nitrogen-13: historical review and future perspectives
TRANSCRIPT

Special Issue Review
Received 16 July 2013, Accepted 29 October 2013 Published online 14 January 2014 in Wiley Online Library
(wileyonlinelibrary.com) DOI: 10.1002/jlcr.3163
244
Nitrogen-13: historical review and futureperspectives†
Vanessa Gómez-Vallejo, Vijay Gaja, Kiran B. Gona, and Jordi Llop*
Positron emission tomography is an ultra-sensitive, in vivo molecular imaging technique that allows the determination ofthe spatiotemporal distribution of a positron emitter labeled radiotracer after administration into living organisms. Amongall existing positron emitters, 18F has been by far the most widely used both in clinical diagnosis and in preclinicalinvestigation, while the use of 11C significantly increased after the 1980s because of the widespread installation ofbiomedical cyclotrons. The use of other shorter-lived positron emitters such as 13N (T1/2 = 9.97min) has been historicallymore restricted. Paradoxically, its stable isotope (14N) is present in many biological active molecules; consequently, thedevelopment of strategies for the efficient incorporation of 13N into radiotracers would represent an interesting alternativeto 11C- and 18F-labeling. In the current paper, the developments related to 13N chemistry are reviewed, including differentproduction routes of primary precursors and their applications to the preparation of more complex 13N-labeled molecules.The current situation and future perspectives are also briefly discussed. Copyright © 2014 John Wiley & Sons, Ltd.
Keywords: nitrogen-13; positron emission tomography; radiotracer; radiochemistry
Radiochemistry Department, Molecular Imaging Unit, CIC biomaGUNE; ParqueTecnológico de Miramón, San Sebastián, 20009, Guipúzcoa, Spain
*Correspondence to: Jordi Llop, Radiochemistry Department, CIC-biomaGUNE, PºMiramón 182, Parque Tecnológico San Sebastián, 20009 San Sebastián,Guipúzcoa, Spain.E-mail: [email protected]
† This article is published in the Journal of Labelled Compounds andRadiopharmaceuticals as a special issue on ‘Current Developments in PET andSPECT Imaging’, edited by Jonathan R. Dilworth, University of Oxford and SofiaI. Pascu, University of Bath.
Introduction
Nitrogen-13 (13N), together with 18F, 15O and 11C, is one of thepositron emitters that can be produced on the multi-gigabecquerel scale in biomedical cyclotrons. Currently, it isusually generated via the 16O(p,α)13N nuclear reaction byirradiation of 5mM ethanol aqueous solution with high-energyprotons.1 Ethanol acts as a scavenger and creates a reducingenvironment; under these conditions, the major 13N-labeledspecies is [13N]NH3, which is used as produced and has beenextensively applied in the preclinical and clinical evaluation ofmyocardium2,3 and brain4 blood flow and/or perfusion.
Because of the short half-life of 13N (9.97min), the distribution of13N-labeled radiotracers from large production facilities tosurrounding clinical centers or research laboratories is unrealisticor, at least, very challenging. In addition, the high positron range(maximum energy of 1.19MeV, maximum range in water of5.4mm) usually leads to low resolution images, especially whencompared with those obtained with 18F-labeled radiotracers(maximum energy of 0.69MeV, maximum range in water of2.4mm). As a result, the use of 13N has been historically veryrestricted, and only a few studies describing new synthetic strategiesfor the incorporation of this radioisotope into bioactive moleculeshave been reported to date. However, 13N can be produced indifferent chemical forms in the cyclotron; thus, the potentialapplications and synthetic opportunities offered by this interestingisotope go far beyond the production and use of [13N]NH3. Inaddition, the stable isotope of nitrogen (14N) is present in themajorityof biological active molecules; therefore, the incorporation of 13N inthe toolbox of positron emission tomography (PET) chemists mightbe a valuable option for the preparation of new labeled compoundsor incorporation of the label in different positions, providing furtherinformation related to the in vivo behavior of radiotracers andbecoming an interesting alternative to 11C and 18F.
J. Label Compd. Radiopharm 2014, 57 244–254
Finally, the new generation of smaller cyclotrons that are capableof producing reasonable amounts of positron emitters in acceptableirradiation times, together with the emergent technology ofmicrofluidics, might facilitate the progressive implementation of anew PET scenario, in which radiotracers could be prepared in situ,in small batches and under the concept of ‘dose on demand’ (oneproduction for one study or patient). Therefore, short-lived isotopessuch as 13N might become in the coming years a valuable tool forradiochemists, biologists and clinicians. Consequently, it is probablythe right time to review what has been developed to date and todiscuss potential future perspectives of this valuable (and oftenunderestimated) positron emitter.
A brief piece of history: the origin ofnitrogen-13
Nitrogen-13 was one of the earliest positron emitters to beproduced; it was discovered in 1934 by Joliot and Curie,5 whoirradiated boron nitride with α particles produced in a poloniumpreparation and produced 13N via the 10B(α,n)13N nuclearreaction. As textually reported by the authors in the paperpublished in Nature,
Copyright © 2014 John Wiley & Sons, Ltd.

Biography
Vanessa Gómez-Vallejo was born in1980 in Barcelona, Spain. She got herdegree in Organic Chemistry at IQS-URL (Barcelona) in 2005 and herdegree in Chemical Engineering alsoin 2005. Between 2005 and 2010, sheworked on her PhD Thesis at IAT-PRBB(Barcelona) and CIC biomaGUNE (SanSebastián) under the supervision ofDr. Jordi Llop and Dr. José IgnacioBorrell. Between 2005 and 2007, sheworked as Quality Control Manager of the RadiopharmaceuticalLaboratory at IAT-PRBB. Since October 2007, she is the PlatformManager of Radiochemistry at CIC biomaGUNE.
Biography
Vijay Gaja received his MSc degree inDrug Chemistry from the University ofPune, India, in 2006. Then, he workedas research associate for three years inthe pharmaceutical industry. In 2009,he moved to CIC biomaGUNE (SanSebastián, Spain) to pursue his PhD inRadiochemistry andMolecular Imagingunder the supervision of Dr. Jordi Llop.His PhD work focused on thedevelopment of 13N-labeling methodsand novel PET radiopharmaceuticalswith diagnostic application in Alzheimer’s disease.
Biography
Kiran Babu Gona was born in AndhraPradesh, India. He received hisMSc degree from Sri VenkateswaraUniversity, India, in 2006 before movingto Avra Labs as a synthetic chemist. In2008, he joined Biocon India Pvt Ltd asa senior research associate and didresearch on benzodiazepine moleculestargeting notch receptors. In 2011, hejoined CIC biomaGUNE to carry out hisPhD studies under the supervision ofDr. Jordi Llop.
Biography
Jordi Llop was born in 1973 inBarcelona, Spain. He got his degreein Chemistry at IQS-URL (Barcelona)in 1997, and one year later, hegraduated as a chemical engineer. In1999, he became a predoctoral fellowat ICMAB-CSIC, and in 2002, he startedtwo postdoctoral stays, one at ClínicaUniversitaria de Navarra and one atUppsala University PET Center. In 2004,he became the Production Managerat IAT-PRBB. In 2007 he moved to CICbiomaGUNE, where he is Principal Investigator and Head ofRadiochemistry. His research interests include radiolabelingof nanomaterials and synthesis of 13N-labeled compounds.
