onco-nephrology: renal toxicities of chemotherapeutic agents
TRANSCRIPT

Onco-Nephrology: Renal Toxicities of ChemotherapeuticAgents
Mark A. Perazella
SummaryDespite dramatic improvements in patient survival and drug tolerability, nephrotoxicity remains an importantcomplication of chemotherapy. Adverse renal effects occur because of innate drug toxicity and a number ofpatient- and drug-related factors. To provide cutting edge care for these patients, nephrologists and oncologistsmust be familiar with the nephrotoxicity of these drugs, particularly their associated clinical and laboratorymanifestations. Rapid diagnosis, targeted treatment, and supportive care are critical to improving care for thesepatients. Unfortunately, some patients who develop nephrotoxicity will be left with long-term complications suchas chronic tubulopathies and CKD. Onco-Nephrology is a new area that is rapidly expanding and requires aclose working relationship between oncologists and nephrologists.
Clin J Am Soc Nephrol 7: 1713–1721, 2012. doi: 10.2215/CJN.02780312
IntroductionTreatment of cancer has undergone many significantadvances in recent times. As such, the oncology land-scape has changed and improved dramatically formany patients. Those patients previously consideredtherapy failures are now deriving significant benefitwith decreased tumor progression and increased sur-vival, often with less severe adverse systemic drugeffects. Despite this positive advancement in chemo-therapeutics for various malignancies, drug nephro-toxicity remains a complication and sometimes limitslife-saving therapy (1–3).
As a number of effective but potentially nephrotoxicchemotherapeutic regimens are released into clinicalpractice, oncologists and consulting nephrologistsshould be familiar with their adverse renal effects. Theseeffects include the clinical and histopathologic manifes-tations of renal toxicity (1–6). Clinicians should also bewell versed in preventive measures available for thevarious chemotherapy regimens, as well as effectivetreatment options for nephrotoxic consequences (1–6).The relationship of nephrologists and oncologists in thecare of these patients has given rise to the nascent butgrowing area of Onco-Nephrology.
Renal Susceptibility to ChemotherapeuticAgents
Not all patients exposed to nephrotoxic chemother-apeutic agents develop kidney injury, suggesting thepresence of several factors that enhance patient risk fornephrotoxicity. In addition to innate drug toxicity, certainhost characteristics and renal handling of the drug in-crease renal injury. In general, one ormore of these factorscombine to increase risk for kidney injury (Table 1).
Many cancers involve the kidneys either directly orindirectly—heightening the risk for kidney injury
with exposure to a potential nephrotoxin. In fact,nearly 60% of patients with cancer have some formof renal disease (1,2). Direct malignant effects includemyeloma-related kidney injury, infiltration of the re-nal parenchyma as seen with leukemias and lympho-mas, urinary tract obstruction from various cancers,and secondary glomerulopathies (2). Indirect effects in-clude true or effective volume depletion from nausea/vomiting, diarrhea, overdiuresis, malignant ascites orpleural effusions, sepsis, and cardiac involvement,which sensitizes the kidney to nephrotoxins byinducing a prerenal state (2). Also, susceptibility todrug toxicity occurs with metabolic disturbances suchas hyperuricemia and hypercalcemia.Undoubtedly, the toxicity of the chemotherapeutic
agent used importantly determines both the develop-ment and type of kidney injury sustained. High dosesand prolonged therapy increase the chance of renalinjury developing, regardless of the absence of otherrisk factors (2–6). Furthermore, combined exposure ofchemotherapeutic agents with other nephrotoxinswill raise the risk for kidney injury (7–9).There are a number of patient-specific risk factors
that must be considered with chemotherapy-associatednephrotoxicity. Many patients are elderly—possessingreduced total body water and an unrecognized de-pressed GFR, higher rates of renal oxidative stress,and excessive levels of angiotensin-II/endothelin, allof which increase drug nephrotoxicity (10,11). Anothernonmodifiable risk factor is the host’s underlying ge-netic makeup, which is likely a powerful explanationfor the heterogeneous response to chemotherapeuticagents (12–14). Gene polymorphisms in the renal cy-tochrome P450 enzyme system, which favor reducedmetabolism and renal excretion, enhance nephrotoxicrisk. Other examples are loss of function mutations inapical secretory transporters and mutations in kinases
Section ofNephrology,Department ofMedicine, YaleUniversity, NewHaven, Connecticut
Correspondence: Dr.Mark A. Perazella,Section ofNephrology,Department ofMedicine, YaleUniversity, 330 CedarStreet, New Haven,CT 06520-8029.Email: [email protected]
www.cjasn.org Vol 7 October, 2012 Copyright © 2012 by the American Society of Nephrology 1713

that regulate drug carrier proteins, which can impair drugexcretion and induce nephrotoxicity by increasing intracel-lular drug concentrations (13,15).Last, the renal handling of drugs is another risk factor for
the development of nephrotoxicity. The kidney is exposedto considerable drug concentrations based on the high renalblood flow rate—approximately 25% of cardiac output. Sig-nificant drug uptake occurs in the proximal tubular cellsthrough both apical uptake and basolateral transport (16,17).Trafficking of these agents through tubular cells explains, inpart, their nephrotoxicity. The high metabolic rate and hyp-oxic environment of loop of Henle and medullary collectingduct cells impart nephrotoxic drug risk (18,19). Metabolism
of drugs by several enzyme systems present in the kidneyfavors toxic metabolite and reactive oxygen species formation.Renal injury may occur, because drug byproducts cause harmthrough lipid peroxidation, protein damage, nucleic acidalkylation or oxidation, and DNA strand breaks (18–20).
