nitric oxide and octreotide in retinal ischemia–reperfusion injury*
TRANSCRIPT

Documenta Ophthalmologica 105: 327–338, 2002.© 2002 Kluwer Academic Publishers. Printed in the Netherlands.
Nitric oxide and octreotide in retinalischemia–reperfusion injury∗
ULKU CELIKER1 and NECIP ILHAN2
1Departments of Ophthalmology and 2Biochemistry, University Medical School, Elazig,Turkey
Accepted 28 December 2001
Abstract. This experimental study was performed to investigate the role of ischemia–reper-fusion injury on retinal nitric oxide activity and to determine whether octreotide, the syntheticanalogue of natural somatostatin, modifies the nitric oxide activity during retinal ischemia–reperfusion in a quinea pig model. Three groups of seven pigmented male quinea pigs wereformed; Control, Ischemia and the Ischemia/Octreotide groups. 90 minutes of pressure-inducedretinal ischemia and 24 h of reperfusion were established in the ischemia and ischemia/octreo-tide groups. Saline for the ischemia group and 50 µg/kg of octreotide for the ischemia/octreo-tide group were administered intraperitoneally five times with 6-h intervals. At the end of thereperfusion period both eyes of the animals of the three groups were enucleated. One eye ofeach animal was randomly selected for biochemical assay and the other for histopathologicalanalysis. Retinal nitrate levels were measured and histopathological changes were evaluatedin the groups. The mean retinal nitrate levels of the control, ischemia and ischemia/octreotidegroups were 157.6±25.2, 106.4±20.1 and 96.4±17.7 µmol/l, respectively. Nitrate levels de-creased significantly both in the ischemia (p<0.01) and ischemia/octreotide (p<0.01) groupsversus control. In the ischemia group, retinal histopathological changes, which were differentfrom the control group, were prominent edema, polymorphonucleated leukocytes infiltrationand vacuolated spaces in the inner retina. No significant change was observed in the histo-pathological specimens of the ischemia/octreotide group. Significant increase in the thicknessof the inner plexiform layer of the retina of the ischemia group was observed versus the con-trol and ischemia/octreotide groups (p<0.01 and p<0.01, respectively).The thickness of theinner plexiform layer of the retina of the ischemia/octreotide group did not change versus thecontrol group. It was concluded that nitric oxide activity decreased during retinal ischemia–reperfusion and, although octreotide prevented the histopathological damage, it could notameliorate the nitric oxide activity of the retina.
Key words: nitric oxide, octreotide, retina, ischemia, reperfusion
∗ This study was presented in part at the 23rd Congress of the European Society ofOphthalmology.

328
Introduction
Retinal ischemia, which occurs as the consequence of a primary ocular dis-ease or as the ocular complication of a systemic disease, produces cell deathby destruction of cellular elements such as DNA, protein and cell membrane[1].
Nitric oxide (NO), which has diverse physiological functions, has alsobeen known to take part in pathological events [2]. It is an important mes-senger and is synthesized by three isoforms of NO synthase (NOS). Neuronaland endothelial isoforms are termed as constitutive NOS (cNOS) and they arecalcium dependent. A third form is inducible NOS (iNOS) which is calciumindependent [3]. Neuronal NOS immunoreactivity is present in amacrine, ho-rizontal and photoreceptor cells, while endothelial NOS immunoreactivity ispresent in the vascular endothelium of the retina and the choroidea. On theother hand, inducible NOS is expressed in retina pigment epithelium, Müllercells and pericytes [4–9]. NO plays an important role in the control of ocularvascular tone and blood flow as it is a potent signaling molecule in bloodvessels, where a continuous formation from endothelial cells maintains vas-odilatation and blood flow and it also affects the vascular system through itsability to inhibit platelet aggregation and adhesion [10, 11] There are contraryreports pointing out whether the level of nitric oxide of the tissue increases ordecreases during ischemia–reperfusion (I/R) [12–14].
