m inmunologia
DESCRIPTION
MedicinaTRANSCRIPT


IntroducciónLa comprensión de las bases del recono-cimiento antigénico por los linfocitos B yT específicos es clave para entender la res-puesta inmune. El sistema inmune gene-ra un vasto elenco de complejos recepto-res distintos, cuyos dominios variables lespermite reconocer todos los posibles agen-tes patógenos o antígenos. Por ello se diceque los repertorios de reconocimientos in-mune son completos.Estos receptores también poseen regio-nes invariantes con dominios constantesque están involucradas en procesos deactivación de las células que los expre-san, y provocan respuestas efectorascomo la fijación de complemento, la op-soinización y otros mecanismos de de-fensa inmune.El receptor de antígeno de los linfocitos B(BCR) está compuesto por moléculas deinmunoglobulina (Ig) ancladas en las su-perficie celular. Estas moléculas tienen unaespecificación idéntica a los anticuerpossecretados por los linfocitos B después desu activación por el antígeno.Los linfocitos B activan la secreción de anticuerpos tras ser estimulados por su an-tígeno específico, al que reconocen pormedio de su receptor de membrana. Launión del antígeno al BCR es un paso cru-cial en la inducción de los linfocitos B aproliferar y diferenciarse en células se-cretoras de Igs. Un individuo puede pro-ducir cientos de millones de moléculas di-ferentes de anticuerpos en sus respuestas
inmunes, siendo cada una de ellas pro-ducto de sólo dos genes.En este capítulo se describirán las pro-piedades estructurales y funcionales de lasIgs y del BCR. Se explicará el especializa-do proceso genético que genera la enor-me diversidad de anticuerpos a partir deun grupo muy limitado de genes, los me-canismos por los que la unión del antíge-no al BCR señaliza los linfocitos B y la ad-quisición de los receptores de antígenodurante el proceso del desarrollo del lina-je B humano, y su mutación y edición du-rante la respuesta inmune.
Las inmunoglobulinas:dominios estructuralesy funcionales
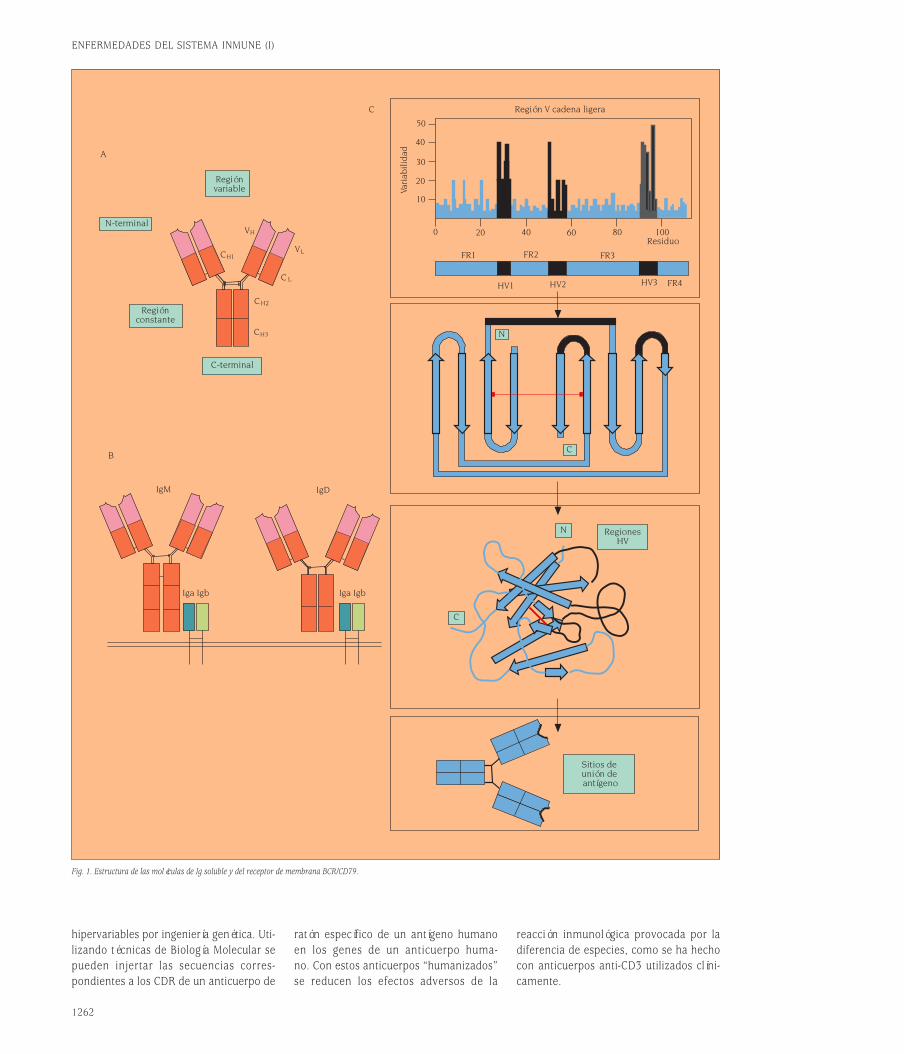
Los anticuerpos forman una familia de pro-teínas plasmáticas conocida colectiva-mente como inmunoglobulinas. Las Igs están constituidas por cuatro cadenas po-lipeptídicas, dos cadenas pesadas (H) y doscadenas ligeras (L). Son por tanto hetero-tetrámeros (H+L)2. Las dos cadenas pe-sadas están unidas covalentemente por unpuente disulfuro y cada cadena H está uni-da a una cadena L a través de otro puen-te disulfuro (fig. 1A)1.Las Igs tienen dos funciones separablesdesde el punto de vista de su estructura.Una es unir específicamente las molécu-las de antígeno para desarrollar una res-puesta inmune; la otra es reclutar otrasmoléculas y células para destruir el antí-geno unido. Estas dos funciones están es-tructuralmente separadas en la moléculade Ig. El dominio que reconoce y une el
antígeno se denomina región variable (V).El dominio de la molécula de Ig que asu-me las funciones efectoras se denominaregión constante (C). Existen cinco clasesde isotipos de Igs (IgM, IgD, IgG, IgA eIgE) definidas por la región constante desu cadena H (µ,δ,γ,α y ε). Sin embargo,existen sólo dos clases de cadena L, kap-pa (κ) y lambda (λ).La estructura terciaria de las cadenas po-lipeptídicas de las Igs forma dominios globulares característicos que son com-partidos por muchas otras estructuras relevantes en el sistema inmune. A todasestas estructuras con dominios globularessemejantes a los de las Igs se les englo-ba en la llamada superfamilia de las inmunoglobulinas. Como se ve en la fi-gura 1A, la asociación entre las cadenasH y L es tal que los dominios V respecti-vos (VH y VL) están emparejados y lo mis-mo ocurre con los dominios constantesCH1 y CL.La variabilidad de secuencia a lo largo delos dominios V no está distribuida regu-larmente. Muchos aminoácidos están con-servados, particularmente aquéllos im-portantes para el mantenimiento de laestructura de plegamiento del dominio.La distribución de residuos variables pue-de establecerse a través de un plot devariabilidad (fig. 1C). Tanto para las ca-
denas H como para las L pueden identi-ficarse tres regiones de alta variabilidadde secuencia que se han denominado re-giones hipervariables (HV1, HV2 y HV3).Los segmentos de secuencia entre las re-giones hipervariables son relativamenteinvariantes y se denominan regiones flan-queantes (FR). Las regiones HV se en-cuentran alejadas en la secuencia lineal,pero quedan muy próximas cuando la se-cuencia se pliega para formar el dominioV tridimensional. Además, cuando los do-minios VH y VL se emparejan, las regionesHV de cada dominio quedan muy próxi-mas creando un sólo sitio hipervariableen el extremo del dominio V, que consti-tuye el sitio de unión al antígeno (fig. 1C).Las regiones HV también se denominanregiones determinantes de complemen-tariedad (CDR) porque forman una su-perficie complementaria al antígeno. Porotra parte, la secuencia del antígeno quees reconocida por el anticuerpo se deno-mina epítopo antigénico.Es posible manipular la especificidad an-tigénica de una molécula de Ig simple-mente cambiando sus CDR o regiones
1261
RECEPTORES DEINMUNOGLOBULINASEN LINFOCITOS BY SUS PRECURSORESE. Sanz*,** y A. de la Hera*,**,****Departamento de Medicina. Universidad de Alcalá. Alcalá de Henares.**Unidad I+D Asociada al Consejo Superior de Investigaciones Científicas.***Centro de Investigaciones Biológicas. Madrid.
Medicine 2000; 8(25): 1261-1270
hipervariables por ingeniería genética. Uti-lizando técnicas de Biología Molecular sepueden injertar las secuencias corres-pondientes a los CDR de un anticuerpo de
ratón específico de un antígeno humano en los genes de un anticuerpo huma-no. Con estos anticuerpos “humanizados”se reducen los efectos adversos de la
reacción inmunológica provocada por ladiferencia de especies, como se ha hechocon anticuerpos anti-CD3 utilizados clíni-camente.
1262
ENFERMEDADES DEL SISTEMA INMUNE (I)
Iga IgbIga Igb
IgDIgM
B
C-terminal
Regiónconstante
C
C
C
V
VN-terminal
Regiónvariable
A
Sitios deunión de antígeno
Región V cadena ligera
50
40
30
20
10
0 20 40 60 80 100
FR1 FR2 FR3
Residuo
HV1 HV2 HV3 FR4
Vari
abil
idad
N
C
N RegionesHV
C
C
H
CH1L
L
H2
H3
Fig. 1. Estructura de las moléculas de Ig soluble y del receptor de membrana BCR/CD79.

