organophosphorus compounds. xii. a new synthesis of phosphindolines and phosphindoles
TRANSCRIPT

Aust. J. Chem., 1974, 27, 831-9
Organophosphorus Compounds. XII* A New Synthesis of Phosphindolines and Phosphindoles
D. J. Collins, L. E. Rowley and J. M. Swan
Chemistry Department, Monash University, Clayton, Vic. 3168,
Abstract
The reaction of 2-phenylethylphosphonous dichloride with zinc chloride at 170' followed by hydrolysis with concentrated hydrochloric acid, and oxidation, gave 72 % of 1-hydroxyphosphindoline 1-oxide. Treatment of the derived phosphinic chloride with vinylmagnesium bromide at -40' afforded 1-vinylphosphindoline 1-oxide which underwent addition of dimethylamine to yield 1-(2'-dimethylaminoethy1)phosphindoline 1-oxide. The conversions of 1-chlorophosphindoline - . - -
1-bxide intol-ethyl- and 1-phenyl-phosphindoline 1-oxide are described. Bromination of l-methoxy- phosphindoline I-oxide with N-bromosuccinimide followed by dehydrobromination afforded 1-methoxyphosphindole 1-oxide.
1-(2'-Dimethylaminoethy1)phosphindoline 1-oxide and the corresponding phosphine sulphide were assayed for analgetic activity.
Introduction
In a preceding paper1 we described a new synthesis of several 1,2,3,4-tetra- hydrophosphinolines (3a) which were prepared en route to some 1,2,3,4,5,6-hexa- hydro-l,5-methano-4,1-benzazaphosphocines (2).' The latter compounds are phos-
(1) R = alkyl or sub- (2) R = alkyl stituted alkyl X =lone electron pair,
0, or S
(3a) R = H, OH, CI, OR, alkyl, etc. (4a) R = CH2CH,NMe, X = lone electron pair, 0, or S X=O
(3b) R =CH,CH,NMe, (4b) R = CH2CH2NMe2 X=S X=S
*Part XI, Aust. J. Chem., 1974, 27, 815.
Rowley, L. E., and Swan, J. M., Aust. J. Chem., 1974, 27, 801. Collins, D. J., Rowley, L. E., and Swan, J. M., Aust. J. Chem., 1974, 27, 815.

D. J. Collins, L. E. Rowley and J. M. Swan
phorus isosteres of the morphine-like analgetics (benzomorphans) (1). Success in the synthesis of the 1,2,3,4-tetrahydrophosphinolines (3a) led us to investigate whether phosphindolines could be synthesized by the same method of ring closure of the phosphorus heterocycle. We also wished to synthesize the 1-(2'-dimethylamino- ethy1)phosphindoline 1-oxide (4a) and 1-sulphide (4b), which were of interest because in initial tests the tetrahydrophosphinoline 1-sulphide (3b) appeared to show appreci- able analgesic activity, whilst the benzomorphan isosteres (2) were inactive.
This paper describes the synthesis of several phosphindolines and phosphindoles, including the amino compounds (4a) and (4b).
Synthesis
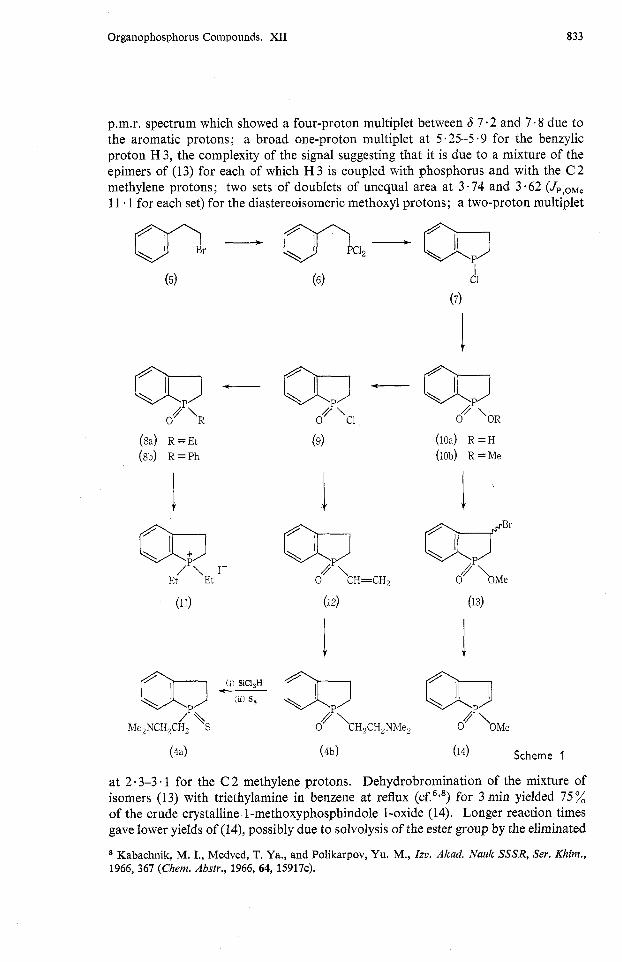
None of the relatively few methods of ring closure which have been ~ s e d ~ - ~ for the synthesis of phosphindolines and phosphindoles is readily applicable to the synthesis of the desired 1-(2'-aminoethyl)phosphindolines (4a) and (4b). These were obtained by an adaptation of our synthesis of 1,2,3,4-tetrahydrophosphinolines.'
