monascin and ankaflavin act as natural ampk activators with pparα agonist activity to down-regulate...
TRANSCRIPT

Food and Chemical Toxicology 64 (2014) 94–103
Contents lists available at ScienceDirect
Food and Chemical Toxicology
journal homepage: www.elsevier .com/locate/ foodchemtox
Monascin and ankaflavin act as natural AMPK activators with PPARaagonist activity to down-regulate nonalcoholic steatohepatitisin high-fat diet-fed C57BL/6 mice
0278-6915/$ - see front matter � 2013 Elsevier Ltd. All rights reserved.http://dx.doi.org/10.1016/j.fct.2013.11.015
Abbreviations: ACC, acetyl-CoA carboxylase; ACOX, acyl-CoA oxidase; ACS, acyl-CoA synthetase; AK, ankaflavin; ALT, alanine aminotranferase; AMPK, AMP-activated kinase; AST, aspartate transaminase; BSA, bovine serum albumin; CPT-1, carnitine palmitoyl transferase I; CV, cardiovascular; DMSO, dimethyl sulfoxide;ELISA, enzyme-linked immunosorbent assay; FABP, fatty acid-binding protein; FAS,fatty acid synthase; FAT, fatty acid transporter; FBS, fetal bovine serum; FFA, freefatty acid; FXR, farnesoid X receptor; GLUT, glucose transporter; H&E, hematoxylinand eosin; HAECs, human aortic endothelial cells; HDL-C, high-density lipoproteincholesterol; HPLC, high-performance liquid chromatography; HUVECs, humanumbilical vein endothelial cells; IACUC, Institutional Animal Care and UseCommittee; ICAM-1, intercellular adhesion molecule-1; IL-6, interleukin-6; iNOS,inducible nitric oxide synthase; JNK, c-Jun NH2-terminal kinase; LDL-C, low densitylipoprotein-cholesterol; MS, monascin; MTP, microsomal triglyceride transferprotein; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepa-titis; NCBI, National Center for Biotechnology Information; NF-jB, nuclear factor-kappaB; Nrf2, nuclear factor erythroid 2-related factor 2; OA, oleic acid; PCR,polymerase chain reaction; PGC-1a, peroxisome proliferator-activated receptorgamma co-activator 1-alpha; PPARa, peroxisome proliferator activated receptoralpha; PPARc, peroxisome proliferator activated receptor gamma; PPREs, peroxi-some-proliferator-response elements; PTP1B, protein tyrosine phosphatase 1B;SDS, sodium dodecyl sulfate; SPPARMs, selective PPAR modulators; SREBP-1c, sterolregulatory element-binding protein; TC, total cholesterol; TG, triaceylglycerol;TGFb, transformation growth factor beta; TLC, thin layer chromatography; TNF-a,tumor necrosis factor-alpha; TR-FRET, time-resolved fluorescence resonance energytransfer; Tyr, tyrosine; TZDs, thiazolidinediones; VCAM-1, vascular cell adhesionmolecule-1.⇑ Corresponding author. Tel.: +886 2 33664519x10; fax: +886 2 33663838.
E-mail address: [email protected] (T.-M. Pan).
Wei-Hsuan Hsu a, Ting-Hung Chen a, Bao-Hong Lee a, Ya-Wen Hsu b, Tzu-Ming Pan a,⇑a Department of Biochemical Science & Technology, College of Life Science, National Taiwan University, No. 1, Sec. 4, Roosevelt Road, Taipei 10617, Taiwanb R&D Division, SunWay Biotechnology Company Limited, Taipei, Taiwan
a r t i c l e i n f o a b s t r a c t
Article history:Received 5 March 2013Accepted 13 November 2013Available online 22 November 2013
Keywords:Ankaflavin (AK)Monascin (MS)Nonalcoholic fatty liver disease (NAFLD)Oleic acid (OA)Peroxisome proliferator-activated receptor(PPAR)-a
Yellow pigments monascin (MS) and ankaflavin (AK) are secondary metabolites derived from Monascus-fermented products. The hypolipidemic and anti-inflammatory effects of MS and AK indicate that theyhave potential on preventing or curing nonalcoholic fatty liver disease (NAFLD). Oleic acid (OA) andhigh-fat diet were used to induce steatosis in FL83B hepatocytes and NAFLD in mice, respectively. Wefound that both MS and AK prevented fatty acid accumulation in hepatocytes by inhibiting fatty aciduptake, lipogenesis, and promoting fatty acid beta-oxidation mediated by activating peroxisomeproliferator-activated receptor (PPAR)-a and AMP-activated kinase (AMPK). Furthermore, MS and AKsignificantly attenuated high-fat diet-induced elevation of total cholesterol (TC), triaceylglycerol (TG),free fatty acid (FFA), and low density lipoprotein-cholesterol (LDL-C) in plasma. MS and AK promotedAMPK phosphorylation, suppressed the steatosis-related mRNA expression and inflammatory cytokinessecretion, as well as upregulated farnesoid X receptor (FXR), peroxisome proliferator-activated receptorgamma co-activator (PGC)-1a, and PPARa expression to induce fatty acid oxidation in the liver of mice.We provided evidence that MS and AK act as PPARa agonists to upregulate AMPK activity and attenuateNAFLD. MS and AK may be supplied in food supplements or developed as functional foods to reduce therisk of diabetes and obesity.
� 2013 Elsevier Ltd. All rights reserved.
1. Introduction
Nonalcoholic fatty liver disease (NAFLD) is defined as fatty infil-tration of the liver that causes a 5–10% increase in liver weight(Salt, 2004). NAFLD represents a spectrum of conditions character-ized microvesicular and macrovesicular hepatic steatosis, whichoccurs in people who do not consume alcohol in amounts generallyconsidered harmful for the liver (Sanyal, 2002). The spectrum ofNAFLD disorders ranges from simple fatty liver (steatosis withoutliver injury), to nonalcoholic steatohepatitis (NASH) and fibrosis/cirrhosis (Matteoni et al., 1999; Adiels et al., 2006).
AMP-activated kinase (AMPK) has been investigated as a poten-tial therapeutic target for the treatment of type 2 diabetes, mostlybecause of AMPK-mediated inhibition of gluconeogenesis in theliver during feeding (Shaw et al., 2005). AMPK activity is regulatedby nutrients, hormones, calcium, and cellular stress levels (Leclercand Rutter, 2004). Activation of AMPK is modulated by changes inATP, ADP, and AMP concentrations as well as phosphorylation atThr172 by an upstream AMPK kinase (Hawley et al., 1996). AMPKacts primarily by directly affecting the activity of enzymes in-volved in carbohydrate, lipid, and protein biosyntheses and sec-ondarily by long-term transcriptional control of key components

W.-H. Hsu et al. / Food and Chemical Toxicology 64 (2014) 94–103 95
of metabolic pathways. AMPK is a major regulator of glucose andlipid metabolism and represents an attractive target for the treat-ment of hepatic disorders (Viollet et al., 2009). Activation of hepa-tic AMPK leads to increased fatty acid oxidation with simultaneousinhibition of hepatic lipogenesis, cholesterol synthesis, and glucoseproduction. In addition to short-term effects on specific enzymes,AMPK also modulates the transcription of the genes involved inlipogenesis and mitochondrial biogenesis. The identification ofAMPK targets in hepatic metabolism should be useful for develop-ing treatments to reverse metabolic abnormalities related to type 2diabetes and metabolic syndrome.
There are three peroxisome proliferator-activated receptor(PPAR) subtypes: PPARa, PPARc, and PPARd. All three subtypescan modulate transcription by binding to specific peroxisome-proliferator-response elements (PPREs) on target genes. PPARais highly expressed in the liver, kidney, and skeletal muscle.Fibrates, which are weak PPARa agonists, alter hepatic energymetabolism that, in part, lowers plasma triaceylglycerol (TG)concentrations and slightly increases high-density lipoproteincholesterol (HDL-C) (Staels and Fruchart, 2005; Tenenbaumet al., 2005). PPARa agonists such as fenofibrate and PPARcagonists such as pioglitazone activate AMPK to stimulate thepathways that increase energy production, such as glucose trans-port and fatty acid oxidation, switching off the pathways thatconsume energy, such as lipogenesis (Chen et al., 2012; Lageet al., 2008).
Monascus-fermented rice is known for its ability to dramaticallyimprove dyslipidemia (Lee et al., 2010). In addition to blood lipid-lowering effects, Monascus-fermented rice also has the potential toregulate blood glucose levels (Shi and Pan, 2010), preventatherosclerosis (Lee et al., 2010), reduce inflammation, act as ananti-oxidant (Hsu et al., 2010a), and attenuate alcoholic fatty liverdisease (Cheng and Pan, 2011). Recent studies have shown thatmonascin (MS) and ankaflavin (AK) isolated from Monascus-fer-mented rice lowers total cholesterol (TC), TG, and low density lipo-protein-cholesterol (LDL-C) levels in blood (Lee et al., 2010) whilereducing inflammation (Cheng and Pan, 2011) and amelioratingthe capacity for lipolysis in mature 3T3-L1 cells (Hsu et al.,2012a; Jou et al., 2010). The high-fat diet has been reported toresult in NAFLD (Kusunoki et al., 2005). Nevertheless, the mecha-nisms underlying MS and AK on NAFLD in vitro and in vivo remainsunclear. In this study, we examined the oleic acid-induced steato-sis effect in FL83B hepatocytes treated with MS and AK. In in vivostudies, mice were fed a high-fat diet to induce NAFLD and werethen treated with MS and AK to examine their potential therapeu-tic effects. We demonstrated that both MS and AK increased AMPKactivity and beta-oxidation in hepatocytes. We provided evidencethat MS and AK act as PPARa agonists to upregulate AMPK activityand attenuate NAFLD.