El receptor B para antígeno (BCR):inmunoglobulinasy módulos de transducciónparalela CD79
Los linfocitos B activan la secreción deIgs tras la unión del antígeno al BCR es-pecífico. El BCR es un complejo receptorconstituido por una Ig anclada a la mem-brana celular (mIg) y asociada a un he-terodímero formado por dos proteínas, Igαe Igβ (o CD79), unidas por un puente di-sulfuro (BCR/CD79, fig. 1B). CD79 sirve auna función homóloga a CD3 en el com-plejo TCR/CD3 de las células T. PrimeroCD79a y CD79b son esenciales durante elensamblaje intracelular del complejo re-ceptor, las Igs y CD79 se tienen que aso-ciar para que el complejo pueda ser trans-portado a la superficie. Defectos en uno delos componentes pueden provocar la re-tención del resto de las subunidades en elcitoplasma de las células. Segundo, las Igstienen colas citoplásmicas muy cortas, incapaces de transducir señales por sí mismas al interior de las células, es el he-terodímero CD79 el encargado de la trans-ducción de señales de activación/aner-gia/apoptosis después de la unión BCR-an-tígeno, por ello se denomina módulo detransducción paralela. Estudios recientes de la estequiometría de estos módulosCD79ab realizados por Michael Reth et alhan demostrado que existe uno por com-plejo BCR, y no dos como se especulabahasta ahora. Los complejos que contienenIgM o IgD que coexpresan la mayoría delas células vírgenes de la sangre y órganoslinfoides periféricos son físicamente inde-pendientes, es decir IgM e IgD forman par-te de complejos BCR distintos cada uno consu módulo de transducción paralela Igα-Igβy no coexisten en un complejo único, comotambién han demostrado esos autores.
Genes deinmunoglobulinas:estructura, maquinariade recombinacióny mecanismosde generación de diversidadDurante los tres primeros cuartos de estesiglo fue un misterio saber cómo los anti-cuerpos eran capaces de reconocer espe-
cíficamente cualquier estructura molecu-lar. El descubrimiento de que la secuen-cia de nucleótidos de los genes determi-na la secuencia de las proteínas aumentóla expectativa, al ser, paradójicamente, mi-llones de veces mayor el número de re-giones variables de las proteínas (anti-cuerpos) que el número total de genes delgenoma humano. El clonaje por el equipodel Premio Nobel Susumu Tonegawa delos genes que codifican las cadenas pesa-das y ligeras de las Igs ha demostrado queel repertorio de reconocimiento antigéni-co se origina a partir de un número limi-tado de genes que generan diversidad através de un proceso de recombinaciónsomática, diversidad que posteriormentese complementa con un fenómeno de hi-permutación somática y de edición de re-ceptores.Los dominios V de las Igs están codifica-dos en vez de por un solo gen heredado,por la recombinación de tres segmentosgénicos llamados variables (V), de diver-sidad (D) y de unión (J)2. Los loci génicosde las cadenas de Igs en líneas germinal(tal como las heredamos de nuestros pa-dres a través de las células germinales:óvulos y espermatozoides) están consti-tuidos por un número elevado de seg-mentos V, varios segmentos D y variossegmentos J. Durante la recombinación so-mática un segmento V de todos los dis-ponibles se une a uno de los segmentosD y a uno de los segmentos J (fig. 2A).La secuenciación de esta región del geno-ma humano se ha completado hace tiem-po y se conoce el mapa cromosómico deesta región con precisión. Por ejemplo, enla cadena pesada el número de genes Ves 44 genes funcionales, agrupados en sie-te familias (se definen como miembros deuna familia los genes con más de 70% deidentidad de secuencia), y 79 seudogenes,hay 27 minigenes D agrupados en cuatrogrupos D1-4, y el número de genes J es 6.La selección de estos segmentos se haceal azar permitiendo un gran número decombinaciones posibles lo que genera unalto grado de diversidad, del orden de do-cenas de miles. Conviene subrayar que elmecanismo combinatorial es sólo uno en-tre los que contribuyen al polimorfismode los anticuerpos.El mecanismo de recombinación se des-cribe en detalle en las figuras 2B y 2C. Elconocimiento de la estructura de los locide inmunoglobulina va a facilitar los es-tudios de patología molecular humana.
El mecanismo de recombinación es, pues,similar al descrito para las células T. Pri-mero tiene lugar la recombinación DJ yposteriormente un segmento V se une aDJ. El proceso es idéntico para las cade-nas L (fig. 2C) excepto que hay un sólopaso de recombinación por la ausencia deelementos de diversidad (D) en las regio-nes que codifican las cadenas ligeras κy λ. En el proceso de recombinación in-tervienen al menos dos recombinasas,RAG-1 y RAG-2, que presentan similitudescon topoisomerasas bacterianas que cata-lizan la ruptura y unión del ADN. Los ge-nes RAG se expresan en dos oleadas en eldesarrollo B. Primero, inmediatamente an-tes de los reordenamientos DJH y VHDJH yluego con anterioridad al reordenamientoVLJL. También se han encontrado en célu-las B cultivadas in vitro con interleucina 4(IL-4) y en los centros germinales de gan-glios linfáticos de ratones inmunizados cé-lulas que expresan recombinasas RAG-1 yRAG-2 y suplantadores de cadena ligera.En estas células se producen los fenóme-nos de “editar” y “revisar” receptores porlos que se pueden cambiar los dominiosvariables de la cadena pesada o ligera deinmunoglobulina, de un clon que antes te-nía otra especificidad. Dichos mecanismosserán objeto de discusión en otros apar-tados de esta revisión.El proceso de recombinación es impreci-so en la elección del lugar exacto de cor-te y empalme entre genes V(D)J. Además,durante la recombinación del gen de ca-dena pesada, enzimas como la deoxinu-cleotidil transferasa terminal (TdT) –y qui-zá otras homólogas descubiertas muyrecientemente como la polimerasa µ– adi-cionan nucleótidos al azar en los extremosde los segmentos recombinados. La in-corporación de estos nucleótidos repre-senta un incremento en el grado de di-versidad además de una informacióngénica que no se hereda y constituye unmarcador clonal de interés terapéutico (porejemplo, en el diagnóstico de monoclona-lidad y de la enfermedad mínima residual).Este mecanismo de diversidad es cuanti-tativamente mucho más importante queel combinatorial puro, y es el responsablede que la región HV3 sea mucho más po-limórfica que las HV1 y HV2, que estáncodificadas por aminoácidos presentes enlos segmentos V, que existen en un nú-mero limitado. Hay millones a billones deposibilidades de “empalme” V(D)J, que ha-cen que puedan emplearse como marca-
1263
RECEPTORES DE INMUNOGLOBULINAS EN LINFOCITOS B Y SUS PRECURSORES

dor clonal ya que se estima que no se haceuna secuencia igual más que una vez du-rante la vida del individuo.Un mecanismo adicional de generación dediversidad sobre los dominios V es el dehipermutación somática, que opera des-pués de que los genes funcionales han sidoensamblados. La hipermutación somáticaocurre en los centros germinales y con-siste en la introducción de mutacionespuntuales al azar en los dominios VH y VL
cuando los linfocitos B responden a un an-tígeno. Si se producen cambios de basesque alteran la secuencia de aminoácidos
en los tres CDR que contactan con el an-tígeno éstas modifican la afinidad conaquél. El patrón de mutación selecciona-do o favorecido indica que el antígeno se-lecciona aquellos clones B que habiendomutado sus receptores en las regiones HVunen mejor el antígeno. Las mutacionesdesfavorables en los HV son contraselec-cionadas, y aquellas que ocurren fuera delos CDR pueden destruir la estructura delanticuerpo y difícilmente, salvo efectosalostéricos, pueden favorecer la unión alantígeno. Este mecanismo de hipermuta-ción somática ayuda a “madurar” la afi-
nidad de las regiones HV1 y HV2 por elantígeno, y compensar su baja diversidad,ya que heredamos en un número bajo ycombinación fija, al residir en uno de los44 genes VH funcionales.La tasa de mutación somática en los ge-nes de inmunoglobulina es tan alta comoen genes del virus de la gripe o del sín-drome de inmunodeficiencia adquirida(SIDA), millones de veces más alta que enotros genes humanos, y se piensa que ayu-da a generar repertorios completos encada momento de la vida del individuo.Esto ocurre con una apariencia adaptati-
1264
ENFERMEDADES DEL SISTEMA INMUNE (I)
Fig. 2. El reordenamiento de las cadenas H y L de las inmunoglubulinas genera diversidad.
Cadenapesada
Traducción
Splicing mARN
Transcritoprimario ARN
Transcripción
ADNreordenado
Recombinaciónsomática
ReordenamientoD-J
Recombinaciónsomática
ADNgerminal
12.000-60.000 combinaciones diferentes
B C
V D JA
Cadenaligera
Traducción
Splicing mARN
Transcritoprimario ARN
Transcripción
ADNreordenado
Recombinaciónsomática
ADNgerminal
L V J C
L V J C
L V J C
AAA
LV J C
AAA
VL
CL
VH
CH1
CH2
CH3
AAA
AAA
L V D J C
L D JV D C
L V D J C
L
V D J C
V D JC
Se selecciona1 de 200-1000
1 de 15 1 de 4
va, ya que como hemos discutido es el fru-to de mecanismos fuertemente selectivos.¿Por qué se necesitan métodos adiciona-les de generación de diversidad adicio-nales a la recombinación si el repertoriopotencial es de billones a trillones de Igsdistintas? (El conjunto de todas las espe-cificidades antigénicas de un individuoconstituyen su repertorio.) Aunque cadaindividuo cuenta con 1012 –un billón– delinfocitos B, sin embargo se estima que eltamaño de cada clon podría ser de media103-104 células, por lo que el número declones en un individuo “sólo” sería 108-109. Por la limitación de la cantidad de te-jido dedicado al sistema inmune, de unoa dos kilos, aunque el repertorio potencialde un individuo se estima en un trillón deespecificidades distintas, sólo se utiliza unaparte muy pequeña de aquél en el reper-torio actual en cada momento dado de lavida; simplemente porque no hay célulassuficientes para más diversidad.Por ello, podría ser muy recomendableevolutivamente para el sistema inmune delos vertebrados el desarrollo de mecanis-mos de rediversificación rápida del reper-torio que se usa activamente en la res-puesta inmune en marcha en cadainstante, para reseleccionar un anticuerpooptimizado para aclarar muy eficiente-mente –por su alta afinidad detectan muybajas concentraciones– los patógenos; yalmacenar luego ese subclón en la pobla-ción de células memoria. De acuerdo conla predicción de esta hipótesis, se ha de-mostrado experimentalmente que las cé-
lulas B activadas/memoria tienen una altafrecuencia de mutaciones somáticas, mien-tras que las células vírgenes tienen la secuencia que heredamos de la línea ger-minal. De esta forma, el repertorio utili-zado se optimiza excepcionalmente biena las circunstancias de la presión am-biental que imponen organismos que evo-lucionan individualmente mucho más rápido que la media del genoma, pero noque los genes de los receptores de antí-genos de la población de linfocitos que nos defiende. La obtención biotecnológi-ca de anticuerpos así optimizados de pa-cientes que se defienden con éxito de diversos cánceres, o enfermedades infec-ciosas como el SIDA, es una avenida deinvestigación en curso que puede ofertarinmunoterapia pasiva de nueva generaciónen procesos para los que el arsenal tera-péutico es hoy muy limitado.
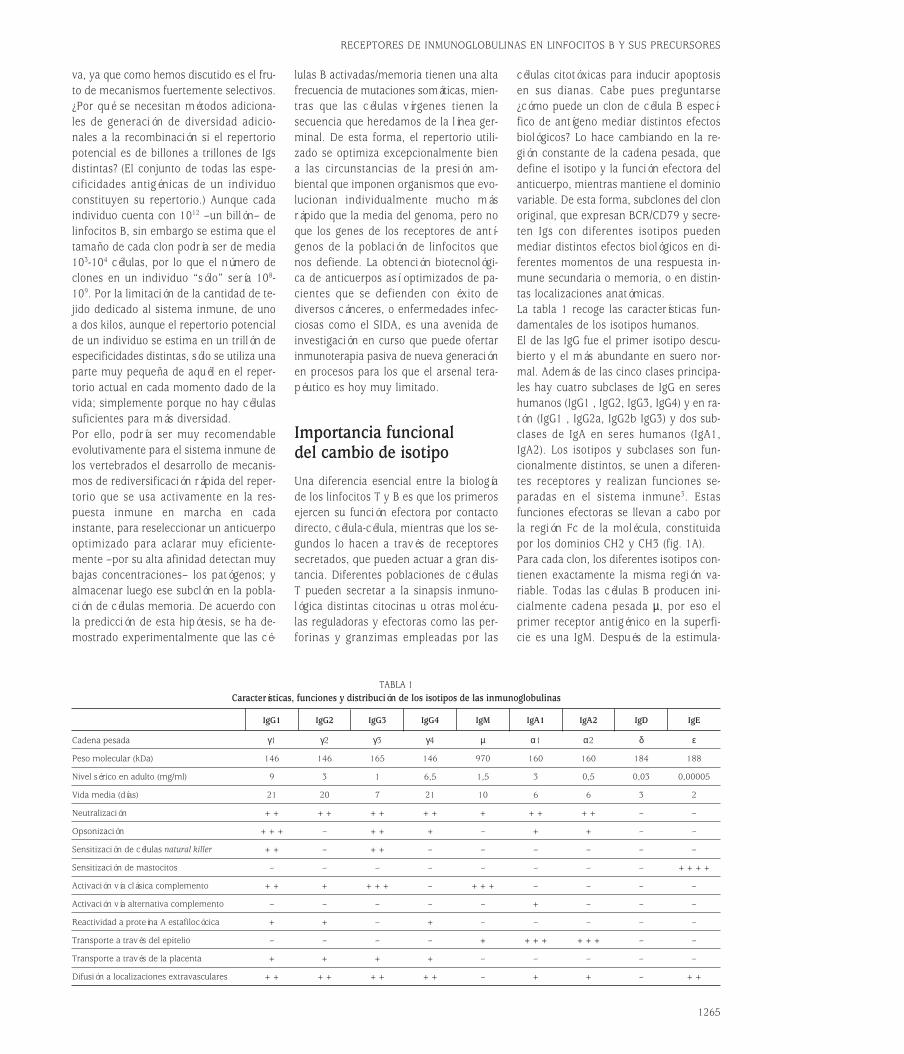
Importancia funcionaldel cambio de isotipoUna diferencia esencial entre la biologíade los linfocitos T y B es que los primerosejercen su función efectora por contactodirecto, célula-célula, mientras que los se-gundos lo hacen a través de receptores secretados, que pueden actuar a gran dis-tancia. Diferentes poblaciones de célulasT pueden secretar a la sinapsis inmuno-lógica distintas citocinas u otras molécu-las reguladoras y efectoras como las per-forinas y granzimas empleadas por las
células citotóxicas para inducir apoptosisen sus dianas. Cabe pues preguntarse¿cómo puede un clon de célula B especí-fico de antígeno mediar distintos efectosbiológicos? Lo hace cambiando en la re-gión constante de la cadena pesada, quedefine el isotipo y la función efectora delanticuerpo, mientras mantiene el dominiovariable. De esta forma, subclones del clonoriginal, que expresan BCR/CD79 y secre-ten Igs con diferentes isotipos pueden mediar distintos efectos biológicos en di-ferentes momentos de una respuesta in-mune secundaria o memoria, o en distin-tas localizaciones anatómicas.La tabla 1 recoge las características fun-damentales de los isotipos humanos.El de las IgG fue el primer isotipo descu-bierto y el más abundante en suero nor-mal. Además de las cinco clases principa-les hay cuatro subclases de IgG en sereshumanos (IgG1 , IgG2, IgG3, IgG4) y en ra-tón (IgG1 , IgG2a, IgG2b IgG3) y dos sub-clases de IgA en seres humanos (IgA1,IgA2). Los isotipos y subclases son fun-cionalmente distintos, se unen a diferen-tes receptores y realizan funciones se-paradas en el sistema inmune3. Estas funciones efectoras se llevan a cabo porla región Fc de la molécula, constituidapor los dominios CH2 y CH3 (fig. 1A).Para cada clon, los diferentes isotipos con-tienen exactamente la misma región va-riable. Todas las células B producen ini-cialmente cadena pesada µ, por eso elprimer receptor antigénico en la superfi-cie es una IgM. Después de la estimula-
1265
RECEPTORES DE INMUNOGLOBULINAS EN LINFOCITOS B Y SUS PRECURSORES
TABLA 1Características, funciones y distribución de los isotipos de las inmunoglobulinas
IgG1 IgG2 IgG3 IgG4 IgM IgA1 IgA2 IgD IgE
Cadena pesada γ1 γ2 γ3 γ4 µ α1 α2 δ ε
Peso molecular (kDa) 146 146 165 146 970 160 160 184 188
Nivel sérico en adulto (mg/ml) 9 3 1 6,5 1,5 3 0,5 0,03 0,00005
Vida media (días) 21 20 7 21 10 6 6 3 2
Neutralización ++ ++ ++ ++ + ++ ++ – –
Opsonización +++ – ++ + – + + – –
Sensitización de células natural killer ++ – ++ – – – – – –
Sensitización de mastocitos – – – – – – – – ++++
Activación vía clásica complemento ++ + +++ – +++ – – – –
Activación vía alternativa complemento – – – – – + – – –
Reactividad a proteína A estafilocócica + + – + – – – – –
Transporte a través del epitelio – – – – + +++ +++ – –
Transporte a través de la placenta + + + + – – – – –
Difusión a localizaciones extravasculares ++ ++ ++ ++ – + + – ++