2-Phenylethyl bromide (5) (Scheme 1) was converted into 2-phenylethylphosphonous dichloride (6) in 45 % yield by reaction of bis(2-phenylethy1)cadmium with phosphorus trichloride at -20". Although the g.1.c. of the crude phosphonous dichloride (6) showed only one peak, the p.m.r. spectrum revealed the presence of less than 5 % of an impurity which could, however, be removed in the next step. The phosphonous dichloride (6) was heated with zinc chloride at 170' for 4.5 h to give, after the hydrolysis-oxidation procedure,' 72% of the crystalline phosphinic acid (10a). It was recently reported7 that 3-(2'-naphthyl)propylphosphonous dichloride did not undergo Friedel-Crafts cyclization. In the light of our present and earlier experience,' it seems more likely that cyclization did occur, but the relatively stable complex of the product with the Lewis acid was not hydrolysed. Esterification of the phosphinic acid (10a) with diazomethane gave a quantitative yield of the methyl ester (lob). The once-distilled phosphinic acid (10a) was sufficiently pure for the transformations described below. Treatment of the derived acid chloride (9) with ethylmagnesium bromide gave 1-ethylphosphindoline 1-oxide @a). Reduction of this with trichloro- silane and reaction of the phosphine with ethyl iodide gave the known 1,l-diethyl- phosphindolinium iodide (ll), the melting point of which was identical with that reported for material obtained by a different route.3 Similarly, the acid chloride (9) was converted into 1-phenylphosphindoline 1-oxide (8b), for which the melting point, 107-log0, is somewhat higher than that reported (97-101") by Chan and Wong6 for material synthesized by another route. This difference is probably due to slight hydration of the sample described by Chan and Wong.
Even with a large excess of the reagent, bromination of the phosphinic acid (10a) with N-bromosuccinimide in chloroform did not proceed to completion. However, reaction of the methyl ester (lob) with one mole equivalent of N-bromosuccinimide proceeded smoothly to give a mixture of the diastereoisomers of 3-bromo-1-methoxy- phosphindoline 1-oxide (13). This was not purified, but characterized by the 60-MHz
Mann, F. G., and Millar, I. T., J. Chem. Soc., 1951, 2205; cf. Egan, W., Tang, R., Zon, G., and Mislow, K., J. Amer. Chem. Soc., 1971, 93, 6205.
Markl, G., Z. Naturforsch. B, 1963, 18, 84. Rausch, M. D., and Klemann, L. P., J. Amev. Chem. Soc., 1967, 89, 5732. Chan, T. H., and Wong, L. T. L., Can. J. Chem., 1971,49, 530. Chen, C. H., and Berlin, K. D., J. Org. Chem., 1971, 36, 2791.
Organophosphorus Compounds. XI1
p.m.r. spectrum which showed a four-proton multiplet between 6 7.2 and 7.8 due to the aromatic protons; a broad one-proton multiplet at 5.25-5.9 for the benzylic proton H 3, the complexity of the signal suggesting that it is due to a mixture of the epimers of (13) for each of which H 3 is coupled with phosphorus and with the C 2 methylene protons; two sets of doublets of unequal area at 3.74 and 3.62 (J,,,,, 11 - 1 for each set) for the diastereoisomeric methoxyl protons; a two-proton multiplet
at 2.3-3.1 for the C 2 methylene protons. Dehydrobromination of the mixture of isomers (13) with triethylamine in benzene at reflux (~ f .~ , ' ) for 3 min yielded 75% of the crude crystalline 1-methoxyphosphindole 1-oxide (14). Longer reaction times gave Iower yields of (Id), possibly due to solvolysis of the ester group by the eliminated
Kabachnik, M. I., Medved, T. Ya., and Polikarpov, Yu. M., Zzv. Akad. Nauk SSSR, Seu. Khim., 1966, 367 (Chem. Abstr., 1966,64, 15917~).
D. J. Collins, L. E. Rowley and J. M. Swan
bromide ions. The i.r. spectrum of (14) showed a medium intensity band at 1450 cm-l characteristic of an Ar-P group, and strong bands at 1230 and 1050 cm-I for the P=O and POCH,, respectively. The 100-MHz p.m.r. spectrum of (14) showed a complex multiplet for the four aromatic protons between 6 7.1 and 7 65, on which was superimposed a doublet of doublets for H 3 centred at 7-28, with splittings J,,, 46, J, ,, 9.0 and J,,, 0.9 Hz; H 2 appeared as a doublet of doublets at 6.11, J2,, 23.8, Jz , , 9.0 Hz; the methoxyl protons showed a doublet, 3-67, J,,,,, 11.7 Hz. The respective assignments to H 2 and H 3 are based on both their chemical shifts and
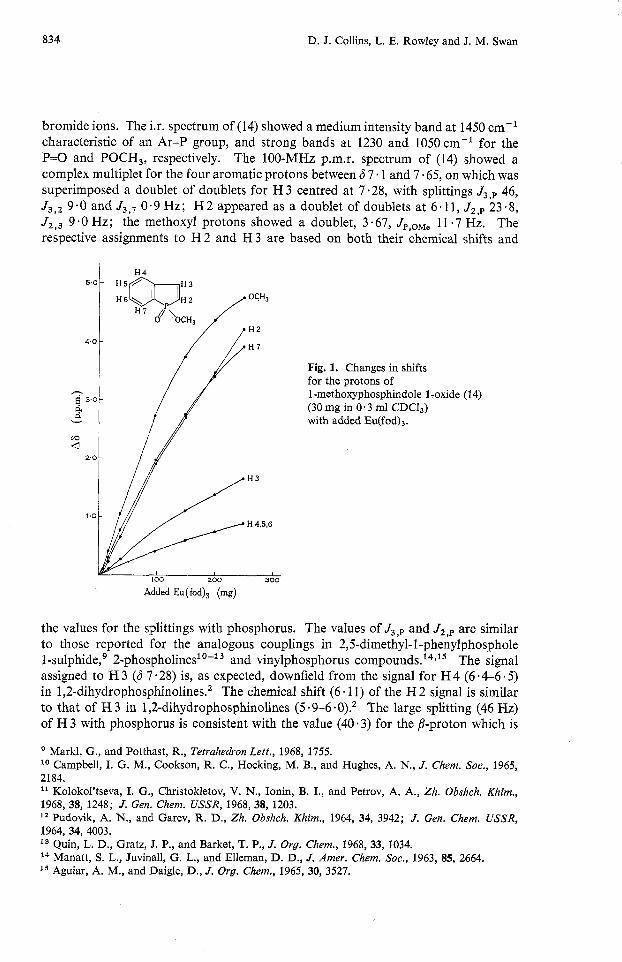
Fig. 1. Changes in shifts for the protons of 1-methoxyphosphindole 1-oxide (14) (30 mg in 0.3 ml CDCI,) with added Eu(f0d)a.