V. Gómez-Vallejo et al.
Copyright © 2014 JohJ. Label Compd. Radiopharm 2014, 57 244–254
Our latest experiments have shown a very striking fact: whenan aluminum foil is irradiated on a polonium preparation,the emission of positrons does not cease immediately, whenthe active preparation is removed. The foil remains radioactiveand the emission of radiation decays exponentially as for anordinary radio-element. We observed the same phenomenonwith boron and magnesium. The half-life period of the activityis 14 min. for boron, 2 min. 30 sec. for magnesium, 3 min. 15sec. for aluminum.
By further treatment of the irradiated boron nitride with sodiumhydroxide and subsequent distillation, a gas was generated,which in contact with a paper soaked in hydrochloric acidproduced a radioactive white deposit, which was identified asNH4Cl. Despite the inaccurate estimation of the half-life, Joliotand Curie were awarded with the Nobel prize in 1935 ‘for theirsynthesis of new radioactive elements’. Also in 1934, theproduction of 13N by irradiation of 13C-enriched graphite with400–500 KeV protons via the nuclear reaction 13C(p,n)13N andby irradiation of natural graphite with deuterons via the nuclearreaction 12C(d,n)13N was reported by Cockcroft et al.6; Cockcroftand Walton had to wait until 1951 to get the Nobel price awardfor ‘their pioneer work on the transmutation of atomic nuclei byartificially accelerated atomic particles’.
Since these early investigations, efforts have been directedtoward the design and development of new targets in order toimprove 13N production yields using accelerators, takingadvantage of the different nuclear reactions suitable for theproduction of 13N using proton and deuteron irradiation ofdifferent materials. Concurrently, new synthetic strategies for thepreparation of 13N-labeled radiotracers were developed, including[13N]N2, [
13N]NH3, [13N]NO�
3 , [13N]NO�2 , [13N]amino acids, [13N]
amides, [13N]amines, [13N]nitroso compounds and more recently[13N]azo derivatives and 13N-labeled nanoparticles (NPs). All thework performed in the period 1934–2002 was collated anddiscussed in a book chapter7 and will be revisited in this review.A revision of the advances occurred during the last 10 years, andfuture perspectives will be also discussed.
13N-labeled primary precursors: differentchemical forms for different applications
Several ways to produce 13N by irradiation of stable materialswith accelerated particles can be encountered in the literature(Table 1). Mainly three nuclear reactions using accelerated ionscan be utilized: 12C(d,n)13N, 13C(p,n)13N and 16O(p,α)13N. Inaddition, irradiation of natural nitrogen (14N) with neutrons alsoproduces 13N via an (n,2n) reaction. Among these, the nuclearreaction 16O(p,α)13N is by far the most commonly utilizednowadays. However, by modifying the incident particle and thechemical and physical form of the irradiated material, different13N-labeled species produced in the target can be easily isolatedand/or transformed into other labeling agents, which might bemore convenient depending on the final application.
24
Production of 13N[N2]
[13N]N2 is an inert gas that has a few potential applications.Because of its inertness, it is not a convenient labeling agentfor the production of more complex 13N-labeled compounds.However, it has been applied in nitrogen fixation8,9 andventilation studies in animals10 and humans.11
www.jlcr.orgn Wiley & Sons, Ltd.
5

Table 1. Methods for the production of 13N using nuclear reactions
Target material Nuclear reaction In-target product
CO2 (trace N2)12C(d,n)13N [13N]N2
Graphite 12C(d,n)13N [13N]CNCharcoal 12C(d,n)13N [13N]N2 + trapped [13N]CN13C-enriched charcoal 13C(p,n)13N Trapped [13N]CNH2O/ethanol
16O(p,α)13N [13N]NH3
H2O16O(p,α)13N [13N]NH3 + [13N]NO�
3 + [13N]NO�2
NaNO3 (aq)14N(n,2n)13N [13N]NH3
Al4C312C(d,n)13N Matrix trapped 13N
CH4 (flowing)12C(d,n)13N [13N]NH3 + [13N]HCN+ [13N]CH3NH2
V. Gómez-Vallejo et al.
246
Historically, after the pioneering work by Cockcroft et al.,6 13Nwas first produced as [13N]N2 in a particle accelerator byirradiation of charcoal in a gas-tight chamber using 8MeVdeuterons.12 The irradiated material containing 13N wasintroduced in a combustion tube containing CuO in a streamof O2. The resulting gas, containing a mixture of [13N]CN, [13N]NH3, [13N]NO and [13N]N2, was purified by slowly passingthrough a series of five traps and spirals immersed in liquid air,a solution of HNO3 and a solution of NH4OH. The resulting[13N]N2 was subsequently used for the assessment of nitrogenfixation in non-leguminous plants. The purification process waslater improved by Nicholas and co-workers,13 who, using acontinuous flow process, swept 13N produced after irradiationof charcoal with 15MeV deuterons with a stream of argonthrough a tube packed with copper at 750 °C and then throughdilute H2SO4 and alkaline sulfite solutions to remove traces ofammonia and nitrogen oxides. Interestingly, 555–1110MBq/min could be produced with a beam intensity of 40μA.Bombardment of 13C-enriched charcoal with 11MeV protons(intensity on target = 0.8–4.0μA, irradiation time= 30min) wasalso used for the production of [13N]N2.
9 The irradiated targetwas thoroughly mixed with fine CuO powder and KNO3 (thelatter to form carrier N2) and subjected to Dumas combustion.The combustion products were passed through CuO and Cu at500 °C using CO2 as a carrier, and after purification with a liquidnitrogen trap, pure [13N]N2 was obtained.
An alternative approach based on a liquid recirculation systemwas proposed by Parks and Krohn (Figure 1).14 When an ammoniaaqueous solution was irradiated with 18MeV protons, 75% [13N]N2
Figure 1. Schematic representation of the liquid recirculation system for the productcyclotron vault. The whole target solution (volume ~ 60mL) was typically circulated at 1
www.jlcr.org Copyright © 2014 Joh
was produced and was collected and accumulated online in aspecially designed reservoir situated outside the cyclotron vault.However, radiolytic decomposition of aqueous NH3 duringirradiation producing carrier N2 led to low specific radioactivities.
In-target production of [13N]N2 has been also reported byirradiation of 13C-enriched charcoal (pre-heated to 800 °C) with19MeV protons at 10μA (Figure 2a).15 Interestingly, under theseconditions, 99.7% of the generated activity was [13N]N2, whilesignificant amounts of [13N]NO and [13N]N2O (22.6% and 44.9%,respectively) were produced when a 1μA beam intensity was used.The radioactive species were transferred using helium gas andtrapped in a liquid cooled charcoal trap. Further conversion oftrapped [13N]N2 into [13N]NH3 was achieved by recirculation underhydrogen pressure through amicrowave discharge cavity (Figure 2b).
More recently, a gas phase target has been also reported as analternative for the production of [13N]N2 by irradiation of amixture N2/0.1% O2 with 18MeV protons (14N(p, pn)13N nuclearreaction) using a standard target for the production of[11C]CO2.
16 Indeed, considerable amounts of [14O]O2, [15O]O2,
[11C]CO and [11C]CO2 were simultaneously produced, and thus,inclusion of a CuO trap for oxidation of [11C]CO and purificationwith soda lime traps were required. Radioactive oxygen specieswere eliminated by exchange on CuO and by radioactive decay.