ción antigénica, los linfocitos B proliferany pueden diferenciarse para producir otrosisotipos; así, la misma región V puede serutilizada por distintas regiones constantespermitiendo a los anticuerpos de una es-pecificidad determinada cambiar su fun-ción efectora. Después de IgM los linfoci-tos B coexpresan IgD en su superficie yposteriores pasos de maduración induci-rán el cambio a alguno de los otros isoti-pos existentes.El proceso de cambio de clase o cambiode isotipo no es el azar sino que está re-gulado por citocinas secretadas por linfo-citos T. Por ejemplo, se sabe que la IL-4 yla IL-13 son críticas para la producción deIgE. El cambio de isotipo no tiene lugar enindividuos deficientes en ligando de CD40(CD40-L), una molécula implicada en in-teracciones entre células B y T coopera-doras. Estos individuos sólo producen IgMy cuentan con niveles anormalmente al-tos de esta Ig en su plasma.Finalmente, las IgG pueden transportarsea través de la placenta directamente al to-rrente sanguíneo del feto durante la vidaintrauterina. Neonatos humanos presen-tan altos niveles de IgG en plasma con elmismo rango de especificidades que susmadres. Además, las IgA son secretadasen la leche materna y transferidas al ne-onato en la ingesta. Estas dos formas detransferir protección al recién nacido sonmuy importantes en un momento en elque el individuo es especialmente vulne-rable puesto que no ha tenido una expo-sición previa a patógenos externos y aúnno ha sintetizado su propio repertorio deanticuerpos.
Importancia de lasmoléculas correceptorasy coestimuladoras en laactivación de los linfocitos BEl receptor de antígeno no es suficientepara desencadenar por sí solo la activa-ción eficiente de los linfocitos B a funcio-nes efectoras. Se precisan, por una parte,los llamados correceptores para hacer queel reconocimiento antigénico sea sensibley, por otra, la participación de moléculascoestimuladoras, tanto solubles como re-ceptores de membrana celular, para lograrevitar los mecanismos de anergia y apop-tosis y poder desencadenar una respues-ta inmune efectora. En la actualidad, los
inmunólogos tratan de agrupar las seña-les de reconocimiento antigénico –dadaspor el receptor de antígeno y sus corre-ceptores– bajo el concepto de “señal 1”.Las señales coestimuladoras solubles,como las citocinas o el sistema de recep-tores y ligandos de membrana CD40/CD40L de las células B se agrupan bajo elconcepto de “señal 2”. La teoría de acti-vación por “dos señales” se desarrolla enla revisión realizada por Prieto et al, queaparecerá en la Unidad Temática n.º 26de esta 8.ª serie de Medicine; aquí revi-saremos la estructura de los receptores ycorreceptores implicados.Recordemos que el receptor de antígenode los linfocitos B es el complejo BCR/CD79 (fig. 1A), y que este complejo no setransporta a la membrana si no existe laasociación previa de la molécula de Ig conCD79. Además, aunque las Igs pudieranexpresarse en ausencia de CD79a y CD79ben la superficie de las células B antes delreconocimiento de antígeno, ni la formade IgM de membrana (mIgM) ni mIgD, ex-presadas en la superficie de linfocitos Bvírgenes o novatos, presentan secuenciasde aminoácidos en su corta cola intrace-lular capaces de proporcionarles inte-racciones con proteínas tirosina cinasas.Cada cadena H posee una región intraci-toplásmica de sólo tres aminoácidos, KVK.Como ocurre con el CD3 en el complejoreceptor de antígeno T, el CD79 actúacomo módulo de transducción paralelapues, aunque no reconoce directamenteel antígeno, transduce señales tras la ocu-pación del sitio de unión del BCR por elligando específico. Los dominios intraci-toplásmicos del CD79 contienen motivosITAM descritos en el capítulo de células T.A estos motivos se acoplan las proteínastirosina cinasas que transmiten las seña-les que se describen en la figura 3. Estu-dios muy recientes indican que las IgG po-seen dominios intracitoplásmicos mayoresque los de IgM, capaces de transducir se-ñales per se, además de aquellas realiza-das a través de las proteínas que se aso-cian a los ITAM de CD79. Esta diferencia,objeto de investigación en la actualidad,puede contribuir a explicar los distintosrequerimientos de activación entre célu-las en respuestas primarias y secundariaso memoria, en las que hay un cambio deisotipo.La actividad de las cinasas asociadas alBCR/CD79 está regulada por otras proteí-nas de superficie llamadas correceptores4.
Las células B activadas producen cantida-des muy limitadas de inmunoglobulinasen ausencia de otras señales originadaspor dichos correceptores. Las células dendríticas foliculares aumentan la sensi-bilidad de los linfocitos B al antígeno a tra-vés de la presentación de inmunocomple-jos. Los inmunocomplejos u antígenossolubles que hayan activado el fragmentoC3d del sistema de complemento puedenamplificar la “señal 1”. C3d es una molé-cula que se une a la superficie de los linfocitos B a través de un complejo co-rreceptor formado por al menos tres componentes, CD19/TAPA-1/CR2. El CR2,también llamado CD21, media la capaci-dad del sistema de complemento para aumentar la producción de anticuerpos enrespuesta a bajas concentraciones de an-tígenos in vivo. El mecanismo es eviden-te, CR2 es un receptor específico para elfragmento C3d que aumenta la avidez dela Ig por los antígenos recubiertos porcomplemento. La utilidad del CR2 paraayudar a las células B a discriminar entreantígenos propios y extraños tiene granimportancia ya que el sistema de com-plemento tiene sistemas de inactivaciónen células autólogas –para complementode la propia especie– pero no en orga-nismos patógenos extraños. De esta for-ma, los “receptores de reconocimiento depatrones”, como los de complemento o li-popolisacaridos (LPS) no sólo son esen-ciales en la inmunidad innata sino clavesen el correcto funcionamiento de la res-puesta inmune específica, mal llamadaadaptativa. Su importancia en patologíaestá teniendo un reconocimiento cre-ciente, no sólo en inmunodeficiencia sinoen otros procesos como los autoinmunes.Una consecuencia, indeseada médica-mente, de la capacidad del complementopara ayudar a discriminar material bioló-gico de otras especies es la activación decomplemento propio sobre la superficiecelular de xenotrasplantes, como discu-ten Ramón Merino y sus colaboradores enla Unidad Temática n.º 26 de esta 8ª se-rie de Medicine. Es importante subrayarque CR2/CD21 es una molécula que noseñaliza por sí misma sino a través deotras. CR2 cumple la función de unión aligando del complejo correceptor B, la fun-ción de señalización corresponde a mo-léculas asociadas a CR2 como CD19 yCD81. El efecto combinado del aumentode avidez por el antígeno y la confluen-cia de la ruta de señalización del corre-
1266
ENFERMEDADES DEL SISTEMA INMUNE (I)
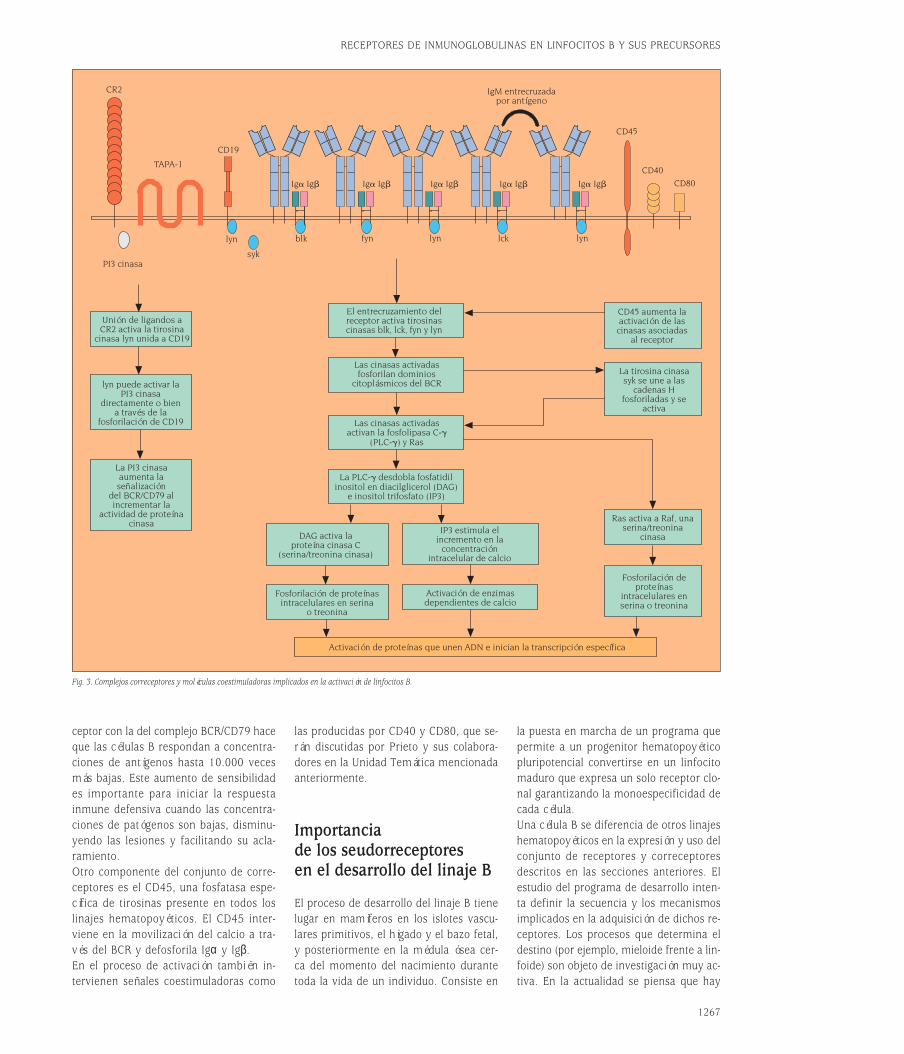
ceptor con la del complejo BCR/CD79 haceque las células B respondan a concentra-ciones de antígenos hasta 10.000 vecesmás bajas. Este aumento de sensibilidades importante para iniciar la respuesta inmune defensiva cuando las concentra-ciones de patógenos son bajas, disminu-yendo las lesiones y facilitando su acla-ramiento.Otro componente del conjunto de corre-ceptores es el CD45, una fosfatasa espe-cífica de tirosinas presente en todos loslinajes hematopoyéticos. El CD45 inter-viene en la movilización del calcio a tra-vés del BCR y defosforila Igα y Igβ.En el proceso de activación también in-tervienen señales coestimuladoras como
las producidas por CD40 y CD80, que se-rán discutidas por Prieto y sus colabora-dores en la Unidad Temática mencionadaanteriormente.
Importanciade los seudorreceptoresen el desarrollo del linaje B
El proceso de desarrollo del linaje B tienelugar en mamíferos en los islotes vascu-lares primitivos, el hígado y el bazo fetal,y posteriormente en la médula ósea cer-ca del momento del nacimiento durantetoda la vida de un individuo. Consiste en
la puesta en marcha de un programa quepermite a un progenitor hematopoyéticopluripotencial convertirse en un linfocitomaduro que expresa un solo receptor clo-nal garantizando la monoespecificidad decada célula.Una célula B se diferencia de otros linajeshematopoyéticos en la expresión y uso delconjunto de receptores y correceptoresdescritos en las secciones anteriores. Elestudio del programa de desarrollo inten-ta definir la secuencia y los mecanismosimplicados en la adquisición de dichos re-ceptores. Los procesos que determina eldestino (por ejemplo, mieloide frente a lin-foide) son objeto de investigación muy ac-tiva. En la actualidad se piensa que hay
1267
RECEPTORES DE INMUNOGLOBULINAS EN LINFOCITOS B Y SUS PRECURSORES
IgM entrecruzadapor antígeno
CD45
Iga IgbIga Igb
CD40
CD80
CD19
TAPA-1
CR2
lyn
PI3 cinasasyk
blk lyn
Unión de ligandos aCR2 activa la tirosina
cinasa lyn unida a CD19
lyn puede activar laPI3 cinasa
directamente o biena través de la
fosforilación de CD19
La PI3 cinasaaumenta laseñalización
del BCR/CD79 alincrementar la
actividad de proteínacinasa
DAG activa laproteína cinasa C
(serina/treonina cinasa)
Fosforilación de proteínasintracelulares en serina
o treonina
Activación de proteínas que unen ADN e inician la transcripción específica
Activación de enzimasdependientes de calcio
Fosforilación deproteínas
intracelulares enserina o treonina
Ras activa a Raf, unaserina/treonina
cinasaIP3 estimula el
incremento en laconcentración
intracelular de calcio
La PLC-g desdobla fosfatidilinositol en diacilglicerol (DAG)
e inositol trifosfato (IP3)
Las cinasas activadasactivan la fosfolipasa C-g
(PLC-g) y Ras
Las cinasas activadasfosforilan dominios
citoplásmicos del BCR
El entrecruzamiento delreceptor activa tirosinascinasas blk, lck, fyn y lyn
CD45 aumenta laactivación de las
cinasas asociadasal receptor
La tirosina cinasasyk se une a las
cadenas Hfosforiladas y se
activa
Iga Igb
lck
Iga Igb
lyn
Iga Igb
fyn
Fig. 3. Complejos correceptores y moléculas coestimuladoras implicados en la activación de linfocitos B.