Added E u ( f ~ d ) ~ (mg)
the values for the splittings with phosphorus. The values of J,,, and J2,, are similar to those reported for the analogous couplings in 2,5-dimethyl-1-phenylphosphole 1-~ulphide,~ 2-phospholine~'~-~~ and vinylphosphorus compounds.14J5 The signal assigned to H 3 (6 7.28) is, as expected, downfield from the signal for H 4 (6.4-6 5) in 1,2-dihydrophosphinolines.2 The chemical shift (6.11) of the H 2 signal is similar to that of H 3 in 1,2-dihydrophosphinolines (5-9-6-0).2 The large splitting (46 Hz) of H 3 with phosphorus is consistent with the value (40.3) for the P-proton which is
Markl, G., and Potthast, R., Tetrahedron Lett., 1968, 1755. lo Campbell, I. G. M., Cookson, R. C., Hocking, M. B., and Hughes, A. N., J. Chem. Soc., 1965, 2184. l1 Kolokol'tseva, I. G., Christokletov, V. N., Ionin, B. I., and Petrov, A. A., Zh. Obshch. Khim., 1968, 38, 1248; J. Gen. Chem. USSR, 1968,38, 1203. IZ Pudovik, A. N., and Garev, R. D., Zh. Obshch. Khim., 1964, 34, 3942; J. Gen. Chem. USSR, 1964,34, 4003. l 3 Quin, L. D., Gratz, J. P., and Barket, T. P., J. Org. Chem., 1968, 33, 1034. l4 Manatt, S. L., Juvinall, G. L., and Elleman, D. D., J. Arner. Chem. Soc., 1963, 85, 2664. '"guiar, A. M., and Daigle, D., J. Org. Chem., 1965, 30, 3527.

Organophosphorus Compounds. XI1
trans related to phosphorus in diphenylstyrylphosphine oxide; the a-proton showed J,,p 19-3 Hz.15 There appears to be some confusion in the assignments made to H 2 and H 3 in the 220-MHz spectrum of 1-phenylph~sphindole.~ The assignments given to H 2 and H 3 in (14) were confirmed by the use of the shift reagent Eu(fod),. The methoxyl protons of (14) suffered the largest downfield shift. This was followed closely by H 2 and H7, while the signal for H 3 showed a much lower shift (see Fig. 1).
The reaction of the phosphinic chloride (9) with vinylmagnesium bromide in tetrahydrofuran at - 40' afforded 74 % of 1-vinylphosphindoline 1-oxide (12) which, upon treatment with 40% aqueous dimethylamine at reflux for 3 h, gave 93% of 1-(2'-dimethylaminoethyl)phosphindoline 1-oxide ('la), an extremely hygroscopic oil. The p.m.r. and mass spectra of (4a) are in accord with this structure. Reduction of (4a) with trichlorosilane followed by oxidation with sulphur gave the phosphine sulphide (4b) which was characterized as the crystalline hydrochloride.
In contrast with the corresponding 1,2,3,Ctetrahydrophosphinoline (3b), the aminophosphindoline sulphide (4b) showed no analgesic activity in the McKenzie- Beechy tail stimulation test. The amino phosphine oxide (4a) was also inactive.16
Experimental
General experimental procedures and spectroscopic methods are the same as given in Part X, except that only one glass column-6 ft by 114 in. packed with 20% w/w SE-30 silicone rubber on silanized Chromosorb W (80-100 mesh)-was used for g.1.c.
