Sakoda et al. 2007, Saris et al. 2006).

63
(P)RR expression is increased in rodent models of HF and in the failing
human heart (Mahmud et al. 2011). Increased levels of (P)RR mRNA and protein
have been found in post-myocardial infarcted rats hearts. (P)RR mRNA gene
expression was elevated in transgenic rats with overexpression of the renin gene.
Furthermore, (P)RR was found to be upregulated in dilated cardiomyopathy
patients (Mahmud et al. 2011).
The underlying cellular mechanisms for direct (P)RR–mediated actions are
still unclear. Experimental models suggest that increased (P)RR synthesis and
activation may be involved in certain diseases e.g high blood pressure, cardiac
fibrosis associated with hypertension and diabetic nephropathy (Burckle et al. 2006, Ichihara et al. 2006b). In a rat model overexpression of the (P)RR gene in
smooth muscle cells, including vascular smooth muscle cells suggested that
(P)RR resulted in elevated blood pressure and increased intraadrenal Ang II
concentrations thereby enhancing plasma aldosterone levels. These alterations
increased progressively with age (Burckle et al. 2006). (P)RR overexpression also
directly or indirectly contributed to the regulation of cyclooxygenase-2 expression
in the renal cortex of the transgenic rats. These animals remained normotensive,
but developed proteinuria and glomerulosis associated with increased ERK1/2,
p38 MAPK and JNK activity, but not epidermal growth factor receptor
phosphorylation. Moreover, the level of transforming growth factor (TGF) β1
expression was increased (Kaneshiro et al. 2007).
Recent studies have reported that most of endogenous (Advani et al. 2009,
Cousin et al. 2009) or transfected (Schefe et al. 2008) (P)RR protein is
intracellular. Cousin et al. (2009) have shown that (P)RR accumulates in the
trans-golgi, where it is cleaved by furin to generate a 10 kDa
transmembrane/cytoplasmic fragment that represents the truncated (P)RR, which
remains associated with the vacuolar H+-adenosine trisphosphatase (V-ATPase).
The 28 kDa soluble form of (P)RR is secreted into the conditioned medium of
cultured cells and it is also found in plasma of humans and rats. Part of the (P)RR
remains intact and can be detected from the plasma membrane (Fig. 6).

64
Fig. 6. Intracellular processing of (pro)renin receptor ([P]RR). Part of (P)RR is cleaved
by furin in the trans-golgi to generate soluble (P)RR, which is secreted and
transmembrane (P)RR, which remains associated with V-ATPase. Part of (P)RR
remains intact and is addressed to the plasma membrane. Modified from Nguyen &
Muller 2010.
2.5.8 Other potential gene targets for the treatment of heart failure
Regulatory proteins of ryanodine receptors
RyR is regulated via several proteins such as PKA, muscle A-kinase anchor
protein, PP1 and PP2, sorcin, calmodulin, S100 protein and FKBP12.6 (also
known as calstabin2) (Marks 2002). Moreover, in the junctional SR, RyR
interacts with calsequestrin, triadin and junctin (Davis et al. 2008, Gyorke et al. 2004). RyR has a large open reading frame, which creates challenges to the use of
viral–mediated gene transfer methods. Therefore several studies have
concentrated on the molecular mechanisms of RyR function by modulating the
regulatory protein of RyRs (Davis et al. 2008). For instance, FKBP12.6 (Marx et al. 2000) and junctional SR proteins such as calsequestrin (Kubalova et al. 2004),
junctin (Gergs et al. 2007), triadin (Terentyev et al. 2005) and histidine-rich Ca2+-
binding protein (Fan et al. 2004) are believed to act as regulators of Ca2+ release

65
from the SR. Moreover, sarcolipin (Babu et al. 2005), Na+/Ca2+ exchanger (Bolck
et al. 2004) and phospholemman (Song et al. 2002) are regulators of cytoplasmic
Ca2+ removal. A few gene transfer studies have also reported the effects of
parvalbumin (Wahr et al. 1999) and sorcin (Frank et al. 2005), which are Ca2+
binding protein modulating cardiac performance.
Sarcomeric proteins
The sarcomere is the major structural and functional unit of striated muscle. It is
composed of overlapping thick and thin filaments (Davis et al. 2008). Sarcomeres
play an important role in cardiac performance and thus sarcomeres have been
described as an attractive target for genetic engineering (Day et al. 2006, Herron
et al. 2007). Several thin filament proteins have been studied via acute genetic
engineering technology by gene transfer. For instance, the troponin subunits,
troponin I, troponin T and troponin C are well-known targets of thin filament
regulation. Cardiac troponin I has a crucial role in cardiac function under
physiological and pathophysiological conditions (Westfall & Metzger 2003) and
cardiac troponin T has a structural and functional role in Ca2+–mediated
regulation of cross-bridge cycling (Sweeney et al. 1998) and troponin C is the
Ca2+ sensor of the thin filament regulatory system (Babu et al. 1992).
Acute gene transfer has been only occasionally utilized for studying thick
filament proteins even though it offers the possibility for directly assessing the
role of the thick filament in cardiac muscle physiology (Davis et al. 2008). In
contrast, thick filament proteins, such as cardiac myosin (Marian et al. 1995), the
myosin essential light chains and myosin regulatory light chains (Fewell et al. 1998, Sanbe et al. 2000), myosin binding protein C (Gautel et al. 1998) and titin
(Granzier & Labeit 2004) have been well studied. One limitation to applying
acute gene transfer for modulating the thick filament is the turnover rate of thick
filament proteins.
Cytoskeletal proteins
Cardiac muscle cytoskeletal proteins such as dystrophin (Townsend et al. 2007),
sarcoglycans (Zhu et al. 2005), desmin (Haubold et al. 2003) and microtubule–
associated proteins (Takahashi et al. 2003) are important for normal physiology
and are involved in certain related disease states. The understanding of
cytoskeletal proteins is incomplete even though it is clear that they are an

66
important group of molecules in the development of heart disease. Gene transfer
techniques have shown some promising results in understanding cardiac disease
caused by defective cytoskeletal proteins and for the treatment of cytoskeletal-
based cardiac disease (Davis et al. 2008).
Cardiac signalling pathways
Cardiac gene transfer techniques have offered the ability to study the temporal
and dose-dependent modulation of cardiac contractile function by a signalling
pathway which may otherwise be demanding to investigate e.g. in transgenic
animals. Moreover, gene deliveries have made it possible to identify cellular
targets within a signalling cascade (Davis et al. 2008, Huq et al. 2002, MacNeill
et al. 2003).
Cardiac gene transfer techniques have been used to study the MAPK
signalling cascades, which are one of the major system involved in the
transduction of the signal from the cell membrane to nucleus as well as their
participation in other intracellular targets. The members of the MAPK family are
involved in the regulation of many cellular processes, such as development,
differentiation, cell growth, cell cycle, cell death and survival (Ravingerova et al. 2003). Cardiac tissue contains mainly ERKs, the stress-activated protein
kinase/JNKs, p38 MAPK and ERK5/big MAPK 1 (Davis et al. 2008,
Ravingerova et al. 2003). Most of gene transfer studies have focused on p38
MAPK and JNKs within the MAPK family. ERK1/2 and JNK studies have
indicated that these cascades primarily influence cardiac function via
transcriptional regulation (Choukroun et al. 1999, Lebeche et al. 2006). In the
heart, mitogenic growth factors activate ERK1/2, while cellular stressors activate
the stress-activated protein kinases, which include JNK and p38 MAPK. Several
studies have described the activation of the MAPKs in myocardial hypertrophy,
and its transition to HF, in ischemic and reperfusion injury and in the
cardioprotection conferred by ischemia- or pharmacologically–induced
preconditioning and thus, it is believed that MAPKs play an essential role in
pathogenic cardiac events (Davis et al. 2008, Ravingerova et al. 2003).
Moreover, several studies have investigated on nitric oxide signalling in the
myocardium. Nitric oxide synthase activity is important during certain
pathophysiological conditions including myocardial ischemia, preconditioning
and heart failure. Most of the myocardial nitric oxide synthase signalling gene
transfer studies have concentrated on investigating the direct effects of nitric

67
oxide synthase on contractile function using nitric oxide synthase-2 or 3 gene
transfers (Davis et al. 2008). Inducible nitric oxide synthase 2 activity increases in
response to pathophysiological conditions, such as MI. Elevating the levels of
nitric oxide synthase 2 in failing hearts has only a weak effect on basal contractile
function and the contractile response to β-AR stimulation is markedly reduced
(Davis et al. 2008, Ziolo et al. 2004). Nitric oxide synthase 3 overexpression has
also been reported to improve LV function after MI (Janssens et al. 2004).
Table 3. Overview of gene targets for the treatment of HF.
System Gene therapy target
The calcium handling proteins SERCA2a, PLB, PP1, I-1, S100A1
β-adrenergic system β-AR, AC6, GRK
Angiogenic factors VEGF
Anti-apoptotic genes BCL-2, Akt
Stem cell homing factors SDF-1
Cardiac natriuretic peptides BNP
RAA system (P)RR
Regulatory proteins of RyR calsequestrin, triadin, junctin, FKBP12.6, histidine-rich
Ca2+-binding protein, sarcolipin, Na+/Ca2+ exchanger,
phospholemman, parvalbumin, sorcin
Sarcomeric proteins troponin I, troponin T, troponin C, thick filament proteins
Cytoskeletal proteins dystrophin, sarcoglycans, desmin, microtubules–
associated proteins
Cardiac signalling pathways MAPK signalling, NO signalling
AC6, adenylyl cyclase 6; BCL-2, B-cell lymphoma-2; BNP, B-type natriuretic peptide; GRK, G-protein-
coupled protein kinase; I-1, inhibitor type-1; MAPK, mitogen-activated protein kinase; NO, nitric oxide;
PLB, phospholamban; PP1, protein phosphatase type 1; (P)RR, (pro)renin receptor; RAA, renin-
angiotensin-aldosterone; SDF-1, stromal derived growth factor-1; SERCA2a, sarcoplasmic reticulum Ca2+-
ATPase; VEGF, vascular endothelial growth factor; β-AR, β-adrenergic receptor
2.6 Clinical trials in gene therapy for heart failure
There are several gene transfer trials currently ongoing or in the planning stages
for the treatment of cardiovascular diseases. SERCA2a, AC6 and SDF-1 gene
transfers have progressed towards clinical trial studies (Table 4.) (Kawase et al. 2011). Moreover, VEGF have been used in clinical trials for cardiovascular
diseases, mainly associated with coronary heart diseases. For instance, a trial of
endocardial VEGF-D gene therapy for the treatment of severe coronary heart
disease is currently recruiting participants. The goal of this study will be to

68
evaluate the safety and efficacy of catheter mediated endocardial adenovirus
VEGF-D gene therapy in patients with severe coronary heart disease
(ClinicalTrials.gov identifier: NCT01002430). The focus of this chapter is on the
clinical trials of target genes, which have direct and interactive effects on
myocytes.
Adenoviral–mediated gene transfers are the most frequently used techniques
in clinical gene transfer studies targeting to the heart. Adenoviral vectors enable
highly efficient cardiac gene delivery and can be produced in sufficient quantities.
Nevertheless, adenoviral–mediated gene transfer induces only transient gene
expression and can evoke an immune response against viral gene products
resulting in the clearance of transduced cells (Hedman et al. 2011, Jooss et al. 1998, Yang et al. 1994). AAVs are a promising alternative to adenovirus because
of their better safety profile. AAV is non-pathogenic and cannot be amplified
without co-infection with a helper virus. AAV vectors transduce the myocardium
as efficiently as adenoviral vectors and AAVs allow stable expression of
transferred genes (Chu et al. 2003, Hedman et al. 2011).
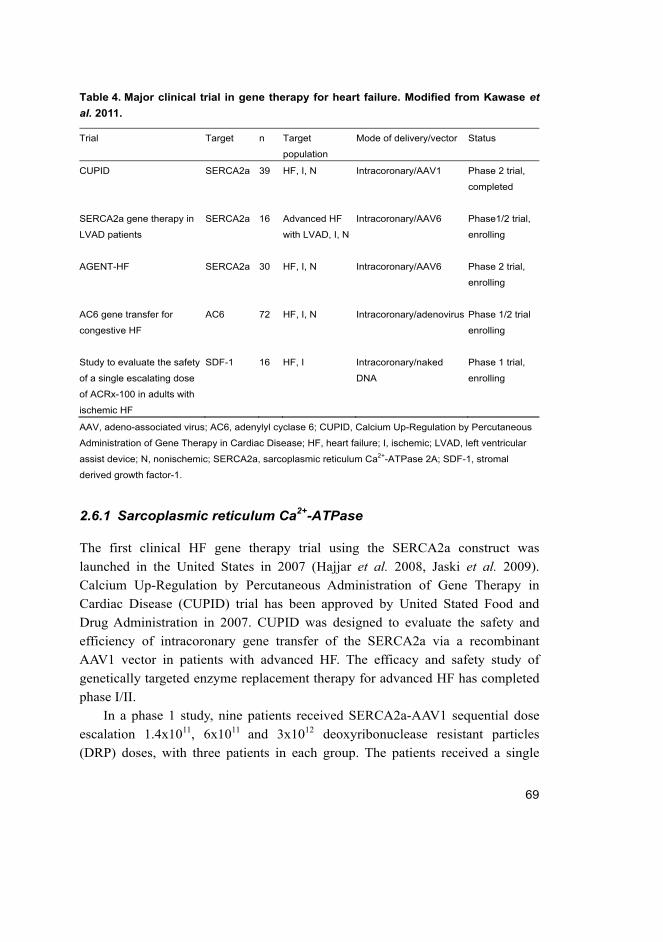
69
Table 4. Major clinical trial in gene therapy for heart failure. Modified from Kawase et
al. 2011.
Trial Target n Target
population
Mode of delivery/vector Status
CUPID SERCA2a 39 HF, I, N Intracoronary/AAV1 Phase 2 trial,
completed
SERCA2a gene therapy in
LVAD patients
SERCA2a 16 Advanced HF
with LVAD, I, N
Intracoronary/AAV6 Phase1/2 trial,
enrolling
AGENT-HF SERCA2a 30 HF, I, N Intracoronary/AAV6 Phase 2 trial,
enrolling
AC6 gene transfer for
congestive HF
AC6 72 HF, I, N Intracoronary/adenovirus Phase 1/2 trial
enrolling
Study to evaluate the safety
of a single escalating dose
of ACRx-100 in adults with
ischemic HF
SDF-1 16 HF, I Intracoronary/naked
DNA
Phase 1 trial,
enrolling
AAV, adeno-associated virus; AC6, adenylyl cyclase 6; CUPID, Calcium Up-Regulation by Percutaneous
Administration of Gene Therapy in Cardiac Disease; HF, heart failure; I, ischemic; LVAD, left ventricular
assist device; N, nonischemic; SERCA2a, sarcoplasmic reticulum Ca2+-ATPase 2A; SDF-1, stromal
derived growth factor-1.
2.6.1 Sarcoplasmic reticulum Ca2+-ATPase
The first clinical HF gene therapy trial using the SERCA2a construct was
launched in the United States in 2007 (Hajjar et al. 2008, Jaski et al. 2009).
Calcium Up-Regulation by Percutaneous Administration of Gene Therapy in
Cardiac Disease (CUPID) trial has been approved by United Stated Food and
Drug Administration in 2007. CUPID was designed to evaluate the safety and
efficiency of intracoronary gene transfer of the SERCA2a via a recombinant
AAV1 vector in patients with advanced HF. The efficacy and safety study of
genetically targeted enzyme replacement therapy for advanced HF has completed
phase I/II.
In a phase 1 study, nine patients received SERCA2a-AAV1 sequential dose
escalation 1.4x1011, 6x1011 and 3x1012 deoxyribonuclease resistant particles
(DRP) doses, with three patients in each group. The patients received a single

70
intracoronary infusion of SERCA2a-AAV1. All the patients in the phase 1 and
phase 2 trials were required to have an implantable cardioverter defibrillator,
because there was concern that inhomogeneous SERCA2a overexpression may
lead to consequent ventricular arrhythmias (Hajjar et al. 2008, Jaski et al. 2009).
At the 6 to 12‒month follow-up of patients showed biological efficacy and an
acceptable safety profile, even if the phase 1 trial was open label and results were
difficult to interpret (Jaski et al. 2009). Improvement was reflected by functional
(4 patients), NT-proBNP-biomarker (2 patients), LV function and remodelling
such as ejection fraction and end-systolic volume (6 patients) and symptom (5
patients) parameters. In a phase 1 trial, two patients who failed to improve had
pre-existing anti-AAV1 neutralizing antibodies (Jaski et al. 2009).
Phase 2 trial was assessed in a randomized, double-blind, placebo-controlled
study in patients with advanced HF. SERCA2a-AAV1 was delivered by the
intracoronary route to the 39 patients at the dose of 6x1011, 3x1012 and 1x1013
DRP and saline control as placebo (Jessup et al. 2011). Functional studies at 6
months post-AAV therapy revealed an improvement or stabilization in NYHA
class, Minnesota Living With Heart Failure Questionnaire, 6-minute walk test,
maximum oxygen consumption, NT-proBNP-biomarker and LV ejection fraction
end-systolic function volume. In the phase 2 trial, no evidence of a cellular
immune response to the viral vector was observed. Furthermore, macromolecules
including AAV exhibit cooperative binding but no clear dose response to the gene
therapy was observed (Jessup et al. 2011). Larger studies will be needed to
establish SERCA2a-AAV2 as a treatment of HF and enrollment for phase 3 has
started (ClinicalTrials.gov identifier: NCT00454818).
Two other separate clinical trials targeting SERCA2a are enrolling patients.
The first trial is being conducted in the United Kindom. SERCA2a-AAV6 or
saline solution is to be administered to patients with advanced HF. The patients
will receive SERCA2a gene by direct injection into the myocardium during
implantation of an LV assist device for an accepted clinical indication
(ClinicalTrials.gov identifier: NCT00534703, Gwathmey et al. 2011, Kawase et al. 2011).
A second phase 2 trial is investigating the efficacy of SERCA2a-AAV6-CMV
on cardiac remodelling parameters in patients with severe HF. This trial is 2
single-center, double-blind, randomized, placebo-controlled, parallel study
(Gwathmey et al. 2011, Kawase et al. 2011).

71
2.6.2 Adenylyl cyclase 6
A clinical trial of AC6 gene transfer in patients with congestive HF started
enrolling patients in May 2010. The adenovirus-5–construct encoding human
AC6 is to be delivered by intracoronary injection in patients with stable but
severe congestive HF with sucrose solution being used as a control
(ClinicalTrials.gov identifier: NCT00787059). Nitroprusside will be delivered
simultaneously to increase gene transfer efficiency. Nitroprusside has been used
safely by intracoronary infusion in patients with heart disease (Hillegass et al. 2001, Roth et al. 2004). The trial will be a randomized, double blinded and
placebo controlled study with 72 patients being enrolled in a 3:1 randomization
ratio (54 patients are received increasing doses of human AC6-adenovirus and 18
patients will receive placebo). The patients are randomized in dose-dependent
manner starting at 3.2x109 to 3.2x1012 viral particles in 6 dose groups
(ClinicalTrials.gov identifier: NCT00787059, Gao & Hammond 2011, Kawase et al. 2011).
2.6.3 Stromal derived growth factor-1
A clinical trial on SDF-1 will examine the effects of SDF-1 naked DNA injection
in patients with ischemic heart disease. SDF-1 naked DNA will be injected
directly into the myocardium at multiple sites via a percutaneous, LV approach
using a needle injection catheter. This trial is currently enrolling patients; it is an
open-label dose escalation study for evaluating the safety of a single escalating
dose of SDF-1 administered by endomyocardial injection. Three cohorts (16 total)
will be studied and the dose will be escalated by increasing the total amount of
SDF-1 delivered per subject from low dose (cohort 1) to middle dose (cohort 2) to
high dose (cohort 3). The impact of cardiac function will be measured with
cardiac perfusion conducted via single-photon emission computed tomography
(SPECT) imaging and assessment of NYHA classification (ClinicalTrials.gov
identifier: NCT01082094, Kawase et al. 2011).

72

73
3 Aims of the research
The aim of the study was to characterize the molecular mechanisms of cardiac
hypertrophy and LV remodelling after MI and in hypertension, and to identify the
novel drug target genes for the treatment of HF. The effects of BNP and (P)RR on
cardiac function and remodelling by direct recombinant adenoviral gene delivery
into the anterior wall of the LV were investigated. The objectives were:
1. To investigate direct structural and functional effects of BNP on LV
remodelling.
2. To study the effects of BNP gene transfer on the underlying signalling
pathways.
3. To characterize the heterogeneity of circulating immunoreactive human NT-
proBNP after adenoviral transfer of human BNP.
4. To examine the Ang II-dependent and Ang II-independent signalling
pathways triggered by (P)RR gene transfer.

74

75
4 Materials and methods
Summary of experimental models and methods is presented in Table 5.
Table 5. Summary of experimental protocols.
Study Experimental model Method
I Adenovirus–mediated BNP gene transfer in vivo Echocardiography
Normal heart Telemetric monitoring
Experimental myocardial infarction Protein extraction and western blotting
Ang II–induced hypertension RNA isolation and real time RT-qPCR
Radioimmunoassay
HPLC
BNP antibody determination
EIA (cGMP)
Immunohistochemistry
Masson’s trichrome staining
TUNEL staining
Lectin staining
II Adenovirus–mediated BNP gene transfer in vivo Protein extraction and western blotting
Normal heart Radioimmunoassay
HPLC
III Adenovirus–mediated (P)RRgene transfer in vivo Echocardiography
Normal heart Protein extraction and western blotting
Losartan infusion
Experimental myocardial infarction1
RNA isolation and real time RT-qPCR
Gelatin zymography
Ang II–induced hypertension1 Immunohistochemistry
Immunofluorescence
X-gal staining
Masson’s trichrome staining
TUNEL staining
Pecam staining
Ang II, angiotensin II; BNP, B-type natriuretic peptide; cGMP, cyclic guanosine monophosphate; EIA,
enzyme immunoassay; HPLC, high–pressure liquid chromatography; (P)RR, (pro)renin receptor; RT-
qPCR, reverse transcriptase quantitative polymerase chain reaction; TUNEL, Terminal deoxynucleotidyl
transferase dUTP nick end labeling. 1methods were not described in original publication III.

76
4.1 Recombinant adenoviral vectors (I, II and III)
For constructions of hBNP and (P)RR a full-length coding regions of cDNA was
cloned into the SalI and HindIII sites of the pShuttle-CMV vector (Qbiogene Inc.,
Illkirch, Cedex-France). The sequences for the cloning primers used were as
follows; hBNP forward 5'-GCG TCG ACT CCA GAG ACA TGG ATC CCC AG-
3' and reverse 5'-CCC AAG CTT TTA ATG CCG CCT CAG CAC-3' and rat
(P)RR forward 5'-GCG TCG ACC GTG GCA CCA TGG CTG TGC-3' and
reverse 5'-CCC AAG CTT TCA ATC CAT TCG AAT CTT CTG GTT TG-3'.
The vector pShuttle-CMV contains the CMV promoter and the SV40
polyadenylation signal. Deletion of E1 and E3 genes in adenoviral plasmid
creates space for foreign DNA and prohibits virus replication and thus, eliminates
any self-replication capabilities. The presence of the insert was confirmed by
restriction digestion and sequence analysis. The shuttle plasmids were linearized
using PmeI and complete digestions were evaluated by electrophoresis.
PmeI-linearized pShuttle-CMV-vector (including gene of interest) was
transformed into BJ5183-AD-1 electroporation competent cells (Stratagene, La
Jolla, CA, USA) using Gene Pulser Transfection Apparatus (Bio-Rad, Hercules,
CA, USA) at 2500 V, 200 Ω and 25 µFD. BJ5183-AD-1 electroporation
competent cells are recombination proficient bacteria carrying the adenoviral
plasmid that encodes the adenovirus-genome. The transformants were selected for
kanamycin resistance and the homologous recombination was confirmed by PacI
digestion. The recombinant adenoviral plasmids were transformed into DH5α
competent cells. Recombinant DNAs were purified with a maxiprep kit
(QIAGEN, Valencia, CA, USA) and with ethanol-sodium acetate precipitation.
The recombined adenoviral backbones were checked by BstXI and PacI
digestions.
Recombinant adenoviral constructs were linearized with PacI and transfected
into AD-293A cells (Qbiogene Inc. Illkirch, Cedex-France). Primary adenovirus
stocks with recombinant adenoviral plasmid were prepared by Lipofectamine
2000 (Invitrogen, Carlsbad, CA, USA) transfection. Once the majority of the cells
had started become to round and detach, the cells were harvested. Adenoviruses
were released by freeze‒thaw cycles and fresh AD-293A cells were infected with
the extracted virus. Adenoviruses were amplified by four rounds of amplification
of adenovirus and concentrated and purified using 15%:30%:40% iodixanol
(OptiPrep, Axis-Shield PoC AS, Oslo, Norway) density gradient centrifugation
100 000 g, at +4 °C overnight. The purified viruses were diluted in 1 x PBS and

77
stored at ‒70 °C. The adenoviral titers were determined by AdEasy Viral Titer Kit
(Stratagene, La Jolla, CA, USA).
LacZ control virus
Adenovirus containing the Escherichia coli LacZ gene (coding β-galactosidase
protein) was used as a control virus. The pShuttle-CMV‒LacZ was a commercial
plasmid (Stratagene, La Jolla, CA, USA). Homologous recombination, production
and purification of the LacZ control virus was performed using the same
technique as hBNP and (P)RR‒adenoviruses.
4.2 Animal studies (I, II and III)
All experimental protocols were approved by the Animal Use and Care
Committee of the University of Oulu and conformed to the Guide for the Care
and Use of Laboratory Animals published by the US National Institutes of Health.
4.2.1 Intramyocardial gene transfer (I, II and III)
Cardiac gene transfer of recombinant adenoviruses hBNP, (P)RR and LacZ-
control into the LV free wall was performed as previously described (Tenhunen et al. 2006a).
8-week old male Sprague-Dawley (SD) rats weighing 250–300 g were
anesthetized with medetomidine hydrochloride (Domitor, 250 μg/kg
intraperitoneally (i.p.)) and ketamine hydrochloride (Ketamine, 50 mg/kg i.p).
Rats were connected to the respirator through a tracheotomy. A left thoracotomy
and pericardial incision was performed to expose the heart and single injections of
recombinant adenovirus, 1x109 infectious units (ifu), in a 100 μl volume, were
injected using a Hamilton precision syringe directly into the anterior wall of the
LV. Ifu is biologically equivalent to plaque forming unit (PFU). The syringe was
inserted into one site of the LV free wall (apex to base), and then slowly the
solution was injected while withdrawing the syringe. The heart was repositioned,
the rat was briefly hyperventilated and the incision closed. After the operation,
anaesthesia was partially antagonized with atipamezole hydrochloride (Antisedan,
1.5 mg/kg i.p.) and rats were hydrated with physiological saline solution (5 ml
subcutaneous (s.c.)). For postoperative analgesia, buprenorphine hydrochloride

78
(Temgesic, 0.05–0.2 mg/kg s.c.) and carprofen (Rimadyl, 5 mg/kg s.c.) were
administered.
4.2.2 Acute myocardial infarction (I and III)
Acute MI was produced by ligation of the LAD during medetomidine
hydrochloride and ketamine hydrochloride anaesthesia as previously described
(Pfeffer et al. 1979, Tenhunen et al. 2006b). The rat was connected to the
respirator through a tracheotomy and ventilated at a rate of 55–60 breaths per
minute. A left thoracotomy and pericardial incision were performed and the LAD
was ligated about 3 mm from its origin. After ligation of the LAD, the heart was
repositioned in the chest and the incision was closed. The anaesthetic effects were
antagonized and the rats were hydrated with 10 ml physiological saline solution.
Buprenorphine hydrochloride and carprofen were administered for postoperative
analgesia. The sham‒operated rats underwent the same surgical procedure
without the ligation of LAD. Recombinant adenovirus was injected into the
anterior wall of the LV before the ligation of LAD. The adenoviral gene delivery
to the sham–operated hearts was performed using the same technique without the
ligation of LAD.
4.2.3 Angiotensin II–mediated hypertension (I and III)
Ang II–mediated hypertension has been used extensively in key studies in the
development of antihypertensive agents (Cataliotti et al. 2008, d’Uscio et al. 1997, Huelsemann et al. 1985, Smits et al. 1991). Using this experimental model
of hypertension, mean arterial pressure increases rapidly (within 3 hours) and
remains significantly elevated throughout the 2‒week period (Suo et al. 2002).
Ang II (33.3 μg/kg/h) was administered via s.c. implanted osmotic
minipumps (Alzet model 2002, Scanbur BK AB, Sollentuna, Sweden), as
described previously (Lako-Futo et al. 2003, Suo et al. 2002). Minipumps were
implanted subcutaneously before the gene delivery.
4.2.4 Losartan treatments with osmotic minipumps (III)
Losartan (400 µg/kg/h) was administered via s.c. implanted osmotic minipumps
(Alzet model 2001 for 1 week and Alzet model 2002 for 2 weeks, Scanbur BK
AB, Sollentuna, Sweden), as described previously (Serpi et al. 2009). Minipumps

79
were implanted before the gene delivery. This dose of losartan was previously
shown to abolish completely Ang II–induced increase in mean arterial pressure,
LV weight/body weight ratio and elevation of skeletal α-actin and β-myosin
heavy chain (MHC) mRNA levels (Lako-Futo et al. 2003).
4.3 Echocardiographic measurements (I and III)
Transthoracic echocardiography was performed using the Acuson Ultrasound
System (SequoiaTM 512) and a 15-MHz linear transducer (15L8) (Acuson,
MountainView, CA, USA) as previously described (Rysa et al. 2010, Tenhunen et al. 2006b). Before examination, rats were sedated with ketamine (50 mg/kg i.p.)
and xylazine (10 mg/kg i.p.). Using two-dimensional imaging, a short axis view
of the LV at the level of the papillary muscles was obtained, and a two
dimensionally guided M-mode recording through the anterior and posterior walls
of the LV was obtained. LV end-systolic and end-diastolic dimensions as well as
thethickness of the interventricular septum and posterior wall were measured
from the M-mode tracings. LV fractional shortening and ejection fraction were
calculated from the M-mode left ventricular dimensions using the following
equations:
Fractional shortening (%) = (LVEDD -LVESD) / LVEDD x 100
Ejection fraction (%) = (LVEDD)3 – (LVESD)3 / LVEDD3 x 100
(LVEDD, left ventricular end diastolic diameter; LVESD, left ventricular end
systolic diameter)
An average of 3 measurements of each variable was used. All
echocardiographical measurements were performed blinded by a person who was
not aware of the treatments. After echocardiography, the animals were sacrificed
and blood samples were collected into pre-cooled tubes containing
ethylenediamine tetra-acetic acid (1.5 mg/1ml blood), hearts were weighed and
the ventricles were immersed in liquid nitrogen and stored at –70 °C for later
analysis.
4.4 Telemetric monitoring (I)
Rats were anesthetized with medetomidine hydrochloride and ketamine
hydrochloride anaesthesia combined with preoperatively administered
buprenorphine (0.05 mg/kg s.c.) and carprofen (Rimadyl, 5 mg/kg s.c.). For
telemetric monitoring of hemodynamics, rats were instrumented with a catheter in

80
the left carotid artery coupled with a sensor and transmitter (TA11PA-C40; Data
Sciences, St. Paul, MN, USA) implanted under the skin (Lako-Futo et al. 2003,
Suo et al. 2002). After the operation, the anaesthesia was partially antagonized
and the rats were given buprenorphine hydrochloride for analgesia. Twelve days
after implantion, adenovirus–mediated local intramyocardial hBNP gene transfer
into the LV was performed. Mean arterial pressure and heart rate were
continuously measured through out the experiment. Hemodynamics were
recorded every 3 min and averaged for every hour. Results represent 24 h average
every day, except on day 0 due to the intramyocardial gene transfer.
4.5 Protein analysis (I, II and III)
4.5.1 Extraction of cytoplasmic protein (I, II and III)
In order to extract the cytoplasmic protein, the LV tissue was broken and reduced
to a powder in liquid nitrogen. The thawed powder was homogenized in a lysis
buffer (20 mmol/l Tris-HCl [pH 7.5], 10 mmol/l NaCl, 0.1 mmol/l EDTA, 0.1
mmol/l EGTA, 1 mmol/l β-glycerophosphate, 1 mmol/l Na3VO4, 2 mmol/l
benzamidine, 1 mmol/l phenylmethylsulfoxide, 50 mmol/l NaF, 1 mmol/l
dithiothreitol and 10 µg/ml each of leupeptin, pepstatin and aprotinin). The
cytosolic fraction was separated out by centrifugation at 2000 revolutions per
minute (rpm) in +4˚C for 1 min. To separate the cytoplasmic protein fraction, 5 x
nuclear extraction buffer (NEB) (100 mM Tris-HCl [pH 7.5], 750 mM NaCl, 5
mM EDTA, 5 mM EGTA, 5% Triton X 100, 12 mM sodium pyrophosphate, 5
mM β-glycerophosphate, 5 mM Na3VO4) were added to the tissue homogenate
followed by centrifugation at 12500 rpm in +4 ˚C for 20 min. The supernatant
was frozen in liquid nitrogen and stored in –70 °C until assayed. Protein
concentrations were determined by Bio-Rad Laboratories Protein Assay.
4.5.2 Western blot analyses (I, II and III)
In the western blot analysis, 30 µg total protein was subjected to SDS-PAGE and
the separated proteins were electrically transferred to Optitran BA-S 85
nitrocellulose membranes (Scleicher & Schuell BioScience, Dassel, Germany).
After blocking the nonspecific background in 5% non-fat milk or a mixture (1:1)
of Odyssey Blocking Buffer – Tris–buffered saline (TBS), nitrocellulose

81
membranes were incubated with appropriate primary antibody in 0.5–1% milk in
a solution of TBS-0.05% Tween-20 or in Odyssey Blocking Buffer– TBS
overnight at +4 °C. The antibody dilution varied from 1:200 to 1:2000, depending
on the signal strength. After washing, antibody binding was detected with specific
secondary antibodies. HRP-conjugated antibodies (dilution 1:2000) used with
enhanced chemiluminescence (ECL) PlusTM Western Blotting Detection System
and fluorescent (Alexa Fluor) antibodies (1:3000–1:5000) used with Odyssey
Infrared Imaging System. The antibodies used in western blot analyses are
presented in Table 6.
For a second western blot, the membranes were stripped in buffer containing
62.5 mmol/l tris (pH 6.8), 2% sodium dodecyl sulfate, and 100 mmol/l
mercapthoethanol. The protein amounts were detected by enhanced
chemiluminescence by a Fujifilm LAS-3000 Imager (Fujifilm, Tokyo, Japan)
when blots were incubated with ECL Plustm-reagents, and the detection was based
on fluorescence measured by the Odyssey Infrared Imaging System. The bands
were scanned and analyzed with Quantity One software (Bio-Rad Laboratories,
Hercules, CA, USA).
Table 6. Summary of the primary and secondary antibodies used for Western blot
analyses.
Manufacturer Antibody Type of antibody
Cell Signalling Technology ERK1/2, phospho-ERK1/2 (Thr202/Tyr204) Primary antibody
p38 Primary antibody
phospho-HSP27 (Ser82) Primary antibody
Akt, phospho-Akt (Ser473) Primary antibody
HRP- linked anti rabbit IgG, Secondary antibody
HRP- linked anti-mouse IgG Secondary antibody
Millipore Phospho-p38 (Thr180/Tyr182) Primary antibody
GAPDH Primary antibody
HSP27 Primary antibody
Santa Cruz Biotechnology SERCA2 Primary antibody
CTGF Primary antibody
Wnt-3 Primary antibody
Frizzled-8 Primary antibody
V-ATPase A1 Primary antibody
PLZF Primary antibody
PLB-Ser16 Primary antibody
Novus Biologicals (P)RR Primary antibody
Badrilla Ltd. PLB, PLB-Thr17 Primary antibody

