modified allele dosage (mad) genotyping by hi-res melting of whole amplicons jason t. mckinney...
TRANSCRIPT

Modified Allele Dosage Modified Allele Dosage (MAD) Genotyping by Hi-(MAD) Genotyping by Hi-Res MeltingRes Melting of Whole of Whole
AmpliconsAmplicons
Jason T. McKinneyJason T. McKinney
ScientistScientist
Human Genetics ApplicationsHuman Genetics Applications

Modified what? … Who’s Modified what? … Who’s MAD?MAD?
• MModified odified AAllele llele DDosage = Pre-PCR DNA osage = Pre-PCR DNA mixingmixing
• ““MAD”? Well, … PHENCODE=ENCODE+GenPhen, MAD”? Well, … PHENCODE=ENCODE+GenPhen, HuGENet,LSDB’s, …HuGENet,LSDB’s, …– Seems as though “MAD” is a MUSTSeems as though “MAD” is a MUST
• Genotype by Hi-Res Melting (HRM) of Genotype by Hi-Res Melting (HRM) of whole ampliconswhole amplicons
• Detection of heteroduplexesDetection of heteroduplexes– ““Core” application of Hi-Res MeltingCore” application of Hi-Res Melting
• Force heteroduplex formation in Force heteroduplex formation in homozygous samples by mixing DNA pre-homozygous samples by mixing DNA pre-PCRPCR– Typically done 1:1 volume:volumeTypically done 1:1 volume:volume

Stealing from Peter?Stealing from Peter?
• Must not compromise ability to Must not compromise ability to distinguish “true” heterozygous distinguish “true” heterozygous samplessamples
• Homozygous “heteroduplexes” = Homozygous “heteroduplexes” = – ““intermediate” quantity of intermediate” quantity of
heteroduplex molecules relative to heteroduplex molecules relative to “true” heterozygous samples and “true” heterozygous samples and wild type sampleswild type samples
