muscular dystrophies in childhood
TRANSCRIPT

Muscular Dystrophies In Childhood
Dr Alpana SKDept of Pediatrics

Dr Alpana SK, www.peditips.com

Dr Alpana SK, www.peditips.com

Dr Alpana SK, www.peditips.com
What is muscular dystrophy?
Increasing weakness and loss of muscle mass and function until the patient is confined to a wheelchair
A problem with the synthesis of proteins in the muscle fibers causes deterioration
Four forms of the disease are recognized, based on: Pattern of inheritance Age when symptoms are first noted Distribution of the muscles first involved

Dystrophinopathy: disorders involving dystrophin
Duchenne MD and Becker MD are the muscular disorders – the two most common and severe dystrophies
Dystrophin is a very large gene on the X-chromosome, ubiquitous in the human body
Dystrophin-Associated Protein (DAP) Complex – composed of the extracellular, transmembrane, and intracellular components

Dr Alpana SK, www.peditips.com
Classification of types of MD
Heritable MDs include the following:
Sex-linked MDs Duchenne Becker Emery-Dreifuss
Autosomal dominant MDs Facioscapulohumeral Distal Ocular Oculopharyngeal
Autosomal recessive MD – limb-girdle form

Dr Alpana SK, www.peditips.com

Dr Alpana SK, www.peditips.com
What is Duchenne’s Muscular Dystrophy?
The disease is characterized Early onset often before school years By progressive muscular deterioration
and death by 14 to 18 years of age A defect of a large gene on the X

Dr Alpana SK, www.peditips.com
Prevalence of DMD
Affects one in 3500 to 5000 newborn males
1/3 of these with previous family history.
2/3 sporadic

Dr Alpana SK, www.peditips.com
What is the pattern of inheritance?
X-linked recessive inheritance

Dr Alpana SK, www.peditips.com
Duchenne Muscular Dystrophy Inheritance.
Mother carries the recessive gene and passes it to her child
Trait is usually expressed in males only
Dr Alpana SK, www.peditips.com
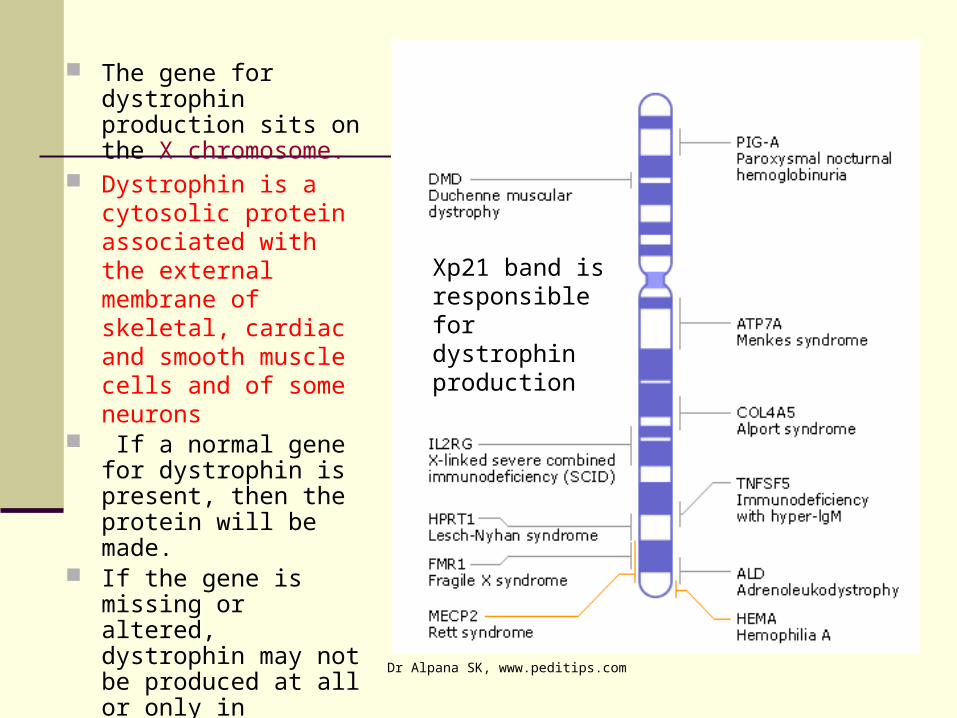
The gene for dystrophin production sits on the X chromosome.
Dystrophin is a cytosolic protein associated with the external membrane of skeletal, cardiac and smooth muscle cells and of some neurons
If a normal gene for dystrophin is present, then the protein will be made.
If the gene is missing or altered, dystrophin may not be produced at all or only in abnormal forms, resulting in Duchenne muscular dystrophy
Xp21 band is responsible for dystrophin production