82
Manufacturer Antibody Type of antibody
BD Transduction Laboratories β-catenin Primary antibody
Ala-Kopsala et al. 2004 human NT-proBNP10–29 Primary antibody
Invitrogen Alexa Fluor 680 goat anti-mouse Secondary antibody
Alexa Fluor 680 goat anti-rabbit Secondary antibody
CTGF, connective tissue growth factor; ERK1/2, extracellular signal regulated kinase; GAPDH,
glyceraldehyde 3-phosphate dehydrogenase; HRP, horseradish peroxidase; HSP27, heat shock protein
27; NT-proBNP, N-terminal profragment of B-type natriuretic peptide; PLB, phospholamban; PLZF,
promyelocytic zinc finger protein; (P)RR, (pro)renin receptor; Ser, serine; SERCA2, sarcoplasmic
reticulum Ca2+-ATPase 2; Thr, threonine
4.5.3 Immunoprecipitation (III)
Samples with 300 μg of total protein of (P)RR or LacZ-treated animals were
incubated with 10 μl of PLZF antibody (Santa Cruz Biotechnology, Santa Cruz,
CA, USA) overnight with continuous rocking at +4 °C and conjugated with
protein G-agarose beads (30 μl/sample) (Santa Cruz Biotechnology, Santa Cruz,
CA, USA) for 3 h with continuous rocking at +4 °C. The absence of primary
antibody in a parallel reaction mix served as a negative control. The beads were
collected by centrifugation, washed 5 times in the lysis buffer, and boiled for 5
min in sodium dodecyl sulphate, resolved by SDS-PAGE and transferred to
Optitran BAS 85 nitrocellulose membranes. The membranes were blocked in
Odyssey Blocking Buffer– TBS (1:1) and then incubated with specific polyclonal
anti-(P)RR antibody (Novus Biologicals, Littleton, CO, USA) at a concentration
of 1:100 in Odyssey Blocking Buffer – TBS solution overnight at +4 °C. On the
following day, (P)RR antibody binding was detected with Alexa Fluor goat anti-
mouse IgG (Invitrogen, Carlsbad, CA, USA) at a 1:3000 dilution. The
chemiluminescence was detected using Odyssey Infrared Detection. The bands
were quantified with Quantity One software.
4.5.4 cGMP assay (I)
LV tissue samples were homogenized in 10 volumes of 5% trichloroacetic acid at
4 °C. Samples were centrifuged and trichloroacetic acid was extracted from the
supernatant by adding 5 volumes of water-saturated ether for 3 times. Residual
ether was removed from the aqueous layer by heating at +70 °C. cGMP was
detected in non-acetylated samples using a cGMP enzyme immunoassay (EIA)

83
Kit (Cayman Chemical Company, Ann Arbor, MI, USA) according to the
manufacturer's directions.
4.5.5 Gelatin zymography (III)
Gelatin zymography was used to detect the gelatinases (MMP-2 and MMP-9) in
total protein samples. Zymography was performed as previously described
(Nyberg et al. 2003). 17 µg total proteins were separated on 10% SDS-PAGE
containing fluorescently labeled gelatin. After electrophoresis, the gelatinases
were renatured by removing sodium dodecyl sulphate with Triton X-100. Gels
were then incubated in Tris-HCl activation buffer overnight at +37 °C to allow the
MMPs to digest the substrate. The degradation of gelatin was visualized under
long wave UV light and the proteins were stained with 0.5% Coomassie Blue R-
250. The films were analyzed with Quantity One software (Bio-Rad Laboratories,
Hercules, CA, USA).
4.5.6 Immunohistochemistry (I and III)
In the histological analysis, the LVs were fixed in 10% buffered formalin solution.
Transversal sections of the LV were embedded in paraffin, and 5 μm sections
were cut. Sections were cut from the mid-section of the heart at the level of the
papillary muscles. Samples from different animals were obtained in an identical
way and from the corresponding sites in order to make the samples fully
comparable. All measurements were performed blinded by persons who were not
aware of the treatments.
Efficiency and localization of gene deliveries (I and III)
To examine the efficiency and localization of the hBNP and (P)RR gene
deliveries, the sections were incubated with specific polyclonal anti-human NT-
proBNP antibody (Ala-Kopsala et al. 2004) at the dilution of 1:2000 or with
specific polyclonal anti-(P)RR antibody (Novus Biological, Littleton, CO, USA)
at the dilution 1:200 3 days after gene transfer. In (P)RR-treated hearts, a red
color was produced by chromogen, amino-9-ethylcarbazole (Zymed, South San
Francisco, CA, USA). Specific polyclonal anti-(P)RR antibody was raised against
a peptide mapping with an extracellular domain of (P)RR.

84
Immunofluorescence staining was performed to analyse the expression of
(P)RR in cardiac cells. The antibodies used were NB100-1318 (Novus
Biologicals, Littleton, CO, USA) for (P)RR and anti-prolyl 4-hydroxylase β
(MAB2073, Millipore, Temecula, CA, USA) for fibroblasts. Nuclei were stained
blue with diamidinophenylindole dihydrochloride (DAPI) (Sigma, St Louis, MO,
USA).
Stem cells (I)
Primary antibody for c-kit (sc-168, Santa Cruz Biotechnology, CA, USA) was
used to stain stem cells. The number of c-kit–positive cells in the anterior wall of
LV was counted. The area of counted section was examined by computerized
methods and the number of positively staining cells was related to the area
(cells/35 mm2).
Cells undergoing division (I and III)
To identify cells undergoing division, immunohistochemical labelling of nuclear
Ki-67 antigen was performed by using monoclonal mouse anti-rat Ki-67 antigen
antibody (DakoCytomation, Glostrup, Denmark). The whole LV was scanned and
stained cells were counted from high power fields (40×) choosing 5 hot spot areas
in each sample.
Inflammatory cells (I)
Primary antibody for CD43 (ab22351, Abcam, UK) was used to detect
inflammatory cells in the LV. The number of positively stained cells was counted
from the whole LV of all samples and related to area (cells/35 mm2) in order to
make the samples comparable. The primary antibodies were detected by
peroxidase conjugated EnVision Detection Kit system (DakoCytomation,
Glostrup, Denmark) and the samples were counterstained with haematoxylin.

85
4.6 RNA analysis (I and III)
4.6.1 Isolation and analysis of RNA (I and III)
The RNA was extracted from the LV tissue by using the guanidine-thiocyanate–
CsCl method (Tenhunen et al. 2006b)
Real time RT-qPCR (I and III)
mRNA levels were analyzed by the reverse transcriptase quantitative polymerase
chain reaction (RT-qPCR) using TaqMan chemistry on an ABI 7300 Sequence
Detection System (Applied Biosystems) as previously described (Tenhunen et al. 2004). The cDNA first strand was synthesized from total RNA with a First-Strand
cDNA Synthesis Kit for RT-qPCR (GE Healthcare/GE Life Sciences). The
sequences of the forward and reverse primers and for fluorogenic probes for RNA
used in RT-qPCR are presented in Table 7. The results were normalized to
ribosomal 18S (18S) RNA quantified from the same samples.
Table 7. Forward and reverse primer and fluorogenic probe sequences used for real
time quantitative RT-qPCR–analysis.
Gene Forward primer Reverse primer Fluorogenic probe
hBNP AAGATGGTGCAAGGGTCT
GG
GGCCACTGGAGGAGCTGA TGCTTTGGGAGGAAGATGGA
CCGG
rBNP TGGGCAGAAGATAGACCG
GA
ACAACCTCAGCCCGTCACAG CGGCGCAGTCAGTCGCTTG
G
ANP GAAAAGCAAACTGAGGGC
TCTG
CCTACCCCCGAAGCAGCT TCGCTGGCCCTCGGAGCCT
Skα TCCTCCGCCGTTGGCT AATCTATGTACACGTCAAAA
ACAGGC
CATCGCCGCCACTGCAGCC
β-MHC GCTACCCAACCCTAAGGAT
GC
TCTGCCTAAGGTGCTGTTTC
AA
TGTGAAGCCCTGAGACCTG
GAGCC
Col Iα1 CCCCTTGGTCTTGGAGGA
A
GCACGGAAACTCCAGCTGAT CTTTGCTTCCCAGATGTCCT
ATGGCTATGATG
Col IIIα1 AGCTGGCCTTCCTCAGACT
TC
GCTGTTTTTGCAGTGGTATG
TAATG
TTCCAGCCGGGCCTCCCAG
ET-1 ATGGACAAGGAGTGTGTCT
ACTTCTG
GGGACGACGCGCTCG CACCTGGACATCATCTGGGT
CAACACTC
FGF-2 CCCGGCCACTTCAAGGAT GATGCGCAGGAAGAAGCC CCAAGCGGCTCTACTGCAAG
AACGG

86
Gene Forward primer Reverse primer Fluorogenic probe
TNF-α GACAAGGCTGCCCCGACT
A
CTCCTGGTATGAAGTGGCAA
ATC
TGCTCCTCACCCACACCGTC
AGC
Fn-1 GCGAGGCAGGATCAGCTG CCAATCTTGTAGGACTGACC
CC
ACCATTGCAAATCGCTGCCA
TGAA
TGFβ1 CATCGACATGGAGCTGGT
GA
TTGGACAGGATCTGGCCAC ACGGAAGCGCATCGAAGCC
ATC
TGFβ2 ACCTTTTTGCTCCTGCATC
TG
GTCGAGGGTGCTGCAGGTA TCCCGGTGGCGCTCAGTCT
GT
Caα-A GGGCCCTCCATTGTCCA GCACAATACTGTCGTCCTGA
GTG
CGCAAGTGCTTCTGAGGCG
GCTAC
PAI-1 GCTGACCACAGCAGGGAA
A
GTGCCCCTCTCACTGATATT
GAA
CCCGGCAGCAGATCCAAGA
TGCTAT
(P)RR CTTGCTGTGGGCAACCTAT
TC
CTACCCCCTTCACTGTCACC
AT
ACCGGCCCCGGGCTACCAT
Serca2a CAGCCATGGAGAACGCTC
A
CGTTGACGCCGAAGTGG ACAAAGACCGTGGAGGAGG
TGCTGG
VEGF-A GATCCGCAGACGTGTAAAT
GTTC
TTAACTCAAGCTGCCTCGCC TGCAAAAACACAGACTCGCG
TTGCA
18S TGGTTGCAAAGCTGAAACT
TAAAG
AGTCAAATTAAGCCGCAGGC CCTGGTGGTGCCCTTCCGTC
A
ANP, atrial natriuretic peptide; Caα-A, cardiac α-actin; ColIα1, collagen Iα1; ColIIIα1, collagen IIIα1; FGF-
2, fibroblast growth factor-2; Fn-1, fibronectin-1; hBNP, human B-type natriuretic peptide; PAI-1,
plasminogen activator inhibitor-1; (P)RR, (pro)renin receptor; rBNP, rat BNP; SERCA2a, sarcoplasmic
reticulum Ca2+-ATPase 2a; Skα, skeletal α-actin; TGFβ, transforming growth factor-β; TNF-α, tumor
necrosis factor-α; VEGF-A, vascular endothelial growth factor-A; 18S, ribosomal 18S; β-MHC, β-myosin
heavy chain
4.7 Immunoassays (I and II)
4.7.1 Radioimmunoassay and HPLC (I and II)
Rat BNP, rat ANP and hBNP peptide levels from plasma and tissue samples were
measured by radioimmunoassay (RIA). The plasma and tissue samples for human
immunoreactive amino-terminal-proBNP10‒29 (ir-NT-proBNP10‒29), human ir-
BNP-32, rat ir-BNP22‒42 and rat ir-NT-proANP79‒98 were extracted with SepPak
C18 cartridges before the measurements. The cardiac tissue peptide levels were
measured from diluted aliquots of the guanidine thiocyanate extracts prepared for
RNA determination. The extraction buffer did not interfere with assay at the
dilutions used. It is known that hBNP assays do not cross-react (<0.01%) with

87
peptides derived from rat proANP (Ala-Kopsala et al. 2004, Ala-Kopsala et al. 2005).
Analysis of the molecular form of hBNP peptides in plasma and in LV
samples was performed by gel filtration high–pressure liquid chromatography
(HPLC). Synthetic hBNP77-108 and recombinant human NT-proBNP1‒76 peptides
were used as calibrators. The samples were applied to a Biosuite 125 HR column
[7.8 (i.d.) x 300 mm; Waters] and eluted with 10% acetonitrile in aqueous
trifluoroacetic acid (1ml/l trifluoroacetic acid in water). The flow rate was
1ml/min, and 0.5 ml fractions were collected.
4.7.2 Determination of BNP antibodies
The presence of BNP antibodies in the circulation was determined using methods
described previously (Ala-Kopsala et al. 2004) with the following modifications:
HPLC-purified recombinant 125I-Tyrosine0-proBNP77–108 was incubated with 1:100
dilution of the serum samples in a volume of 200 µl overnight at +4 °C. The
bound and free fractions were then separated by the addition of 0.5 mg bovine
gammaglobulin in 1 ml 20% polyethylene glycol 6000 followed by
centrifugation. The supernatants were discarded and the pellets were counted for
radioactivity in a Wallac CliniGamma counter. Specific rabbit antiserum against
hBNP (Ala-Kopsala et al. 2004) was used the positive control. The results are
expressed as the percentage of radioiodinated Tyrosine0-BNP bound.
4.8 Histology and image analysis (I and III)
The hearts were treated as described in chapter 4.5.6.
Analysis of fibrotic area (I and III)
Sections were stained with hematoxylin and eosin or Massons's trichrome to
examine the fibrotic area of the LV. In the study of the local response to
adenovirus–mediated gene transfer, fibrotic area in the LV was measured at 2
weeks after intramyocardial injection of adenoviral construct expressing LacZ
and PBS-based buffer (3% iodixanol-PBS) as well as from the hearts with needle-
prick (no injection of fluid) and non-injected hearts.

88
Infarct size (I)
The infarcted area was measured from LV circumference in the Masson’s
trichrome–stained sections using a digital image analysis system (Nikon
NIS_Elements BR 2.30 software). One block and section per heart was taken
from the same site in all samples and used for staining.
Size of cardiomyocytes (I and III)
To assess cardiomyocyte hypertrophy, the cross-sectional area of cardiomyocytes
was calculated from five correspondingly located fields per sample (3 from
epicardial and 2 from endocardial side of the LV). The-cross sectional area of 10
cells per field was measured using Nikon NIS_Elements BR 2.30 software.
Angiogenesis (I and III)
Biotinylated Griffonia simplicifolia lectin-1 (GSL-1) (B-1205, Vector
laboratories, Burlingame, CA, USA) or Pecam-1 (sc-1506-R, Santa Cruz
Biotechnology, Santa Cruz, CA, USA) was used to stain endothelial cells. The
number of capillaries was calculated from 5 representative high power fields
(40×) from the LV of each section; 3 from the epicardial and 2 from the
endocardial side of the LV were selected.
Apoptotic cells (I and III)
Apoptotic cells were detected by in situ labeling of the 3'-ends of the DNA
fragments generated by apoptosis‒associated endonucleases (TUNEL staining)
performed using the ApopTag in situ apoptosis detection kit (Oncor, Gaithersburg,
MD, USA), as previously described (Soini et al. 1996). DNA fragmentation was
identified by applying terminal transferase enzyme with digoxigenin-labeled
nucleotides. Anti-digoxigenin antibody was used to recognize the digoxigenin-
labeled nucleotide chains attached to the 3'-ends of sample DNA. A colour
reaction was produced with diaminobenzidine and the sections were
counterstained with hematoxylin. The apoptotic cells and bodies were counted in
5 high power fields (40× objective) choosing hot spot areas in each sample in
order to make the results comparable.

89
X-gal-staining (III)
To confirm the efficiency of adenovirus–mediated gene transfer, X-gal staining of
the LacZ-treated hearts was performed. Hearts were rinsed in PBS and fixed in
PBS containing 4% paraformaldehyde for 1 h at 4 °C, washed 3 times in rinsing
buffer (100 mM sodium phosphate pH 7.3, 2 mM MgCl2, 0.01% sodium
deoxycholate, 0.02% NP-40) for 30 min, and incubated with 1 mg/ml X-gal (5-
bromo-4-chloro-3-indolyl-β-D-galactopyranoside) in reaction buffer (5 mM
K3Fe(CN)6, 5 mM K4Fe(CN)6xH2O, 100 mM sodium phosphate pH 7.3, 2 mM
MgCl2, 0.01% sodium deoxycholate, 0.02% NP-40) at +37 °C for 5 h, and post-
fixed overnight in 10% formalin. The hearts were photographed, and for frozen
sections, hearts were embedded in OCT compound, frozen and sectioned.
Sections were counterstained with hematoxylin-eosin.
4.9 Statistical analysis
The results are expressed as means ± standard error of the mean (SEM).
Statistical analyses were performed using SPSS version 16.0.1 (SPSS Inc.,
Chicago, IL, USA). Statistical significance was evaluated by one-way analysis of
variance (ANOVA) followed by a least significant difference (LSD) post hoc test
for multiple comparisons. The hemodynamic variables were analysed with two-
way repeated-measures ANOVA. Student's t-test was used for comparison
between two groups. A P value of < 0.05 was considered statistically significant.

90

91
5 Results
5.1 Augmentation of left ventricular gene expression by adenoviral
gene delivery (I, II and III)
hBNP and (P)RR gene transfers protocols were established to locally increase
their protein levels in the normal adult rat LV and in hearts during the remodelling
process after MI and in Ang II–induced hypertension.
5.1.1 Human BNP gene delivery (I and II)
Increased hBNP gene expression was noted when hBNP expressing adenoviral
constructs were injected into the LV free wall at 1x109 ifu. hBNP mRNA and
peptide levels were highest at day 3 after the injections and declined significantly
thereafter during the follow-up period.
A 22–fold hBNP mRNA increase was observed at 3 days (P < 0.001) and 12–
fold increase at 1 week (P < 0.001) after hBNP gene transfer when normalized to
hBNP levels at 2 weeks. hBNP mRNA levels in the LV at 1 and 2 weeks’ post-
infarction were quantitatively equal to the hBNP levels in normal adult rat hearts
at 1 and 2 weeks after hBNP gene transfer.
LV human NT-proBNP peptide levels increased to 78 fmol/mg (P < 0.001 vs.
human NT-proBNP at 2 weeks) at day 3 by gene transfer and decreased during
the follow-up period, so that at 1 week, the human NT-pro-BNP peptide level was
47.7 fmol/mg (P < 0.05 vs. human NT-proBNP at 2 weeks). In LacZ-treated
hearts, human NT-proBNP was not detectable.
The efficiency and localization of the hBNP gene delivery was further
confirmed by immunohistochemistry. Immunohistochemical analysis showed
local and segmental hBNP staining in the anterior wall of the LV of hBNP-treated
hearts whereas LacZ-treated hearts were virtually negative. BNP localized
subcellularly to granules, nearly all of it being in the perinuclear zone (Fig. 7).
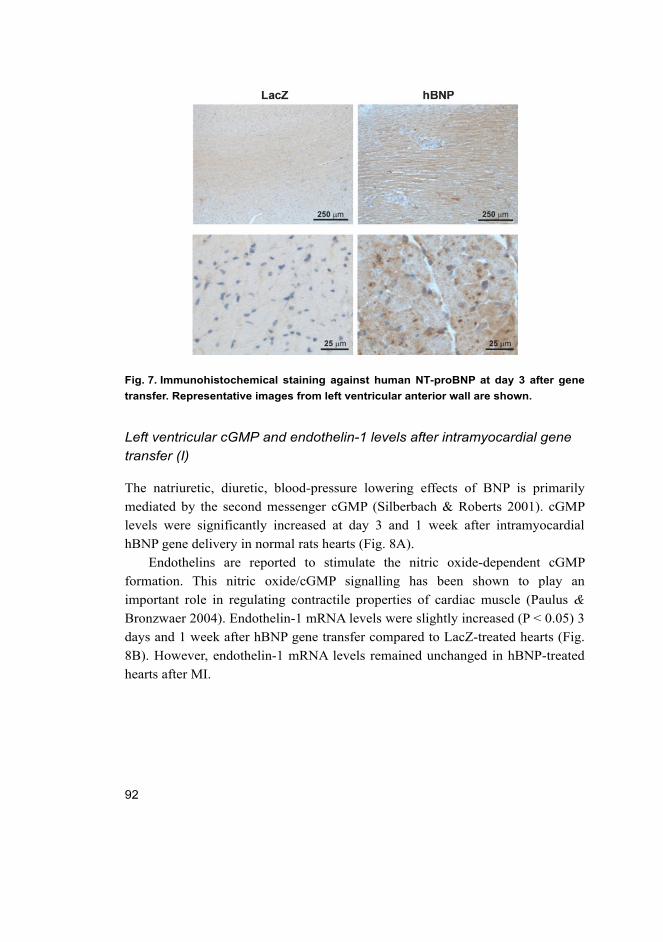
92
Fig. 7. Immunohistochemical staining against human NT-proBNP at day 3 after gene
transfer. Representative images from left ventricular anterior wall are shown.
Left ventricular cGMP and endothelin-1 levels after intramyocardial gene
transfer (I)
The natriuretic, diuretic, blood-pressure lowering effects of BNP is primarily
mediated by the second messenger cGMP (Silberbach & Roberts 2001). cGMP
levels were significantly increased at day 3 and 1 week after intramyocardial
hBNP gene delivery in normal rats hearts (Fig. 8A).
Endothelins are reported to stimulate the nitric oxide-dependent cGMP
formation. This nitric oxide/cGMP signalling has been shown to play an
important role in regulating contractile properties of cardiac muscle (Paulus &
Bronzwaer 2004). Endothelin-1 mRNA levels were slightly increased (P < 0.05) 3
days and 1 week after hBNP gene transfer compared to LacZ-treated hearts (Fig.
8B). However, endothelin-1 mRNA levels remained unchanged in hBNP-treated
hearts after MI.
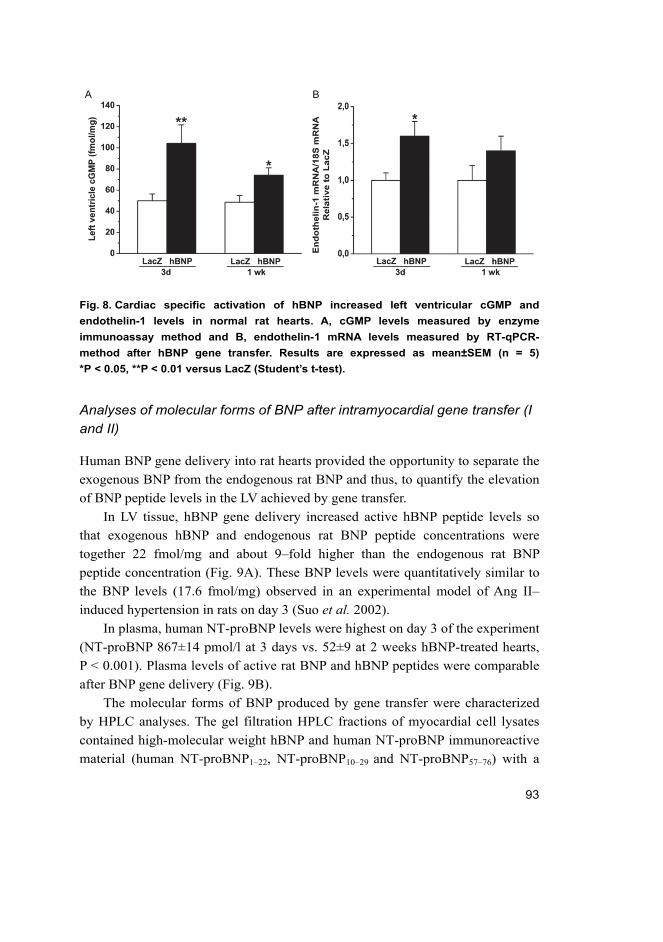
93
Fig. 8. Cardiac specific activation of hBNP increased left ventricular cGMP and
endothelin-1 levels in normal rat hearts. A, cGMP levels measured by enzyme
immunoassay method and B, endothelin-1 mRNA levels measured by RT-qPCR-
method after hBNP gene transfer. Results are expressed as mean±SEM (n = 5)
*P < 0.05, **P < 0.01 versus LacZ (Student’s t-test).
Analyses of molecular forms of BNP after intramyocardial gene transfer (I
and II)
Human BNP gene delivery into rat hearts provided the opportunity to separate the
exogenous BNP from the endogenous rat BNP and thus, to quantify the elevation
of BNP peptide levels in the LV achieved by gene transfer.
In LV tissue, hBNP gene delivery increased active hBNP peptide levels so
that exogenous hBNP and endogenous rat BNP peptide concentrations were
together 22 fmol/mg and about 9–fold higher than the endogenous rat BNP
peptide concentration (Fig. 9A). These BNP levels were quantitatively similar to
the BNP levels (17.6 fmol/mg) observed in an experimental model of Ang II–
induced hypertension in rats on day 3 (Suo et al. 2002).
In plasma, human NT-proBNP levels were highest on day 3 of the experiment
(NT-proBNP 867±14 pmol/l at 3 days vs. 52±9 at 2 weeks hBNP-treated hearts,
P < 0.001). Plasma levels of active rat BNP and hBNP peptides were comparable
after BNP gene delivery (Fig. 9B).
The molecular forms of BNP produced by gene transfer were characterized
by HPLC analyses. The gel filtration HPLC fractions of myocardial cell lysates
contained high-molecular weight hBNP and human NT-proBNP immunoreactive
material (human NT-proBNP1‒22, NT-proBNP10‒29 and NT-proBNP57‒76) with a
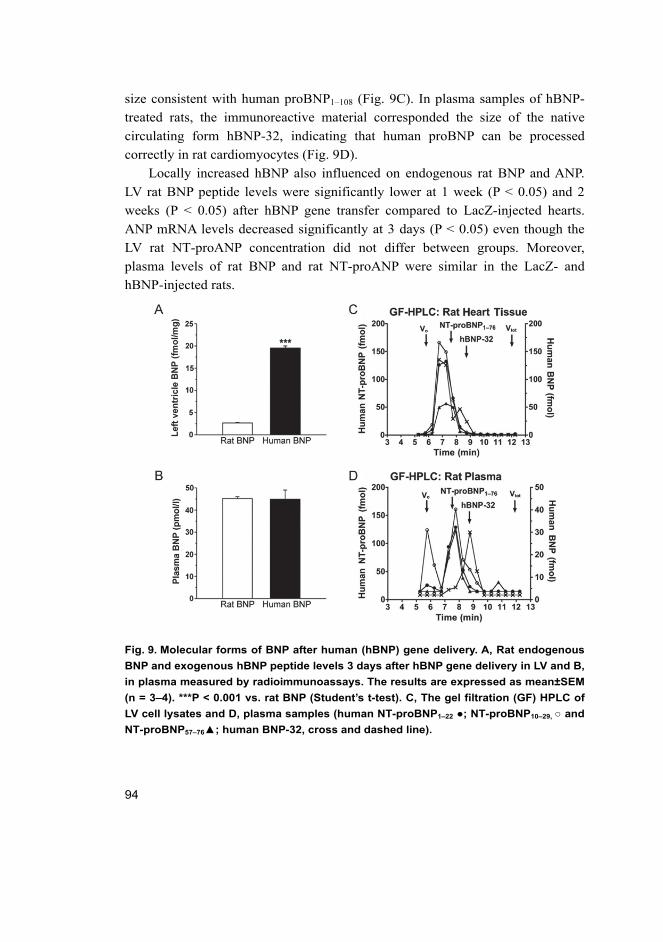
94
size consistent with human proBNP1‒108 (Fig. 9C). In plasma samples of hBNP-
treated rats, the immunoreactive material corresponded the size of the native
circulating form hBNP-32, indicating that human proBNP can be processed
correctly in rat cardiomyocytes (Fig. 9D).
Locally increased hBNP also influenced on endogenous rat BNP and ANP.
LV rat BNP peptide levels were significantly lower at 1 week (P < 0.05) and 2
weeks (P < 0.05) after hBNP gene transfer compared to LacZ-injected hearts.
ANP mRNA levels decreased significantly at 3 days (P < 0.05) even though the
LV rat NT-proANP concentration did not differ between groups. Moreover,
plasma levels of rat BNP and rat NT-proANP were similar in the LacZ- and
hBNP-injected rats.
Fig. 9. Molecular forms of BNP after human (hBNP) gene delivery. A, Rat endogenous
BNP and exogenous hBNP peptide levels 3 days after hBNP gene delivery in LV and B,
in plasma measured by radioimmunoassays. The results are expressed as mean±SEM
(n = 3–4). ***P < 0.001 vs. rat BNP (Student’s t-test). C, The gel filtration (GF) HPLC of
LV cell lysates and D, plasma samples (human NT-proBNP1‒22 ; NT-proBNP10‒29, and
NT-proBNP57‒76; human BNP-32, cross and dashed line).

95
Immune response and infiltration of inflammatory cells following hBNP gene transfer (I)
Earlier studies demonstrated that the short-term expression of adenoviral–
mediated gene transfers was associated with the intense immune reaction (Dai et al. 1995, Schagen 2004). Therefore, antibody formation against hBNP was
examined in plasma samples at the 2‒weeks’ follow-up period. hBNP antibody
formation expression was noted when hBNP expressing adenoviral constructs
were injected into the LV free wall at 1x109 ifu. hBNP gene transfer increased
significantly the production of neutralizing antibodies against the transferred
hBNP. The antibody formation against hBNP was highest at 2 weeks after
injections (Fig. 10A). This immune response probably attenuated the effect of
hBNP gene delivery on cardiac function.
In order to study the cardiac infiltration of inflammatory cells after adenoviral
hBNP gene transfer histological section were stained against CD43. As shown in
fig. 10B, the number of infiltrating cells in hBNP injected hearts was similar to
that in LacZ-injected hearts. After MI CD43 positive cells were increased,
compared to noninfarcted groups. No differences were noted between hBNP and
LacZ post-infarction (Fig. 10B). These findings support the concept that it is
direct and not possible proinflammatory effects, that play a role in angiogenesis
and antifibrosis of NPs.
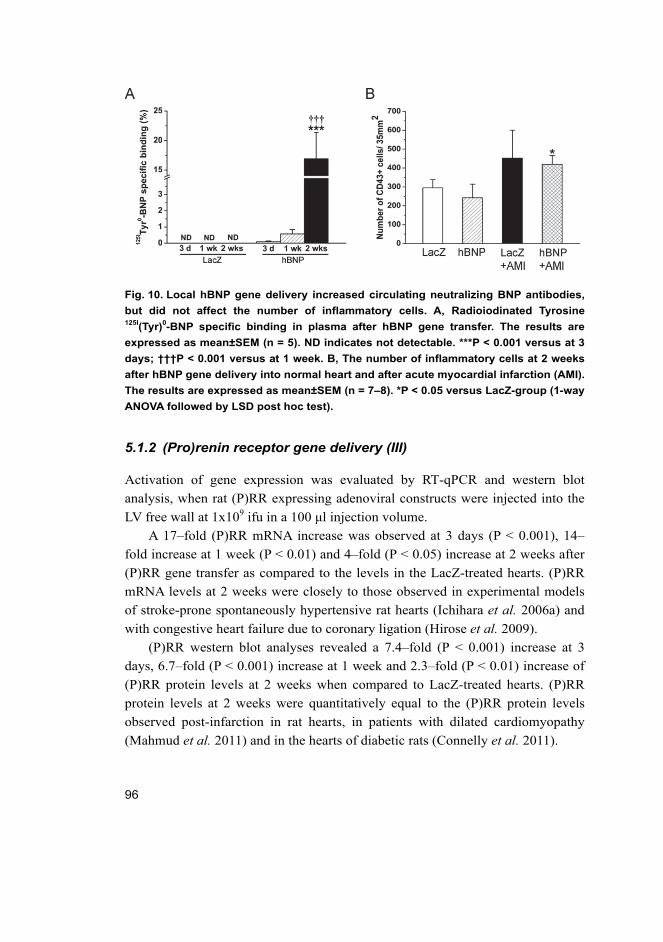
96
Fig. 10. Local hBNP gene delivery increased circulating neutralizing BNP antibodies,
but did not affect the number of inflammatory cells. A, Radioiodinated Tyrosine 125I(Tyr)0-BNP specific binding in plasma after hBNP gene transfer. The results are
expressed as mean±SEM (n = 5). ND indicates not detectable. ***P < 0.001 versus at 3
days; †††P < 0.001 versus at 1 week. B, The number of inflammatory cells at 2 weeks
after hBNP gene delivery into normal heart and after acute myocardial infarction (AMI).
The results are expressed as mean±SEM (n = 7–8). *P < 0.05 versus LacZ-group (1-way
ANOVA followed by LSD post hoc test).
5.1.2 (Pro)renin receptor gene delivery (III)
Activation of gene expression was evaluated by RT-qPCR and western blot
analysis, when rat (P)RR expressing adenoviral constructs were injected into the
LV free wall at 1x109 ifu in a 100 μl injection volume.
A 17–fold (P)RR mRNA increase was observed at 3 days (P < 0.001), 14–
fold increase at 1 week (P < 0.01) and 4–fold (P < 0.05) increase at 2 weeks after
(P)RR gene transfer as compared to the levels in the LacZ-treated hearts. (P)RR
mRNA levels at 2 weeks were closely to those observed in experimental models
of stroke-prone spontaneously hypertensive rat hearts (Ichihara et al. 2006a) and
with congestive heart failure due to coronary ligation (Hirose et al. 2009).
(P)RR western blot analyses revealed a 7.4–fold (P < 0.001) increase at 3
days, 6.7–fold (P < 0.001) increase at 1 week and 2.3–fold (P < 0.01) increase of
(P)RR protein levels at 2 weeks when compared to LacZ-treated hearts. (P)RR
protein levels at 2 weeks were quantitatively equal to the (P)RR protein levels
observed post-infarction in rat hearts, in patients with dilated cardiomyopathy
(Mahmud et al. 2011) and in the hearts of diabetic rats (Connelly et al. 2011).
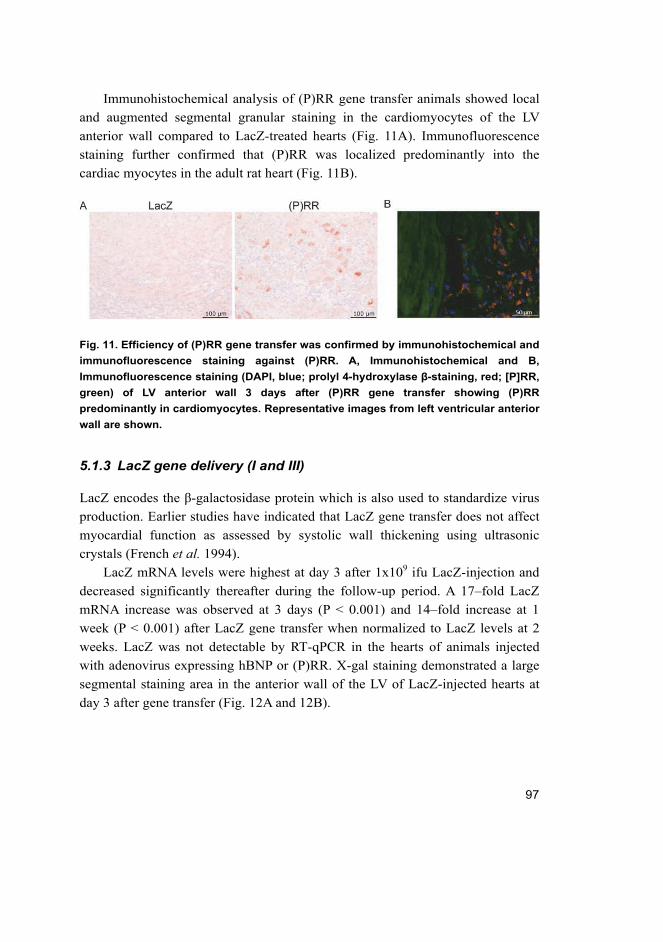
97
Immunohistochemical analysis of (P)RR gene transfer animals showed local
and augmented segmental granular staining in the cardiomyocytes of the LV
anterior wall compared to LacZ-treated hearts (Fig. 11A). Immunofluorescence
staining further confirmed that (P)RR was localized predominantly into the
cardiac myocytes in the adult rat heart (Fig. 11B).
Fig. 11. Efficiency of (P)RR gene transfer was confirmed by immunohistochemical and
immunofluorescence staining against (P)RR. A, Immunohistochemical and B,
Immunofluorescence staining (DAPI, blue; prolyl 4-hydroxylase β-staining, red; [P]RR,
green) of LV anterior wall 3 days after (P)RR gene transfer showing (P)RR
predominantly in cardiomyocytes. Representative images from left ventricular anterior
wall are shown.
5.1.3 LacZ gene delivery (I and III)
LacZ encodes the β-galactosidase protein which is also used to standardize virus
production. Earlier studies have indicated that LacZ gene transfer does not affect
myocardial function as assessed by systolic wall thickening using ultrasonic
crystals (French et al. 1994).
LacZ mRNA levels were highest at day 3 after 1x109 ifu LacZ-injection and
decreased significantly thereafter during the follow-up period. A 17–fold LacZ
mRNA increase was observed at 3 days (P < 0.001) and 14–fold increase at 1
week (P < 0.001) after LacZ gene transfer when normalized to LacZ levels at 2
weeks. LacZ was not detectable by RT-qPCR in the hearts of animals injected
with adenovirus expressing hBNP or (P)RR. X-gal staining demonstrated a large
segmental staining area in the anterior wall of the LV of LacZ-injected hearts at
day 3 after gene transfer (Fig. 12A and 12B).