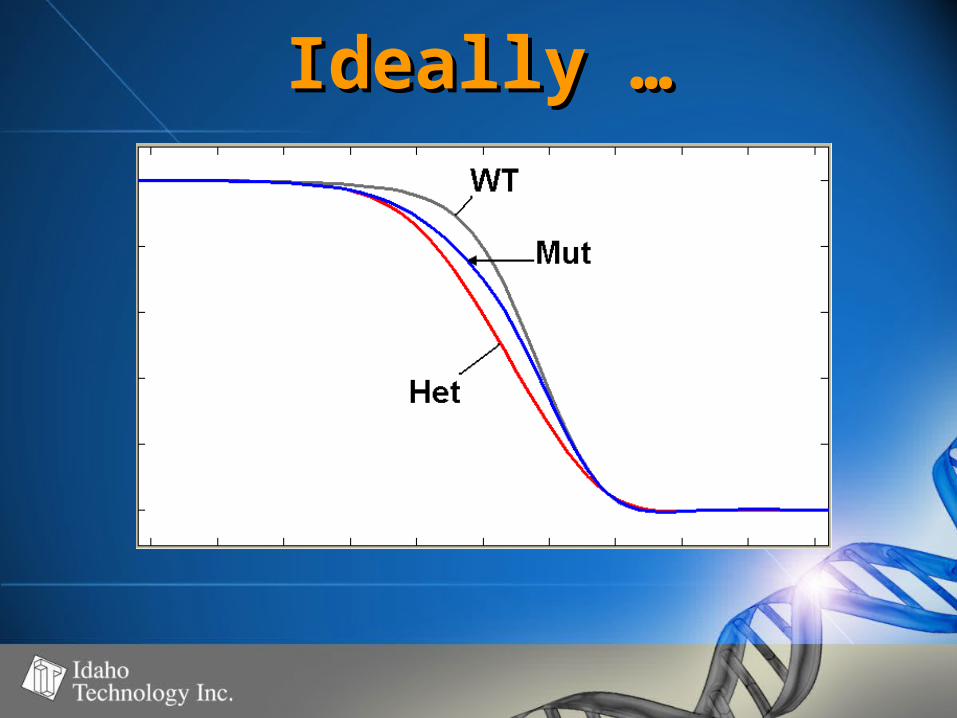
Ideally …Ideally …
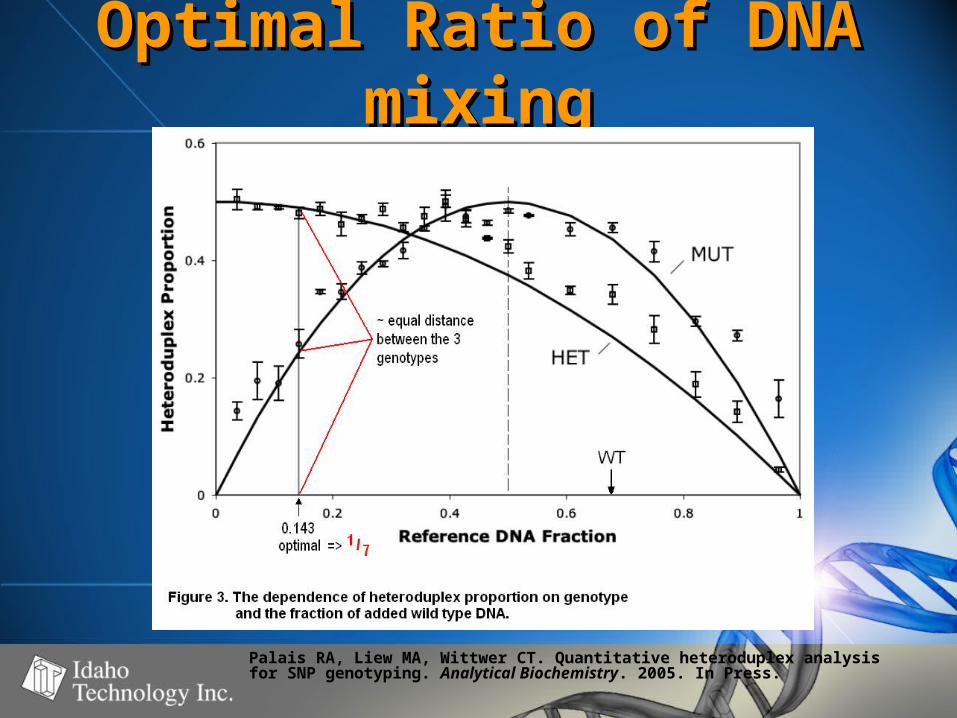
Optimal Ratio of DNA Optimal Ratio of DNA mixingmixing
Palais RA, Liew MA, Wittwer CT. Quantitative heteroduplex analysisfor SNP genotyping. Analytical Biochemistry. 2005. In Press.

Optimal ratio, … in English Optimal ratio, … in English please?please?
• 1X WT DNA + 6X Unknown DNA yields 1X WT DNA + 6X Unknown DNA yields the following heteroduplex ratio’s:the following heteroduplex ratio’s:
• Given HRM detection of ~5-10% Given HRM detection of ~5-10% variant DNA, these ratio’s should be variant DNA, these ratio’s should be easily discriminatedeasily discriminated

BenefitsBenefits• Genotype sans probe Genotype sans probe (labeled or (labeled or
