membranoproliferative glomerulonephritis, adult - springer · membranoproliferative...
TRANSCRIPT

MembranoproliferativeGlomerulonephritis, Adult
Mariam P. Alexander and Sanjeev Sethi
AbstractMembranoproliferative glomerulonephritis(MPGN) is a pattern of glomerular injuryseen in varied disease conditions, and in itselfdoes not refer to a specific disease entity. Pre-viously it was classified according to the ultra-structural location of deposits as MPGN type I,II or III. Since then, we have moved onto aclassification based on etiology and pathogen-esis. The two broad pathogenetic pathwaysinclude either glomerular injury secondary toan immune complex/monoclonal immuno-globulin deposition or consequent to comple-ment deposition in the setting of dysregulatedabnormalities of the complement system.Appropriate classification based on immuno-fluorescence permits a detailed and tailoredevaluation for underlying infections, autoim-mune diseases, monoclonal gammopathy and/or abnormalities of the complement system.Recent use of ancillary diagnostic techniquessuch as pronase immunflourescence and massspectrometry promote accurate diagnosis inchallenging cases.
KeywordsMPGN • Immune complex • Complement • C3glomerulopathy • Monoclonal immunoglobu-lin • Infections • Autoimmune diseases
ContentsDefinition . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
Former Classification . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
Newer Classification Based on Etiology andPathogenesis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
Immune Complex/Monoclonal Ig-MediatedMPGN . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3
Infection-Associated MPGN . . . . . . . . . . . . . . . . . . . . . . . . 3
Autoimmune Disease-Associated MPGN . . . . . . . . . . 4
Monoclonal Ig-Associated MPGN . . . . . . . . . . . . . . . . . . 4Cryoglobulins and MPGN . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
Complement-Mediated MPGN (C3Glomerulopathy) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7
Pathogenesis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9MPGN with Masked Immune Deposits . . . . . . . . . . . . . . 11MPGN Without Immunoglobulins or Complement . . 11
Evaluation of MPGN . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11Immune Complex-Mediated MPGN . . . . . . . . . . . . . . . . . . 12Complement-Mediated MPGN (C3 Glomerulopathy) 12
Uncommon Causes of MPGN . . . . . . . . . . . . . . . . . . . . . . . 13
Summary . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
M.P. Alexander • S. Sethi (*)Department of Laboratory Medicine and Pathology, MayoClinic, Rochester, MN, USAe-mail: [email protected]
# Springer International Publishing AG 2017H. Trachtman et al. (eds.), Glomerulonephritis,DOI 10.1007/978-3-319-27334-1_22-1
1

Definition
Membranoproliferative glomerulonephritis(MPGN) is a pattern of glomerular injury seen invaried disease conditions (Sethi and Fervenza2012). The name in itself does not refer to aspecific disease entity. The characteristic mor-phology includes a proliferative componentrepresented by mesangial and endocapillaryhypercellularity and resolving membrano-compo-nent represented by thickened glomerular base-ment membranes. These changes impart alobular appearance of the glomerular capillarytufts. Generally speaking, MPGN reflects anactive-chronic disease process with both an activecomponent represented by the hypercellularityand the chronic component represented by thedouble-contour formation. Other names forMPGN include lobular glomerulonephritis andmesangiocapillary glomerulonephritis.
Former Classification
Historically MPGN has been classified intoMPGN type I, MPGN type II, and MPGN typeIII based on electron microscopy. It should bepointed out at the outset that this classification isnot based on pathophysiology but rather on ultra-structural characteristics and location of the elec-tron-dense deposits.
MPGN type I, the most common type, is char-acterized histologically bymesangial proliferationwith cellular interposition and double contoursresulting in the typical MPGN pattern of injury.Immunofluorescence microscopy reveals immu-noglobulins (Ig) and C3 or predominantly C3deposits. Electron microscopy (EM) showsmesangial and subendothelial deposits. MPGNtype II is characterized by an MPGN pattern ofinjury, bright staining for C3 on IF, and mesangialand intramembranous highly electron-densedeposits on electron microscopy. MPGN II isalso called dense deposit disease (DDD). MPGNtype III is characterized histologically by anMPGN pattern of injury. Immunofluorescencemicroscopy reveals Ig and C3 or predominantlyC3 deposits. Electron microscopy of MPGN type
III is characterized by subendothelial deposits andintramembranous and subepithelial electron-dense deposits. It is further classified into twotypes: the Burkholder and Anders/Strife type.The Burkholder type (Burkholder et al. 1970) ischaracterized by many of the features of MPGNtype I but with the additional presence of numer-ous subepithelial electron-dense deposits. TheAnders/Strife type of MPGN type III (Anders etal. 1977) has large variably denseintramembranous electron-dense deposits whichconnect subepithelial and intramembranousdeposits.
The disadvantage of this classification is itslack of clinical significance. This classificationbears no relation to etiopathogenesis and thereforedoes not guide therapy. A clearer understanding ofetiology has evolved since the original classifica-tion, particularly in relation to the role of comple-ment pathways involved in glomerulonephritis.This has led to a more practical approach andreclassification of MPGN termed the “Mayo clas-sification” of MPGN (Sethi and Fervenza 2011,2012).
Newer Classification Based on Etiologyand Pathogenesis
There are largely two broad pathogenetic path-ways in the evolution of MPGN based on thenew “Mayo Clinic” classification of MPGN(Sethi and Fervenza 2011, 2012):
(a) Immune complex or monoclonal Ig deposi-tion in the glomeruli with or without comple-ment deposition
(b) Complement deposition subsequent todysregulated abnormalities of the comple-ment system
A third pathogenic pathway which leads tocapillary wall remodeling and thickened glomer-ular basement membranes in the absence ofimmune complex or complement deposition isseen in the setting of chronic endothelial injuryor chronic thrombotic microangiopathy. Otheruncommon causes such as cryofibrinogen
2 M.P. Alexander and S. Sethi

glomerulopathy, lipoprotein glomerulopathy, etc.that result in endothelial injury and deposition ofincreased or abnormal proteins along the capillarywalls can also result in anMPGN pattern of injury.
Immune Complex/Monoclonal Ig-Mediated MPGN
TheMPGN pattern of injury is likely secondary to(1) persistent antigenemia and circulating immunecomplexes or (2) monoclonal Ig. Typically, in ourexperience immune complex/monoclonal Ig-mediated MPGN results in the setting of infec-tions and autoimmune disease, while monoclonalIg-mediatedMPGN results in the setting of mono-clonal gammopathy (although in some cases MIgis not detected on routing SPEP/IFE studies). Theimmune complexes and monoclonal Ig deposit inthe mesangial and subendothelial areas of theglomerulus. There is subsequent complementactivation, which accounts for the hypo-complementemia. A proliferative glomerulone-phritis is noted in the acute phase with influx ofneutrophils initially, followed by mononuclearinflammatory cells. In the reparative phase, theinjured mesangial cells and endothelium laydown new basement membrane, and themesangial matrix expands. The complexremodeling of the glomerular basement mem-brane results in the thickened membranes. Takentogether, these changes result in anMPGN patternof injury. The immune complex and monoclonalIg-mediated MPGN is characterized by the pres-ence of Igs on IF studies; in immune complex-mediated MPGN, the deposits are polyclonal,while in monoclonal Ig-mediated MPGN, the Igis monotypic. C3 is most often present as well,indicating activation of complement pathway.
Infection-Associated MPGN
There are several infections, mostly chronic, asso-ciated with an MPGN pattern of injury. The mostcommon infection associated with an MPGN pat-tern of injury is hepatitis C infection. Hepatitis Cis associated with varied renal diseases including
membranous nephropathy, focal segmentalglomerulosclerosis (Stehman-Breen et al. 1999),thrombotic microangiopathy (Baid et al. 1999),and fibrillary glomerulonephritis (Markowitz etal. 1998). The exact mechanism of HCV-relatedglomerular disease is unclear. Toll-like receptors,particularly TLR3, might have a role to play inHCV-related MPGN (Wornle et al. 2006).