Octreotide, the synthetic derivative of somatostatin has been reported totake part in the protection from oxygen free radicals and octreotide and so-matostatin have also been previously reported to have modulatory effect onNO levels [15–19].
The objective of this study is to investigate the role of I/R on retinal NOactivity and to determine whether octreotide modulates the retinal activity ofNO during I/R injury of the retinal tissue in a guinea pig model.
Materials and methods
Three groups were formed from 21 pigmented male guinea pigs weighingbetween 470 and 640 g and each of the groups included seven animals:
Control group: no drug was used and I/R was not inducedIschemia group: I/R was induced and normal saline was administered.Ischemia /octreotide group: I/R was induced and octreotide was admin-istered.
Intramuscular ketamine HCl (50 mg/kg) and xylazine HCl (5 mg/kg) wereused for the anesthesia of the animals. Proparacaine HCl (0.05%) was admin-istered as topical anesthetic to both eyes of the animals. Pressure-induced ret-

329
inal ischemia was performed in the ischemia and ischemia/octreotide groups;both anterior chambers of each animals were cannulated with a 27-gaugeneedle connected to a bottle of normal saline and the bottle was rapidly liftedto a height of 205 cm in order to raise the intraocular pressure to 150 mmHg.This lasted for 90 min and reperfusion was established by lowering the sa-line bottle to the eye level. Then the eyes were decanulated. The reperfusionperiod lasted for 24 h. One ml of normal saline was injected intraperiton-eally 15 min prior the ischemic insult to the animals of the ischemia groupand it was repeated four times with 6-h intervals and 50 µ/kg of octreotidewas administered via the same route five times with 6-h intervals, simil-arly the first dose being injected 15 min before the ischemic insult in theischemia/octreotide group. After the reperfusion period of the ischemia andischemia/octreotide groups, the animals were reanesthetized and both eyesof all the animals including the controls were rapidly enucleated and theanimals were killed by intracardiac thiopenthal sodium (50 mg/kg). One eyeof each animal was randomly selected for biochemical assay and the otherfor histopathological evaluation. The enucleated eyes, which were put on iceslices, were immediately dissected coronally through pars plana for biochem-ical assay. After removing the vitreous, the retinal tissue was dissected fromthe choroidea under the operating microscope and stored at −80◦C coveredin aluminum foils until the biochemical assay. The NO activity of the retinaltissue was assessed indirectly by measuring retinal nitrate, a the metabolite ofNO. One hundred µl of homogenizated tissue sample were deproteinizatedby adding 200 µl of 10% zinc sulphate and 200 µl of 0.5 N sodium hydroxidesolution. Then it was centrifugated at 2500 ×g and +4◦C for 5 min. Spectro-photometric measurement of nitrate level was obtained from this supernatantas µmol/l according to the method described by Borries and Borries [20].
The eyes selected for histopathological examination were fixed in 10%formaline immediately after enucleation and transverse sections passing th-rough the optic nerve were obtained. The samples were embedded in paraffinwax and 5-µm micrometer thick paraffin sections were prepared and thespecimens were stained with hematoxylin and eosin. An Olympus BX50light microscope was used for the histopathological evaluation of the tissuesections of three groups and prominent histological changes, which were dif-ferent from the control group, were noted for the I/R induced groups.Thequantification of the retinal ischemic damage was made by measuring thethickness of the inner plexiform layer of the retina of the groups. The meas-urements were made with an ocular micrometer ×400 magnification within0.5 mm from the optic nerve. Three measurements from adjacent locationsin each nasal and temporal hemispheres were obtained and an average retinalthickness for each eye was obtained by averaging the six measurements. All

330
Figure 1. The mean retinal nitrate levels of the groups: ∗ p<0.01. Isc, Ischemia; Oct,Octreotide.