unlabeled)unlabeled)– Cost and technical benefitCost and technical benefit
• No asymmetric amplificationNo asymmetric amplification– Technical benefitTechnical benefit
• Use “scanning” primers to Use “scanning” primers to genotypegenotype
Potential to observe variants Potential to observe variants other thanother than the SNP of interest the SNP of interest– Relative to a probe-based genotyping methodRelative to a probe-based genotyping method

System System RequirementsRequirements
• Sequence characterized reference Sequence characterized reference sample (wildtype or variant)sample (wildtype or variant)
• Accurately quantified DNA samplesAccurately quantified DNA samples• dsDNA “saturation” dye (LCGreen dsDNA “saturation” dye (LCGreen
PlusPlus))• High-resolution instrumentation for High-resolution instrumentation for
melting curve analysis melting curve analysis (HR-1, (HR-1, LightScanner)LightScanner)
• Well characterized region around Well characterized region around SNP is beneficialSNP is beneficial

Validation of MAD Validation of MAD genotypinggenotyping
• Risk burden of HDL haplotypes – 6 Risk burden of HDL haplotypes – 6 genes regulating HDL cholesterol genes regulating HDL cholesterol metabolismmetabolism1.1. Primer designPrimer design
a.a. Area around SNP’s interrogated in Area around SNP’s interrogated in 100 100 chromosomeschromosomes
b.b. Primers designed to avoid observed variantsPrimers designed to avoid observed variants
2.2. PCR qualityPCR qualitya.a. Primer concentration across annealing Primer concentration across annealing