2. Materials and methods
2.1. Chemicals and reagents
F-12K medium was purchased from Gibco BRL Life Technologies, Inc. (Gaithers-burg, MD, USA). Trypsin–EDTA and fetal bovine serum (FBS) were purchased fromInvitrogen Life Technologies (Carlsbad, CA, USA). Bovine serum albumin (BSA), so-dium dodecyl sulfate (SDS), KCl, fenofibrate, NaCl, crystal violet, oil-red O, and oleicacid were purchased from Sigma Chem. Co. (St. Louis, MO, USA). Dimethyl sulfoxide(DMSO), formaldehyde, and KH2PO4 were purchased from Wako Pure Chem. (Sai-tama, Japan). Chloroform, methanol, and isopropanol purchased from Merck. Co.,Inc. (Rahway, NJ, USA). High-capacity cDNA reverse transcription kit was purchasedfrom Applied Biosystems (Foster City, CA, USA). MK886 (PPARa antagonist) andWY14643 (PPARa agonist) were purchased from Cayman Chemical (Ann Arbor,MI, USA). Cytokines assay kits were purchased from Peprotech (Rocky hill, NJ,USA). For MS and AK preparation, the crude extracts of Monascus-fermented ricewere obtained after filtering and concentrating under reduced pressure, and thencoated on silica gel and subjected to dry flash chromatography. Sufficient n-hexanewas passed through the column to remove the oily hydrophobic materials.
Extensive gradient elution was then employed using different ethyl acetate in n-hexane ratios to yield numerous fractions. Similar fractions were combined accord-ing to thin layer chromatography (TLC), and the solvent was removed under re-duced pressure. These fractions were further analyzed by high performance liquidchromatography (HPLC), and then fractions with a similar single peak profile werecombined, respectively. Finally, the fraction with the desired compound was con-centrated to dryness. Preparation of MS and AK (>95% purity) was confirmed by nu-clear magnetic resonance (NMR, Varian Gemini, 200 MHz, FT-NMR, Varian Inc., PaloAlto, CA, USA) and electrospray ionization-mass spectrometry (ESI-MS, ThermoElectron Co., Waltham, MA, USA) analysis. Monascin (C21H26O5) (SupplementalFig. 1A): ESIMS m/z 359 [M+H]+. 1H NMR (CDCl3, 200 MHz) d: 0.88 (3H, t, J = 6.6,H-19), 1.30 (4H, m, H-17, H-18), 1.43 (3H, s, H-12), 1.61 (2H, m, H-16), 1.84 (3H,d, J = 6.0, H-11), 2.49 (1H, m, H-15a), 2.64 (1H, m, H-5a), 2.70 (1H, m, H-15b),2.99 (1H, m, H-5b), 3.14 (1H, m, H-6), 3.64 (1H, d, J = 9.0, H-13), 4.67 (1H, d,J = 12.6, H-1a), 5.02 (1H, d, J = 12.6, H-1b), 5.26 (1H, s, H-4), 5.85 (1H, d, J = 15.4,H-9), 6.47 (1H, dt, J = 15.4, 6.0, H-10); 13C NMR (CDCl3, 50 MHz) d: 12.9 (C-19),16.7 (C-12), 17.5 (C-11), 21.4 (C-18), 21.8 (C-16), 28.4 (C-17), 30.1 (C-5), 41.9 (C-6), 53.9 (C-13), 62.8 (C-1), 82.2 (C-7), 102.3 (C-4), 113.0 (C-8a), 123.4 (C-9), 134.4(C-10), 149.8 (C-4a), 159.5 (C-3), 168.5 (C-13a), 188.8 (C-8), 201.5 (C-14). Ankafla-vin (C23H30O5) (Supplemental Fig. 1B): ESIMS m/z 409 [M+Na]+. 1H NMR (d6-ace-tone, 400 MHz) d: 0.87 (3H, t, J = 6.8, H-21), 1.29 (8H, m, H-17–20), 1.45 (3H, s,H-12), 1.60 (2H, m, H-16), 1.82 (3H, d, J = 7.2, H-11), 2.66 (2H, m, H-5), 2.70 (1H,m, H-15a), 2.92 (1H, m, H-15b), 3.15 (1H, m, H-6), 4.27 (1H, d, J = 13.2, H-13),4.68 (1H, d, J = 12.4, H-1a), 4.90 (1H, d, J = 12.4, H-1b), 5.50 (1H, s, H-4), 6.01 (1H,d, J = 15.6, H-9), 6.41 (1H, dq, J = 15.6, 7.2, H-10); 13C NMR (d6-acetone, 100 MHz)d: 14.2 (C-21), 17.7 (C-12), 18.3 (C-11), 23.2 (C-20), 23.6 (C-16), 29.2 (C-5), 29.9(C-17, C-18), 32.3 (C-19), 43.4 (C-15), 44.4 (C-6), 55.3 (C-13), 64.2 (C-1), 84.0 (C-7), 104.6 (C-4), 115.1 (C-8a), 125.5 (C-9), 134.6 (C-10), 151.6 (C-4a), 160.2 (C-3),171.3 (C-13a), 190.6 (C-8), 203.6 (C-14).
2.2. Cell culture
Mouse liver cell FL83B is a hepatocyte cell line isolated from a normal liver ta-ken from a 15 to 17 day old fetal mouse. FL83B cells were cultured in a humidifiedatmosphere of 95% air and 5% CO2 at 37 �C in F-12K medium containing 10% FBS.The medium was renewed every 2–3 days and subcultured every 4 days.
2.3. Oil red O stain
FL83B hepatocytes (1 � 105 cells) were seeded in 24-well plates and treatedwith MS (10 lM), AK (10 lM), or WY14643 (10 lM; PPARa agonist) with or with-out MK886 (10 lM; PPARa antagonist) solved in medium containing BSA (0.5 mM)and oleic acid (OA; 2 mM) as model-1 (Fig. 1A). In model-2, FL83B hepatocytes weretreated in medium containing BSA (0.5 mM) and OA (2 mM) for 12 h to induce fattyacid accumulation. And then, MS (10 lM), AK (10 lM), or WY14643 (10 lM) withor without MK886 (10 lM) were added (Fig. 1B). The cells were incubated with10% formaldehyde for 1 h. Subsequently, oil red O working solution was added toeach well and stained for 15 min. The staining photos were taken under the micro-scope. Data were expressed by resolving the oil red O working solution with 100%isopropanol then as percentage of change in absorbance at 490 nm by using the ELI-SA reader (Jou et al., 2010).
2.4. Immunoblot analysis
FL83B cells were lysed and the cell lysates were centrifuged (10,000g for10 min) to recover the supernatant. The supernatant was taken as the cell extract.The protein concentration in the cell extract was determined using a Bio-Rad pro-tein assay kit. The samples were subjected to 10% SDS–polyacrylamide gel electro-phoresis (PAGE). The protein spots were electrotransferred to a polyvinyldienedifluoride (PVDF) membrane. The membrane was incubated with block bufferand then probed with primary antibody overnight at 4 �C. The membrane waswashed, shaken in a solution of HRP-linked anti-rabbit IgG secondary antibody.The expressions of proteins were detected by enhanced chemiluminescent (ECL) re-agent (Millipore, Billerica, MA, USA).
2.5. Real-time polymerase chain reaction (PCR)
Total RNA from liver tissue and hepatocytes was obtained using the Trizolreagent (Gibco BRL Life Technologies, Inc., Gaithersburg, MD, USA) according tothe manufacturer’s instructions. Primers were blasted according to National Centerfor Biotechnology Information (NCBI) primer database to estimate their specificitiesand then synthesized by MD-Bio, Inc. (Taipei, Taiwan). Primers were shown inSupplemental Table 1. The gene expression level was determined by relativequantitative real-time PCR (CFX Cycler System, Bio Rad Laboratories, Inc., Hercules,CA, USA).

Fig. 1. Flow chart of MS and AK attenuated fatty acid accumulation. Model-1:FL83B hepatocytes were treated with MS, AK, or WY with or without MK first, andthen OA was added for 20 h as the preventive model. Model-2: FL83B hepatocyteswere treated with OA for 12 h first, and then the cells were treated with MK, and/orMS, AK, or WY as the therapeutic model. MS: monascin; AK: ankaflavin; WY:WY14643; MK: MK886; OA: oleic acid.
96 W.-H. Hsu et al. / Food and Chemical Toxicology 64 (2014) 94–103
2.6. Animal induction
Male C57BL/6J male mice (5 weeks old) were kept and used for the experiment,which were purchased from BioLASCO Co., Ltd. (Taipei, Taiwan). The mice werehoused in a temperature-controlled room (25 ± 1 �C) and kept on a 12-h light/darkcycle (light on at 08:00). (Protocol complied with guidelines described in the ‘‘Ani-mal Protection Law’’, amended on January 17, 2001 Hua-Zong-(1)-Yi-Tzi-9000007530, Council of Agriculture, Executive Yuan, Taiwan, ROC). The animalstudy was approved by Institutional Animal Care and Use Committee (IACUC) ofNational Taiwan University (Permit Number: 100-48).
The experiments were carried out in a qualified animal breeding room in theAnimal Center at our institute. The mice were fed with chow diet (Rodent Labora-tory Chow 5001, Ralston Purina, St. Louis, MO, USA) for 1 week in order to accom-modate them to the environment. Animals were randomly assigned to (A) control,(B) high-fat diet, (C) high-fat diet + MS, and (D) high-fat diet + AK. Mice of controlgroup are continuously consumed chow diet, and others were fed with high-fatdiet. The normal Rodent Laboratory Chow 5001 contained 4.5% fat and calories wereabout 3.34 kcal/g. The preparation of high-fat diet was adding 26.7% butter powderinto the Rodent Laboratory Chow 5001, This diet contained 30% fat and the calories
was about 4.85 kcal/g (Kusunoki et al., 2005). All animals were provided ad libitumaccess to the diets for 16 weeks. Both MS (5 mg/kg bw/day) and AK (5 mg/kg bw/day) were orally administered from week-11 to week-16. Animals were sacrificedby CO2 asphyxia after fasting for 12 h, and plasma was collected for biochemicalanalysis. The liver was dissected out and storage at �80 �C for further experiments.
2.7. Biochemical analysis
Blood glucose, aspartate transaminase (AST), alanine transaminase (ALT), TG,TC, HDL-C, and LDL-C were measured. Blood glucose was immediately determinedafter the blood was drawn from the mice by the glucose analyzer (Eumed Biotech-nology Co., Ltd., Hsinchu, Taiwan). AST and ALT were measured in triplicate usingan automatic biochemical analyzer (Beckman-700, Fullerton, CA, USA). TG, TC,HDL-C, and LDL-C were measured by the commercial analyzing kit (Randox Inc., An-trim, UK).