genes, como Pax-5 para células B, que ha-cen irreversible el desarrollo hacia un li-naje a la vez que bloquean el desarrollohacia otro, fijando los patrones de desa-rrollo de las células pluripotenciales. El conocimiento de estos genes homeóti-cos puede ser de gran interés médico yaque puede permitir alterar el destino delas células, con aplicación en diversas cir-cunstancias como el cáncer o la terapiacelular y génica.Desde hace tiempo se conoce que las ca-denas H se recombinan antes que las L yen un alto porcentaje de clones precurso-res lo hacen de manera no productiva. Haconstituido un reto entender cómo las ca-denas H controlaban el proceso de desa-rrollo antes de la recombinación de las ca-denas L y cómo se discriminaba qué
clones de precursores habían realizado unarecombinación productiva. La solución aesta pregunta ha sido posible gracias aldescubrimiento de las cadenas suplanta-doras de H y L.Por un lado, se identificaron dos cadenaspolipeptídicas, VpreB y λ5, cuya asocia-ción forma una seudocadena ligera (ψL)que sustituye a la cadena L convencionalcuando ésta aún no se ha reordenado. Porotro, el complejo ψL se asocia con cade-nas ψH en linfocitos proB formando unprorreceptor y con cadenas H convencio-nales en linfocitos pre-B formando un pre-rreceptor. Estos seudorreceptores desem-peñan un papel importante, en lasuperficie de los precursores de los linfo-citos B en el tránsito de un estadio del de-sarrollo al siguiente5.
El estadio más temprano en el linaje B sedenomina pro-B/pre-BI (fig. 4) y se carac-teriza por progenitores que se están divi-diendo activamente, están reordenandoDHJH, expresan TdT y RAG-1 y -2 y en lasuperficie presentan VpreB y λ5 asociadasa una o varias proteínas, entre ellas unade aproximadamente 120 kDa de pesomolecular denominada ψH. Esta asocia-ción constituye el complejo ψHψL. Lacomposición final del prorreceptor no estácompletamente definida pues no se co-noce el número de cadenas asociadas aψL ni se ha clonado aún ψH. También sedesconoce el modo en que CD79 está asociado a este receptor o si transduce señales. Las células pre-BII grandes sonprecursores B que también se dividen ac-tivamente y están reordenando VHDJH.
1268
ENFERMEDADES DEL SISTEMA INMUNE (I)
Reordenamientos Marcadores Estadio
Cadenapesada
Cadenaligera
Intracelulares
Líneagerminal
Líneagerminal TdT
TdT
RAG-1
RAG-2
CD34
CD34 VpreB
CD10 l5
CD19 cH
CD79D JH H
Líneagerminal
V DJH H
Líneagerminal
TdT
RAG-1
RAG-2
Cm
CD19
CD79
VpreB
l5
TdT
RAG-1
RAG-2
CD19
CD79
VpreB
l5V DJH H
V JL L
TdT
RAG-1
RAG-2
CD19
CD79
sIgMV DJ V JH H L L
TdT
RAG-1
RAG-2
CD19
CD79
sIgM
sIgDV DJ V JH H L L
?
ProB/preBI
Precursorhematopoyético
Reordenamientono productivocadena pesada
Reordenamientoproductivo
cadena pesada
CD79
CD79
PreB II pequeñas
Reordenamientono productivocadena ligera
Reordenamientoproductivo
cadena ligera
sIgMCD79
B madura
sIgM
CD79
sIgDCD79
c c
cc
Superficie
+
++
+
+
+
++
+
+
-
+
+
+
+
_
_
_
_
_
_
+
+
_
_
_
_
_
_
+
+
+
+
+
+
+
PreBII grandes
B inmadura
+
+
_
+
Igd
CD79
Receptor Bcreditado
Fig. 4. Esquema de diferenciación de células B humanas en médula ósea. +: se expresa proteína; –: no se expresa proteína.

1269
RECEPTORES DE INMUNOGLOBULINAS EN LINFOCITOS B Y SUS PRECURSORES
Aquéllas en las que este reordenamientohaya sido productivo expresan en super-ficie un prerreceptor HψL asociado a Igα-Igβ y por tanto capaz de transmitir seña-les, aunque se desconoce si puede unirantígeno.En el siguiente estadio del desarrollo, elde las células pre-BII pequeñas, los linfo-citos han dejado de dividirse y han co-menzado el reordenamiento VLJL; por tan-to, en este estadio vuelve a expresarseRAG-1 y RAG-2. La célula ya está prepa-rada para producir una mIg funcional y sepierde la expresión de VpreB y λ5.La expresión en superficie de una IgM con-vencional (sIgM) define el estadio de cé-lula B inmadura, que se convierte en unlinfocito B maduro cuando coexpresa sIgMy sIgD.Se han encontrado pacientes SCID (seve-re combined immunodeficiency) humanosque no producen linfocitos B, en los queexiste una mutación en RAG-1 o en RAG-2 o en ambos. También se han diagnos-ti–cado inmunodeficiencias asociadas a lafalta de función de uno o varios de loscomponentes de los seudorreceptores descritas previamente en modelos ex-perimentales animales. Recientemente,nosotros y otros laboratorios hemos de-sarrollado sistemas para reproducir el de-sarrollo de linfocitos B humanos a partirde células madre de la sangre en sistemasin vitro6. Estos modelos pueden facilitar elestudio de los mecanismos de desarrollode las células productoras de anticuerposy su fracaso en distintas patologías hu-manas. Éstas no se limitan a la inmuno-deficiencia, sino a otros procesos más co-munes como la autoinmunidad en las quepueden ocurrir fallos autónomos celularesde los progenitores B en los que fracasanlos mecanismos de selección para tole-rancia a lo propio. Una posibilidad tera-péutica objeto en la actualidad de ensayoclínico es el trasplante de médula ósea. Elconocimiento de los defectos subyacentespuede permitir en un futuro más lejanotratamientos más selectivos e individuali-zados de terapia génica.
Procesos para editary revisar los receptoresde inmunoblobulinas
Hasta hace poco se pensaba que una vezadquirido el complejo BCR/CD79 cada clon
mantenía esa especificidad durante el res-to de su vida, salvo la mutación somáticapara maduración de afinidad ya discutida.Sin embargo, el proceso de recombinaciónse puede reactivar en tres entornos dife-rentes en la vida de un linfocito B. Uno,en los linfocitos vírgenes IgM+IgD– “reciénnacidos” en la médula ósea y emigrantesrecientes a los órganos linfoides periféri-cos; los linfocitos autorreactivos antes desufrir apoptosis para lograr tolerancia a lopropio pueden delecionar la recombina-ción VJ de su cadena ligera y hacer unanueva recombinación con un gen VL másdistal. La pareja (H+L)2 cambia de espe-cificidad que ahora puede no ser autoa-gresiva y permitir “rescatar” ese clon. Esteproceso se activa por el reconocimientoantigénico y pretende lograr tolerancia ala vez que optimiza el aprovechamientode los clones generados de nuevo cada díaal compensar un exceso de deleción clo-nal. En este proceso se reexpresan ade-más de RAG-1 y RAG-2 los suplantadoresVpreB y λ5, para permitir el recambio delas cadenas ligeras. Este proceso se ha des-crito recientemente también para la ca-dena pesada, y permite un cambio brus-co de la especificidad clonal, distinto dela deriva genética progresiva, con madu-ración que permite la mutación puntual.El poder generar nuevas especificidadespor los dos mecanismos tiene ventajas deaumentar la diversidad y evitar la autoin-munidad por edición de receptores auto-rreactivos. Sin embargo, el mecanismo decambio de la región variable de la cadenapesada recientemente descubierto operabajo un mecanismo distinto llamado revi-sión. En la revisión es la ausencia de es-tímulo antigénico en un clon lo que acti-varía de nuevo la recombinación, y elconsecuente cambio de especificidad. Ellaboratorio de Rajewsky descubrió re-cientemente que los linfocitos B precisande un estímulo constante, basal, a travésdel BCR/CD79 para sobrevivir. Dicha se-ñal no provoca expansión clonal o una res-puesta inmune efectora, pero es necesa-ria para evitar la apoptosis del clon. En elcurso de las respuestas inmunes, los clo-nes que dejan de ser estimulados parecen,de acuerdo a los resultados de Capra y suscolaboradores, estar sometidos a una pre-sión selectiva para revisar los receptoresrecombinando sus regiones VH de nuevo7.Merece la pena subrayar que a diferenciade la edición en que es el estímulo del re-ceptor BCR lo que “dispara” la recombi-
nación aquí es la ausencia de estímulo. Es-tos mecanismos de deriva genética no sóloofrecen ventajas al aumentar la diversidady reducir el aborto clonal, sino que supo-nen importantes riesgos biológicos con re-percusiones biomédicas. El cambio de laespecificidad clonal supone un riesgo ale-atorio de generar clones autoagresivos, yde hecho en enfermos con patología autoinmune se ha demostrado que estaposibilidad no es sólo teórica sino que ocu-rre en la práctica. Se postula en la actua-lidad que la estimulación crónica, prolon-gada, del sistema inmune favorece lagénesis de clones de tamaños inusual-mente grandes de los que existe una po-sibilidad estadísticamente significativa demutaciones autoagresivas, por mutaciónsomática y revisión de los dominios va-riables de los receptores de antígeno. Estateoría subraya la importancia de la infla-mación crónica como mecanismo subya-cente a múltiples patologías inmunológi-cas, y la de actuar en etapas tempranasde desencadenamiento inicial de la infla-mación para evitar procesos clásicos quede forma recidivante y progresiva provo-can cuadros prolongados de difícil cura-ción.
Complejos receptores deantígeno B y T: similitudesy diferencias
Los receptores de antígeno de las célulasT y B son homólogos en términos de suestructura y organización genética, y enlos medios por los que generan el diversorepertorio de cada tipo de receptor. Losmódulos de transducción de señales, CD3y CD79 en cada caso, presentan tambiénciertas homologías. En la tabla 2 se reco-gen comparativamente las característicasprincipales de ambos receptores.Una de las distinciones estructurales másimportantes reside en sus dominios cons-tantes. Por medio de un procesamiento al-ternativo de mensaje o splicing, la regiónC de una Ig puede variarse de manera quese produzca una mIg o una Ig secretada,mientras que los receptores T son siem-pre proteínas de membrana.La diferencia funcional más significativaentre los receptores T y B reside en la na-turaleza de los ligandos que reconocen.Las células B pueden unir moléculas solu-bles o de membrana de, virtualmente,