Dr Alpana SK, www.peditips.com

Dr Alpana SK, www.peditips.com

Dr Alpana SK, www.peditips.com
In Duchenne muscular dystrophy (DMD) patients with a deletion of exons 45–54, an out-of-frame transcript is generated in which exon 44 is spliced to exon 55. Owing to the frame shift, a stop codon occurs in exon 55, which prematurely aborts dystrophin synthesis. b | Using an exon-internal antisense oligonucleotide (AON) in exon 44, the skipping of this exon can be induced in cultured muscle cells. Accordingly, the transcript is back in-frame and a Becker muscular dystrophy (BMD)-like dystrophin can be synthesized

Dr Alpana SK, www.peditips.com


Dr Alpana SK, www.peditips.com
In the Duchenne muscular dystrophy, the attachment of muscle fibers to their surrounding endomysium (extracellular matrix) becomes weakened due to mutations in the dystrophin gene.
(Image courtesy of the National Cancer Institute)

Dr Alpana SK, www.peditips.com
What are the symptoms?
difficulty in walking at about the
age of four years increased size in calf muscles Loss of the ability to walk at about
the age of 11 Death at about the age of 18 or
later, usually because of respiratory failure.

Dr Alpana SK, www.peditips.com

Dr Alpana SK, www.peditips.com
Symptoms of DMD
Awkward gait (patients tend to walk on their forefeet, because of an increased calve tonus)
Frequent falls Progressive difficulty walking , Eventual loss of ability to walk (usually
by the age of 12) Difficulty with motor skills (running, hopping, jumping) Fatigue Higher risk of behaviour and learning difficulties. Skeletal deformities (including scoliosis in some cases) Muscle deformities of tongue and calf muscles. The enlarged muscle
tissue is eventually replaced by fat and connective tissue, hence the term pseudohypertrophy.
of heels and legs, rendering them unusable because the muscle fibers shorten and fibrosis occurs in connective tissue

Dr Alpana SK, www.peditips.com

Dr Alpana SK, www.peditips.com
Posture changes during progression of Duchenne muscular dystrophy.

Dr Alpana SK, www.peditips.com
Possible Complications
Deformities Permanent, progressive disability
Decreased mobility Decreased ability for self-care
Mental impairment (varies, usually minimal) Pneumonia or other respiratory infections Respiratory failure Cardiomyopathy Congestive heart failure (rare) Heart arrhythmias (rare)