Other reported strategies for the generation of [13N]N2
comprise production of different species in the cyclotronfollowed by a post-processing step. One example is thetreatment of a mixture of [13N]NHþ
4 , [13N]NO�
3 and [13N]NO�2
(produced by irradiation of water with 27MeV protons) withDevarda’s alloy to yield [13N]NHþ
4 , which was reacted with NaOBr
ion and accumulation of [13N]N2. All components except the target were outside the
cm/s by using a variable speed pump. Adapted from the work of Parks and Krohn.14
n Wiley & Sons, Ltd. J. Label Compd. Radiopharm 2014, 57 244–254

Figure 2. (a) Schematic representation of the13C-enriched charcoal solid target. Approximately 0.8 g of 97% enriched
13C powder was packed into a
12C cylinder, which
was spring loaded into a quartz tube. The target allowed for the on-line production of13N in assorted chemical forms, which could be trapped in liquid nitrogen cooled
activated charcoal. (b) Schematic view of the recycling microwave discharge apparatus for the conversion of [13N]N2 to [
13N]NH3. Trapped [
13N]N2 was desorbed into a
closed recycling loop, the system was then pressurized with hydrogen and the gases were recycled through a microwave discharge cavity. The [13N]NH3 was removed
by condensation as it was produced. Adapted from the work of Ferrieri et al.15.
V. Gómez-Vallejo et al.
24
and carrier ammonia to give [13N]N2.17,18 The method was later
on refined to achieve higher specific activities.8
Production of [13N]NH3
As mentioned earlier, [13N]NH3 was first produced as early as1934 by Joliot and Curie5 by alpha particle irradiation of boronnitride and further heating with sodium hydroxide. A similarsynthetic strategy was used by Hunter and Monahan.19 However,in this occasion, 13N was produced by irradiation of an Al4C3 solidstate target with 8–12MeV deuterons. The activated target wasthen treated with KOH aqueous solution, and [13N]NH3 wasdistilled and trapped in an acidic solution as [13N]NH4OH. Welchand Lifton studied the formation of 13N-labeled species indifferent inorganic carbides after irradiation with 7MeVdeuterons.20 Among all carbides tested, Al4C3 yielded maximumpercentage of [13N]NH3 (75–90% depending on integratedcurrent in the target). During irradiation of aluminum carbidewith deuterons,28Al is also produced (2.4min half-life, highenergy gamma and beta radiation); to prevent the formation ofthis by-product, an alternative method based on the irradiationof continuously flowing methane with 8MeV deuterons wasproposed.21 The irradiated gas was directly bubbled throughwater or isotonic solution in which [13N]NH3 (radiochemicalpurity = 95%) was dissolved; 740MBq was produced in 5min ofbeamtime.
Krizek and co-workers described 1 year later that irradiation ofwater in the liquid phase with protons yielded a mixture of [13N]NO�
3 , [13N]NH3, and [13N]NO�
2 .22 [13N]NH3 is initially produced by
hydrogen abstraction from the target water matrix, and there isan increasing production of oxo anions of nitrogen withincreased target dose due to radiolytic oxidation. Using a 10μAbeam for 15min, 7.95 GBq of 13N was produced at the end ofbombardment (EOB); the percentage of [13N]NO�
3 was >85%,and treatment with Ti(OH)3 yielded 3.515GBq of [13N]NH3.
Copyright © 2014 JohJ. Label Compd. Radiopharm 2014, 57 244–254
Interestingly, when a cryogenic target containing frozen waterwas used, beam currents from 1 to 20μA produced [13N]NH3
directly because low temperature decreased radiolysis.23 Theradiochemical purity of the [13N]NH3 produced was >95%.However, the use of liquid targets is more convenient, and Idoand Iwata developed a fully automated system for theproduction of [13N]NH3 using proton-irradiated liquid water as13N source and titanium chloride as reducing agent24;radiochemical purities >99.9% were obtained with excellentradiochemical yields (87–91%).
In 1991, Wieland and co-workers discovered that the additionof scavengers to the irradiated water modified the percentage ofdifferent 13N-labeled species ([13N]NO�
3 , [13N]NHþ
4 , [13N]NO�
2 and[13N]2).
1 By addition of 5mM ethanol to the target, more than96% of the radioactivity was produced as [13N]NH3, irrespectiveof the beam intensity (15–30μA). Addition of acetic acid at aconcentration >5mM also yielded more than 95% of theradioactivity as [13N]NH3. The mechanisms by which smallamounts of scavenger react in the target to promote in-targetammonia production are not very well understood, but it is clearthat they prevent radiolytic oxidation of ammonia. As mentionedearlier, the irradiation of ethanol aqueous solution is currentlythe most widely used approach for the preparation of [13N]NH3
(see Figure 3 for typical target design). Alternatively,pressurization of the water target with hydrogen25–27 andmethane28 has also been successfully used for the productionof [13N]NH3. The combination of hydrogen and ethanol wasmore efficient than either alone at high beam doses.26
Because of the cross section of the nuclear reaction 16O(p,α)13N, the irradiation of water or aqueous solutions withprotons requires energies >10MeV for the production ofsignificant amounts of 13N. Thus, despite that solid targets areusually less convenient and more difficult to be remotelycontrolled, efforts have been focused to the development ofalternative production routes for small cyclotrons. Ferrieri and
www.jlcr.orgn Wiley & Sons, Ltd.
7

Figure 3. Typical target design for the production of [13N]NH3 by irradiation of 5mM ethanol aqueous solution with protons, based on the target Nirta® Liquid provided
by IBA Molecular.
V. Gómez-Vallejo et al.
248
co-workers15 developed a method for the conversion of the 13N2
produced in the cyclotron using the 13C(p,n)13N reaction into[13N]NH3 using microwave radiation as an external excitationsource to dissociate the nitrogen gas in a hydrogen plasma with65% conversion (Figure 2b). Irradiation of 13C-enriched graphitewas also used by Bida et al.29 In this case, continuous elution ofthe target with water was used to extract the ammonia. Verylow deuteron energies (0.8–3.2MeV) were successfully used byShefer et al.30 and Dence et al.31 who irradiated a windowlesssolid target and produced [13N]NH3 by the 12C(d,n)13N reaction.Initially, the graphite was heated in a stream of pure oxygen at800 °C to yield [13N]NO�
2 , which was eluted with water andconverted to [13N]NH3 with Raney nickel.
Production of [13N]NO�3 and [13N]NO�
2
[13N]NO�3 is the major species generated when pure water is
irradiated with protons;22 the formation of this species increaseswith increased target dose due to radiolytic oxidation.14
However, the presence of minor amounts of [13N]NO�2 , [13N]
NHþ4 and impurities coming from the target window (e.g., 48V)
can be detected. Cationic species can be removed by solid phaseextraction using a cation exchange resin,32 while removal of[13N]NO�
2 can be achieved by addition of 10% H2SO4 and boilingto decompose nitrous acid, which is expelled as NOx.
32
If [13N]NO�2 is the desired species, it can be obtained by
reduction of [13N]NO�3 produced after water irradiation. First
attempts consisted of using a cadmium–copper amalgam,providing a maximal ratio of [13N]NO�
2 to [13N]NO�3 of 6.14 Almost
quantitative reduction was achieved later by circulation of theirradiated solution through a copper-plated cadmium column,prepared by sequential treatment of cadmium with hydrochloricacid solution, purified water, copper (II) sulfate, ammoniumchloride solution and purified water.33 Radiochemical purity over97% was obtained after elimination of residual [13N]NH3 byaddition of NaOH, evaporation to dryness and reconstitution.The reduction using a copper-plated column has beensuccessfully applied by other research groups.34–38 Alternatively,the in situ generation of [13N]NO�
2 by treatment of [13N]NO�3 with
copper dust and application to N-nitrosation reactions has alsobeen reported.39
www.jlcr.org Copyright © 2014 Joh
13N-labeled compounds: in the pursuit ofefficient labeling methods
The primary precursors described earlier might be used asradiotracers with different applications. [13N]NH3 has beenextensively applied in the preclinical and clinical evaluation ofmyocardium and brain blood flow and perfusion.2–4 [13N]N2
has been used in investigations of nitrogen fixation8,9 andventilation studies in animals10 and humans,11 while [13N]NO�
3has been applied to the determination of [13N]NO�
3 transportvelocity in plants40 and to denitrification studies.41 However, manyother more complex and biologically active molecules can besynthesized using 13N. In this section, the chemical routes for thepreparation of such molecules will be examined. The particularapplications of the radiotracers are out of the scope of this review.