Hepatitis B is also implicated inMPGN (Johnsonand Couser 1990; Knieser et al. 1974; Myers et al.1973). The pathology is likely related to the trappingof circulating immune complexes in the mesangiumand subendothelial regions. It is also possible thatmesangial cells are directly infected by hepatitis Bvirions (Knieser et al. 1974).
Rarely, MPGN may be seen with acute viralinfections such as Puumala hantavirus (Mustonenet al. 2001).
Chronic bacterial infections are well-recog-nized causes of MPGN. Deep-seated abscesses,such as Pott’s abscess from tuberculosis ornocardial abscesses, (Elmaci et al. 2007; Ram etal. 2014) infected indwelling catheters, andshunts, are causes of MPGN due to continuouslow-dose antigenemia (Marini et al. 1976; Okadaet al. 2016; Takaki et al. 2012; Yared et al. 1999).“Shunt nephritis” as may be seen with ventricu-loatrial or ventriculocaval shunts for hydrocepha-lus is caused most commonly by coagulase-negative staphylococcal infections (Black et al.1965). Other organisms include coagulase-posi-tive staphylococci or Propionibacterium acnes.Catheter infections are often seen in the settingof total parenteral nutrition. Brucellosis maycause anMPGN pattern of renal injury in endemicregions (Ceylan et al. 2009). Filariasis and leprosyhave been recognized as causes of MPGN in thetropical environment (Date et al. 1983). Fungaland parasitic infections are less commonly asso-ciated with an MPGN pattern of injury(Altiparmak et al. 2002; Ubesie et al. 2013).Leishmaniasis has been associated with MPGN(Ortiz et al. 2015). Sethi et al. recently described acase of leishmaniasis with MPGN and abundantcomplement deposition within the glomeruli(Sethi et al. 2016a).
Infections resulting in MPGN are often char-acterized by the presence of mesangial and
Membranoproliferative Glomerulonephritis, Adult 3

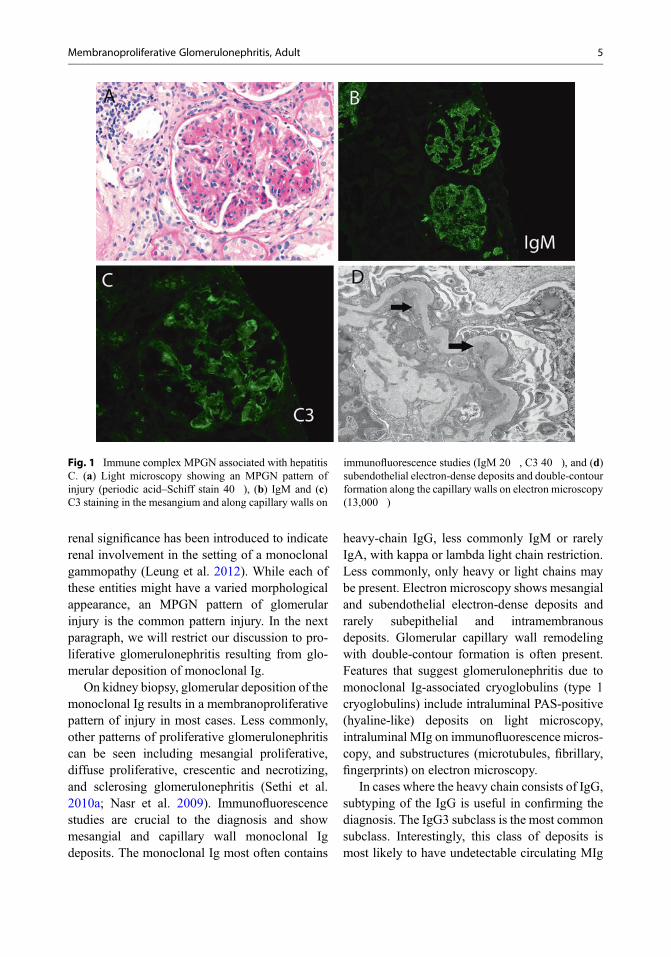
capillary wall staining for IgM and C3 in thesetting of viral infections; small amounts of IgGmay also be present. The presence of IgG (withlesser intensity of IgM) and C3 is more often seenin bacterial infections. In some cases with chronicinfections, the Ig staining may be weak comparedto the C3 staining. Electron microscopy showsmesangial and subendothelial deposits. The cap-illary walls are thickened with entrapment of cel-lular elements, subendothelial electron-densedeposits, matrix like material, and new basementmembrane material often resulting in the forma-tion of double contours. A representative figurefrom MPGN secondary to hepatitis C is shown inFig. 1. The figure shows an MPGN pattern onlight microscopy, IgM and C3 staining in themesangium and along capillary walls on immu-nofluorescence studies, and subendothelial elec-tron-dense deposits and double-contour formationalong the capillary walls on electron microscopy.
Autoimmune Disease-AssociatedMPGN
Membranoproliferative glomerulonephritis ismost commonly seen in systemic lupuserythematosus, as the expression of chroniclupus nephritis (Weening et al. 2004). Other auto-immune diseases known to be associated withMPGN include rheumatoid arthritis, primarySjögren’s syndrome, undifferentiated connectivetissue disease, primary sclerosing cholangitis, andGraves’ disease (Zand et al. 2014; Cortez et al.1995; Goules et al. 2000). Ghoules et al. reportedrenal disease in approximately 4% of patients withprimary SS. Interstitial nephritis and glomerulo-nephritis were the most common causes of renaldisease. Glomerulonephritis, which includedMPGN and mesangioproliferative GN, occurredlater in the course of the disease and was associ-ated with less favorable outcomes. The majorityof patients with glomerulonephritis (80%) hadmixed monoclonal cryoglobulinemia IgM kappa(type II) and lower complement C4 levels (Gouleset al. 2000). Sjögren’s syndrome is reportedly themost common cause of non-HCV-related
cryoglobulinemia (Khan et al. 1988; Anand et al.2015).