2.8. Cytokine analysis
Liver tissues were immediately removed and homogenized with ice-cold10 mM Tris–HCl buffer (pH 7.4). After centrifugation at 12,000g at 4 �C for30 min, the supernatant was collected for the measurement of cytokines. The levelsof tumor necrosis factor-a (TNF-a), transformation growth factor (TGF)-b, andinterleukin (IL)-6 were measured by ELISA kits (Rocky hill, NJ, USA).
2.9. Histological evaluation
Small piece of liver tissues taken from the experimental animals were fixed in10% neutral formalin, alcohol-dehydrated, paraffin-embedded and the sectionedto mean thickness of 5 lm. The histological examination by above conventionalmethods was evaluated by hematoxylin and eosin (H&E) stain.
2.10. Statistical analysis
Results were expressed as the mean ± SD in vitro and mean ± SEM in vivo. Com-parisons among groups were made using one-way ANOVA. The differences betweenmean values in all groups were tested through the Duncan’s multiple-range test(SPSS statistical software package, version 17.0, SPSS, Chicago, IL, USA). A possibilityof p value less than 0.05 was considered as significantly different between means.
3. Results
3.1. Inhibition of fatty acid accumulation by MS and AK in OA-treatedFL83B hepatocytes
MS and AK were prepared to >95% purity and analyzed accord-ing to Hsu et al. (2010b) by using nuclear magnetic resonance(NMR, Varian Gemini, 200 MHz, FT-NMR, Varian Inc., Palo Alto,CA, USA) and electrospray ionization-mass spectrometry (ESI-MS,Thermo Electron Co., Waltham, MA, USA) (Hsu et al., 2012b; Leeet al., 2013a). MS and AK were not cytotoxic for FL83B hepatocytesat a concentration range of 5–20 lM (cell survival rate was >85%;data not shown). We first measured inhibition of fatty acid accu-mulation in OA-induced FL83B hepatocytes co-treated with MS,AK, or the PPARa agonist WY14643 in the presence of absence ofthe PPARa antagonist MK886. The anti-steatohepatitis effects ofthese treatments are shown in model-1 (Fig. 2A). Staining withoil red O revealed significantly increased fatty acid accumulationdue to OA induction in FL83B hepatocytes; this effect was blockedby treatment with MS, AK, or WY14643. In addition, co-treatmentwith the PPARa antagonist MK886 did not reverse the inhibition offatty acid accumulation by MS, AK, or WY14643. However, theanti-steatohepatitis effect of MS, AK, and WY14643 was impairedwhen cells were co-treated with MK886. These results suggestedthat both MS and AK prevented fatty acid accumulation by inhib-iting fatty acid uptake, lipogenesis, or promoting fatty acid beta-oxidation mediated by PPARa.
In order to determine whether MS and AK increased beta-oxida-tion, we investigated inhibition of fatty acid accumulation by MSand AK in FL83B hepatocytes after induction with OA for 12 h,depicted in model-2. Our results suggested that MS, AK, andWY14643 significantly abated fatty acid accumulation, but the
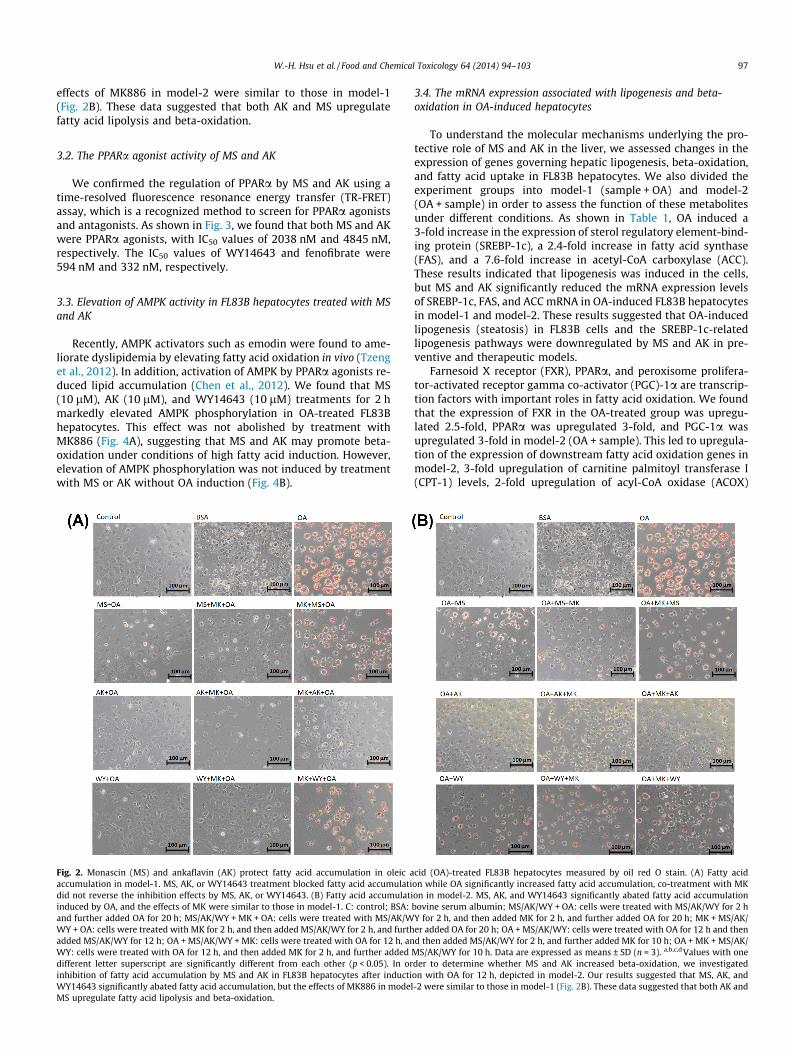
W.-H. Hsu et al. / Food and Chemical Toxicology 64 (2014) 94–103 97
effects of MK886 in model-2 were similar to those in model-1(Fig. 2B). These data suggested that both AK and MS upregulatefatty acid lipolysis and beta-oxidation.
3.2. The PPARa agonist activity of MS and AK
We confirmed the regulation of PPARa by MS and AK using atime-resolved fluorescence resonance energy transfer (TR-FRET)assay, which is a recognized method to screen for PPARa agonistsand antagonists. As shown in Fig. 3, we found that both MS and AKwere PPARa agonists, with IC50 values of 2038 nM and 4845 nM,respectively. The IC50 values of WY14643 and fenofibrate were594 nM and 332 nM, respectively.
3.3. Elevation of AMPK activity in FL83B hepatocytes treated with MSand AK
Recently, AMPK activators such as emodin were found to ame-liorate dyslipidemia by elevating fatty acid oxidation in vivo (Tzenget al., 2012). In addition, activation of AMPK by PPARa agonists re-duced lipid accumulation (Chen et al., 2012). We found that MS(10 lM), AK (10 lM), and WY14643 (10 lM) treatments for 2 hmarkedly elevated AMPK phosphorylation in OA-treated FL83Bhepatocytes. This effect was not abolished by treatment withMK886 (Fig. 4A), suggesting that MS and AK may promote beta-oxidation under conditions of high fatty acid induction. However,elevation of AMPK phosphorylation was not induced by treatmentwith MS or AK without OA induction (Fig. 4B).
Fig. 2. Monascin (MS) and ankaflavin (AK) protect fatty acid accumulation in oleic aaccumulation in model-1. MS, AK, or WY14643 treatment blocked fatty acid accumulatidid not reverse the inhibition effects by MS, AK, or WY14643. (B) Fatty acid accumulatiinduced by OA, and the effects of MK were similar to those in model-1. C: control; BSA:and further added OA for 20 h; MS/AK/WY + MK + OA: cells were treated with MS/AK/WWY + OA: cells were treated with MK for 2 h, and then added MS/AK/WY for 2 h, and furthadded MS/AK/WY for 12 h; OA + MS/AK/WY + MK: cells were treated with OA for 12 h, anWY: cells were treated with OA for 12 h, and then added MK for 2 h, and further addeddifferent letter superscript are significantly different from each other (p < 0.05). In orinhibition of fatty acid accumulation by MS and AK in FL83B hepatocytes after inductiWY14643 significantly abated fatty acid accumulation, but the effects of MK886 in modelMS upregulate fatty acid lipolysis and beta-oxidation.
3.4. The mRNA expression associated with lipogenesis and beta-oxidation in OA-induced hepatocytes
To understand the molecular mechanisms underlying the pro-tective role of MS and AK in the liver, we assessed changes in theexpression of genes governing hepatic lipogenesis, beta-oxidation,and fatty acid uptake in FL83B hepatocytes. We also divided theexperiment groups into model-1 (sample + OA) and model-2(OA + sample) in order to assess the function of these metabolitesunder different conditions. As shown in Table 1, OA induced a3-fold increase in the expression of sterol regulatory element-bind-ing protein (SREBP-1c), a 2.4-fold increase in fatty acid synthase(FAS), and a 7.6-fold increase in acetyl-CoA carboxylase (ACC).These results indicated that lipogenesis was induced in the cells,but MS and AK significantly reduced the mRNA expression levelsof SREBP-1c, FAS, and ACC mRNA in OA-induced FL83B hepatocytesin model-1 and model-2. These results suggested that OA-inducedlipogenesis (steatosis) in FL83B cells and the SREBP-1c-relatedlipogenesis pathways were downregulated by MS and AK in pre-ventive and therapeutic models.