Classification of Chemotherapy-Associated RenalLesionsThere are a number of ways that one can approach
classifying the kidney lesions caused by the various chemo-therapeutic agents. For example, one could categorize thechemotherapy-related kidney lesions based on the nephronsites primarily affected by the drug. Agents that injure therenal vasculature, glomerulus, proximal and distal tubularsegments, and collecting ducts are described (Table 2), recog-nizing that all nephrotoxic drugs cannot possibly be covered.
Renal Vasculature: Thrombotic MicroangiopathyChemotherapeutic agents, such as bevacizumab and
gemcitabine, can injure the renal vasculature and causethrombotic microangiopathy (TMA). TMA presents clinicallyas microangiopathic hemolytic anemia, thrombocytopenia,hypertension, and AKI with hematuria and proteinuria,although renal-limited TMA does occur.
BevacizumabRecognition that tumor growth was highly dependent on
pathologic angiogenesis induced by local production ofvascular endothelial growth factor (VEGF) paved the wayfor the development of drugs targeting this pathway (21).This pathway was a logical point of attack to supplementother tumor-directed therapies—and turned out to be a ben-eficial addition to the therapeutic armamentarium. Althoughthere are numerous drugs that target VEGF effects, the anti-VEGF antibody bevacizumab will be the focus of discussion,recognizing that there are differences among the agents.VEGF importantly regulates vasculogenesis and angio-
genesis during development and in disease through reg-ulation of vascular permeability, endothelial cell migration,proliferation, and survival (21). This regulation raises thepossibility that these drugs may be associated with ad-verse effects. In fact, this finding is the case, because anumber of adverse systemic end-organ effects have beendescribed, including kidney injury. This finding is not sur-prising, because VEGF is produced by renal visceral epi-thelial cells and binds to VEGF receptors located onglomerular endothelium and mesangium, as well as peri-tubular capillaries (21). Local VEGF production maintainsnormal functioning of all of these cells, including injuryrepair and cell turnover. Importantly, there is crosstalkbetween the glomerular endothelium and epithelium,maintaining the integrity of the filtration barrier.The most important renal effects described with anti-
angiogenesis therapy are new or worsened hypertensionand kidney-specific injury, including proteinuria and AKI.Importantly, the development of hypertension in patientspredicts a better tumor response to therapy (22), and itshould prompt clinicians to continue therapy and controlBP with antihypertensive agents rather than discontinuingantiangiogenic therapy. Animal experiments documented a
Table 1. Risk factors for chemotherapy-inducednephrotoxicity
Tumor-related kidney effectsdirect renal involvementmyeloma-related kidney injuryrenal infiltration (lymphoma and leukemia)urinary obstructionneoplasia-associated glomerulopathies
indirect renal involvementtrue volume depletion (N/V, diarrhea, andoverdiuresis)
effective volume depletion (cardiomyopathy,malignant ascites, and pleural effusions)
metabolic effects (hyperuricemia and hypercalcemia)Innate drug toxicityhigh-dose drug exposure and prolonged course oftherapy
insoluble drug or metabolite form crystals withinintratubular lumens
potent direct nephrotoxic effects of the drug or toxindrug combinations enhance nephrotoxicityNSAIDs, aminoglycosides, and radiocontrast
Patient factorsolder ageunderlying AKI or CKDimmune response genesincreased allergic reactions to drugs
pharmacogenetics favoring drug/toxin toxicitygene mutations in hepatic and renal CYP450enzyme systems
gene mutations in transport proteins and renaltransporters
Renal drug handlinghigh blood (and drug) delivery rate to the kidneysproximal tubular uptake of toxinsapical tubular uptake by endocytosisor another pathway
basolateral tubular transport through OATand OCT pathways
relatively hypoxic renal environmenthigh metabolic rate of tubular cells in the loop of Henleincreased drug/toxin concentration in renal medullaand interstitium
biotransformation of substances to ROS causingoxidative stress
N/V, nausea/vomiting; NSAIDs, nonsteroidal anti-inflammatory drugs; OAT, organic anion transporter; OCT,organic cation transporters; ROS, reactive oxygen species.
1714 Clinical Journal of the American Society of Nephrology
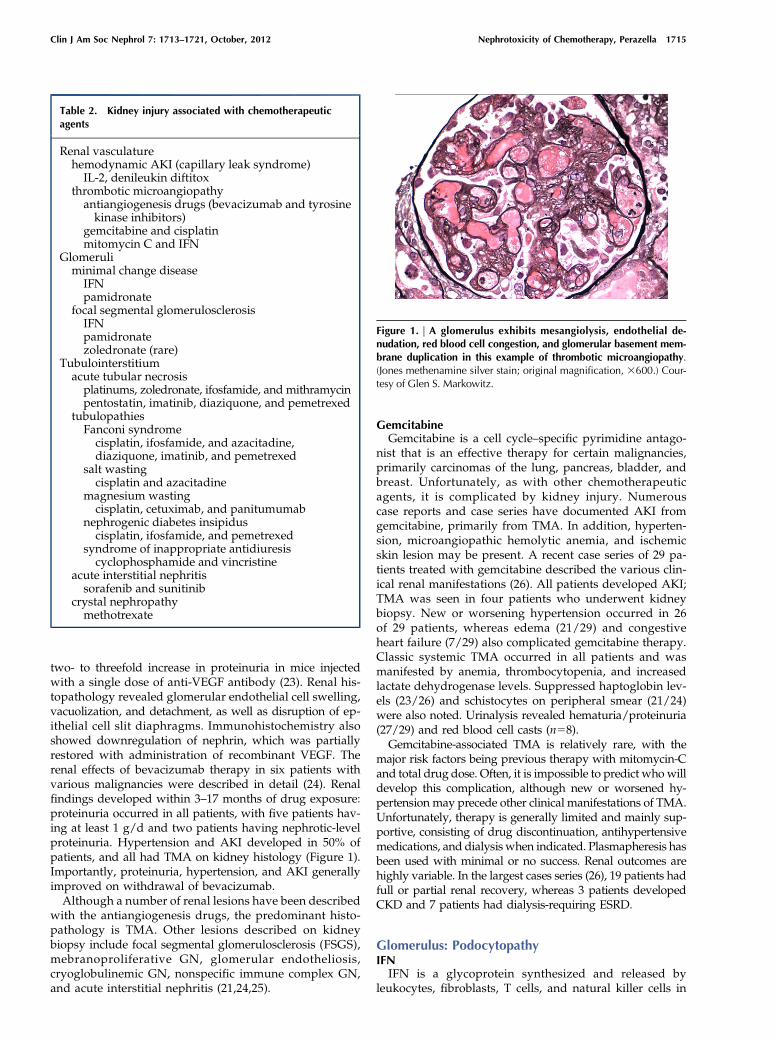