98
Fig. 12. X-gal-staining demonstrated localization and efficiency of cardiac specific
activation of LacZ control virus by adenoviral gene delivery into the left ventricle at
day 3 after gene transfer. A, Whole mount x-gal staining showed a large segmental
staining area in anterior wall of left ventricle of LacZ injected hearts. B, Microscopic
image of x-gal–stained LV anterior wall.
Quantification of left ventricular fibrosis in LacZ-control hearts after adenovirus–mediated gene transfer
Quantification of fibrosis in LVs with no injections, needle-prick (no injection of
fluid), injected with PBS based buffer alone (dilution of adenoviruses for
injection) and injected with LacZ-adenovirus were examined in paraffin-
embedded histological sections stained with Masson’s trichrome. The degree of
fibrosis did not significantly differ between PBS based buffer (fibrotic area of LV
8.5%) and LacZ-adenovirus (fibrotic area of LV 9%) injected hearts, but tended to
be higher in these groups than needle-prick (fibrotic area of LV 5%) and
particularly with the no injection group (fibrotic area of LV 3%).
5.2 Cardiac effects of intramyocardial BNP gene delivery (I)
The effects of hBNP gene delivery on myocardial fibrosis, angiogenesis and
hemodynamic parameters were studied in normal rat hearts and in hearts during
the remodelling process after infarction and in an experimental model of Ang II–
mediated hypertension. In an attempt to evaluate the potential mechanisms
triggering the effects of local hBNP gene delivery, changes in the SERCA2
protein, MAPK signalling pathways and Akt signalling were evaluated.

99
5.2.1 BNP as a regulator of cardiac fibrosis
Cardiac fibrosis in normal adult rat heart
Fibrosis decreased significantly by hBNP gene delivery at 2 weeks (Fig. 13A and
13B). Consistent with this, hBNP overexpression decreased collagen IIIα1 mRNA
levels at 2 weeks (Fig. 13C). Moreover, collagen Iα1 gene expression was
slightly, but nonsignificantly reduced (Fig. 13E). No significant changes were
detected in other fibrosis-related genes such as collagen Iα1, TGFβ1 and TGFβ2,
fibronectin-1, MMP-2 and MMP-9 gene expressions.
Fig. 13. Human BNP (hBNP) gene delivery decreased myocardial fibrosis in normal rat
heart at 2 weeks after gene delivery. A, Fibrotic area of left ventricle (LV) were defined
with Masson’s trichrome–stained sections B, Representative images are shown, C,
Collagen IIIα1 (ColIIIα1) and E, Collagen Iα1 (ColIα1) mRNA levels. Results are
expressed as mean±SEM (n = 7–8). **P < 0.01 versus LacZ (Student’s t-test).

100
Cardiac fibrosis after myocardial infarction
The percentage of fibrotic area measured from infarct border zone was 3.1–fold
higher in the infarcted animals than in sham-operated animals (P < 0.001). The
size of the infarcted area remained unchanged by BNP gene delivery when hBNP
treated hearts were compared to LacZ-treated heart at 2 weeks’ post-infarction.
The fibrotic area of infarcted area (Fig. 14A and 14B) or border zone (Fig.
14C and 14D) did not differ between the infarcted hBNP- and LacZ-treated hearts
at 2 weeks’ post-infarction. Collagen gene expressions were similarly up-
regulated in the hBNP- and LacZ-treated hearts post-infarction (Fig. 14E and
14F). Furthermore, there was no significant difference between TNF-α and
endothelin-1 mRNA levels between groups, together indicating that hBNP gene
delivery had no effect of myocardial fibrosis post-infarction.
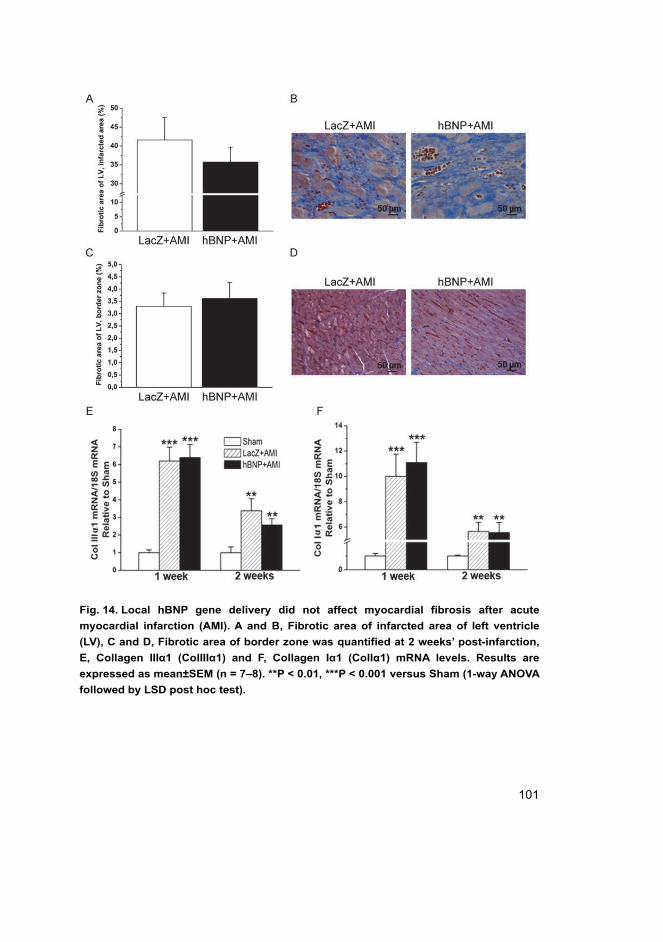
101
Fig. 14. Local hBNP gene delivery did not affect myocardial fibrosis after acute
myocardial infarction (AMI). A and B, Fibrotic area of infarcted area of left ventricle
(LV), C and D, Fibrotic area of border zone was quantified at 2 weeks’ post-infarction,
E, Collagen IIIα1 (ColIIIα1) and F, Collagen Iα1 (ColIα1) mRNA levels. Results are
expressed as mean±SEM (n = 7–8). **P < 0.01, ***P < 0.001 versus Sham (1-way ANOVA
followed by LSD post hoc test).

102
Cardiac fibrosis in angiotensin II–induced hypertension
Ang II–induced fibrosis was significantly attenuated by hBNP gene transfer at 2
weeks (Fig. 15A and 15B). Furthermore, a significant decrease in collagen IIIα1
(Fig. 15C) and collagen Iα1 (Fig. 15D) mRNA levels, up-regulated by Ang II, was
observed by hBNP gene transfer.
Fig. 15. Human BNP (hBNP) gene delivery decreased myocardial fibrosis in
angiotensin II (Ang II)–induced hypertension at 2 weeks. A, Fibrotic area of left
ventricle (LV), B, Representative images. C, Collagen IIIα1 (ColIIIα1) mRNA and E,
Collagen Iα1 (ColIα1) mRNA levels. Results are expressed as mean±SEM (n = 6–11).
*P < 0.05 versus LacZ; ††P < 0.01, †††P < 0.001 versus LacZ; ‡P < 0.05, ‡‡P < 0.01
versus LacZ with Ang II (1-way ANOVA followed by LSD post hoc test).

103
5.2.2 Coronary angiogenesis after BNP gene transfer
Histological sections were immunohistochemically stained against lectin in order
to investigate the effect of hBNP gene delivery on coronary angiogenesis. Local
BNP gene delivery resulted in a statistically significant increase in capillary
density (94.7±1.8 capillaries/field hBNP-treated hearts vs. 86.3±5.1
capillaries/field LacZ-treated hearts, P < 0.05), whereas BNP gene transfer had no
effect on capillary area (767±54 capillary area μm2/field hBNP-treated hearts vs.
735±74 capillary area μm2/field LacZ-treated hearts). FGF-2 is cardioprotective
and angiogenic mediator (Virag et al. 2007), and the enhanced angiogenesis by
BNP gene transfer was associated with the increased gene expression of FGF-2 at
3 days (P < 0.05) and 1 week (P < 0.05).
Angiogenesis contributes to the LV remodelling post-infarction. However, no
differences in the capillary density and the capillary area in the anterior wall of
the LV between hBNP- and LacZ-treated groups at 2 weeks’ post-infarction were
detected. Furthermore, hBNP gene delivery had no effect on coronary
angiogenesis in Ang II–mediated hypertension.
5.2.3 Hemodynamics in rats overexpressing BNP in the heart
The functional consequences of hBNP gene transfer were studied by
echocardiography. Ejection fraction, fractional shortening and LV dimensions of
the hBNP-treated animals were similar to those of LacZ-treated group, indicating
that hBNP overexpression had no apparent effect on cardiac function in normal
rats during the 2‒weeks’ follow-up period (Table 8).
Table 8. Echocardiographic parameters 2 weeks after intramyocardial hBNP gene
delivery.
Variable LacZ hBNP
LV diastole (mm) 7.6±0.1 7.7±0.1
LV systole (mm) 5.5±0.3 5.2±0.1
Ejection fraction (%) 60±3 66±2
Fractional shortening (%) 28±2 33±1
hBNP, human B-type natriuretic peptide; LV, left ventricle
Ligation of LAD caused a marked decrease in ejection fraction and fractional
shortening within 2 weeks. hBNP gene delivery significantly improved LV
ejection fraction (Fig. 16A) and fractional shortening (Fig. 16B) at 2 weeks’ post-
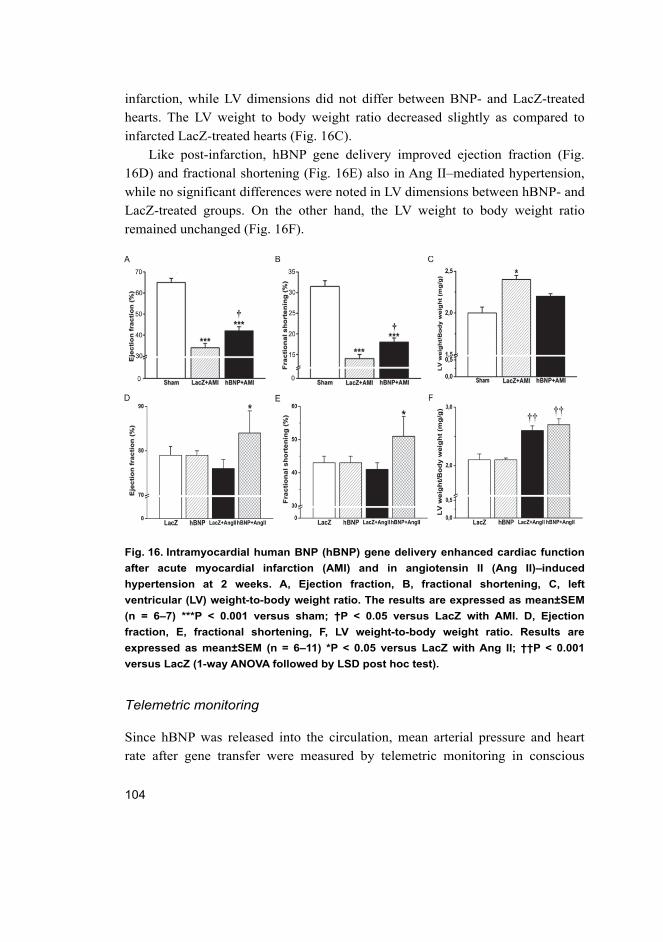
104
infarction, while LV dimensions did not differ between BNP- and LacZ-treated
hearts. The LV weight to body weight ratio decreased slightly as compared to
infarcted LacZ-treated hearts (Fig. 16C).
Like post-infarction, hBNP gene delivery improved ejection fraction (Fig.
16D) and fractional shortening (Fig. 16E) also in Ang II–mediated hypertension,
while no significant differences were noted in LV dimensions between hBNP- and
LacZ-treated groups. On the other hand, the LV weight to body weight ratio
remained unchanged (Fig. 16F).
Fig. 16. Intramyocardial human BNP (hBNP) gene delivery enhanced cardiac function
after acute myocardial infarction (AMI) and in angiotensin II (Ang II)–induced
hypertension at 2 weeks. A, Ejection fraction, B, fractional shortening, C, left
ventricular (LV) weight-to-body weight ratio. The results are expressed as mean±SEM
(n = 6–7) ***P < 0.001 versus sham; †P < 0.05 versus LacZ with AMI. D, Ejection
fraction, E, fractional shortening, F, LV weight-to-body weight ratio. Results are
expressed as mean±SEM (n = 6–11) *P < 0.05 versus LacZ with Ang II; ††P < 0.001
versus LacZ (1-way ANOVA followed by LSD post hoc test).
Telemetric monitoring
Since hBNP was released into the circulation, mean arterial pressure and heart
rate after gene transfer were measured by telemetric monitoring in conscious

105
animals. Intramyocardial hBNP gene delivery had no statistically significant
effect on hemodynamics, although there was a trend for mean arterial pressure to
decrease at 2 day after gene delivery (106.6±3.4 mmHg hBNP treated hearts vs.
122.1±6.6 mmHg LacZ treated hearts). Mean arterial pressure and heart rate were
similar in LacZ- and hBNP-injected rats prior gene transfer.
5.2.4 Activation of cardiac gene expressions and signalling pathways by BNP gene
Changes in the SERCA2 mRNA and protein levels, PLB phosphorylations and
MAPK and Akt signalling pathways were analysed to investigate the potential
mechanisms triggering the improvement of systolic function post-infarction by
local hBNP gene delivery. Moreover, gene expression of β-MHC and skeletal α-
actin were analyzed.
BNP gene transfer resulted normalization of SERCA2 and PLB levels after myocardial infarction
BNP gene delivery normalized SERCA2 mRNA and protein levels (Fig. 17A) at 1
week post-infarction. Since, SERCA activity is regulated in the heart by
interaction with PLB (Bers et al. 2002) Ser16 and Thr17 specific antibodies were
used to determine the phosphorylated residues. hBNP gene transfer increased
PLB phosphorylation at Thr17 (Fig. 17B), but not at Ser16. In agreement with
changes in contractile function, β-MHC (Fig. 17C) mRNA levels increased
significantly at 1 weeks’ post-infarction and skeletal α-actin (Fig. 17D) mRNA
levels increased significantly at 2 weeks’ post-infarction by hBNP gene transfer.
SERCA2 and PLB levels remained unchanged in Ang II–induced hypertrophy by
BNP gene transfer.
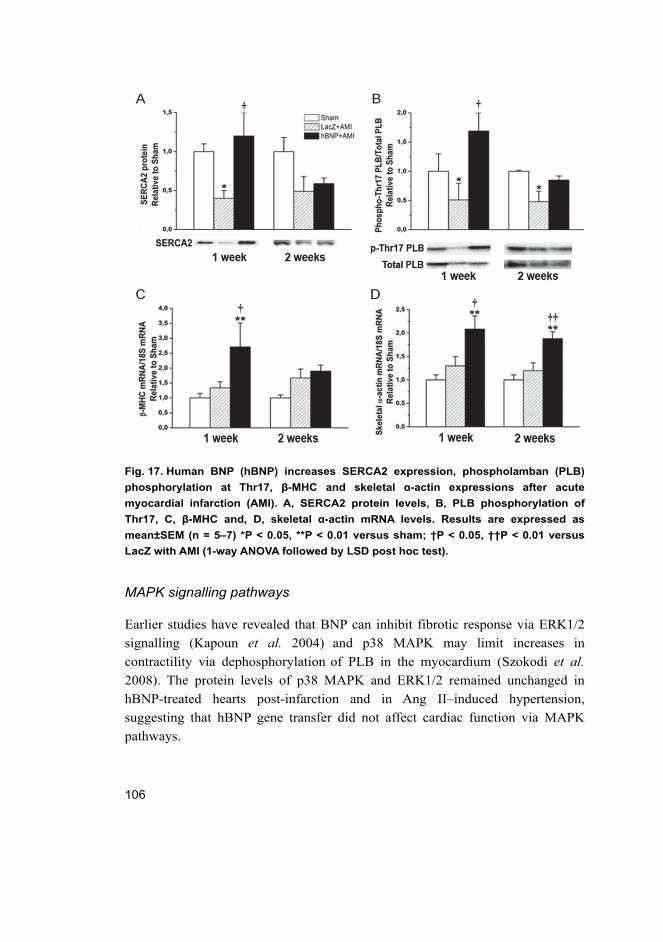
106
Fig. 17. Human BNP (hBNP) increases SERCA2 expression, phospholamban (PLB)
phosphorylation at Thr17, β-MHC and skeletal α-actin expressions after acute
myocardial infarction (AMI). A, SERCA2 protein levels, B, PLB phosphorylation of
Thr17, C, β-MHC and, D, skeletal α-actin mRNA levels. Results are expressed as
mean±SEM (n = 5–7) *P < 0.05, **P < 0.01 versus sham; †P < 0.05, ††P < 0.01 versus
LacZ with AMI (1-way ANOVA followed by LSD post hoc test).
MAPK signalling pathways
Earlier studies have revealed that BNP can inhibit fibrotic response via ERK1/2
signalling (Kapoun et al. 2004) and p38 MAPK may limit increases in
contractility via dephosphorylation of PLB in the myocardium (Szokodi et al. 2008). The protein levels of p38 MAPK and ERK1/2 remained unchanged in
hBNP-treated hearts post-infarction and in Ang II–induced hypertension,
suggesting that hBNP gene transfer did not affect cardiac function via MAPK
pathways.
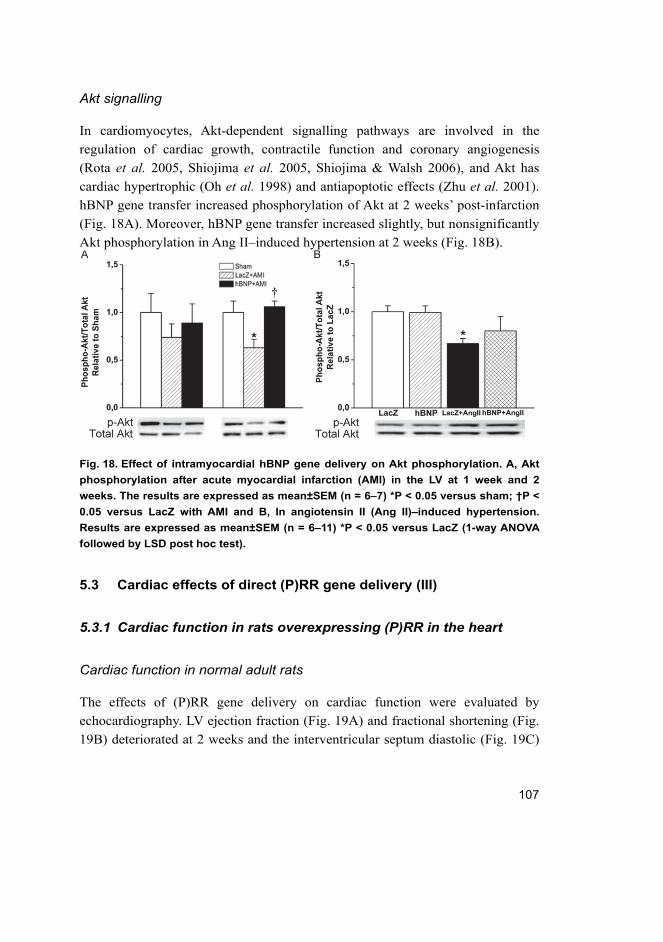
107
Akt signalling
In cardiomyocytes, Akt-dependent signalling pathways are involved in the
regulation of cardiac growth, contractile function and coronary angiogenesis
(Rota et al. 2005, Shiojima et al. 2005, Shiojima & Walsh 2006), and Akt has
cardiac hypertrophic (Oh et al. 1998) and antiapoptotic effects (Zhu et al. 2001).
hBNP gene transfer increased phosphorylation of Akt at 2 weeks’ post-infarction
(Fig. 18A). Moreover, hBNP gene transfer increased slightly, but nonsignificantly
Akt phosphorylation in Ang II–induced hypertension at 2 weeks (Fig. 18B).
Fig. 18. Effect of intramyocardial hBNP gene delivery on Akt phosphorylation. A, Akt
phosphorylation after acute myocardial infarction (AMI) in the LV at 1 week and 2
weeks. The results are expressed as mean±SEM (n = 6–7) *P < 0.05 versus sham; †P <
0.05 versus LacZ with AMI and B, In angiotensin II (Ang II)–induced hypertension.
Results are expressed as mean±SEM (n = 6–11) *P < 0.05 versus LacZ (1-way ANOVA
followed by LSD post hoc test).
5.3 Cardiac effects of direct (P)RR gene delivery (III)
5.3.1 Cardiac function in rats overexpressing (P)RR in the heart
Cardiac function in normal adult rats
The effects of (P)RR gene delivery on cardiac function were evaluated by
echocardiography. LV ejection fraction (Fig. 19A) and fractional shortening (Fig.
19B) deteriorated at 2 weeks and the interventricular septum diastolic (Fig. 19C)
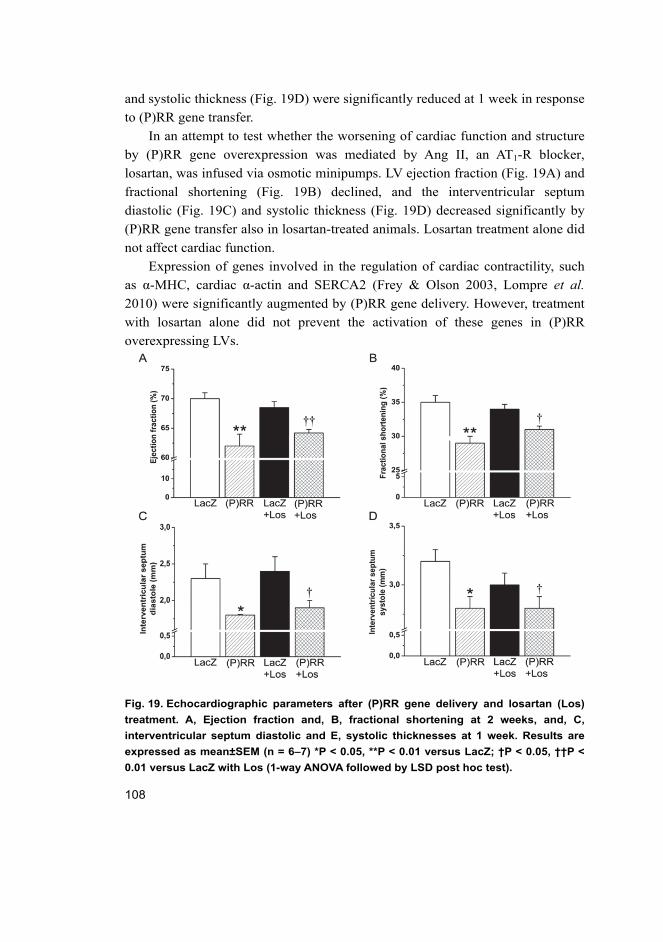
108
and systolic thickness (Fig. 19D) were significantly reduced at 1 week in response
to (P)RR gene transfer.
In an attempt to test whether the worsening of cardiac function and structure
by (P)RR gene overexpression was mediated by Ang II, an AT1-R blocker,
losartan, was infused via osmotic minipumps. LV ejection fraction (Fig. 19A) and
fractional shortening (Fig. 19B) declined, and the interventricular septum
diastolic (Fig. 19C) and systolic thickness (Fig. 19D) decreased significantly by
(P)RR gene transfer also in losartan-treated animals. Losartan treatment alone did
not affect cardiac function.
Expression of genes involved in the regulation of cardiac contractility, such
as α-MHC, cardiac α-actin and SERCA2 (Frey & Olson 2003, Lompre et al. 2010) were significantly augmented by (P)RR gene delivery. However, treatment
with losartan alone did not prevent the activation of these genes in (P)RR
overexpressing LVs.
Fig. 19. Echocardiographic parameters after (P)RR gene delivery and losartan (Los)
treatment. A, Ejection fraction and, B, fractional shortening at 2 weeks, and, C,
interventricular septum diastolic and E, systolic thicknesses at 1 week. Results are
expressed as mean±SEM (n = 6–7) *P < 0.05, **P < 0.01 versus LacZ; †P < 0.05, ††P <
0.01 versus LacZ with Los (1-way ANOVA followed by LSD post hoc test).

109
Cardiac function in post-infarction and in angiotensin II–induced hypertension in rats
Ligation of LAD caused a marked decrease in the ejection fraction (Fig. 20A) and
fractional shortening (Fig. 20B) within 2 weeks. (P)RR gene delivery had no
significant effect on LV ejection fraction and fractional shortening at 2 weeks’
post-infarction (Fig. 20A and 20B), and also LV versus body weight ratio did not
differ between (P)RR- and LacZ-treated hearts (Fig. 20C).
As post-infarction, (P)RR gene delivery had no effect on ejection fraction
(Fig. 20D) and fractional shortening (Fig. 20E) in Ang II–mediated hypertension,
and no significant differences in LV vs. body weight ratio between (P)RR- and
LacZ-treated groups were noted (Fig. 20F).
Fig. 20. Echocardiographic measurements were performed at 2 weeks’ post-infarction
and in angiotensin II (Ang II)‒induced hypertension in (P)RR‒treated hearts A, Ejection
fraction, B, fractional shortening and C, left ventricular (LV) weight-to-body weight
ratio. The results are expressed as mean±SEM (n = 5–7) **P < 0.01, ***P < 0.001 versus
sham. D, Ejection fraction, E, fractional shortening, F, LV weight-to-body weight ratio
in Ang II–induced hypertension. Results are expressed as mean±SEM (n = 7–9)
**P < 0.01, ***P < 0.001 versus LacZ (1-way ANOVA followed by LSD post hoc test).

110
5.3.2 Angiotensin II-independent and dependent effects triggered by (P)RR
Extracellular matrix remodelling in normal adult rat heart
Myocardial fibrosis increased significantly by (P)RR gene delivery at 1 week
(P < 0.05) and 2 weeks (P < 0.001) (Fig. 21A). Furthermore, (P)RR
overexpression significantly increased local LV expression of TGFβ1 (Fig. 21B)
and connective tissue growth factor (CTGF) (Fig. 21C). (P)RR gene transfer also
augmented the expression of other genes related to extracellular matrix
remodelling such as plasminogen inhibitor activator-1, collagen Iα1, fibronectin-
1, MMP-2 and MMP-9. Gelatin zymography analysis showed that MMP-2
protein levels (pro and active form) (Fig. 21D and 21F) were significantly
increased and MMP-9 (pro form) (Fig. 21E and 21F) slightly, but nonsignificantly
increased by (P)RR. Losartan treatment on its own did not prevent fibrosis or the
activation of pro-fibrotic and fibrosis–related genes in (P)RR overexpressing
hearts. However, the increased MMP-2 protein levels in losartan-treated (P)RR
overexpressing hearts were noted. In addition to pathological fibrosis, cell
proliferation and the size of cardiomyocytes were determined, but no differences
in the number of Ki-67+ cells and cardiomyocyte cross sectional area between
(P)RR and LacZ-treated groups were observed.
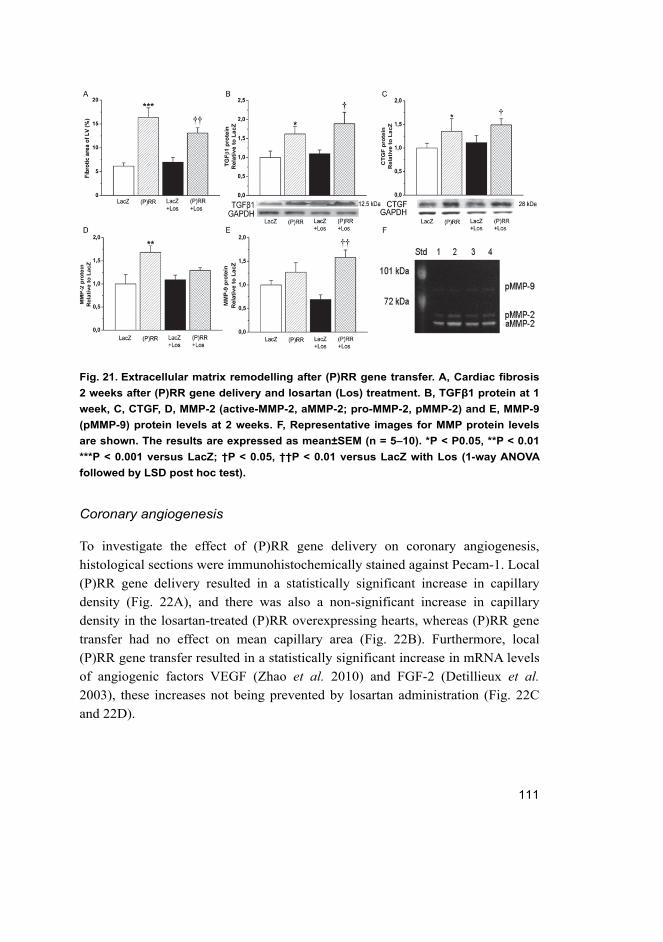
111
Fig. 21. Extracellular matrix remodelling after (P)RR gene transfer. A, Cardiac fibrosis
2 weeks after (P)RR gene delivery and losartan (Los) treatment. B, TGFβ1 protein at 1
week, C, CTGF, D, MMP-2 (active-MMP-2, aMMP-2; pro-MMP-2, pMMP-2) and E, MMP-9
(pMMP-9) protein levels at 2 weeks. F, Representative images for MMP protein levels
are shown. The results are expressed as mean±SEM (n = 5–10). *P < P0.05, **P < 0.01
***P < 0.001 versus LacZ; †P < 0.05, ††P < 0.01 versus LacZ with Los (1-way ANOVA
followed by LSD post hoc test).
Coronary angiogenesis
To investigate the effect of (P)RR gene delivery on coronary angiogenesis,
histological sections were immunohistochemically stained against Pecam-1. Local
(P)RR gene delivery resulted in a statistically significant increase in capillary
density (Fig. 22A), and there was also a non-significant increase in capillary
density in the losartan-treated (P)RR overexpressing hearts, whereas (P)RR gene
transfer had no effect on mean capillary area (Fig. 22B). Furthermore, local
(P)RR gene transfer resulted in a statistically significant increase in mRNA levels
of angiogenic factors VEGF (Zhao et al. 2010) and FGF-2 (Detillieux et al. 2003), these increases not being prevented by losartan administration (Fig. 22C
and 22D).
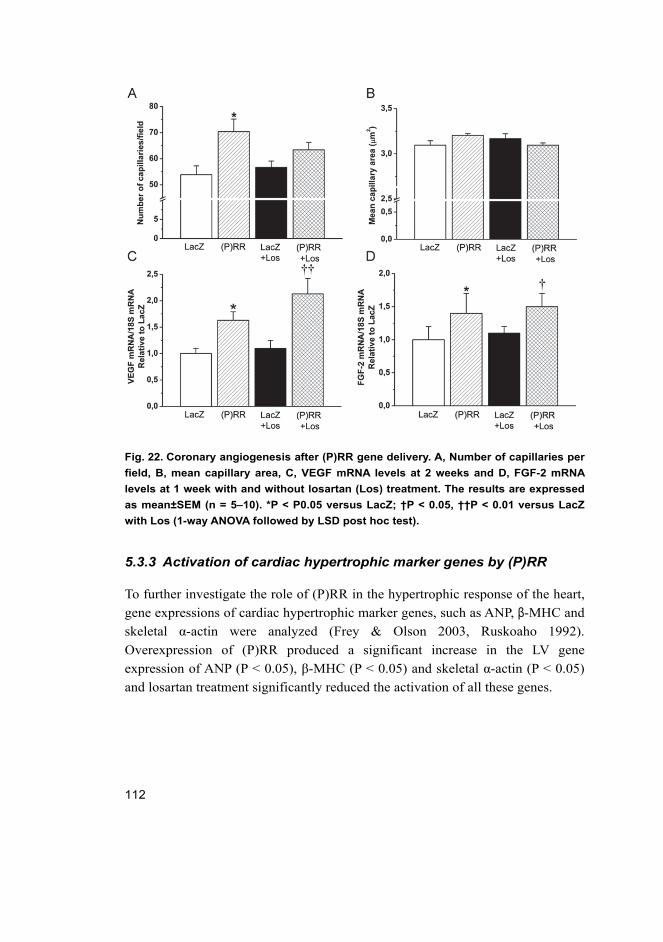
112
Fig. 22. Coronary angiogenesis after (P)RR gene delivery. A, Number of capillaries per
field, B, mean capillary area, C, VEGF mRNA levels at 2 weeks and D, FGF-2 mRNA
levels at 1 week with and without losartan (Los) treatment. The results are expressed
as mean±SEM (n = 5–10). *P < P0.05 versus LacZ; †P < 0.05, ††P < 0.01 versus LacZ
with Los (1-way ANOVA followed by LSD post hoc test).
5.3.3 Activation of cardiac hypertrophic marker genes by (P)RR
To further investigate the role of (P)RR in the hypertrophic response of the heart,
gene expressions of cardiac hypertrophic marker genes, such as ANP, β-MHC and
skeletal α-actin were analyzed (Frey & Olson 2003, Ruskoaho 1992).
Overexpression of (P)RR produced a significant increase in the LV gene
expression of ANP (P < 0.05), β-MHC (P < 0.05) and skeletal α-actin (P < 0.05)
and losartan treatment significantly reduced the activation of all these genes.