Figure 2. Normal retina of the control group (hematoxylin and eosin, ×400).
experiments performed in this investigation conformed to the ARVO Resolu-tion on the Use of Animals in Research.
The mean nitrate levels and the thickness of the retinal tissues were calcu-lated as the mean±SD and statistical analysis was made by Mann–WhitneyU -test. Differences were considered significant at p<0.05.
Results
The mean retinal nitrate levels of the control, ischemia and ischemia/octreotidegroups were 157.6±25.2, 106.4±20.1 and 96.4±17.7 µmol/l, respectively.

331
Figure 3. The retinal tissue section of the ischemia group. Prominent edema in the innerplexiform layer (thick arrow), PMNLs (long arrows) in the inner nuclear layer and vacuolatedspace (short arrow) in the ganglion cell layer (hematoxylin and eosin, ×400).
Figure 4. Mild edema (arrow) in the ganglion cell layer of the retina of the ischemia/octreotidegroup (hematoxylin and eosin, ×400).
332
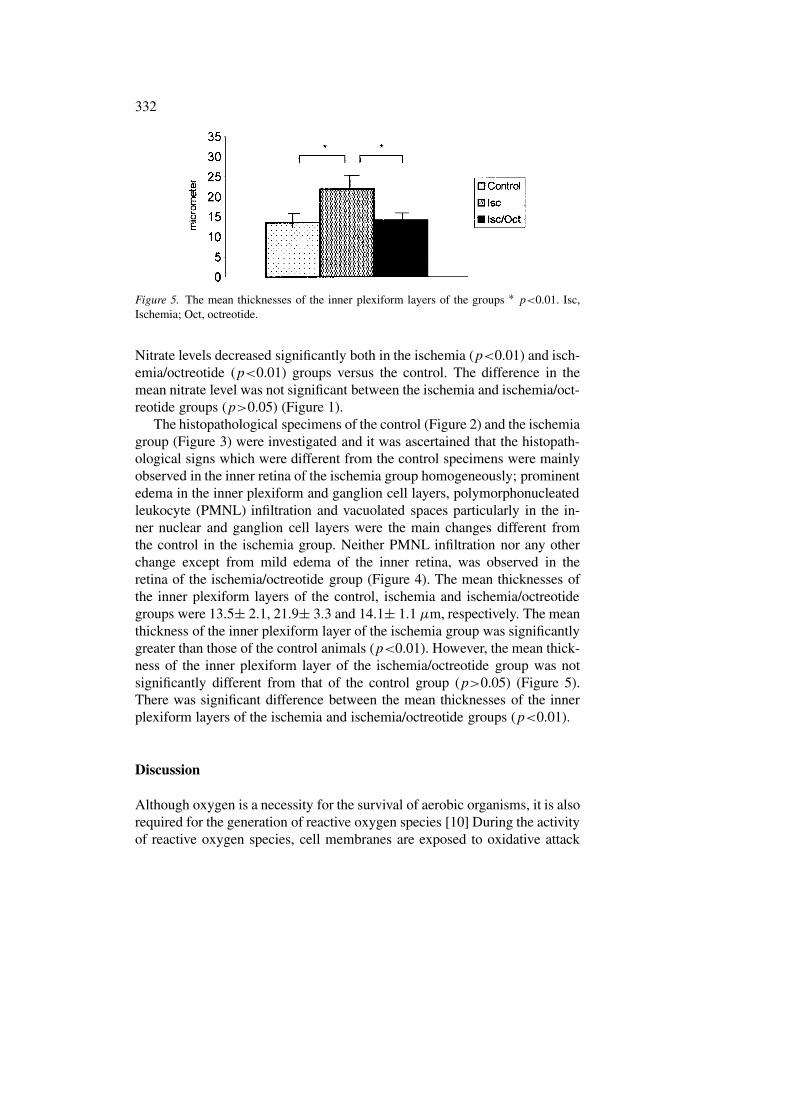
Figure 5. The mean thicknesses of the inner plexiform layers of the groups ∗ p<0.01. Isc,Ischemia; Oct, octreotide.