(a) 2-Phenylethylphosphonous Dichlovide (6)
A solution of 2-phenylethylmagnesium bromide was prepared from 2-phenylethyl bromide (230 g, 1.24 mol; b.p. 98-100°/20 mm, lit.17 92"/11 mm) in anhydrous ether (1.2 1.) and magnesium turnings (45 g, 1.72 mol). The excess of magnesium was removed by filtration through glass wool with a positive pressure of nitrogen. The filtrate was cooled in an ice bath and stirred vigorously under nitrogen whilst powdered anhydrous cadmium chloride (120 g, 0.65 mol, redried at 150°/1 mm for 24 h over P2O5) was added rapidly. The suspension was stirred at 0' for 2 h, then added during 30 min to a rapidly stirred solution of freshly distilled phosphorus trichloride (250 g, 1.83 mol) in anhydrous ether (300 ml). During the addition the temperature of the reaction mixture was kept below -20". After the addition was complete, the mixture was stirred at room temperature for 6 h. Most of the ethereal supernatant was decanted from the cooled mixture and the thick white precipitate was filtered from the remainder with a sintered glass Buchner funnel. Ether washings of the filter cake were combined with the main ether solution, and evaporated. Distillation gave a pungent colourless liquid (115 g, 4573, b.p. 64-66°/0.001 mm. G.1.c. (80-250" at 24"/min, 30 ml/min nitrogen) indicated that this liquid contained one major component (R, 6.1 min), and approximately 5% of an impurity (R, 4 .2 min). A p.m.r. spectrum (60 MHz, CDCI,) showed a weak quartet at 6 4.4 with integration equivalent to 0.2 protons, attributable only to an impurity. P.m.r, spectra of other fractions collected during the distillation also showed this signal. A middle fraction from the distillation, which showed no sign of the impurities by g.l.c., gave pure 2-phenyF ethylphosphonous dichlovide (Found: C, 45.8; H, 4.4; P, 14.9. C8H,Cl,P requires C, 46.3; H,4 .4 ; P, 15.0%). v,,. (film): 3080, 3060, 3025, 1605, 1590, 1495, 1450, 1375, 1335, 1320, 1190, 1165, 1080, 1035, 920, 765, 720 cm-l. P.m.r. (60 MHz, CDCI,): 7.0-7.5, m, 5H (aromatic); 2.2-3.3, m, 4H (aliphatic). Mass spectrum (mle > 2%): 206 (8), 135 (2), 133 (2), 107 (2), 106 (ll), 105 (loo), 104 (13), 103 (15), 102 (3), 101 (5), 92 (3), 91 (24), 89 (2), 80 (2), 79 (16), 77 (18), 76 (2), 75 (2), 74 (2), 66 (3), 65 (lo), 63 (6), 62 (2), 53 (2), 52 (4), 51 (15), 50 (7%); m*: 59.4 (105 -t 79).
l6 Bentley, G. A,, and Newman, Kathleen, unpublished data. l7 Perlman, D., Davidson, D., and Bogert, M. T., J. Oug. Chem., 1936, 1, 288.

D. J. Collins, L. E. Rowley and J. M. Swan
(b) I-Hydroxyphosphindoline I-Oxide (IOU)
A mixture of 2-phenylethylphosphonous dichloride (50 g, 0.24 mol) and freshly fused, powdered zinc chloride (50 g, 0.37 mol) was stirred rapidly and heated under a nitrogen atmosphere in an oil bath kept at 170". A vigorous evolution of hydrogen chloride ensued. After the mixture had been heated at 170" (oil bath temp.) for 4 h the temperature of the oil bath was reduced to 150' and concentrated hydrochloric acid (150 rnl) was added cautiously during 10 min. The mixture was heated under reflux for 15 min, then the hydrochloric acid was removed in vacuum (water pump). The thick orange-red residue was dissolved in 2~ hydrochloric acid (250 ml) and stirred at 0" whilst bromine was added cautiously until the bromine colour persisted. The solution was stirred for a further 1 h, then extracted with chloroform. The extract was washed several times with dilute hydrochloric acid, then the phosphinic acid was extracted with 2~ sodium hydroxide. Acidi- fication and extraction with chloroform gave the crude crystalline product (31 g, 72%), m.p. 141-142". Recrystallization from cyclohexane-benzene followed by sublimation at 135°/0.001 mm gave the analytical sample of I-hydroxyphosphindoline I-oxide, m.p. 144-145" (Found: C, 56.8; H, 5.4; P, 18.4. C8H902P requires C, 57.1 ; H, 5.4; P, 18.4%). v,., (Nujol): 2600, 2250, 2050, 1670 (all of these broad absorptions are attributable to P(O)OH), 1595, 1460 (Ar-P), 1375, 1260, 1140, 1080, 1050, 980, 835, 790, 760 cm-l. A,,, ( e ) : 261 (719), 267 (1110), 274 nm (1120). P.m.r. (100 MHz, CDCI,): 12.7, s, 1H (: P(=)OH); 7.78, dm, J7,, 9.9, 1H (H 7), when the multiplet at 6 7.1-7.5 was irradiated this signal collapsed to a d with splitting 8.7 Hz; 7.1-7.5, m, 3H (H4,5,6); 3.10, dt, with additional small splittings, J3,P 13.5, JZ,3 7.5, 2H (H3,3), this signal collapsed to a distorted d with splitting J3,, as above upon irradiation of the signal at 6 2.17; 2.17, dt with additional small splittings, J Z , p 14.9, 2H (H2,2), this signal collapsed to a distorted d with splitting J2,, as above upon irradiation of the signal at 6 3.10. Mass spectrum (mle, peaks > 5 %): 168 (loo), 169 (12), 167 (78), 166 (8), 150 (14), 149 (34), 105 (8), 104 (39), 103 (21), 102 (lo), 91 (6), 78 (21), 77 (30), 76 (7), 75 (7), 74 (7), 69 (5), 65 (7), 63 (9), 52 (5), 51 (19), 50 (13), 47 (9%); m*: 132.9 (167 + 149), 58.5, 57.6 (104 + 78, 103 -t 77).