113
5.3.4 Activation of ERK1/2 and p38 MAPK/HSP27 pathways by (P)RR
Phosphorylation of ERK1/2 and p38 MAPK/HSP27 in normal adult rat
heart
In mesangial and vascular smooth muscle cells, the binding of prorenin to (P)RR
induced the phosphorylation of the ERK1/2 (Nguyen et al. 2002). (P)RR gene
delivery significantly increased ERK1/2 phosphorylation at 1 week (P < 0.05) and
at 2 weeks (P < 0.05). Infusion of losartan had no effect on the (P)RR gene
delivery–induced increase in ERK1/2 phosphorylation. (P)RR gene transfer also
increased p38 MAPK (P < 0.05) and HSP27 (P < 0.05) phosphorylation, the latter
being significantly attenuated by losartan treatment. (P)RR gene transfer
increased apoptotic cell death at 2 weeks (P < 0.01), which was significantly
reduced by losartan in (P)RR-overexpressing hearts.
Phosphorylation of ERK1/2 and p38 MAPK/HSP27 post-infarction and in angiotensin II–induced hypertension
The extents of phosphorylation of p38 MAPK, HSP27 and ERK1/2 were
significantly increased in (P)RR-treated hearts post-infarction (Fig. 23A through
23C). In contrast, in Ang II–induced hypertension, the phosphorylation levels of
p38 MAPK, HSP27 and ERK1/2 remained unchanged in (P)RR-treated hearts
(Fig. 23D through 23F).
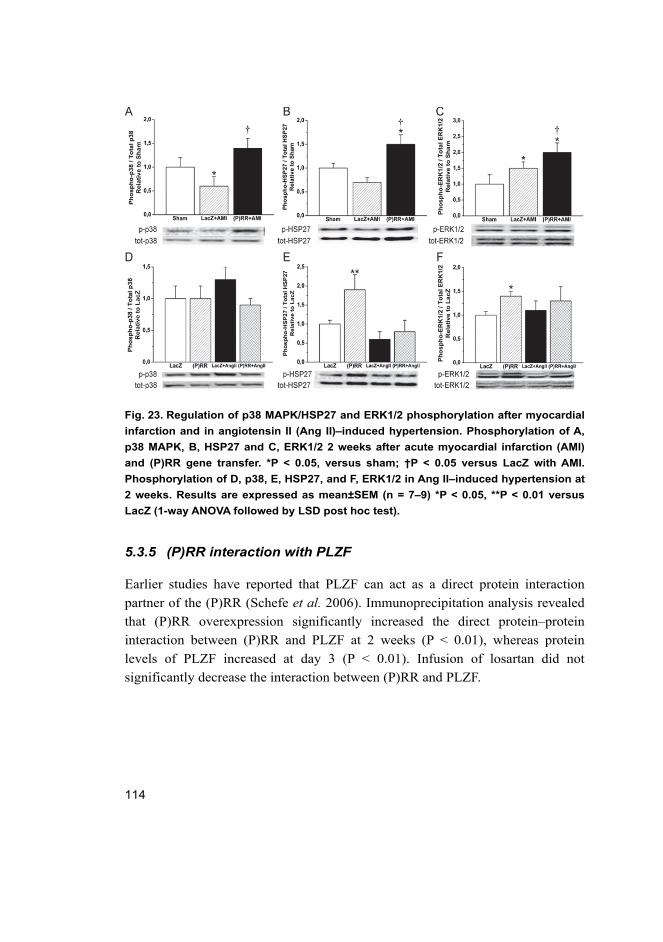
114
Fig. 23. Regulation of p38 MAPK/HSP27 and ERK1/2 phosphorylation after myocardial
infarction and in angiotensin II (Ang II)–induced hypertension. Phosphorylation of A,
p38 MAPK, B, HSP27 and C, ERK1/2 2 weeks after acute myocardial infarction (AMI)
and (P)RR gene transfer. *P < 0.05, versus sham; †P < 0.05 versus LacZ with AMI.
Phosphorylation of D, p38, E, HSP27, and F, ERK1/2 in Ang II–induced hypertension at
2 weeks. Results are expressed as mean±SEM (n = 7–9) *P < 0.05, **P < 0.01 versus
LacZ (1-way ANOVA followed by LSD post hoc test).
5.3.5 (P)RR interaction with PLZF
Earlier studies have reported that PLZF can act as a direct protein interaction
partner of the (P)RR (Schefe et al. 2006). Immunoprecipitation analysis revealed
that (P)RR overexpression significantly increased the direct protein–protein
interaction between (P)RR and PLZF at 2 weeks (P < 0.01), whereas protein
levels of PLZF increased at day 3 (P < 0.01). Infusion of losartan did not
significantly decrease the interaction between (P)RR and PLZF.

115
5.3.6 Wnt signalling and V-ATPase pathway after (P)RR gene transfer
(P)RR has been linked to Wnt signalling and V-ATPase (Advani et al. 2009,
Cruciat et al. 2010). However myocardial (P)RR gene delivery had no effects on
the protein levels of Wnt-3, β-catenin, Frizzled-8 and V-ATPase A1 in (P)RR-
treated hearts suggesting that (P)RR gene transfer did not influence Wnt
signalling or the V-ATPase pathways.

116

117
6 Discussion
6.1 Characterization of the efficiency of adenoviral–mediated gene
delivery (I and III)
Control genes can be used to assess the efficiency of gene delivery and to
standardize virus production. Commonly used controls for cardiac gene transfer
studies are an empty adenovirus, PBS-solution, or adenovirus carrying cDNA for
eGFP, LacZ or scrambled cDNA (O'Donnell 2012). eGFP has not been generally
used as a control for studies of gene transfer into the heart, because it is difficult
to distinguish authentic eGFP fluorescence from the endogenous background
fluorescence (O'Donnell 2012, Prasad et al. 2011). Moreover, Izumo & Shioi
(1998) showed that eGFP can evoke a contractile dysfunction in transgenic mouse
hearts in which eGFP has been expressed.
LacZ is the most commonly used control vector although some organs have a
low level of endogenous LacZ expression. Therefore it is important to separate
the endogenous signal in animals without viral gene transfer. In the heart, the
endogenous LacZ signal is insignificant. Hence, O’Donnell et al. (2008) observed
no signs of LacZ expression in the PBS-treated control rats.
Earlier studies have reported that adenoviral controls can affect cardiac
function. Weisser-Thomas et al. (2005) showed a significant reduction in
contraction amplitude in human cardiomyocytes after adenovirus–mediated LacZ
gene transfer when compared to the noninfarcted myocytes. Similarly Lafont et al. (1997) observed vasomotor dysfunction after adenoviral LacZ gene delivery in
rabbit arteries. On the other hand, others have reported that the LacZ vector does
not affect myocardial function such as systolic wall thickening assessed using
ultrasonic crystals (French et al. 1994).
In the present study, the efficiency of direct cardiac gene transfer was
confirmed by following the delivery and expression of the recombinant
adenovirus construct expressing LacZ gene under the control of the CMV
promoter. Infection with the adenovirus construct expressing LacZ resulted in a
highly efficient and homogenous expression of β-galactosidase throughout the LV.
Importantly, an earlier study had demonstrated that infarcted rat hearts showed the
same relative levels of expression as normal rat hearts (Tenhunen et al. 2006b).
Moreover, intramyocardial injection of the adenoviral LacZ construct resulted in a
nonsignificant local response with increased myocardial fibrosis. Although

118
intramyocardial LacZ injection was associated with fibrosis close to the injection
site, the present results indicate that the effects of LacZ transfection did not differ
from injection of PBS-based buffer, suggesting that the mechanical distension
caused by fluid is mainly responsible for the fibrotic effect seen at the injection
site.
6.1.1 Immune reaction and inflammatory response following adenovirus–mediated gene delivery (I)
Adenoviral vectors are highly immunogenic and these immune reactions pose a
significant obstacle to the use of adenoviral vectors in cardiovascular gene
therapy (Tilemann et al. 2012). The immunogenity of adenoviral vectors limits
the effective period of adenoviral based gene delivery techniques (Schagen et al. 2004). Approximately 97% of the population forms neutralizing antibodies
against type C adenoviruses, which include the commonly used adenovirus
serotype 2 and serotype 5 (Nayak & Herzog 2010). AAV-delivery vectors have
less cytotoxic effects (Tilemann et al. 2012). However, a less cytotoxic effects
also implies that the infection efficiency remains lower and higher doses of AAVs
are required for gene delivery, which increases the possibility of toxic effects
(Weisser-Thomas et al. 2005).
Adenovirus infection leads to a dose-dependent inflammatory reaction
followed by cell death. The inflammatory response is dependent on species and
the development stage of the myocyte (Schagen et al. 2004). In the present study,
adenoviral gene transfer resulted a notable inflammatory response within the
normal heart. This indicates that for functional and cytotoxic reasons, the virus
dose needs to be optimized to maintain normal cell function and survival. MI also
evokes a significant inflammatory response and neurohumoral activation (Sutton
& Sharpe 2000). However, in the present study, the number of infiltrating cells in
hBNP injected hearts was similar to that in LacZ treated normal hearts and in
hearts after MI.
In addition to adenovirus specific antibodies, neutralizing antibodies might be
generated against the transgene product. Transgene specific antibodies neutralize
the transgene product when it enters the circulation and thus the effects of gene
transfer are attenuated (Schagen et al. 2004). Several studies have reported that
the immunogenicity of transgene proteins is a primary determinant of the
temporal transgene expression (Connelly et al. 1996, Fields et al. 2000, Tripathy
et al. 1996). In the present study, local myocardial BNP gene transfer increased

119
the levels of neutralizing BNP antibodies by about 17–fold at 2 weeks. This
increase probably attenuated the effect and persistence of BNP expression. The
strong immune response could, at least partly, be a consequence of differences in
the molecular structure of BNP between species. It has been postulated that the
immune response against the transgene product is dependent on the nature of the
mutation that affects the endogenous state (Goodeve & Peake 2003, Schagen et al. 2004). Thus, although the human BNP gene transfer into the rat hearts made it
possible to separate the exogenous BNP from endogenous rat BNP, it is possible
that structural differences between hBNP and rat BNP were responsible for the
immune response and this attenuated the effect of BNP gene delivery. An earlier
study has also demonstrated that persistent expression of a foreign antigen may
even induce tolerance to the therapeutic antigen (Mingozzi et al. 2003), which is a
major potential problem, particularly in human gene therapy studies and clinical
trials.
Although hBNP gene transfer resulted in a significant immune response
against the transgene product, it is interesting to note that in plasma of hBNP
treated rats, immunoreactive material corresponding exactly to the size of native
circulating form human BNP-32 was observed, indicating that human proBNP
can be processed correctly in the rat cardiomyocytes. A recent study in rats has
also demonstrated that injected human proBNP was effectively processed in the
circulation into BNP-32 (Semenov et al. 2011). Mature hBNP consists of 32
amino acids (Sudoh et al. 1988) whereas rat BNP has 45 amino acids (Aburaya et al. 1989, Kambayashi et al. 1989). Nevertheless, the sites of proBNP processing
are rather similar in humans and rats. In both cases, arginine residues are located
in the same positions and these arginines are considered to be essential for
substrate recognition by subtilisin-like proprotein convertases. Human proBNP is
cleaved in rats at the site which is recognized by furin, pointing to the
involvement of subtilisin-like proprotein convertases (Remacle et al. 2008,
Semenov et al. 2011). Hence, the data from study I indicate that in rats BNP-
specific receptors are able to recognize exogenous human BNP, mainly because of
the high homology between human and rat BNPs.
6.2 BNP as a therapeutic target for the treatment of heart failure (I)
BNP has been used as a marker of ventricular loading, which results from volume
overload and stretch of myocytes. BNP levels are elevated during heart failure

120
and a reduction of these levels in patients with decompensated heart failure is
considered as a marker of clinical improvement (Boerrigter & Burnett 2004).
Synthetic natriuretic peptides have been developed for use in HF. Human
recombinant ANP was first approved for the clinical management of acute
decompensated congestive HF in Japan in the year 1995. Exogenous natriuretic
peptide in the form of recombinant human BNP, nesiritide, has been approved for
the treatment of decompensated heart failure since 2001. Exogenous BNP
improves hemodynamic properties in patients with HF, although its effect on
clinical outcomes is still unknown. The use of nesiritide has also been limited by
the appearance of hypotension and its tendency to worse renal function (Chen &
Burnett 2006, Moe 2006). Thus, a meta-analysis of the clinical trials suggested
that nesiritide might be detrimental to renal function in patients with acute
decompensated HF (Sackner-Bernstein et al. 2005). Earlier studies have reported
that nesiritide could decrease levels of some endogenous hormones, such as
noradrenaline, renin, aldosterone and endothelin-1, which are abnormally
elevated in cases of heart failure (Abraham et al. 1998, Yoshimura et al. 1991).
NPs elicit their biological effects by activation of the cGMP-cGMP‒
dependent protein kinase pathway. The effects of NPs are mediated through their
GC-A receptor (Lee & Burnett 2007, Potter et al. 2009). The major target receptor
for BNP is GC-A, the rank order of GC-A activation by NPs being
ANP≥BNP>>CNP (Koller et al. 1991, Suga et al. 1992) in homologous assay
systems with endogenous ligands and receptors of the same species. In the heart,
GC-A is expressed in cardiac myocytes, fibroblast and endothelial cells (Cao &
Gardner 1995, Lin et al. 1995), and the potency of BNPs for cGMP production
varies from cell to cell. Marked species differences exist in the potencies of the
BNP to activate for cGMP production e.g. the potency of BNP to stimulate cGMP
production via the biologically active receptor depends not only on subtypes of
the biologically activated receptor, but also on a species difference in the
molecular structure of the receptors (Potter et al. 2009, Suga et al. 1992).
Importantly, cGMP production can be activated by hBNP in rat cells; for
example, hBNP-32 added to purified rat ventricular myocytes resulted in a
marked accumulation of cGMP, similar to rat ANP (Lin et al. 1995). In contrast,
in rat aortic smooth muscle cells (where the majority of the receptors are GC-B),
hBNP was 6–fold less potent than rat BNP in evoking cGMP production (Suga et al. 1992). In view of the fact that in frozen cardiac sections, BNP binding sites are
localized to the endothelium of the endomural channels and endocardium
(Oehlenschlager et al. 1989), it is noteworthy that in bovine endothelial cells,

121
hBNP is an even more potent activator of cGMP production than rat BNP (Suga et al. 1992). In primary cultures of neonatal rat cardiac fibroblasts, in which both
GG-A and GC-B are expressed, porcine BNP-32 (a structure very similar to
hBNP-32) inhibited growth factor dependent [3H]thymidine incorporation, this
effect being enhanced by phosphodiesterase inhibition (Cao & Gardner 1995).
The ability of hBNP to stimulate cGMP production in rat cells has been explained
by the presence of a minimum bioactive unit homologous to hBNP-32 at the
carboxy-terminus of rat BNP (Aburaya et al. 1989, Kambayashi et al. 1989).
In the present study, increased cGMP production and decreased synthesis of
endogenous NPs in the LV of rat heart demonstrated that BNP was acting locally
within the heart. Although, hBNP levels were significantly upregulated for up to 2
weeks, both hBNP mRNA and peptide levels were highest at day 3 after
injections and declined thereafter during the 2‒week follow-up period. Thus, it is
possible that the upregulation of hBNP expression preceded the decreased
endogenous rat BNP peptide levels, reflecting the local action of BNP gene
transfer. As discussed above, this could be mediated via GC-A–induced cGMP
production.
Transgenic models have suggested that, in part, the cardiac effects of BNP are
mediated through its systemic hemodynamic actions (Kawakami et al. 2004,
Lopez et al. 1995, Oliver et al. 1997, Tamura et al. 2000). In the present study,
plasma levels of BNP doubled following BNP gene delivery and exogenous
human proBNP was processed correctly in rat cardiomyocytes. Nevertheless,
BNP gene delivery did not affect blood pressure.
Study I detected the presence of numerous BNP granula–like structures after
BNP gene delivery as compared to control hearts. In contrast to ANP, cardiac
BNP does not appear to be stored to the same extent and ventricular myocytes
secrete BNP mainly in a constitutive fashion. However, the presence of BNP
containing granules has been described in surgical and autoptic tissue specimens
of human heart examined with a double immunogold technique (Nakamura et al. 1991). In the atrial myocytes, ANP was localized in almost all of the secretory
granules, whereas BNP was colocalized with ANP in some of the granules.
Although, very few secretory granules were observed in ventricular myocytes, the
colocalization of ANP and BNP was basically the same as in atrial myocytes
(Nakamura et al. 1991). Tanaka et al. (1994) also reported that ANP-positive
myocytes in the LV also showed BNP immunoreactivity and the distributions of
ANP- and BNP-positive myocytes were almost identical in all dilated
cardiomyopathy cases. The number of ANP- and BNP-positive myocytes was

122
smaller in the LV than in the right atria, and the positively stained granules in the
LV were scattered throughout the myocyte cytosol. Interestingly, neither ANP nor
BNP immunoreactivity was found in the LV myocytes of control hearts. These
results suggest that BNP is stored in some of the granules with ANP both in atrial
and ventricular tissues, particularly when peptide levels are high (Tanaka et al. 1994). In agreement with this hypothesis, in the present study, BNP containing
granules were detected in ventricles after overexpression of hBNP, while staining
was diffuse in LacZ-injected hearts, suggesting that because of the high BNP
concentration following BNP gene delivery, hBNP was stored to some extent in
granules.
Endothelin-1 is a well-characterized vasoactive peptide, which has inotropic
and chronotropic effects in the heart (Yamada & Yoshida 1991). Endothelin-1 has
been reported to stimulate the production of VEGF and FGF-2 and to be involved
in the effects of TGFβ and platelet-derived growth factor (Luscher & Barton
2000). In the current study, the level of endothelin-1 gene expression was
increased, reflecting its neurohumoral activation following BNP gene delivery.
6.2.1 Antifibrotic and angiogenic effects of BNP gene delivery in
normal heart
Myocardial fibrosis is of crucial importance in HF, because it results in decreased
capillary density and an increased oxygen diffusion distance which in turn causes
hypoxia and dysfunction of the myocytes surrounded by collagen (Petrovic 2004).
In the present study, the local increase in LV BNP peptide levels resulted in a
reduction in myocardial fibrosis in the healthy heart. Earlier studies have revealed
that BNP can influence the regulation of myocardial fibrosis. In cultured
fibroblasts, BNP decreased collagen synthesis and increased MMPs (Tsuruda et al. 2002). Transgenic mice overexpressing BNP targeted to the liver increased
MMP-9 expression in the infarcted area after MI. Moreover, targeted deletion of
the GC-A gene resulted in cardiac hypertrophy and fibrosis (Lopez et al. 1995,
Oliver et al. 1997), whereas BNP knockout mice had a normal heart size, but
increased amounts of ventricular fibrosis (Tamura et al. 2000).
Previous studies have also associated the NP/GC-A system to the stimulation
of angiogenesis. BNP/GC-A stimulates the proliferation and migration of cultured
microvascular endothelial cells by activating cGMP-dependent protein kinase I
(Kuhn et al. 2009). Yamahara et al. (2003) demonstrated that in transgenic mice
overexpressing BNP in response to hindlimb ischemia, neovascularization with

123
the appropriate mural cell coating was accelerated. Moreover, a selective
disruption of the endothelial GC-A evoked diminished angiogenesis in a model of
pressure overload–induced hypertrophy (Kuhn et al. 2009). The present study
identified BNP as a coronary angiogenic factor. BNP gene delivery resulted in an
increase in capillary density, but the capillary area remained unchanged. BNP is
induced in response to hemodynamic load (Ruskoaho 2003) and this stretch–
induced activation of BNP expression is mediated by GATA (Pikkarainen et al. 2003). GATA-4 has also been identified as a regulator of coronary angiogenesis
(Heineke et al. 2007, Rysa et al. 2010). One can hypothesize that BNP is acting
locally as a major mechanical load–activated and GATA-4–activated regulator of
angiogenesis.
The angiogenesis induced by BNP was associated with increased FGF-2 gene
expression. FGF-2 is activated by inflammation, but has long been known also to
stimulate proliferation of cultured mesenchymal cells such as fibroblasts,
endothelial cells, smooth muscle cells, and skeletal myoblasts. This growth factor
is also involved in the regulation of cell survival, migration, and matrix
production and degradation (Detillieux et al. 2003). Furthermore, FGF-2 has been
studied extensively for its ability to promote angiogenesis in models of chronic
ischemia. Virag et al. 2007 used FGF-2 knockout and overexpressing transgenic
FGF-2 mice to determine the role of FGF-2 in myocardial infarct repair. Vascular
density declined significantly after infarction in the hearts of both wild type and
FGF-2 knockout animals, but this decline was more dramatic in the FGF-2
knockout group. In the hearts of mice overexpressing FGF-2, there was
significantly increased endothelial proliferation detected at 2 days and 4 days
after MI. Thus, FGF-2 is also a strong cardioprotective and angiogenic mediator.
As discussed in chapter 6.1.2, in the present study the number of infiltrating cells
in hBNP injected hearts was similar to that found in LacZ-injected hearts. This
finding supports the concept that it is direct effects of NPs, and not secondary
proinflammatory effects, that are involved in the angiogenesis.
6.2.2 Functional role of BNP after myocardial infarction
Study I demonstrated that BNP gene delivery improved LV systolic function after
MI. The loss of functional capillaries and microvessels is a critical determinant of
myocardial remodelling (Walsh & Shiojima 2007). However, in the present study,
the capillary density and capillary area remained unchanged. Furthermore, several
other factors such as cardiac fibrosis, apoptotic cell death, cell proliferation and

124
cardiac stem cell recruitment contributing to the structural and functional
remodelling of infarcted myocardium (Dorn 2009, Yi et al. 2010) remained
unchanged in BNP treated hearts. Therefore, a number of potential mechanisms
triggering the improvement of LV function by BNP gene transfer after MI were
studied. It has been proposed that the members of MAPK family are involved in
several pathophysiological processes, such as hypertrophy, heart failure,
ischemia/reperfusion and cardioprotective responses in the heart (Ravingerova et al. 2003). Earlier studies have reported that BNP inhibits fibrotic response
through ERK signalling (Kapoun et al. 2004) and that p38 MAPK is the limiting
factor to increase contractility through phosphorylation of PLB in the
myocardium (Szokodi et al. 2008). In the present study, BNP overexpression did
not influence p38 MAPK and ERK1/2 phosphorylations suggesting that BNP
gene transfer did not influence cardiac function via MAPK pathways.
SERCA2 plays a crucial and central role by being the core of Ca2+ cycling in
the cardiomyocyte (Chaanine et al. 2010). Decreased SERCA2 expression is
observed consistently in HF (Hasenfuss 1998). SERCA2 enzymatic activity is
controlled by the inhibitory peptide, PLB (Rapti et al. 2011). In failing hearts, the
levels of phosphorylation of PLB at Ser16 and Thr17 are reduced (Dash et al. 2001, Schwinger et al. 1999). Data from study I demonstrated that the
improvement of cardiac function may be related to the normalization of SERCA2
levels. As a consequence, BNP increased PLB phosphorylation of Thr17 but not
Ser16. Earlier studies have revealed that SERCA2 overexpression restores cardiac
contractile function in the rat ischemic HF model (del Monte et al. 2004).
Furthermore, a previous study has shown that in intact cardiac myocytes Thr17
phosphorylation by CamKII occurs in the absence of Ser16 phosphorylation
(Hagemann et al. 2000) and inhibition of PLB resulted in improved contractility,
reversal of adverse remodelling and decrease in fibrosis in HF model (Suckau et al. 2009). The effect of hBNP gene delivery on phosphorylation of Thr17 residue
of PLB may be indirect, because phosphorylation of PLB by cGMP-dependent
protein kinase occurs at Ser16 (Lompre et al. 2010). There is no evidence that
BNP can activate CaMKII directly in cardiac myocytes. The upstream signalling
pathways which ultimately activate CaMKII remain an important question for
future studies.
In the heart, Akt activity regulates cardiac growth, contractile function and
coronary angiogenesis. A recent study of Akt-/- knockout mice revealed defective
exercise–induced hypertrophy but Akt2-/- knockout mice did not show this
phenotype (Hers et al. 2011). Transgenic animal models using cardiac specific

125
inducible Akt, demonstrated that short-term expression of Akt1 led to
physiological hypertrophy and longer term Akt activation resulted in hypertrophy
with contractile dysfunction (Hers et al. 2011, Shiojima et al. 2005). In the
present study, BNP overexpression normalized Akt phosphorylation after MI.
Moreover, β-MHC and skeletal α-actin gene expressions were increased by BNP
gene delivery after MI supporting the improvement of contractile function.
To summarize the data of study I, the favourable effects of BNP
overexpression on cardiac function after MI appears to be mediated through
normalization of SERCA2 expression and phosphorylation of PLB restoring
cardiomyocyte function by improving Ca2+ uptake into the SR. Moreover,
normalization of Akt signalling may also have favourable effects on cardiac
function after MI.
6.2.3 Effects of BNP gene delivery in an experimental model of angiotensin II–mediated hypertension
The RAA system, through the production Ang II and activation of its AT1-R
triggers collagen production and cardiac fibroblast proliferation, thus enhancing
fibrotic myocardial remodelling in the failing heart (Brown et al. 2005). In an
experimental model of Ang II–mediated hypertension, BNP gene transfer
significantly improved systolic function. Moreover, myocardial fibrosis as well as
collagen gene expression were decreased, whereas no differences in angiogenesis
were observed between BNP-treated hearts and control hearts. Furthermore,
SERCA2, PLB, MAPKs or Akt signalling were not changed by BNP gene
delivery, suggesting that antifibrosis is the main factor that mediates its
favourable effects in Ang II–mediated hypertension.
6.2.4 Context-dependent effects of BNP in the heart
The finding that BNP exerts unique, context-dependent (i.e. differences between
experimental models) favourable actions in the heart is very interesting. For
example, because overexpression of BNP improved LV fractional shortening and
ejection fraction post-infarction, then one can postulate that numerous potential
mechanisms could contribute to the structural and functional remodelling of
infarcted myocardium. However, myocardial angiogenesis, cardiac fibrosis,
apoptotic cell death, cell proliferation and cardiac stem cell recruitment remained
unchanged after BNP gene delivery, whereas in healthy heart and Ang II–induced

126
hypertension the local increase in LV BNP peptide levels by gene delivery caused
antifibrotic effects (Fig. 24).
The context-dependent effects might be dependent on distinct
pathophysiological processes in MI and hypertension–induced heart disease. In
agreement with this hypothesis, an earlier study reported that in the normal adult
rat heart, the physiological consequences of p38 MAPK overexpression were
cardiac cell proliferation and myocardial inflammation associated with fibrosis
(Tenhunen et al. 2006a), while normalization of the reduced p38 MAPK activity
prevented the adverse post-infarction remodelling through a distinct angiogenic
and antiapoptotic mechanism (Tenhunen et al. 2006b). Accordingly, the activation
or inhibition of cGMP-dependent protein kinase substrates by BNP gene transfer
may be context-dependent. Further studies, perhaps involving systematic
silencing of BNP-activated signalling molecules via lentiviral vectors, antibodies
or small molecule inhibitors in rats and conditional, doxycyline-inducible
transgenic mice in combination with intramyocardial BNP gene transfer, will be
needed to clarify this possibility and to reveal other potential mechanisms.
The aim of study I was to examine the effect of BNP gene delivery during the
early LV remodelling period (0‒2 weeks) after MI and in Ang II–induced
hypertension. The major structural and functional changes encpuntered in the
remodelling process occurred between day 0 and 14. Moreover, measurements of
fibrosis, angiogenesis, cell proliferation and c-kit+ cells as well as gene
expression and protein analyses revealed significant changes within 2 weeks.
Therefore, the functional and structural consequences of BNP gene delivery were
only studied up to 2 weeks. Although the effects at 2 weeks may represent long-
term functional and structural effects, no longer-term effects were evaluated.
Therefore, it is not known if survival after infarction can be improved by
intramyocardial BNP gene delivery.
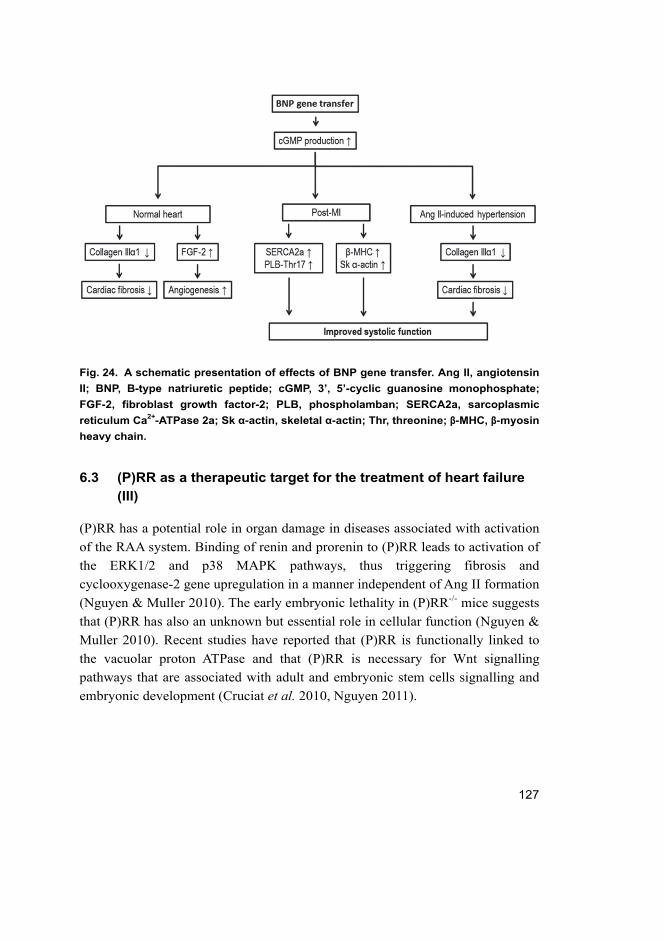
127
Fig. 24. A schematic presentation of effects of BNP gene transfer. Ang II, angiotensin
II; BNP, B-type natriuretic peptide; cGMP, 3’, 5’-cyclic guanosine monophosphate;
FGF-2, fibroblast growth factor-2; PLB, phospholamban; SERCA2a, sarcoplasmic
reticulum Ca2+-ATPase 2a; Sk α-actin, skeletal α-actin; Thr, threonine; β-MHC, β-myosin
heavy chain.
6.3 (P)RR as a therapeutic target for the treatment of heart failure (III)
(P)RR has a potential role in organ damage in diseases associated with activation
of the RAA system. Binding of renin and prorenin to (P)RR leads to activation of
the ERK1/2 and p38 MAPK pathways, thus triggering fibrosis and
cyclooxygenase-2 gene upregulation in a manner independent of Ang II formation
(Nguyen & Muller 2010). The early embryonic lethality in (P)RR-/- mice suggests
that (P)RR has also an unknown but essential role in cellular function (Nguyen &
Muller 2010). Recent studies have reported that (P)RR is functionally linked to
the vacuolar proton ATPase and that (P)RR is necessary for Wnt signalling
pathways that are associated with adult and embryonic stem cells signalling and
embryonic development (Cruciat et al. 2010, Nguyen 2011).

128
6.3.1 Functional effects in normal heart triggered by (P)RR
Data from study III revealed a deterioration of cardiac function and a reduction in
the interventricular septum diastolic and systolic thicknesses triggered by (P)RR
gene delivery. Infusion of an AT1-R blocker, losartan, did not prevent the
deterioration in cardiac function or the decrease in interventricular septum
systolic and diastolic thicknesses indicating that (P)RR induced an impairment of
LV function independent of Ang II generation. Cardiac contractility markers (Frey
& Olson 2003, Lompre et al. 2010) α-MHC, cardiac α-actin and SERCA2 gene
expressions were augmented by (P)RR treatment.
6.3.2 Activation of extracellular matrix remodelling
The cardiac remodelling process can be considered to consist of several
molecular, cellular and extracellular responses that lead to changes in LV size,
shape, dilatation and function. The major constituents of the extracellular matrix
are the fibrillar collagens I and III with lesser amounts of other compounds e.g.
collagens IV, V, VI, elastin, laminin, proteoglycans and glycosaminoglycans.
Collagens are degraded by specific collagenases, MMPs, which are activated by
extracellular Ser proteinases (Brown et al. 2005).
In study III, (P)RR gene delivery in the normal adult rat heart induced
myocardial fibrosis associated with increased TGFβ1 and CTGF expression.
Furthermore, collagen I, PAI-1 and fibronectin-1 and collagenolytic enzymes,
such as MMP-2 and MMP-9 gene expressions were increased indicating that the
hearts were undergoing a fibrotic remodelling process. Activation of MMPs is
associated with the appearance LV dilatation (Brown et al. 2005), which possibly
explains the reduced interventricular septum diastolic and systolic thicknesses.
Myocardial fibrosis and increased fibrotic and MMP gene expression were not
prevented by an infusion of losartan indicating that PRR is stimulating the
myocardial extracellular matrix remodelling independent of Ang II generation. In
summary, study III suggested that Ang II-independent alteration in the
extracellular matrix and contractile genes are mainly responsible for the
deterioration of cardiac function by (P)RR gene delivery.