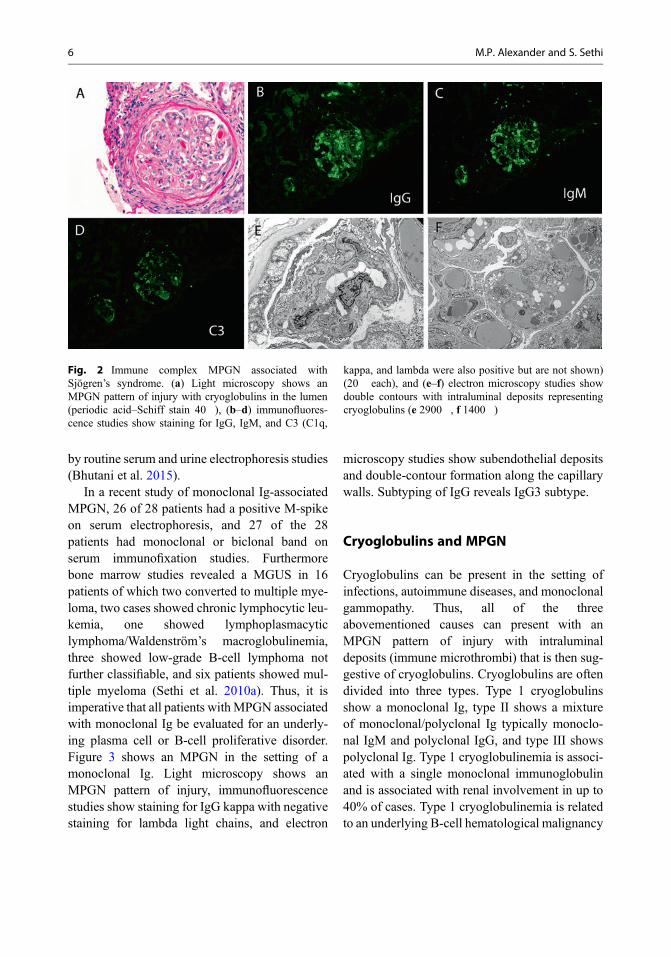
MPGN associated with autoimmune diseasesis often characterized by a full-house pattern of IFstaining, i.e., positive staining for IgG, IgA, IgM,C1q, C3, kappa, and lambda light chains, partic-ularly in the setting of systemic lupuserythematosus. On the other hand, IgM may bethe dominant Ig in MPGN associated with rheu-matoid arthritis and primary Sjögren’s syndrome.Electron microscopy shows mesangial and capil-lary wall electron-dense deposits. With regard tocapillary wall deposits, subendothelial depositsare most common. Subepithelial deposits mayalso be present. In such cases, a membranouscomponent of the disease should be considered.Tubuloreticular inclusions are often present in theendothelial cells. Figure 2 shows an MPGN in thesetting of an autoimmune disease (Sjögren’s syn-drome). Light microscopy shows an MPGN pat-tern of injury with cryoglobulins in the lumen,immunofluorescence studies show staining forIgG, IgM, and C3 (C1q, kappa, and lambda werealso positive but are not shown), and electronmicroscopy studies show double contours withintraluminal deposits representing cryoglobulins.
Monoclonal Ig-Associated MPGN
Monoclonal gammopathies encompass a heterog-enous spectrum of disorders characterized byclonal proliferation of Ig-producing B-lympho-cytes or plasma cell clone proteins that results ina monoclonal Ig that can be detected in the bloodor urine (M-protein) (Kyle et al. 2002). The phys-icochemical properties of the monoclonal Ig oftenresult in renal disease, even in the absence of overtmalignancies such as lymphoma, multiple mye-loma, or Waldenström’s macroglobulinemia. Theglomerular diseases included in this group includeproliferative glomerulonephritis with monoclonalIg deposits, amyloidosis, fibrillary glomerulone-phritis, immunotactoid glomerulopathy, andmonoclonal immunoglobulin deposition disease(Sethi et al. 2010a, 2016b; Sethi and Rajkumar2013). The term monoclonal gammopathy of
4 M.P. Alexander and S. Sethi
renal significance has been introduced to indicaterenal involvement in the setting of a monoclonalgammopathy (Leung et al. 2012). While each ofthese entities might have a varied morphologicalappearance, an MPGN pattern of glomerularinjury is the common pattern injury. In the nextparagraph, we will restrict our discussion to pro-liferative glomerulonephritis resulting from glo-merular deposition of monoclonal Ig.
On kidney biopsy, glomerular deposition of themonoclonal Ig results in a membranoproliferativepattern of injury in most cases. Less commonly,other patterns of proliferative glomerulonephritiscan be seen including mesangial proliferative,diffuse proliferative, crescentic and necrotizing,and sclerosing glomerulonephritis (Sethi et al.2010a; Nasr et al. 2009). Immunofluorescencestudies are crucial to the diagnosis and showmesangial and capillary wall monoclonal Igdeposits. The monoclonal Ig most often contains
heavy-chain IgG, less commonly IgM or rarelyIgA, with kappa or lambda light chain restriction.Less commonly, only heavy or light chains maybe present. Electron microscopy shows mesangialand subendothelial electron-dense deposits andrarely subepithelial and intramembranousdeposits. Glomerular capillary wall remodelingwith double-contour formation is often present.Features that suggest glomerulonephritis due tomonoclonal Ig-associated cryoglobulins (type 1cryoglobulins) include intraluminal PAS-positive(hyaline-like) deposits on light microscopy,intraluminal MIg on immunofluorescence micros-copy, and substructures (microtubules, fibrillary,fingerprints) on electron microscopy.
In cases where the heavy chain consists of IgG,subtyping of the IgG is useful in confirming thediagnosis. The IgG3 subclass is the most commonsubclass. Interestingly, this class of deposits ismost likely to have undetectable circulating MIg
Fig. 1 Immune complex MPGN associated with hepatitisC. (a) Light microscopy showing an MPGN pattern ofinjury (periodic acid–Schiff stain 40�), (b) IgM and (c)C3 staining in the mesangium and along capillary walls on
immunofluorescence studies (IgM 20�, C3 40�), and (d)subendothelial electron-dense deposits and double-contourformation along the capillary walls on electron microscopy(13,000�)
Membranoproliferative Glomerulonephritis, Adult 5
by routine serum and urine electrophoresis studies(Bhutani et al. 2015).
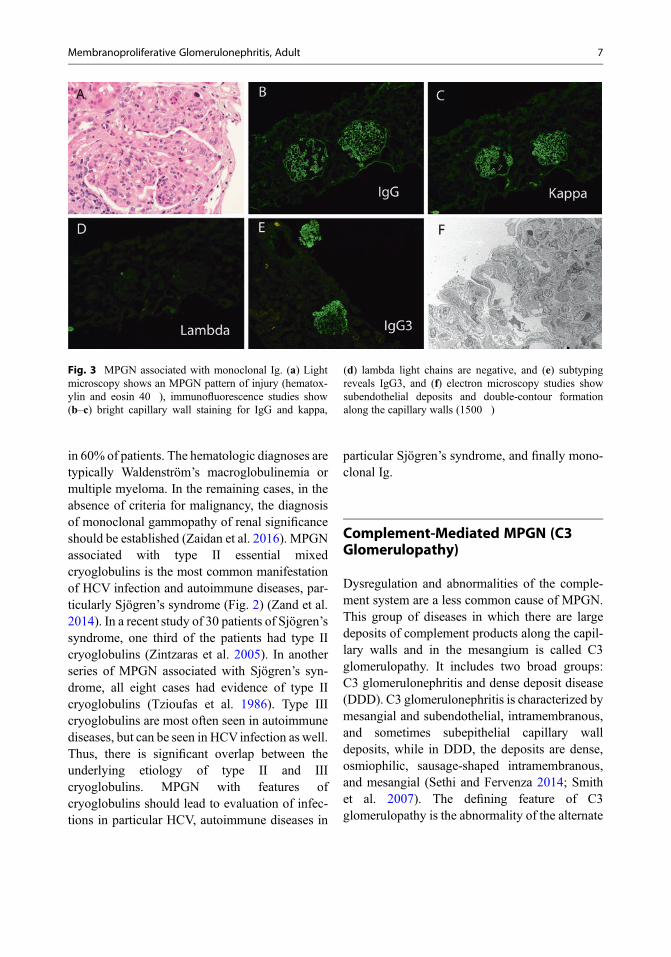
In a recent study of monoclonal Ig-associatedMPGN, 26 of 28 patients had a positive M-spikeon serum electrophoresis, and 27 of the 28patients had monoclonal or biclonal band onserum immunofixation studies. Furthermorebone marrow studies revealed a MGUS in 16patients of which two converted to multiple mye-loma, two cases showed chronic lymphocytic leu-kemia, one showed lymphoplasmacyticlymphoma/Waldenström’s macroglobulinemia,three showed low-grade B-cell lymphoma notfurther classifiable, and six patients showed mul-tiple myeloma (Sethi et al. 2010a). Thus, it isimperative that all patients withMPGN associatedwith monoclonal Ig be evaluated for an underly-ing plasma cell or B-cell proliferative disorder.Figure 3 shows an MPGN in the setting of amonoclonal Ig. Light microscopy shows anMPGN pattern of injury, immunofluorescencestudies show staining for IgG kappa with negativestaining for lambda light chains, and electron
microscopy studies show subendothelial depositsand double-contour formation along the capillarywalls. Subtyping of IgG reveals IgG3 subtype.