cualquier conformación. Las células T, sinembargo, reconocen péptidos y otras mo-léculas pequeñas en el contexto del MHC.Esta diferencia en el reconocimiento ase-gura que los anticuerpos interaccionen conagentes infecciosos extracelulares mien-tras que las células T detectan antígenosintracelulares procesados por las célulaspresentadoras de antígeno y presentadospor el MHC propio.Otra diferencia importante entre los dosreceptores es que el TCR posee varios mó-dulos de transducción paralela mientrasque el BCR sólo tiene uno. La capacidadde las IgG para señalizar a través de sudominio intracitoplásmico, cualidad hastaahora no descrita para el TCR podría jus-tificar la necesidad de más módulos detransducción paralela.
Los linfocitos B sonheterogéneos: células B1 y B2 o convencionales
Aunque no existan en el linaje B distin-tos receptores como el TCRγδ y TCRαβo subpoblaciones con distintos correcep-tores como CD4 y CD8 en las células T,no todas las células B se ajustan al esquema de desarrollo que se acaba dedescribir en esta revisión. Una poblaciónpequeña en el adulto pero funcional-mente muy significativa de células B de-nominadas B1a, aparecen muy tempra-no en la ontogenia y tienen un repertorioy unas propiedades funcionales diferen-tes. El resto de las células B se conocencomo B2. Las células B1a son la pobla-
ción predominante en los islotes vascu-lares primitivos y en cavidades meso-teliales como el peritoneo, expresan elmarcador CD5 y sIgM con poco o nadasIgD.Los receptores de estas células presentanpocas inserciones de nucleótidos, ya quela TdT no se expresa tan temprano en laontogenia. Además, las uniones V(D)J delas células B1a son mucho menos diver-sas que las de las B2 y los receptores yanticuerpos que producen tienden a serpoliespecíficos y con frecuencia autorre-activos.A medida que un individuo se desarrolla,las células B1a dejan de originarse a par-tir de precursores hematopoyéticos de lamédula ósea. En individuos adultos semantienen por autorreplicación en tejidosperiféricos donde son la diana de trans-formación tumoral en la leucemia más fre-cuente, la leucemia linfocítica crónica decélulas B.
BIBLIOGRAFÍA
1. Janeway C, Travers P, Walport M, Capra JD, eds. Immu-nología (1.a ed.). Barcelona: Masson, 2000.2. Paul W, ed. Fundamental Immunologiy (4.a ed). Filadel-fia: Lippincott-Raven, 1999.3. Natvig JB, Kunkel HG. Human immunoglobulin: classes.subclasses, genetic variants and idiotypes. Adv Immunol1973; 16: 1-59.4. Fearon DT. The CD79-CR2-TAPA-1 complex, CD45 andsignaling by the antigen receptor of B lymphocytes. CurrOpin Immunol 1993; 5: 341-348.5. Ghia P, ten Boekel E, Sanz E, de la Hera A, Rolink A,Melchers F. Ordering of human bone marrow B lymphocy-te precursors by single cell PCR analyses of the rearrange-ment status of the IgH and L chain gene loci. J Exp Med1996; 184: 2.217-2.229.6. Fluckiger AC, Sanz E, García-Lloret M, Su T, Hao Q-L,Kato R, et al. In vitro reconstitution of human B cells onto-geny: from CD34+ multipotent progenitors to Ig secreting.Blood, 1998; 92 (12): 4.509-4.520.7. Nemazee D, Weigert M. Revising B cell receptors. J ExpMed 2000; 191: 1.813-1.817.
1270
ENFERMEDADES DEL SISTEMA INMUNE (I)
TABLA 2Similitudes y diferencias entre los receptores de antígeno de las células T y B
BCR αβTCR
Origen Médula ósea Timo
Formas secretadas Sí No
Formas de membrana Sí Sí
Isotipos con funciones diferentes Sí No (variación CD3)
Superfamilia de las inmunoglobulinas Sí Sí
Múltiples sitios de unión Sí Sí
Regiones constantes distinta función Muchas Una
Reordenamiento VDJ o VJ Sí Sí
Hipermutación somática Sí, extensa Limitada
Editar y revisar receptores Sí, habitual Limitado
Uso de seudorreceptores Sí Sí
Uso de correceptores Sí Sí
Uso de módulos de señalización paralela Sí Sí
Reconoce antígeno soluble Sí No
Reconoce antígeno presentado por MHC No Sí
Activación por entrecruzamiento Sí Sí

IntroducciónLos linfocitos T constituyen un brazo efec-tor esencial en las respuestas inmunes hu-manas, y son responsables del control yeliminación de patógenos intracelularescomo virus y micobacterias, y de célulaspropias alteradas como células neoplási-cas. Sin embargo, las células T son unarma de “doble-filo”, ya que también sonresponsables de la patogenia de síndro-mes alérgicos, autoinmunes y del rechazode trasplantes. Las células T deben, pues,discriminar el contexto para desarrollarrespuestas efectoras inflamatorias y cito-tóxicas únicamente frente a antígenos in-deseables, mientras toleran o no respon-den en el contexto de los tejidos normales.La especificidad de las respuestas de lascélulas T la determina el reconocimientopor el receptor de antígeno, el complejoTCR/CD3. El ligando de este receptor sonfragmentos de antígeno presentados pormoléculas de histocompatibilidad (MHC).Es decir, a diferencia del complejo recep-tor de inmunoglobulinas en las células B,que reconoce antígenos líbres en el espa-cio extracelular, las células T reconocenlos antígenos de una forma “restringida”por el MHC; y, el complejo TCR/CD3 re-conoce no solo “antígeno” sino también ala molécula de histocompatibilidad. Ge-neralmente, existen dos rutas indepen-dientes de procesamiento de antígenos–extracelulares e intracelulares– que tie-nen un profundo impacto en la regula-ción del sistema inmune al afectar a la ac-tividad de dos subpoblaciones de linfo-citos funcionalmente distintas, las célulasT CD4+, cooperadoras (Th), y las células TCD8+, citóxico/supresoras (Tc).
En este capítulo consideraremos el estadoactual del conocimiento sobre el procesa-miento, presentación, y reconocimientode antígeno por las células T fruto de avan-ces recientes de la investigación en esteárea.
El receptor T para antígenoes un complejo compuestode módulos dereconocimiento y módulosde transducción paralela de señalesComo se observa en la figura 1, el recep-tor T para antígeno es un complejo de pro-teínas denominado TCR/CD3, compuestode módulos de reconocimiento TCR y detransducción de señales CD3/ζ . Existendos poblaciones de células T clasificadassegún los módulos de reconocimiento TCRque empleen. La mayoría (por ejemplo,>95%) de las células T presentes en losórganos linfoides convencionales empleareceptores con módulos αβTCR, y con-cordantemente se denominan células Tαβ.Una minoría de células T, aunque más fre-cuentes en las puertas de entrada en el or-ganismo como epitelios de superficies mu-cosas y piel, emplean receptores γδTCR.El descubrimiento más reciente de las cé-lulas Tγδ hace mayor nuestra ignoranciasobre la estructura y función de su siste-ma de reconocimiento, por lo que nos re-feriremos preferentemente a la biología delas células Tαβ, haciendo alguna observa-ción más puntual respecto a las célulasTγδ. Los módulos de recocimiento son he-terodímeros. Las cadenas αTCR y βTCRson proteínas integrales de membrana ycada una cuenta con dos dominios extra-
celulares de la superfamilia de las inmu-noglobulinas, que están conectados a laregión transmembrana a través de un“péptido de conexión” en el que existeuna cisteína para la formación de un en-lace disulfuro con la otra cadena del he-terodímero. Es importante observar quelas colas intracitoplásmicas que siguen ala región transmembrana son muy cortas,y no tienen per se capacidad de transdu-cir señales.Notablemente, los dominios del TCR másalejados de la membrana son de tipo va-riable (Vα y Vβ), por ser distintos de clona clon de células T, mientras que los máspróximos son de tipo constante. En la ac-tualidad se considera que existen dos mó-dulos de reconocimiento αβTCR en cadacomplejo receptor αβTCR/CD3, en lugarde uno como se suponía hasta ahora. Porconsiguiente, el receptor T para antígenosería multivalente –con varios sitios deunión para antígeno–, como lo es el de in-munoglobulinas en las células B.Mientras la función de unión específica alligando, el complejo antígeno-MHC, recaeen los módulos de reconocimiento TCR, latransducción de señales se realiza de for-ma paralela por tres módulos de trans-ducción de señales distintos (fig. 1). Dosson heterodímeros formados por proteí-nas codificadas por el locus génico CD3,los módulos CD3εγ y CD3εδ, el tercero esun homodímero codificado por el gen ζ(zeta). En los módulos de tipo CD3, en losque CD3ε se asocia de forma no covalen-te y mutuamente excluyente o alternativacon la subunidad CD3γ o CD3δ, cada ca-dena consta de un dominio extracelularde tipo constante de la superfamilia de in-munoglobulinas, que se continúa a travésde un péptido de conexión con la regióntransmembrana, y una extensa región ci-toplásmica que contiene los dominios res-ponsables de la señalización. Los domi-nios de señalización mejor caracterizadosson los motivos ITAM (Inmuno ThyrosineActivation Motif.), señalados como cajasazules en el esquema. Cada cadena CD3contiene un motivo ITAM. Es algo notableque, cada cadena del tercer módulo detransducción, el homodímero ζζ , tiene uncorto péptido de conexión extracelular, se-guido de la región transmembrana y unalarga cola intracelular que contiene tresdominios ITAM.Los motivos ITAM son secuencias comu-nes a otros receptores de activación de cé-lulas inmunes que se caracterizan por con-
1271
EL RECEPTOR DE ANTÍGENOEN LOS LINFOCITOS T Y LASMOLÉCULAS DE HISTOCOMPABILIDAD: PAREJAS INSEPARABLESA. de la Hera, G. Fernández-Miguel, E. Calvo-Alcocer y E. SanzDepartamento de Inmunología, Centro de Investigaciones Biológicas (CSIC) y Departamento de Medicina.Universidad de Alcalá. Madrid.
Medicine 2000; 8(25): 1271-1289