Farnesoid X receptor (FXR), PPARa, and peroxisome prolifera-tor-activated receptor gamma co-activator (PGC)-1a are transcrip-tion factors with important roles in fatty acid oxidation. We foundthat the expression of FXR in the OA-treated group was upregu-lated 2.5-fold, PPARa was upregulated 3-fold, and PGC-1a wasupregulated 3-fold in model-2 (OA + sample). This led to upregula-tion of the expression of downstream fatty acid oxidation genes inmodel-2, 3-fold upregulation of carnitine palmitoyl transferase I(CPT-1) levels, 2-fold upregulation of acyl-CoA oxidase (ACOX)
cid (OA)-treated FL83B hepatocytes measured by oil red O stain. (A) Fatty acidon while OA significantly increased fatty acid accumulation, co-treatment with MKon in model-2. MS, AK, and WY14643 significantly abated fatty acid accumulationbovine serum albumin; MS/AK/WY + OA: cells were treated with MS/AK/WY for 2 hY for 2 h, and then added MK for 2 h, and further added OA for 20 h; MK + MS/AK/er added OA for 20 h; OA + MS/AK/WY: cells were treated with OA for 12 h and thend then added MS/AK/WY for 2 h, and further added MK for 10 h; OA + MK + MS/AK/
MS/AK/WY for 10 h. Data are expressed as means ± SD (n = 3). a,b,c,d Values with oneder to determine whether MS and AK increased beta-oxidation, we investigatedon with OA for 12 h, depicted in model-2. Our results suggested that MS, AK, and-2 were similar to those in model-1 (Fig. 2B). These data suggested that both AK and
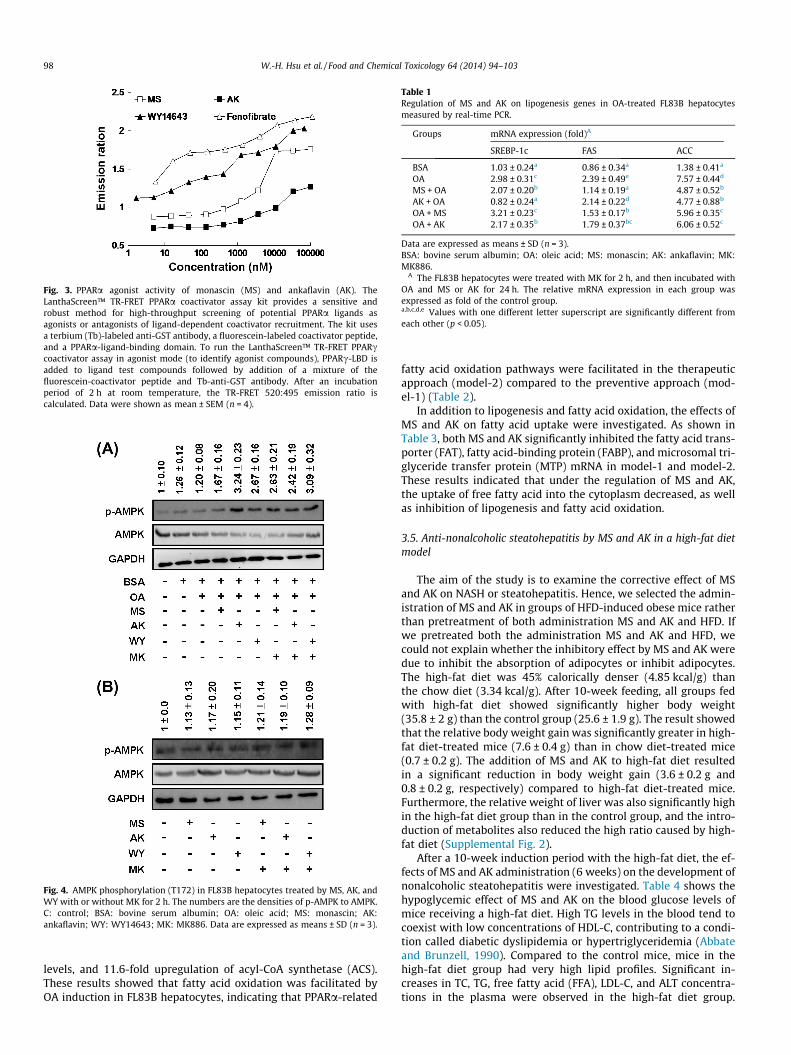
Fig. 3. PPARa agonist activity of monascin (MS) and ankaflavin (AK). TheLanthaScreen™ TR-FRET PPARa coactivator assay kit provides a sensitive androbust method for high-throughput screening of potential PPARa ligands asagonists or antagonists of ligand-dependent coactivator recruitment. The kit usesa terbium (Tb)-labeled anti-GST antibody, a fluorescein-labeled coactivator peptide,and a PPARa-ligand-binding domain. To run the LanthaScreen™ TR-FRET PPARccoactivator assay in agonist mode (to identify agonist compounds), PPARc-LBD isadded to ligand test compounds followed by addition of a mixture of thefluorescein-coactivator peptide and Tb-anti-GST antibody. After an incubationperiod of 2 h at room temperature, the TR-FRET 520:495 emission ratio iscalculated. Data were shown as mean ± SEM (n = 4).
Fig. 4. AMPK phosphorylation (T172) in FL83B hepatocytes treated by MS, AK, andWY with or without MK for 2 h. The numbers are the densities of p-AMPK to AMPK.C: control; BSA: bovine serum albumin; OA: oleic acid; MS: monascin; AK:ankaflavin; WY: WY14643; MK: MK886. Data are expressed as means ± SD (n = 3).
Table 1Regulation of MS and AK on lipogenesis genes in OA-treated FL83B hepatocytesmeasured by real-time PCR.
Groups mRNA expression (fold)A
SREBP-1c FAS ACC
BSA 1.03 ± 0.24a 0.86 ± 0.34a 1.38 ± 0.41a
OA 2.98 ± 0.31c 2.39 ± 0.49e 7.57 ± 0.44d
MS + OA 2.07 ± 0.20b 1.14 ± 0.19a 4.87 ± 0.52b
AK + OA 0.82 ± 0.24a 2.14 ± 0.22d 4.77 ± 0.88b
OA + MS 3.21 ± 0.23c 1.53 ± 0.17b 5.96 ± 0.35c
OA + AK 2.17 ± 0.35b 1.79 ± 0.37bc 6.06 ± 0.52c
Data are expressed as means ± SD (n = 3).BSA: bovine serum albumin; OA: oleic acid; MS: monascin; AK: ankaflavin; MK:MK886.
A The FL83B hepatocytes were treated with MK for 2 h, and then incubated withOA and MS or AK for 24 h. The relative mRNA expression in each group wasexpressed as fold of the control group.a,b,c,d,e Values with one different letter superscript are significantly different fromeach other (p < 0.05).
98 W.-H. Hsu et al. / Food and Chemical Toxicology 64 (2014) 94–103
levels, and 11.6-fold upregulation of acyl-CoA synthetase (ACS).These results showed that fatty acid oxidation was facilitated byOA induction in FL83B hepatocytes, indicating that PPARa-related
fatty acid oxidation pathways were facilitated in the therapeuticapproach (model-2) compared to the preventive approach (mod-el-1) (Table 2).
In addition to lipogenesis and fatty acid oxidation, the effects ofMS and AK on fatty acid uptake were investigated. As shown inTable 3, both MS and AK significantly inhibited the fatty acid trans-porter (FAT), fatty acid-binding protein (FABP), and microsomal tri-glyceride transfer protein (MTP) mRNA in model-1 and model-2.These results indicated that under the regulation of MS and AK,the uptake of free fatty acid into the cytoplasm decreased, as wellas inhibition of lipogenesis and fatty acid oxidation.
3.5. Anti-nonalcoholic steatohepatitis by MS and AK in a high-fat dietmodel
The aim of the study is to examine the corrective effect of MSand AK on NASH or steatohepatitis. Hence, we selected the admin-istration of MS and AK in groups of HFD-induced obese mice ratherthan pretreatment of both administration MS and AK and HFD. Ifwe pretreated both the administration MS and AK and HFD, wecould not explain whether the inhibitory effect by MS and AK weredue to inhibit the absorption of adipocytes or inhibit adipocytes.The high-fat diet was 45% calorically denser (4.85 kcal/g) thanthe chow diet (3.34 kcal/g). After 10-week feeding, all groups fedwith high-fat diet showed significantly higher body weight(35.8 ± 2 g) than the control group (25.6 ± 1.9 g). The result showedthat the relative body weight gain was significantly greater in high-fat diet-treated mice (7.6 ± 0.4 g) than in chow diet-treated mice(0.7 ± 0.2 g). The addition of MS and AK to high-fat diet resultedin a significant reduction in body weight gain (3.6 ± 0.2 g and0.8 ± 0.2 g, respectively) compared to high-fat diet-treated mice.Furthermore, the relative weight of liver was also significantly highin the high-fat diet group than in the control group, and the intro-duction of metabolites also reduced the high ratio caused by high-fat diet (Supplemental Fig. 2).
After a 10-week induction period with the high-fat diet, the ef-fects of MS and AK administration (6 weeks) on the development ofnonalcoholic steatohepatitis were investigated. Table 4 shows thehypoglycemic effect of MS and AK on the blood glucose levels ofmice receiving a high-fat diet. High TG levels in the blood tend tocoexist with low concentrations of HDL-C, contributing to a condi-tion called diabetic dyslipidemia or hypertriglyceridemia (Abbateand Brunzell, 1990). Compared to the control mice, mice in thehigh-fat diet group had very high lipid profiles. Significant in-creases in TC, TG, free fatty acid (FFA), LDL-C, and ALT concentra-tions in the plasma were observed in the high-fat diet group.