Production of [13N]amino acids
Amino acids have been labeled with 13N at the R group usingchemical methods and at the amino group using enzymaticmethods. Chemical methods are usually time consuming andless efficient, and only a few examples can be found in theliterature. The synthesis of L-[13N]aspargine was reported byreaction of L-α-N-Boc-aspartate with [13N]NH3 and furtherhydrolysis (Scheme 1).42 However, synthetic chemistry techniquesare relatively slow, produce intermediate compounds and yieldracemic mixtures of compounds having optical isomers. On theother hand, enzymatic syntheses are rapid, produce nointermediate compounds and yield the specific end product. Thus,biosynthetic methods are usually more convenient.
The first attempts to synthesize 13N-labeled amino acids usingbiosynthetic methods consisted of direct incubation of [13N]NH3
with the appropriate precursor and the enzyme (with cofactors)in buffered aqueous solution. By using this approach, L-[13N]glutamate was rapidly synthesized by incubation ofα-ketoglutarate with [13N]NH3 in the presence of L-glutamatedehydrogenase, which catalyses the reductive amination ofalpha-keto acids to amino acids, and nicotinamide adeninedinucleotide phosphate as cofactor in phosphate buffer at 25 °Cfor 5min (Scheme 2a). The crude reaction mixture was boiled toremove unreacted [13N]NH3 and filtered to yield 8mCi of L-[13N]glutamate (radiochemical yield = 15%).43 Following a parallel
n Wiley & Sons, Ltd. J. Label Compd. Radiopharm 2014, 57 244–254

Scheme 1. Synthesis of L-[13N]aspargine by a synthetic chemistry method.
Scheme 2. Synthesis of L-[13N]glutamate and L-[
13N]glutamine via biosynthetic routes.
V. Gómez-Vallejo et al.
strategy, L-[13N]glutamine was synthesized by incubation at of[13N]NH3 with glutamic acid and glutamine synthetase in thepresence of cysteine and adenosine triphosphate and MgSO4 ascofactors at 37 °C for 10min (Scheme 2b).44 Straatman and Welchdemonstrated the feasibility of utilizing glutamate dehydrogenaseto incorporate 13N into amino acids other than glutamate bysynthesizing L-[13N]valine, L-[13N]alanine and L-[13N]leucine fromthe corresponding α-keto acids and [13N]-ammonia withdecay-corrected yields ranging from 6.8% to 26%.45
Despite the fact that amino acids prepared as describedearlier43–45 were appropriate for animal studies, furtherpurification for complete elimination of the enzyme wasrequired for human applications. Thus, a purification methodbased on column chromatography was explored for thesynthesis of L-[13N]glutamic acid and L-[13N]alanine.46 When thecolumn was permitted to elute by gravity, the enzyme appearedto be bound to the top of the column. However, because of theshort half-life of 13N, pressure had to be applied, and thisresulted in lower separation efficiency and the presence of theenzyme in the final product. Thus, an alternative syntheticstrategy based on the use of ‘immobilized enzymes’ (enzymesthat are synthetically attached to a water-insoluble support byeither physical or chemical means) was developed. This was firstsuccessfully employed in the preparation of L-[13N]glutamic acidby dissolving [13N]NH3, alpha-ketoglutarate and nicotinamideadenine dinucleotide phosphate in buffered solution. Thesolution was passed through a reaction column containingglutamic acid dehydrogenase immobilized by covalent binding
Scheme 3. Synthesis of L-[13N]glutamate and L-[
13N]alanine using immobilized enzy
Copyright © 2014 JohJ. Label Compd. Radiopharm 2014, 57 244–254
to derivatized silica beads. Interestingly, addition of pyruvic acidand elution through a second reaction column containingimmobilized glutamate-pyruvate transaminase yielded L-[13N]alanine, which was purified using a solid exchange column(Scheme 3).46
Immobilization of the enzyme (glutamic acid dehydrogenase)on an activated CNBr-Sepharose support was also applied to thepreparation of L-[13N]glutamic acid.47 A parallel strategy wasused for the preparation of the branched-chain L-amino acidsL-[13N]valine and L-[13N]leucine, eluting a mixture of thecorresponding alpha-keto acids with [13N]NH3 through a columncontaining glutamic acid dehydrogenase immobilized on thesame support;48 other amino acids, for example, L-[13N]alanine,L-α-[13N]aminobutirate or L-[13N]methionine, were preparedunder similar experimental conditions with radiochemicalconversions of 16%, 58% and 41%, respectively.49
The synthesis of aromatic amino acids has also been reported bytwo different methods. The first method involves the glutamatedehydrogenase-catalyzed conversion of [13N]ammonia to L-[13N]glutamate and transamination using aspartate aminotransferase(which has some inherent tyrosine aminotransferase activity) withp-hydroxyphenylpyruvate to yield L-[13N]tyrosine. The secondmethod utilizes phenylalanine dehydrogenase to catalyze thereversible reductive 13N-amination of either phenylpyruvate orp-hydroxyphenylpyruvate to form L-[13N]phenylalanine or L-[13N]tyrosine, respectively.50
Production of [13N]amines and amides
[13N]amines and [13N]amides have been prepared using differentstrategies, the amides usually occurring as reaction intermediatesfor the preparation of amines via Hoffman rearrangement.
13N-labeled amphetamine was prepared via formation of the[13N]imine by reacting phenylacetone and [13N]NH3 along withaluminum, mercuric chloride and concentrated ammoniumhydroxide (Scheme 4).51 However, radiochemical yields (3.5%)were pretty low. In addition, an excess of carrier ammonia wasrequired to avoid the formation of the secondary amine, and thuslow specific radioactivity values (296MBq/mmol) were obtained.
13N-labeled β-phenethylamine (PEA), an importantneuromodulator, was prepared by the reaction of phenylpropionylchloride with [13N]NH3-containing water, carrier ammonia andsodium hydroxide.52 The labeled amide was rearranged (Hofmannrearrangement) to the corresponding amine by heating the
mes.
www.jlcr.orgn Wiley & Sons, Ltd.
249

Scheme 6. Synthesis of [13N]amines using polymeric borane reagents.
Scheme 7. Synthesis of [13N]adenosine by ammonolysis of 6-chloro and 6-fluoro-
9-β-D-ribo-furanosyl purine.
Scheme 4. Synthesis of [13N]amphetamine via formation of the imine.
Scheme 5. Synthesis of [13N]β-phenethylamine via Hofmann rearrangement (a)
and reduction of the amide (b).