Dr Alpana SK, www.peditips.com
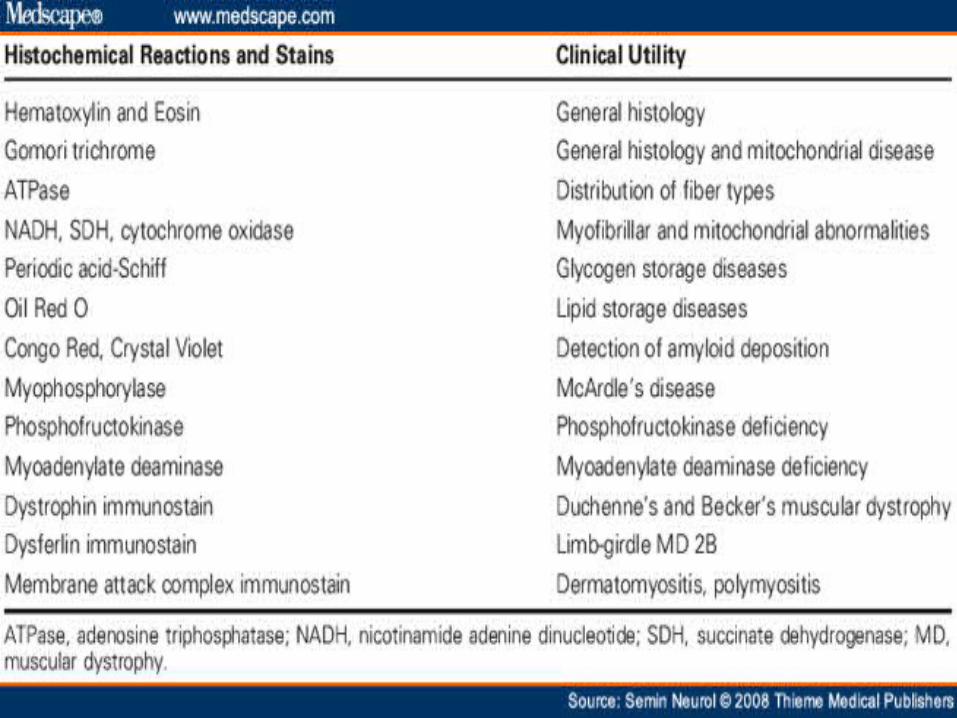
Diagnosing Muscular Dystrophy Detailed patient and family history (3 generation pedigree)
Determine the source of the muscle weakness (nerve or muscle
Laboratory tests: Serum CK is very elevated Troponin I is elevated above normal but not to levels in cardiac
ischemia Liver enzymes show high AST & ALT Muscle biopsy - Endomysial fibrosis Variable fiber size: Small
fibers rounded Dystrophic muscle Hypercontracted (opaque) muscle fibers
Myopathic grouping Muscle fiber degeneration & regeneration Muscle fiber internal architecture: Normal or immature Dystrophin: Absent staining Other membrane proteins: Sarcoglycans and Aquaporin 4 are
reduced

Dr Alpana SK, www.peditips.com
Diagnosing Muscular Dystrophy CPK test In DMD patients CPK leaks out of the muscle
cell into the bloodstream, so a high level (nearly 50 to 100 times more) confirms that there is muscle damage. Affected individuals may have a value as high as 15,000 to 35,000iu/l (normal = 60iu/l).
DNA test The dystrophin gene is composed of 79 exons,
and DNA testing and analysis can usually identify the specific type of mutation and the exon or exons that are affected. DNA testing confirms the diagnosis in most cases.[3]