129
6.3.3 Angiogenetic and apoptotic responses
Prorenin and the (P)RR may be involved in the development of some serious
ocular diseases, since nonproteolytic activation of protein mediated (P)RR is
associated with retinal neovascularization in animal models (Satofuka et al. 2007). In the current study, coronary angiogenesis was increased after (P)RR gene
delivery. The myocardial capillary density was increased and a slight but
statistically nonsignificant increase was observed also in the presence of losartan
treatment. (P)RR–induced angiogenesis was associated with an increase in the
expressions of angiogenic factors, FGF-2 and VEGF genes, which were also
independent of Ang II generation.
The primary substrate of p38 MAPK is MAPK activated protein kinase,
which phosphorylates HSP27. HSP27 promotes the polymerization of actin
filaments and maintains the integrity of the cytoskeleton, and thus it influences
cell growth, motility, survival and death (Ravingerova et al. 2003, Whelan et al. 2010). In the present study, (P)RR gene transfer increased apoptotic cell death, an
effect which could be reduced by losartan treatment. Consistent with this
observation, HSP27 phosphorylation was activated by (P)RR and losartan
significantly attenuated the phosphorylation of HSP27, indicating that the
HSP27–mediated apoptosis triggered by (P)RR is at least partly dependent on
Ang II.
6.3.4 Hypertrophic stimuli activated by (P)RR gene transfer
Several genes for transcriptional factors, NPs (ANP and BNP), sarcomeric
proteins (β-MHC, smooth muscle and skeletal α-actins and myosin light chains 1a
and 2a) and growth factors are induced and regulated by hypertrophic stimuli
(Sutton & Sharpe 2000). In the present study, (P)RR gene delivery induced a
distinct activation of the downstream genes involved in cardiomyocyte
hypertrophy. Ang II-dependent activation was demonstrated in several
pathological hypertrophy associated genes: ANP, β-MHC and skeletal α-actin.
6.3.5 ERK1/2 pathway activation by (P)RR
Activation of the ERK subfamily occurs in response to mitogenic and growth
factors acting through receptor protein kinases or GPCR. Moreover, this
activation process is associated with physical stress (Ravingerova et al. 2003). In

130
most cell types (P)RR activates ERK1/2 phosphorylation. Earlier studies reported
that ERK1/2 phosphorylation can be observed in the presence of AT1-R and AT2-
R antagonist, indicating that the ERK activation is independent of Ang II
generation (Huang et al. 2006, Huang et al. 2007, Nguyen & Contrepas 2008). In
the present study, infusion of losartan had no effect on the (P)RR gene delivery
induced increase in ERK1/2 phosphorylation supporting the findings that
activation of the ERK1/2 pathway by (P)RR is independent of Ang II generation.
6.3.6 PLZF interaction with (P)RR
The (P)RR signal transduction pathway involves a direct protein–protein
interaction between (P)RR and the transcription factor PLZF and the nuclear
translocation of PLZF upon renin stimulation. This interaction is believed to exert
cellular effects, because renin stimulation induced an increase in proliferation and
a decrease in apoptotic activity in rat cardiomyocytes. PLZF is also described as
an adaptor protein of AT2-R, mediating the antifibrotic and antiproliferative
effects of Ang II (Schefe et al. 2006, Schefe et al. 2008). In the present study,
PLZF protein levels were increased by (P)RR gene delivery and the interaction
between (P)RR and PLZF was accentuated, suggesting stronger repression of
(P)RR transcription. The interaction between (P)RR and PLZF was not attenuated
by losartan infusion, indicating that this interaction is independent of Ang II
generation.
6.3.7 Effects of (P)RR gene delivery to Wnt signalling
(P)RR is a component of the Wnt receptor complex. Cruciat et al. (2010) reported
that (P)RR acts in a renin-independent manner as an adaptor between Wnt-
receptors and the V-ATPase complex. The present study demonstrated that local
(P)RR gene transfer did not affect Wnt-3, β-catenin, Frizzled-8 and V-ATPase A1
protein levels, evidence that the (P)RR induced myocardial effects are not
mediated via the Wnt/β-catenin signalling pathway.
6.3.8 Role of (P)RR gene delivery in experimental models of MI and
angiotensin II–mediated hypertension
Increased (P)RR expression has been reported in rodent models of HF and in the
failing human heart (Mahmud et al. 2011). In the present study (P)RR gene

131
delivery did not affect cardiac function after MI, even though the level of
phosphorylation of p38 MAPK/HSP27 as well as ERK1/2 pathways was
increased. In Ang II–induced hypertension, there were no differences in the
hemodynamic parameters between (P)RR-treated and LacZ-treated hearts.
Moreover, phosphorylation of p38 MAPK/HSP27 and ERK1/2 pathways did not
change after (P)RR gene transfer.
It has been suggested that different degrees of (P)RR transgene expression
might account for distinct phenotypes (Nguyen & Muller 2010). It is possible that
the dose of (P)RR used in the present experiments was not sufficiently large to
trigger functional changes after MI or in Ang II–induced hypertension, although
significant changes in phosphorylation of MAPKs were observed. In a transgenic
model overexpressing human (P)RR gene in smooth muscle cells, it was
demonstrated that the vascular expression of the transgene was barely elevated in
the kidney and thus, proteinuria and glomerulosclerosis were not induced
(Burckle et al. 2006). Moreover, significant Ang II generation resulting from
(pro)renin-(P)RR interaction occurred at lower (pro)renin levels than direct
ERK1/2 activation suggesting that signalling derived from (pro)renin–(P)RR
interaction may be concentration-dependent (Batenburg et al. 2011).
6.3.9 (P)RR as a multifunctional protein
Recent studies of (P)RR have allowed a better understanding of the complex
biochemical mechanisms of (P)RR, although the role of (P)RR in diseases is
unclear because of the absence of specific (P)RR antagonist and (P)RR knockout
mice. Moreover, the unexpected properties of (P)RR have hinted that some (P)RR
functions might not be mediated via the RAA system. For instance, it has been
revealed that (P)RR gene expression starts very early in development, whereas the
expression of renin is detected in large intrarenal arteries only at 15.5 days of
gestation (Cousin et al. 2010). Study III revealed that (P)RR gene delivery caused
deleterious effects on cardiac function. Local cardiac (P)RR gene delivery
resulted in Ang II-independent activation of ERK1/2 phosphorylation and
myocardial fibrosis. In contrast, apoptotic cell death seen in that experiment was
Ang II-dependent (Fig. 25). These results implicate that (P)RR blockers may
possess significant cardiac effects in addition to effective RAA system blockade,
because activation of (P)RR induced a distinct Ang II-independent extracellular
matrix remodelling and evoked a deterioration of cardiac function. It is notable
that (P)RR triggered activations could also be mediated through AT2-R, which is

132
expressed at low levels in the normal heart and thus the importance of this
receptor in mediating (P)RR related actions remains to be clarified.
Fig. 25. A schematic presentation of angiotensin II (Ang II) dependent and Ang II-
independent (P)RR pathways. ANP, atrial natriuretic peptide; Caα-A, cardiac α-actin;
Col I, Collagen Iα1; CTGF, connective tissue growth factor; ERK1/2, extracellular
signal regulated kinase; FGF, fibroblast growth factor; Fn-1, fibronectin-1; HSP27, heat
shock protein 27; MHC, myosin heavy chain; MMP, matrix metalloproteinase; p,
phosphorylated; PAI-1, plasminogen activator inhibitor-1; SERCA2, sarcoplasmic
reticulum Ca2+-ATPase 2; Sk α-actin, skeletal α-actin; TGFβ1, transforming growth
factor β1; VEGF, vascular endothelial growth factor.

133
7 Summary and conclusions
The present results indicate that adenoviral gene transfer is an efficient method to
identify novel targets for the treatment of heart failure and can help to elucidate
the molecular basis of cardiac diseases. Direct myocardial effects of BNP and
(P)RR on cardiac function were examined by using adenovirus–mediated gene
delivery in normal heart, during increased blood pressure and post-infarction
remodelling. In addition, the mechanisms underlying the actions of BNP and
(P)RR delivery were evaluated and the downstream targets of these genes
investigated. The main findings of the study can be summarized as follows:
1. Local hBNP gene delivery into the rat adult heart improved LV contractility
during the remodelling process both after infarction and in a model of
pressure overload. The favourable effect of BNP on cardiac function after
infarction was associated with normalization of SERCA2 expression and
Thr17 phosphorylation of PLB, and Akt signalling.
2. In normal heart, the local increase in LV hBNP peptide levels after gene
delivery was antifibrotic and angiogenic without affecting systolic function.
Moreover, Ang II–induced fibrosis was attenuated by hBNP overexpression.
Consistent with this proposal, collagen expression was decreased by hBNP
gene transfer.
3. Analyses of molecular forms of BNP after intramyocardial gene transfer
revealed that there was immunoreactive material in the circulation
corresponding to the size of the native circulating form of human BNP-32.
LV tissue contained high-molecular-weight hBNP and human NT-proBNP
immunoreactive material showing that human proBNP can be processed
correctly in rat cardiomyocytes.
4. Local (P)RR gene delivery caused a deterioration in cardiac function and
administration of an AT1-R blocker, losartan, did not prevent the attenuation
of cardiac function, indicating that (P)RR–induced worsening of LV function
was independent of Ang II generation. Moreover, the expressions of cardiac
contractility genes, such as α-MHC, cardiac α-actin and SERCA2, were
increased in a manner independent of Ang II generation.
5. (P)RR overexpression triggered Ang II-independent extracellular matrix
remodelling in normal adult rat heart. Myocardial fibrosis was increased by
(P)RR gene delivery and was accompanied by increased LV expression of

134
TGFβ1 and CTGF, plasminogen activator inhibitor-1, collagen Iα1,
fibronectin-1, MMP-2 and MMP-9.
6. Local (P)RR gene transfer increased capillary density, which was slightly, but
nonsignificantly increased in losartan-treated (P)RR overexpressing hearts. In
agreement with this hypothesis, (P)RR resulted in augmentation of VEGF
and FGF-2 gene expressions also in losartan-treated animals, suggesting that
the (P)RR–mediated angiogenesis was an Ang II-independent process.
7. (P)RR gene delivery increased ERK1/2 phosphorylation and losartan
treatment had no effect on the (P)RR gene delivery induced increase in
ERK1/2 phosphorylation, indicating that activation of the ERK1/2 pathway
by (P)RR was Ang II-independent. (P)RR gene transfer increased apoptotic
cell death, this being reduced by losartan-treatment. Furthermore, the extent
of HSP27 phosphorylation was increased by (P)RR and this was attenuated
by losartan indicating that HSP27 activation by (P)RR was partly dependent
on Ang II.
8. (P)RR increased the expression of the pathological hypertrophy associated
genes (ANP, β-MHC and skeletal α-actin), all these being attenuated by
losartan administration, suggesting that the hypertrophic process mediated by
(P)RR activation is an Ang II-dependent mechanism.

135
References
Abbate A, Biondi-Zoccai GGL, Bussani R, Dobrina A, Camilot D, Feroce F, Rossiello R, Baldi F, Silvestri F, Biasucci LM & Baldi A (2003) Increased myocardial apoptosis in patients with unfavorable left ventricular remodeling and early symptomatic post-infarction heart failure. J Am Coll Cardiol 41(5): 753–760.
Abbate A, Biondi-Zoccai GG & Baldi A (2002) Pathophysiologic role of myocardial apoptosis in post-infarction left ventricular remodeling. J Cell Physiol 193(2): 145–153.
Abbott JD, Huang Y, Liu D, Hickey R, Krause DS & Giordano FJ (2004) Stromal cell–derived factor-1α plays a critical role in stem cell recruitment to the heart after myocardial infarction but is not sufficient to induce homing in the absence of injury. Circulation 110(21): 3300–3305.
Abraham W, Lowes B, Ferguson D, Odom J, Kim J, Robertson A, Bristow M & Schrier RW (1998) Systemic hemodynamic, neurohormonal, and renal effects of a steady-state infusion of human brain natriuretic peptide in patients with hemodynamically decompensated heart failure. J Card Fail 4(1): 37–44.
Aburaya M, Hino J, Minamino N, Kangawa K & Matsuo H (1989) Isolation and identification of rat brain natriuretic peptides in cardiac atrium. Biochem Biophys Res Commun 163(1): 226–232.
Advani A, Kelly DJ, Cox AJ, White KE, Advani SL, Thai K, Connelly KA, Yuen D, Trogadis J, Herzenberg AM, Kuliszewski MA, Leong-Poi H & Gilbert RE (2009) The (Pro)renin receptor. Hypertension 54(2): 261–269.
Ala-Kopsala M, Magga J, Peuhkurinen K, Leipala J, Ruskoaho H, Leppaluoto J & Vuolteenaho O (2004) Molecular heterogeneity has a major impact on the measurement of circulating N-terminal fragments of A- and B-type natriuretic peptides. Clin Chem 50(9): 1576–1588.
Ala-Kopsala M, Ruskoaho H, Leppaluoto J, Seres L, Skoumal R, Toth M, Horkay F & Vuolteenaho O (2005) Single assay for amino-terminal fragments of cardiac A- and B-type natriuretic peptides. Clin Chem 51(4): 708–718.
Aoki M, Morishita R, Muraishi A, Moriguchi A, Sugimoto T, Maeda K, Dzau VJ, Kaneda Y, Higaki J & Ogihara T (1997) Efficient in vivo gene transfer into the heart in the rat myocardial infarction model using the HVJ (Hemagglutinating Virus of Japan)—liposome method. J Mol Cell Cardiol 29(3): 949–959.
Aragay AM, Ruiz-Gomez A, Penela P, Sarnago S, Elorza A, Jimenez-Sainz MC & Mayor F, Jr (1998) G protein-coupled receptor kinase 2 (GRK2): mechanisms of regulation and physiological functions. FEBS Lett 430(1–2): 37–40.
Arai M, Alpert N, MacLennan D, Barton P & Periasamy M (1993) Alterations in sarcoplasmic reticulum gene expression in human heart failure. A possible mechanism for alterations in systolic and diastolic properties of the failing myocardium. Circ Res 72(2): 463–469.

136
Arai M, Otsu K, MacLennan DH & Periasamy M (1992) Regulation of sarcoplasmic reticulum gene expression during cardiac and skeletal muscle development. Am J Physiol Cell Physiol 262(3): C614–C620.
Askari AT, Unzek S, Popovic ZB, Goldman CK, Forudi F, Kiedrowski M, Rovner A, Ellis SG, Thomas JD, DiCorleto PE, Topol EJ & Penn MS (2003) Effect of stromal-cell-derived factor 1 on stem-cell homing and tissue regeneration in ischaemic cardiomyopathy. Lancet 362(9385): 697–703.
Atlas SA (2007) The renin-angiotensin aldosterone system: pathophysiological role and pharmacologic inhibition. J Manag Care Pharm 2007 13(8 Suppl B): 9–20.
Babu A, Su H, Ryu Y & Gulati J (1992) Determination of residue specificity in the EF-hand of troponin C for Ca2+ coordination, by genetic engineering. J Biol Chem 267(22): 15469–15474.
Babu GJ, Zheng Z, Natarajan P, Wheeler D, Janssen PM & Periasamy M (2005) Overexpression of sarcolipin decreases myocyte contractility and calcium transient. Cardiovasc Res 65(1): 177–186.
Balabanian K, Lagane B, Infantino S, Chow KYC, Harriague J, Moepps B, Arenzana-Seisdedos F, Thelen M & Bachelerie F (2005) The chemokine SDF-1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J Biol Chem 280(42): 35760–35766.
Barr E, Carroll J, Kalynych A, Tripathy S, Kozarsky K, Wilson J & Leiden J (1994) Efficient catheter–mediated gene transfer into the heart using replication-defective adenovirus. Gene Ther 1(1): 51–58.
Bartel S, Vetter D, Schlegel W, Wallukat G, Krause E & Karczewski P (2000) Phosphorylation of phospholamban at threonine-17 in the absence and presence of β-adrenergic stimulation in neonatal rat cardiomyocytes. J Mol Cell Cardiol 32(12): 2173–2185.
Batenburg WW, Lu X, Leijten F, Maschke U, Muller DN & Danser AHJ (2011) Renin- and prorenin–induced effects in rat vascular smooth muscle cells overexpressing the human (pro)renin receptor. Hypertension 58(6): 1111–1119.
Belke DD, Swanson E, Suarez J, Scott BT, Stenbit AE & Dillmann WH (2007) Increased expression of SERCA in the hearts of transgenic mice results in increased oxidation of glucose. Am J Physiol Heart Circ Physiol 292(4): H1755–H1763.
Bennett JA, Riegel B, Bittner V & Nichols J (2002) Validity and reliability of the NYHA classes for measuring research outcomes in patients with cardiac disease. Heart Lung 31(4): 262–270.
Berenji K, Drazner MH, Rothermel BA & Hill JA (2005) Does load–induced ventricular hypertrophy progress to systolic heart failure? Am J Physiol Heart Circ Physiol 289(1): H8–H16.
Berridge MJ, Bootman MD & Roderick HL (2003) Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4(7): 517–529.
Bers DM & Despa S (2006) Cardiac myocytes Ca2+ and Na+ regulation in normal and failing hearts. J Pharmacol Sci 100(5): 315–322.
Bers DM (2002) Cardiac excitation-contraction coupling. Nature 415(6868): 198–205.

137
Bing O, Brooks W, Conrad C, Sen S, Perreault C & Morgan J (1991) Intracellular calcium transients in myocardium from spontaneously hypertensive rats during the transition to heart failure. Circ Res 68(5): 1390–1400.
Bish LT, Morine K, Sleeper MM, Sanmiguel J, Wu D, Gao G, Wilson JM & Sweeney HL (2008a) Adeno-associated virus (AAV) serotype 9 provides global cardiac gene transfer superior to AAV1, AAV6, AAV7, and AAV8 in the mouse and rat. Hum Gene Ther 19(12): 1359–1368.
Bish LT, Sleeper MM, Brainard B, Cole S, Russell N, Withnall E, Arndt J, Reynolds C, Davison E, Sanmiguel J, Wu D, Gao G, Wilson JM & Lee Sweeney H (2008b) Percutaneous transendocardial delivery of self-complementary adeno-associated virus 6 achieves global cardiac gene transfer in canines. Mol Ther 16(12): 1953–1959.
Blomer U, Naldini L, Kafri T, Trono D, Verma IM & Gage FH (1997) Highly efficient and sustained gene transfer in adult neurons with a lentivirus vector. J Virol 71(9): 6641–6649.
Boerrigter G & Burnett JC,Jr (2004) Recent advances in natriuretic peptides in congestive heart failure. Expert Opin Investig Drugs 13(6): 643–652.
Boerrigter G, Lapp H & Burnett JC,Jr (2009) Modulation of cGMP in heart failure: a new therapeutic paradigm. Handb Exp Pharmacol 191: 485–506.
Bohm M, Reiger B, Schwinger RHG & Erdmann E (1994) cAMP concentrations, cAMP dependent protein kinase activity, and phospholamban in non-failing and failing myocardium. Cardiovasc Res 28(11): 1713–1719.
Bolck B, Munch G, Mackenstein P, Hellmich M, Hirsch I, Reuter H, Hattebuhr N, Weig H, Ungerer M, Brixius K & Schwinger RHG (2004) Na+/Ca2+ exchanger overexpression impairs frequency- and ouabain-dependent cell shortening in adult rat cardiomyocytes. Am J Physiol Heart Circ Physiol 287(4): H1435–H1445.
Bonci D, Cittadini A, Latronico MVG, Borello U, Aycock JK, Drusco A, Innocenzi A, Follenzi A, Lavitrano ML, Monti MG, Ross J, Naldini L, Peschle C, Cossu G & Condorelli G (2003) Advanced generation lentiviruses as efficient vectors for cardiomyocyte gene transduction in vitro and in vivo. Gene Ther 10(8): 630–636.
Boutin S, Monteilhet V, Veron P, Leborgne C, Benveniste O, Montus M & Masurier C (2010) Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum Gene Ther 21(6): 704–712.
Boyle AJ, Yeghiazarians Y, Shih H, Hwang J, Ye J, Sievers R, Zheng D, Palasubramaniam J, Palasubramaniam D, Karschimkus C, Whitbourn R, Jenkins A & Wilson A (2011) Myocardial production and release of MCP-1 and SDF-1following myocardial infarction: differences between mice and man. J Transl Med 12(9): 150.
Bridges CR, Gopal K, Holt DE, Yarnall C, Cole S, Anderson RB, Yin X, Nelson A, Kozyak BW, Wang Z, Lesniewski J, Su LT, Thesier DM, Sundar H & Stedman HH (2005) Efficient myocyte gene delivery with complete cardiac surgical isolation in situ. J Thorac Cardiovasc Surg 130(5): 1364.e1–1364.e8.

138
Brinks H, Rohde D, Voelkers M, Qiu G, Pleger ST, Herzog N, Rabinowitz J, Ruhparwar A, Silvestry S, Lerchenmuller C, Mather PJ, Eckhart AD, Katus HA, Carrel T, Koch WJ & Most P (2011) S100A1 genetically targeted therapy reverses dysfunction of human failing cardiomyocytes. J Am Coll Cardiol 58(9): 966–973.
Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, Lurie K, Billingham ME, Harrison DC & Stinson EB (1982) Decreased catecholamine sensitivity and β-adrenergic-receptor density in failing human hearts. N Engl J Med 307(4): 205–211.
Brocheriou V, Hagege AA, Oubenaïssa A, Lambert M, Mallet VO, Duriez M, Wassef M, Kahn A, Menasche P & Gilgenkrantz H (2000) Cardiac functional improvement by a human Bcl-2 transgene in a mouse model of ischemia/reperfusion injury. J Gene Med 2(5): 326–333.
Brodde OE (1991) β1- and β2-adrenoceptors in the human heart: properties, function, and alterations in chronic heart failure. Pharmacol Rev 43(2): 203–242.
Brodde O, Bruck H & Leineweber K (2006) Cardiac adrenoceptors: physiological and pathophysiological relevance. J Pharmacol Sci 100(5): 323–337.
Brown RD, Ambler SK, Mitchell MD & Long CS (2005) The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Annu Rev Pharmacol Toxicol 45(1): 657–687.
Buerke M, Murohara T, Skurk C, Nuss C, Tomaselli K & Lefer AM (1995) Cardioprotective effect of insulin-like growth factor I in myocardial ischemia followed by reperfusion. Proc Natl Acad Sci U S A 92(17): 8031–8035.
Bull DA, Bailey SH, Rentz JJ, Zebrack JS, Lee M, Litwin SE & Kim SW (2003) Effect of Terplex/VEGF-165 gene therapy on left ventricular function and structure following myocardial infarction: VEGF gene therapy for myocardial infarction. J Controlled Release 93(2): 175–181.
Burckle CA, Jan Danser AH, Muller DN, Garrelds IM, Gasc J, Popova E, Plehm R, Peters J, Bader M & Nguyen G (2006) Elevated blood pressure and heart rate in human renin receptor transgenic rats. Hypertension 47(3): 552–556.
Burger AJ, Horton DP, LeJemtel T, Ghali JK, Torre G, Dennish G, Koren M, Dinerman J, Silver M, Cheng ML & Elkayam U (2002) Effect of nesiritide (B-type natriuretic peptide) and dobutamine on ventricular arrhythmias in the treatment of patients with acutely decompensated congestive heart failure: the precedent study. Am Heart J 144(6): 1102–1108.
Buttrick P, Kass A, Kitsis R, Kaplan M & Leinwand L (1992) Behavior of genes directly injected into the rat heart in vivo. Circ Res 70(1): 193–198.
Calcedo R, Vandenberghe LH, Gao G, Lin J & Wilson JM (2009) Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J Infect Dis 199(3): 381–390.
Camper-Kirby D, Welch S, Walker A, Shiraishi I, Setchell KDR, Schaefer E, Kajstura J, Anversa P & Sussman MA (2001) Myocardial Akt activation and gender: increased nuclear activity in females versus males. Circ Res 88(10): 1020–1027.
Cao L & Gardner DG (1995) Natriuretic peptides inhibit DNA synthesis in cardiac fibroblasts. Hypertension 25(2): 227–234.

139
Carey RM & Siragy HM (2003) Newly recognized components of the renin-angiotensin system: potential roles in cardiovascular and renal regulation. Endocr Rev 24(3): 261–271.
Carr A, Schmidt A, Suzuki Y, del Monte F, Sato Y, Lanner C, Breeden K, Jing S, Allen P, Greengard P, Yatani A, Hoit B, Grupp I, Hajjar R, DePaoli-Roach A & Kranias E (2002) Type 1 phosphatase, a negative regulator of cardiac function. Mol Cell Biol 22(12): 4124–4135.
Cataliotti A, Chen HH, Schirger JA, Martin FL, Boerrigter G, Costello-Boerrigter LC, James KD, Polowy K, Miller MA, Malkar NB, Bailey KR & Burnett JC,Jr (2008) Chronic actions of a novel oral B-type natriuretic peptide conjugate in normal dogs and acute actions in angiotensin II–mediated hypertension. Circulation 118(17): 1729–1736.
Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, Capla JM, Galiano RD, Levine JP & Gurtner GC (2004) Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med 10(8): 858–864.
Chaanine AH, Kalman J & Hajjar RJ (2010) Cardiac gene therapy. Semin Thorac Cardiovasc Surg 22(2): 127–139.
Chatterjee K & Massie B (2007) Systolic and diastolic heart failure: differences and similarities. J Card Fail 13(7): 569–576.
Chen HH & Burnett JC (2006) Clinical application of the natriuretic peptides in heart failure. Eur Heart J Suppl 8(suppl E): E18–E25.
Chen Y, Escoubet B, Prunier F, Amour J, Simonides WS, Vivien B, Lenoir C, Heimburger M, Choqueux C, Gellen B, Riou B, Michel J, Franz WM & Mercadier J (2004) Constitutive cardiac overexpression of sarcoplasmic/endoplasmic reticulum Ca2+-ATPase delays myocardial failure after myocardial infarction in rats at a cost of increased acute arrhythmias. Circulation 109(15): 1898–1903.
Chen Z, Chua CC, Ho Y, Hamdy RC & Chua BHL (2001) Overexpression of Bcl-2 attenuates apoptosis and protects against myocardial I/R injury in transgenic mice. Am J Physiol Heart Circ Physiol 280(5): H2313–H2320.
Choi D, Koch WJ, Hunter JJ & Rockman HA (1997) Mechanism of β-adrenergic receptor desensitization in cardiac hypertrophy is increased β-adrenergic receptor kinase. J Biol Chem 272(27): 17223–17229.
Choukroun G, Hajjar R, Fry S, del Monte F, Haq S, Guerrero J, Picard M, Rosenzweig A & Force T (1999) Regulation of cardiac hypertrophy in vivo by the stress-activated protein kinases/c-Jun NH2-terminal kinases. J Clin Invest 104(4): 391–398.
Chu G & Kranias E (2002) Functional interplay between dual site phospholambam phosphorylation: insights from genetically altered mouse models. Basic Res Cardiol 97(Suppl. 1): I/43–I/48.
Chu D, Sullivan CC, Weitzman MD, Du L, Wolf PL, Jamieson SW & Thistlethwaite PA (2003) Direct comparison of efficiency and stability of gene transfer into the mammalian heart using adeno-associated virus versus adenovirus vectors. J Thorac Cardiovasc Surg 126(3): 671–679.

140
Cleutjens JPM, Kandala JC, Guarda E, Guntaka RV & Weber KT (1995) Regulation of collagen degradation in the rat myocardium after infarction. J Mol Cell Cardiol 27(6): 1281–1292.
Condorelli G, Drusco A, Stassi G, Bellacosa A, Roncarati R, Iaccarino G, Russo MA, Gu Y, Dalton N, Chung C, Latronico MVG, Napoli C, Sadoshima J, Croce CM & Ross J (2002) Akt induces enhanced myocardial contractility and cell size in vivo in transgenic mice. Proc Natl Acad Sci U S A 99(19): 12333–12338.
Cong L, Chen H, Li Y, Zhou L, McGibbon MA, Taylor SI & Quon MJ (1997) Physiological role of Akt in insulin-stimulated translocation of GLUT4 in transfected rat adipose cells. Mol Endocrinol 11(13): 1881–1890.
Connelly KA, Advani A, Kim S, Advani SL, Zhang M, White KE, Kim YM, Parker C, Thai K, Krum H, Kelly DJ & Gilbert RE (2011) The cardiac (pro)renin receptor is primarily expressed in myocyte transverse tubules and is increased in experimental diabetic cardiomyopathy. J Hypertens 29(6): 1175–1184.
Connelly S, Mount J, Mauser A, Gardner J, Kaleko M, McClelland A & Lothrop CJ (1996) Complete short-term correction of canine hemophilia A by in vivo gene therapy. Blood 88(10): 3846–3853.
Cousin C, Bracquart D, Contrepas A & Nguyen G (2010) Potential role of the (pro)renin receptor in cardiovascular and kidney diseases. J Nephrol 23(5): 508–513.
Cousin C, Bracquart D, Contrepas A, Corvol P, Muller L & Nguyen G (2009) Soluble form of the (pro)renin receptor generated by intracellular cleavage by furin Is secreted in plasma. Hypertension 53(6): 1077–1082.
Creemers EE, Cleutjens JP, Smits JF & Daemen MJ (2001) Matrix metalloproteinase inhibition after myocardial infarction. Circ Res 89(3): 201–210.
Cruciat C, Ohkawara B, Acebron SP, Karaulanov E, Reinhard C, Ingelfinger D, Boutros M & Niehrs C (2010) Requirement of prorenin receptor and vacuolar H+-ATPase–mediated acidification for Wnt signaling. Science 327(5964): 459–463.
Dai Y, Schwarz E, Gu D, Zhang W, Sarvetnick N & Verma IM (1995) Cellular and humoral immune responses to adenoviral vectors containing factor IX gene: tolerization of factor IX and vector antigens allows for long-term expression. Proc Natl Acad Sci U S A 92(5): 1401–1405.
Dally S, Bredoux R, Corvazier E, Andersen J, Clausen J, Dode L, Fanchaouy M, Gelebart P, Monceau V, del Monte F, Gwathmey J, Hajjar R, Chaabane C, Bobe R, Raies A & Enouf J (2006) Ca2+-ATPases in non-failing and failing heart: evidence for a novel cardiac sarco/endoplasmic reticulum Ca2+-ATPase 2 isoform (SERCA2c). Biochem J 395(2): 249–258.
Dargie H, McMurray J & McDonagh T (1996) Heart failure-implications of the true size of the problem. J Intern Med 239(4): 309–315.
Dash R, Kadambi VJ, Schmidt AG, Tepe NM, Biniakiewicz D, Gerst MJ, Canning AM, Abraham WT, Hoit BD, Liggett SB, Lorenz JN, Dorn GW & Kranias EG (2001) Interactions between phospholamban and β-adrenergic drive may lead to cardiomyopathy and early mortality. Circulation 103(6): 889–896.

141
Davidson MJ, Jones JM, Emani SM, Wilson KH, Jaggers J, Koch WJ & Milano CA (2001) Cardiac gene delivery with cardiopulmonary bypass. Circulation 104(2): 131–133.
Davis J, Westfall MV, Townsend D, Blankinship M, Herron TJ, Guerrero-Serna G, Wang W, Devaney E & Metzger JM (2008) Designing heart performance by gene transfer. Physiol Rev. 88(4): 1567–1651.
Day S, Davis J, Westfall M & Metzger J (2006) Genetic engineering and therapy for inherited and acquired cardiomyopathies. Ann N Y Acad Sci 1080(1): 437–450.
De La Luz Sierra M, Yang F, Narazaki M, Salvucci O, Davis D, Yarchoan R, Zhang HH, Fales H & Tosato G (2004) Differential processing of stromal-derived factor-1α and stromal-derived factor-1β explains functional diversity. Blood 103(7): 2452–2459.
del Monte F, Harding SE, Dec GW, Gwathmey JK & Hajjar RJ (2002) Targeting phospholamban by gene transfer in human heart failure. Circulation 105(8): 904–907.
del Monte F, Harding SE, Schmidt U, Matsui T, Kang ZB, Dec GW, Gwathmey JK, Rosenzweig A & Hajjar RJ (1999) Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation 100(23): 2308–2311.
del Monte F, Lebeche D, Guerrero JL, Tsuji T, Doye AA, Gwathmey JK & Hajjar RJ (2004) Abrogation of ventricular arrhythmias in a model of ischemia and reperfusion by targeting myocardial calcium cycling. Proc Natl Acad Sci U S A 101(15): 5622–5627.
del Monte F, Williams E, Lebeche D, Schmidt U, Rosenzweig A, Gwathmey JK, Lewandowski ED & Hajjar RJ (2001) Improvement in survival and cardiac metabolism after gene transfer of sarcoplasmic reticulum Ca2+-ATPase in a rat model of heart failure. Circulation 104(12): 1424–1429.
Detillieux KA, Sheikh F, Kardami E & Cattini PA (2003) Biological activities of fibroblast growth factor-2 in the adult myocardium. Cardiovasc Res 57(1): 8–19.
Di Pasquale G & Chiorini JA (2006) AAV transcytosis through barrier epithelia and endothelium. Mol Ther 13(3): 506–516.
Dickstein K, Cohen-Solal A, Filippatos G, McMurray JJV, Ponikowski P, Poole-Wilson PA, Stromberg A, van Veldhuisen DJ, Atar D, Hoes AW, Keren A, Mebazaa A, Nieminen M, Priori SG, Swedberg K, ESC committee for practice guidelines (CPG) & document reviewers (2008) ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2008. The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2008 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association of the ESC (HFA) and endorsed by the European Society of Intensive Care Medicine (ESICM). Eur Heart J 29(19): 2388–2442.
Dorn GW 2nd & Force T (2005) Protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Invest 115(3): 527–537.
Dorn GW 2nd (2009) Novel pharmacotherapies to abrogate postinfarction ventricular remodeling. Nat Rev Cardiol 6(4): 283–291.
Douglas JT (2007) Adenoviralvectors for gene therapy. Mol Biotechnol 36(1): 71–80.