(c) I-Methoxyphosphindoline I-Oxide (lob)
Esterification of a sample of crude 1-hydroxyphosphindoline 1-oxide (1.0 g, 5 .9 mmol) with diazomethane and bulb-to-bulb distillation of the product at 120°/0.01 mm afforded a colourless hygroscopic oil (1.1 g, 98 %). This material was shown to be essentially homogeneous by g.1.c. (80-250' at 24O/min, 30 ml/min nitrogen, Rt 7.2 min), t.1.c. (R, 0.48, chloroform/methanol 30/1), and the p.m.r. spectrum which showed only one set of doublets, 6 3.71, attributable to a :P(=)OCH3 group. Preparative t.1.c. and three successive bulb-to-bulb distillations of the residue gave an analyt- ical sample of I-methoxyphosphindoline I-oxide as a colourless hygroscopic oil, which was sealed for analysis without exposure to the atmosphere (Found: C, 59.1 ; H, 6.1 ; P, 17.4. C9H1102P requires C, 59.3; H, 6.1; P, 17.0%). v,,, (film): 3060, 2990, 2950, 2850, 1600, 1460 (Ar-P), 1410, 1265, 1235 (P=O), 1190, 1150, 1085, 1040 (P-0-CH,), 950, 860, 815, 775, 755, 700 cm-I. A,,,,, (8 ) : 262sh (670), 268 (1085), 276 nm (1100). P.m.r. (100 MHz, CDCl,): 7-70, dm, J7,, 9.1, 1H (H7), this signal collapsed to a d with splitting 8.0 Hz when the multiplet at 6 7.2-7.5 was irradiated; 7.2-7.5, m, 3H (H4,5,6); 3.71, d, J p , o M e 11.0, 3H (:P(=)OCH3); 2.9-3.2, m, 2H (H3,3); 1.95-2.35, m, 2H (H2,2). Mass spectrum (mle, peaks >2%): 182 (loo), 183 (13), 181 (35), 167 (8), 154 (3), 153 (9), 152 (76), 151 (23), 150 (18), 149 (48), 137 (3), 134 (13), 133 (14), 121 (6), 118 (4), 117 (3), 107 (3), 105 (lo), 104 (la), 103 (17), 95 (2), 91 (5), 89 (4), 79 (7), 78 (15), 77 (27), 76 (7), 75 (5), 74 (4), 69 (3), 65 (3), 63 (8), 62 (2), 52 (4), 51 (13), 50 (8), 47 (9%); m*: 126.9 (182 + 152), 122.7 (181 + 149), 117.1 (151 -+ 133), 58.3, 57.6 (104 -t 78, 103 -, 77).
(d) I-Ethylphosphindoline I-Oxide (8a)
A solution of crude 1-hydroxyphosphindoline 1-oxide (1.0 g, 5.9 mmol) in thionyl chloride (7 ml) was heated under reflux for 30 min. The excess of thionyl chloride was removed in vacuum, then toluene was added and evaporated to remove the last traces of thionyl chloride. To a vigorously stirred solution of this in dry tetrahydrofuran was added under nitrogen a filtered solution of ethyl- magnesium bromide prepared from ethyl bromide (0.9 g, 8.0 mmol), magnesium (0.33 g, 13.9 mmol) and dry tetrahydrofuran (10 rnl). The mixture was stirred and heated under reflux for 20 min, cooled and treated with saturated ammonium chloride. Isolation with chloroform and bulb-to-bulb distillation of the residue at 160°/0. 1 mm gave a colourless hygroscopic liquid (2.3 g, 72%). Pre-

Organophosphorus Compounds. XI1
parative t.1.c. (RF 0.38, chlor~forrn/methan~l 15/1), and bulb-to-bulb distillation of the major component gave 'pure' I-ethylphosphindoline I-oxide as a colourless hygroscopic liquid. This was shown to be pure by g.1.c. (80-250" at 24O/min, 30 ml/min nitrogen, R, 8.4 min). A sample was redistilled twice and the second distillate sealed without exposure to the atmosphere (Found: C, 65.6; H, 7.6; P, 17.0. C10H130P requires C, 66.6; H, 7.3; P, 17.2. CloHl,0P,0~2H20 requires C, 65.4; H, 7.4; P, 16.9%). The low carbon value indicates that water was absorbed when the sample was opened. v,,, (film): 3050, 2980, 2930, 2780, 1595, 1455 (Ar-P), 1410, 1270, 1255, 1190 (P=O), 1165, 1145, 1125, 1080, 1045, 800, 770 cm-l. A,,, (8): 263sh (640), 269 (1030), 276 nm (1030). P.m.r. (100 MHz, CDCI,): 7.70, distorted dm, J7,P 8.1, 1H (H7), this signal collapsed to a distorted d (incomplete decoupling) with splitting 8.0 Hz when the signal at 6 7.2-7.6 was irradiated; 7.2-7.6, m, 3H (H4,5,6); 2.6-3.5, m, 2H (H3,3); 1.7-2.4, m, 4H (H2,2, :P(=)CH2CH3); 1.14, dt, J(P,CH2CH3) 17.8, J(CH2,CH3) 7,3H :(P(=)CH2CH3). Mass spectrum (mle, peaks >5%): 180 (20), 181 (5), 153 (7), 152 (63), 151 (loo), 149 (8), 136 (7), 134 (13), 133 (67), 121 (8), 107 (14), 105 (7), 104 (19), 103 (20), 102 (lo), 95 (7), 91 (8), 89 (7), 79 (6), 78 (24), 77 (35), 76 (8), 75 (8), 69 (7), 65 (7), 63 (12), 52 (7), 51 (20), 50 (10%); m*: 128.3 (180 -+ 152), 117.1 (151 -+ 133), 86.1 (133 -+ 107), 58.5, 57.6 (104 -+ 78, 103 -+ 77). An i.r. spectrum of a sample which was exposed to the atmosphere showed additional broad absorption bands at 3300 and 1630 cm-l, consistent with the hygroscopic nature of the compound.