Cryoglobulins and MPGN
Cryoglobulins can be present in the setting ofinfections, autoimmune diseases, and monoclonalgammopathy. Thus, all of the threeabovementioned causes can present with anMPGN pattern of injury with intraluminaldeposits (immune microthrombi) that is then sug-gestive of cryoglobulins. Cryoglobulins are oftendivided into three types. Type 1 cryoglobulinsshow a monoclonal Ig, type II shows a mixtureof monoclonal/polyclonal Ig typically monoclo-nal IgM and polyclonal IgG, and type III showspolyclonal Ig. Type 1 cryoglobulinemia is associ-ated with a single monoclonal immunoglobulinand is associated with renal involvement in up to40% of cases. Type 1 cryoglobulinemia is relatedto an underlying B-cell hematological malignancy
Fig. 2 Immune complex MPGN associated withSjögren’s syndrome. (a) Light microscopy shows anMPGN pattern of injury with cryoglobulins in the lumen(periodic acid–Schiff stain 40�), (b–d) immunofluores-cence studies show staining for IgG, IgM, and C3 (C1q,
kappa, and lambda were also positive but are not shown)(20� each), and (e–f) electron microscopy studies showdouble contours with intraluminal deposits representingcryoglobulins (e 2900�, f 1400�)
6 M.P. Alexander and S. Sethi
in 60% of patients. The hematologic diagnoses aretypically Waldenström’s macroglobulinemia ormultiple myeloma. In the remaining cases, in theabsence of criteria for malignancy, the diagnosisof monoclonal gammopathy of renal significanceshould be established (Zaidan et al. 2016). MPGNassociated with type II essential mixedcryoglobulins is the most common manifestationof HCV infection and autoimmune diseases, par-ticularly Sjögren’s syndrome (Fig. 2) (Zand et al.2014). In a recent study of 30 patients of Sjögren’ssyndrome, one third of the patients had type IIcryoglobulins (Zintzaras et al. 2005). In anotherseries of MPGN associated with Sjögren’s syn-drome, all eight cases had evidence of type IIcryoglobulins (Tzioufas et al. 1986). Type IIIcryoglobulins are most often seen in autoimmunediseases, but can be seen in HCVinfection as well.Thus, there is significant overlap between theunderlying etiology of type II and IIIcryoglobulins. MPGN with features ofcryoglobulins should lead to evaluation of infec-tions in particular HCV, autoimmune diseases in
particular Sjögren’s syndrome, and finally mono-clonal Ig.
Complement-Mediated MPGN (C3Glomerulopathy)
Dysregulation and abnormalities of the comple-ment system are a less common cause of MPGN.This group of diseases in which there are largedeposits of complement products along the capil-lary walls and in the mesangium is called C3glomerulopathy. It includes two broad groups:C3 glomerulonephritis and dense deposit disease(DDD). C3 glomerulonephritis is characterized bymesangial and subendothelial, intramembranous,and sometimes subepithelial capillary walldeposits, while in DDD, the deposits are dense,osmiophilic, sausage-shaped intramembranous,and mesangial (Sethi and Fervenza 2014; Smithet al. 2007). The defining feature of C3glomerulopathy is the abnormality of the alternate
Fig. 3 MPGN associated with monoclonal Ig. (a) Lightmicroscopy shows an MPGN pattern of injury (hematox-ylin and eosin 40�), immunofluorescence studies show(b–c) bright capillary wall staining for IgG and kappa,
(d) lambda light chains are negative, and (e) subtypingreveals IgG3, and (f) electron microscopy studies showsubendothelial deposits and double-contour formationalong the capillary walls (1500�)
Membranoproliferative Glomerulonephritis, Adult 7

complement pathway (Fakhouri et al. 2010; Bar-bour et al. 2013a; Pickering et al. 2013).
On light microscopy the pattern of injury isquite variable and includes mesangial prolifera-tive, membranoproliferative, and endocapillaryproliferative, and, in rare circumstance with min-imal abnormalities, crescents or sclerosing lesionsmay also be seen (Fervenza et al. 2012; Nasr et al.2009; Sethi et al. 2011, 2012a). In DDD thicken-ing of the glomerular basement membranes, par-ticularly on the periodic acid–Schiff stain, canoften be appreciated.
Immunofluorescence microscopy demon-strates a predominance of C3 deposition in theglomeruli which is largely granular staining ofthe mesangium and peripheral capillary walls.Nasr et al. in their study of 32 patients withDDD demonstrated C3 in the mesangium as gran-ular or ringlike deposits and along glomerularcapillary walls in a linear to semi-linear pattern.Sometimes the linear staining appeared as narrowtram tracks outlining the inner and outer aspects ofthe thickened glomerular capillary walls. Focallinear or semi-linear C3 staining was also seenalong tubular basement membranes in 60% ofpatients and along Bowman’s capsule in 30% ofpatients (Nasr et al. 2009). Similar staining patternof C3 in the mesangium and along glomerularcapillary walls is noted in C3 glomerulonephritis,although the ringlike deposits are not a typicalfinding. Tubular staining for C3 may be seen inDDD; it is not a feature of C3 glomerulonephritis.
In most laboratories C3 is evaluated by C3ccomponent. However the C3 breakdown prod-ucts, which include iC3b, C3c, and C3dg, arelikely differentially deposited in glomeruli, andtheir interactions with complement receptorsmight contribute to the pathophysiology of thedisease. The C3GN consensus report suggests itwould be ideal if antibodies to the different C3breakdown products could be employed toenhance our understanding of the disease (Picker-ing et al. 2013; Sethi et al. 2016c).
Immunoglobulins are often deposited alongwith C3 in C3 glomerulopathy. It has beenobserved that if the most restrictive criteria of
“C3 only” was utilized, it would capture onlyhalf of the cases with DDD (compared with 8%of type I and 10% of type III). Adding the mostliberal definition (C3-dominant staining of at leasttwo orders of intensity stronger than any combi-nation of IgG, IgM, IgA, and C1q) identified 88%of those with DDD (compared with 31% of type Iand 39% of type III) (Hou et al. 2014). Thiswarrants a more liberal allowance for minimalimmunoglobulin deposition to avoid missingcases with alternate complement pathway abnor-malities (Fakhouri et al. 2010). The significanceof finding immunoglobulins is not elucidated. Aninitial activation of complement via the classicalpathway may trigger or unmask a dysregulation ofthe alternate complement pathway. On the otherhand, the small amounts of Ig may represent non-specific entrapment of immunoglobulins orpodocyte protein reabsorption droplets (Pickeringet al. 2013). A useful test to add to the immuno-fluorescence panel is C4d (Sethi et al. 2015). C4dis a by-product of activation of the classic andlectin pathways. A recent study by Sethi et al.used glomerular staining of C4d to differentiatebetween immune complex GN and C3glomerulopathy. C4d represents activation of theclassical and/or lectin pathway. The premise of thehypothesis was that patients with immune com-plex GN likely activate the classical pathway ofcomplement and thus would stain positively forC4d. On the other hand, C4d would be negative inC3 glomerulopathy that were purely a result ofactivation of alternative pathway. The studyshowed that specimens of immune complex-mediated GN, except two specimens ofIgA nephropathy and one specimen of sclerosingmembranoproliferative GN, showed bright(2–3+) C4d staining. C4d staining was completelynegative in 80% of C3 glomerulopathy, and trace/1+ C4d staining was detected in 20% specimens(Sethi et al. 2015).