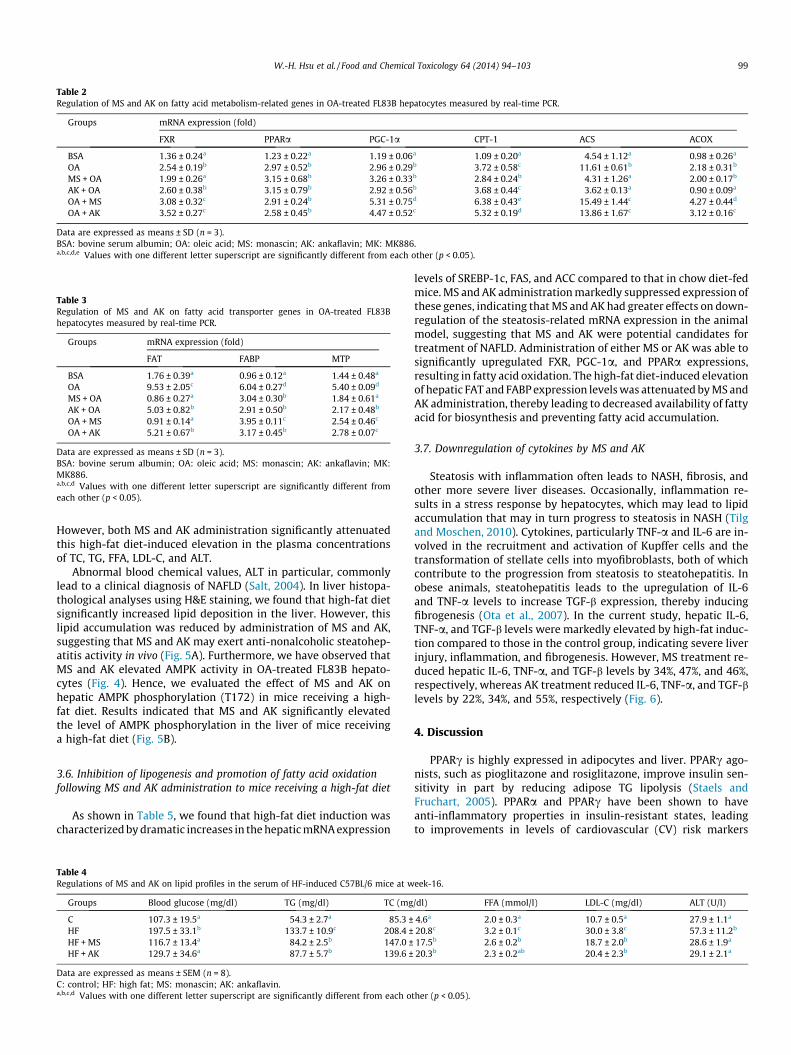
Table 2Regulation of MS and AK on fatty acid metabolism-related genes in OA-treated FL83B hepatocytes measured by real-time PCR.
Groups mRNA expression (fold)
FXR PPARa PGC-1a CPT-1 ACS ACOX
BSA 1.36 ± 0.24a 1.23 ± 0.22a 1.19 ± 0.06a 1.09 ± 0.20a 4.54 ± 1.12a 0.98 ± 0.26a
OA 2.54 ± 0.19b 2.97 ± 0.52b 2.96 ± 0.29b 3.72 ± 0.58c 11.61 ± 0.61b 2.18 ± 0.31b
MS + OA 1.99 ± 0.26a 3.15 ± 0.68b 3.26 ± 0.33b 2.84 ± 0.24b 4.31 ± 1.26a 2.00 ± 0.17b
AK + OA 2.60 ± 0.38b 3.15 ± 0.79b 2.92 ± 0.56b 3.68 ± 0.44c 3.62 ± 0.13a 0.90 ± 0.09a
OA + MS 3.08 ± 0.32c 2.91 ± 0.24b 5.31 ± 0.75d 6.38 ± 0.43e 15.49 ± 1.44c 4.27 ± 0.44d
OA + AK 3.52 ± 0.27c 2.58 ± 0.45b 4.47 ± 0.52c 5.32 ± 0.19d 13.86 ± 1.67c 3.12 ± 0.16c
Data are expressed as means ± SD (n = 3).BSA: bovine serum albumin; OA: oleic acid; MS: monascin; AK: ankaflavin; MK: MK886.a,b,c,d,e Values with one different letter superscript are significantly different from each other (p < 0.05).
Table 3Regulation of MS and AK on fatty acid transporter genes in OA-treated FL83Bhepatocytes measured by real-time PCR.
Groups mRNA expression (fold)
FAT FABP MTP
BSA 1.76 ± 0.39a 0.96 ± 0.12a 1.44 ± 0.48a
OA 9.53 ± 2.05c 6.04 ± 0.27d 5.40 ± 0.09d
MS + OA 0.86 ± 0.27a 3.04 ± 0.30b 1.84 ± 0.61a
AK + OA 5.03 ± 0.82b 2.91 ± 0.50b 2.17 ± 0.48b
OA + MS 0.91 ± 0.14a 3.95 ± 0.11c 2.54 ± 0.46c
OA + AK 5.21 ± 0.67b 3.17 ± 0.45b 2.78 ± 0.07c
Data are expressed as means ± SD (n = 3).BSA: bovine serum albumin; OA: oleic acid; MS: monascin; AK: ankaflavin; MK:MK886.a,b,c,d Values with one different letter superscript are significantly different fromeach other (p < 0.05).
W.-H. Hsu et al. / Food and Chemical Toxicology 64 (2014) 94–103 99
However, both MS and AK administration significantly attenuatedthis high-fat diet-induced elevation in the plasma concentrationsof TC, TG, FFA, LDL-C, and ALT.
Abnormal blood chemical values, ALT in particular, commonlylead to a clinical diagnosis of NAFLD (Salt, 2004). In liver histopa-thological analyses using H&E staining, we found that high-fat dietsignificantly increased lipid deposition in the liver. However, thislipid accumulation was reduced by administration of MS and AK,suggesting that MS and AK may exert anti-nonalcoholic steatohep-atitis activity in vivo (Fig. 5A). Furthermore, we have observed thatMS and AK elevated AMPK activity in OA-treated FL83B hepato-cytes (Fig. 4). Hence, we evaluated the effect of MS and AK onhepatic AMPK phosphorylation (T172) in mice receiving a high-fat diet. Results indicated that MS and AK significantly elevatedthe level of AMPK phosphorylation in the liver of mice receivinga high-fat diet (Fig. 5B).
3.6. Inhibition of lipogenesis and promotion of fatty acid oxidationfollowing MS and AK administration to mice receiving a high-fat diet
As shown in Table 5, we found that high-fat diet induction wascharacterized by dramatic increases in the hepatic mRNA expression
Table 4Regulations of MS and AK on lipid profiles in the serum of HF-induced C57BL/6 mice at w
Groups Blood glucose (mg/dl) TG (mg/dl) TC (mg
C 107.3 ± 19.5a 54.3 ± 2.7a 85.3 ±HF 197.5 ± 33.1b 133.7 ± 10.9c 208.4 ±HF + MS 116.7 ± 13.4a 84.2 ± 2.5b 147.0 ±HF + AK 129.7 ± 34.6a 87.7 ± 5.7b 139.6 ±
Data are expressed as means ± SEM (n = 8).C: control; HF: high fat; MS: monascin; AK: ankaflavin.a,b,c,d Values with one different letter superscript are significantly different from each o
levels of SREBP-1c, FAS, and ACC compared to that in chow diet-fedmice. MS and AK administration markedly suppressed expression ofthese genes, indicating that MS and AK had greater effects on down-regulation of the steatosis-related mRNA expression in the animalmodel, suggesting that MS and AK were potential candidates fortreatment of NAFLD. Administration of either MS or AK was able tosignificantly upregulated FXR, PGC-1a, and PPARa expressions,resulting in fatty acid oxidation. The high-fat diet-induced elevationof hepatic FAT and FABP expression levels was attenuated by MS andAK administration, thereby leading to decreased availability of fattyacid for biosynthesis and preventing fatty acid accumulation.
3.7. Downregulation of cytokines by MS and AK
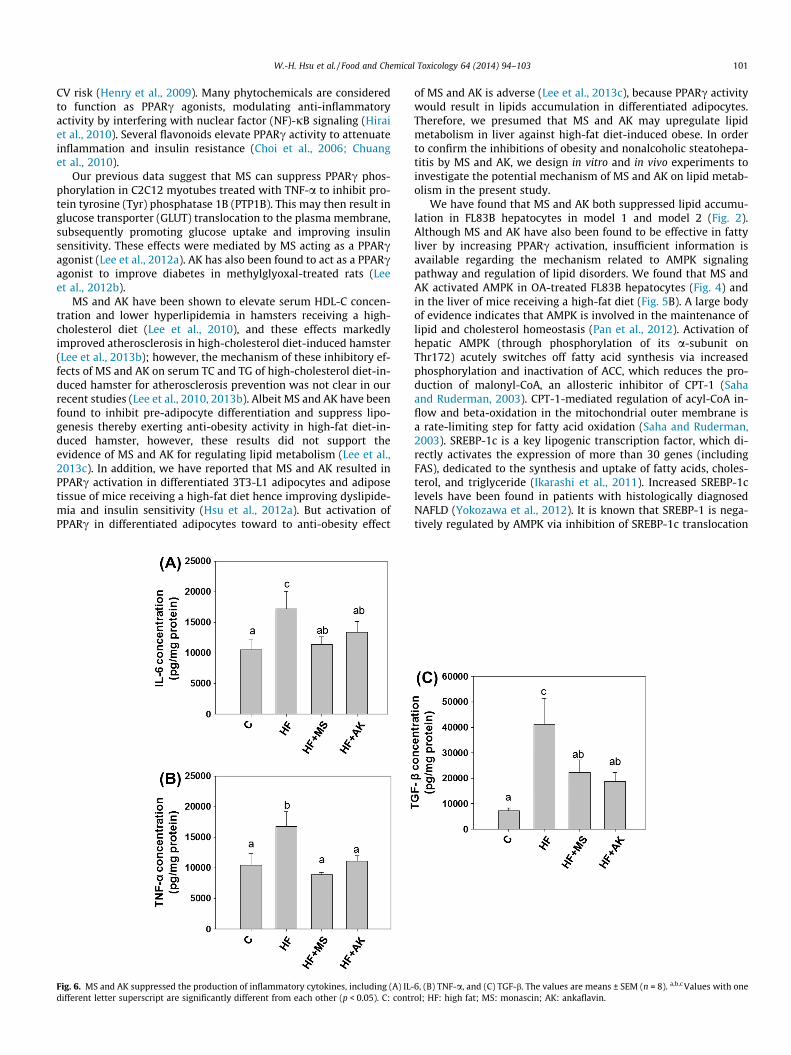
Steatosis with inflammation often leads to NASH, fibrosis, andother more severe liver diseases. Occasionally, inflammation re-sults in a stress response by hepatocytes, which may lead to lipidaccumulation that may in turn progress to steatosis in NASH (Tilgand Moschen, 2010). Cytokines, particularly TNF-a and IL-6 are in-volved in the recruitment and activation of Kupffer cells and thetransformation of stellate cells into myofibroblasts, both of whichcontribute to the progression from steatosis to steatohepatitis. Inobese animals, steatohepatitis leads to the upregulation of IL-6and TNF-a levels to increase TGF-b expression, thereby inducingfibrogenesis (Ota et al., 2007). In the current study, hepatic IL-6,TNF-a, and TGF-b levels were markedly elevated by high-fat induc-tion compared to those in the control group, indicating severe liverinjury, inflammation, and fibrogenesis. However, MS treatment re-duced hepatic IL-6, TNF-a, and TGF-b levels by 34%, 47%, and 46%,respectively, whereas AK treatment reduced IL-6, TNF-a, and TGF-blevels by 22%, 34%, and 55%, respectively (Fig. 6).