tener motivos del aminoácido tirosina conun espaciamiento definido entre sí (...YXX[L/V]XX7-11YXX[L/V]..., donde Y es tirosi-na, L es leucina, V valina y X un aminoá-cido cualquiera en código de una letra).Los ITAM son reconocidos específicamen-te por los dominios adaptadores de pro-teínas tirosina quinasa (PTK) implicadasen transadución de señales (por ejemplo,ZAP70, lck, etc), como será consideradopor Prieto y sus colaboradores en el capí-tulo de activación de linfocitos, que apa-recerá publicado en la Unidad Temáticanúmero 26 de esta serie de Medicine.
Un proceso de controlarquitectónico garantiza elensamblaje de los módulosde reconocimiento ytransducción de señales del complejo TCR/CD3La figura 2 resume el proceso de cons-trucción de los complejos TCR/CD3 ex-presados en la superficie celular. Los dis-tintos módulos se ensamblan primero porseparado y luego entre sí de una forma or-
denada, para crear complejos receptores“completos” como se ve a la izquierda delesquema. Existen sistemas moleculares dereconocimiento entre subunidades que fa-vorecen el apareamiento selectivo de cadamódulo primero, y de los módulos entresí después. Además, hay sistemas de re-tención intracelular de las cadenas aisla-das por secuencias aminoacídicas que seocultan con el ensamblaje, que impidenque los complejos “incompletos” seantransportados a la membrana plasmática,evitando la expresión de receptores nofuncionales.La expresión del resto de los componen-tes del complejo TCR/CD3 en la superficiecelular se reduce de forma importante, enlas inmunodeficiencias de una subunidad,cuando no se pierde de forma absoluta.Así, se ha demostrado que en células Tmaduras la expresión de αTCR, βTCR yCD3ε es estructuralmente esencial para laexpresión del complejo TCR/CD3 en la su-perficie celular. Por ello, las células congenes CD3ε, αTCR o βTCR defectuosos noexpresan el resto de las subunidades delcomplejo TCR/CD3 en su superficie. Sinembargo, en los últimos años también seha descubierto que se pueden transportar
a la superficie celular complejos TCR/CD3carentes de una de las siguientes cadenasCD3δ, CD3γ o ζ, aunque la cantidad de receptores presentes en la membrana plas-mática se reduce de 6-100 veces. Las células T normales expresan unos 30.000-45.000 complejos receptores TCR/CD3“completos” por linfocito T, siendo el nivel citado: normal “completo” > CD3δ–
> CD3γ− > ζ–. Aunque la carencia ab-soluta de una de dichas subunidades también provoca cuadros de inmunodefi-ciencia, al producirse bloqueos en la dife-renciación de la mayoría de las células T,una pequeña fracción de células T “esca-pan al control” y se diferencian en los in-dividuos inmunodeficientes para CD3δ,CD3γ y CD3ζ.Resultados recientes de nuestro laborato-rio, y otros, indican que los individuos nor-males –no inmunodeficientes- también tenemos pequeñas subpoblaciones de cé-lulas T que carecen de una de dichas subunidades, y que CD3δ y CD3γ se ex-presan de forma mutuamente excluyenteen una porción de los receptores de la ma-yoría de las células. Estos resultados im-plican que los receptores TCR/CD3 de lascélulas T no son homogéneos en su com-posición de módulos de transducción; yque existen receptores sin módulo CD3εγo sin módulo CD3εδ en células funciona-les y con un número normal de comple-jos TCR/CD3 en su superficie. Se sabe queCD3γ y CD3δ desempeñan un papel fun-cional distinto durante el desarrollo de loslinfocitos T en el timo, como detallaremosmás tarde, pero no se conoce si tienenuna función también distinta o específicaen células T maduras. Caso de que CD3γy CD3δ tengan funciones especializadas,como presumimos, la regulación diferen-cial de su expresión tendría consecuenciasfuncionales relevantes en medicina. Des-graciadamente, los anticuerpos anti-CD3humano disponibles en la actualidad re-conocen el dominio de inmunoglobulinasde CD3ε, por lo que no discriminan entrelos dos módulos CD3: CD3εγ y CD3εδ alreconocer el componente común. Los an-ticuerpos anti-CD3 provocan activación ysupresión de las respuestas T, en funciónde que se empleen dosis bajas o altas, res-pectivamente, de anticuerpo. Además seunen a ambos módulos con distinta afini-dad. Aunque sea tentador pensar que am-bas actividades funcionales se pueden se-parar a través del empleo de reactivos–anticuerpos- específicos de módulo, se
1272
ENFERMEDADES DEL SISTEMA INMUNE (I)
e d a b b
Va Vb
S-S
S-S
aCa C
z z
g
Vb Va
e
+
S-S
-
-+
+ ++
+- ---
b C C
Fig. 1. Modelo mínimo de receptor T bivalente con una estructura decamérica que permite una unión multivalente y el aparea-miento y neutralización de los aminoácidos cargdos positiva y negativamente presentes en las regiones transmembrana de cadasubunidad del complejo TCR/CD3.

necesitan trabajos de investigación adi-cionales para substanciar esta hipótesis, ycaracterizar y producir dichos reactivos.Durante la diferenciación de las células Ten el timo hay dos excepciones a la reglaque hace esencial las cadenas TCR en elensamblaje del complejo TCR/CD3, que seesquematizan a la derecha de la figura 2.Una es la expresión en la superficie de lascélulas pre-T del llamado “complejo pre-rreceptor” que tiene todos los componen-tes a excepción de αTCR. Ello es posi-ble por la existencia de proteínas, comopreTα, suplantadoras de la “necesidad” deαTCR en el complejo prerreceptor. Dichasproteínas se expresan selectivamente enprecursores T, y no requieren recombina-ción somática para su expresión comoocurre con las cadenas TCR. La expresiónde este prerreceptor permite seleccionaraquella subpoblación de células pre-T queexpresan una cadena βTCR funcional eneste estadio del desarrollo; y constituye un
“punto de control” de la diferenciación detimocitos, como detallaremos más ade-lante.La otra excepción, recogida en la esquinasuperior derecha del esquema de la fi-gura 2, ocurre en precursores más tem-panos, células pro-T, que no expresan nicadenas αTCR ni cadenas βTCR, aunquesí tienen pequeñas cantidades, casi inde-tectables, de CD3 en su superficie asocia-das a la chaperonina calnexina, que par-ticipa en la retención de complejosincompletos en el retículo endoplásmico.Se está investigando si este último recep-tor, prorreceptor T con sólo CD3, tiene al-guna significación fisiológica.Es importante subrayar que las secuenciaso motivos de contacto entre subunidadesque permiten el apareamiento ordenadode los componentes del complejo en lasparejas que constituyen los módulos, pri-mero, y el ensamblaje del complejoTCR/CD3, después, no sólo son importan-
tes en el ensamblaje sino también en latransducción de señales per se. Recorde-mos que los módulos αβTCR reconocen alantígeno-MHC pero no transducen seña-les. En los últimos años se están caracte-rizando motivos en los péptidos de cone-xión, regiones transmembrana y dominiosextracelulares cuya función capital seríatransferir la información obtenida por elTCR de que el receptor está ocupado porsus ligandos a los módulos de transduc-ción paralela, para que éstos señalicen. Seha demostrado que mutaciones en esosmotivos (por ejemplo, péptidos de cone-xión) pueden afectar a la señalización, in-terrumpiendo parte de la cascada de transducción de señales o eventos de ac-tivación (por ejemplo, secreción de inter-leucina-2), sin afectar a otros eventos deseñalización/activación y manteniendo losniveles normales de receptor expresadoen la superficie. La caracterización de losmotivos que permiten la transducción pa-
1273
EL RECEPTOR DE ANTÍGENO EN LOS LINFOCITOS T Y LAS MOLÉCULAS DE HISTOCOMPATIBILIDAD: PAREJAS INSEPARABLES
b
InmunoincompetentesComplejo (TCR) /CD3inmunocompetente Complejo clonotípico
con pseudocadenaspre-TCR/CD3
Complejo CICno clonotípicopro-TCR/CD3
Calnexina
Células d Células g Células z
caa b
e d g e
a b
g e
a b
e d
a
e d g e
zz
MP
zz zz
b
g ee d
¿
MP
e d g e
Expresión reducida Lisosoma
ed(ab) gezz
ed(ab) ge
Golgi
CD3CD3
dg
TCR
TCR
a
b
CD3 e
Degradaciónprelisosomal
ed(ab) gezz
Complejosdeficientes en z
Compartimentointermedio
ed(ab) gezz
Complejos TCR/CD3 "incompletos"(deficientes en una cadena)
Calnexina
Calnexina
t= 40 min t= 2 min
ca**ca*
ed
eg g
e
d
a z b
Retículoendoplásmico
n
nn
n
n
zz
Fig. 2. Representación esquemática y resumida del proceso de ensamblaje intracelular del complejo receptor TCR/CD3. También engloba complejos presentes en la superficie de precursores de célu-las T en diferentes estadios de su maduración (zona superior derecha) y complejos presentes en la superficie de células T maduras (zona superior izquierda). Los asteriscos indican el inicio (*) y laterminación (**) de la expresión de ψα. MP indica membrana plasmática y t indica los tiempos promedio de duración de las etapas de biosíntesis.

ralela de diversas señales entre las subu-nidades del complejo TCR/CD3 puede abrirvías insospechadas para el diagnóstico mo-lecular de la patología de la activación Ten pacientes con niveles de expresión nor-mal del receptor, y facilitar el desarrollode nuevas estrategias de intervención te-rapéutica.Finalmente, haremos una breve referen-cia a los receptores de las células Tγδ. Lasinvestigaciones realizadas hasta ahora in-dican que la estructura general del com-plejo TCR/CD3 es similar, con módulos dereconocimiento γδTCR a los que se aso-cian tres módulos de transducción para-lela de señales: CD3εγ, CD3εδ y ζζ . Se des-conoce, sin embargo si el receptor Tγδ esmono o multivalente como el de las célu-las Tαβ. Un descubrimiento interesante esque los motivos de los péptidos de cone-xión (CPM) de las cadenas αβTCR y γδTCRson distintos, y no intercambiables. Comodichos motivos están implicados en latransducción de señales a los módulosCD3, el descubrimiento sugiere diferen-cias en los mecanismos de señalización deambos receptores. En este sentido, noso-tros y otros autores hemos demostradoque la cadena CD3δ es prescindible parael desarrollo y funcionamiento de las cé-lulas Tγδ, aunque forme estructuralmenteparte de su receptor T, mientras que esesencial para la expresión y función nor-mal de la mayoría de las células Tαβ. Denuevo se requieren estudios adicionalespara substanciar estas diferencias y podertomar ventaja médica de ellas en el diag-nóstico y terapia. Sin embargo, el descu-brimiento de diferencias tan profundas enla biología de las células Tαβ y Tγδ puedeayudar a diferenciar las funciones inmu-nológicas que puedan desarrollar selec-tivamente ambas subpoblaciones de células T.
Los dominios variablesVαTCR y VβTCR tienen tresgiros hipervariablesresponsables de la uniónespecífica, y los dominiosconstantes Cα y Cβ creanuna cavidad que aloja los módulos CD3Como se muestra en la figura 3, se cono-ce la estructura tridimensional a nivel ató-
mico del módulo de reconocimientoαβTCR que ha sido cristalizado reciente-mente. Es interesante destacar que cadauno de los dominios variables tiene tresgiros hipervariables (HV, en gris) que mi-ran hacia arriba, donde se situa la zonade contacto con el complejo antígeno-MHC. Las regiones marco, con hebras betaantiparalelas –en amarillo en el esquema–,están conservadas aunque se trate de undominio variable, y sirven a una funciónde soporte estructural que es colocar losseis giros hipervariables, tres en αTCR ytres en βTCR, en un área plana muy pe-queña contiguos en el espacio. Por lo tan-to, a nivel de estructura de proteínas losreceptores T expresan su diversidad entres segmentos discontinuos de su se-cuencia –estructura primaria- en cada unade dos cadenas distintas αTCR y βTCR. Alparticipar dos cadenas en la creación delsitio de unión se dice que éste es combi-natorial. El que dichos segmentos HV es-tén en giros implica que toleran muchasposibilidades de secuencia y conformación–estructura secundaria- (fig. 3, arriba).Como hemos recalcado, a nivel espacial–estructura terciaria y cuaternaria- se dis-ponen contiguamente en un área planaelíptica, y en este sentido es importantecontemplar que los dos giros HV3, deαTCR y βTCR, ocupan una posición cen-tral en dicha área de contacto, mientrasque los correspondientes HV1 y HV2 ocu-pan una posición períférica.Los módulos de reconocimiento TCR notransducen señales por sí mismos, lo ha-cen a través de su asociación con los mó-dulos de transducción paralela. La flechaazul en la figura 3 indica una cavidad -en-tre el dominio Cβ y Cα, bajo el “codo” oprotrusión de βTCR– donde se propone enla actualidad que se aloja el dominio ex-tracelular de la subunidad CD3ε de los mó-
dulos de tipo CD3: CD3εγ o CD3εδ. Ade-más de esta asociación entre los dominiosextracelulares de CD3 y TCR existen inte-racciones entre los péptidos de conexióny las regiones transmembrana de las su-bunidades TCR y CD3/ζ. En este sentido,se ha postulado que los dos péptidos deconexión del heterodímero ζ-ζ se situa-rían engarzando las dos cadenas TCRβ delsuperdímero (αβTCR)2 (fig. 1). Todo ellopermite la transducción paralela de seña-les a los tres módulos de reconocimientotras el reconocimiento del complejo antí-geno-MHC específico por el TCR.
Los dominios variables segeneran por recombinaciónsomática de minigenes V,(D) y JLos genes que codifican los dominios va-riables del módulo de reconocimientoαβTCR, y el γδTCR no se heredan en unaconformación susceptible de ser expresa-da, en contraste con lo que ocurre con lamayoría de los genes humanos. Los genesque codifican los dominios V de proteínase generan tras una recombinación so-mática de minigenes V, (D) y J durante ladiferenciación de los progenitores de cé-lulas T a partir de células madre de la san-gre (fig. 4). El sistema es combinatorial,existen varios elementos de variabilidad(V), diversidad (D) y unión (J) lo que per-mite escoger diferentes combinaciones dedichos elementos. En el genoma humano,se estima que existen unos 52 genes quecodifican segmentos génicos Vβ, 2 Dβ y 13segmentos Jβ, que son los responsables decodificar los dominios TCRVβ, por recom-binación V-D-Jβ. En el caso de los domi-nios TCRVα no hay elementos D, y se
1274
ENFERMEDADES DEL SISTEMA INMUNE (I)
Fig. 3. La flecha celeste apuntahacia la “cueva” del cristal delmódulo αβTCR que representala localización propuesta paralos módulos CD3. La oquedadtiene el tamaño de un dominiode tipo inmunoglobulina redu-cido y está situada entre elcodo de TCRβ por arriba, lascaras internas del dominio Cβpor delante [la situación lejosdel lector] y Cα por detrás [la si-tuada cerca del lector]. TCRβestá en primer plano y TCRα,en segundo plano.