Dr Alpana SK, www.peditips.com
a. Hematoxylin and eosin staining of control tissue
b. Hematoxylin and eosin staining of DMD patient, which shows abnormal variation in fiber size, degenerating and regenerating fibers, immune cell infiltration, and increased fibrosis
c. Immunofluorescence analysis of dystrophin in control tissue biopsy
d. Immunofluorescence analysis of dystrophin in a young DMD patient biopsy, illustrating the loss of sarcolemmal staining in DMD
Dr Alpana SK, www.peditips.com
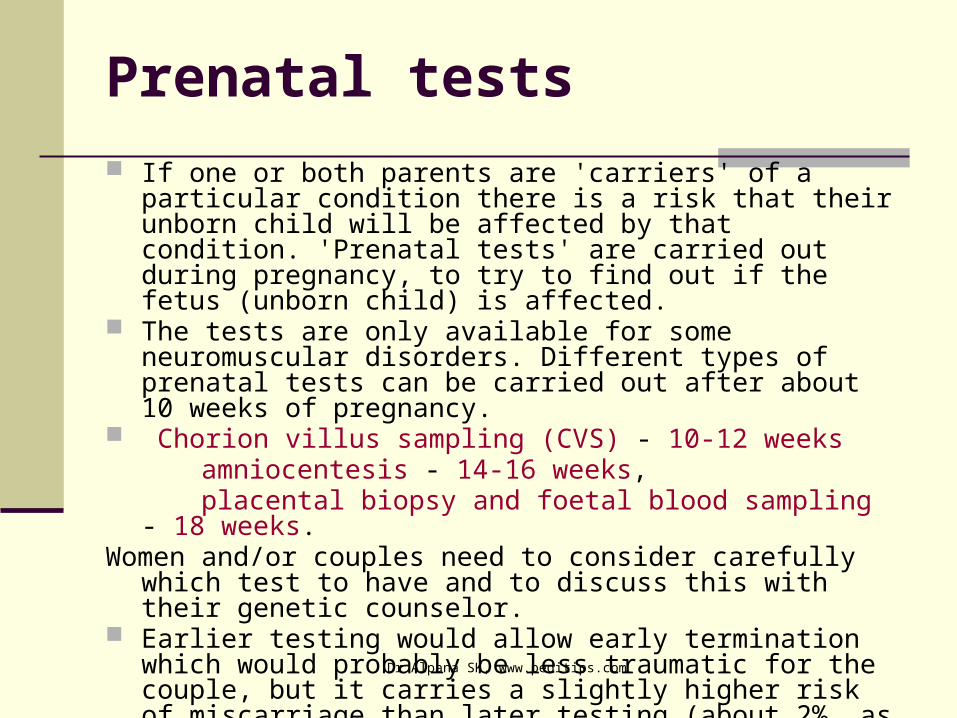
Prenatal tests If one or both parents are 'carriers' of a particular
condition there is a risk that their unborn child will be affected by that condition. 'Prenatal tests' are carried out during pregnancy, to try to find out if the fetus (unborn child) is affected.
The tests are only available for some neuromuscular disorders. Different types of prenatal tests can be carried out after about 10 weeks of pregnancy.
Chorion villus sampling (CVS) - 10-12 weeks amniocentesis - 14-16 weeks, placental biopsy and foetal blood sampling - 18 weeks. Women and/or couples need to consider carefully which
test to have and to discuss this with their genetic counselor.
Earlier testing would allow early termination which would probably be less traumatic for the couple, but it carries a slightly higher risk of miscarriage than later testing (about 2%, as opposed to 0.5%).

Dr Alpana SK, www.peditips.com
Approach to Diagnosis of Childhood Muscular Dystrophy
No DeletionDetected
Diagnosis of Dystrophinopathy (DM D or BM D)
Deletion NotInform ativefor Severity
Deletion Inform ativefor Severity.
Positive Fam ily History
DeletionDetected
DNA Analysisfor Dystrophin
Gene Abnorm ality
M ale
Abnorm alDystrophin
M erosin andAdhalin Analysis
Norm alDystrophin
M uscle Biopsy
Female
Elevated
M uscular DystrophyUnlikely
Norm al
C K
M uscular W eakness
Dr Alpana SK, www.peditips.com

Dr Alpana SK, www.peditips.com
Therapy
Rehabilitation Therapy:
Physical Occupational Respiratory
Promising Therapies:
Pharmacologic Therapy Cell Therapy Gene Transfer Therapy Newer Targets

Dr Alpana SK, www.peditips.com
Treatment
There is no known cure for Duchenne muscular dystrophy
Treatment is generally aimed at control of
symptoms to maximize the quality of life.
Corticosteroids such as prednisone and deflazacort increase energy and strength and defer severity of some symptoms.
Mild, non-jarring physical activity such as swimming is encouraged. Inactivity (such as bed rest) can worsen the muscle disease.

Dr Alpana SK, www.peditips.com
Treatment Physical therapy is helpful to maintain muscle strength,
flexibility, and function.
Orthopedic appliances (such as braces and wheelchairs) may improve mobility and the ability for self-care. Form-fitting removable leg braces that hold the ankle in place during sleep can defer the onset of contractures.
Appropriate respiratory support as the disease
progresses is important Idebenone is a new drug currently being actively
researched and is expected to be approved in the US and Europe by Santhera Pharmaceuticals. Meanwhile it is sold in the US as a nutritional products mainly by Cognitive Nutrition. www.cognitivenutrition.com/ The effective dose is 450 mg daily.