4. Discussion
PPARc is highly expressed in adipocytes and liver. PPARc ago-nists, such as pioglitazone and rosiglitazone, improve insulin sen-sitivity in part by reducing adipose TG lipolysis (Staels andFruchart, 2005). PPARa and PPARc have been shown to haveanti-inflammatory properties in insulin-resistant states, leadingto improvements in levels of cardiovascular (CV) risk markers
eek-16.
/dl) FFA (mmol/l) LDL-C (mg/dl) ALT (U/l)
4.6a 2.0 ± 0.3a 10.7 ± 0.5a 27.9 ± 1.1a
20.8c 3.2 ± 0.1c 30.0 ± 3.8c 57.3 ± 11.2b
17.5b 2.6 ± 0.2b 18.7 ± 2.0b 28.6 ± 1.9a
20.3b 2.3 ± 0.2ab 20.4 ± 2.3b 29.1 ± 2.1a
ther (p < 0.05).
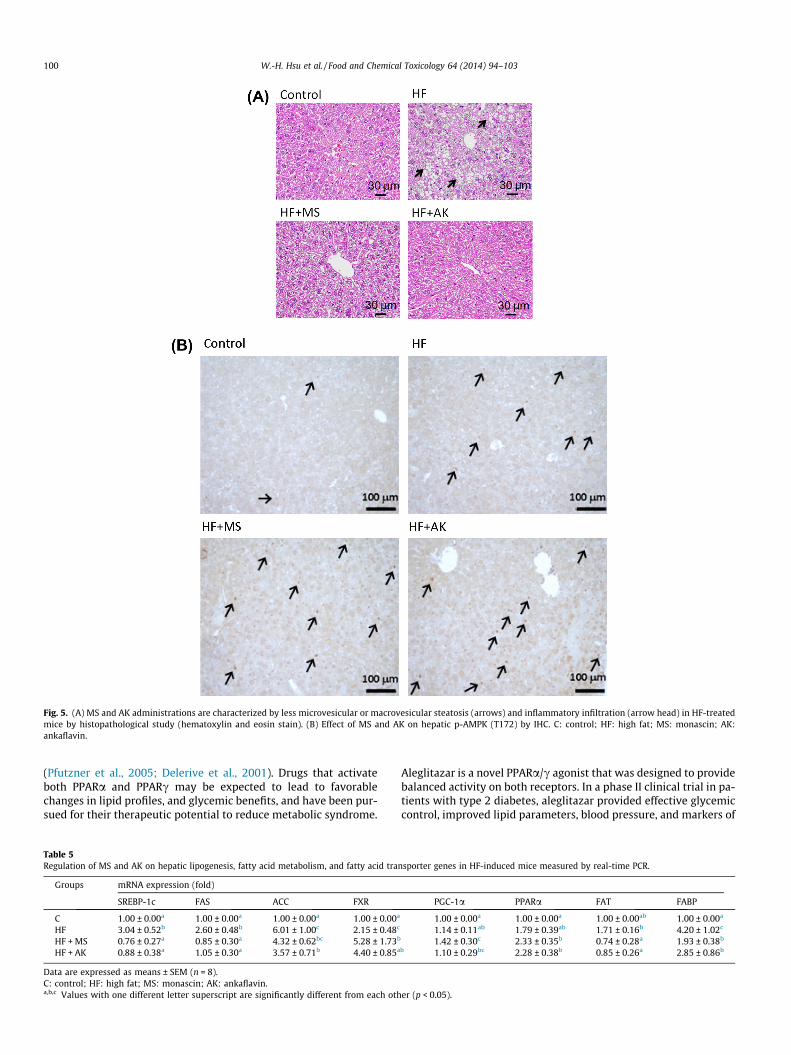
Fig. 5. (A) MS and AK administrations are characterized by less microvesicular or macrovesicular steatosis (arrows) and inflammatory infiltration (arrow head) in HF-treatedmice by histopathological study (hematoxylin and eosin stain). (B) Effect of MS and AK on hepatic p-AMPK (T172) by IHC. C: control; HF: high fat; MS: monascin; AK:ankaflavin.
100 W.-H. Hsu et al. / Food and Chemical Toxicology 64 (2014) 94–103
(Pfutzner et al., 2005; Delerive et al., 2001). Drugs that activateboth PPARa and PPARc may be expected to lead to favorablechanges in lipid profiles, and glycemic benefits, and have been pur-sued for their therapeutic potential to reduce metabolic syndrome.
Table 5Regulation of MS and AK on hepatic lipogenesis, fatty acid metabolism, and fatty acid tra
Groups mRNA expression (fold)
SREBP-1c FAS ACC FXR
C 1.00 ± 0.00a 1.00 ± 0.00a 1.00 ± 0.00a 1.00 ± 0.00a
HF 3.04 ± 0.52b 2.60 ± 0.48b 6.01 ± 1.00c 2.15 ± 0.48c
HF + MS 0.76 ± 0.27a 0.85 ± 0.30a 4.32 ± 0.62bc 5.28 ± 1.73b
HF + AK 0.88 ± 0.38a 1.05 ± 0.30a 3.57 ± 0.71b 4.40 ± 0.85a
Data are expressed as means ± SEM (n = 8).C: control; HF: high fat; MS: monascin; AK: ankaflavin.a,b,c Values with one different letter superscript are significantly different from each oth
Aleglitazar is a novel PPARa/c agonist that was designed to providebalanced activity on both receptors. In a phase II clinical trial in pa-tients with type 2 diabetes, aleglitazar provided effective glycemiccontrol, improved lipid parameters, blood pressure, and markers of
nsporter genes in HF-induced mice measured by real-time PCR.
PGC-1a PPARa FAT FABP
1.00 ± 0.00a 1.00 ± 0.00a 1.00 ± 0.00ab 1.00 ± 0.00a
1.14 ± 0.11ab 1.79 ± 0.39ab 1.71 ± 0.16b 4.20 ± 1.02c
1.42 ± 0.30c 2.33 ± 0.35b 0.74 ± 0.28a 1.93 ± 0.38b
b 1.10 ± 0.29bc 2.28 ± 0.38b 0.85 ± 0.26a 2.85 ± 0.86b
er (p < 0.05).
W.-H. Hsu et al. / Food and Chemical Toxicology 64 (2014) 94–103 101
CV risk (Henry et al., 2009). Many phytochemicals are consideredto function as PPARc agonists, modulating anti-inflammatoryactivity by interfering with nuclear factor (NF)-jB signaling (Hiraiet al., 2010). Several flavonoids elevate PPARc activity to attenuateinflammation and insulin resistance (Choi et al., 2006; Chuanget al., 2010).
Our previous data suggest that MS can suppress PPARc phos-phorylation in C2C12 myotubes treated with TNF-a to inhibit pro-tein tyrosine (Tyr) phosphatase 1B (PTP1B). This may then result inglucose transporter (GLUT) translocation to the plasma membrane,subsequently promoting glucose uptake and improving insulinsensitivity. These effects were mediated by MS acting as a PPARcagonist (Lee et al., 2012a). AK has also been found to act as a PPARcagonist to improve diabetes in methylglyoxal-treated rats (Leeet al., 2012b).
MS and AK have been shown to elevate serum HDL-C concen-tration and lower hyperlipidemia in hamsters receiving a high-cholesterol diet (Lee et al., 2010), and these effects markedlyimproved atherosclerosis in high-cholesterol diet-induced hamster(Lee et al., 2013b); however, the mechanism of these inhibitory ef-fects of MS and AK on serum TC and TG of high-cholesterol diet-in-duced hamster for atherosclerosis prevention was not clear in ourrecent studies (Lee et al., 2010, 2013b). Albeit MS and AK have beenfound to inhibit pre-adipocyte differentiation and suppress lipo-genesis thereby exerting anti-obesity activity in high-fat diet-in-duced hamster, however, these results did not support theevidence of MS and AK for regulating lipid metabolism (Lee et al.,2013c). In addition, we have reported that MS and AK resulted inPPARc activation in differentiated 3T3-L1 adipocytes and adiposetissue of mice receiving a high-fat diet hence improving dyslipide-mia and insulin sensitivity (Hsu et al., 2012a). But activation ofPPARc in differentiated adipocytes toward to anti-obesity effect
Fig. 6. MS and AK suppressed the production of inflammatory cytokines, including (A) IL-different letter superscript are significantly different from each other (p < 0.05). C: contr
of MS and AK is adverse (Lee et al., 2013c), because PPARc activitywould result in lipids accumulation in differentiated adipocytes.Therefore, we presumed that MS and AK may upregulate lipidmetabolism in liver against high-fat diet-induced obese. In orderto confirm the inhibitions of obesity and nonalcoholic steatohepa-titis by MS and AK, we design in vitro and in vivo experiments toinvestigate the potential mechanism of MS and AK on lipid metab-olism in the present study.