Ultrastructural findings commonly includemesangial expansion and capillary wall thicken-ing. In C3 glomerulonephritis, electron-densedeposits found in the mesangium, in the sub-endothelium, and often in the subepithelial
8 M.P. Alexander and S. Sethi

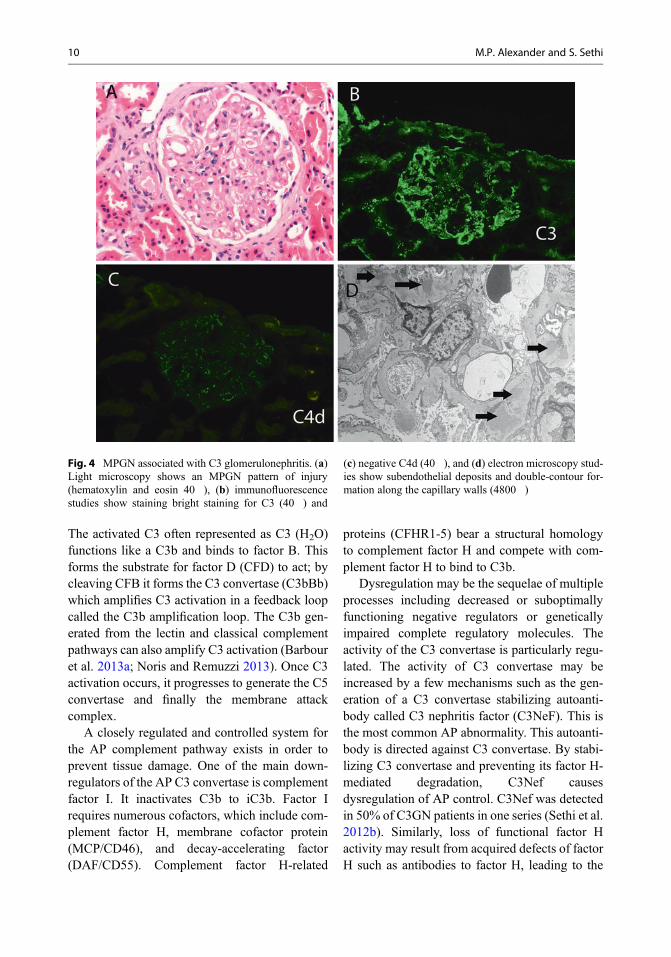
regions. The deposits vary in appearance. Theyare often intramembranous but commonly lackthe dense osmiophilic characteristics of thoseseen in DDD. They are often ill-defined withvague borders, and they may rarely resembleimmune complex-type discrete deposits. Theylack substructure. Subepithelial deposits areoften hump shaped, resembling postinfectiousglomerulonephritis (Sethi et al. 2011; Servais etal. 2007). In some cases, there is complex multi-layering of the deposits with intervening base-ment membrane material. In all likelihood, mostcases of MPGN type III of Strife type representedC3GN (Barbour et al. 2013b; Sethi et al. 2012b,c). In DDD the deposits are typically large andhighly electron-dense intramembranous depositswith irregularly thickened lamina densa. Theintramembranous deposits might be interruptedgiving it a “sausage-string” appearance. Promi-nent involvement of the glomerular basementmembrane reflection over the mesangium withless involvement of the peripheral glomerular cap-illary walls may be noted in a few cases of DDD.Large hump-shaped, subepithelial electron-densedeposits may be noted. Similar electron-densedeposits may be seen in the Bowman’s capsuleand tubular basement membranes in DDD.Mesangial deposits forming large rounded nod-ules (“ring forms”) or small and granular depositsmay also be noted (Nasr et al. 2009). Despite theapparently distinctive ultrastructural featuresbetween C3 glomerulonephritis and DDD, casesoften demonstrate an overlap of these ultrastruc-tural features, reflecting the common pathogenicpathway in both these diseases (Sethi et al. 2015).Figure 4 shows an MPGN in a patient with C3glomerulonephritis. Light microscopy shows anMPGN pattern of injury with thickened glomeru-lar basement membranes, immunofluorescencestudies show staining bright staining for C3 (negative staining for all Ig including kappa andlambda light chains) and negative C4d, and elec-tron microscopy studies show subendothelialdeposits and double-contour formation along thecapillary walls. Figure 5 shows an MPGN in apatient with DDD. Light microscopy shows an
MPGN pattern of injury, immunofluorescencestudies show staining bright staining for C3, andelectron microscopy studies showsintramembranous dense deposits.
The proteomic profile in this subset of diseaseshas been evaluated by mass spectrometry. In astudy by Sethi et al. (2012b), the proteomic profileof patients with C3GN showed accumulation ofalternate complement pathway and terminal com-plement complex proteins. The deposition of C3and C9 was extensive in all cases. C5, C6, C7, andC8 were also present in all cases. Complementregulating proteins vitronectin, clusterin, and apo-lipoprotein E were present in abundance. CFHR-1was present in all cases. Sethi et al. evaluated theproteomic profile of the glomerular deposits inconfirmed cases of DDD. They found that all ofthe glomeruli contained components of the alter-native pathway and terminal complement com-plex. Factor C9 was also uniformly present.Clusterin and vitronectin, two fluid-phase regula-tors of terminal complement complex, were alsoidentified. Their study demonstrated that in addi-tion to fluid-phase dysregulation of the alternativepathway, soluble components of the terminalcomplement complex contribute to glomerularlesions found in DDD (Sethi et al. 2009). Morerecently, on further analysis, the C3 deposits werecomposed predominantly of C3dg and smalleramounts of iC3b. C3a and C3f were absent indi-cating that that deposition of C3 in C3glomerulopathy is an active process with accumu-lation of mostly the terminal breakdown frag-ments, i.e., C3dg that remains bound to theglycocalyx over the endothelial cells and glomer-ular basement membrane via the thioester bond(Sethi et al. 2016c). The proteomic profile of C3breakdown products was similar in C3 glomeru-lonephritis and DDD.
Pathogenesis
The alternate complement pathway is constantlyactivated at a low level by the spontaneous hydro-lysis of C3 by a process called the “C3 tick over.”
Membranoproliferative Glomerulonephritis, Adult 9
The activated C3 often represented as C3 (H2O)functions like a C3b and binds to factor B. Thisforms the substrate for factor D (CFD) to act; bycleaving CFB it forms the C3 convertase (C3bBb)which amplifies C3 activation in a feedback loopcalled the C3b amplification loop. The C3b gen-erated from the lectin and classical complementpathways can also amplify C3 activation (Barbouret al. 2013a; Noris and Remuzzi 2013). Once C3activation occurs, it progresses to generate the C5convertase and finally the membrane attackcomplex.
A closely regulated and controlled system forthe AP complement pathway exists in order toprevent tissue damage. One of the main down-regulators of the AP C3 convertase is complementfactor I. It inactivates C3b to iC3b. Factor Irequires numerous cofactors, which include com-plement factor H, membrane cofactor protein(MCP/CD46), and decay-accelerating factor(DAF/CD55). Complement factor H-related
proteins (CFHR1-5) bear a structural homologyto complement factor H and compete with com-plement factor H to bind to C3b.