Nitrate levels decreased significantly both in the ischemia (p<0.01) and isch-emia/octreotide (p<0.01) groups versus the control. The difference in themean nitrate level was not significant between the ischemia and ischemia/oct-reotide groups (p>0.05) (Figure 1).
The histopathological specimens of the control (Figure 2) and the ischemiagroup (Figure 3) were investigated and it was ascertained that the histopath-ological signs which were different from the control specimens were mainlyobserved in the inner retina of the ischemia group homogeneously; prominentedema in the inner plexiform and ganglion cell layers, polymorphonucleatedleukocyte (PMNL) infiltration and vacuolated spaces particularly in the in-ner nuclear and ganglion cell layers were the main changes different fromthe control in the ischemia group. Neither PMNL infiltration nor any otherchange except from mild edema of the inner retina, was observed in theretina of the ischemia/octreotide group (Figure 4). The mean thicknesses ofthe inner plexiform layers of the control, ischemia and ischemia/octreotidegroups were 13.5± 2.1, 21.9± 3.3 and 14.1± 1.1 µm, respectively. The meanthickness of the inner plexiform layer of the ischemia group was significantlygreater than those of the control animals (p<0.01). However, the mean thick-ness of the inner plexiform layer of the ischemia/octreotide group was notsignificantly different from that of the control group (p>0.05) (Figure 5).There was significant difference between the mean thicknesses of the innerplexiform layers of the ischemia and ischemia/octreotide groups (p<0.01).
Discussion
Although oxygen is a necessity for the survival of aerobic organisms, it is alsorequired for the generation of reactive oxygen species [10] During the activityof reactive oxygen species, cell membranes are exposed to oxidative attack

333
and lipid hydroperoxide production because of their high polyunsaturatedfatty acid content. Peroxidation products cause cell death by producing edemaand inflammation and increasing vascular permeability [21-26]. Octreotidehas been previously reported to stimulate cellular radical scavenger systemsalthough the exact mechanism has not been elucidated and to inhibit therelease of superoxide anion from stimulated monocytes [15, 16]. It is a longacting somatostatin analogue [27] and somatostatin has been known to reducethe generation of free radicals and infiltration of PMNLs [18, 28]. One of theeffector systems of free radicals is the PMNLs and they release free radicalsinto the extracellular space [18, 28–31]. Szabo et al. has reported PMNLinfiltration of the retinal tissue after I/R of the rat eye previously [32]. Theinhibitory properties of octreotide on leukocyte functions have also been pre-viously reported [33]. In the present study we have observed that octreotidehas prevented PMNL infiltration during I/R as PMNL infiltration was onlyseen in the histopathological specimens of the ischemia group.