We have found that MS and AK both suppressed lipid accumu-lation in FL83B hepatocytes in model 1 and model 2 (Fig. 2).Although MS and AK have also been found to be effective in fattyliver by increasing PPARc activation, insufficient information isavailable regarding the mechanism related to AMPK signalingpathway and regulation of lipid disorders. We found that MS andAK activated AMPK in OA-treated FL83B hepatocytes (Fig. 4) andin the liver of mice receiving a high-fat diet (Fig. 5B). A large bodyof evidence indicates that AMPK is involved in the maintenance oflipid and cholesterol homeostasis (Pan et al., 2012). Activation ofhepatic AMPK (through phosphorylation of its a-subunit onThr172) acutely switches off fatty acid synthesis via increasedphosphorylation and inactivation of ACC, which reduces the pro-duction of malonyl-CoA, an allosteric inhibitor of CPT-1 (Sahaand Ruderman, 2003). CPT-1-mediated regulation of acyl-CoA in-flow and beta-oxidation in the mitochondrial outer membrane isa rate-limiting step for fatty acid oxidation (Saha and Ruderman,2003). SREBP-1c is a key lipogenic transcription factor, which di-rectly activates the expression of more than 30 genes (includingFAS), dedicated to the synthesis and uptake of fatty acids, choles-terol, and triglyceride (Ikarashi et al., 2011). Increased SREBP-1clevels have been found in patients with histologically diagnosedNAFLD (Yokozawa et al., 2012). It is known that SREBP-1 is nega-tively regulated by AMPK via inhibition of SREBP-1c translocation
6, (B) TNF-a, and (C) TGF-b. The values are means ± SEM (n = 8). a,b,c Values with oneol; HF: high fat; MS: monascin; AK: ankaflavin.

Fig. 7. The potential mechanisms of MS and AK protect NAFLD by down-regulatinglipogenesis.
102 W.-H. Hsu et al. / Food and Chemical Toxicology 64 (2014) 94–103
into nuclei (Li et al., 2011). We found that MS and AK could activateAMPK and suppress SREBP-1c and FAS expressions in hepatocytesin model-1 and model-2. These data implied that MS- and AK-mediated activation of AMPK might prevent SREBP-1c transloca-tion to the nucleus and downregulate SREBP-1c, FAS, and ACCexpression, leading to inhibition of hepatic lipogenesis in vitroand in vivo (Tables 1 and 5). FXR, PPARa, and PGC-1a are importanttranscription factors in fatty acid oxidation. Both MS and AK in-creased FXR, PPARa, and PGC-1a levels in model-2 in vitro, causingan increase in CPT-1, ACOX, and ACS levels and resulting in fattyacid beta-oxidation. Elevation of FXR, PPARa, and PGC-1a levelsalso increased CPT-1, ACOX, and ACS levels in the liver of micereceiving a high-fat diet (Tables 2 and 5). Moreover, expressionsof FAT and FABP were attenuated by MS and AK administration,leading to low availability of fatty acid for biosynthesis and pre-venting fatty acid accumulation in vitro and in vivo (Tables 3 and 5).
The degree of hypercholesterolemia due to TC is directly pro-portional to the severity of obesity. High TG and TC concentrationsin the blood tend to decrease concentrations of HDL-C, contribut-ing to a condition called dyslipidemia or hypertriglyceridemia(Abbate and Brunzell, 1990). In this study, we showed that admin-istration of MS and AK significantly reduced serum TC and TG con-centrations and increased HDL-C concentration (Table 4). As wellas AMPK activity, a study has reported that PPARa activation isable to enhance peroxisomal fatty acid beta-oxidation, therebylowering lipid levels, including TC and TG (Kondo et al., 2010).We found that both MS and AK were PPARa agonists. As shownin Fig. 3, the IC50 values of MS and AK for PPARa agonist activitywere 2038 nM and 4845 nM, respectively. In addition, the IC50 val-ues of WY14643 and fenofibrate (PPARa agonists) were 594 nMand 332 nM, respectively. PPARa agonists such as fenofibrate andPPARc agonists such as pioglitazone activate AMPK to stimulatethe pathways that increase energy production, such as glucosetransport and fatty acid oxidation, switching off the pathways thatconsume energy, such as lipogenesis (Chen et al., 2012; Lage et al.,2008). Taken together, these findings indicated that MS and AKmight have protective effects against dyslipidemia mediated byPPARa agonist activity and AMPK up-regulation (Fig. 7).
Tesaglitazar (PPARc agonists) was suspected of causing irre-versible impairment of renal function (Bays et al., 2007; Ratneret al., 2007), while concerns about muraglitazar (PPARc agonists)were related to increased cardiovascular risk (Nissen et al., 2005).Selective recruitment of co-activators or co-repressors may
provide additional mechanisms whereby different PPAR-targeteddrugs can differentially activate target pathways. Since emergingevidence suggests that each PPAR drug has a unique efficacy andsafety profile, the molecular effects of co-administering two differ-ent dual PPARa and PPARc agonists (aleglitazar and tesaglitazar)and a combination of a PPARa and PPARc agonist (pioglitazoneplus fenofibrate) have been investigated (Deehan et al., 2012). An-other term used to describe PPARa/c agonists is selective PPARmodulators (SPPARMs), which can be divided into two groups:PPARa/c dual partial agonists and PPARc modulators. PPARa/cagonists have lower affinity for both PPAR isoforms than the spe-cific agonists, and exert beneficial effects on serum lipid concentra-tions and insulin sensitivity and may have fewer side effects (Balintand Nagy, 2006). We have found that MS and AK act PPARa andPPARc agonist and exert SPPARMs activity, revealing that theyare safer than other PPARa or PPARc agonists. Although the IC50
of PPARa activation by MS or AK was lower than that ofWY14643 and fenofibrate, but MS and AK may exert fewer side ef-fects by acting as PPARc agonists (Hsu et al., 2012a; Lee et al.,2012a and b). These SPPARMs (MS and AK) exert hypoglycemicand hypolipidemic abilities, and are safer than synthetic thiazolid-inediones (TZDs). In addition, the liver or kidney impairments werenot found by serum biochemical values and histological evaluationdescription in our recent studies (Hsu et al., 2012a,b; Lee et al.,2013a,b,c).
We have found that MS and AK can suppress adhesion mole-cules expression induced by TNF-a in human umbilical vein endo-thelial cells (HUVECs) thereby preventing atherogenesis (Hsu et al.,2012c). In Caenorhabditis elegans model, MS regulate the FOXO/DAF-16-dependent insulin signaling pathway and induction ofstress response/anti-oxidant genes to enhance oxidative stressresistance (Shi et al., 2012). The anti-oxidation of AK mediatedby activating nuclear factor erythroid 2-related factor 2 (Nrf2) toattenuate airway inflammation was also investigated in our recentstudy (Hsu et al., 2012b).
In conclusion, similar to other PPARa and PPARc agonists, MSand AK have potential anti-inflammatory, anti-diabetic, and anti-obesity effects. In this study, we hypothesized that MS and AKmay exert fewer side effects by acting as SPPARMs. However, theirbenefits still need to be investigated further in clinical studies. Inthe future, we anticipate that MS and AK may be supplied in foodsupplements or developed as a ‘‘functional foods’’ to reduce therisk of diabetes and obesity.
Conflict of Interest
The authors declared no conflicts of interest with respect to theauthorship and/or the publication of this article.
Appendix A. Supplementary material
Supplementary data associated with this article can be found, inthe online version, at http://dx.doi.org/10.1016/j.fct.2013.11.015.
References
Abbate, S.L., Brunzell, J.D., 1990. Pathophysiology of hyperlipidemia in diabetesmellitus. J. Cardiovasc. Pharmacol. 9, S1–S7.
Adiels, M., Taskinen, M.R., Packard, C., Caslake, M.J., Soro-Paavonen, A., 2006.Overproduction of large VLDL particles is driven by increased liver fat content inman. Diabetologia 49, 755–765.
Balint, B.L., Nagy, L., 2006. Selective modulators of PPAR activity as new therapeutictools in metabolic diseases. Endocr. Metab. Immune Disord. Drug Targets 6, 33–43.
Bays, H., McElhattan, J., Bryzinski, B.S., 2007. A double-blind, randomised trial oftesaglitazar versus pioglitazone in patients with type 2 diabetes mellitus. Diab.Vasc. Dis. Res. 4, 181–193.

W.-H. Hsu et al. / Food and Chemical Toxicology 64 (2014) 94–103 103
Chen, W.L., Chen, Y.L., Chiang, Y.M., Wang, S.G., Lee, H.M., 2012. Fenofibrate lowerslipid accumulation in myotubes by modulating the PPARa/AMPK/FoxO1/ATGLpathway. Biochem. Pharmacol. 84, 522–531.
Cheng, C.F., Pan, T.M., 2011. Protective effect of Monascus-fermented red mold riceagainst alcoholic liver disease by attenuating oxidative stress and inflammatoryresponse. J. Agric. Food Chem. 59, 9950–9957.
Choi, I., Park, Y., Choi, H., Lee, E.H., 2006. Anti-adipogenic activity of rutin in 3T3-L1cells and mice fed with high-fat diet. BioFactors 26, 273–281.
Chuang, C.C., Martinez, K., Xie, G., Kennedy, A., Bumrungpert, A., Overman, A., Jia,W., Mclntosh, M.K., 2010. Quercetin is equally or more effective than resveratrolin attenuating tumor necrosis factor-a-mediated inflammation and insulinresistance in primary human adipocytes. J. Am. Clin. Nutr. 92, 1511–1521.
Deehan, R., Maerz-Weiss, P., Catlett, N.L., Steiner, G., Wong, B., Wright, M.B., Blander,G., Elliston, K.O., Ladd, W., Bobadilla, M., Mizrahi, J., Haefliger, C., Edgar, A., 2012.Comparative transcriptional network modeling of three PPARa/c co-agonistsreveals distinct metabolic gene signatures in primary human hepatocytes. PLoSONE 7, e35012.
Delerive, P., Fruchart, J.C., Staels, B., 2001. Peroxisome proliferator-activatedreceptors in inflammation control. J. Endocrinol. 169, 453–459.
Hawley, S.A., Davison, M., Woods, A., Davies, S.P., Beri, R.K., Carling, D., Hardie, D.G.,1996. Characterization of the AMP-activated protein kinase kinase from rat liverand identification of threonine 172 as the major site at which it phosphorylatesAMP-activated protein kinase. J. Biol. Chem. 271, 27879–27887.
Henry, R.R., Lincoff, A.M., Mudaliar, S., Rabbia, M., Chognot, C., Herz, M., 2009. Effectof the dual peroxisome proliferator-activated receptor-alpha/gamma agonistaleglitazar on risk of cardiovascular disease in patients with type 2 diabetes(SYNCHRONY): a phase II, randomised, dose-ranging study. Lancet 374, 126–135.
Hirai, S., Takahashi, N., Goto, T., Lin, S., Uemura, T., Yu, R., Kawada, T., 2010.Functional food targeting the regulation of obesity-induced inflammatoryresponses and pathologies. Mediat. Inflamm. http://dx.doi.org/10.1155/2010/367838.