two- to threefold increase in proteinuria in mice injectedwith a single dose of anti-VEGF antibody (23). Renal his-topathology revealed glomerular endothelial cell swelling,vacuolization, and detachment, as well as disruption of ep-ithelial cell slit diaphragms. Immunohistochemistry alsoshowed downregulation of nephrin, which was partiallyrestored with administration of recombinant VEGF. Therenal effects of bevacizumab therapy in six patients withvarious malignancies were described in detail (24). Renalfindings developed within 3–17 months of drug exposure:proteinuria occurred in all patients, with five patients hav-ing at least 1 g/d and two patients having nephrotic-levelproteinuria. Hypertension and AKI developed in 50% ofpatients, and all had TMA on kidney histology (Figure 1).Importantly, proteinuria, hypertension, and AKI generallyimproved on withdrawal of bevacizumab.Although a number of renal lesions have been described
with the antiangiogenesis drugs, the predominant histo-pathology is TMA. Other lesions described on kidneybiopsy include focal segmental glomerulosclerosis (FSGS),mebranoproliferative GN, glomerular endotheliosis,cryoglobulinemic GN, nonspecific immune complex GN,and acute interstitial nephritis (21,24,25).
GemcitabineGemcitabine is a cell cycle–specific pyrimidine antago-
nist that is an effective therapy for certain malignancies,primarily carcinomas of the lung, pancreas, bladder, andbreast. Unfortunately, as with other chemotherapeuticagents, it is complicated by kidney injury. Numerouscase reports and case series have documented AKI fromgemcitabine, primarily from TMA. In addition, hyperten-sion, microangiopathic hemolytic anemia, and ischemicskin lesion may be present. A recent case series of 29 pa-tients treated with gemcitabine described the various clin-ical renal manifestations (26). All patients developed AKI;TMA was seen in four patients who underwent kidneybiopsy. New or worsening hypertension occurred in 26of 29 patients, whereas edema (21/29) and congestiveheart failure (7/29) also complicated gemcitabine therapy.Classic systemic TMA occurred in all patients and wasmanifested by anemia, thrombocytopenia, and increasedlactate dehydrogenase levels. Suppressed haptoglobin lev-els (23/26) and schistocytes on peripheral smear (21/24)were also noted. Urinalysis revealed hematuria/proteinuria(27/29) and red blood cell casts (n58).Gemcitabine-associated TMA is relatively rare, with the
major risk factors being previous therapy with mitomycin-Cand total drug dose. Often, it is impossible to predict whowilldevelop this complication, although new or worsened hy-pertension may precede other clinical manifestations of TMA.Unfortunately, therapy is generally limited and mainly sup-portive, consisting of drug discontinuation, antihypertensivemedications, and dialysis when indicated. Plasmapheresis hasbeen used with minimal or no success. Renal outcomes arehighly variable. In the largest cases series (26), 19 patients hadfull or partial renal recovery, whereas 3 patients developedCKD and 7 patients had dialysis-requiring ESRD.
Glomerulus: PodocytopathyIFNIFN is a glycoprotein synthesized and released by
leukocytes, fibroblasts, T cells, and natural killer cells in
Table 2. Kidney injury associated with chemotherapeuticagents
Renal vasculaturehemodynamic AKI (capillary leak syndrome)IL-2, denileukin diftitox
thrombotic microangiopathyantiangiogenesis drugs (bevacizumab and tyrosinekinase inhibitors)
gemcitabine and cisplatinmitomycin C and IFN
Glomeruliminimal change diseaseIFNpamidronate
focal segmental glomerulosclerosisIFNpamidronatezoledronate (rare)
Tubulointerstitiumacute tubular necrosisplatinums, zoledronate, ifosfamide, and mithramycinpentostatin, imatinib, diaziquone, and pemetrexed
tubulopathiesFanconi syndromecisplatin, ifosfamide, and azacitadine,diaziquone, imatinib, and pemetrexed
salt wastingcisplatin and azacitadine
magnesium wastingcisplatin, cetuximab, and panitumumab
nephrogenic diabetes insipiduscisplatin, ifosfamide, and pemetrexed
syndrome of inappropriate antidiuresiscyclophosphamide and vincristine
acute interstitial nephritissorafenib and sunitinib
crystal nephropathymethotrexate
Figure 1. | A glomerulus exhibits mesangiolysis, endothelial de-nudation, red blood cell congestion, and glomerular basement mem-brane duplication in this example of thrombotic microangiopathy.(Jones methenamine silver stain; original magnification, 3600.) Cour-tesy of Glen S. Markowitz.