temperature gradienttemperature gradient
b.b. Definitive criteria for Hi-Res Melting successDefinitive criteria for Hi-Res Melting success

MethodsMethods
• Quantify DNA samples (~10ng/Quantify DNA samples (~10ng/l)l)• Add Add referencereference DNA (wildtype) to DNA (wildtype) to
master mix at master mix at 11//77 total DNA total DNA
• Add Add unknownunknown DNA at DNA at 66//77 total total
• Amplification – 96 well thermal Amplification – 96 well thermal blockblock
• HRM on LightScannerHRM on LightScanner• Automated calling of genotypesAutomated calling of genotypes
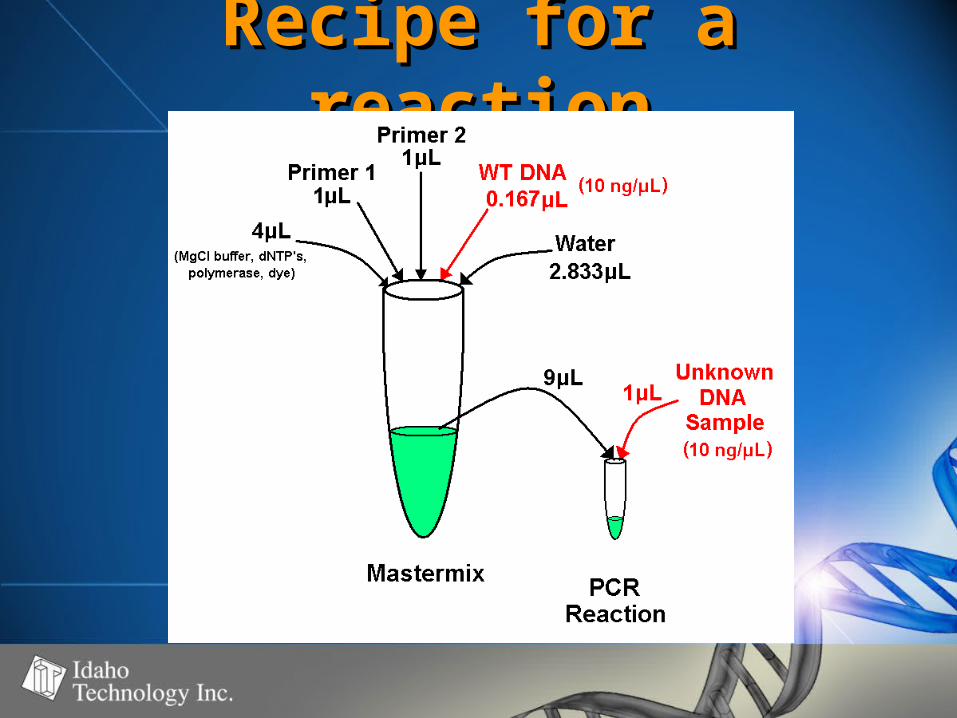
Recipe for a Recipe for a reactionreaction
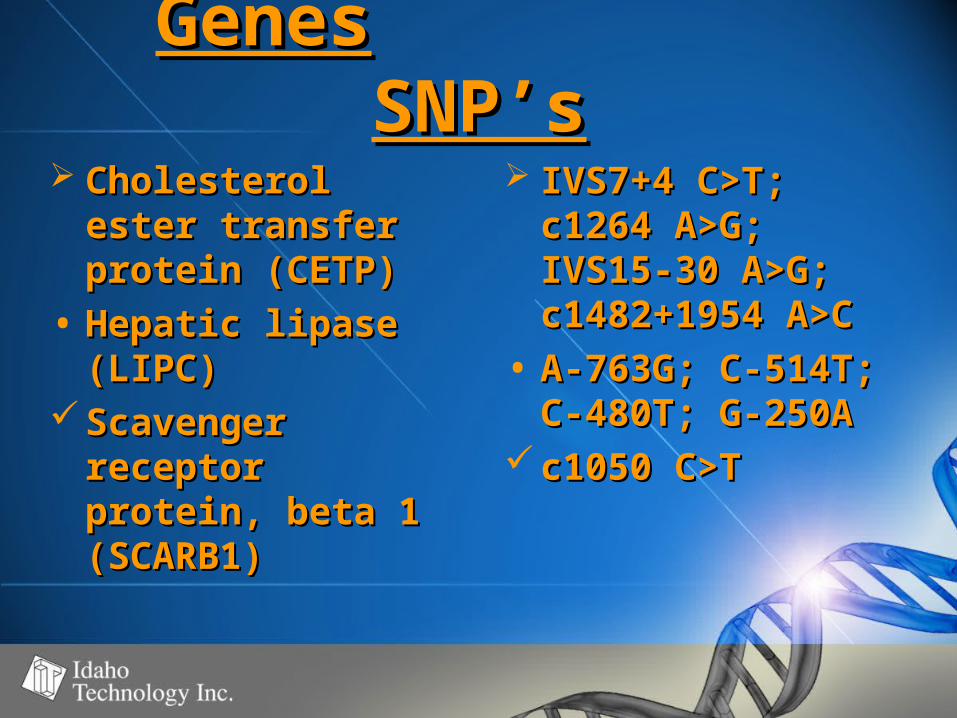
GenesGenes SNP’sSNP’s Cholesterol Cholesterol
ester transfer ester transfer protein (CETP)protein (CETP)