(e) 1,I- Diethylphosphindolinium Iodide (I I )
I-Ethylphosphindoline 1-oxide (1.0 g, 5.56 mmol) was dissolved in benzene (40 ml) and half of the solvent distilled to remove traces of water. To the cooled solution under nitrogen was added trichlorosilane (1 ml), and the mixture was heated under reflux for 2 h. The excess of trichlorosilane was removed by evaporation of half of the benzene, and the remaining silicon compounds were hydrolysed by washing the benzene suspension with several portions of 4M sodium hydroxide. The organic layer was washed with water, dried (Na2S0,), and ethyl iodide was added. Crystalliza- tion of the solid from light petroleum-anhydrous ethanol afforded pure 1,l-diethylphosphindol- inium iodide (0.8 g, 45 %), m.p. 1 2 2 5-123" (lit.3 123").
(f) I-Phenylphosphindoline I-Oxide (86)
This compound was prepared from 1-hydroxyphosphindoline 1-oxide (2.0 g, 11.9 mmol) using a method analogous to that described in (d). Isolation with chloroform, and bulb-to-bulb distillation of the residue at 150°/0.005 mm, followed by recrystallization of the distillate from cyclohexane- benzene afforded 1-phenylphosphindoline 1-oxide as colourless, slightly hygroscopic crystals (2.3 g, 85 %), m.p. 107-108" (lk6 97-101°), the purity of which was confirmed by t.1.c. (RF 0.67, chloroform/ methanol 1511) (Found: C, 74.0; H, 6.0; P, 13.4. Calc. for C14H130P: C, 73.7; H, 5.8; P, 13.6%). v,,, (Nujol): 1440 (Ar-P), 1260, 1205 (P=O), 1175, 1145, 1120, 1075, 790, 780, 755, 730, 710 cm-'. A,,, (8): 227 (6300), 261sh (1920), 270 (2130), 277 nm (1740). P.m.r. (100 MHz, CDCl,): 7.59, dm, JTSP 9.4, 1H (H7); 7.2-7.5, m, 8H (4,5,6, :P(=)Ph); 3.0-3.6, m, 2H (H3,3); 2.2-2.6, m, 2H (H2,2). Mass spectrum (mle, peaks 5%): 228 (loo), 229 (19), 227 (73), 201 (7), 200 (19), 199 (8), 183 (7), 179 (5), 178 (9), 165 (lo), 152 (ll), 151 (19), 150 (lo), 149 (32), 135 (16), 134 (7), 133 (40), 121 (ll), 107 (17), 104 (13), 103 (15), 102 (9), 91 (9), 89 (5), 78 (21), 77 (44), 76 (7), 75 (7), 74 (7), 65 (7), 63 (9), 52 (7), 51 (34), 50 (1 I), 47 (22 %); m* : 226 (228 -+ 227), 175.4 (228 -+ 200), 148 (152 -+ 150), 131.1 (135 -+ 133), 117.1 (151 + 133), 98.3 (149 -+ 121), 86.2 (133 -+ 107), 58.5, 57.6 (104 -+ 78, 103 -+ 77).
(g) I-Vinylphosphindoline I-Oxide (12)
A solution of I-chlorophosphindoline 1-oxide prepared as in (d ) from 1-hydroxyphosphindoline 1-oxide (19.0 g, 0.113 mol) in dry tetrahydrofuran (200 ml) was cooled to -40' and treated with vinylmagnesium bromide in tetrahydrofuran (140 ml of l ~ , 14.0 mmol), added during 1 h. While still cold the mixture was poured into chilled 2~ ammonium chloride solution. Extraction with chloroform yielded a liquid which was distilled through a short Vigreux column to give a colourless hygroscopic liquid (14.9 g, 74 %), b.p. 116-118°/0~01 mm. Attempted crystallization of this material from anhydrous cyclohexanebenzene gave a small amount of an extremely deliquescent crystalline solid which separated slowly over 4 days, m.p. 35-36", after prolonged drying over phosphorus pentoxide at 0.1 mm. Preparative t.1.c. of a portion of this (RF 0.18 chloroform/methanol 3011)

D. J. Collins, L. E. Rowley and J. M. Swan
gave, after bulb-to-bulb distillation at 130°/0.01 mm, a pure sample of 1-vinylphosphindoline I-oxide as a colourless hygroscopic liquid. This was shown to be pure by g.1.c. (80-250' at 24O/min, 30 ml/min nitrogen, Rt 8.2 min). A sample was redistilled twice more and sealed without exposure to the atmosphere. The analytical results indicate that the sample absorbed water when it was opened for analysis (Found: C, 66.5; H, 6.5. CloHllOP requires C, 67.4; H, 6.2. CloHl10P,0~2H20 requires C, 66.2; H, 6.3 %). v,,, (film): 3050, 2990, 2920, 2850, 1595, 1460, 1455 (Ar-P), 1405 1265, 1250, 1210 (P=O), 1165, 1145, 1125, 1075, 995, 945, 850, 795, 770, 735, 710 cm-l, A,,, (6): 264sh (645), 269 (1024), 277 nm (990). P.m.r. (100 MHz, CDCI,): 7.64, dm, J,,, 9.2, 1H (H7); 7.15-7.5, m, 3H (H4,5,6); 5.7-6.7, m, 3H (olefinic); 2.7-3.6, m, 2H (H3,3); 1 +8-2.5, m, 2H (H2,2). Mass spectrum (mle, peaks > 10%): 178 (loo), 179 (15), 177 (18), 152 (27), 151 (57), 150 (34), 149 (19), 136 (16), 135 (98), 134 (24), 133 (97), 129 (12), 121 (lo), 116 (ll), 115 (ll), 107 (18), 104 (23), 103 (26), 102 (17), 91 (la), 89 (lo), 78 (28), 77 (47), 76 (12), 75 (14), 74 (13), 69 (lo), 63 (20), 52 (lo), 51 (34), 50 (18), 47 (34%); m*: 131.1 (135 -+ 133), 126.4 (178 + 150), 117.1 (151 -+ 133), 86.2 (133 -+ 107), 58.5, 57.6 (104 4 78, 103 -+ 77). A sample which was exposed to the atmosphere developed additional broad absorption bands at 3400 and 1630 cm-l, due to uptake of water.