Nitrite and nitrate, the degradation products of NO are commonly used forthe detection of NO activity as the direct measurement of NO activity in bio-logical system is limited because of its scant release and rapid breakdown [34,35]. NO levels have been reported to decrease in the presence of oxygen freeradicals during I/R of the tissue and this decrease has been proposed to actas an additional factor in tissue dysfunction [12,14,36–39]. It has been sug-gested that the NO pathway fails after I/R because of increased binding andbreakdown by superoxide anion [36,37]. In our study, the decreased nitratelevels of the ischemia induced groups (the ischemia and ischemia/octreotidegroups) are consistent with these previous data. The endothelium, which linesthe lumen of the blood vessels has many functions including the releaseof endothelium-derived agents such as NO [38]. The decreased levels ofnitrate in the I/R induced groups of this study might be affected from thevascular endothelial damage induced by I/R itself in the present study. L-NAME, an inhibitor of NOS has been reported to increase lipid peroxidationand consequently this increase in lipid peroxidation has been accompaniedby a decrease in NO levels [39]. In a recent study, octreotide has been re-ported to suppress lipid peroxidation [40]. Under the light of these previ-ous data, our expectation has been towards to observe an amelioration inthe nitrate levels of the ischemia/octreotide group because of the presumedconsequence of the effect of octreotide on lipid peroxidation. However, thisexpected amelioration in the nitrate level of the ischemia/octreotide grouphas not been observed in our study. Moreover, although there has been nosignificance, the nitrate level of the ischemia/octreotide group has been muchlower than that of the ischemia group. NO when produced by a Ca-dependentNOS takes a physiological role, but when a Ca-independent NOS produces

334
NO, then NO plays a part in pathological conditions such as tissue injury[41]. The Ca-independent iNOS has been reported to be induced by mac-rophages, neutrophils and other cells responding to inflammatory stimuli andPMNLs generate NO via a pathway in which L-arginine has been metabolized[3,42]. PMNL infiltration has been observed only in the ischemia group in thisstudy. Although we can not discuss the interaction of Ca-dependent and Ca-independent NOS in our study, we may speculate that the nitrate level of theischemia group probably would have been lower than the ischemia/octreotidegroup if PMNL infiltration had not taken place and the insignificant decreasein the nitrate level of the ischemia/octreotide group versus the ischemia groupmight be caused by the suppressing effect of octreotide on leukocyte func-tions. Moreover, it has been reported previously that somatostatin and oct-reotide suppress NO release by peritoneal macrophages in vitro [19]. In an ex-perimental study of Le, octreotide has also been reported to decrease plasmaand gastric mucosal NO in rats with portal hypertensive gastropathy [17].Although the precise mechanism by which NO exerts its effect remains un-defined, it acts as an antiadhesive molecule in leukocyte–endothelial cellinteraction and superoxide which is produced by PMNLs and endothelialcells rapidly inactivates NO and superoxide anion has been implicated as amediator of I/R induced leukocyte adhesion [37,38,44]. Therefore, our ex-pectation has been towards a decrease in NO activity and because of itsantiadhesive property for the leukocytes, PMNL infiltration is expected whenNO activity decreases. As seen in the previous data, the interaction of NOwith leukocytes is somewhat complex. In the ischemia group, apart from theprimary effect of ischemia itself, the interaction of decreased NO activitymight lead to an increase in PMNL infiltration. In the ischemia/octreotidegroup, we have observed the inability of octreotide to raise the nitrate level tothat of the control group besides its the ability of inhibiting PMNL infiltration.
Previously, the inner retina has been reported to be more susceptible toIOP-induced ischemia than the outer retina and this sensitivity of the innerretina has been proposed to be more likely a consequence of failure of thecentral retinal microcirculation than an indication of thresholds of differentretinal neurons to damage [45, 46]. The histopathological changes such assevere edema, vacuolated spaces and PMNL infiltration have been observedmainly in the inner retina of the ischemia group in the present study. However,we cannot speculate that high intraocular pressure selectively affects onlyinner retina in limits of the present study. In a previous report inner retinalneurons have been reported to be more vulnerable than the outer retinal cellsin the ischemic diseases of the retina [47]. Hughes has reported that, althoughthe inner retina has gone under extensive damage, the outer retina was, forthe most part, intact after 90 min of ischemia but when the ischemic period

335
prolonged to 120 or 180 min prominent damage in the outer retina has beenobserved in rat retina [46]. A limitation of our study was not to performthe experiment with longer ischemic periods other than 90 min. Probably,prominent histopathological changes would be observed in the outer retinaas well with an ischemic period longer than 90 min. Thickness of the innerplexiform layer has been selected for the quantification of ischemic retinaldamage in the present study as it has been reported previously that, ischemiaand reperfusion induced damage has been well recognized and documentedin the inner plexiform layer of the retina [32]. The thickness of the innerplexiform layer has significantly increased in the ischemia group versus thecontrol and ischemia/octreotide groups.This increase in the thickness hasbeen interpreted as the consequence of retinal edema in the ischemia group.In rat retina, retinal edema and PMNL infiltration have been observed after90 min of ischemia and 24 h of reperfusion [32]. The antiedema effect ofoctreotide has also been reported previously [48, 49]. Indeed, the thicknessof the inner plexiform layer of the ischemia/octreotide group has not changewhen compared with the control group .