Clin J Am Soc Nephrol 7: 1713–1721, October, 2012 Nephrotoxicity of Chemotherapy, Perazella 1715

response to pathogens, such as viruses, parasites, and bac-teria, as well as tumor cells. It is a protective defense thatallows communication between cells to eradicate infectionor malignant cells. In general, IFN-a and -b reduce viralreplication and protein synthesis in neighboring cells,whereas IFN-g activates macrophage and MHC expression(27,28). Based on these characteristics, exogenous IFN has anumber of therapeutic uses. The most commonly usedagent is IFN-a, which is used to treat hepatitis C and Bviruses and various malignancies. IFN-b is used to treatmultiple sclerosis, whereas IFN-g was studied as a treatmentfor chronic granulomatous disease but has been abandoned.Chronic IFN therapy is associated with clinical renal disease,
which seems to be associated primarily with podocyte injury(27,28). Based on published cases, minimal change diseasehas been described with both IFN-a (n56) and -b (n52). Thelesion developed anywhere from 5 days to 22 months aftertherapy (27). Nephrotic syndrome with urinary protein lev-els of 2.3–28 g/d occurred in all patients, whereas AKI com-plicated the course of two patients. In follow-up rangingfrom 53 days to 12 months, complete remission was notedin all patients, with discontinuation of IFN and steroid ther-apy in three patients (27,28).FSGS constitutes another form of podocytopathy that can
complicate IFN therapy. The lesion FSGS-not otherwisespecified has been described in 10 patients treated with IFNtherapy (IFN-a in 9 patients and -g in 1 patient) (27,28). Inthese cases, IFN therapy ranged from 19 days to 20 monthsand was associated with nephrotic syndrome in all 10 patients,with proteinuria of 6.3–42 g/d and AKI in 8 patients. Withfollow-up ranging from 1 to 16 months in 9 of 10 patients,complete or partial remission was noted in 4 of 9 patientswith discontinuation of IFN. Six patients received steroids,of which only two patients benefited with remission.Collapsing FSGS (Figure 2) also complicates IFN ther-
apy. In 14 patients with this lesion, IFN-a was the causa-tive agent in 9 patients, whereas IFN-b and -g therapy wasused in 3 and 2 patients, respectively (27,28). IFN exposureranged from 1 to 48 months; nephrotic syndrome was
present in 12 of 13 patients, with proteinuria rangingfrom 1.9 to 27 g/d. AKI occurred in 11 patients. Urinesediment was bland in nearly one-half of the patients,whereas red and white blood cells were seen in five andone patients, respectively. Remission was inconsistent andincomplete in most patients after IFN discontinuation.With follow-up in 10 patients, which ranged from 2 to54 months, complete remission was noted in 1 patientand partial remission was noted in 3 patients, whereassome improvement in proteinuria and kidney functionwas described in 5 patients. Of these 10 patients, 8 patientsreceived steroids, and 1 patient received cyclophospha-mide. Thus, although IFN discontinuation sometimeshelps, it is not always associated with remission. Steroidsseem unhelpful, especially in FSGS.The mechanism of podocyte injury with IFN is incom-
pletely understood. A number of putative mechanismshave been put forward (27). A direct IFN effect on thepodocyte is possible through receptor binding and activa-tion that promotes two potential injurious effects: (1) al-tered cellular proliferation and metabolism of thepododyte and (2) increased podocyte oxidative capacityand increased MHC class II antigen expression. IndirectIFN effects on the podocyte may also contribute to thedevelopment of FSGS. IFNs activate adaptive immunemechanisms that increase macrophage activation—an ex-ample is hemophagocytic syndrome, which is associatedwith collapsing FSGS. Viral diseases such as HIV and par-vovirus B19, which increase IFN production, are also as-sociated with collapsing FSGS. Finally, IFN may enhancesynthesis of pathogenic cytokines such as IL-6 and -13,which are permeability factors in FSGS and minimalchange disease.
Tubules: Acute Tubular InjuryCisplatinCisplatin is a platinum compound that is an effective
therapy for many carcinomas and sarcomas, as well aslymphomas. Its major adverse effect is nephrotoxicity,although ototoxicity also occurs. Both are dose-relatedtoxicities, causing apoptosis and necrosis of cells (29–32).Cisplatin injures multiple renal compartments, includingblood vessels, glomeruli, and most commonly, the tubules(29). Nephrotoxicity is generally reversible, but it can bepermanent. Tubular injury as manifested by AKI and tu-bular dysfunction syndromes will be described.Cisplatin’s mechanism of nephrotoxicity is related to its
drug characteristics, its renal handling, and the kidney re-sponse to the cisplatin molecule (29–32). Chloride at thecis-position of the molecule is one such factor that pro-motes kidney injury, whereas the pathway of excretionthrough the cell (in through organic anion transporter 1and out through efflux transporters) potentially increasesintracellular concentrations (Figure 3). After it is inside thetubular cell, a number of intracellular injury pathways oc-cur. These pathways include caspase activation, cyclin-dependent kinases, mitogen-activated protein kinaseactivation, and p53 signaling. In addition, cellular injuryalso develops from inflammation and oxidative stress,whereas vascular injury and decreased GFR with ischemicinjury also occur (29–32). These pathways of injury result
Figure 2. | A glomerulus exhibits global wrinkling and retraction ofthe glomerular basement membrane and diffuse swelling and hy-perplasia of overlying visceral epithelial cells in this example ofcollapsing focal segmental glomerulosclerosis. (Jones methenaminesilver stain; original magnification, 3600.) Courtesy of Glen S.Markowitz.