(h) I-(2'-Dimethylaminoethyl)phosphindoline I-Oxide (4a)
A solution of crude 1-vinylphosphindoline 1-oxide (5.0 g, 28.1 mmol) in aqueous dimethylamine (40 ml of 40%) was heated under reflux for 3 h, then cooled and extracted with chloroform. Bulb-to- bulb distillation of the residue at 150°/0.005 mm gave a colourless hygroscopic liquid (5.83 g, 93 %). T.1.c. (chloroform/methanol 311) indicated that this liquid contained a significant amount of an impurity (5-10%) which could not be removed by recrystallization of the derived hydrochloride, or by chromatography of the free amine on an alumina column (400 g of activity I, chloroform/ methanol 3011 as eluent). A sample of the crude product was subjected to preparative t.l.c., followed by three successive bulb-to-bulb distillations to give 'pure' I-(2'-dimethylaminoethy1)phosphindoline I-oxide. The final distillate was sealed without exposure to the atmosphere; however, water was evidently absorbed during handling for analysis (Found: C, 63.9; H, 8.3. C12H18NOP requires C, 64.6; H, 8.1. C12H18NOP,0. 15Hz0 requires C, 63.9; H, 8.2%). v,,, (film): 3040, 2920, 2850,2810, 2760 (CH3-N and CH2-N), 1695, 1450 (C6H5-P), 1375, 1260, 1205 (P=O), 1195, 1170, 1140, 1075, 1055, 890, 865, 795, 765 cm-l. A,,, (6): 263 (634), 269 (1030), 276 nm (1020). P.m.r. (100 MHz, CDCl,): 7.72, dm, J7,, 8.4, 1H (H7), this signal collapsed to a distorted d with splitting 7.5 Hz when the multiplet at 6 7.15-7.5 was irradiated; 7.15-75, m, 3H (H4,5,6); 2.8-3.4, m, 2H (H3,3); 1.8-2.8, m, 6H (H2,2, >P(=)CHzCHzN); 2.12, s, 6H (2x CH,). Mass spectrum (mle, peaks >2%): 223 (3), 179 (2), 153 (2), 152 (14), 151 (17), 149 (3), 135 (3), 134 (lo), 133 (24), 121 (3), 117 (2), 116 (2), 115 (4), 107 (4), 105 (5), 104 (7), 103 (8), 101 (3), 95 (2), 91 (6), 89 (2), 79 (3), 78 (8), 77 (13), 76 (2), 73 (2), 72 (33), 71 (loo), 70 (81, 69 (21, 65 (3), 63 (3), 59 (3), 58 (2), 57 (5), 56 (27), 55 (3), 52 (2), 51 (6), 50 (2), 47 (8), 44 (8), 43 (17), 42 (30), 41 (3 %); m*: 131.1 (135 -+ 133), 117.1 (151 -+ 133), 86.2 (133 -+ 107). The hydroscopic nature of this compound was shown by the observation that a sample which had been exposed to the atmosphere developed additional broad bands at 3400 and 1630 cm-I in the i.r. spectrum and an additional sharp singlet at 6 3.52 (exchanged by D20) in the p.m.r. spectrum.