V. Gómez-Vallejo et al.
250
reaction mixture containing [13N]phenylpropionamide withsodium hypobromite (Scheme 5a). The fairly good yields, 50%, ofthis reaction reduced drastically to 5% when no carrier-addedconditions were applied to obtain high specific activities. Thepreparation of [13N]PEA was later on improved by amination ofphenylacetyl chloride and reduction of the amide using lithiumaluminum hydride; higher radiochemical yields (60–70%) couldbe obtained even when carrier ammonia was not used(Scheme 5b).53
A method for the synthesis of 13N-labeled alkylamines undermild conditions via amination of organoboranes was reportedin 1986.54 The method was based on the in situ preparation of[13N]chloramine, using [13N]NH3 and sodium hypochlorite, whichwas reacted with trialkylboranes at room temperature. Thesemild conditions also enabled the use of this method for thepreparation of functionalized amines such as γ-aminobutyricacid;55 the use of polymeric borane reagents simplified thepurification of the final labeled product (Scheme 6).56
The preparation of [13N]adenosine by ammonolysis of two halo-substrates, 6-chloro and 6-fluoro-9-β-D-ribo-furanosylpurine, wasalso reported (Scheme 7).57 Radiochemical yield for the 6-fluoroderivative was close to 20%when carrier ammonia was used, whileonly 11% was obtained in non-carrier added experiments.Interestingly, the 6-chloro derivative gave a much lower yield,probably due to the lower inductive effect of the chlorine atom.
Scheme 8. Synthesis of13N-labeled 1,3-bis(2-chloroethyl)-1-nitrosourea by in situ re
Scheme 9. Synthesis of S-[13N]-nitrosoglutathione.
www.jlcr.org Copyright © 2014 Joh
[13N]nitroso compounds
The synthesis of [13N]nitrosamines and [13N]nitrosothiols was firstreported by Vavrek and Mulholland.34 Cyclotron-produced [13N]NO�
3 was reduced to [13N]NO�2 using a cadmium column; [13N]
NO�2 was trapped using an anion exchange resin, which was
further eluted with a solution of the corresponding thiol orsecondary amine in acidic media to form the corresponding [13N]nitrosothiol (90% yield) or [13N]nitrosoamine (30% yield),respectively.
The generation of [13N]nitrous acid by treatment [13N]NO�2
with acids has been also applied to the preparation of 13N-labeled 1-(2-chloroethyl)-3-cyclohexyl-1-nitrosourea.58 Becauseof the very low yield (0.3–1.5%), an improved method basedon in situ generation of [13N]NO�
2 from [13N]NO�3 using copper
dust was developed and applied to the preparation of 13N-labeled 1,3-bis(2-chloroethyl)-1-nitrosourea (Scheme 8).39 Higherradiochemical yields (20–40%) were obtained. Its carbamateanalog N-nitroso-N-chloroethyl-1-chloroethyl carbamate wasalso successfully prepared a few years later.59
On the basis of themethod reported by Vavrek andMulholland,34
a fast and simple strategy for the production of S-[13N]-nitrosoglutathione ([13N]GSNO) via the radio-nitrosation ofglutathione was reported (Scheme 9).35 The production of theradioactive precursor [13N] NO�
2 was achieved by reduction of
duction of [13N]NO�
3 .
n Wiley & Sons, Ltd. J. Label Compd. Radiopharm 2014, 57 244–254

Scheme 10. Synthesis of [13N]nitrosamines by previous activation of the
secondary amine using Ph3P/Br2; example for the [13N]nitrosation of
diisopropylamine.
V. Gómez-Vallejo et al.
cyclotron-produced [13N] NO�3 with pre-treated cadmium. The
reaction of [13N]NO�2 with the non-radioactive precursor glutathione
in a vial in acidic media led to the formation of [13N]GSNO in highradiochemical yield (24.2±2.0%, EOS) in 3min production time,including a purification step based on solid phase extraction.
This methodology was further extended to the preparation ofother 13N-labeled nitrosothiols. With this purpose, a methodologyto trap [13N]NO�
2 on an anion exchange resin and perform thelabeling reaction in the cartridge was developed. The full processwas automated, and five different [13N]nitrosothiols weresynthesized with decay-corrected radiochemical yields in the range33.8–60.6%.36 The preparation of 13N-labeled nitrosothiols wasrecently reported using microfluidics technology.60 In generalterms, under similar experimental conditions, higher radiochemicalconversion values were obtained when compared with solid phasesupport synthesis in similar overall reaction times.
[13N]nitrosation was also applied to the preparation of 13N-labeled nitrosamines.38 However, because of the lowernucleophilic character of amines, and contrarily to the resultsreported by Vavrek and Mulholland, previous activation usingPh3P/Br2 was required (Scheme 10). Four different 13N-nitrosamines were obtained with decay corrected radiochemicalyields in the range 34–40.7% in overall production times below10min, including purification by HPLC.
Other 13N-labeled compounds
[13N]ammonia has been used for the preparation of differentlabeled compounds that do not fall into one of theaforementioned categories. One example is [13N]cisplatin;61 in afirst step, potassium iodide was reacted with K2PtCl4 underheating for 2min to yield K2PtI4. A mixture of water trapped[13N]NH3 and carrier NH3 was pumped into the solution, whichwas heated after complete trapping to form cis-Pt([13N]NH3)2I2.After addition of AgNO3, the solution was filtered and collectedin a vial containing NaCl to yield cis-Pt([13N]NH3)2Cl2, which waspurified using an anion exchange column. Overall radiochemicalyield and specific radioactivity were 27.1% and 11GBq/μmole(EOB), respectively. Significant improvement of the radiochemicalyield (47.6%, decay corrected) was achieved by directly sparging
Scheme 11. One-pot synthesis of [13N]urea (a) and [
13N]carbamate (b,c) analogues u
Copyright © 2014 JohJ. Label Compd. Radiopharm 2014, 57 244–254
[13N]NH3 into K2PtI4 instead of trapping of [13N]NH3 in water.62
The radiochemical yield was substantially improved up to 80%using a strong anion exchange resin as the solid state supportfor the tetraiodo platinum salt.63
[13N]NH3 has been also applied to the preparation of13N-labeled
peptides. The opioid tetrapeptide H-Tyr-D-Met(O)-Phe-Gly-[13N]NH2 (SD-62) was synthesized by amination of its activatedp-nitrophenol ester with [13N]NH3 with decay-correctedradiochemical yield of 48%.64 The same tetrapeptide was labeledwith 13N by treating its active ester precursor with [13N]NH3.
65
[13N]urea was prepared by boiling [13N]NH3 in presence ofammonium cyanate.66 The reaction solution was purified usingan ion exchange resin to obtain [13N]urea in 56% decay-corrected radiochemical yield in 10min. Non-carrier added[13N]urea and [13N]carbamate analogues were recentlysynthesized using one-pot synthesis and anhydrous [13N]NH3,
67
which was prepared according to the method reported bySuzuki and Yoshida.68 In this approach, a primary amine, asecondary amine or an alcohol was treated with triphosgene toyield the intermediate isocyanate, carbamoyl chloride orchloroformate, respectively; further reaction with [13N]NH3
yielded the corresponding [13N]urea and [13N]carbamateanalogues (Scheme 11).
A similar strategy was used to synthesize [13N]carbamazepine (a promising PET ligand for brain imaging)with high radiochemical yield (56–74%, decay corrected andbased on [13N]NH3) and high specific radioactivity (22–33 GBq/μmol).