1716 Clinical Journal of the American Society of Nephrology

in renal tubular cell apoptosis and necrosis—resulting inclinical AKI and/or a tubulopathy.Tubular injury without AKI also complicates cisplatin
therapy and manifests as a number of tubulopathies, whichinclude isolated proximal tubulopathy (proteinuria andphosphate wasting) or full-blown Fanconi syndrome (2,29).Sodium wasting is another consequence of cisplatin therapy,which can be associated with hypovolemia, orthostasis,and prerenal AKI. In the distal nephron, cisplatin can im-pair reabsorption of magnesium, causing refractory hypo-magnesemia. Finally, water absorption in the collectingduct can be disturbed, resulting in a form of nephrogenicdiabetes insipidus.AKI is a dose-related complication of cisplatin—it can be a
functional decline in GFR or caused by TMA, but mostcommonly, it is a result of acute tubular injury/necrosis.Although AKI can recover, renal outcomes such as pro-
gressive CKD from chronic tubulointerstitial fibrosis andirreversible chronic tubulopathies may result (2,29).A focus of care for patients receiving cisplatin is pre-
vention of kidney injury (2,29). Forced diuresis with intra-venous normal saline or hypertonic saline is used tocounteract the toxic effect of chloride on the cis-positionof the molecule. The addition of mannitol to induce aforced diuresis is sometimes used, but evidence of benefitis lacking; in some cases, this approach may cause wors-ened kidney function (33). Amifostine is a glutathione an-alog taken up by normal cells that blunts cisplatin-inducedcellular injury. It is, however, complicated by nausea andvomiting. A number of other agents have been used toreduce nephrotoxicity, such as sodium thiosulfate,
nucleophilic sulfur thiols, neurotrophins, phosphonic acid,melanocortins, and free oxygen radical scavengers, but theirutility is unclear. In high-risk patients, other platinums suchas carboplatin and oxalaplatin are used based on their lessnephrotoxic profile compared with cisplatin. Two potentialexplanations for reduced nephrotoxicity exist. Neither ofthese molecules is transported by organic cation transporter2 (OCT-2), thereby reducing proximal tubular intracellularconcentrations (29–32). In addition, the chloride at cis-positionin cisplatin is replaced by carboxylate and cyclobutane in car-boplatin and oxalaplatin, respectively, which may further re-duce toxicity (29–32).Treatment of toxic renal manifestations is primarily
supportive (2,28). There are no effective therapies to reverseAKI or tubular dysfunction. Dialysis is reserved for ad-vanced AKI as manifested by uremia, metabolic disturban-ces, and hypervolemia. There is no role for dialytic removalof cisplatin. Maneuvers to correct hypovolemia from saltwasting (intravenous normal saline or oral sodium chloride)and address symptomatic hypomagnesemia with intrave-nous and/or oral magnesium are required. Fanconi syn-drome is notoriously difficult to treat.
IfosfamideIfosfamide is an alkylating agent used for certain cancers,
including sarcomas, testicular cancer, and some forms oflymphoma. Ifosfamide’s major adverse effect is kidney in-jury. Nephrotoxic manifestations include tubulopathiessuch as proximal tubular injury or Fanconi syndromeand nephrogenic diabetes insipidus; additionally, AKI is oftenreversible but can be permanent (2,34,35). Histopathology
Figure 3. | Cisplatin (Cis) nephrotoxicity is, in part, related to its uptake by proximal tubular cells. Cis enters cells through organic cationtransporters (OCTs), andwhen it accumulateswithin cells, it causes cell injury throughmultiplemechanisms. Apoptosis and necrosis of tubularcells result and cause clinical AKI and tubulopathy. CDKs, cyclin-dependent kinases; MAPK, mitogen-activated protein kinase; MRP, multidrug-resistant protein; NaDC, sodium dicarboxylate; OAT, organic anion transporter; P53, protein 53; Pgp, P glycoprotein; ROS, reactive oxygenspecies.
Clin J Am Soc Nephrol 7: 1713–1721, October, 2012 Nephrotoxicity of Chemotherapy, Perazella 1717

reveals features of tubular cell injury/necrosis with swollen,dysmorphic mitochondria.The difference in adverse effects for ifosfamide (neph-
rotoxicity) and cyclophosphamide (hemorrhagic cystitis),which are related compounds, is caused by the major toxicmetabolite that they produce. Acrolein produced by cyclo-phosphamide is non-nephrotoxic, whereas chloracetaldehydeproduced by ifosfamide injures kidney tissue. At equivalentdoses, ifosfamide produces 40 timesmore chloroacetaldehydethan cyclophosphamide (2,34,35). Furthermore, ifosfamideenters proximal tubular cells through OCT2, whereas cy-clophosphamide does not (2,35). Risk factors for adverserenal effects include previous cisplatin exposure, cumulativedose .90 g/m2, and underlying CKD.Preventive measures are limited for ifosfamide. Mesna,
which is effective for hemorrhagic cystitis, is of limitedvalue for ifosfamide-induced kidney injury. Dose reductionhelps but also limits the efficacy of tumor killing. Becausethis agent is transported into cells through OCT2, compet-itive inhibition of this pathway with cimetidine is beingevaluated (35). Treatment, as with many of these agents, issupportive. Attention to supplementing electrolyte defi-ciencies, monitoring for progressive CKD, and dialysis asindicated are important. In addition to CKD and ESRD,long-term complications include a permanent proximal tu-bulopathy (1%) and isolated renal phosphaturia in up to20%. This latter complication may cause osteomalacia orgrowth problems in children and exacerbate osteoporosisin the elderly (2,34,35).