(i) I-(2'-Dimethy1aminoethyl)phosphindoline I-Sulphide (4b)
A solution of 1-(2'-dimethylaminoethyl)phosphindoline 1-oxide (3.0 g, 13.4 mmol) in benzene (120 ml) was reduced with trichlorosilane (3 ml, 30 mmol) by a procedure similar to that described in (e). After removal of the excess of trichlorosilane by distillation of some of the solvent, sulphur (3 g) was added and the mixture was stirred at room temperature for 12 h. The silicon compounds were hydrolysed by careful addition of 4~ sodium hydroxide and the aqueous layer was separated and extracted with benzene. The combined benzene extract was washed with 2M sodium hydroxide, then with water, dried (Na2S0,) and concentrated to half volume. Passage of dry hydrogen chloride through this solution and crystallization of the solid product from absolute ethanol gave colourless crystals of I-(2'-dimethylaminoethy1)phosphindoline I-sulphide hydrochloride (2.65 g, 72%), m.p. 245-246' (Found: C, 52.4; H, 7.1 ; P, 11.5; S, 11 ~ 8 . ClzHl9ClNPS requires C, 52.3; H, 7.0; P, 11.3; S, 11 a 7 %). The free base was obtained from the hydrochloride in the usual way, and bulb- to-bulb distillation at 150°/0.005 rnm afforded an analytical sample of I-(2'-dimethylaminoethyl)- phosphindoline I-sulphide as a colourless liquid, the purity of which was confirmed by t.1.c. (RF 0.54

Organophosphorus Compounds. XI1
chloroform/methanol 1511) (Found: C, 59.9; H, 7.5; N, 5.9; P, 13.0; S, 13.5. C12H18NPS requires C, 60.2; H, 7.6; N, 5.9; P, 13.0; S, 13.4%). v,,, (film): 3050, 2960,2930, 2860, 2820, 2780 (CH3-N and CH2-N), 2730, 1595, 1450 (Ar-P), 1405, 1375, 1310, 1265, 1220, 1165, 1140, 1045, 1000, 980, 880, 865, 840, 785, 765, 730, 695 cm-l. A,,, (8 ) : 247 (2070), 260 (1740), 269 (1680), 276nm(1340). P.m.r. (100 MHz, CDC13): 7.68, dm, J7,P 9.6, lH(H7); 7.15-7+45,m, 3H(H4,5,6); 3.0-4.4, m, 2H (H 3,3); 2.0-2.9, m, 6H (H2,2, :P(=)CH2CH2N); 2.13, s, 6H (2 x CH,). Mass spectrum (mle, peaks >2%): 239 (8), 167 (5), 152 (6), 139 (3), 136 (2), 135 (14), 134 (lo), 133 (24), 115 (2), 109 (3), 108 (2), 107 (7), 103 (2), 102 (2), 91 (9), 89 (3), 78 (3), 77 (6), 72 (23), 71 (loo), 70 (8), 65 (3), 63 (7), 59 (2), 58 (42), 57 (5), 56 (26), 51 (4), 44 (5), 43 (13), 41 (22), 40 (2%); m*: 131.1 (135 -+ 133), 86.2 (133 -+ 107).
( j ) I-Methoxyphosphindole I-Oxide (14)
A solution of 1-methoxyphosphindoline 1-oxide (2.7 g, 14.8 mmol) in carbon tetrachloride (50 ml) was stirred rapidly under irradiation with a 500-W mercury arc lamp, and N-bromosuccinimide (2.9 g, 1 6 3 mmol) was added cautiously in small portions during 10 min. The mixture was irradiated for an additional 20 min then cooled and filtered to remove the precipitated succinimide. The filtrate was washed with water, dried (Na2S0,) and evaporated to give a colourless oil (3.5 g) which gave a p.m.r. spectrum consistent with 3-bromo-1-methoxyphosphindoline 1-oxide (60 MHz, CDCI,): 7.2-7.8, m, 4H (H4,5,6,7); 5.25-5.9, m, 1H (H 3); two sets of d of unequal area at S 3.74 and 3.62, Jp,OMe 11.1 HZ for both sets, 3H (:P(=)OCH,); 2.3-3.1, m, 2H (H2,2). The crude bromo ester was dissolved in benzene (30 ml), triethylamine was added and the solution heated under reflux with stirring for 3 min. The precipitated triethylamine hydrobromide was filtered off, the filtrate concentrated and the residue distilled (bulb-to-bulb) at 140°/0.01 mm to give colourless deliquescent plates (2.0 g, 75%). Recrystallization from carbon tetrachloride gave I-methoxyphosphindole I-oxide, m.p. 89-90". The purity of this sample was confirmed by g.1.c. (80-250" at 24"/min, 30 ml/min nitrogen, R, 7.4 min), and t.1.c. (R, 0.53, chloroform/methanol 30/1) (Found: C, 59.6; H, 5.1 ; P, 17.2. C9H902P requires C, 59.9; H, 5.0; P, 17.2 %). v,,, (film): 3050, 2950, 2850, 1600, 1450 (Ar-P), 1355, 1285, 1230 (P=O), 1200, 1140, 1110, 1050 (P-0-CH,), 915, 880, 825, 805, 775, 730, 710 cm-l. A,,, (8): 228 (5670), 280sh (1280), 312 nm (2138). P.m.r. (100 MHz, CDCI,): 7.1-7.65, m, 4H (H4,5,6,7); 7.28, ddd, J3,P 46, J 3 , 2 9.0, J,,, 0.9, 1H (H 3); 6.11, dd, J2,, 23.8 Hz, 1H (H2); 3.67, d, JPSOMe 11.7, 3H (>P(=)OCH3). Mass spectrum (mle, peaks > 10%): 180 (loo), 181 (IS), 165 (54), 162 (39), 152 (lo), 151 (ll), 150 (87), 149 (81), 137 (36), 134 (17), 121 (28), 118 (lo), 116 (16), 109 (16), 103 (13), 102 (27), 101 (lo), 95 (]I), 90 (lo), 89 (13), 78 (18), 77 (33), 76 (28), 75 (33), 74 (23), 69 (13), 63 (22), 52 (ll), 51 (32), 50 (32), 47 (32%); m*: 148 (150 -, 149), 145.7 (180 -+ 162), 125 (180 -+ 150), 118.7 (165 + 137), 110.9 (162 -+ 134), 97.6 (150 -+ 211), 56.6 (102 -+ 76).
The effects of the shift reagent tris(1,1,1,2,2,3,3-heptafluoro-7,7-dimethyloctane-4,6-dione)- europium(~~~), Eu(fod),, on the 100 MHz p.m.r, spectrum confirmed the assignments to H 2 and H 3 (see Fig. 1).
Acknowledgments
One of us (L.E.R.) is grateful to Monash University for the award of a Monash Graduate Scholarship. We thank Associate Professor G. A. Bentley and Mrs Kathleen Newman for biological testing. We are grateful to the Australian Research Grants Committee and Merck Sharp & Dohme (Aust.) for generous support.
Manuscript received 8 November 1973