calcula que hay ,70 elementos Vα y 61elementos Jα, que crean productos de re-combinación V-Jα.Se sabe que TCRδ tiene una estructura si-milar a TCRβ, y TCRγ a TCRα. Es impor-tante observar (fig. 4) que el locus génicoque codifica TCRδ está “incluido” entre Vα
y Jα. De esta forma puede operar uno delos mecanismos que garantizan la llama-da “exclusión alélica” entre el uso de re-ceptores αβTCR y γδTCR por distintas sub-poblaciones de células T. Si una célula
precursora T recombina Vα con Jα para darlugar a una cadena αTCR, elimina el ADNgenómico del locus TCRδ de ese cromo-soma impidiendo así la coexpresión deambos genes –TCRα y TCRδ–, aun en elcaso de que δTCR estuviera ya recombi-nado y se pudiese expresar previamente.El proceso de recombinación es ordena-do, en células Tαβ ocurre primero en ellocus TCRβ, y en primer lugar se recom-bina uno de los genes Dβ con Jβ. A conti-nuación se produce la recombinación de
un elemento Vβ con el segmento D-Jβ an-tes recombinado. Finalmente, una vez re-combinado TCRβ se procede a realizar larecombinación Vα-Jα que permite generaruna cadena TCRα.El proceso de recombinación somática quegenera los dominios variables del TCR esposible gracias a la existencia de “se-cuencias de reconocimiento de señales”(SRS) que flanquean a los genes V, (D) y Jen el lugar que se corta y empalma el ADNexistente entre ellos. Las SRS son se-
1275
EL RECEPTOR DE ANTÍGENO EN LOS LINFOCITOS T Y LAS MOLÉCULAS DE HISTOCOMPATIBILIDAD: PAREJAS INSEPARABLES
Configuracióngénica
germinal
V V D J C
V V J C
b
a
CD3 se expresaen superficie
d
V V J Ca
a
V V J C
V D J C
b
b
D
V V J Ca
d
Reordenamiento
Vb
DJb
Reordenamiento
Db
Jb
b se expresaen superficie
Detenciónreordenamiento
b si esproductivo
V D J Cb
InducciónCD4-CD8
Comienzatranscripción a
Reordenamiento
Va Ja
a bse expresanen superficie
HSC/Protimocito CD3
a
bca cb?
CD3?
CD3
Timocito CD4 8
Timocito CD4 8CD3
a
b
CD3
ca b
b productivo b no productivo
Muerte programada
b b
Timocito CD4 +8
+
CD3+
a bs+
CD4
CD8
Comienzan eventos de selección positiva y negativa del TCR/CD3 y correceptores
por MHC
+
+
VJ J C
Calnexina
CD3
ca b
:
:
Fig. 4. La adquisición del módulo de reconocimiento αβTCR es un proceso ordenado que requiere recombinación génica.

cuencias consenso de bases o motivosconservados en todos los genes V, (D) y J.Los SRS están compuestos por heptáme-ros y nonámeros de bases de ADN sepa-radas entre sí por espaciadores de 12 ó23 bases, y están situadas en los intronesinmediantamente vecinos a los genes ci-tados, es decir 3’ de V, 5’ y 3’ de D y 5’de J. Estas secuencias tienen una especi-ficidad que impide una recombinación di-recta V → J en el locus TCRβ y favorecenla recombinación ordenada facilitando la“accesibilidad” secuencial de las regionescorrespondientes a los sitios de recombi-nación.En los ultimos años se ha demostrado quedichas secuencias son reconocidas espe-cíficamente por dos enzimas con acti-vidad recombinasa, llamadas RAG-1 yRAG-2, que son las responsables de la re-combinación somática, al azar, con múl-tiples combinaciones, de los genes V, D yJ. Esta maquinaria de recombinación escomún a los receptores γδTCR y las in-munoglobulinas. Sin embargo, las célulasnatural killer (NK) no precisan esta ma-quinaria enzimática para expresar sus re-ceptores KIR y KAR, capaces de recono-cer el MHC de forma análoga a comohacen los TCR.Cuando las enzimas RAG-1 y RAG-2 reco-nocen las SRS torsionan el ADN para for-mar un complejo en el que una pareja degenes D y J o V y D de entre el conjuntode los que se pueden recombinar se apro-ximan, en lugar de estar muy distantescomo ocurre en la configuración que sehereda a través de las células germinales,y se mantiene en el resto de células delorganismo. Todo el ADN intermedio for-ma un bucle o lazo hacia fuera del cro-mosoma para permitir que los genes a recombinar queden íntimamente vecinos.A continuación, RAG-1 y RAG-2 cortan ladoble cadena de ADN entre cada uno delos dos genes a recombinar y la secuen-cia SRS reconocida, realizando dos cortescompletos a la doble hebra del ADN (DSB).Este proceso genera por lo tanto dos frag-mentos cuyos extremos contienen las secuencias codificantes de los genes a recombinar –D y Jβ, V y Dβ o V y Jα– (lla-mados en inglés coding ends), y un tercerfragmento de ADN cuyos dos extremosterminan con las SRS que han “marcado”el lugar de corte (los llamados signal ends).Este último fragmento contiene todo elADN que ocupaba en el cromosoma unaposición intermedia o interpuesta entre los
segmentos génicos V(D)J que se van a re-combinar. A continuación, los dos extre-mos codificantes se unen entre sí em-pleando la maquinaria celular de repara-ción de roturas de doble cadena (DSB),con lo que se repara el cromosoma intro-duciendo una recombinación somáticaV(D)J. Estas enzimas son comunes a la re-paración de roturas DSB por RAG-1/2 yotras causas comunes de rotura como irra-diación. Finalmente, se unen los extremoscon las SRS correspondiente a los signalends formando un anillo de ADN que que-da excluido del cromosoma, y sin posibi-lidad de replicación con la división celu-lar al ser una estructura cromosómica sincentrómeros ni telómeros.Hasta la fecha no se han demostrado re-combinaciones intercromosómicas poreste mecanismo en células normales, gra-cias a la estructura del complejo de re-combinación que tiende a evitarlas. Sinembargo, esta posibilidad existe, aunquecon una frecuencia muy baja de error, yse especula que la actividad de recombi-nasas en células tumorales pudiera tenerun papel en la existencia frecuente detranslocaciones cromosómicas con valorpatogénico en diversas neoplasias. En al-gunos casos como en el oncogén SCL (stemcell leukemia) se ha propuesto que las re-giones translocadas contienen secuenciashomólogas a las SRS típicas de los genesV, (D) y J, lo que podría favorecer la trans-locación aberrante más frecuente a dichosloci protooncogénicos.El conocimiento de este mecanismo tieneinterés médico por varios motivos. Por unaparte, los defectos de la maquinaria de recombinación y reparación de ADN implicada en la recombinación V-(D)-J pro-vocan cuadros de inmunodeficiencia com-binada severa (SCID), al ser necesariospara formar células T y B. Estas últimasno pueden diferenciarse a partir de célu-las madre de la sangre, ni vivir una vezdiferenciadas, sin sus receptores de antí-geno. Se han descrito mutaciones de losgenes RAG que provocan SCID en sereshumanos, bien con fenotipos clásicos (conbloqueo completo de la recombinación yausencia total de linfocitos T y B), bienatípicos como el síndrome de Omenn. Enéste, el déficit de linfocitos es parcial de-bido a una pérdida de eficiencia de la re-combinación; aunque ésta ocurre en me-nor grado no es nula. En estos pacientesse forman células T y B en cantidades re-ducidas que subsecuentemente sufren pro-
cesos de selección anormal con anergia yactivación de las células T oligoclonales, ehiperprodución de IgE e hipereosinofiliaque son reponsables de las especificida-des del cuadro clínico en dichos pacientesinmunodeficientes con síndrome deOmenn.Este ejemplo ilustra cómo además de lasmutaciones con pérdida total de función–que son las que en la actualidad se es-tudian habitualmente en modelos anima-les con destrución génica intencionada,también llamados KO, de knock out– seobservan con frecuencia en la clínica hu-mana diversas mutaciones con pérdidaparcial de función que dan manifestacio-nes clínicas distintas entre sí, y a vecescontradictorias con los modelos animalesKO. En cualquier caso, el conocimiento ac-tual de la maquinaria de recombinación yreparación ya permite el diagnóstico mo-lecular de algunas de estas patologías, quepuede ser realizado a nivel prenatal encaso de riesgo familiar. Hoy en día se pueden estudiar a nivel genético clínicoRAG-1 y RAG-2 además del complejo dela proteína quinasa dependiente de ADN(DNA-PK) implicada en reparación de DSB,y que consta de una subunidad catalítica(DNA-PKcs) y un heterodímero (Ku70/Ku80) que reconoce el ADN en los extre-mos cortados. Un hecho interesante es quela investigación de pacientes humanos conSCID con sensibilidad aumentada a la irra-diación está conduciendo al descubri-miento de nuevos componentes de la ma-quinaria de recombinación/ reparación, enpacientes con actividad normal de loscomponentes conocidos.Ello demuestra, una vez más, que la in-vestigación clínica también tiene interésfundamental, y reafirma la importancia delestudio de la heterogeneidad de los me-canismos moleculares de enfermedad.Desde un punto de vista clínico, esta he-terogeneidad molecular puede provocardistintas manifestaciones clínicas, por loque tiene indudable importancia aplicada.En un futuro, los avances del conocimientoposibilitarán un diagnóstico individual ytratamientos de terapia génica de formauniversal en los sistemas nacionales de sa-lud, y no solo en los centros de élite enlos que se realizan hoy los ensayos. Estefuturo es casi presente, ya que el desa-rrollo de tecnología que está acompañan-do al programa de genoma humano estápermitiendo que accedamos acelerada-mente a una serie de aplicaciones biomé-
1276
ENFERMEDADES DEL SISTEMA INMUNE (I)