Anhydrous [13N]NH3 was also used by the same group for thesynthesis of [13N]thalidomide (Scheme 12).69 The approach wasbased on the reaction of N-phthaloylglutamic anhydride with [13N]NH3, followed by cyclization using carbonyldiimidazole. Goodradiochemical yield (56±12%, decay corrected) and specificradioactivity at the end of the synthesis (49±24GBq/μmol, EOS)were obtained. Also, the same group reported recently the synthesisof [13N]dantrolene, a skeletal muscle relaxant (Scheme 13).70
The use of the radioactive precursor [13N] NO�2 has been
recently used for the preparation of 13N-labeled azocompounds.37 The synthetic route was based on a two-stepprocess, namely (i) reaction of [13N]NO�
2 with aromatic primaryamines to generate the corresponding 13N-labeled diazoniumsalts and (ii) further reaction of these salts with aromatic aminesor phenols (Scheme 14). Good radiochemical yields (20.4–47.2%,decay corrected) were achieved. These compounds are currentlybeing evaluated as potential β-amyloid markers. This processwas also deployed using microfluidics technology, resulting inimproved radiochemical conversion values.60
sing non-carrier added [13N]NH3.
www.jlcr.orgn Wiley & Sons, Ltd.
251

Scheme 13. Synthesis of [13N]dantrolene using non-carrier added [
13N]NH3.
Scheme 12. Synthesis of [13N]thalidomide using non-carrier added [
13N]NH3.
Scheme 14. Synthesis of [13N]azo derivatives.
V. Gómez-Vallejo et al.
252
Very recently, a straightforward method for the direct protonbeam activation of commercially available aluminum oxide NPs(Al2O3 NPs) via the 16O(p,α)13N nuclear reaction was reported(Figure 4).71 NPs were placed in an in-house designed aluminumcapsule, which was then placed in a solid target holder andirradiated with 16MeV protons. The radiolabeling of the NPsdid not alter the surface or structural properties of NPs, and theirbiodistribution pattern could be studied in rats after intravenousadministration using PET. This unprecedented activation strategymight be applied to other synthetic or commercially availablemetal oxide particles.
Future perspectives of nitrogen-13 labeling
Currently, only a few groups in the world are working on thedevelopment of new strategies for the preparation of 13N-labeled compounds. 13N has a short half-life, which creates threemain difficulties: (i) its use is restricted to those clinical and
Figure 4. Activation of Al2O3 nanoparticles by proton irradiation via the16O
(p,α)13N nuclear reaction. Reprinted with permission from Pérez-Campaña et al.71
Copyright 2013 American Chemical Society.
www.jlcr.org Copyright © 2014 Joh
research organizations with access to a nearby cyclotron,because distribution from centralized production centers tosurrounding facilities is unfeasible; (ii) rapid and efficientmethods have to be applied to the preparation of 13N-labeledradiotracers, to prevent decay-associated radioactivity loss; and(iii) even when the preparation of the radiotracer is feasible,the amount of radioactivity has to be sufficient for subsequentimaging studies and the biological half-life of the radiotracershould match the physical half-life of the isotope, that is,biological or physiological processes with slow kinetics cannotbe investigated. On top of that, the high positron range resultsin poor resolution images, which makes quantification and datainterpretation challenging, especially in small animal studies. Onthe other hand, as mentioned in the introduction, the stableisotope of nitrogen (14N) is present in the majority of biologicalactive molecules, and thus a wide range of molecules might belabeled with 13N as an alternative to 11C or 18F, while anadvantage of the short half-life is that repeated studies may beperformed on the same individual within a short period.
Inconveniences (i) and (ii) might be overcome in the comingyears. First, there is a growing interest in the development of anew generation of smaller (and hopefully cheaper and withlower investment requirements) cyclotrons. If this promisebecomes a fact, then more and more centers might own aparticle accelerator, facilitating the progressive implementationof a ‘dose on demand’ PET scenario, and short-lived isotopesmay gain significance. Of course, such small cyclotrons will haveparticle energy limitations, and this can become a new problem.The most established method for the production of 13N inbiomedical cyclotrons is the irradiation with protons of purewater or ethanol/water solutions, using the 16O(p,α)13N nuclearreaction. Because of the cross section values of this reaction(Figure 5a), energies above 10MeV are usually required to obtain
n Wiley & Sons, Ltd. J. Label Compd. Radiopharm 2014, 57 244–254
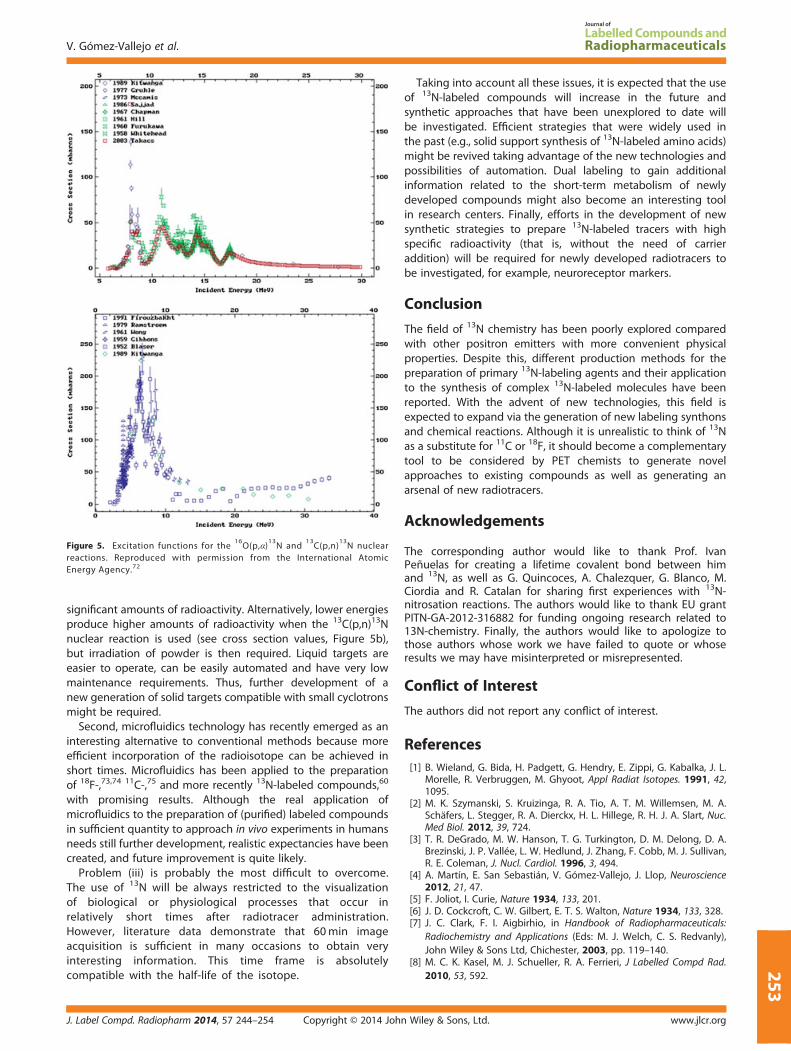
Figure 5. Excitation functions for the16O(p,α)
13N and
13C(p,n)
13N nuclear
reactions. Reproduced with permission from the International AtomicEnergy Agency.72
V. Gómez-Vallejo et al.
25
significant amounts of radioactivity. Alternatively, lower energiesproduce higher amounts of radioactivity when the 13C(p,n)13Nnuclear reaction is used (see cross section values, Figure 5b),but irradiation of powder is then required. Liquid targets areeasier to operate, can be easily automated and have very lowmaintenance requirements. Thus, further development of anew generation of solid targets compatible with small cyclotronsmight be required.
Second, microfluidics technology has recently emerged as aninteresting alternative to conventional methods because moreefficient incorporation of the radioisotope can be achieved inshort times. Microfluidics has been applied to the preparationof 18F-,73,74 11C-,75 and more recently 13N-labeled compounds,60
with promising results. Although the real application ofmicrofluidics to the preparation of (purified) labeled compoundsin sufficient quantity to approach in vivo experiments in humansneeds still further development, realistic expectancies have beencreated, and future improvement is quite likely.