Hsu, W.H., Lee, B.H., Pan, T.M., 2010a. Protection of Monascus-fermented dioscoreaagainst DMBA-induced oral injury in hamster by anti-inflammatory andantioxidative potentials. J. Agric. Food Chem. 58, 6715–6720.
Hsu, Y.W., Hsu, L.C., Liang, Y.H., Kuo, Y.H., Pan, T.M., 2010b. Monaphilones A–C,three new antiproliferative azaphilone derivatives from Monascus purpureusNTU 568. J. Agric. Food Chem. 58, 8211–8216.
Hsu, W.H., Liao, T.H., Lee, B.H., Hsu, Y.W., Pan, T.M., 2012a. Ankaflavin regulatesadipocyte function and attenuates hyperglycemia caused by high-fat diet viaPPAR-gamma activation. J. Funct. Foods 5, 124–132.
Hsu, W.H., Lee, B.H., Huang, Y.C., Hsu, Y.W., Pan, T.M., 2012b. Ankaflavin, a novelNrf-2 activator for attenuating allergic airway inflammation. Free Radic. Biol.Med. 53, 1643–1651.
Hsu, W.H., Lee, B.H., Lu, I.J., Pan, T.M., 2012c. Ankaflavin and monascin regulateendothelial adhesion molecules and endothelial NO synthase (eNOS) expressioninduced by tumor necrosis factor-a (TNF-a) in human umbilical veinendothelial cells (HUVECs). J. Agric. Food Chem. 60, 1666–1672.
Ikarashi, N., Toda, T., Okaniwa, T., Ito, K., Ochiai, W., Sugiyama, K., 2011. Anti-obesityand anti-diabetic effects of Acacia polyphenol in obese diabetic KKAy mice fedhigh-fat diet. Evid. Based Complement. Alternat. Med. http://dx.doi.org/10.1093/ecam/nep241.
Jou, P.C., Ho, B.Y., Hsu, Y.W., Pan, T.M., 2010. The effect of Monascus secondarypolyketide metabolites, monascin and ankaflavin, on adipogenesis and lipolysisactivity in 3T3-L1. J. Agric. Food Chem. 58, 12703–12709.
Kondo, K., Sugioka, T., Tsukada, K., Aizawa, M., Takizawa, M., Shimizu, K., Morimoto,M., Suematsu, M., Goda, N., 2010. Fenofibrate, a peroxisome proliferator-activated receptor alpha agonist, improves hepatic microcirculatory patencyand oxygen availability in a high-fat-diet-induced fatty liver in mice. Adv. Exp.Med. Biol. 662, 77–82.
Kusunoki, M., Tsutsumi, K., Iwata, K., Yin, W., Nakamura, T., 2005. NO-1886(ibrolipim), a lipoprotein lipase activator, increases the expression ofuncoupling protein 3 in skeletal muscle and suppresses fat accumulation inhigh-fat diet-induced obesity in rats. Metabolism 54, 1587–1592.
Lage, R., Dieguez, C., Vidal-Puig, A., Lopez, M., 2008. AMPK: a metabolic gaugeregulating whole-body energy homeostasis. Trends Mol. Med. 14, 539–549.
Leclerc, I., Rutter, G.A., 2004. AMP-activated protein kinase: a new beta-cell glucosesensor? Regulation by amino acids and calcium ions. Diabetes 53, S67–S74.
Lee, C.L., Kung, Y.H., Wu, C.L., Hsu, Y.W., Pan, T.M., 2010. Monascin and ankaflavinact as novel hypolipidemic and high-density lipoprotein cholesterol-raisingagents in red mold dioscorea. J. Agric. Food Chem. 58, 9013–9019.
Lee, B.H., Hsu, W.H., Liao, T.H., Pan, T.M., 2012a. The Monascus metabolite monascinagainst TNF-a-induced insulin resistance via suppressing PPARcphosphorylation in C2C12 myotubes. Food Chem. Toxicol. 49, 2609–2617.
Lee, B.H., Hsu, W.H., Chang, Y.Y., Kuo, H.F., Hsu, Y.W., Pan, T.M., 2012b. Ankaflavin: anatural novel PPARc agonist upregulates Nrf2 to attenuate methylglyoxal-induced diabetes in vivo. Free Radic. Biol. Med. 53, 2008–2016.
Lee, B.H., Hsu, W.H., Huang, Tao., Chang, Y.Y., Hsu, Y.W., Pan, T.M., 2013a. Effects ofmonascin on anti-inflammation mediated by Nrf2 activation in advancedglycation end product-treated THP-1 monocytes and methylglyoxal-treatedWistar rats. J. Agric. Food Chem. 61, 1288–1298.
Lee, C.L., Hung, Y.P., Hsu, Y.P., Pan, T.M., 2013b. Monascin and ankaflavin have moreanti-atherosclerosis effect and less side effect involving increasing creatininephosphokinase activity than monacolin K under the same dosages. J. Agric. FoodChem. 61, 143–150.
Lee, C.L., Wen, J.Y., Hsu, Y.W., Pan, T.M., 2013c. Monascus-fermented yellowpigments monascin and ankaflavin showed antiobesity effect via thesuppression of differentiation and lipogenesis in obese rats fed a high-fat diet.J. Agric. Food Chem. 61, 1493–1500.
Li, Y., Xu, S., Mihaylova, M.M., Zheng, B., Hou, X., Jiang, B., Park, O., Luo, Z., Lefai, E.,Shyy, J.Y., Gao, B., Wierzbicki, M., Verbeuren, T.J., Shaw, R.J., Cohen, R.A., Zang,M., 2011. AMPK phosphorylates and inhibits SREBP activity to attenuate hepaticsteatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab.13, 376–388.
Matteoni, C.A., Younossi, Z.M., Gramlich, T., Boparai, N., Liu, Y.C., 1999. Nonalcoholicfatty liver disease: a spectrum of clinical and pathological severity.Gastroenterology 116, 1413–1419.
Nissen, S.E., Wolski, K., Topol, E.J., 2005. Effect of muraglitazar on death and majoradverse cardiovascular events in patients with type 2 diabetes mellitus. JAMA294, 2581–2586.
Ota, T., Takamura, T., Kurita, S., Matsuzawa, N., Kita, Y., Uno, M., Akahori, H., Misu,H., Sakurai, M., Zen, Y., Nakanuma, Y., Kaneko, S., 2007. Insulin resistanceaccelerates a dietary rat model of nonalcoholic steatohepatitis.Gastroenterology 132, 282–293.
Pan, C.H., Tsai, C.H., Lin, W.H., Chen, G.Y., Wu, C.H., 2012. Ethanolic extract of Vitisthunbergii exhibits lipid lowering properties via modulation of the AMPK-ACCpathway in hypercholesterolemic rabbits. Evid. Based Complement. Alternat.Med. http://dx.doi.org/10.1155/2012/436786.
Pfutzner, A., Marx, N., Lubben, G., Langenfeld, M., Walcher, D., 2005. Improvement ofcardiovascular risk markers by pioglitazone is independent from glycemiccontrol: results from the pioneer study. J. Am. Coll. Cardiol. 45, 1925–1931.
Ratner, R.E., Parikh, S., Tou, C., 2007. Efficacy, safety and tolerability of tesaglitazarwhen added to the therapeutic regimen of poorly controlled insulin treatedpatients with type 2 diabetes. Diab. Vasc. Dis. Res. 4, 214–221.
Saha, A.K., Ruderman, N.B., 2003. Malonyl-CoA and AMP activated protein kinase:and expanding partnership. Mol. Cell. Biochem. 253, 65–70.
Salt, W.B., 2004. Nonalcoholic fatty liver disease (NAFLD): a comprehensive review.J. Insur. Med. 36, 27–41.
Sanyal, A.J., 2002. AGA technical review on nonalcoholic fatty liver disease.Gastroenterology 123, 1705–1725.
Shaw, R.J., Lamia, K.A., Vasquez, D., Koo, S.H., Bardeesy, N., Depinho, A., Montminy,M., Cantley, L.C., 2005. The kinase LKB1 mediates glucose homeostasis in liverand therapeutic effects of metformin. Science 310, 1642–1646.
Shi, Y.C., Liao, H.C., Pan, T.M., 2012. Monascin from red mold dioscorea as a novelantidiabetic and antioxidative stress agent in rats and Caenorhabditis elegans.Free Radic. Biol. Med. 52, 109–117.
Shi, Y.C., Pan, T.M., 2010. Anti-diabetic effects of Monascus purpureus NTU 568fermented products on streptozotocin-induced diabetic rats. J. Agric. FoodChem. 58, 7634–7640.
Staels, B., Fruchart, J.C., 2005. Therapeutic roles of peroxisome proliferator activatedreceptor agonists. Diabetes 54, 2460–2470.
Tenenbaum, A., Motro, M., Fisman, E.Z., 2005. Dual and pan-peroxisomeproliferator-activated receptors (PPAR) co-agonism: the bezafibrate lessons.Cardiovasc. Diabetol. 4, 14.
Tilg, H., Moschen, A.R., 2010. Evolution of inflammation in nonalcoholic fatty liverdisease: the multiple parallel hits hypothesis. Hepatology 52, 1836–1846.
Tzeng, T.F., Lu, H.J., Liou, S.S., Chang, C.J., Liu, I.M., 2012. Emodin, a naturallyoccurring anthraquinone derivative, ameliorates dyslipidmia by activatingAMP-activated protein kinase in high-fat-diet fed rats. Evid. BasedComplement. Alternat. Med. http://dx.doi.org/10.1155/2012/781812.
Viollet, B., Guigas, B., Leclerc, J., Hebrard, S., Lantier, L., Mounier, R., Andreelli, F.,Foretz, M., 2009. AMP-activated protein kinase in the regulation of hepaticenergy metabolism: from physiology to therapeutic perspectives. Acta Physiol.196, 81–89.
Yokozawa, T., Cho, E.J., Park, C.H., Kim, J.H., 2012. Protective effect ofproanthocyanidin against diabetic oxidative stress. Evid. Based Complement.Alternat. Med. http://dx.doi.org/10.1155/2012/623879.