PemetrexedPemetrexed is an antifolate agent that inhibits enzymes
involved in purine/pyrimidine metabolism, thereby im-pairing RNA/DNA synthesis in tumors such as malignant
mesothelioma and non-small cell lung cancer. This agent isexcreted unchanged by the kidneys (70%–90% in 24 hours),with a half-life of 3.5 hours (36). Its renal handling mayexplain some of the drug’s nephrotoxicity. It is postulatedthat pemetrexed enters proximal tubular cells through twoseparate pathways. One cell entry site is the basolateralmembrane, where pemetrexed is transported through re-duced folate carrier, whereas the other is apical drug uptakethrough the folate receptor-a transport pathway (Figure 4).After inside the cell, pemetrexed is polyglutamylated withtwo resulting effects. First, with polyglutamylation, thedrug can no longer be transported out of the cell, increasingintracellular concentrations. Second, the higher drug con-centrations more fully inhibit folate metabolism enzymesand impair cellular RNA/DNA synthesis.Although its major adverse effects are myelosuppression
and neutropenia, reversible AKI has been noted with high-dose therapy (600 mg/m2). Pemetrexed nephrotoxicity hasbeen described in several case reports: acute tubular necrosis(n55), acute interstitial nephritis (n52), nephrogenic diabe-tes insipidus/renal tubular acidosis (n51), and nephrogenicdiabetes insipidus (n51) (36). A decline in CrCl from 88 to77 ml/min was reported after four cycles of pemetrexed in amesothelioma trial (36). Most patients present with AKI andminimal proteinuria, which stabilizes with drug discontinu-ation but can lead to permanent CKD. Renal histology re-veals primarily chronic tubulointerstitial fibrosis and tubularatrophy, consistent with a toxic tubular injury (36).
Tubules: Magnesium WastingCetuximabCetuximab is a chimeric monoclonal antibody against EGF
receptor (EGFR) used to treat various epithelial malignancies,
Figure 4. | Pemetrexed (Pmx) nephrotoxicity may result from proximal tubular cell uptake of drug through apical (folate receptor-a [FR-a])and basolateral (reduced folate carrier [RFC]) transporters. After it is inside the cell, Pmx is polyglutamylated, which inhibits drug transportout of the cell and enhances drug inhibition of folate metabolism. Impaired synthesis of RNA and DNA by cells causes clinical AKI andtubulopathy. Pmx-glt, polyglutamylated pemetrexed.
1718 Clinical Journal of the American Society of Nephrology

including colorectal, head/neck, breast, and lung cancers.EGF overexpression present in these malignancies reducesapoptosis and enhances tumor cell growth. Cetuximab hasa 10-fold greater affinity for EGFR than natural ligand, makingit an effective targeted therapy in these cancers. However, oneof the drug’s adverse effects is hypomagnesemia (37–41).Cetuximab-induced hypomagnesemia results from a renal
leak of magnesium. Magnesium reabsorption in the distalconvoluted tubule is, in part, dependent on EGF binding itsreceptor on the basolateral membrane (37,38,41). Activationof the EGFR sets in motion intracellular signals that stimu-late the movement of the cation channel transient receptorpotential M6 into the apical membrane, which facilitates thereabsorption of magnesium from the urinary space into thecell (37,38,41). Cetuximab competitively inhibits EGF bind-ing to its receptor (Figure 5), thereby blunting the placementof transient receptor potential M6 into the apical membraneand causing renal magnesium wasting (37,38,41).The incidence of hypomagnesemia with cetuximab in
initial colorectal cancer trials was only 1.8%–5.8% (37,38).However, a higher incidence was noted when magnesiumlevels were measured more rigorously. More than one-halfof patients develop hypomagnesemia with cetuximab, andnearly 100% of patients have some decline in serum mag-nesium concentrations (37,38,41). In fact, a meta-analysisof randomized controlled trials of cetuximab versus othertherapies showed an odds ratio of 4.7 for all-grade hypo-magnesemia and 5.3 for grade 3/4 hypomagnesemia (42).In addition, hypokalemia and hypocalcemia occur withsevere hypomagnesemia. Risk factors for hypomagnesemiainclude duration of cetuximab therapy, older age, and base-line magnesium concentration. Panitumumab is also com-plicated by hypomagnesemia (36%) but of less severegrade, because only 3% developed grade 3/4 hypomag-nesemia (1,2).
Treatment of hypomagnesemia requires intravenousrepletion, because oral magnesium is ineffective and oftencomplicated by diarrhea. Along with magnesium sup-plementation, calcium and potassium repletion are alsocommonly required. In general, renal magnesium wastingimproves and eventually resolves approximately 4–6 weeksafter discontinuation of cetuximab.
Tubules: Crystal NephropathyMethotrexateThe dihydrofolate reductase inhibitor methotrexate is
widely used, especially in high dose, to treat malignanciessuch as high-grade lymphomas. Nephrotoxicity is a knowncomplication of high-dose therapy (1–12 g/m2) but rarelyoccurs with long-term conventional dosing (1,2). The in-cidence is highly variable depending on the patient’s riskfactors and the appropriate employment of preventivemeasures, such as intravenous fluids (1,2,43).Methotrexate and its major metabolite, 7-OH metho-
trexate, are filtered by the glomerulus and secreted intothe urinary space by proximal tubules. AKI is primarilythe result of acute tubular injury from precipitation ofmethotrexate/7-OH methotrexate in distal tubular lumens(2,43). However, tubular apoptosis/necrosis may also de-velop from oxygen radicals associated with decreasedadenosine deaminase activity (44). AKI incidence, whendefined as grade .2 nephrotoxicity, ranges from 1.8% to12% (2,43).Risk factors for nephrotoxicity include intravascular
volume depletion with sluggish urinary flow, acid urinepH, and underlying kidney disease (GFR,60 ml/min)(2,42). Based on these factors, prevention is focused onvolume repletion before/during drug infusion, appropri-ate drug dosing, and alkalinization of the urine (pH.7.1).