142
Drago GA & Colyer J (1994) Discrimination between two sites of phosphorylation on adjacent amino acids by phosphorylation site-specific antibodies to phospholamban. J Biol Chem 269(40): 25073–25077.
D'Souza SP, Davis M & Baxter GF (2004) Autocrine and paracrine actions of natriuretic peptides in the heart. Pharmacol Ther 101(2): 113–129.
Du X, Cole TJ, Tenis N, Gao X, Kontgen F, Kemp BE & Heierhorst J (2002) Impaired cardiac contractility response to hemodynamic stress in S100A1-deficient mice. Mol Cell Biol 22(8): 2821–2829.
Du X, Gao X, Jennings GL, Dart AM & Woodcock EA (2000) Preserved ventricular contractility in infarcted mouse heart overexpressing β2-adrenergic receptors. Am J Physiol Heart Circ Physiol 279(5): H2456–H2463.
d’Uscio LV, Moreau P, Shaw S, Takase H, Barton M & Luscher TF (1997) Effects of chronic ETA-receptor blockade in angiotensin II–induced hypertension. Hypertension 29(1): 435–441.
Edes I & Kranias EG (1990) Phospholamban and troponin I are substrates for protein kinase C in vitro but not in intact beating guinea pig hearts. Circ Res 67(2): 394–400.
Eizema K, Fechner H, Bezstarosti K, Schneider-Rasp S, van der Laarse A, Wang H, Schultheiss H, Poller WC & Lamers JMJ (2000) Adenovirus-based phospholamban antisense expression as a novel approach to improve cardiac contractile dysfunction: comparison of a constitutive viral versus an endothelin-1–responsive cardiac promoter. Circulation 101(18): 2193–2199.
El-Armouche A, Wittkopper K, Degenhardt F, Weinberger F, Didie M, Melnychenko I, Grimm M, Peeck M, Zimmermann WH, Unsold B, Hasenfuss G, Dobrev D & Eschenhagen T (2008) Phosphatase inhibitor-1-deficient mice are protected from catecholamine–induced arrhythmias and myocardial hypertrophy. Cardiovasc Res 80(3): 396–406.
Elmadbouh I, Haider HK, Jiang S, Idris NM, Lu G & Ashraf M (2007) Ex vivo delivered stromal cell-derived factor-1α promotes stem cell homing and induces angiomyogenesis in the infarcted myocardium. J Mol Cell Cardiol 42(4): 792–803.
Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35(4): 495–516.
Engelhardt S, Grimmer Y, Fan G & Lohse MJ (2001) Constitutive activity of the human β1-adrenergic receptor in β1-receptor transgenic mice. Mol Pharmacol 60(4): 712–717.
Engelhardt S, Hein L, Dyachenkow V, Kranias EG, Isenberg G & Lohse MJ (2004) Altered calcium handling is critically involved in the cardiotoxic effects of chronic β-adrenergic stimulation. Circulation 109(9): 1154–1160.
Eriksson H, Svardsudd K, Larsson B, Ohlson L, Tibblin G, Welin L & Wilhelmsen L (1989) Risk factors for heart failure in the general population: the study of men born in 1913. Eur Heart J 10(7): 647–656.
Fan G, Gregory KN, Zhao W, Park WJ & Kranias EG (2004) Regulation of myocardial function by histidine-rich, calcium-binding protein. Am J Physiol Heart Circ Physiol 287(4): H1705–H1711.

143
Feldman A, Ray P, Silan C, Mercer J, Minobe W & Bristow M (1991) Selective gene expression in failing human heart. Quantification of steady-state levels of messenger RNA in endomyocardial biopsies using the polymerase chain reaction. Circulation 83(6): 1866–1872.
Feldt S, Batenburg WW, Mazak I, Maschke U, Wellner M, Kvakan H, Dechend R, Fiebeler A, Burckle C, Contrepas A, Jan Danser AH, Bader M, Nguyen G, Luft FC & Muller DN (2008) Prorenin and renin–induced extracellular signal-regulated kinase 1/2 activation in monocytes is not blocked by Aliskiren or the handle-region peptide. Hypertension 51(3): 682–688.
Ferrarini M, Arsic N, Recchia FA, Zentilin L, Zacchigna S, Xu X, Linke A, Giacca M & Hintze TH (2006) Adeno-associated virus–mediated transduction of VEGF165 improves cardiac tissue viability and functional recovery after permanent coronary occlusion in conscious dogs. Circ Res 98(7): 954–961.
Fewell J, Hewett T, Sanbe A, Klevitsky R, Hayes E, Warshaw D, Maughan D & Robbins J (1998) Functional significance of cardiac myosin essential light chain isoform switching in transgenic mice. J Clin Invest 101(12): 2630–2639.
Fields PA, Kowalczyk DW, Arruda VR, Armstrong E, McCleland ML, Hagstrom JN, Pasi KJ, Ertl HCJ, Herzog RW & High KA (2000) Role of vector in activation of T cell subsets in immune responses against the secreted transgene product factor IX. Mol Ther 1(3): 225–235.
Flaherty JD, Bax JJ, De Luca L, Rossi JS, Davidson CJ, Filippatos G, Liu PP, Konstam MA, Greenberg B, Mehra MR, Breithardt G, Pang PS, Young JB, Fonarow GC, Bonow RO, Gheorghiade M & for the Acute Heart Failure Syndromes International Working Group (2009) Acute heart failure syndromes in patients with coronary artery disease: early assessment and treatment. J Am Coll Cardiol 53(3): 254–263.
Flesch M, Schwinger RH, Schnabel P, Schiffer F, van Gelder I, Bavendiek U, Sudkamp M, Kuhn-Regnier F & Bohm M (1996) Sarcoplasmic reticulum Ca2+ATPase and phospholamban mRNA and protein levels in end-stage heart failure due to ischemic or dilated cardiomyopathy. J Mol Med (Berl) 74(6): 321–332.
Fleury S, Simeoni E, Zuppinger C, Deglon N, von Segesser LK, Kappenberger L & Vassalli G (2003) Multiply attenuated, self-inactivating lentiviral vectors efficiently deliver and express genes for extended periods of time in adult rat cardiomyocytes in vivo. Circulation 107(18): 2375–2382.
Francis GS (2001) Pathophysiology of chronic heart failure. Am J Med 110 Suppl7A: 37–46.
Frank KF, Bolck B, Ding Z, Krause D, Hattebuhr N, Malik A, Brixius K, Hajjar RJ, Schrader J & Schwinger RHG (2005) Overexpression of sorcin enhances cardiac contractility in vivo and in vitro. J Mol Cell Cardiol 38(4): 607–615.
Freeman K, Lerman I, Kranias EG, Bohlmeyer T, Bristow MR, Lefkowitz RJ, Iaccarino G, Koch WJ & Leinwand LA (2001) Alterations in cardiac adrenergic signaling and calcium cycling differentially affect the progression of cardiomyopathy. J Clin Invest 107(8): 967–974.

144
French B, Mazur W, Geske R & Bolli R (1994) Direct in vivo gene transfer into porcine myocardium using replication- deficient adenoviral vectors. Circulation 90(5): 2414–2424.
Frey N & Olson EN (2003) Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol 65(1): 45–79.
Fromes Y, Salmon A, Wang X, Collin H, Rouche A, Hagege A, Schwartz K & Fiszman M (1999) Gene delivery to the myocardium by intrapericardial injection. Gene Ther 6(4): 683–688.
Fujio Y, Nguyen T, Wencker D, Kitsis RN & Walsh K (2000) Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-reperfusion injury in mouse heart. Circulation 101(6): 660–667.
Gao M, Ping P, Post S, Insel P, Tang R & Hammond HK (1998) Increased expression of adenylyl cyclase type VI proportionately increases β-adrenergic receptor-stimulated production of cAMP in neonatal rat cardiac myocytes. Proc Natl Acad Sci U S A Feb 95(3): 1038–1043.
Gao MH & Hammond HK (2011) Unanticipated signaling events associated with cardiac adenylyl cyclase gene transfer. J Mol Cell Cardiol 50(5): 751–758.
Gao MH, Lai NC, Roth DM, Zhou J, Zhu J, Anzai T, Dalton N & Hammond HK (1999) Adenylylcyclase increases responsiveness to catecholamine stimulation in transgenic mice. Circulation 99(12): 1618–1622.
Gautel M, Furst DO, Cocco A & Schiaffino S (1998) Isoform transitions of the myosin binding protein C family in developing human and mouse muscles: lack of isoform transcomplementation in cardiac muscle. Circ Res 82(1): 124–129.
Gergs U, Berndt T, Buskase J, Jones LR, Kirchhefer U, Muller FU, Schluter K, Schmitz W & Neumann J (2007) On the role of junctin in cardiac Ca2+ handling, contractility, and heart failure. Am J Physiol Heart Circ Physiol 293(1): H728–H734.
Ghadge SK, Muhlstedt S, Ozcelik C & Bader M (2011) SDF-1α as a therapeutic stem cell homing factor in myocardial infarction. Pharmacol Ther 129(1): 97–108.
Giordano FJ, Gerber H, Williams S, VanBruggen N, Bunting S, Ruiz-Lozano P, Gu Y, Nath AK, Huang Y, Hickey R, Dalton N, Peterson KL, Ross J, Chien KR & Ferrara N (2001) A cardiac myocyte vascular endothelial growth factor paracrine pathway is required to maintain cardiac function. Proc Natl Acad Sci U S A 98(10): 5780–5785.
Giordano FJ, Ping P, McKirnan MD, Nozaki S, DeMaria AN, Dillmann WH, Mathieu-Costello O & Hammond HK (1996) Intracoronary gene transfer of fibroblast growth factor-5 increases blood flow and contractile function in an ischemic region of the heart. Nat Med 2(5): 534–539.
Go L, Moschella M, Watras J, Handa K, Fyfe BS & Marks A (1995) Differential regulation of two types of intracellular calcium release channels during end-stage heart failure. J Clin Invest 95(2): 888–894.
Goodeve AC & Peake IR (2003) The molecular basis of hemophilia A: genotype-phenotype relationships and inhibitor development. Semin Thromb Hemost 29(01): 23–30.
Granzier HL & Labeit S (2004) The giant protein titin. Circ Res 94(3): 284–295.

145
Gregorevic P, Blankinship MJ, Allen JM, Crawford RW, Meuse L, Miller DG, Russell DW & Chamberlain JS (2004) Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat Med 10(8): 828–834.
Griendling K, Murphy T & Alexander R (1993) Molecular biology of the renin-angiotensin system. Circulation 87(6): 1816–1828.
Griscelli F, Opolon P, Chianale C, Di Falco N, Franz W, Perricaudet M & Ragot T (1997) Expression from cardiomyocyte-specific promoter after adenovirus–mediated gene transfer in vitro and in vivo. C R Acad Sci III. 320(2): 103–112.
Grossman PM, Han Z, Palasis M, Barry JJ & Lederman RJ (2002) Incomplete retention after direct myocardial injection. Catheter Cardiovasc Interv 55(3): 392–397.
Gupta R, Tongers J & Losordo DW (2009) Human studies of angiogenic gene therapy. Circ Res 105(8): 724–736.
Guzman R, Lemarchand P, Crystal R, Epstein S & Finkel T (1993) Efficient gene transfer into myocardium by direct injection of adenovirus vectors. Circ Res 73(6): 1202–1207.
Gwathmey J, Copelas L, MacKinnon R, Schoen F, Feldman M, Grossman W & Morgan J (1987) Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ Res 61(1): 70–76.
Gwathmey JK, Yerevanian AI & Hajjar RJ (2011) Cardiac gene therapy with SERCA2a: from bench to bedside. J Mol Cell Cardiol 50(5): 803–812.
Gyorke I, Hester N, Jones LR & Gyorke S (2004) The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys J 86(4): 2121–2128.
Hagemann D, Kuschel M, Kuramochi T, Zhu W, Cheng H & Xiao R (2000) Frequency-encoding Thr17 phospholamban phosphorylation is independent of Ser16 phosphorylation in cardiac myocytes. J Biol Chem 275(29): 22532–22536.
Haghighi K, Schmidt AG, Hoit BD, Brittsan AG, Yatani A, Lester JW, Zhai J, Kimura Y, Dorn GW, MacLennan DH & Kranias EG (2001) Superinhibition of sarcoplasmic reticulum function by phospholamban induces cardiac contractile failure. J Biol Chem 276(26): 24145–24152.
Hajjar RJ, Schmidt U, Matsui T, Guerrero JL, Lee K, Gwathmey JK, Dec GW, Semigran MJ & Rosenzweig A (1998) Modulation of ventricular function through gene transfer in vivo. Proc Natl Acad Sci U S A 95(9): 5251–5256.
Hajjar RJ, Zsebo K, Deckelbaum L, Thompson C, Rudy J, Yaroshinsky A, Ly H, Kawase Y, Wagner K, Borow K, Jaski B, London B, Greenberg B, Pauly DF, Patten R, Starling R, Mancini D & Jessup M (2008) Design of a phase 1/2 trial of intracoronary administration of AAV1/SERCA2a in patients with heart failure. J Card Fail 14(5): 355–367.
Halbert C, Miller A, McNamara S, Emerson J, Gibson R, Ramsey B & Aitken M (2006) Prevalence of neutralizing antibodies against adeno-associated virus (AAV) types 2, 5, and 6 in cystic fibrosis and normal populations: implications for gene therapy using AAV vectors. Hum Gene Ther 17(4): 440–447.

146
Hall RA & Lefkowitz RJ (2002) Regulation of G protein–coupled receptor signaling by scaffold proteins. Circ Res 91(8): 672–680.
Hanada M, Aime-Sempe C, Sato T & Reed JC (1995) Structure-function analysis of Bcl-2 protein. J Biol Chem 270(20): 11962–11969.
Harding VB, Jones LR, Lefkowitz RJ, Koch WJ & Rockman HA (2001) Cardiac βARK1 inhibition prolongs survival and augments β blocker therapy in a mouse model of severe heart failure. Proc Natl Acad Sci U S A 98(10): 5809–5814.
Hasenfuss G, Reinecke H, Studer R, Meyer M, Pieske B, Holtz J, Holubarsch C, Posival H, Just H & Drexler H (1994) Relation between myocardial function and expression of sarcoplasmic reticulum Ca2+-ATPase in failing and nonfailing human myocardium. Circ Res 75(3): 434–442.
Hasenfuss G (1998) Alterations of calcium-regulatory proteins in heart failure. Cardiovasc Res 37(2): 279–289.
Haubold K, Herrmann H, Langer SJ, Evans RM, Leinwand LA & Klymkowsky MW (2003) Acute effects of desmin mutations on cytoskeletal and cellular integrity in cardiac myocytes. Cell Motil Cytoskeleton 54(2): 105–121.
Hayase M, del Monte F, Kawase Y, MacNeill BD, McGregor J, Yoneyama R, Hoshino K, Tsuji T, De Grand AM, Gwathmey JK, Frangioni JV & Hajjar RJ (2005) Catheter-based antegrade intracoronary viral gene delivery with coronary venous blockade. Am J Physiol Heart Circ Physiol 288(6): H2995–H3000.
He H, Giordano F, Hilal-Dandan R, Choi D, Rockman HA, McDonough P, Bluhm W, Meyer M, Sayen M, Swanson E & Dillmann W (1997) Overexpression of the rat sarcoplasmic reticulum Ca2+ ATPase gene in the heart of transgenic mice accelerates calcium transients and cardiac relaxation. J Clin Invest 100(2): 380–389.
He H, Meyer M, Martin JL, McDonough PM, Ho P, Lou X, Lew WYW, Hilal-Dandan R & Dillmann WH (1999) Effects of mutant and antisense RNA of phospholamban on SR Ca2+-ATPase activity and cardiac myocyte contractility. Circulation 100(9): 974–980.
Hedman M, Hartikainen J & Yla-Herttuala S (2011) Progress and prospects: hurdles to cardiovascular gene therapy clinical trials. Gene Ther 18(8): 743–749.
Heineke J, Auger-Messier M, Xu J, Oka T, Sargent MA, York A, Klevitsky R, Vaikunth S, Duncan SA, Aronow BJ, Robbins J, Crombleholm TM & Molkentin JG (2007) Cardiomyocyte GATA4 functions as a stress-responsive regulator of angiogenesis in the murine heart. J Clin Invest 117(11): 3198–3210.
Heineke J & Molkentin JD (2006) Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol 7(8): 589–600.
Hengartner MO & Horvitz HR (1994) C. elegans cell survival gene ced-9 encodes a functional homolog of the mammalian proto-oncogene bcl-2. Cell 76(4): 665–676.
Herron TJ, Vandenboom R, Fomicheva E, Mundada L, Edwards T & Metzger JM (2007) Calcium-independent negative inotropy by β-myosin heavy chain gene transfer in cardiac myocytes. Circ Res 100(8): 1182–1190.
Hers I, Vincent EE & Tavare JM (2011) Akt signalling in health and disease. Cell Signal 23(10): 1515–1527.

147
Hillegass WB, Dean NA, Liao L, Rhinehart RG & Myers PR (2001) Treatment of no-reflow and impaired flow with the nitric oxide donor nitroprusside following percutaneous coronary interventions: initial human clinical experience. J Am Coll Cardiol 37(5): 1335–1343.
Hirose T, Mori N, Totsune K, Morimoto R, Maejima T, Kawamura T, Metoki H, Asayama K, Kikuya M, Ohkubo T, Kohzuki M, Takahashi K & Imai Y (2009) Gene expression of (pro)renin receptor is upregulated in hearts and kidneys of rats with congestive heart failure. Peptides 30(12): 2316–2322.
Hirsch JC, Borton AR, Albayya FP, Russell MW, Ohye RG & Metzger JM (2004) Comparative analysis of parvalbumin and SERCA2a cardiac myocyte gene transfer in a large animal model of diastolic dysfunction. Am J Physiol Heart Circ Physiol 286(6): H2314–H2321.
Hockenbery DM, Oltvai ZN, Yin X, Milliman CL & Korsmeyer SJ (1993) Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell 75(2): 241–251.
Hoeben A, Landuyt B, Highley MS, Wildiers H, Van Oosterom AT & De Bruijn EA (2004) Vascular endothelial growth factor and angiogenesis. Pharmacol Rev 56(4): 549–580.
Hofland HE, Nagy D, Liu J, Spratt K, Lee Y, Danos O & Sullivan SM (1997) In vivo gene transfer by intravenous administration of stable cationic lipid/DNA complex. Pharm Res 14(6): 742–749.
Holland R, Rechel B, Stepien K, Harvey I & Brooksby I (2010) Patients' self-assessed functional status in heart failure by New York Heart Association class: a prognostic predictor of hospitalizations, quality of life and death. J Card Fail 16(2): 150–156.
Holubarsch C, Hasenfuss G, Schmidt-Schweda S, Knorr A, Pieske B, Ruf T, Fasol R & Just H (1993) Angiotensin I and II exert inotropic effects in atrial but not in ventricular human myocardium. An in vitro study under physiological experimental conditions. Circulation 88(3): 1228–1237.
Hoshijima M, Ikeda Y, Iwanaga Y, Minamisawa S, Date M, Gu Y, Iwatate M, Li M, Wang L, Wilson J, Wang Y, Ross JJ & Chien K (2002) Chronic suppression of heart-failure progression by a pseudophosphorylated mutant of phospholamban via in vivo cardiac rAAV gene delivery. Nat Med 8(8): 864–871.
Hu X, Dai S, Wu W, Tan W, Zhu X, Mu J, Guo Y, Bolli R & Rokosh G (2007) Stromal cell–derived factor-1α confers protection against myocardial ischemia/reperfusion injury. Circulation 116(6): 654–663.
Huang Y, Wongamorntham S, Kasting J, McQuillan D, Owens RT, Yu L, Noble NA & Border W (2006) Renin increases mesangial cell transforming growth factor-β1 and matrix proteins through receptor–mediated, angiotensin II-independent mechanisms. Kidney Int 69(1): 105–113.
Huang Y, Noble NA, Zhang J, Xu C & Border WA (2007) Renin-stimulated TGF-β1 expression is regulated by a mitogen-activated protein kinase in mesangial cells. Kidney Int 72(1): 45–52.

148
Huelsemann J, Sterzel R, McKenzie D & Wilcox C (1985) Effects of a calcium entry blocker on blood pressure and renal function during angiotensin–induced hypertension. Hypertension 7(3): 374–379.
Humpl T, Zaidi SHE, Coe JY, Russell J, Kaneda Y, Massaeli H, Benson LN & Rabinovitch M (2005) Gene transfer of prostaglandin synthase maintains patency of the newborn lamb arterial duct. Pediatr Res 58(5): 976–980.
Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG, Jessup M, Konstam MA, Mancini DM, Michl K, Oates JA, Rahko PS, Silver MA, Stevenson LW, Yancy CW, Antman EM, Smith SC, Adams CD, Anderson JL, Faxon DP, Fuster V, Halperin JL, Hiratzka LF, Hunt SA, Jacobs AK, Nishimura R, Ornato JP, Page RL & Riegel B (2005) ACC/AHA 2005 guideline update for the diagnosis and management of chronic heart failure in the adult. Circulation 112(12): e154–e235.
Huq F, del Monte F & Hajjar RJ (2002) Modulating signaling pathways in hypertrophy and heart failure by gene transfer. J Card Fail 8(Suppl6): S389–S400.
Ichihara A, Kaneshiro Y, Takemitsu T, Sakoda M, Suzuki F, Nakagawa T, Nishiyama A, Inagami T & Hayashi M (2006a) Nonproteolytic activation of prorenin contributes to development of cardiac fibrosis in genetic hypertension. Hypertension 47(5): 894–900.
Ichihara A, Suzuki F, Nakagawa T, Kaneshiro Y, Takemitsu T, Sakoda M, Nabi AH, Nishiyama A, Sugaya T, Hayashi M & Inagami T (2006b) Prorenin receptor blockade inhibits development of glomerulosclerosis in diabetic angiotensin II type 1a receptor–deficient mice. J Am Soc Nephrol 17(7): 1950–1961.
Ikeda Y, Gu Y, Iwanaga Y, Hoshijima M, Oh SS, Giordano FJ, Chen J, Nigro V, Peterson KL, Chien KR & Ross J (2002) Restoration of deficient membrane proteins in the cardiomyopathic hamster by in vivo cardiac gene transfer. Circulation 105(4): 502–508.
Inagaki K, Fuess S, Storm TA, Gibson GA, Mctiernan CF, Kay MA & Nakai H (2006) Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol Ther 14(1): 45–53.
Inglese J, Luttrell LM, Iñiguez-Lluhi JA, Touhara K, Koch WJ & Lefkowitz RJ (1994) Functionally active targeting domain of the β-adrenergic receptor kinase: an inhibitor of Gβγ–mediated stimulation of type II adenylyl cyclase. Proc Natl Acad Sci U S A 91(9): 3637–3641.
Ishikawa Y, Katsushika S, Chen L, Halnon NJ, Kawabe J & Homcy CJ (1992) Isolation and characterization of a novel cardiac adenylylcyclase cDNA. J Biol Chem 267(19): 13553–13557.
Ishikawa Y, Sorota S, Kiuchi K, Shannon R, Komamura K, Katsushika S, Vatner D, Vatner S & Homcy C (1994) Downregulation of adenylylcyclase types V and VI mRNA levels in pacing–induced heart failure in dogs. J Clin Invest 93(5): 2224–2229.
Ito K, Yan X, Feng X, Manning WJ, Dillmann WH & Lorell BH (2001) Transgenic expression of sarcoplasmic reticulum Ca2+ ATPase modifies the transition from hypertrophy to early heart failure. Circ Res 89(5): 422–429.

149
Iwanaga Y, Hoshijima M, Gu Y, Iwatate M, Dieterle T, Ikeda Y, Date M, Chrast J, Matsuzaki M, Peterson KL, Chien KR & Ross J (2004) Chronic phospholamban inhibition prevents progressive cardiac dysfunction and pathological remodeling after infarction in rats. J Clin Invest 113(5): 727–736.
Izumo S & Shioi T (1998) Cardiac transgenic and gene-targeted mice as models of cardiac hypertrophy and failure: a problem of (new) riches. J Card Fail 4(4): 263–270.
James P, Inui M, Tada M, Chiesi M & Carafoli E (1989) Nature and site of phospholamban regulation of the Ca2+ pump of sarcoplasmic reticulum. Nature 342(6245): 90–92.
Janssens S, Pokreisz P, Schoonjans L, Pellens M, Vermeersch P, Tjwa M, Jans P, Scherrer-Crosbie M, Picard MH, Szelid Z, Gillijns H, Van de Werf F, Collen D & Bloch KD (2004) Cardiomyocyte-specific overexpression of nitric oxide synthase 3 improves left ventricular performance and reduces compensatory hypertrophy after myocardial infarction. Circ Res 94(9): 1256–1262.
Jaski BE, Jessup ML, Mancini DM, Cappola TP, Pauly DF, Greenberg B, Borow K, Dittrich H, Zsebo KM & Hajjar RJ (2009) Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID trial), a first-in-human phase 1/2 clinical trial. J Card Fail 15(3): 171–181.
Jessup M & Brozena S (2003) Heart failure. N Engl J Med 348(20): 2007–2018. Jessup M, Greenberg B, Mancini D, Cappola T, Pauly DF, Jaski B, Yaroshinsky A, Zsebo
KM, Dittrich H & Hajjar RJ (2011) Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID). Circulation 124(3): 304–313.
Jones JM, Wilson KH, Koch WJ & Milano CA (2002) Adenoviral gene transfer to the heart during cardiopulmonary bypass: effect of myocardial protection technique on transgene expression. Eur J Cardiothorac Surg 21(5): 847–852.
Jooss K, Yang Y, Fisher KJ & Wilson JM (1998) Transduction of dendritic cells by DNA viral vectors directs the immune response to transgene products in muscle fibers. J Virol 72(5): 4212–4223.
Jooss K & Chirmule N (2003) Immunity to adenovirus and adeno-associated viral vectors: implications for gene therapy. Gene Ther 10(11): 955–963.
Kadambi V, Ponniah S, Harrer J, Hoit B, Dorn G, Walsh R & Kranias EG (1996) Cardiac-specific overexpression of phospholamban alters calcium kinetics and resultant cardiomyocyte mechanics in transgenic mice. J Clin Invest 97(2): 533–539.
Kambayashi Y, Nakao K, Itoh H, Hosoda K, Saito Y, Yamada T, Mukoyama M, Arai H, Shirakami G, Suga S, Ogawa Y, Jougasaki M, Minamino N, Kangawa K, Matsuo H, Inouye K & Imura H (1989) Isolation and sequence determination of rat cardiac natriuretic peptide. Biochem Biophys Res Commun 163(1): 233–240.
Kaneda Y (1999) Development of a novel fusogenic viral liposome system (HVJ-liposomes) and its applications to the treatment of acquired diseases. Mol Membr Biol 16(1): 119–122.

150
Kaneshiro Y, Ichihara A, Sakoda M, Takemitsu T, Nabi AHMN, Uddin MN, Nakagawa T, Nishiyama A, Suzuki F, Inagami T & Itoh H (2007) Slowly progressive, angiotensin II–independent glomerulosclerosis in human (pro)renin receptor–transgenic rats. J Am Soc Nephrol 18(6): 1789–1795.
Kang PM & Izumo S (2000) Apoptosis and heart failure: a critical review of the literature. Circ Res 86(11): 1107–1113.
Kapoun AM, Liang F, O'Young G, Damm DL, Quon D, White RT, Munson K, Lam A, Schreiner GF & Protter AA (2004) B-Type natriuretic peptide exerts broad functional opposition to transforming growth factor-β in primary human cardiac fibroblasts: fibrosis, myofibroblast conversion, proliferation, and inflammation. Circ Res 94(4): 453–461.
Kaspar BK, Roth DM, Chin Lai N, Drumm JD, Erickson DA, McKirnan MD & Hammond HK (2005) Myocardial gene transfer and long-term expression following intracoronary delivery of adeno-associated virus. J Gene Med 7(3): 316–324.
Kass-Eisler A, Falck-Pedersen E, Alvira M, Rivera J, Buttrick PM, Wittenberg BA, Cipriani L & Leinwand LA (1993) Quantitative determination of adenovirus–mediated gene delivery to rat cardiac myocytes in vitro and in vivo. Proc Natl Acad Sci U S A 90(24): 11498–11502.
Kato K & Kimura S (1985) S100ao (alpha alpha) protein is mainly located in the heart and striated muscles. Biochim Biophys Acta 842(2–3): 146–150.
Katz MG, Swain JD, White JD, Low D, Stedman H & Bridges CR (2010) Cardiac gene therapy: optimization of gene delivery techniques in vivo. Hum Gene Ther 21(4): 371–380.
Katz MG, Fargnoli AS, Swain JD, Tomasulo CE, Ciccarelli M, Huang ZM, Rabinowitz JE & Bridges CR (2012) AAV6-βARKct gene delivery mediated by molecular cardiac surgery with recirculating delivery (MCARD) in sheep results in robust gene expression and increased adrenergic reserve. J Thorac Cardiovasc Surg 143(3): 720.e3–726.e3.
Katz MG, Fargnoli AS, Tomasulo CE, Pritchette LA & Bridges CR (2011) Model-specific selection of molecular targets for heart failure gene therapy. J Gene Med 13(10): 573–586.
Kaumann AJ, Hall JA, Murray KJ, Wells FC & Brown MJ (1989) A comparison of the effects of adrenaline and noradrenaline on human heart: the role of β1- and β2-adrenoceptors in the stimulation of adenylate cyclase and contractile force. Eur Heart J 10(suppl B): 29–37.
Kawakami R, Saito Y, Kishimoto I, Harada M, Kuwahara K, Takahashi N, Nakagawa Y, Nakanishi M, Tanimoto K, Usami S, Yasuno S, Kinoshita H, Chusho H, Tamura N, Ogawa Y & Nakao K (2004) Overexpression of brain natriuretic peptide facilitates neutrophil infiltration and cardiac matrix metalloproteinase-9 expression after acute myocardial infarction. Circulation 110(21): 3306–3312.
Kawase Y, Ladage D & Hajjar RJ (2011) Rescuing the failing heart by targeted gene transfer. J Am Coll Cardiol 57(10): 1169–1180.

151
Kawase Y, Ly HQ, Prunier F, Lebeche D, Shi Y, Jin H, Hadri L, Yoneyama R, Hoshino K, Takewa Y, Sakata S, Peluso R, Zsebo K, Gwathmey JK, Tardif J, Tanguay J & Hajjar RJ (2008) Reversal of cardiac dysfunction after long-term expression of SERCA2a by gene transfer in a pre-clinical model of heart failure. J Am Coll Cardiol 51(11): 1112–1119.
Kaye DM, Preovolos A, Marshall T, Byrne M, Hoshijima M, Hajjar R, Mariani JA, Pepe S, Chien KR & Power JM (2007) Percutaneous cardiac recirculation–mediated gene transfer of an inhibitory phospholamban peptide reverses advanced heart failure in large animals. J Am Coll Cardiol 50(3): 253–260.
Kiriazis H, Sato Y, Kadambi VJ, Schmidt AG, Gerst MJ, Hoit BD & Kranias EG (2002) Hypertrophy and functional alterations in hyperdynamic phospholamban-knockout mouse hearts under chronic aortic stenosis. Cardiovasc Res 53(2): 372–381.
Kirshenbaum LA & de Moissac D (1997) The bcl-2 gene product prevents programmed cell death of ventricular myocytes. Circulation 96(5): 1580–1585.
Kishimoto I, Tokudome T, Horio T, Garbers D, Nakao K & Kangawa K (2009) Natriuretic peptide signaling via guanylyl cyclase (GC)-A: an endogenous protective mechanism of the heart. Curr Cardiol Rev 5(1): 45–51.
Koch K, Schaefer W, Liehn E, Rammos C, Mueller D, Schroeder J, Dimassi T, Stopinski T & Weber C (2006) Effect of catheter-based transendocardial delivery of stromal cell-derived factor 1α on left ventricular function and perfusion in a porcine model of myocardial infarction. Basic Res Cardiol 101(1): 69–77.
Koller K, Lowe, DG, Bennett, GL, Minamino N, Kangawa K, Matsuo H & Goeddel D (1991) Selective activation of the B natriuretic peptide receptor by C-type natriuretic peptide (CNP). Science 252(5002): 120–123.
Korpisalo P & Yla-Herttuala S (2010) Stimulation of functional vessel growth by gene therapy. Integr Biol (Camb) 2(2–3): 102–112.
Kubalova Z, Gyorke I, Terentyeva R, Viatchenko-Karpinski S, Terentyev D, Williams SC & Gyorke S (2004) Modulation of cytosolic and intra-sarcoplasmic reticulum calcium waves by calsequestrin in rat cardiac myocytes. J Physiol 561(2): 515–524.
Kucia M, Dawn B, Hunt G, Guo Y, Wysoczynski M, Majka M, Ratajczak J, Rezzoug F, Ildstad ST, Bolli R & Ratajczak MZ (2004) Cells expressing early cardiac markers reside in the bone marrow and are mobilized into the peripheral blood after myocardial infarction. Circ Res 95(12): 1191–1199.
Kuhn M, Volker K, Schwarz K, Carbajo-Lozoya J, Flogel U, Jacoby C, Stypmann J, van Eickels M, Gambaryan S, Hartmann M, Werner M, Wieland T, Schrader J & Baba HA (2009) The natriuretic peptide/guanylyl cyclase–A system functions as a stress-responsive regulator of angiogenesis in mice. 119(7): 2019–2030.
Kulik G, Klippel A & Weber MJ (1997) Antiapoptotic signalling by the insulin-like growth factor I receptor, phosphatidylinositol 3-kinase, and Akt. Mol Cell Biol 17(3): 1595–1606.
Kuriyama N, Kuriyama H, Julin C, Lamborn K & Israel M (2000) Pretreatment with protease is a useful experimental strategy for enhancing adenovirus–mediated cancer gene therapy. Hum Gene Ther 11(16): 2219–2230.

152
Kypson A, Hendrickson S, Akhter S, Wilson K, McDonald P, Lilly R, Dolber P, Glower D, Lefkowitz R & Koch W (1999) Adenovirus–mediated gene transfer of the β2-adrenergic receptor to donor hearts enhances cardiac function. Gene Ther 6(7): 1298–1304.
Lafont A, Loirand G, Pacaud P, Vilde F, Lemarchand P & Escande D (1997) Vasomotor dysfunction early after exposure of normal rabbit arteries to an adenoviral vector. Hum Gene Ther 8(9): 1033–1040.
Laguens R, Cabeza Meckert P, Vera Janavel G, De Lorenzi A, Lascano E, Negroni J, del Valle H, Cuniberti L, Martínez V, Dulbecco E, Melo C, Fernandez N, Criscuolo M & Crottogini A (2004) Cardiomyocyte hyperplasia after plasmid–mediated vascular endothelial growth factor gene transfer in pigs with chronic myocardial ischemia. J Gene Med 6(2): 222–227.
Lahteenvuo JE, Lahteenvuo MT, Kivela A, Rosenlew C, Falkevall A, Klar J, Heikura T, Rissanen TT, Vahakangas E, Korpisalo P, Enholm B, Carmeliet P, Alitalo K, Eriksson U & Yla-Herttuala S (2009) Vascular endothelial growth factor-B induces myocardium-specific angiogenesis and arteriogenesis via vascular endothelial growth factor receptor-1– and neuropilin receptor-1–dependent mechanisms. Circulation 119(6): 845–856.
Lai NC, Roth DM, Gao MH, Fine S, Head BP, Zhu J, McKirnan MD, Kwong C, Dalton N, Urasawa K, Roth DA & Hammond HK (2000) Intracoronary delivery of adenovirus encoding adenylyl cyclase VI increases left ventricular function and cAMP-generating capacity. Circulation 102(19): 2396–2401.
Lai NC, Roth DM, Gao MH, Tang T, Dalton N, Lai YY, Spellman M, Clopton P & Hammond HK (2004) Intracoronary adenovirus encoding adenylyl cyclase VI increases left ventricular function in heart failure. Circulation 110(3): 330–336.
Lako-Futo Z, Szokodi I, Sarman B, Foldes G, Tokola H, Ilves M, Leskinen H, Vuolteenaho O, Skoumal R, deChatel R, Ruskoaho H & Toth M (2003) Evidence for a functional role of angiotensin II type 2 receptor in the cardiac hypertrophic process in vivo in the rat heart. Circulation 108(19): 2414–2422.
Lang RE, Tholken H, Ganten D, Luft FC, Ruskoaho H & Unger T (1985) Atrial natriuretic factor-a circulating hormone stimulated by volume loading. Nature 314(6008): 264–266.
Lapidot T & Petit I (2002) Current understanding of stem cell mobilization: the roles of chemokines, proteolytic enzymes, adhesion molecules, cytokines, and stromal cells. Exp Hematol 30(9): 973–981.
Lebeche D, Kaprielian R & Hajjar R (2006) Modulation of action potential duration on myocyte hypertrophic pathways. J Mol Cell Cardiol 40(5): 725–735.
Lee C & Burnett JJ (2007) Natriuretic peptides and therapeutic applications. Heart Fail Rev 12(2): 131–142.
Leineweber K, Brandt K, Wludyka B, Beilfuß A, Ponicke K, Heinroth-Hoffmann I & Brodde O (2002) Ventricular hypertrophy plus neurohumoral activation is necessary to alter the cardiac β-adrenoceptor system in experimental heart failure. Circ Res 91(11): 1056–1062.