Dysregulation may be the sequelae of multipleprocesses including decreased or suboptimallyfunctioning negative regulators or geneticallyimpaired complete regulatory molecules. Theactivity of the C3 convertase is particularly regu-lated. The activity of C3 convertase may beincreased by a few mechanisms such as the gen-eration of a C3 convertase stabilizing autoanti-body called C3 nephritis factor (C3NeF). This isthe most common AP abnormality. This autoanti-body is directed against C3 convertase. By stabi-lizing C3 convertase and preventing its factor H-mediated degradation, C3Nef causesdysregulation of AP control. C3Nef was detectedin 50% of C3GN patients in one series (Sethi et al.2012b). Similarly, loss of functional factor Hactivity may result from acquired defects of factorH such as antibodies to factor H, leading to the
Fig. 4 MPGN associated with C3 glomerulonephritis. (a)Light microscopy shows an MPGN pattern of injury(hematoxylin and eosin 40�), (b) immunofluorescencestudies show staining bright staining for C3 (40�) and
(c) negative C4d (40�), and (d) electron microscopy stud-ies show subendothelial deposits and double-contour for-mation along the capillary walls (4800�)
10 M.P. Alexander and S. Sethi

uninhibited activity of C3 convertase. A back-ground of autoimmune disease is thus often seenin C3 glomerulonephritis (Alexander et al. 2016).Dysregulation may also involve mutations incomplement genes (such as those encoding factorH, factor B, factor I, or even C3) that preventnormal C3 convertase control. Monoclonalgammopathy is often seen in both C3 glomerulo-nephritis and DDD (Sethi and Rajkumar 2013;Sethi et al. 2010b; Zand et al. 2013). MonoclonalIg may stabilize C3 convertase and thus behave asa C3NeF. In cases without C3Nef, the monoclonalIg may interfere with alternative complement reg-ulation via other mechanisms. For example,monoclonal Ig may act as an autoantibody tocomplement factor H, resulting in functionalabnormality of factor H, resulting in over activityof the alternative pathway of complement (Meri etal. 1992). The existence of limited reports show-ing clinical improvement in the glomerular dis-ease following chemotherapy for the underlyingB-cell neoplasia supports a pathogenic role for themonoclonal Ig.
MPGN with Masked Immune Deposits
This is a recently described entity (Larsen et al.2015). The pathology is that of a membranoproli-ferative glomerulonephritis with isolated C3deposits (by routine IF) in the setting of a mono-clonal gammopathy. Paraffin IF unmasks mono-clonal immunoglobulin glomerular deposits. Theimportance of recognizing this entity is to avoidmisdiagnosis of these cases as C3GN due to
monoclonal gammopathy (Barbour et al. 2013b).Also, as most of these cases are associated with alow-grade lymphoma or plasma cell dyscrasia, anaccurate diagnosis will permit treatment of theunderlying hematological disorder. A C4d stainis also helpful in detecting masked immunedeposits (Sethi et al. 2016d).
MPGN Without Immunoglobulins orComplement
Glomeruli may demonstrate membranoproli-ferative-like changes in the absence of immuno-globulin or complement deposits in chronicthrombotic microangiopathy. The differentialdiagnosis includes hemolytic uremic syndrome(HUS), thrombotic thrombocytopenic purpura(TTP), antiphospholipid antibody syndrome(Bienaime et al. 2016; Gigante et al. 2009), sclero-derma (Mouthon et al. 2014), and malignanthypertension (Nzerue et al. 2014) and certainmedications such as gemcitabine (Richmond etal. 2013) radiation treatment, or stem cell trans-plant (Laskin et al. 2011).
Evaluation of MPGN
The evaluation for MPGN is determined based onthe suspected etiology. The diagnostic workupshould be guided by clinical presentation andbiopsy findings. A standard workup for MPGNshould include complement evaluation. Low C3
Fig. 5 MPGN associated with DDD. (a) Light micros-copy shows an MPGN pattern of injury (periodicacid–Schiff stain 40�), (b) immunofluorescence studies
show staining bright staining for C3 (40�), and (c) electronmicroscopy studies shows intramembranous densedeposits (4400�)
Membranoproliferative Glomerulonephritis, Adult 11

and low C4 are more typical of classical pathwayactivation as one might see in immune complex-mediated glomerulonephritis. A low C3 with anormal C4 suggests abnormalities of the alternatecomplement pathway. These findings along withthe biopsy findings; particularly immunofluores-cence and electron microscopy can further guidedetailed evaluation.
Immune Complex-Mediated MPGN
Infections Workup in the evaluation of infec-tion-related MPGN depends on the suspectedpathogen. In the case of hepatitis-related MPGN,tests should include viral serology and quantifica-tion of viral load by PCR studies. The workup forparasitic infection-related MPGN should includeblood tests for malaria, urine and stool test forschistosomiasis, and serological tests for schisto-somiasis and leishmaniasis.
Blood cultures, cultures of indwelling cathetertips, imaging studies for deep-seated abscesses,and transthoracic echocardiograms for valvularvegetations of fungal and bacterial infections mustbe part of the comprehensive work upto excludean infectious etiology.
Of note, parasitic and fungal infections areonly investigated in the appropriate clinical situa-tion (history of recent travel to endemic regions,prolonged fever of unknown origin, atypical pul-monary infiltrates).
Autoimmune Diseases The evaluation is basedon clinical presentation. In cases of suspectedsystemic lupus erythematosus, the workup shouldinclude ANA and ds DNA. In suspected Sjögren’ssyndrome, a positive anti-Ro/SSA and/or anti-La/SSB and a positive lip biopsy help confirm thediagnosis. The diagnostic tests of the autoimmunediseases should follow the established criteria, e.g., American College of Rheumatology, forSjögren’s syndrome and rheumatoid arthritis(Neogi et al. 2010; Shiboski et al. 2012).
Monoclonal Gammopathy The workup shouldinclude serum protein electrophoresis, urine
protein electrophoresis, serum and urineimmunofixation, and serum-free light chainassays. Bone marrow evaluation should beperformed to confirm an underlying plasma celldyscrasia and/or lymphoproliferative disorder.
Complement-Mediated MPGN (C3Glomerulopathy)
If the biopsy suggests complement-mediatedMPGN, the workup should trigger an evaluationof abnormalities of the complement pathway. Theworkup for abnormalities of the alternate pathwayof complement includes (a) functional assays, (b)quantification of complement components andregulators, (c) measurement of complement acti-vation markers, (d) genetic evaluation, (e) autoan-tibodies, and (f) testing for monoclonalimmunoglobulins (Angioi et al. 2016).
(a) Functional assays: These are based on theability of complements to lyse sheep erythro-cytes. This is an important screening test todetermine whether the complement pathwayis preserved or disrupted. The two tests avail-able for this purpose include the total hemo-lytic component (CH50) assay and thecomplement factor H functional assay.
(b) Quantification of complement components:C3 and C4 are commonly measured. C3 islowered in 40–75% of C3GN.
(c) Measurement of complication activationmarkers: C3 decay products including C3d,C4 decay products including C4d, and termi-nal complement pathway are often measured.
(d) Autoantibodies: C3 nephritic factor is anautoantibody to C3 convertase; C4 nephriticfactor is an antibody to C4 convertase. Anti-bodies to FH should be studied and ideallyantibodies to factor B should also bereviewed.
(e) Genetic analysis: This should be initiated forall the known complement-related genes: C3,CFH, CFI, CFB, and CFHR1–5 (Pickering etal. 2013).
(f) Serum protein electrophoresis,immunofixation electrophoresis, and serum-
12 M.P. Alexander and S. Sethi

free light chains – A paraprotein may beresponsible for activation of the alternativecomplement cascade (Zand et al. 2013). If amonoclonal gammopathy is discovered, spe-cialized tests are required to determinewhether or not the protein could be responsi-ble for the C3 glomerulopathy (Noris andRemuzzi 2013).
Uncommon Causes of MPGN
1. Cryofibrinogen-related membranoproliferativeglomerulonephritis: Cryofibrinogen is a rela-tively rare resulting in an MPGN pattern ofinjury. Cryofibrinogen is a cryoprecipitatethat develops after refrigeration of plasma butdoes not occur in cold serum. Cryofibrinogenmay be asymptomatic, but can be associatedwith thromboembolic disease, particularlyaffecting the skin. We recently published areport of two cases of MPGN with prominentdeposits of cryofibrinogen within the glomer-uli (Sethi 2017). The deposits did not showimmunoreactivity to immunoglobulins. Ultra-structural studies showed the deposits werecharacterized by haphazardly arranged largefibrils with tubular structures with variabledimensions. The tubules had large centralbore, and some had double or triple layering.In addition there were randomly distributedfibers in a matrix. The luminal diameter rangedfrom 121 to 211 with a mean diameter of158 nm.