Figure 5. | Cetuximab (C) is an EGF receptor (EGFR) antibody that causes renal magnesium wasting by competing with EGF for its receptor.Normally, EGF binds its receptor (EGFR) and stimulates magnesium reabsorption in the distal convoluted cell. EGFR activation is associatedwith magnesium absorption through transient receptor potential M6 (TRPM6) in the apical membrane. NCC, sodium chloride cotransporter.
Clin J Am Soc Nephrol 7: 1713–1721, October, 2012 Nephrotoxicity of Chemotherapy, Perazella 1719

Treatment is comprised of leucovorin rescue at 24–36hours of methotrexate therapy to reduce nonmalignantcell injury (2,43,45). Glucarbidase cleaves methotrexate tononcytotoxic metabolites. It is reserved for use when meth-otrexate levels are toxic, and there is significant risk forsystemic toxicity (45). High-flux hemodialysis clears theplasma of methotrexate fairly well (76%) but is associatedwith immediate postdialysis plasma rebound (1,2). It mayhave a role when severe AKI is present, but it may becomeunnecessary with the availability of glucarbidase.
ConclusionChemotherapeutic agents have improved cancer patient
survival; however, nephrotoxicity remains an importantcomplication. A number of patient- and drug-related fac-tors increase risk for adverse renal events; some events aremodifiable, and others are not. Clinicians must be familiarwith the nephrotoxicity of these drugs, particularly the as-sociated clinical and laboratory manifestations. Preventivemeasures should be used when possible, as well as support-ive care and available therapies. Some patients will be leftwith long-term complications such as chronic tubulopathiesand CKD. Onco-Nephrology is a rapidly expanding area thatrequires a close working relationship between oncologistsand nephrologists.
DisclosuresNone.
References1. Sahni V, Choudhury D, Ahmed Z: Chemotherapy-associated re-
nal dysfunction. Nat Rev Nephrol 5: 450–462, 20092. Perazella MA, Moeckel GW: Nephrotoxicity from chemothera-
peutic agents: Clinical manifestations, pathobiology, andprevention/therapy. Semin Nephrol 30: 570–581, 2010
3. Finkel KW, Foringer JR: Renal disease in patientswith cancer.NatClin Pract Nephrol 3: 669–678, 2007
4. Lameire NH, Flombaum CD, Moreau D, Ronco C: Acute renalfailure in cancer patients. Ann Med 37: 13–25, 2005
5. de JongeMJA, Verweij J: Renal toxicities of chemotherapy. SeminOncol 33: 68–73, 2006
6. Humphreys BD, Soiffer RJ, Magee CC: Renal failure associatedwith cancer and its treatment: An update. J Am Soc Nephrol16: 151–161, 2005
7. Evenepoel P: Acute toxic renal failure. Best Pract Res Clin An-aesthesiol 18: 37–52, 2004
8. Singh NP, Ganguli A, Prakash A: Drug-induced kidney diseases.J Assoc Physicians India 51: 970–979, 2003
9. Guo X, Nzerue C: How to prevent, recognize, and treat drug-induced nephrotoxicity. Cleve Clin J Med 69: 289–297, 2002
10. Jerki�c M, Vojvodi�c S, Lopez-Novoa JM: The mechanism of in-creased renal susceptibility to toxic substances in the elderly. Part I.The role of increased vasoconstriction. Int Urol Nephrol 32: 539–547, 2001
11. Perazella MA: Renal vulnerability to drug toxicity. Clin J Am SocNephrol 4: 1275–1283, 2009
12. Harty L, Johnson K, Power A: Race and ethnicity in the era ofemerging pharmacogenomics. J Clin Pharmacol 46: 405–407,2006
13. Ciarimboli G, Koepsell H, Iordanova M, Gorboulev V, Durner B,Lang D, Edemir B, Schroter R, Van Le T, Schlatter E: IndividualPKC-phosphorylation sites in organic cation transporter 1 de-termine substrate selectivity and transport regulation. J Am SocNephrol 16: 1562–1570, 2005
14. Ulrich CM, Bigler J, Potter JD: Non-steroidal anti-inflammatorydrugs for cancer prevention: Promise, perils and pharmacogenetics.Nat Rev Cancer 6: 130–140, 2006
15. Lang F: Regulating renal drug elimination? J Am Soc Nephrol 16:1535–1536, 2005
16. Enomoto A, Endou H: Roles of organic anion transporters (OATs)and a urate transporter (URAT1) in the pathophysiology of humandisease. Clin Exp Nephrol 9: 195–205, 2005
17. Ciarimboli G, Ludwig T, Lang D, Pavenstadt H, Koepsell H,Piechota HJ, Haier J, Jaehde U, Zisowsky J, Schlatter E: Cisplatinnephrotoxicity is critically mediated via the human organiccation transporter 2. Am J Pathol 167: 1477–1484, 2005
18. Cummings BS, Schnellmann RG: Pathophysiology of nephro-toxic cell injury. In:Diseases of the Kidney and Urogenital Tract,edited by Schrier RW, Philadelphia, PA, Lippincott Williams &Wilkinson, 2001, pp 1071–1136
19. Kaloyanides GJ, Bosmans J-L, DeBroe ME: Antibiotic andimmunosuppression-related renal failure. In: Diseases of theKidney andUrogenital Tract, edited by Schrier RW, Philadelphia,PA, Lippincott Williams & Wilkinson, 2001, pp 1137–1174