153
Leone AM, Rutella S, Bonanno G, Abbate A, Rebuzzi AG, Giovannini S, Lombardi M, Galiuto L, Liuzzo G, Andreotti F, Lanza GA, Contemi AM, Leone G & Crea F (2005) Mobilization of bone marrow-derived stem cells after myocardial infarction and left ventricular function. Eur Heart J 26(12): 1196–1204.
Leotta E, Patejunas G, Murphy G, Szokol J, McGregor L, Carbray J, Hamawy A, Winchester D, Hackett N, Crystal R & Rosengart T (2002) Gene therapy with adenovirus–mediated myocardial transfer of vascular endothelial growth factor 121 improves cardiac performance in a pacing model of congestive heart failure. J Thorac Cardiovasc Surg 123(6): 1101–1113.
Lew W, Chen Z, Guth B & Covell J (1985) Mechanisms of augmented segment shortening in nonischemic areas during acute ischemia of the canine left ventricle. Circ Res 56(3): 351–358.
Li J, Wang D, Qian S, Chen Z, Zhu T & Xiao X (2003) Efficient and long-term intracardiac gene transfer in δ-sarcoglycan-deficiency hamster by adeno-associated virus-2 vectors. Gene Ther 10: 1807–1813.
Li Q, Li B, Wang X, Leri A, Jana KP, Liu Y, Kajstura J, Baserga R & Anversa P (1997) Overexpression of insulin-like growth factor-1 in mice protects from myocyte death after infarction, attenuating ventricular dilation, wall stress, and cardiac hypertrophy. J Clin Invest 100(8): 1991–1999.
Liggett SB, Tepe NM, Lorenz JN, Canning AM, Jantz TD, Mitarai S, Yatani A & Dorn GW (2000) Early and delayed consequences of β2-adrenergic receptor overexpression in mouse hearts: critical role for expression level. Circulation 101(14): 1707–1714.
Lin H, Parmacek M, Morle G, Bolling S & Leiden J (1990) Expression of recombinant genes in myocardium in vivo after direct injection of DNA. Circulation 82(6): 2217–2221.
Lin X, Hanze J, Heese F, Sodmann R & Lang RE (1995) Gene expression of natriuretic peptide receptors in myocardial cells. Circ Res 77(4): 750–758.
Linck B, Bokník P, Eschenhagen T, Muller FU, Neumann J, Nose M, Jones LR, Schmitz W & Scholz H (1996) Messenger RNA expression and immunological quantification of phospholamban and SR-Ca2+-ATPase in failing and nonfailing human hearts. Cardiovasc Res 31(4): 625–632.
Lindemann JP & Watanabe AM (1985) Phosphorylation of phospholamban in intact myocardium. Role of Ca2+-calmodulin-dependent mechanisms. J Biol Chem 260(7): 4516–4525.
Lindemann J, Jones L, Hathaway D, Henry B & Watanabe A (1983) β-Adrenergic stimulation of phospholamban phosphorylation and Ca2+-ATPase activity in guinea pig ventricles. J Biol Chem 258(1): 464–471.
Lips DJ, deWindt LJ, van Kraaij DJW & Doevendans PA (2003) Molecular determinants of myocardial hypertrophy and failure: alternative pathways for beneficial and maladaptive hypertrophy. Eur Heart J 24(10): 883–896.

154
Logeart D, Vinet L, Ragot T, Heimburger M, Louedec L, Michel J, Escoubet B & Mercadier J (2006) Percutaneous intracoronary delivery of SERCA gene increases myocardial function: a tissue doppler imaging echocardiographic study. Am J Physiol Heart Circ Physiol 291(4): H1773–H1779.
Lohse MJ (1995) G-protein-coupled receptor kinases and the heart. Trends Cardiovasc Med 5(2): 63–68.
Lohse MJ, Engelhardt S & Eschenhagen T (2003) What is the role of β-adrenergic signaling in heart failure? Circ Res 93(10): 896–906.
Lompre A, Hajjar RJ, Harding SE, Kranias EG, Lohse MJ & Marks AR (2010) Ca2+ cycling and new therapeutic approaches for heart failure. Circulation 121(6): 822–830.
Lopez M, Wong S, Kishimoto I, Dubois S, Mach V, Friesen J, Garbers D & Beuve A (1995) Salt-resistant hypertension in mice lacking the guanylyl cyclase-A receptor for atrial natriuretic peptide. Nature 378(6552): 65–68.
Luo W, Grupp I, Harrer J, Ponniah S, Grupp G, Duffy J, Doetschman T & Kranias E (1994) Targeted ablation of the phospholamban gene is associated with markedly enhanced myocardial contractility and loss of beta-agonist stimulation. Circ Res 75(3): 401–409.
Luscher TF & Barton M (2000) Endothelins and endothelin receptor antagonists: therapeutic considerations for a novel class of cardiovascular drugs. Circulation 102(19): 2434–2440.
Ly H, Kawase Y, Yoneyama R & Hajjar RJ (2007) Gene therapy in the treatment of heart failure. Physiology 22(2): 81–96.
MacNeill B, Hayase M & Hajjar R (2003) Targeting signaling pathways in heart failure by gene transfer. Curr Atheroscler Rep 5(3): 178–185.
Mahmud H, Sillje HH, Cannon MV, van Gilst WH & de Boer RA (2011) Regulation of the (pro)renin-renin receptor in cardiac remodeling. J Cell Mol Med 16(4): 722–729.
Makinen PI, Koponen JK, Karkkainen AM, Malm TM, Pulkkinen KH, Koistinaho J, Turunen MP, Yla-Herttuala S (2006) Stable RNA interference: comparison of U6 and H1 promoters in endothelial cells and in mouse brain. J Gene Med 8(4): 433–441.
Mann DL & Bristow MR (2005) Mechanisms and models in heart failure. Circulation 111(21): 2837–2849.
Marian AJ, Yu QT, Mann DL, Graham FL & Roberts R (1995) Expression of a mutation causing hypertrophic cardiomyopathy disrupts sarcomere assembly in adult feline cardiac myocytes. Circ Res 77(1): 98–106.
Marks AR (2002) Ryanodine receptors, FKBP12, and heart failure. Front Biosci 7: d970–d979.
Maron BJ (1997) Hypertrophic cardiomyopathy. Lancet 350(9071): 127–133. Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N & Marks AR
(2000) PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101(4): 365–376.
Matsui T, Li L, del Monte F, Fukui Y, Franke TF, Hajjar RJ & Rosenzweig A (1999) Adenoviral gene transfer of activated phosphatidylinositol 3'-kinase and Akt inhibits apoptosis of hypoxic cardiomyocytes in vitro. Circulation 100(23): 2373–2379.

155
Matsui T, Nagoshi T & Rosenzweig A (2003) Akt and PI 3-kinase signaling in cardiomyocyte hypertrophy and survival. Cell Cycle 2(3): 219–222.
Matsui T, Tao J, del Monte F, Lee K, Li L, Picard M, Force TL, Franke TF, Hajjar RJ & Rosenzweig A (2001) Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation 104(3): 330–335.
Matsuno Y, Iwata H, Umeda Y, Takagi H, Mori Y, Miyazaki J, Kosugi A & Hirose H (2003) Nonviral gene gun mediated transfer into the beating heart. ASAIO Journal 49(6): 641–644.
Mattiazzi A, Mundina-Weilenmann C, Guoxiang C, Vittone L & Kranias E (2005) Role of phospholamban phosphorylation on Thr17 in cardiac physiological and pathological conditions. Cardiovasc Res 68(3): 366–375.
Maurice J, Hata J, Shah A, White D, McDonald P, Dolber P, Wilson K, Lefkowitz R, Glower D & Koch W (1999) Enhancement of cardiac function after adenoviral–mediated in vivo intracoronary β2–adrenergic receptor gene delivery. J Clin Invest 104(1): 21–29.
McDonagh TA, Morrison CE, Lawrence A, Ford I, Tunstall-Pedoe H, McMurray JJ & Dargie HJ (1997) Symptomatic and asymptomatic left-ventricular systolic dysfunction in an urban population. Lancet 350(9081): 829–833.
McKee PA, Castelli WP, McNamara PM & Kannel WB (1971) The natural history of congestive heart failure: the framingham study. N Engl J Med 285(26): 1441–1446.
McMurray JJ & Pfeffer MA (2005) Heart failure. Lancet 365(9474): 1877–1889. Mercadier J, Lompre A, Duc P, Boheler K, Fraysse J, Wisnewsky C, Allen P, Komajda M
& Schwartz K (1990) Altered sarcoplasmic reticulum Ca2+-ATPase gene expression in the human ventricle during end-stage heart failure. J Clin Invest 85(1): 305–309.
Mergia A & Heinkelein M (2003) Foamy virus vectors. Curr Top Microbiol Immunol 277: 131–159.
Meyer M, Schillinger W, Pieske B, Holubarsch C, Heilmann C, Posival H, Kuwajima G, Mikoshiba K, Just H & Hasenfuss G (1995) Alterations of sarcoplasmic reticulum proteins in failing human dilated cardiomyopathy. Circulation 92(4): 778–784.
Milano C, Allen L, Rockman H, Dolber P, McMinn T, Chien K, Johnson T, Bond R & Lefkowitz R (1994) Enhanced myocardial function in transgenic mice overexpressing the β2-adrenergic receptor. Science 264(5158): 582–586.
Minamisawa S, Hoshijima M, Chu G, Ward CA, Frank K, Gu Y, Martone ME, Wang Y, Ross J,Jr, Kranias EG, Giles WR & Chien KR (1999) Chronic phospholamban–sarcoplasmic reticulum calcium ATPase interaction is the critical calcium cycling defect in dilated cardiomyopathy. Cell 99(3): 313–322.
Mingozzi F, Liu Y, Dobrzynski E, Kaufhold A, Liu JH, Wang Y, Arruda VR, High KA & Herzog RW (2003) Induction of immune tolerance to coagulation factor IX antigen by in vivo hepatic gene transfer. J Clin Invest 111(9): 1347–1356.
Miyamoto MI, del Monte F, Schmidt U, DiSalvo TS, Kang ZB, Matsui T, Guerrero JL, Gwathmey JK, Rosenzweig A & Hajjar RJ (2000) Adenoviral gene transfer of SERCA2a improves left-ventricular function in aortic-banded rats in transition to heart failure. Proc Natl Acad Sci U S A 97(2): 793–798.

156
Miyoshi H, Takahashi M, Gage FH & Verma IM (1997) Stable and efficient gene transfer into the retina using an HIV-based lentiviral vector. Proc Natl Acad Sci U S A 94(19): 10319–10323.
Moe GW (2006) B-type natriuretic peptide in heart failure. Curr Opin Cardiol 21(3): 208–214.
Molkentin JD (2004) Calcineurin–NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc Res 63(3): 467–475.
Morita E, Yasue H, Yoshimura M, Ogawa H, Jougasaki M, Matsumura T, Mukoyama M & Nakao K (1993) Increased plasma levels of brain natriuretic peptide in patients with acute myocardial infarction. Circulation 88(1): 82–91.
Most P, Bernotat J, Ehlermann P, Pleger ST, Reppel M, Borries M, Niroomand F, Pieske B, Janssen PML, Eschenhagen T, Karczewski P, Smith GL, Koch WJ, Katus HA & Remppis A (2001) S100A1: a regulator of myocardial contractility. Proc Natl Acad Sci U S A 98(24): 13889–13894.
Most P, Boerries M, Eicher C, Schweda C, Volkers M, Wedel T, Sollner S, Katus HA, Remppis A, Aebi U, Koch WJ & Schoenenberger C (2005) Distinct subcellular location of the Ca2+-binding protein S100A1 differentially modulates Ca2+-cycling in ventricular rat cardiomyocytes. J Cell Sci 118(2): 421–431.
Most P, Pleger ST, Volkers M, Heidt B, Boerries M, Weichenhan D, Loffler E, Janssen PM, Eckhart AD, Martini J, Williams ML, Katus HA, Remppis A & Koch WJ (2004) Cardiac adenoviral S100A1 gene delivery rescues failing myocardium. J Clin Invest 114(11): 1550–1563.
Most P, Remppis A, Pleger ST, Katus HA & Koch WJ (2007) S100A1: a novel inotropic regulator of cardiac performance. Transition from molecular physiology to pathophysiological relevance. Am J Physiol Regul Integr Comp Physiol. 293(2): R568–R577.
Most P, Remppis A, Pleger ST, Loffler E, Ehlermann P, Bernotat J, Kleuss C, Heierhorst J, Ruiz P, Witt H, Karczewski P, Mao L, Rockman HA, Duncan SJ, Katus HA & Koch WJ (2003) Transgenic overexpression of the Ca2+-binding protein S100A1 in the heart leads to increased in vivo myocardial contractile performance. J Biol Chem 278(36): 33809–33817.
Most P, Seifert H, Gao E, Funakoshi H, Volkers M, Heierhorst J, Remppis A, Pleger ST, DeGeorge BR, Eckhart AD, Feldman AM & Koch WJ (2006) Cardiac S100A1 protein levels determine contractile performance and propensity toward heart failure after myocardial infarction. Circulation 114(12): 1258–1268.
Motomura S, Zerkowski H, Daul A & Brodde O (1990) On the physiologic role of β-2 adrenoceptors in the human heart: in vitro and in vivo studies. Am Heart J 119(3, Part 1): 608–619.
Movsesian MA, Nishikawa M & Adelstein RS (1984) Phosphorylation of phospholamban by calcium-activated, phospholipid-dependent protein kinase. Stimulation of cardiac sarcoplasmic reticulum calcium uptake. J Biol Chem 259(13): 8029–8032.

157
Movsesian M, Karimi M, Green K & Jones L (1994) Ca2+-transporting ATPase, phospholamban, and calsequestrin levels in nonfailing and failing human myocardium. Circulation 90(2): 653–657.
Mukoyama M, Nakao K, Hosoda K, Suga S, Saito Y, Ogawa Y, Shirakami G, Jougasaki M, Obata K, Yasue H, Kambayashi Y, Inouye K & Imura H (1991) Brain natriuretic peptide as a novel cardiac hormone in humans. Evidence for an exquisite dual natriuretic peptide system, atrial natriuretic peptide and brain natriuretic peptide. J Clin Invest 87(4): 1402–1412.
Muller OJ, Katus HA & Bekeredjian R (2007) Targeting the heart with gene therapy-optimized gene delivery methods. Cardiovasc Res 73(3): 453–462.
Muller OJ, Lange M, Rattunde H, Lorenzen H, Muller M, Frey N, Bittner C, Simonides W, Katus HA & Franz W (2003) Transgenic rat hearts overexpressing SERCA2a show improved contractility under baseline conditions and pressure overload. Cardiovasc Res 59(2): 380–389.
Muller OJ, Leuchs B, Pleger ST, Grimm D, Franz W, Katus HA & Kleinschmidt JA (2006) Improved cardiac gene transfer by transcriptional and transductional targeting of adeno-associated viral vectors. Cardiovasc Res 70(1): 70–78.
Munch G, Bolck B, Hoischen S, Brixius K, Bloch W, Reuter H & Schwinger R (1998) Unchanged protein expression of sarcoplasmic reticulum Ca2+-ATPase, phospholamban, and calsequestrin in terminally failing human myocardium. J Mol Med 76(6): 434–441.
Mundina-Weilenmann C, Vittone L, Rinaldi G, Said M, de Cingolani GC & Mattiazzi A (2000) Endoplasmic reticulum contribution to the relaxant effect of cGMP- and cAMP-elevating agents in feline aorta. Am J Physiol Heart Circ Physiol 278(6): H1856–H1865.
Nabel EG (1995) Gene therapy for cardiovascular disease. Circulation 91(2): 541–548. Nagata K, Marban E, Lawrence JH & Donahue KJ (2001) Phosphodiesterase inhibitor–
mediated potentiation of adenovirus delivery to myocardium. J Mol Cell Cardiol 33(3): 575–580.
Nakamura S, Naruse M, Naruse K, Kawana M, Nishikawa T, Hosoda S, Tanaka I, Yoshimi T, Yoshihara I & Inagami T (1991) Atrial natriuretic peptide and brain natriuretic peptide coexist in the secretory granules of human cardiac myocytes. Am J Hypertens 4(11): 909–912.
Nakayama H, Otsu K, Yamaguchi O, Nishida K, Date M, Hongo K, Kusakari Y, Toyofuku T, Hikoso S, Kashiwase K, Takeda T, Matsumura Y, Kurihara S, Hori M & Tada M (2003) Cardiac-specific overexpression of a high Ca2+ affinity mutant of SERCA2a attenuates in vivo pressure overload cardiac hypertrophy. FASEB J 17(1): 61–63.
Narula J, Haider N, Virmani R, DiSalvo TG, Kolodgie FD, Hajjar RJ, Schmidt U, Semigran MJ, Dec GW & Khaw B (1996) Apoptosis in myocytes in end-stage heart failure. N Engl J Med 335(16): 1182–1189.
Nayak S & Herzog RW (2010) Progress and prospects: immune responses to viral vectors. Gene Ther 17(3): 295–304.

158
NCT00454818: Efficacy and safety study of genetically targeted enzyme replacement therapy for advanced heart failure (CUPID). URI: http://www.clinicaltrials.gov/ show/NCT00454818. Cited 2012/4/10.
NCT00534703: SERCA gene therapy trial. URI: http://www.clinicaltrials.gov/ show/NCT00534703. Cited 2012/4/12.
NCT00787059: ad5.hAC6 gene transfer for CHF. URI: http://www.clinicaltrials.gov/ show/NCT00787059. Cited 2012/4/12.
NCT01002430: Endocardial vascular endothelial growth factor D (VEGF-D) gene therapy for the treatment of severe coronary heart disease (KAT301). URI: http://www.clinicaltrials.gov/show/NCT01002430. Cited 2012/5/20.
NCT01082094: Study to evaluate the safety of a single escalating dose of ACRX-100 in adults with ischemic heart failure. URI: http://www.clinicaltrials.gov/show/ NCT01082094. Cited 2012/4/15.
Nguyen G, Delarue F, Burckle C, Bouzhir L, Giller T & Sraer JD (2002) Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest 109(11): 1417–1427.
Nguyen G (2011) Renin and prorenin receptor in hypertension: what's new? Curr Hypertens Rep 13(1): 79–85.
Nguyen G & Contrepas A (2008) Physiology and pharmacology of the (pro)renin receptor. Curr Opin Pharmacol 8(2): 127–132.
Nguyen G & Muller DN (2010) The Biology of the (pro)renin receptor. J Am Soc Nephrol 21(1): 18–23.
Nian M, Lee P, Khaper N & Liu P (2004) Inflammatory cytokines and postmyocardial infarction remodeling. Circ Res 94(12): 1543–1553.
Nicolaou P, Rodriguez P, Ren X, Zhou X, Qian J, Sadayappan S, Mitton B, Pathak A, Robbins J, Hajjar RJ, Jones K & Kranias EG (2009) Inducible expression of active protein phosphatase-1 inhibitor-1 enhances basal cardiac function and protects against ischemia/reperfusion injury. Circ Res 104(8): 1012–1020.
Nishizaki K, Mazda O, Dohi Y, Kawata T, Mizuguchi K, Kitamura S & Taniguchi S (2000) In vivo gene gun–mediated transduction into rat heart with Epstein-Barr virus-based episomal vectors. Ann Thorac Surg 70(4): 1332–1337.
Niwano K, Arai M, Koitabashi N, Watanabe A, Ikeda Y, Miyoshi H & Kurabayashi M (2008) Lentiviral vector–mediated SERCA2 gene transfer protects against heart failure and left ventricular remodeling after myocardial infarction in rats. Mol Ther 16(6): 1026–1032.
Nyberg P, Heikkila P, Sorsa T, Luostarinen J, Heljasvaara R, Stenman U, Pihlajaniemi T & Salo T (2003) Endostatin inhibits human tongue carcinoma cell invasion and intravasation and blocks the activation of matrix metalloprotease-2, -9, and -13. J Biol Chem 278(25): 22404–22411.
O'Donnell JM (2012) Hemodynamics – New diagnostic and therapeutic approaches. Advantages of catheter-based adenoviral delivery of genes to the heart for studies of cardiac disease. Chapter 8. Publisher: InTech, Edited by A Seda Artis (ISBN 978-953-51-0559-6).

159
O'Donnell JM & Lewandowski ED (2005) Efficient, cardiac-specific adenoviral gene transfer in rat heart by isolated retrograde perfusion in vivo. Gene Ther 12(12): 958–964.
O'Donnell JM, Fields A, Xu X, Chowdhury SAK, Geenen DL & Bi J (2008) Limited functional and metabolic improvements in hypertrophic and healthy rat heart overexpressing the skeletal muscle isoform of SERCA1 by adenoviral gene transfer in vivo. Am J Physiol Heart Circ Physiol 295(6): H2483–H2494.
Oehlenschlager W, Baron D, Schomer H & Currie M (1989) Atrial and brain natriuretic peptides share binding sites in the kidney and heart. Eur J Pharmacol 161(2–3): 159–164.
Ogawa Y, Nakao K, Nakagawa O, Komatsu Y, Hosoda K, Suga S, Arai H, Nagata K, Yoshida N & Imura H (1992) Human C-type natriuretic peptide. Characterization of the gene and peptide. Hypertension 19(6): 809–813.
Oh H, Fujio Y, Kunisada K, Hirota H, Matsui H, Kishimoto T & Yamauchi-Takihara K (1998) Activation of phosphatidylinositol 3-kinase through glycoprotein 130 induces protein kinase B and p70 S6 kinase phosphorylation in cardiac myocytes. J Biol Chem 273(16): 9703–9710.
Oikawa S, Imai M, Ueno A, Tanaka S, Noguchi T, Nakazato H, Kangawa K, Fukuda A & Matsuo H (1984) Cloning and sequence analysis of cDNA encoding a precursor for human atrial natriuretic polypeptide. Nature 309(5970): 724–726.
Oliver PM, Fox JE, Kim R, Rockman HA, Kim H, Reddick RL, Pandey KN, Milgram SL, Smithies O & Maeda N (1997) Hypertension, cardiac hypertrophy, and sudden death in mice lacking natriuretic peptide receptor A. Proc Natl Acad Sci U S A 94(26): 14730–14735.
Pathak A, del Monte F, Zhao W, Schultz J, Lorenz JN, Bodi I, Weiser D, Hahn H, Carr AN, Syed F, Mavila N, Jha L, Qian J, Marreez Y, Chen G, McGraw DW, Heist EK, Guerrero JL, DePaoli-Roach AA, Hajjar RJ & Kranias EG (2005) Enhancement of cardiac function and suppression of heart failure progression by inhibition of protein phosphatase 1. Circ Res 96(7): 756–766.
Paul M, Poyan Mehr A & Kreutz R (2006) Physiology of local renin-angiotensin systems. Physiol Rev. 86(3): 747–803.
Paulus WJ & Bronzwaer JG (2004) Nitric oxide's role in the heart: control of beating or breathing? Am J Physiol Heart Circ Physiol 287(1): H8–H13.
Percherancier Y, Berchiche YA, Slight I, Volkmer-Engert R, Tamamura H, Fujii N, Bouvier M & Heveker N (2005) Bioluminescence resonance energy transfer reveals ligand–induced conformational changes in CXCR4 homo- and heterodimers. J Biol Chem 280(11): 9895–9903.
Periasamy M & Kalyanasundaram A (2007) SERCA pump isoforms: their role in calcium transport and disease. Muscle Nerve 35(4): 430–442.
Periasamy M, Reed TD, Liu LH, Ji Y, Loukianov E, Paul RJ, Nieman ML, Riddle T, Duffy JJ, Doetschman T, Lorenz JN & Shull GE (1999) Impaired cardiac performance in heterozygous mice with a null mutation in the sarco(endo)plasmic reticulum Ca2+-ATPase isoform 2 (SERCA2) gene. J Biol Chem 274(4): 2556–2562.

160
Petrovic D (2004) Cytopathological basis of heart failure-cardiomyocyte apoptosis, interstitial fibrosis and inflammatory cell response. Folia Biol (Praha) 50(2): 58–62.
Pfeffer MA, Pfeffer JM, Fishbein MC, Fletcher PJ, Spadaro J, Kloner RA & Braunwald E (1979) Myocardial infarct size and ventricular function in rats. Circ Res 44(4): 503–512.
Pierkes M, Gambaryan S, Bokník P, Lohmann SM, Schmitz W, Potthast R, Holtwick R & Kuhn M (2002) Increased effects of C-type natriuretic peptide on cardiac ventricular contractility and relaxation in guanylyl cyclase A-deficient mice. Cardiovasc Res 53(4): 852–861.
Pieruzzi F, Abassi ZA & Keiser HR (1995) Expression of renin-angiotensin system components in the heart, kidneys, and lungs of rats with experimental heart failure. Circulation 92(10): 3105–3112.
Pikkarainen S, Tokola H, Majalahti-Palviainen T, Kerkela R, Hautala N, Bhalla SS, Charron F, Nemer M, Vuolteenaho O & Ruskoaho H (2003) GATA-4 is a nuclear mediator of mechanical stretch-activated hypertrophic program. J Biol Chem 278(26): 23807–23816.
Ping P, Anzai T, Gao M & Hammond HK (1997) Adenylyl cyclase and G protein receptor kinase expression during development of heart failure. Am J Physiol Heart Circ Physiol 273(2): H707–H717.
Pitard B, Pollard H, Agbulut O, Lambert O, Vilquin J, Cherel Y, Abadie J, Samuel J, Rigaud J, Menoret S, Anegon I & Escande D (2002) A nonionic amphiphile agent promotes gene delivery in vivo to skeletal and cardiac muscles. Hum Gene Ther 13(14): 1767–1775.
Pleger ST, Most P, Boucher M, Soltys S, Chuprun JK, Pleger W, Gao E, Dasgupta A, Rengo G, Remppis A, Katus HA, Eckhart AD, Rabinowitz JE & Koch WJ (2007) Stable myocardial-specific AAV6-S100A1 gene therapy results in chronic functional heart failure rescue. Circulation 115(19): 2506–2515.
Pleger ST, Shan C, Ksienzyk J, Bekeredjian R, Boekstegers P, Hinkel R, Schinkel S, Leuchs B, Ludwig J, Qiu G, Weber C, Raake P, Koch WJ, Katus HA, Muller OJ & Most P (2011) Cardiac AAV9-S100A1 Gene therapy rescues post-ischemic heart failure in a preclinical large animal model. Sci Transl Med 3(92): 92ra64–92ra64.
Ponnazhagan S, Curiel DT, Shaw DR, Alvarez RD & Siegal GP (2001) Adeno-associated virus for cancer gene therapy. Cancer Res 61(17): 6313–6321.
Potter LR, Yoder AR, Flora DR, Antos LK & Dickey DM (2009) Natriuretic peptides: their structures, receptors, physiologic functions and therapeutic applications. 191: 341–366.
Prasad KR, Xu Y, Yang Z, Acton ST & French BA (2011) Robust cardiomyocyte-specific gene expression following systemic injection of AAV: in vivo gene delivery follows a poisson distribution. Gene Ther 18(1): 43–52.
Pyo RT, Sui J, Dhume A, Palomeque J, Blaxall BC, Diaz G, Tunstead J, Logothetis DE, Hajjar RJ & Schecter AD (2006) CXCR4 modulates contractility in adult cardiac myocytes. J Mol Cell Cardiol 41(5): 834–844.

161
Raake P, Tscheschner H, Reinkober J, Ritterhoff J, Katus H, Koch W & Most P (2011) Gene therapy targets in heart failure: the path to translation. Clin Pharmacol Ther 90(4): 542–553.
Raake P, von Degenfeld G, Hinkel R, Vachenauer R, Sandner T, Beller S, Andrees M, Kupatt C, Schuler G & Boekstegers P (2004) Myocardial gene transfer by selective pressure-regulated retroinfusion of coronary veins: comparison with surgical and percutaneous intramyocardial gene delivery. J Am Coll Cardiol 44(5): 1124–1129.
Raper SE, Chirmule N, Lee FS, Wivel NA, Bagg A, Gao G, Wilson JM & Batshaw ML (2003) Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol Genet Metab 80(1–2): 148–158.
Rapti K, Chaanine AH & Hajjar RJ (2011) Targeted gene therapy for the treatment of heart failure. Can J Cardiol 27(3): 265–283.
Ravingerova T, Barancík M & Strniskova M (2003) Mitogen-activated protein kinases: a new therapeutic target in cardiac pathology. Mol Cell Biochem 247(1): 127–138.
Remacle AG, Shiryaev SA, Oh E, Cieplak P, Srinivasan A, Wei G, Liddington RC, Ratnikov BI, Parent A, Desjardins R, Day R, Smith JW, Lebl M & Strongin AY (2008) Substrate cleavage analysis of furin and related proprotein convertases. J Biol Chem 283(30): 20897–20906.
Remppis A, Greten T, Schafer BW, Hunziker P, Erne P, Katus HA & Heizmann CW (1996) Altered expression of the Ca2+-binding protein S100A1 in human cardiomyopathy. Biochim Biophys Acta 1313(3): 253–257.
Rengo G, Lymperopoulos A, Leosco D & Koch WJ (2011) GRK2 as a novel gene therapy target in heart failure. J Mol Cell Cardiol 50(5): 785–792.
Rengo G, Lymperopoulos A, Zincarelli C, Donniacuo M, Soltys S, Rabinowitz JE & Koch WJ (2009) Myocardial adeno-associated virus serotype 6–βARKct gene therapy improves cardiac function and normalizes the neurohormonal axis in chronic heart failure. Circulation 119(1): 89–98.
Rissanen TT & Yla-Herttuala S (2007) Current status of cardiovascular gene therapy. Mol Ther 15(7): 1233–1247.
Rockman HA, Chien KR, Choi D, Iaccarino G, Hunter JJ, Ross J, Lefkowitz RJ & Koch WJ (1998) Expression of a β-adrenergic receptor kinase 1 inhibitor prevents the development of myocardial failure in gene-targeted mice. Proc Natl Acad Sci U S A 95(12): 7000–7005.
Rockman HA, Koch WJ & Lefkowitz RJ (2002) Seven-transmembrane-spanning receptors and heart function. Nature 415(6868): 206–212.
Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D & Turner MB (2012) Executive summary: heart disease and stroke statistics—2012 update. Circulation 125(1): 188–197.

162
Rota M, Boni A, Urbanek K, Padin-Iruegas ME, Kajstura TJ, Fiore G, Kubo H, Sonnenblick EH, Musso E, Houser SR, Leri A, Sussman MA & Anversa P (2005) Nuclear targeting of Akt enhances ventricular function and myocyte contractility. Circ Res 97(12): 1332–1341.
Roth DM, Lai NC, Gao MH, Fine S, McKirnan MD, Roth DA & Hammond HK (2004) Nitroprusside increases gene transfer associated with intracoronary delivery of adenovirus. Hum Gene Ther 15(10): 989–994.
Roth DM, Gao MH, Lai NC, Drumm J, Dalton N, Zhou JY, Zhu J, Entrikin D & Hammond HK (1999) Cardiac-directed adenylyl cyclase expression improves heart function in murine cardiomyopathy. Circulation 99(24): 3099–3102.
Rowe WP, Huebner RJ, Gilmore LK, Parrott RH & Ward TG (1953) Isolation of a cytopathogenic agent from human adenoids undergoing spontaneous degeneration in tissue culture. Proc Soc Exp Biol Med 84(3): 570–573.
Ruskoaho H, Tholken H & Lang RE (1986) Increase in atrial pressure releases atrial natriuretic peptide from isolated perfused rat hearts. Pflugers Arch 407(2): 170–174.
Ruskoaho H (1992) Atrial natriuretic peptide: synthesis, release, and metabolism. Pharmacol Rev 44(4): 479–602.
Ruskoaho H (2003) Cardiac hormones as diagnostic tools in heart failure. Endocr Rev 24(3): 341–356.
Rutledge EA, Halbert CL & Russell DW (1998) Infectious clones and vectors derived from adeno-associated virus (AAV) serotypes other than AAV type 2. J Virol 72(1): 309–319.
Rysa J, Tenhunen O, Serpi R, Soini Y, Nemer M, Leskinen H & Ruskoaho H (2010) GATA-4 is an angiogenic survival factor of the infarcted heart. Circ Hear Fail 3(3): 440–450.
Sackner-Bernstein JD, Skopicki HA & Aaronson KD (2005) Risk of worsening renal function with nesiritide in patients with acutely decompensated heart failure. Circulation 111(12): 1487–1491.
Sadoshima J & Izumo S (1993) Molecular characterization of angiotensin II–induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Critical role of the AT1 receptor subtype. Circ Res 73(3): 413–423.
Sakoda M, Ichihara A, Kaneshiro Y, Takemitsu T, Nakazato Y, Nabi AH, Nakagawa T, Suzuki F, Inagami T & Itoh H (2007) (Pro)renin receptor–mediated activation of mitogen-activated protein kinases in human vascular smooth muscle cells. Hypertens Res 30(11): 1139–1146.
Sam F, Sawyer DB, Chang DL, Eberli FR, Ngoy S, Jain M, Amin J, Apstein CS & Colucci WS (2000) Progressive left ventricular remodeling and apoptosis late after myocardial infarction in mouse heart. Am J Physiol Heart Circ Physiol 279(1): H422–H428.
Sanbe A, Gulick J, Hayes E, Warshaw D, Osinska H, Chan C, Klevitsky R & Robbins J (2000) Myosin light chain replacement in the heart. Am J Physiol Heart Circ Physiol 279(3): H1355–H1364.