The results of this study has shown that, NO activity decreases after I/Rof the retinal tissue and although it has no effect on the amelioration of NOactivity during I/R of the retinal tissue octreotide may reduce some of thehazardous results of decreased NO activity such as PMNL infiltration.
References
1. Choi DW. Glutamate neurotoxicity: A three stage process. In Guidotti A, ed. Neurotox-icity of amino acids. FIDIA Research Foundation Symposium Series. New York: RavenPress, 1991: 235–42
2. Goldstein IM, Ostwald P, Roth S. Nitric oxide: A review of its role in retinal functionand disease. Vision Res 1996; 36: 2979–94.
3. Hangai M, Yoshimura N, Hiroi K, Mandai M, Honda Y. Inducible nitric oxide synthasein retinal ischemia-reperfusion injury. Exp Eye Res 1996; 63: 501–9.
4. Yamamato R, Bredt DS, Snyder SH, Stone RA. The localization of nitric oxide synthasein rat eye and related cranial ganglia. Neuroscience 1993; 54: 19–200.
5. Koch K, Lambrecht H, Haberecht M, Redburn D, Schmith HHHW. Functional couplingof a calcium/calmodulin-dependent nitric oxide synthase an a soluble quanyl cyclase invertebrate photoreceptor cells. EMBO J 1994; 13: 3312–20.
6. Perez MTR, Larsson B, Alm P, Anderson KE, Ehinger B. Localization of neuronal nitricoxide synthase-immunoreactivity in rat and rabbit retinas. Brain Res 1995; 104: 207–17.
7. Chakravarty U, Stitt AW, McNally J, Bailie J, Hoey EM, Duprex P. Nitric oxide synthaseactivity and expression in retinal capillary endothelial cells and pericytes. Curr Eye Res1994; 14: 285–94.
8. Goureau O, Hicks D, Courtois Y, de Kozak Y. Induction and regulation of nitric oxidesynthase in retinal Müller glial cells. J Neurochem 1994; 63: 310–7.

336
9. Meyer P, Champion C, Schlotzer-Schrehardt U, Flammer J, Haefliger IO. Localization ofnitric oxide synthase isoforms in porcine ocular tissues. Curr Eye Res 1999; 18: 375–80.
10. Harrdy P, Dumont I, Bhattacharya M, Hou X, Lachapelle P, Varma D, Chemtob S.Oxidants, nitric oxide and prostanoids in the developing ocular vasculature: a basis forischemic retinopathy. Cardiovasc Res 2000; 47: 489–509.
11. Radomski MW, Palmer RMJ, Moncada S. An L-arginine/nitric oxide pathway present inhuman platelets regulates aggregation. Proc Natl Acad Sci USA 1990 87; 5193–7.
12. Engelman DT, Watanabe M, Engelman RM, Rousou JA, Flack JE, Deaton D, Das DK.Constitutive nitric oxide release is impaired after ischemia and reperfusion. J ThoracCardiovasc Surg 1995; 110: 1047–53.
13. Araki M, Tanaka M, Hasegawa K, Yokota R, Maeda T, Ishikawa M, Yabuuchi Y,Sasayama S. Nitric oxide inhibition improved myocardial metabolism independent oftissue perfusion during ischemia but not during reperfusion. J Mol Cell Cardiol 2000;32: 375–84.
14. Mashiach Elisheva, Shifra Sela, Winaver J, Shasha SM, Kristal B. Renal ischemia–reperfusion injury: contribution of nitric oxide and renal blood flow. Nephron 1998; 80:45–7.