2. C4 glomerulopathy: C4 glomerulopathy is arecently described disease entity characterizedby glomerular deposits of predominantly C4with little or no immunoglobulins or C3 depo-sition. This glomerulopathy encompassesC4DDD and C4 glomerulonephritis (Sethi etal. 2014, 2016e). Sethi et al. described the firstcase of C4DDD in a young woman who firstmanifested with nephrotic-range proteinuria(Sethi et al. 2014). Her biopsy showed amembranoproliferative pattern of injury withextremely thick glomerular capillary walls.
Staining with periodic acid–Schiff showed rib-bonlike material lining the glomerular base-ment membrane. This material was negativeon methenamine silver staining. Immunofluo-rescence microscopy showed bright stainingfor C4d along the capillary walls and no glo-merular staining for IgG, IgA, IgM, C1q, C3,C4c, or kappa or lambda light chains. Electronmicroscopy showed large subendothelialosmiophilic dense deposits lining the glomer-ular basement membrane. There were nodeposits along the Bowman’s capsule or tubu-lar basement membranes. The patient workupshowed no defects in the classical or alternatepathway of complement, but indicated an over-active lectin pathway of complement.
Summary
MPGN is a pattern of injury and not a diagnosticentity in itself. MPGN can essentially be classifiedinto immune complex/monoclonal Ig-mediatedMPGN and complement-mediated MPGN (C3glomerulopathy). Immune complex MPGNshould lead to evaluation for infections and auto-immune diseases. Monoclonal Ig-mediatedMPGN should lead to evaluation for an underly-ing B-cell or plasma cell disorder. Complement-mediated MPGN should lead to evaluation of thealternative pathway of complement. Identificationof the underlying etiology and pathophysiology ofMPGN should then lead to appropriate manage-ment of the patient.
References
Alexander MP et al (2016) C3 glomerulonephritis andautoimmune disease: more than a fortuitous associa-tion? J Nephrol 29:203–209
Altiparmak MR, Pamuk GE, Pamuk ON, Tabak F (2002)Brucella glomerulonephritis: review of the literatureand report on the first patient with brucellosis andmesangiocapillary glomerulonephritis. Scand J InfectDis 34:477–480
Anand A, Krishna GG, Sibley RK, Kambham N (2015)Sjogren syndrome and cryoglobulinemic glomerulone-phritis. Am J Kidney Dis 66:532–535
Membranoproliferative Glomerulonephritis, Adult 13

Anders D, Agricola B, Sippel M, Thoenes W (1977) Base-ment membrane changes in membranoproliferativeglomerulonephritis. II. Characterization of a third typeby silver impregnation of ultra thin sections. VirchowsArch A Pathol Anat Histol 376:1–19
Angioi A et al (2016) Diagnosis of complement alternativepathway disorders. Kidney Int 89:278–288
Baid S et al (1999) Renal thrombotic microangiopathyassociated with anticardiolipin antibodies in hepatitisC-positive renal allograft recipients. J Am Soc Nephrol10:146–153
Barbour TD, Pickering MC, Terence Cook H (2013a)Dense deposit disease and C3 glomerulopathy. SeminNephrol 33:493–507
Barbour TD, Pickering MC, Cook HT (2013b) Recentinsights into C3 glomerulopathy. Nephrol Dial Trans-plant 28:1685–1693
Bhutani G et al (2015) Hematologic characteristics ofproliferative glomerulonephritides with nonorganizedmonoclonal immunoglobulin deposits. Mayo Clin Proc90:587–596
Bienaime F, Legendre C, Terzi F, Canaud G (2017) Anti-phospholipid syndrome and kidney disease. Kidney Int91:34–44
Black JA, Challacombe DN, Ockenden BG (1965)Nephrotic syndrome associated with bacteraemia aftershunt operations for hydrocephalus. Lancet 2:921–924
Burkholder PM, Marchand A, Krueger RP (1970) Mixedmembranous and proliferative glomerulonephritis. Acorrelative light, immunofluorescence, and electronmicroscopic study. Lab Invest 23:459–479
Ceylan K et al (2009) Renal involvement in Brucellainfection. Urology 73:1179–1183
Cortez MS, Sturgill BC, Bolton WK (1995) Membrano-proliferative glomerulonephritis with primarySjogren’s syndrome. Am J Kidney Dis 25:632–636
Date A, Neela P, Shastry JC (1983) Membranoproliferativeglomerulonephritis in a tropical environment. AnnTrop Med Parasitol 77:279–285
Elmaci I et al (2007) Nocardial cerebral abscess associatedwith mycetoma, pneumonia, and membranoproli-ferative glomerulonephritis. J Clin Microbiol45:2072–2074
Fakhouri F, Fremeaux-Bacchi V, Noel LH, Cook HT, Pick-ering MC (2010) C3 glomerulopathy: a new classifica-tion. Nat Rev Nephrol 6:494–499
Fervenza FC, Smith RJ, Sethi S (2012) Association of anovel complement factor H mutation with severe cres-centic and necrotizing glomerulonephritis. Am J Kid-ney Dis 60:126–132
Gigante A et al (2009) Antiphospholipid antibodies andrenal involvement. Am J Nephrol 30:405–412
Goules A et al (2000) Clinically significant and biopsy-documented renal involvement in primary Sjogren syn-drome. Medicine 79:241–249
Hou J et al (2014) Toward a working definition of C3glomerulopathy by immunofluorescence. Kidney Int85:450–456
Johnson RJ, Couser WG (1990) Hepatitis B infection andrenal disease: clinical, immunopathogenetic and thera-peutic considerations. Kidney Int 37:663–676
Khan MA, Akhtar M, Taher SM (1988) Membranoproli-ferative glomerulonephritis in a patient with primarySjogren’s syndrome. Report of a case with review of theliterature. Am J Nephrol 8:235–239
Knieser MR et al (1974) Pathogenesis of renal diseaseassociated with viral hepatitis. Arch Pathol 97:193–200
Kyle RA et al (2002) A long-term study of prognosis inmonoclonal gammopathy of undetermined signifi-cance. N Engl J Med 346:564–569
Larsen CP et al (2015) Membranoproliferative glomerulo-nephritis with masked monotypic immunoglobulindeposits. Kidney Int 88:867–873
Laskin BL, Goebel J, Davies SM, Jodele S (2011) Smallvessels, big trouble in the kidneys and beyond: hema-topoietic stem cell transplantation-associated throm-botic microangiopathy. Blood 118:1452–1462
Leung N et al (2012) Monoclonal gammopathy of renalsignificance: when MGUS is no longer undeterminedor insignificant. Blood 120:4292–4295
Marini G et al (1976) Membranoproliferative glomerulo-nephritis associated with infected ventriculoatrialshunt. Report of two cases recovered after removal ofthe shunt. Mod Probl Paediatr 18:207–210
Markowitz GS, Cheng JT, Colvin RB, Trebbin WM,D’Agati VD (1998) Hepatitis C viral infection is asso-ciated with fibrillary glomerulonephritis andimmunotactoid glomerulopathy. J Am Soc Nephrol9:2244–2252
Meri S, Koistinen V, Miettinen A, Tornroth T, Seppala I(1992) Activation of the alternative pathway of com-plement by monoclonal lambda light chains inmembranoproliferative glomerulonephritis. J ExpMed 175:939–950