• Hepatic lipase Hepatic lipase (LIPC)(LIPC)
Scavenger Scavenger receptor receptor protein, beta 1 protein, beta 1 (SCARB1)(SCARB1)
IVS7+4 C>T; IVS7+4 C>T; c1264 A>G; c1264 A>G; IVS15-30 A>G; IVS15-30 A>G; c1482+1954 c1482+1954 A>CA>C
• A-763G; C-514T; A-763G; C-514T; C-480T; G-250AC-480T; G-250A
c1050 C>Tc1050 C>T

Show me the Show me the Data!Data!
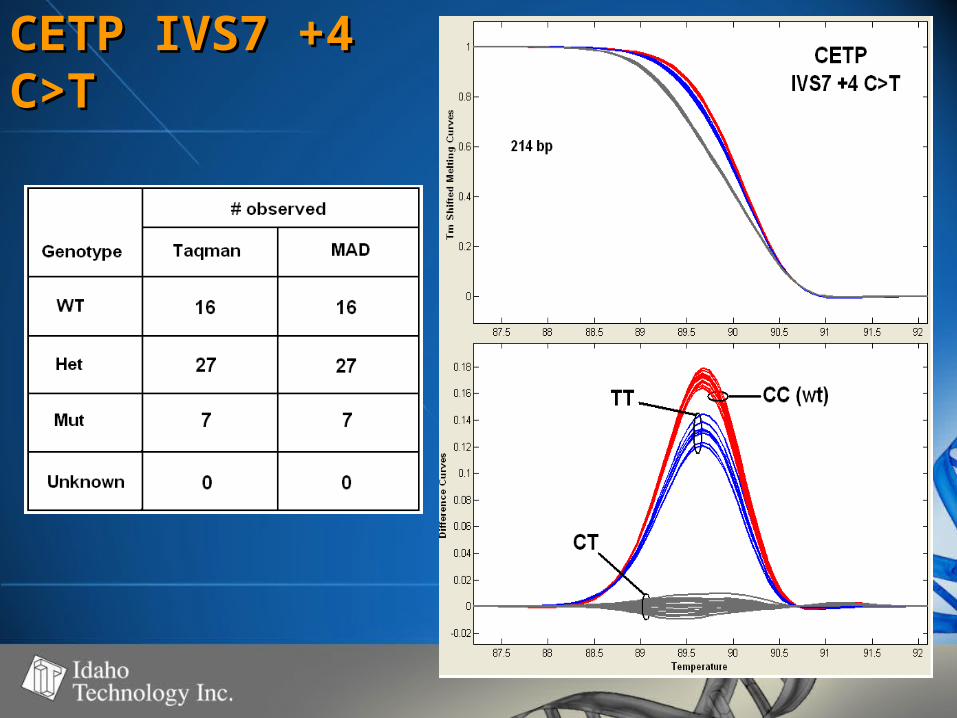
CETP IVS7 +4 CETP IVS7 +4 C>TC>T
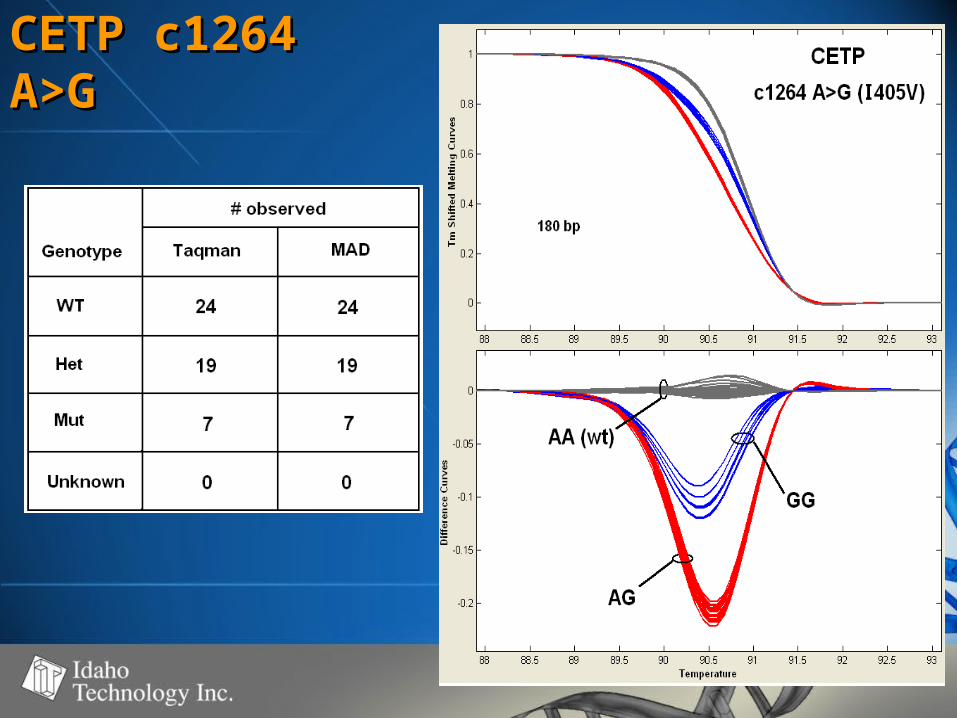
CETP c1264 CETP c1264 A>GA>G
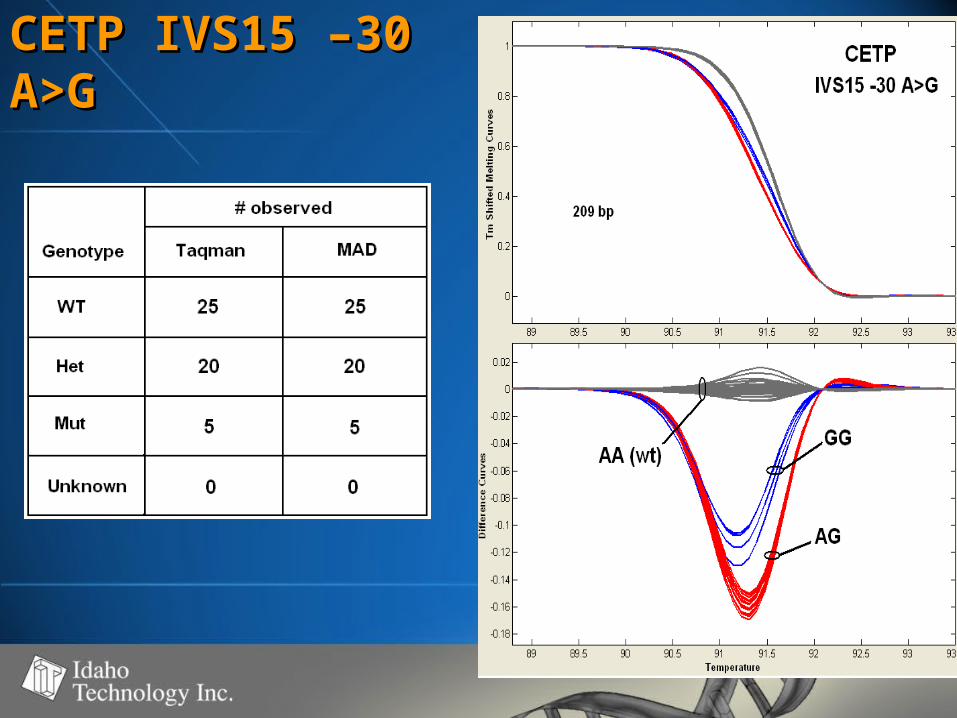
CETP IVS15 –30 CETP IVS15 –30 A>GA>G
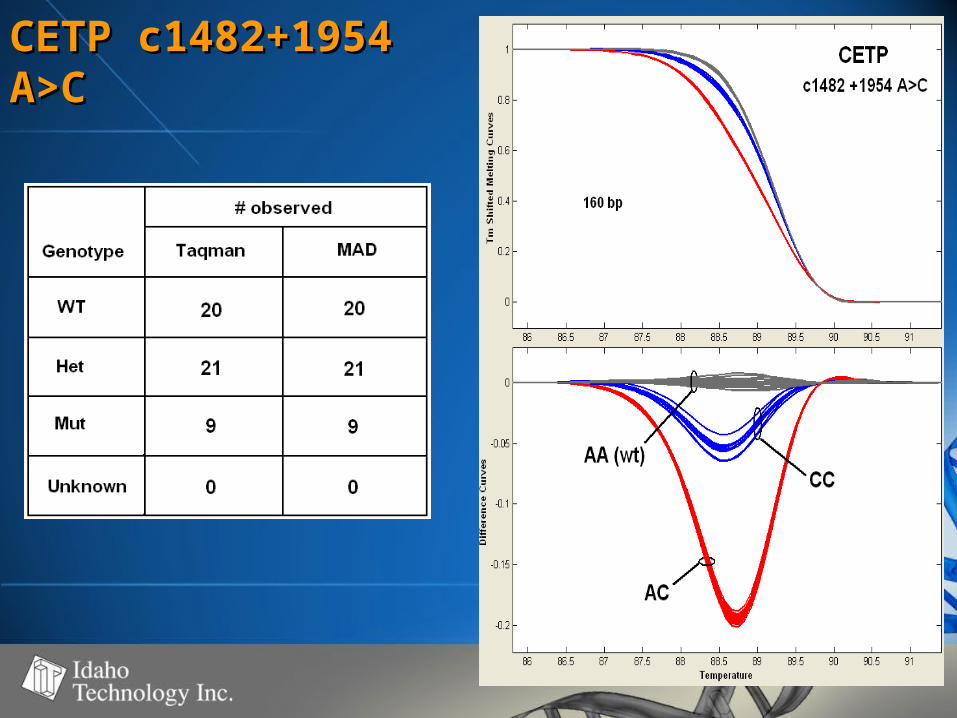
CETP c1482+1954 CETP c1482+1954 A>CA>C
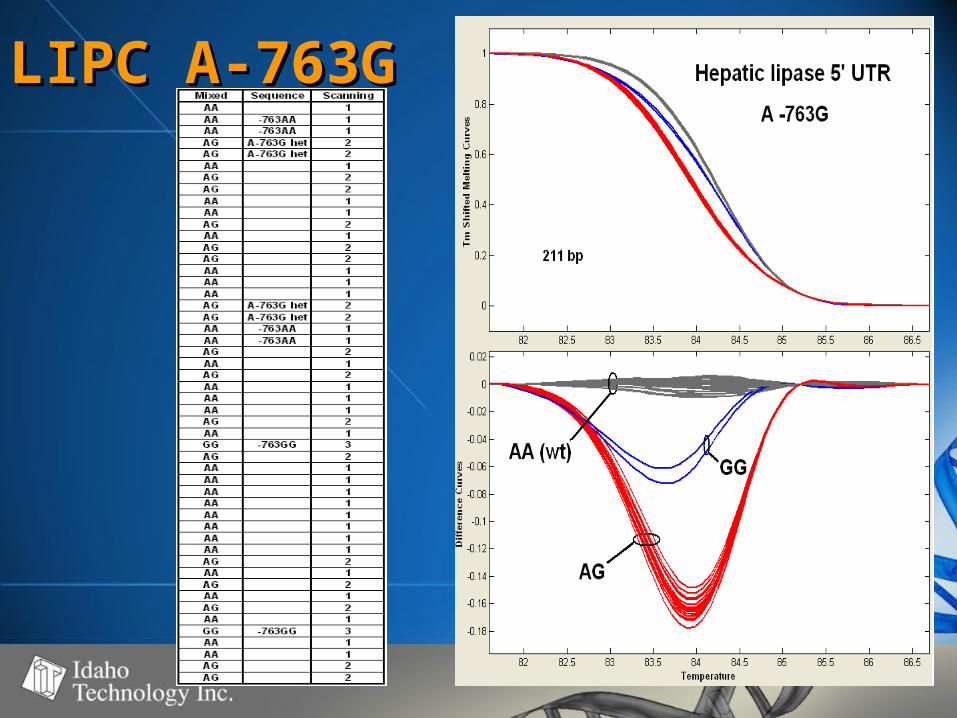
LIPC A-763GLIPC A-763G
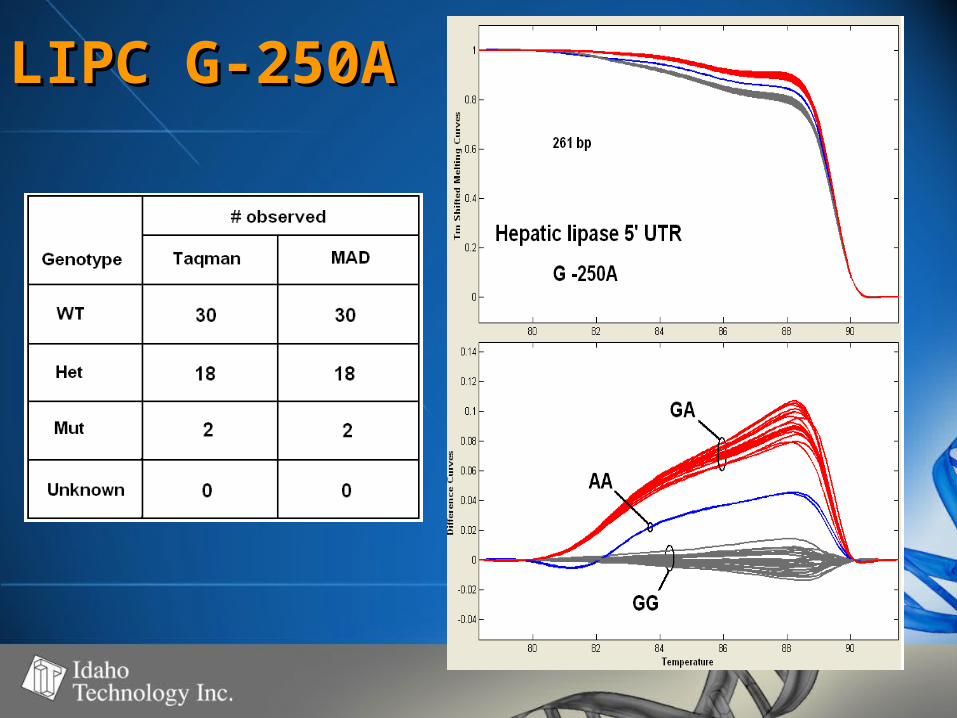