Dr Alpana SK, www.peditips.com
Advances in Gene Therapy(3)
Researches have developed "minigenes," which carry instructions for a slightly smaller version of dystrophin, that can fit inside a virus. Utrophin.. Is similar to dystrophin and can be engineered.
Researchers have also created the so-called gutted virus, a virus that has had its own genes removed so that it is carrying only the dystrophin gene

Dr Alpana SK, www.peditips.com
Prognosis
Duchenne muscular dystrophy eventually affects all voluntary muscles and involves the heart and respioratory muscles in later stages.
Survival is rare beyond the early 30s,although recent advancements in medicine are extending the lives of those afflicted.
Death in Duchenne muscular dystrophy most commonly results from pulmonary insufficiency, respiratory infections.
In about 10% of cases, death is due to cardiac dysfunction..

Congenital Muscular Dystrophy
Presentation: neonatal onset of severe weakness, delayed motor milestones, contractures
Merosin negative/CMD A1 White matter hypodensities on brain scan but
normal mental capacity Diagnosis by muscle biopsy
immunohistochemistry showing loss of α2-laminin (AR-chromosome 6q22-23)

Neuronal Migration Disorders
With neuronal migration disorders get mental retardation, brain malformations, and clinical eye involvement
Fukuyama’s muscular dystrophy – affects fukutin protein (AR – chromosome 9q31)
Muscle-eye-brain disease – affects POMGnT1, (AR – chromosome 1p32-34)
Walker Warburg – affects POMT1 (AR) Glycosyltransferases are also important in
neuronal development

Myotonic Muscular Dystrophy or Steinert’s disease Presentation – adult with multiple systems affected
Primarily distal and facial weakness Facial features: frontal balding in men, ptosis, low-
set ears, hatchet jaw, dysarthria, swan neck, ^ shaped upper lip
Myotonia: worse in cold weather, after age 20 Heart: conduction block – evaluate syncope Smooth muscle: constipation, care with swallowing,
gallstones, problems with childbirth, BP lability Brain: learning disabilities, increased sleep
requirement Ophthalmology: cataracts Endocrine: insulin resistance, hypothyroidism,
testicular atrophy

FascioScapularHumeral Muscular Dystrophy
Presentation: Facial weakness with trouble blowing up a balloon,
sipping through a straw, whistling, trouble closing the eyes at night, scapular winging that may be asymmetric, pain
May have absence of pectoralis, biceps, or brachioradialis
Also affected: mild high pitched hearing loss, retinal abnormalities, mental retardation in early onset
Genetics/Testing Southern blot testing available (chromosome 4q35)
for decrease in repeats normally present Muscle biopsy may show lymphocytic infiltrates

Limb Girdle Muscular Dystrophy
Presentation: variable age of onset with weakness and wasting of the limb-girdle
May have calf hypertrophy, involvement of scapular muscle and deltoid in sarcoglycanopathies
Many types involve dysfunctional sarcoglycans – transmembrane proteins of the DAP that interact with cytoplasmic proteins
Table 2 – types of LGMD

Oculopharyngeal Muscular Dystrophy
Presentation: mid-adult with ptosis, facial muscle weakness with difficulty swallowing, proximal muscle weakness, may have extraocular muscle weakness, more common in French-Canadian and Hispanic population
Genetics Muscle biopsy shows filamentous nuclear inclusions
and ubiquitin containing vacuoles Affects poly A binding protein 2 (PABP2) by
expansion of a GCG repeat without anticipation seen – Southern blot (chromosome 14q11-13)

Emery-Dreifuss Muscular DystrophyScapuloperoneal MD
Presentation: stiff joints, shoulder and upper arm weakness, calf weakness, cardiac conduction defects and arrhythmias, contractures
Genetics X-linked type affects emerin
Diagnose by protein analysis of leukocytes or skin fibroblasts
DNA testing available (chromosome Xq28) AD affects lamin A or lamin C (chromosome 1q21) Nuclear membrane proteins