15. Kusterer K, Blöchle C, Konrad T, Palitzsch KD, Usadel KH. Rat liver injury induced byhypoxic ischemia and reperfusion: protective action by somatostatin and two derivatives.Regul Pept 1993; 44: 251–6.
16. Niedermühlbicher M, Wiedermann CJ. Supression of superoxide release from humanmonocytes by somatostatin related peptides. Regul Pept 1992; 41: 39–47.
17. Le Q, Zhang X, Zhang J. Effects of sandostatin on gastric mucosal perfusion in rats withportal hypertensive gastropathy. Chung Hua Kan Tsang Ping Tsa Chih 2000; 8: 21–3.
18. Arias-Diaz J, Vara E, Torres-Molero J, Garcia C, Hernandez J, Balibrea JL. Local pro-duction of oxygen free radicals and nitric oxide in rat diaphragm during sepsis: effectsof pentoxifylline and somatostatin. Eur J Surg 1997; 163: 619–25.
19. Chao TC, Cheng HP, Walter RJ. Somatostatin and macrophage function: modulation ofhydrogen peroxide, nitric oxide and tumor necrosis factor release. Regul Pept 1995; 58:1–10.
20. Borries PN, Borries C. Nitrate determination in biological fluids by an enzymatic one-step assay with nitrate reductase. Clin Chem 1995; 41: 904–7.
21. Del Maestro RF. An approach to the free radicals in medicine and biology. Acta PhysiolScand 1980; 492: 153–68.
22. Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxinonenal,malondialdehyde and related aldehydes. Free Radic Biol Med 1991; 11: 81–128.
23. Comporti M. Lipid peroxidation and biogenic aldehydes: from the identification of 4-hydroxinonenal to further achievements in biopathology. Free Radic Res 1998; 28: 623–35.
24. Buko VU, Artukevich AA, Ignatenko KV. Aldehydic products of lipid peroxidation inactivate cytochrome P-50. Exp Toxicol Pathol 1999; 51: 294–8.
25. Keller JN, Hanni KB, Markesberry WR. 4-Hydroxinonenal increases neuronal suscept-ibility to oxidative stress. J Neurosci Res 1999; 58: 823–30.
26. Mylonas C, Kouretas D. Lipid peroxidation and tissue damage. In Vivo 1999; 13: 295–309.
27. Grant MB, Wargovich TJ, Ellis EA, Cabellero BS, Mansour M, Pepine CJ. Localizationof insulin-like growth factor I and inhibition of coronary smooth muscle cell growth bysomatostatin analogues in human coronary smooth muscle cells. Circulation 1994; 89:1511–7.

337
28. Grigorevski VP, Korotkina RN, Karelin AA. Effects of several regulatory peptides onthe functional activity of the pancreas in acute experimental pancreatitis. Biull EkspBiol Med 1989; 108: 428–30.
29. Szabo M, Droy-Lefaix MT, Doly M, Braquet P. Modification of ischemia-reperfusioninduced ion shifts (Na, K, Ca and Mg) by free radical scavengers in the rat retina.Ophthalm Res 1993; 25: 1–9.
30. Hinder RA, Stein HJ. Oxygen derived free radicals. Arch Surg 1991; 126: 104–6.31. Wiedermann CJ, Sitte B, Zilian U, Reinisch N, Beimpold H, Finkenstedt G, Braunsteiner
H. Inhibition of superoxide anion release from circulating neutrophils by L-arginine inman. Clin Invest 1993; 71: 985–99
32. Szabo ME, Droy-Lefaix MT, Doly M, Carre C, Braquet P. Ischemia and reperfusioninduced histological changes in rat retina. Invest Ophthalmol Vis Sci 1991; 32: 1471–8.