LIPC G-250ALIPC G-250A
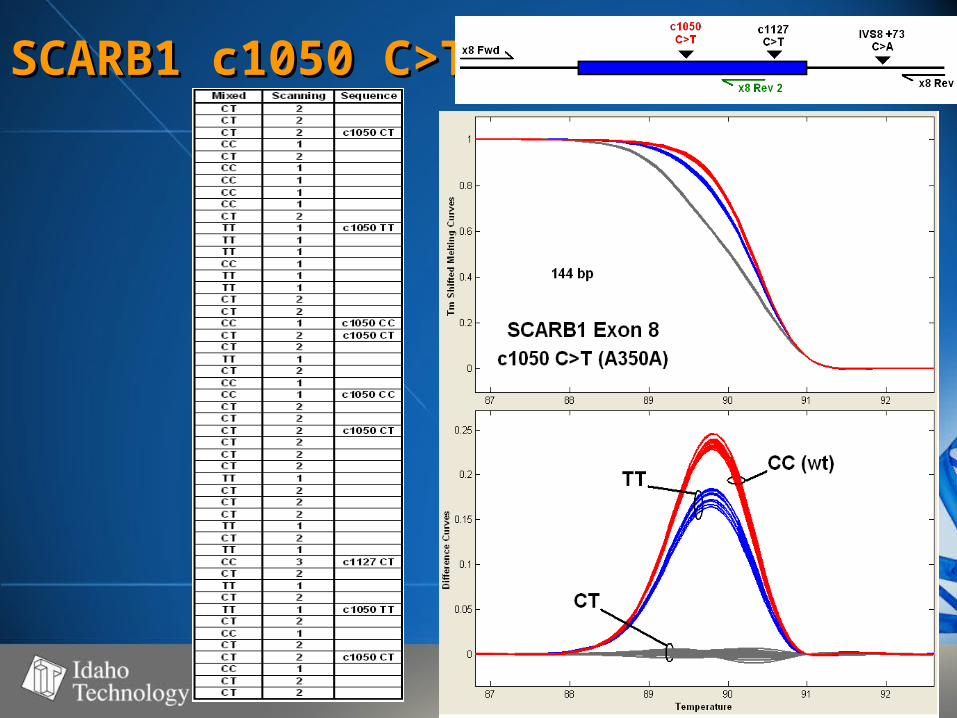
SCARB1 c1050 SCARB1 c1050 C>TC>T
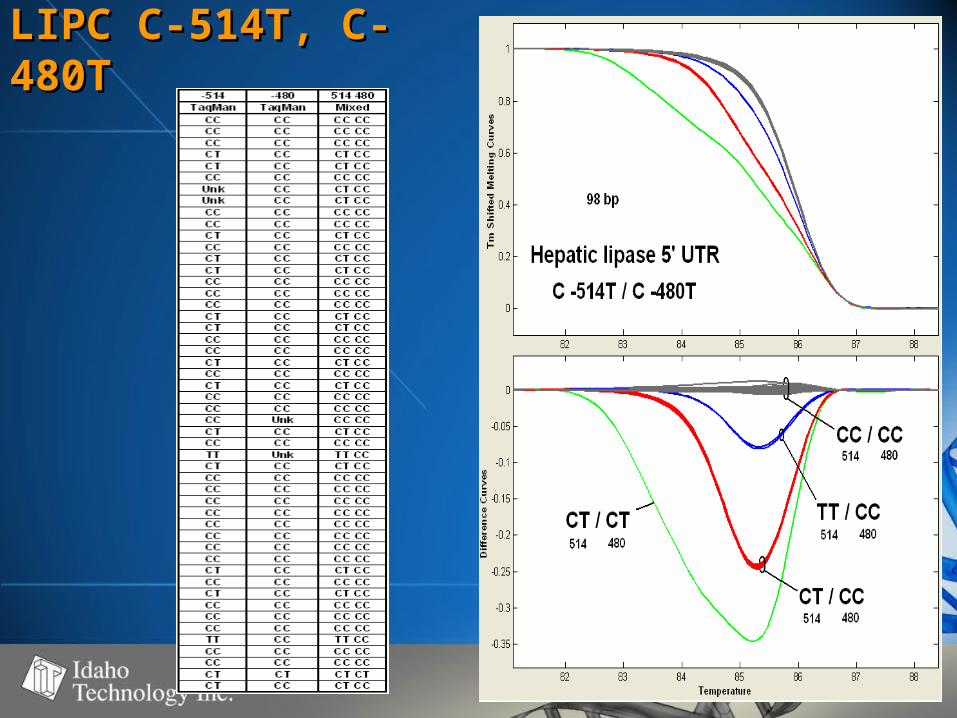
LIPC C-514T, C-LIPC C-514T, C-480T480T

Summary of DataSummary of Data• SNP’s observedSNP’s observed
– C>T (4), A>G (3), A>C (1), G>A (1)C>T (4), A>G (3), A>C (1), G>A (1)
• Fragment size range - 98-261 bpFragment size range - 98-261 bp– Strong correlation between fragment Strong correlation between fragment
size and WT – Het difference (r=0.93)size and WT – Het difference (r=0.93)– No correlation between WT – Mut No correlation between WT – Mut
difference and fragment size (r=0.12)difference and fragment size (r=0.12)
• 50-60% GC50-60% GC• 100% genotype concordance100% genotype concordance
– TaqMan TaqMan (350)(350)
– Sequencing Sequencing (68)(68)