Problem (iii) is probably the most difficult to overcome.The use of 13N will be always restricted to the visualizationof biological or physiological processes that occur inrelatively short times after radiotracer administration.However, literature data demonstrate that 60min imageacquisition is sufficient in many occasions to obtain veryinteresting information. This time frame is absolutelycompatible with the half-life of the isotope.
Copyright © 2014 JohJ. Label Compd. Radiopharm 2014, 57 244–254
Taking into account all these issues, it is expected that the useof 13N-labeled compounds will increase in the future andsynthetic approaches that have been unexplored to date willbe investigated. Efficient strategies that were widely used inthe past (e.g., solid support synthesis of 13N-labeled amino acids)might be revived taking advantage of the new technologies andpossibilities of automation. Dual labeling to gain additionalinformation related to the short-term metabolism of newlydeveloped compounds might also become an interesting toolin research centers. Finally, efforts in the development of newsynthetic strategies to prepare 13N-labeled tracers with highspecific radioactivity (that is, without the need of carrieraddition) will be required for newly developed radiotracers tobe investigated, for example, neuroreceptor markers.
Conclusion
The field of 13N chemistry has been poorly explored comparedwith other positron emitters with more convenient physicalproperties. Despite this, different production methods for thepreparation of primary 13N-labeling agents and their applicationto the synthesis of complex 13N-labeled molecules have beenreported. With the advent of new technologies, this field isexpected to expand via the generation of new labeling synthonsand chemical reactions. Although it is unrealistic to think of 13Nas a substitute for 11C or 18F, it should become a complementarytool to be considered by PET chemists to generate novelapproaches to existing compounds as well as generating anarsenal of new radiotracers.
Acknowledgements
The corresponding author would like to thank Prof. IvanPeñuelas for creating a lifetime covalent bond between himand 13N, as well as G. Quincoces, A. Chalezquer, G. Blanco, M.Ciordia and R. Catalan for sharing first experiences with 13N-nitrosation reactions. The authors would like to thank EU grantPITN-GA-2012-316882 for funding ongoing research related to13N-chemistry. Finally, the authors would like to apologize tothose authors whose work we have failed to quote or whoseresults we may have misinterpreted or misrepresented.
Conflict of Interest
The authors did not report any conflict of interest.
References[1] B. Wieland, G. Bida, H. Padgett, G. Hendry, E. Zippi, G. Kabalka, J. L.
Morelle, R. Verbruggen, M. Ghyoot, Appl Radiat Isotopes. 1991, 42,1095.
[2] M. K. Szymanski, S. Kruizinga, R. A. Tio, A. T. M. Willemsen, M. A.Schäfers, L. Stegger, R. A. Dierckx, H. L. Hillege, R. H. J. A. Slart, Nuc.Med Biol. 2012, 39, 724.
[3] T. R. DeGrado, M. W. Hanson, T. G. Turkington, D. M. Delong, D. A.Brezinski, J. P. Vallée, L. W. Hedlund, J. Zhang, F. Cobb, M. J. Sullivan,R. E. Coleman, J. Nucl. Cardiol. 1996, 3, 494.
[4] A. Martín, E. San Sebastián, V. Gómez-Vallejo, J. Llop, Neuroscience2012, 21, 47.
[5] F. Joliot, I. Curie, Nature 1934, 133, 201.[6] J. D. Cockcroft, C. W. Gilbert, E. T. S. Walton, Nature 1934, 133, 328.[7] J. C. Clark, F. I. Aigbirhio, in Handbook of Radiopharmaceuticals:
Radiochemistry and Applications (Eds: M. J. Welch, C. S. Redvanly),John Wiley & Sons Ltd, Chichester, 2003, pp. 119–140.
[8] M. C. K. Kasel, M. J. Schueller, R. A. Ferrieri, J Labelled Compd Rad.2010, 53, 592.
www.jlcr.orgn Wiley & Sons, Ltd.
3

V. Gómez-Vallejo et al.
254
[9] C. P. Wolk, S. M. Austin, J. Bortins, A. Galonsky, J. Cell Biol. 1974, 61, 440.[10] T. J. Wellman, T. Winkler, E. L. V. Costa, G. Musch, R. S. Harris, J. G.
Venegas, F. M. Vidal Melo, J. Nucl. Med. 2010, 51, 646.[11] R. Greene, C. A. Burnham, J. Nucl. Med. 1972, 13, 433.[12] S. Ruben, W. Z. Hassid, M. D. Kamen, Science 1940, 91, 578.[13] D. J. D. Nicholas, D. J. Silvester, J. F. Fowler, Nature 1961, 189, 634.[14] N. J. Parks, K. A. Krohn, Int J Appl Radiat Isotopes. 1978, 29, 754.[15] R. A. Ferrieri, D. J. Schlyer, B. W. Wieland, A. P. Wolf, Int J Appl Radiat.
Isotopes. 1983, 34, 897.[16] D. Le Bars, J Labelled Compd Rad. 2001, 44, 1.[17] L. Lindner, J. Helmer, G. A. Brinkman, Int J Appl Radiat Isotopes. 1979,
30, 506.[18] W. Vaalburg, A. Steenhoek, A. M. J. Paans, R. Peset, S. Reiffers, M. G.
Woldring. J Labelled Compd Rad. 1981, 18, 303.[19] W. W. Hunter, W. G. Monahan, J. Nucl. Med. 1971, 12, 368.[20] M. J. Welch, J. F. Lifton, J. Am. Chem. Soc. 1971, 93, 3385.[21] W. G. Monahan, R. S. Tilbury, J. S. Laughlin, J. Nucl. Med. 1972, 13, 274.[22] H. Krizek, N. Lembares, R. Dinwoodie, I. Gloria, K. A. Lathrop, P. V.
Harper, J. Nucl. Med. 1973, 14, 629.[23] M. L. Firouzbakht, D. J. Schlyer, A. P. Wolf, J. S. Fowler, Nucl. Med. Biol.
1999, 26:437.[24] T. Ido, R. Iwata, J Labelled Compd. Rad. 1981, 18, 244.[25] G. K. Mulholland, M. R. Kilbourn, J. J. Moskwa, Appl Radiat Isotopes.
1990, 41, 1193.[26] M. S. Berridge, B. J. Landmeier, Appl Radiat Isotopes. 1993, 44, 1433.[27] M. V. Korsakov, R. N. Krasikova, O. S. Fedorova, J Radioanal Nucl Ch.
1996; Articles, 204, 231.[28] R. N. Krasikova, O. S. Fedorova, M. V. Korsakov, B. Landmeier
Bennington, M. S. Berridge, Appl Radiat Isotopes. 1999, 51, 395.[29] G. Bida, B. W. Wieland, T. J. Ruth, D. G. Schmidt, G. O. Hendry, R. E.