Distal Muscular Dystrophy
Presentation: weakness in forearms, hands, and lower legs clinically similar to a neuropathy but NCV normal
Muscle biopsy with autophagocytic vacuoles/ inclusion bodies
Table 3 – Types of DMDWelander distal myopathy AD/2p13 hands first
Anterior tibial/Markesbery-Griggs/Udd
AD/2q31-33
Nonaka/Inclusion body myopathy 2
AR/9p13 Rimmed vacuoles, inclusion bodies, affects GNE
Gowers/Laing distal myopathy AD/14q11
Miyoshi myopathy AR/2p13 Affects dysferlin
Distal myopathy with vocal cord and pharyngeal weakness
AD/5

Myopathies
Central core disease: Ryanodine receptor, Ca channel that mediates
excitation/contraction coupling, (AD – chromosome 19q13) Associated with Malignant Hyperthermia
Myotubular myopathy Myotubularin, important in myogenesis (Xq28)
Nemaline Myopathy Caused by many defects, disorder of thin filaments Rod-like stuctures on muscle biopsy
Inflammatory Juvenile Dermatomyositis Inclusion Body Myositis (usually distal) Adult Polymyositis (associated with malignancy)

Therapy
Contracture prevention Stretching exercises and postural
changing Stretch the most contracture prone
groups (gastrocnemius, hip flexors, iliotibial bands, hamstrings)
AFO at night to supplement

Strengthening/conditioning/endurance Goal is to maintain or improve muscle strength
and maximize functional ability – slight improvement is possible
Additional goal is to avoid muscular damage by overwork or injury
No eccentric contraction or delayed soreness Voluntary active exercise such as
swimming/hydrotherapy or cycling in ambulatory children currently recommended

Mobility aids Walking orthoses – KAFO Standing frames, standing wheelchairs, swivel
walker occasionally used Walkers where arm strength less affected Transfer board Wheelchair – power needed for independence Plan for indoor lift, van with lift, roll in shower
Improving daily activities of daily living Physical and Occupational Therapy – teaching
modified techniques Antigravity orthoses are being developed to assist in
daily living activities Splinting and therapy to prevent hand contractures

comfysplints.com

Surgery note the risk inherent to surgery – malignant
hyperthermia Palliative vs. rehabilitative Tendon releases
Achilles Need KAFO to walk post-op Relieves pain and allow shoe wear
Hamstring and iliotibial band Relieves hip and knee pain or contracture Allows better gait compensation

Scoliosis – spine stabilization Bracing is not effective with progressive
neuromuscular disease Timely correction of scoliosis is important
for patient comfort and respiratory ability Spine and scapular stabilization may aid
function of armsOphthalmology
Deficient eye closure oculomaxillofacial MD and FSH MD may require artificial tears or tarsorrhaphy
Treatment for cataracts in Myotonic MD

Respiratory Patients with morning headache, nightmares,
excessive daytime somnolence, mental dullness, difficulty concentrating, increased colds, coughing, or pneumonia should undergo evaluation
Influenza vaccine and pneumococcal vaccine In-exsufflator for airway clearance, cough assist Pulmonologist, pulmonary function testing

Assisted noninvasive ventilation Oxygen alone does not ventilate! Positive pressure ventilation vs. volume ventilation
with pressure limit Assisted ventilation with tracheostomy
Talk to patient about degree of desired intervention when respiratory status first starts to decline and before an acute event
The goal is home ventilation Cardiology
EKG – pacemaker for conduction defects and arrhythmias
Echocardiogram – afterload reduction, digoxin for cardiomyopathy

Nutrition/GI Overweight and underweight are
common problems Overweight impairs mobility Underweight decreases strength & health
Protein and calorie supplements Assess for dysphagia Intestinal hypomotility in DMD, CMD,
and myotonic dystrophy can require a bowel regimen to prevent constipation

Osteopenia/Osteoporosis Begins before walking stops, fractures may end
walking Worsened by steroids Calcium supplements, Miacalcin may help
Psychology/Neuropsychological Education – aid in planning Special education may not be needed with
accomodation and modifications Progressive loss of function affects patient and
family