Mouthon L, Bussone G, Berezne A, Noel LH, Guillevin L(2014) Scleroderma renal crisis. J Rheumatol41:1040–1048
Mustonen J et al (2001) Mesangiocapillary glomerulone-phritis caused by Puumala hantavirus infection. Neph-ron 89:402–407
Myers BD, Griffel B, Naveh D, Jankielowiiz T, Klajman A(1973) Membrano-proliferative glomerulonephritisassociated with persistent viral hepatitis. Am J ClinPathol 60:222–228
Nasr SH et al (2009) Proliferative glomerulonephritis withmonoclonal IgG deposits. J Am Soc Nephrol20:2055–2064
Neogi T et al (2010) The 2010 American college of rheu-matology/European league against rheumatism classi-fication criteria for rheumatoid arthritis: phase 2methodological report. Arthritis Rheum 62:2582–2591
Noris M, Remuzzi G (2013) Overview of complementactivation and regulation. Semin Nephrol 33:479–492
Nzerue C et al (2014) Malignant hypertension with throm-botic microangiopathy and persistent acute kidneyinjury (AKI). Clin Kidney J 7:586–589
14 M.P. Alexander and S. Sethi

Okada M et al (2016) Central venous catheter infection-related glomerulonephritis under long-term paren-teral nutrition: a report of two cases. BMC ResNotes 9:196
Ortiz M et al (2015) Glomerulonephritis andcryoglobulinemia: first manifestation of visceral leish-maniasis. Clin Nephrol 83:370–377
Pickering MC et al (2013) C3 glomerulopathy: consensusreport. Kidney Int 84:1079–1089
Ram R, Sandeep P, Sridhar AV, Rukumangadha N,Sivakumar V (2014) Membranoproliferative glomeru-lonephritis and Pott’s disease. Clin Kidney J 7:391–393
Richmond J, Gilbar P, Abro E (2013) Gemcitabine-inducedthrombotic microangiopathy. Intern Med J43:1240–1242
Servais A et al (2007) Primary glomerulonephritis withisolated C3 deposits: a new entity which shares com-mon genetic risk factors with haemolytic uraemic syn-drome. J Med Genet 44:193–199
Sethi S, Fervenza FC (2011) Membranoproliferative glo-merulonephritis: pathogenetic heterogeneity and pro-posal for a new classification. Semin Nephrol31:341–348
Sethi S, Fervenza FC (2012) Membranoproliferative glo-merulonephritis: a new look at an old entity. N Engl JMed 366:1119–1131
Sethi S, Fervenza F (2014) Pathology of renal diseasesassociated with dysfunction of the alternative pathwayof complement: C3 glomerulopathy and atypical hemo-lytic uremic syndrome (aHUS). Semin Thromb Hemost40:416–421
Sethi S, Rajkumar SV (2013) Monoclonalgammopathy–associated proliferative glomerulone-phritis. Mayo Clin Proc 88:1284–1293
Sethi S et al (2009) Glomeruli of dense deposit diseasecontain components of the alternative and terminalcomplement pathway. Kidney Int 75:952–960
Sethi S et al (2010a) Membranoproliferative glomerulone-phritis secondary to monoclonal gammopathy. Clin JAm Soc Nephrol 5:770–782
Sethi S et al (2010b) Dense deposit disease associated withmonoclonal gammopathy of undetermined signifi-cance. Am J Kidney Dis 56:977–982
Sethi S et al (2011) Proliferative glomerulonephritis sec-ondary to dysfunction of the alternative pathway ofcomplement. Clin J Am Soc Nephrol 6:1009–1017
Sethi S, Fervenza FC, Zhang Y, Smith RJH (2012a) Sec-ondary focal and segmental glomerulosclerosis associ-ated with single-nucleotide polymorphisms in thegenes encoding complement factor H and C3. Am JKidney Dis 60:316–321
Sethi S et al (2012b) C3 glomerulonephritis: clinicopatho-logical findings, complement abnormalities, glomeru-lar proteomic profile, treatment, and follow-up. KidneyInt 82:465–473
Sethi S, Nester CM, Smith RJH (2012c) Membranoproli-ferative glomerulonephritis and C3 glomerulopathy:resolving the confusion. Kidney Int 81:434–441
Sethi S, Sullivan A, Smith RJ (2014) C4 dense-depositdisease. N Engl J Med 370:784–786
Sethi S, Nasr SH, De Vriese AS, Fervenza FC (2015) C4das a diagnostic tool in proliferative GN. J Am SocNephrol 26:2852–2859
Sethi S, Fervenza FC, Siddiqui A, Quint PS, Pritt BS(2016a) Leishmaniasis-associated membranoproli-ferative glomerulonephritis with massive complementdeposition. Kidney Int Rep 1:125–130
Sethi S, Fervenza FC, Rajkumar SV (2016b) Spectrum ofmanifestations of monoclonal gammopathy-associatedrenal lesions. Curr Opin Nephrol Hypertens25:127–137
Sethi S et al (2016c) Characterization of C3 in C3glomerulopathy. Nephrol Dial Transplant
Sethi S, Hernandez LH, Alexander MP, Fervenza FC(2016d) C4d as a marker for masked immune deposits.Kidney Int 90:223–224
Sethi S et al (2016e) C4 glomerulopathy: a disease entityassociated with C4d deposition. Am J Kidney Dis67:949–953
Sethi S, Yachoui R, Murray DL, Radhakrishnan J, Alex-ander MP (2017) Cryofibrinogen-Associated Glomer-ulonephritis. Am J Kidney Dis 69:302–308
Shiboski SC, Shiboski CH, Criswell L, Baer A et al (2012)American College of Rheumatology classificationcriteria for Sjogren’s syndrome: a data-driven, expertconsensus approach in the Sjogren’s International Col-laborative Clinical Alliance cohort. Arthritis Care Res(Hoboken) 64:475
Smith RJH et al (2007) New approaches to the treatment ofdense deposit disease. J Am Soc Nephrol18:2447–2456
Stehman-Breen C, Alpers CE, Fleet WP, Johnson RJ(1999) Focal segmental glomerular sclerosis amongpatients infected with hepatitis C virus. Nephron81:37–40
Takaki A et al (2012) Peritoneovenous shunting for refrac-tory ascites results in worsening of nephrotic syn-drome. Hepatol Res 42:1048–1053
Tzioufas A et al (1986) Cryoglobulinemia in autoimmunerheumatic diseases. Evidence of circulating monoclo-nal cryoglobulins in patients with primary Sjogren’ssyndrome. Arthritis Rheum 29:1098–1104
Ubesie AC et al (2013) Disseminated Histoplasmosis in a13-year-old girl: a case report. Afr Health Sci13:518–521
Weening JJ et al (2004) The classification of glomerulone-phritis in systemic lupus erythematosus revisited. Kid-ney Int 65:521–530
Wornle M et al (2006) Novel role of toll-like receptor 3 inhepatitis C-associated glomerulonephritis. Am J Pathol168:370–385
Yared G, Seidner DL, Steiger E, Hall PM, Nally JV (1999)Tunneled right atrial catheter infection presenting asrenal failure. JPEN J Parenter Enteral Nutr 23:363–365
Zaidan M et al (2016) Renal involvement during type 1cryoglobulinemia. Nephrol Ther 12(Suppl 1):S71–S81
Membranoproliferative Glomerulonephritis, Adult 15

Zand L et al (2013) C3 glomerulonephritis associated withmonoclonal gammopathy: a case series. Am J KidneyDis 62:506–514
Zand L, Fervenza FC, Nasr SH, Sethi S (2014) Membrano-proliferative glomerulonephritis associated with auto-immune diseases. J Nephrol 27:165–171
Zintzaras E, Voulgarelis M, Moutsopoulos H (2005) Therisk of lymphoma development in autoimmune dis-eases: a meta-analysis. Arch Intern Med165:2337–2344
16 M.P. Alexander and S. Sethi