33. Wiedermann CJ, Niedermühlbichler M, Braunsteiner H. Inhibition of recombinanthuman growth hormone-induced and prolactin-induced activation of neutrophils byoctreotide. Naunyn-Schmiedeberg’s Arch Pharmacol 1992; 347: 336–41.
34. Guarner C, Soriano G, Tomas A, Bulbena O, Novella MT, Balanzo J. Increased serumnitrite and nitrate levels in patients with cirrhosis. Hepatology 1993; 18: 1139–42.
35. Chintila MS, Chiu PJS, Vermulapalli S, Watkins RW, Sybert EJ. Inhibition of EDRFaggravates ischemic acute renal failure in anesthetized rats. Naunyn Schmiedeberg’sArch Pharmacol 1993; 348: 305–10.
36. Pinsky DJ, Oz MC, Koga S. Cardiac preservation is enhanced in a heterotrophic rattransplant model by supplementing the nitric oxide pathway. J Clin Invest 1994; 93:2291–7.
37. Gryglewski RJ, Palmer RMJ, Moncada S. Superoxide anion is involved in the breakdownof endothelium derived relaxing factor. Nature 1986; 320: 454–6.
38. Davidge ST, Ojimba J, McLaughin MK. Vascular function in the vitamin E-deprivedrat. An interaction between nitric oxide and superoxide anions. Hypertension 1998; 31:830–5.
39. Köken T, Inal M. The effect of nitric oxide on ischemia-reperfusion injury in rat liver.Clin Chim Acta 1999; 288: 55–62.
40. Wenger FA, Klian M, Jacobi CA, Mauthsch I, Peter FJ, Guski H, Schimke I, MullerJM. Effects of octreotide on liver metastasis and intrametastatic lipid peroxidation inexperimental pancreatic cancer. Oncology 2001; 60: 282–8.
41. De Belder AJ, Rodomski MW, Why HJF. Nitric oxide synthase activities in humanmyocardium. Lancet 1993; 341: 83–5.
42. McCall T, Broughton-Smith NK, Palmer RMJ, Whittle BJR, Mponcada S. Synthesisof nitric oxide from L-arginine by neutrophils. Release and interaction with superoxideanion. Biochem J 1989; 261: 293–6.
43. Kubes P, Suzuki M, Granger DN. Nitric oxide: An endogenous modulator of leukocytesadhesion. Proc Natl Acad Sci USA 1991; 88: 4651–5.
44. Suzuki M, Inauen W, Kvietya PR, Grisham MB, Meininger C, Schelling ME. Superoxidemediates reperfusion induced leukocyte-endothelial cell interactions. Am J Phsiol 1989;257: 1740–5.
45. Weber M, Mohand S, Hicks D, Dreyfus H, Sahel JA. Monosialoganglioside GM1 re-duces ischemia-reperfusion induced injury in rat retina. Invest Ophthalmol Vis Sci 1996;37: 267–73.
46. Hughes WF. Quantification of ischemic damage in rat retina. Exp Eye Res 1991; 53:573–82.

338
47. Adachi K, Fujita Y, Morizane C, Akaike A, Ueda M, Satoh M, Masai H, Kashii S, HondaY. Inhibition of NMDA receptors and nitric oxide synthase reduces ischemic injury ofthe retina. Eur J Pharmacol 1998; 53–7.
48. Günal Aí, Isìk A, Celiker H, Eren O, Celebi H, Günal SY. Short term reduction of leftventricular mass in primary hypertrophic cardiomyopathy by octreotide injections. Heart1996; 76: 418–21.
49. Toriumi Y, Samuel I, Wilcokson DP, Turkelson CM, Solomon TE, Joehl RJ. Octreotideand cholecystokinin reduce edema in obstruction induced acute pancreatitis. J Lab ClinMed 1993; 122: 450–4.
Address for correspondence: U. Celiker, Firat Universitesi, Firat Tip Merkezi Göz Klinigi,23200 Elazig, TurkeyFax: +90-4242388096; E-mail: [email protected]