163
Saris JJ, ’t Hoen PA, Garrelds IM, Dekkers DH, den Dunnen JT, Lamers JM & Jan Danser AH (2006) Prorenin induces intracellular signaling in cardiomyocytes independently of angiotensin II. Hypertension 48(4): 564–571.
Saris JJ, Derkx FHM, De Bruin RJA, Dekkers DHW, Lamers JMJ, Saxena PR, Schalekamp MADH & Jan Danser AH (2001) High-affinity prorenin binding to cardiac man-6-P/IGF-II receptors precedes proteolytic activation to renin. Am J Physiol Heart Circ Physiol 280(4): H1706–H1715.
Sato Y, Kiriazis H, Yatani A, Schmidt AG, Hahn H, Ferguson DG, Sako H, Mitarai S, Honda R, Mesnard-Rouiller L, Frank KF, Beyermann B, Wu G, Fujimori K, Dorn GW & Kranias EG (2001) Rescue of contractile parameters and myocyte hypertrophy in calsequestrin overexpressing myocardium by phospholamban ablation. J Biol Chem 276(12): 9392–9399.
Satofuka S, Ichihara A, Nagai N, Koto T, Shinoda H, Noda K, Ozawa Y, Inoue M, Tsubota K, Itoh H, Oike Y & Ishida S (2007) Role of nonproteolytically activated prorenin in pathologic, but not physiologic, retinal neovascularization. Invest Ophthalmol Vis Sci 48(1): 422–429.
Sawada Y, Suda M, Yokoyama H, Kanda T, Sakamaki T, Tanaka S, Nagai R, Abe S & Takeuchi T (1997) Stretch–induced hypertrophic growth of cardiocytes and processing of brain-type natriuretic peptide are controlled by proprotein-processing endoprotease furin. J Biol Chem 272(33): 20545–20554.
Saxena A, Fish JE, White MD, Yu S, Smyth JWP, Shaw RM, DiMaio JM & Srivastava D (2008) Stromal cell–derived factor-1α is cardioprotective after myocardial infarction. Circulation 117(17): 2224–2231.
Schagen FH, Ossevoort M, Toes RE & Hoeben RC (2004) Immune responses against adenoviral vectors and their transgene products: a review of strategies for evasion. Crit Rev Oncol 50(1): 51–70.
Schefe JH, Menk M, Reinemund J, Effertz K, Hobbs RM, Pandolfi PP, Ruiz P, Unger T & Funke-Kaiser H (2006) A novel signal transduction cascade involving direct physical interaction of the renin/prorenin receptor with the transcription factor promyelocytic zinc finger protein. Circ Res 99(12): 1355–1366.
Schefe JH, Neumann C, Goebel M, Danser J, Kirsch S, Gust R, Kintscher U, Unger T & Funke-Kaiser H (2008) Prorenin engages the (pro)renin receptor like renin and both ligand activities are unopposed by aliskiren. J Hypertens 26(9): 1789–1794
Schmidt AG, Zhai J, Carr AN, Gerst MJ, Lorenz JN, Pollesello P, Annila A, Hoit BD & Kranias EG (2002) Structural and functional implications of the phospholamban hinge domain: impaired SR Ca2+ uptake as a primary cause of heart failure. Cardiovasc Res 56(2): 248–259.
Schultz JEJ, Glascock BJ, Witt SA, Nieman ML, Nattamai KJ, Liu LH, Lorenz JN, Shull GE, Kimball TR & Periasamy M (2004) Accelerated onset of heart failure in mice during pressure overload with chronically decreased SERCA2 calcium pump activity. Am J Physiol Heart Circ Physiol 286(3): H1146–H1153.

164
Schwinger RHG, Bohm M, Schmidt U, Karczewski P, Bavendiek U, Flesch M, Krause E & Erdmann E (1995) Unchanged protein levels of SERCA II and phospholamban but reduced Ca2+ uptake and Ca2+-ATPase activity of cardiac sarcoplasmic reticulum from dilated cardiomyopathy patients compared with patients with nonfailing hearts. Circulation 92(11): 3220–3228.
Schwinger RHG, Munch G, Bolck B, Karczewski P, Krause E & Erdmann E (1999) Reduced Ca2+-sensitivity of SERCA 2a in failing human myocardium due to reduced serin-16 phospholamban phoshorylation. J Mol Cell Cardiol 31(3): 479–491.
Segers VF, Tokunou T, Higgins LJ, MacGillivray C, Gannon J & Lee RT (2007) Local delivery of protease-resistant stromal cell derived factor-1 for stem cell recruitment after myocardial infarction. Circulation 116(15): 1683–1692.
Semenov AG, Seferian KR, Tamm NN, Artem'eva MM, Postnikov AB, Bereznikova AV, Kara AN, Medvedeva NA & Katrukha AG (2011) Human pro–B-type natriuretic peptide is processed in the circulation in a rat model. Clin Chem 57(6): 883–890.
Seravalle G, Cattaneo B, Giannattasio C, Perondi R, Saino A, Grassi G & Mancia G (1993) RAA system and cardiovascular control in normal subjects, hypertensives and patients with congestive heart failure. J Hum Hypertens Suppl 2: S13–S18.
Serpi R, Tolonen AM, Huusko J, Rysa J, Tenhunen O, Yla-Herttuala S & Ruskoaho H (2011) Vascular endothelial growth factor-B gene transfer prevents angiotensin II–induced diastolic dysfunction via proliferation and capillary dilatation in rats. Cardiovasc Res 89(1): 204–213.
Serpi R, Tolonen A, Tenhunen O, Pievilainen O, Kubin A, Vaskivuo T, Soini Y, Kerkela R, Leskinen H & Ruskoaho H (2009) Divergent effects of losartan and metoprolol on cardiac remodeling, C-kit+ cells, proliferation and apoptosis in the left ventricle after myocardial infarction. Clin Transl Sci 2(6): 422–430.
Shah AS, Lilly RE, Kypson AP, Tai O, Hata JA, Pippen A, Silvestry SC, Lefkowitz RJ, Glower DD & Koch WJ (2000) Intracoronary adenovirus–mediated delivery and overexpression of the β2-adrenergic receptor in the heart: Prospects for molecular ventricular assistance. Circulation 101(4): 408–414.
Shah AS, White DC, Emani S, Kypson AP, Lilly RE, Wilson K, Glower DD, Lefkowitz RJ & Koch WJ (2001) In vivo ventricular gene delivery of a β-adrenergic receptor kinase inhibitor to the failing heart reverses cardiac dysfunction. Circulation 103(9): 1311–1316.
Shamhart PE & Meszaros JG (2010) Non-fibrillar collagens: key mediators of post-infarction cardiac remodeling? J Mol Cell Cardiol 48(3): 530–537.
Shioi T, McMullen JR, Kang PM, Douglas PS, Obata T, Franke TF, Cantley LC & Izumo S (2002) Akt/Protein kinase B promotes organ growth in transgenic mice. Mol Cell Biol 22(8): 2799–2809.
Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci W & Walsh K (2005) Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest 115(8):2108–2118.
Shiojima I & Walsh K (2006) Regulation of cardiac growth and coronary angiogenesis by the Akt/PKB signaling pathway. Genes Dev 20(24): 3347–3365.

165
Shiraishi I, Melendez J, Ahn Y, Skavdahl M, Murphy E, Welch S, Schaefer E, Walsh K, Rosenzweig A, Torella D, Nurzynska D, Kajstura J, Leri A, Anversa P & Sussman MA (2004) Nuclear targeting of Akt enhances kinase activity and survival of cardiomyocytes. Circ Res 94(7): 884–891.
Shizukuda Y & Buttrick PM (2002) Subtype specific roles of β-adrenergic receptors in apoptosis of adult rat ventricular myocytes. J Mol Cell Cardiol 34(7): 823–831.
Sierro F, Biben C, Martínez-Muñoz L, Mellado M, Ransohoff RM, Li M, Woehl B, Leung H, Groom J, Batten M, Harvey RP, Martínez-A C, Mackay CR & Mackay F (2007) Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc Natl Acad Sci U S A 104(37): 14759–14764.
Sigurdsson A & Swedberg K (1996) The role of neurohormonal activation in chronic heart failure and postmyocardial infarction. Am Heart J 132(1 Pt 2 Su): 229–234.
Silberbach M & Roberts CT,Jr (2001) Natriuretic peptide signalling: molecular and cellular pathways to growth regulation. Cell Signal 13(4): 221–231.
Simmerman HK, Collins JH, Theibert JL, Wegener AD & Jones LR (1986) Sequence analysis of phospholamban. Identification of phosphorylation sites and two major structural domains. J Biol Chem 261(28): 13333–13341.
Smits G, Koepke J & Blaine E (1991) Reversal of low dose angiotension hypertension by angiotensin receptor antagonists. Hypertension 18(1): 17–21.
Soini Y, Virkajarvi N, Lehto VP & Paakko P (1996) Hepatocellular carcinomas with a high proliferation index and a low degree of apoptosis and necrosis are associated with a shortened survival. Br J Cancer 73(9): 1025–1030.
Song J, Zhang X, Carl LL, Qureshi A, Rothblum LI & Cheung JY (2002) Overexpression of phospholemman alters contractility and [Ca2+]i transients in adult rat myocytes. Am J Physiol Heart Circ Physiol 283(2): H576–H583.
Srivastava A, Lusby EW & Berns KI (1983) Nucleotide sequence and organization of the adeno-associated virus 2 genome. J Virol 45(2): 555–564.
Steenaart NAE, Ganim JR, Di Salvo J & Kranias EG (1992) The phospholamban phosphatase associated with cardiac sarcoplasmic reticulum is a type 1 enzyme. Arch Biochem Biophys 293(1): 17–24.
Steinberg SF (1999) The Molecular basis for distinct β-adrenergic receptor subtype actions in cardiomyocytes. Circ Res 85(11): 1101–1111.
Stewart MJ, Plautz GE, Del Buono L, Yang ZY, Xu L, Gao X, Huang L, Nabel EG & Nabel GJ (1992) Gene transfer in vivo with DNA–liposome complexes: safety and acute toxicity in mice. Hum Gene Ther. 3(3): 267–275.
Stratford-Perricaudet L, Makeh I, Perricaudet M & Briand P (1992) Widespread long-term gene transfer to mouse skeletal muscles and heart. J Clin Invest 90(2): 626–630.
Strayer D, Agrawal L, Cordelier P, Liu B, Louboutin J, Marusich E, McKee H, Ren C & Strayer M (2006) Long-term gene expression in dividing and nondividing cells using SV40-derived vectors. Mol Biotechnol 34(2): 257–270.

166
Suckau L, Fechner H, Chemaly E, Krohn S, Hadri L, Kockskamper J, Westermann D, Bisping E, Ly H, Wang X, Kawase Y, Chen J, Liang L, Sipo I, Vetter R, Weger S, Kurreck J, Erdmann V, Tschope C, Pieske B, Lebeche D, Schultheiss H, Hajjar RJ & Poller WC (2009) Long-term cardiac-targeted RNA interference for the treatment of heart failure restores cardiac function and reduces pathological hypertrophy. Circulation 119(9): 1241–1252.
Sudoh T, Kangawa K, Minamino N, Matsuo H (1988) A new natriuretic peptide in porcine brain. Nature 332(6159):78–81.
Suga S, Nakao K, Hosoda K, Mukoyama M, Ogawa Y, Shirakami G, Arai H, Saito Y, Kambayashi Y & Inouye K (1992) Receptor selectivity of natriuretic peptide family, atrial natriuretic peptide, brain natriuretic peptide, and C-type natriuretic peptide. Endocrinology 130(1): 229–239.
Sun Y, Cleutjens JPM, Diaz-Arias AA & Weber KT (1994) Cardiac angiotensin converting enzyme and myocardial fibrosis in the rat. Cardiovasc Res 28(9): 1423–1432.
Suo M, Hautala N, Foldes G, Szokodi I, Tóth M, Leskinen H, Uusimaa P, Vuolteenaho O, Nemer M & Ruskoaho H (2002) Posttranscriptional control of BNP gene expression in angiotensin II–induced hypertension. Hypertension 39(3): 803–808.
Sutton MG & Sharpe N (2000) Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation 101(25): 2981–2988.
Svensson EC, Marshall DJ, Woodard K, Lin H, Jiang F, Chu L & Leiden JM (1999) Efficient and stable transduction of cardiomyocytes after intramyocardial injection or intracoronary perfusion with recombinant adeno-associated virus vectors. Circulation 99(2): 201–205.
Swedberg K, Cleland J, Dargie H, Drexler H, Follath F, Komajda M, Tavazzi L, Smiseth OA, Gavazzi A, Haverich A, Hoes A, Jaarsma T, Korewicki J, Levy S, Linde C, Lopez-Sendon J, Nieminen MS, Pierard L & Remme WJ (2005) Guidelines for the diagnosis and treatment of chronic heart failure: executive summary (update 2005). Eur Heart J 26(11): 1115–1140.
Sweeney HL, Feng HS, Yang Z & Watkins H (1998) Functional analyses of troponin T mutations that cause hypertrophic cardiomyopathy: insights into disease pathogenesis and troponin function. Proc Natl Acad Sci U S A 95(24): 14406–14410.
Symes JF (2001) Gene therapy for ischemic heart disease: therapeutic potential. Am J Cardiovasc Drugs 1(3); 159–166.
Szatkowski M, Westfall M, Gomez C, Wahr P, Michele D, DelloRusso C, Turner I, Hong K, Albayya F & Metzger J (2001) In vivo acceleration of heart relaxation performance by parvalbumin gene delivery. J Clin Invest 107(2): 191–198.
Szokodi I, Kerkela R, Kubin A, Sarman B, Pikkarainen S, Konyi A, Horvath IG, Papp L, Toth M, Skoumal R & Ruskoaho H (2008) Functionally opposing Roles of extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase in the regulation of cardiac contractility. Circulation 118(16): 1651–1658.

167
Tada M, Kirchberger MA & Katz AM (1975) Phosphorylation of a 22,000-dalton component of the cardiac sarcoplasmic reticulum by adenosine 3':5'-monophosphate-dependent protein kinase. J Biol Chem 250(7): 2640–2647.
Takahashi M, Shiraishi H, Ishibashi Y, Blade KL, McDermott PJ, Menick DR, Kuppuswamy D & Cooper G (2003) Phenotypic consequences of β1-tubulin expression and MAP4 decoration of microtubules in adult cardiocytes. Am J Physiol Heart Circ Physiol 285(5): H2072–H2083.
Takahashi T, Allen P & Izumo S (1992) Expression of A-, B-, and C-type natriuretic peptide genes in failing and developing human ventricles. Correlation with expression of the Ca2+-ATPase gene. Circ Res 71(1): 9–17.
Takahashi T, Tang T, Lai NC, Roth DM, Rebolledo B, Saito M, Lew WYW, Clopton P & Hammond HK (2006) Increased cardiac adenylyl cyclase expression is associated with increased survival after myocardial infarction. Circulation 114(5): 388–396.
Tamura N, Ogawa Y, Chusho H, Nakamura K, Nakao K, Suda M, Kasahara M, Hashimoto R, Katsuura G, Mukoyama M, Itoh H, Saito Y, Tanaka I, Otani H, Katsuki M & Nakao K (2000) Cardiac fibrosis in mice lacking brain natriuretic peptide. Proc Natl Acad Sci U S A 97(8): 4239–4244.
Tanaka M, Hiroe M, Nishikawa T, Sato T & Marumo F (1994) Cellular localization and structural characterization of natriuretic peptide-expressing ventricular myocytes from patients with dilated cardiomyopathy. J Histochem Cytochem 42(9): 1207–1214.
Tang J, Wang J, Song H, Huang Y, Yang J, Kong X, Guo L, Zheng F & Zhang L (2010) Adenovirus–mediated stromal cell-derived factor-1α gene transfer improves cardiac structure and function after experimental myocardial infarction through angiogenic and antifibrotic actions. Mol Biol Rep 37(4): 1957–1969.
Tang YL, Qian K, Zhang YC, Shen L & Phillips MI (2005) Mobilizing of haematopoietic stem cells to ischemic myocardium by plasmid–mediated stromal-cell-derived factor-1α treatment. Regul Pept 125(1–3): 1–8.
Tawaragi Y, Fuchimura K, Tanaka S, Minamino N, Kangawa K & Matsuo H (1991) Gene and precursor structures of human C-type natriuretic peptide. Biochem Biophys Res Commun 175(2): 645–651.
Tripathy S, Black H, Goldwasser E & Leiden J (1996) Immune responses to the renin-angiotensin system. Nat Med (5):545–550.
Teerlink JR (1996) Neurohumoral mechanisms in heart failure: a central role for the renin-angiotensin system. J Cardiovasc Pharmacol 27 Suppl 2: S1–8.
Tenhunen O, Rysa J, Ilves M, Soini Y, Ruskoaho H & Leskinen H (2006a) Identification of cell cycle regulatory and inflammatory genes as predominant targets of p38 mitogen-activated protein kinase in the heart. Circ Res 99(5): 485–493.
Tenhunen O, Sarman B, Kerkela R, Szokodi I, Papp L, Toth M & Ruskoaho H (2004) Mitogen-activated protein kinases p38 and ERK 1/2 mediate the wall stress–induced activation of GATA-4 binding in adult heart. J Biol Chem 279(23): 24852–24860.

168
Tenhunen O, Soini Y, Ilves M, Rysa J, Tuukkanen J, Serpi R, Pennanen H, Ruskoaho H & Leskinen H (2006b) p38 Kinase rescues failing myocardium after myocardial infarction: evidence for angiogenic and anti-apoptotic mechanisms. FASEB J 20(11): 1907–1909.
Terentyev D, Cala SE, Houle TD, Viatchenko-Karpinski S, Gyorke I, Terentyeva R, Williams SC & Gyorke S (2005) Triadin overexpression stimulates excitation-contraction coupling and increases predisposition to cellular arrhythmia in cardiac myocytes. Circ Res 96(6): 651–658.
Tevaearai HT, Eckhart AD, Walton GB, Keys JR, Wilson K & Koch WJ (2002) Myocardial gene transfer and overexpression of β2-adrenergic receptors potentiates the functional recovery of unloaded failing hearts. Circulation 106(1): 124–129.
Tilemann L, Ishikawa K, Weber T & Hajjar RJ (2012) Gene therapy for heart failure. Circ Res 110(5): 777–793.
Tomiyasu K, Oda Y, Nomura M, Satoh E, Fushiki S, Imanishi J, Kondo M & Mazda O (2000) Direct intra-cardiomuscular transfer of β2-adrenergic receptor gene augments cardiac output in cardiomyopathic hamsters. Gene Ther 7(24): 2087–2093.
Townsend D, Blankinship MJ, Allen JM, Gregorevic P, Chamberlain JS & Metzger JM (2007) Systemic administration of micro-dystrophin restores cardiac geometry and prevents dobutamine–induced cardiac pump failure. Mol Ther 15(6): 1086–1092.
Tripathy S, Black H, Goldwasser E & Leiden J (1996) Immune responses to transgene-encoded proteins limit the stability of gene expression after injection of replication-defective adenovirus vectors. Nat Med 2(5): 545–550.
Trono D (2000) Lentiviral vectors: turning a deadly foe into a therapeutic agent. Gene Ther 71(1): 20–23.
Tsuruda T, Boerrigter G, Huntley BK, Noser JA, Cataliotti A, Costello-Boerrigter LC, Chen HH & Burnett JC,Jr (2002) Brain natriuretic peptide Is produced in cardiac fibroblasts and induces matrix metalloproteinases. Circ Res 91(12): 1127–1134.
Umeda Y, Iwata H, Yoshikawa S, Matsuno Y, Marui T, Nitta T, Idia Y, Takagi H, Mori Y, Miyazaki J, Kosugi A & Hirose H (2002) Gene gun–mediated CTLA4Ig-gene transfer for modification of allogeneic cardiac grafts. Transplant Proc 34(7): 2622–2623.
Ungerer M, Bohm M, Elce J, Erdmann E & Lohse M (1993) Altered expression of β-adrenergic receptor kinase and β1-adrenergic receptors in the failing human heart. Circulation 87(2): 454–463.
Unzek S, Zhang M, Mal N, Mills W, Laurita K & Penn M (2007) SDF-1 recruits cardiac stem cell-like cells that depolarize in vivo. Cell Transplant 16(9): 879–886.
Vailhe B, Vittet D, Feige JJ (2001) In vitro models of vasculogenesis and angiogenesis. Lab Invest 81(4): 439–452.
Ver Heyen M, Heymans S, Antoons G, Reed T, Periasamy M, Awede B, Lebacq J, Vangheluwe P, Dewerchin M, Collen D, Sipido K, Carmeliet P & Wuytack F (2001) Replacement of the muscle-specific sarcoplasmic reticulum Ca2+-ATPase isoform SERCA2a by the nonmuscle SERCA2b homologue causes mild concentric hypertrophy and impairs contraction-relaxation of the heart. Circ Res 89(9): 838–846.

169
Vera Janavel G, Crottogini A, Cabeza Meckert P, Cuniberti L, Mele A, Papouchado M, Fernandez N, Bercovich A, Criscuolo M, Melo C & Laguens R (2006) Plasmid–mediated VEGF gene transfer induces cardiomyogenesis and reduces myocardial infarct size in sheep. Gene Ther 13(15): 1133–1142.
Verboomen H, Wuytack F, Van den Bosch L, Mertens L & Casteels R (1994) The functional importance of the extreme C-terminal tail in the gene 2 organellar Ca2+-transport ATPase (SERCA2a/b). Biochem J 303(Pt 3): 979–984.
Verma IM & Somia N (1997) Gene therapy - promises, problems and prospects. Nature 389(6648): 239–242.
Vinge LE, Raake PW & Koch WJ (2008) Gene therapy in heart failure. Circ Res 102(12): 1458–1470.
Virag JA, Rolle ML, Reece J, Hardouin S, Feigl EO & Murry CE (2007) Fibroblast growth factor-2 regulates myocardial infarct repair: effects on cell proliferation, scar contraction, and ventricular function. Am J Pathol. 171(5): 1431–1440.
Vuolteenaho O, Arjamaa O & Ling N (1985) Atrial natriuretic polypeptides (ANP): rat atria store high molecular weight precursor but secrete processed peptides of 25–35 amino acids. Biochem Biophys Res Commun 129(1): 82–88.
Wahr PA, Michele DE & Metzger JM (1999) Parvalbumin gene transfer corrects diastolic dysfunction in diseased cardiac myocytes. Proc Natl Acad Sci U S A 96(21): 11982–11985.
Walsh K & Shiojima I (2007) Cardiac growth and angiogenesis coordinated by intertissue interactions. J Clin Invest 117(11): 3176–3179.
Wang J, Faust SM & Rabinowitz JE (2011) The next step in gene delivery: molecular engineering of adeno-associated virus serotypes. J Mol Cell Cardiol 50(5): 793–802.
Wang T & Brown MJ (2004) Differential expression of adenylyl cyclase subtypes in human cardiovascular system. Mol Cell Endocrinol 223(1–2): 55–62.
Wang TJ, Evans JC, Benjamin EJ, Levy D, LeRoy EC & Vasan RS (2003) Natural history of asymptomatic left ventricular systolic dysfunction in the community. Circulation 108(8): 977–982.
Wang Z, Zhu T, Qiao C, Zhou L, Wang B, Zhang J, Chen C, Li J & Xiao X (2005) Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat Biotech 23(3): 321–328.
Wasala NB, Shin J & Duan D (2011) The evolution of heart gene delivery vectors. J Gene Med 13(10): 557–565.
Wegener AD, Simmerman HK, Lindemann JP & Jones LR (1989) Phospholamban phosphorylation in intact ventricles. Phosphorylation of serine 16 and threonine 17 in response to β-adrenergic stimulation. J Biol Chem 264(19): 11468–11474.
Weig H, Laugwitz K, Moretti A, Kronsbein K, Stadele C, Bruning S, Seyfarth M, Brill T, Schomig A & Ungerer M (2000) Enhanced cardiac contractility after gene transfer of V2 vasopressin receptors in vivo by ultrasound-guided injection or transcoronary delivery. Circulation 101(13): 1578–1585.
Weisman HF & Healy B (1987) Myocardial infarct expansion, infarct extension, and reinfarction: pathophysiologic concepts. Prog Cardiovasc Dis 30(2): 73–110.

170
Weisser-Thomas J, Dieterich E, Janssen PML, Schmidt-Schweda S, Maier LS, Sumbilla C & Pieske B (2005) Method-related effects of adenovirus–mediated LacZ and SERCA1 gene transfer on contractile behavior of cultured failing human cardiomyocytes. J Pharmacol Toxicol Methods 51(2): 91–103.
Westfall MV & Metzger JM (2003) Gene transfer of troponin I isoforms, mutants, and chimeras. Adv Exp Med Biol 538:169–174
Whelan RS, Kaplinskiy V & Kitsis RN (2010) Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol 72(1): 19–44.
Williams ML, Hata JA, Schroder J, Rampersaud E, Petrofski J, Jakoi A, Milano CA & Koch WJ (2004) Targeted β-adrenergic receptor kinase (βARK1) inhibition by gene transfer in failing human hearts. Circulation 109(13): 1590–1593.
Wu F, Yan W, Pan J, Morser J & Wu Q (2002) Processing of pro-atrial natriuretic peptide by corin in cardiac myocytes. J Biol Chem 277(19): 16900–16905.
Xiao RP, Ji X & Lakatta EG (1995) Functional coupling of the β2-adrenoceptor to a pertussis toxin-sensitive G protein in cardiac myocytes. Mol Pharmacol 47(2): 322–329.
Yamada K & Yoshida S (1991) role of endogenous endothelin in renal function during altered sodium balance. J Cardiovasc Pharmacol 17 Suppl 7: S290–S292.
Yamahara K, Itoh H, Chun T, Ogawa Y, Yamashita J, Sawada N, Fukunaga Y, Sone M, Yurugi-Kobayashi T, Miyashita K, Tsujimoto H, Kook H, Feil R, Garbers DL, Hofmann F & Nakao K (2003) Significance and therapeutic potential of the natriuretic peptides/cGMP/cGMP-dependent protein kinase pathway in vascular regeneration. Proc Natl Acad Sci U S A 100(6): 3404–3409.
Yamani MH, Ratliff NB, Cook DJ, Tuzcu EM, Yu Y, Hobbs R, Rincon G, Bott-Silverman C, Young JB, Smedira N & Starling RC (2005) Peritransplant ischemic injury is associated with up-regulation of stromal cell-derived factor-1. J Am Coll Cardiol 46(6): 1029–1035.
Yang Y, Nunes FA, Berencsi K, Furth EE, Gonczol E & Wilson JM (1994) Cellular immunity to viral antigens limits E1-deleted adenoviruses for gene therapy. Proc Natl Acad Sci U S A 91(10): 4407–4411.
Yasufumi K (2000) Virosomes: evolution of the liposome as a targeted drug delivery system. Adv Drug Deliv Rev 43(2–3): 197–205.
Yi BA, Wernet O & Chien KR (2010) Pregenerative medicine: developmental paradigms in the biology of cardiovascular regeneration. J Clin Invest 120(1): 20–28.
Yla-Herttuala S, Rissanen TT, Vajanto I & Hartikainen J (2007) Vascular endothelial growth factors: biology and current status of clinical applications in cardiovascular medicine. J Am Coll Cardiol 49(10):1015–1026.
Yoshimura M, Yasue H & Ogawa H (2001) Pathophysiological significance and clinical application of ANP and BNP in patients with heart failure. Can J Physiol Pharmacol 79(8): 730–735.

171
Yoshimura M, Yasue H, Morita E, Sakaino N, Jougasaki M, Kurose M, Mukoyama M, Saito Y, Nakao K & Imura H (1991) Hemodynamic, renal, and hormonal responses to brain natriuretic peptide infusion in patients with congestive heart failure. Circulation 84(4): 1581–1588.
Yoshitomi Y, Nishikimi T, Kojima S, Kuramochi M, Takishita S, Kangawa K & Matsuo H (1998) Plasma natriuretic peptides as indicators of left ventricular remodeling after myocardial infarction. Int J Cardiol 64(2): 153–160.
Zaiss AK & Muruve DA (2008) Immunity to adeno-associated virus vectors in animals and humans: a continued challenge. Gene Ther 15(11): 808–816.
Zaugg M, Xu W, Lucchinetti E, Shafiq SA, Jamali NZ & Siddiqui MA (2000) β-adrenergic receptor subtypes differentially affect apoptosis in adult rat ventricular myocytes. Circulation 102(3): 344–350.
Zhai J, Schmidt AG, Hoit BD, Kimura Y, MacLennan DH & Kranias EG (2000) Cardiac-specific overexpression of a superinhibitory pentameric phospholamban mutant enhances Inhibition of cardiac function in vivo. J Biol Chem 275(14): 10538–10544.
Zhang M, Mal N, Kiedrowski M, Chacko M, Askari AT, Popovic ZB, Koc ON & Penn MS (2007) SDF-1 expression by mesenchymal stem cells results in trophic support of cardiac myocytes after myocardial infarction. FASEB J 21(12): 3197–3207.
Zhang T & Brown JH (2004) Role of Ca2+/calmodulin-dependent protein kinase II in cardiac hypertrophy and heart failure. Cardiovasc Res 63(3): 476–486.
Zhao J, Pettigrew GJ, Thomas J, Vandenberg JI, Delriviere L, Bolton EM, Carmichael A, Martin JL, Marber MS & Lever AM (2002) Lentiviral vectors for delivery of genes into neonatal and adult ventricular cardiac myocytes in vitro and in vivo. Basic Res Cardiol 97(5): 348–358.
Zhao T, Zhao W, Chen Y, Ahokas RA & Sun Y (2010) Vascular endothelial growth factor (VEGF)-A: role on cardiac angiogenesis following myocardial infarction. Microvasc Res 80(2): 188–194.
Zhu T, Zhou L, Mori S, Wang Z, McTiernan CF, Qiao C, Chen C, Wang DW, Li J & Xiao X (2005) Sustained whole-body functional rescue in congestive heart failure and muscular dystrophy hamsters by systemic gene transfer. Circulation 112(17): 2650–2659.
Zhu W, Zheng M, Koch WJ, Lefkowitz RJ, Kobilka BK & Xiao R (2001) Dual modulation of cell survival and cell death by β2-adrenergic signaling in adult mouse cardiac myocytes. Proc Natl Acad Sci U S A 98(4): 1607–1612.
Zincarelli C, Soltys S, Rengo G & Rabinowitz JE (2008) Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol Ther 16(6): 1073–1080.
Ziolo MT, Maier LS, Piacentino V, Bossuyt J, Houser SR & Bers DM (2004) Myocyte nitric oxide synthase 2 contributes to blunted β-adrenergic response in failing human hearts by decreasing Ca2+ transients. Circulation 109(15): 1886–1891.

172
Zvaritch E, Backx PH, Jirik F, Kimura Y, de Leon S, Schmidt AG, Hoit BD, Lester JW, Kranias EG & MacLennan DH (2000) The transgenic expression of highly inhibitory monomeric forms of phospholamban in mouse heart impairs cardiac contractility. J Biol Chem 275(20): 14985–14991.

173
Original articles
I Moilanen AM, Rysä J, Mustonen E, Serpi R, Aro J, Tokola H, Leskinen H, Manninen A, Levijoki J, Vuolteenaho O & Ruskoaho H (2011) Intramyocardial BNP gene delivery improves cardiac function through distinct context-dependent mechanisms. Circ Heart Fail 4: 483–95.
II Ala-Kopsala M, Moilanen AM, Rysä J, Ruskoaho H & Vuolteenaho O (2010) Characterization of molecular forms of N-terminal B-type natriuretic peptide in vitro. Clin Chem 56: 1822–9.
III Moilanen AM, Rysä J, Serpi R, Mustonen E, Szabó Z, Aro J, Näpänkangas J, Tenhunen O, Sutinen M, Salo T & Ruskoaho H (2012) (Pro)renin receptor triggers distinct angiotensin II-independent extracellular matrix remodelling and deterioration of cardiac function. PLoS ONE 7(7): e41404.
Reprinted with permission from Lippincott, Williams & Wilkins (I) and American
Association of Community Colleges (II).
Original articles are not included in the electronic version of the dissertation.

174

A C T A U N I V E R S I T A T I S O U L U E N S I S
Book orders:Granum: Virtual book storehttp://granum.uta.fi/granum/
S E R I E S D M E D I C A
1153. Meriläinen, Merja (2012) Tehohoitopotilaan hoitoympäristö : psyykkinenelämänlaatu ja toipuminen
1154. Liisanantti, Janne (2012) Acute drug poisoning: outcome and factors affectingoutcome
1155. Markkanen, Piia (2012) Human δ opioid receptor : The effect of Phe27Cyspolymorphism, N-linked glycosylation and SERCA2b interaction on receptorprocessing and trafficking
1156. Hannula, Annukka (2012) Imaging studies of the urinary tract in children withacute urinary tract infection
1157. Korvala, Johanna (2012) In quest of genetic susceptibility to disorders manifestingin fractures : Assessing the significance of genetic factors in femoral neck stressfractures and childhood non-OI primary osteoporosis
1158. Takala, Heikki (2012) Biomarkers in esophageal cancer
1159. Hyry, Marjo (2012) Lysyl hydroxylases 1 and 2 : Characterization of their in vivoroles in mouse and the molecular level consequences of the lysyl hydroxylase 2mutations found in Bruck syndrome
1160. Laatio, Liisa (2012) In search of new prognostic molecular markers in ovariancancer
1161. Klintrup, Kai (2012) Inflammation and invasive margin in colorectal cancer
1162. Buler, Marcin (2012) Energy sensing factors modulate expression of inflammatorymediators, mitochondria acetylation and drug metabolism in the liver
1163. Salo, Jarmo (2012) Long-term consequences and prevention of urinary tractinfections in childhood
1164. Ronkainen, Veli-Pekka (2012) Effects of chronic hypoxia on myocardial geneexpression and function
1165. Kinnula, Sohvi (2012) Hospital-associated infections and the safety of alcoholhand gels in children
1166. Niemelä, Mika (2012) Structured child-centred interventions to support familieswith a parent suffering from cancer : from practice-based evidence towardsevidence-based practice
1167. Pohjolainen, Virva (2012) Characterization of non-collagenous extracellularmatrix proteins in cardiac and aortic valve remodelling

ABCDEFG
UNIVERS ITY OF OULU P.O.B . 7500 F I -90014 UNIVERS ITY OF OULU F INLAND
A C T A U N I V E R S I T A T I S O U L U E N S I S
S E R I E S E D I T O R S
SCIENTIAE RERUM NATURALIUM
HUMANIORA
TECHNICA
MEDICA
SCIENTIAE RERUM SOCIALIUM
SCRIPTA ACADEMICA
OECONOMICA
EDITOR IN CHIEF
PUBLICATIONS EDITOR
Senior Assistant Jorma Arhippainen
Lecturer Santeri Palviainen
Professor Hannu Heusala
Professor Olli Vuolteenaho
Senior Researcher Eila Estola
Director Sinikka Eskelinen
Professor Jari Juga
Professor Olli Vuolteenaho
Publications Editor Kirsti Nurkkala
ISBN 978-951-42-9912-4 (Paperback)ISBN 978-951-42-9913-1 (PDF)ISSN 0355-3221 (Print)ISSN 1796-2234 (Online)
U N I V E R S I TAT I S O U L U E N S I S
MEDICA
ACTAD
D 1168
ACTA
Anne-M
ari Moilanen
OULU 2012
D 1168
Anne-Mari Moilanen
IDENTIFICATION OF NOVEL DRUG TARGETS FORTHE TREATMENT OF HEART FAILURE
UNIVERSITY OF OULU GRADUATE SCHOOL;UNIVERSITY OF OULU,FACULTY OF MEDICINE, INSTITUTE OF BIOMEDICINE,DEPARTMENT OF PHARMACOLOGY AND TOXICOLOGY;BIOCENTER OULU