20. Aleksa K, Matsell D, Krausz K, Gelboin H, Ito S, Koren G: Cyto-chrome P450 3A and 2B6 in the developing kidney: Implicationsfor ifosfamide nephrotoxicity. Pediatr Nephrol 20: 872–885, 2005
21. Gurevich F, Perazella MA: Renal effects of anti-angiogenesistherapy: Update for the internist. Am J Med 122: 322–328,2009
22. De Stefano A, Carlomagno C, Pepe S, Bianco R, De Placido S:Bevacizumab-related arterial hypertension as a predictivemarker in metastatic colorectal cancer patients. CancerChemother Pharmacol 68: 1207–1213, 2011
23. Sugimoto H, Hamano Y, Charytan D, Cosgrove D, Kieran M,Sudhakar A, Kalluri R: Neutralization of circulating vascularendothelial growth factor (VEGF) by anti-VEGF antibodies andsoluble VEGF receptor 1 (sFlt-1) induces proteinuria. J Biol Chem278: 12605–12608, 2003
24. Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M,Weisstuch J, Richardson C, Kopp JB, Kabir MG, Backx PH,Gerber HP, Ferrara N, Barisoni L, Alpers CE, Quaggin SE: VEGFinhibition and renal thrombotic microangiopathy. N Engl J Med358: 1129–1136, 2008
25. Izzedine H, Rixe O, Billemont B, Baumelou A, Deray G: An-giogenesis inhibitor therapies: Focus on kidney toxicity and hy-pertension. Am J Kidney Dis 50: 203–218, 2007
26. Glezerman IG, Kris MG, Miller V, Seshan S, Flombaum CD:Gemcitabine nephrotoxicity and hemolytic uremic syndrome:Report of 29 cases from a single institution. Clin Nephrol 71:130–139, 2009
27. Markowitz GS, Nasr SH, Stokes MB, D’Agati VD: Treatment withIFN-alpha, -beta, or -gamma is associated with collapsing focalsegmental glomerulosclerosis. Clin J Am Soc Nephrol 5: 607–615, 2010
28. Colovic M, Jurisic V, Jankovic G, Jovanovic D, Nikolic LJ,Dimitrijevic J: Interferon alpha sensitisation induced fatal renalinsufficiency in a patient with chronic myeloid leukaemia: Casereport and review of literature. J Clin Pathol 59: 879–881, 2006
29. Pabla N, Dong Z: Cisplatin nephrotoxicity: Mechanisms andrenoprotective strategies. Kidney Int 73: 994–1007, 2008
30. Kawai Y, Nakao T, KunimuraN, Kohda Y, GembaM: Relationshipof intracellular calcium and oxygen radicals to Cisplatin-relatedrenal cell injury. J Pharmacol Sci 100: 65–72, 2006
31. Faubel S, Ljubanovic D, Reznikov L, Somerset H, Dinarello CA,Edelstein CL: Caspase-1-deficient mice are protected againstcisplatin-induced apoptosis and acute tubular necrosis. KidneyInt 66: 2202–2213, 2004
32. RameshG, ReevesWB: TNFR2-mediated apoptosis and necrosisin cisplatin-induced acute renal failure. Am J Physiol RenalPhysiol 285: F610–F618, 2003
33. Morgan KP, Buie LW, Savage SW: The role of mannitol as anephroprotectant in patients receiving cisplatin therapy. AnnPharmacother 46: 276–281, 2012
34. Zamlauski-Tucker MJ, Morris ME, Springate JE: Ifosfamide metab-olite chloroacetaldehyde causes Fanconi syndrome in the perfusedrat kidney. Toxicol Appl Pharmacol 129: 170–175, 1994
35. Ciarimboli G, Holle SK, Vollenbrocker B, Hagos Y, Reuter S,Burckhardt G, Bierer S, Herrmann E, Pavenstadt H, Rossi R, KletaR, Schlatter E: New clues for nephrotoxicity induced by ifosfamide:Preferential renal uptake via the human organic cation trans-porter 2. Mol Pharm 8: 270–279, 2011
1720 Clinical Journal of the American Society of Nephrology

36. Glezerman IG, PietanzaMC,Miller V, Seshan SV: Kidney tubulartoxicity ofmaintenance pemetrexed therapy.Am J KidneyDis 58:817–820, 2011
37. Glaudemans B, Knoers NV, Hoenderop JG, Bindels RJ: Newmolecular players facilitating Mg(21) reabsorption in the distalconvoluted tubule. Kidney Int 77: 17–22, 2010
38. Schrag D, Chung KY, Flombaum C, Saltz L: Cetuximab therapyand symptomatic hypomagnesemia. J Natl Cancer Inst 97: 1221–1224, 2005
39. Fakih MG, Wilding G, Lombardo J: Cetuximab-induced hypo-magnesemia in patients with colorectal cancer. Clin ColorectalCancer. 6: 152–156, 2006
40. Saif MW: Management of hypomagnesemia in cancer patientsreceiving chemotherapy. J Support Oncol 6: 243–248, 2008
41. Dietrich A, Chubanov V, Gudermann T: Renal TRPathies. J AmSoc Nephrol 21: 736–744, 2010
42. CaoY, LiaoC, Tan A, Liu L,Gao F:Meta-analysis of incidence andrisk of hypomagnesemia with cetuximab for advanced cancer.Chemotherapy 56: 459–465, 2010
43. PerazellaMA:Crystal-induced acute renal failure.Am JMed 106:459–465, 1999
44. Pinheiro FV, Pimentel VC, De Bona KS, Scola G, Salvador M,Funchal C, Moretto MB: Decrease of adenosine deaminase ac-tivity and increase of the lipid peroxidation after acute metho-trexate treatment in young rats: protective effects of grape seedextract. Cell Biochem Funct 28: 89–94, 2010
45. Patterson DM, Lee SM: Glucarbidase following high-dosemethotrexate: Update on development. Expert Opin Biol Ther10: 105–111, 2010
Published online ahead of print. Publication date available at www.cjasn.org.
Clin J Am Soc Nephrol 7: 1713–1721, October, 2012 Nephrotoxicity of Chemotherapy, Perazella 1721