Keen, Radiochim Acta. 1986, 23, 1217.[30] R. E. Shefer, B. J. Hughey, R. E. Klinkowstein, M. J. Welch, C. S. Dence,
Nucl. Med. Biol. 1994, 21, 977.[31] C. S. Dence, M. J. Welch, B. J. Hughey, R. E. Shefer, R. E. Klinkowstein,
Nucl. Med. Biol. 1994, 21, 987.[32] J. Wieneke, B. Nebeling, Z. Pflanzenernähr Bodenk. 1990, 153, 117.[33] M. W. McElfresh, J. C. Meeks, N. J. Parks, J Radioanal Chem. 1979, 53, 337.[34] M. T. Vavrek, G. K. Mulholland, J Labelled Compd Rad. 1995, 37, 118.[35] J. Llop, V. Gómez-Vallejo, M. Bosque, G. Quincoces, I. Peñuelas, Appl
Radiat Isotopes. 2009, 67, 95.[36] V. Gómez-Vallejo, K. Kato, I. Oliden, J. Calvo, Z. Baz, J. I. Borrell, J. Llop,
Tetrahedron Lett. 2010, 51, 2990.[37] V. Gómez-Vallejo, J. I. Borrell, J. Llop, Eur. J. Med. Chem. 2010, 45, 5318.[38] V. Gómez-Vallejo, K. Kato, M. Hanyu, K. Minegishi, J. I. Borrell, J. Llop,
Bioorg. Med. Chem. Lett. 2009, 19, 1913.[39] W. A. Pettit, R. S. Tilbury, G. A. Digenis, R. H. Mortara, J. Labelled
Compd. Rad. 1977, 13, 119.[40] W. Liang, Y. Nie, J. Wang, J. Wu, H. Liu, Q. Wang, L. Huang, H. Guo, B.
Shu, J. Lv, Anal. Chem. 2011, 83, 578.[41] R. Gersberg, K. Krohn, N. Peek, C. R. Goldman, Science 1976, 192, 1229.[42] D. R. Elmalech, D. J. Hnatowitch, S. Kulprathipanja, J Labelled Compd
Rad. 1979, 16, 92.[43] N. Lembares, R. Dinwoodie, I. Gloria, P. Harper, K. Lathrop, J. Nucl.
Med. 1972, 13, 786.[44] M. B. Cohen, L. Spolter, N. S. MacDonald, B. Cassen, J. Nucl. Med.
1972, 13, 422.[45] M. G. Straatman, M. J. Welch, Radiat. Res. 1973, 56, 48.[46] M. B. Cohen, L. Spolter, C. C. Chang, N. S. MacDonald, J. Takahashi, D.
D. Babinet, J. Nucl. Med. 1974, 15, 1192.
www.jlcr.org Copyright © 2014 Joh
[47] A. S. Gelbard, R. S. Bonus, R. E. Reiman, J. M. McDonald, J. J. Vomero,J. S. Laughlin, J. Nucl. Med. 1980, 21, 988.
[48] J. R. Barrio, F. J. Baumgartner, E. Henze, M. S. Stauber, J. E. Egbert, N.S. MacDonald, H. R. Schelbert, M. E. Phelps, F. T. Liu, J. Nucl. Med.1983, 24, 937.
[49] A. J. L. Cooper, A. S. Gelbard. Anal. Biochem. 1981, 111, 42.[50] A. S. Gelbard, A. J. L. Cooper, Y. Asano, E. Nieves, S. Filc-Dericco, K. C.
Rosenpire, Appl Radiat Isotopes. 1990, 41, 229.[51] R. D. Finn, D. R. Christman, A. P. Wolf, J Labelled Compd Rad. 1981,
18, 909.[52] T. Tominaga, O. Inoue, T. Irie, K. Suzuki, T. Yamasaki, M. Hirobe. Int J
Appl Rad Isotopes. 1985, 36, 555.[53] T. Tominaga, O. Inoue, K. Suzuki, T. Yamasaki, M. Hirobe, Appl Rad
Isotopes. 1986, 37, 1209.[54] P. J. Kothari, R. D. Finn, G. W. Kabalka, M. M. Vora, T. E. Boothe, A. M.
Emran, Appl Rad Isotopes. 1986, 37, 469.[55] G. W. Kabalka, Z. Wang, J. F. Green, M. M. Goodman, Appl Radiat
Isotopes. 1992, 43, 389.[56] G. W. Kabalka, M. M. Goodman, J. F. Green, R. Marks, D. Longford, J
Labelled Compd Rad. 1993, 32, 165.[57] T. Irie, O. Inoue, K. Suzuki, T. Tominaga. Int J Appl Radiat Isotopes.
1985, 36, 345.[58] W. A. Pettit, R. H. Mortara, G. A. Digenis, M. F. Reed, J. Med. Chem.
1975, 18, 1029.[59] G. A. Digenis, Y. C. Cheng, R. L. McQuinn, B. R. Freed, R. S. Tilbury,
Advances in Chemistry, 197: Short-Lived Radionuclides in Chemistryand Biology, Chapter 18, 1982, 351.
[60] V. Gaja, V. Gómez-Vallejo, M. Cuadrado-Tejedor, J. I. Borrell, J. Llop, JLabelled Compd Rad. 2012, 55, 332.
[61] B. De Spiegeleer, G. Slegers, C. Vandecasteele, W. Van den Bossche, K.Schelstraete, A. Claeys, P. De Moerloose, J. Nucl. Med. 1986, 27, 399.
[62] J. Z. Ginos, A. J. L. Cooper, V. Dhawan, J. C. K. Lai, S. C. Strother, N.Alcock, D. A. Rottenberg, J. Nucl. Med. 1987, 28,1844.
[63] M. Holschbach, W. Hamkens, A. Steinbach, K. Hamacher, G. Stocklin,Appl Radiat. Isotopes. 1997, 48, 739.
[64] H. Saji, D. Tsutsumi, Y. Kiso, S. Iimuma, T. Mimoto, K. Akaji, Y. Magata, H.Nakamura, A. Kita, J. Konishi, Int. J. Rad. Appl. Instrum. B 1992, 19, 455.
[65] Y. Kiso, S. Iinuma, T. Mimoto, H. Saji, A. Yokoyama, K. Akaji. ChemPharm Bull (Tokyo) 1991, 39, 2734.
[66] H. Krizek, P. V. Harper, B. Mock, J Labelled Compd Rad. 1977, 13, 207.[67] K. Kumata, M. Takei, M. Ogawa, K. Kato, K. Suzuki, M. R. Zhang, J
Labelled Compd Rad. 2009, 52, 166.[68] K. Suzuki, Y. Yoshida, Appl Radiat Isotopes. 1999, 50, 497.[69] K. Kumata, M. Takei, M. Ogawa, J. Yui, A. Hatori, K. Suzuki, M. R.
Zhang. J. Labelled Compd Rad 2010, 53, 53.[70] K. Kumata, M. Ogawa, M. Takei, M. Fujinaga, Y. Yoshida, N. Nengaki, T.
Fukumura, K. Suzuki, M. R. Zhang, Bioorg. Med. Chem. Lett. 2012,20, 305.
[71] C. Pérez-Campaña, V. Gómez-Vallejo, M. Puigivila, A. Martín, T. Calvo-Fernández, S. E. Moya, R. F. Ziolo, T. Reese, J. Llop, ACS Nano 2013, 7, 3498.
[72] Cyclotron Produced Radionuclides: Physical Characteristics andProduction Methods. Technical Reports Series nº 468, InternationalAtomic Energy Agency, Vienna, 2009.
[73] G. Pascali, G. Mazzone, G. Saccomanni, C. Manera, P. A. Salvadori,Nucl. Med. Biol. 2010, 37, 547.
[74] H. Anderson, N. Pillarsetty, M. Cantorias, J. S. Lewis, Nucl. Med. Biol.2010, 37, 439.
[75] S. Kealey, C. Plisson, T. L. Collier, N. J. Long, S. M. Husbands, L.Martarello, A. D. Gee, Org. Biomol. Chem. 2011, 9, 3313.
n Wiley & Sons, Ltd. J. Label Compd. Radiopharm 2014, 57 244–254