dicas que ayer parecían futuristas. Porejemplo, la creación de chips de ADN quehoy “sólo” pueden incluir 10.000 genes,o un 10% del genoma humano, algo im-pensable hace sólo unos años, y que aplicando métodos de nanomecánica yapueden discriminar incluso mutacionespuntuales. La posibilidad de incluir pane-les o paletas de genes (gene arrays) en sis-temas automatizados de diagnóstico pue-de permitir que la idea formulada haceaños por el premio Nobel Jean Dausset deuna Medicina “predictiva” se convierta en una realidad asistencial común en lapróxima década. Este caso aislado lo to-mamos casi como una provocación parailustrar una tendencia común a muchasáreas de la medicina, y que sospechamosmuchos de los lectores jovenes de las Mo-nografías de Actualización Médica MEDI-CINE tendrán la oportunidad de aplicar co-tidianamente en su ejercicio profesional.Otra observación que ya está encontran-do aplicación médica es la generación de“círculos” que contienen el ADN que se haeliminado del cromosoma y que tiene loselementos del locus TCR que ocupaban lasecuencia interpuesta entre los dos seg-mentos recombinados. Este ADN extra-cromosómico carece de capacidad repli-cativa y, consecuentemente, se pierde enuna de las dos células hijas con la divisióncelular. La frecuencia de células con estoscírculos es alta entre las “emigrantes re-cientes” del timo a la perifería, y las cé-lulas vírgenes. Sin embargo, la frecuenciao porcentaje de células con dichos “círcu-los” cae por dilución de forma exponen-cial con la replicación de las células acti-vadas o memoria. Hay situaciones clínicascomo el síndrome de inmunodeficienciaadquirida (SIDA) o el trasplante de médu-la ósea, en las que es importante recupe-rar la produción tímica de células T vír-genes, en las que se toma ventaja de esteconocimiento para valorar cuantitativa-mente la formación de nuevos linfocitoscomo indicador experimental de respues-ta terapeútica de reconstrucción del com-partimento T.Por otra parte, un hecho aparentementetan académico como que el mecanismode recombinación [corte y pegado del ADNentre V-(D)-J] cree dos lugares de “empal-me” en TCRβ y uno en TCRα, tiene tam-bién gran interés para los lectores de ME-DICINE. Se dice que el mecanismo decorte y empalme es “impreciso”. Esta im-precisión se debe a que la maquinaria de
recombinación puede “comer” secuenciasde los exones V, D o J, o, alternativamente,cortar en el intrón que flaquea V, D o J“dejando” unas pocas bases de ADN “ex-tras” a los exones de V, D o J propiamentedichos. Además, hay polimerasas de ADN–como la transferasa terminal de dideo-xinucleotidos (TdT) y la polimerasa µ– queen el momento en que se produce la for-mación de los extremos de ADN codifi-cante son capaces de añadir nucleótidosal azar, sin molde de una hebra comple-mentaria de ADN, en el sitio o “roto” don-de se va a producir el empalme de los dosextremos codificantes con rotura de doblecadena. Esto hace que el lugar de empal-me tenga en cada clon una secuencia úni-ca, y no se repita en un individuo porquese calcula que la maquinaria de diversifi-cación es capaz de generar casi un billónde empalmes distintos en esta región, laHV3, y se estima que el repertorio actualincluye en cada momento unos 100 mi-llones de clones de células T o B.Es importante pues recordar que la regiónhipervariable 3 (HV3) de los TCR coincidecon el lugar de empalme entre V y J enTCRα; y con D y los lugares de empalmeD-J y V-D en TCRβ. Por el contrario, lasregiones HV1 y HV2 residen en el seg-ménto génico V y se heredan, por lo queexisten en un número mucho más limita-do, igual al número de segmentos V. LasHV3 son los giros del receptor que ocupanuna posición central en el heterodímeroαβTCR y son los responsables de contac-tar más extensamente con el antígenoCDR3 presentado por la molécula de his-tocompatibilidad. El hecho de que seanúnicos de cada clon les da un alto valordiagnóstico, por lo que la secuenciaciónde estas regiones es una diana de análisisen procesos linfoproliferativos desde haceaños. Estos estudios permiten evidenciarsi la expansión celular es poli o monoclo-nal, y generar sondas específicas del lugarde empalme, incluyendo en su caso D, quea su vez pueden ser empleadas ulterior-mente en el diagnóstico de enfermedadmínima residual. En la actualidad se pue-den estudiar las recombinaciones V-(D)-Jin situ, y utilizando una sola célula comofuente de ácidos nucleicos. La aplicaciónmédica de estas tecnologías está permi-tiendo demostrar la relación clonal en le-siones tumorales. Así, por ejemplo, se hademostrado recientemente que las célulasde Reed-Sternberg que acompañan en laenfermedad de Hodgkin a las B células tí-
picas también son parte del clon tumoral,y no células no tumorales reactivas, aun-que tengan una anatomía patológica atí-pica para una célula B y una variedad depresentaciones inmunofenotípicas. Este fe-notipo “aberrante” tendría relación con laexpresión de una proteína del virus de Eps-tein-Barr virus (VEB), (latent membrane pro-tein 1, LMP-1), que se expresa en niveleselevados en las células de Hodgkin/Reed-Sternberg. Estos estudios han demostradotambién que las células T presentes en di-chas lesiones de Hodgkin no son mono-clonales.Finalmente, conviene señalar que el co-nocimiento de la diversidad del CDR3/HV3 no sólo tiene aplicaciones en patolo-gía linfoproliferativa. En los últimos añosse han desarrollado diferentes tecnolo-gías para medir la diversidad de la HV3que se están aplicando al estudio de otrasenfermedades, fundamentalmente de tipoinflamatorio/autoinmune. El estudio de losrepertorios de las células presentes en laslesiones inmunológicas, y la posibilidad dediscriminar entre las células patogénicas,las reguladoras, y las que aun estando enla lesión son “mirones inocentes” por sercélulas de paso en el proceso de recircu-lación, puede ayudar en el futuro a mejo-ras en el diagnóstico y monitorización dela terapia al permitir focalizar el objeto deestudio. Algunas de la posibilidades con-templadas en este apartado están de mo-mento recluidas a modelos experimenta-les o son objeto de ensayos clínicos, otrasque ya son practicables se consideran enapartados ulteriores de este capítulo.
Selección intratímica de cadenas TCRβ conrecombinación correcta
El mecanismo de recombinación V-(D)-J esimpreciso, lo que genera una enorme di-versidad en los sitios de unión y hace queD pueda leerse en los tres “marcos de lec-tura abierta” que tiene el ADN. El precioa pagar por la complejidad del mecanis-mo de recombinación y la posibilidad tanenorme de diversificación, es que con fre-cuencia se generan recombinaciones nofuncionales (por ejemplo, por incluir uncodón de detención el TCR reordenado,debido al cambio del marco de lectura detripletes de bases de ADN). Recientemen-te se ha demostrado que la expresión de
1277
EL RECEPTOR DE ANTÍGENO EN LOS LINFOCITOS T Y LAS MOLÉCULAS DE HISTOCOMPATIBILIDAD: PAREJAS INSEPARABLES

los módulos de transducción ocurre antesque dé comienzo la recombinación de losgenes TCR (figs. 2 y 4, arriba a la dere-cha). Como hemos citado antes, en proti-mocitos se pueden expresar en la super-ficie celular módulos CD3 en cantidadesmuy reducidas, encontrándose asociadosa la chaperona calnexina. Inicialmente se pensó que podían estar asociados a un complejo suplantador del αβTCR,ψαψβTCR, hipótesis que hoy está descar-tada. La calnexina retiene fisiológicamen-te los módulos CD3 no asociados a TCRen el retículo endoplásmico. Se sabe quehay recirculación de vesículas entre el re-tículo endoplásmico y el aparato de Gol-gi, y entre éste y la superficie celular. Sesupone que este mecanismo permite la retención que devuelve las proteínas a re-tener en vesículas retrógradamente delaparato de Golgi al retículo endoplásmicotolera pequeños “escapes” que en estecaso hacen que cantidades muy pequeñasdel CD3 retenido intracelularmente (porejemplo, <1%) alcancen la superficie ce-lular sin que dichas moléculas tengan unasignificación fisiológica aceptada.Es importante observar que la expresiónde una cadena TCRβ funcional permite encélulas pre-T la expresión de un prerre-ceptor T, gracias a la presencia de los módulos de transducción y pre-Tα que suplanta a TCRα . Este prerreceptorψαβTCR/CD3 señaliza la entrada en divi-són celular de las células que lo expresane induce la expresión de las moléculas CD4y CD8 (al coexpresar ambos correcepto-res se les denomina habitualmente timo-citos “doble positivos”) por parte de susprecursores “doble negativos” (por care-cer tanto de CD4 como de CD8). La en-trada en división durante cinco a diez ci-clos permite expandir unas 30-1.000 veceslas células que hacen correctamente unacadena βTCR, ya que se suceden unas seisa diez divisiones celulares. De esta formase produce la llamada selección de TCRβ.Gracias a ella se compensa la pérdida ce-lular que corresponde a la mayor fre-cuencia de recombinaciones incorrectascon la expansión de las células que sí lahan hecho con éxito, y se generan un nú-mero suficiente de células T a partir de unnúmero limitado de progenitores T. Lasmutaciones con pérdida de función delprerreceptor T cursan con una inmuno-deficiencia en la que los números de cé-lulas T formadas se reducen más de 20veces. Este proceso de selección de TCRβ
requiere que CD3γ esté integrado en el re-ceptor, pero es prescindible para dichopunto de control la presencia de CD3δ. Yahemos explicado antes que las células querecombinan TCRα son descartadas del li-naje Tγδ, por deleción de TCRδ. En esteproceso, el prerreceptor T desempeñaríaun importante papel de “contraselección”del linaje Tγδ. Hasta la fecha no se ha des-crito un prerreceptor equivalente alψαβTCR/CD3 en el linaje Tγδ, y las célulasTγδ se desarrollan normalmente cuando elindividuo es deficiente en preTα o CD3δ,pero no CD3γ u otras subunidades del re-ceptor γδTCR/CD3.También es importante señalar que la re-combinasa RAG-2 se inactiva con la en-trada en el ciclo celular, lo que junto a lainactivación transcripcional de RAG-1 yRAG-2 detiene el proceso de recombina-ción en TCRβ, favoreciendo la exclusiónalélica, es decir que las células tengan unsólo TCRβ como ocurre en el 99% de lascélulas T. El locus TCRβ queda desde en-tonces inaccesible a la maquinaria de re-combinación, mientras que la activacióndel prerreceptor “abre” o hace accesible ala transcripción el TCRα. Se acepta gene-ralmente que la accesibilidad selectiva uordenada a la transcripción de los locis delas cadenas receptoras de antígeno, y lapresencia simultánea también controladade recombinasas, son mecanismos im-portantes para explicar la determinaciónde linaje y evitar la transformación onco-génica por translocación cromosómica deprotooncogenes.Cuando dichas células pre-T terminan suexpansión, se expresan en las células pre-T CD4+CD8+ de nuevo las recombi-nasas RAG-1 y RAG-2 que ahora intenta-rán recombinar con éxito el TCRα . Enaquellos timocitos que lo logren se ex-presará un receptor maduro αβTCR/CD3y se completará el proceso de diferencia-ción con la selección positiva y negativapara el reconocimiento del MHC propio,que estudiaremos en otro apartado. Re-sultados recientes de nuestro laboratorioy otros laboratorios demuestran que CD3δsí es imprescindible para la selección po-sitiva de la mayoría de las células Tαβ, encontraste con el carácter prescindible desu función en la señalización por el pre-receptor ψαβTCR/CD3.Como hemos dicho, los defectos del pre-receptor causan inmunodeficiencia al re-ducir el número de emigrantes tímicos re-cientes de forma severa (unas 20 veces).
Conviene aquí hacer énfasis en que mu-chos defectos del sistema inmune no sonestructurales sino funcionales, y con unafrecuencia significativa iatrogénicos. Fár-macos de uso creciente como los corti-coides o la ciclosporina A, que bloqueanla proliferación dirigida por el prerrecep-tor T, provocan una importante disminu-ción de la celularidad de células pre-T do-ble positivas y redución del numero deemigrantes. En un adulto con un reperto-rio y número normal de células T perifé-ricas (un billón, 1012) la contribución deestas células emigrantes recientes a la fun-ción T efectora es limitada o “prescindi-ble”. Sin embargo, en tratamientos cróni-cos, y particularmente en situaciones dedepleción periférica severa y reconstitu-ción hematopoietica global o T, este efec-to del prerreceptor puede ser importanteno solo en niños sino en adultos (por ejem-plo, trasplante de médula ósea, SIDA, etc).
El reconocimiento deantígeno por el complejoTCR/CD3 está restringidopor las moléculas de MHCque presentan el antígeno
Como muestra la figura 5, el receptor Treconoce antígenos “presentados” por lasmoléculas de MHC expresadas en la mem-brana de la célula presentadora de antí-geno (APC), en vez de antígenos libresexistentes en el espacio extracelular comohacen las células B. El TCR contacta a tra-vés de sus regiones HV directamente conla molécula de histocompatibilidad, ade-más de con el antígeno presentado porésta. Este reconocimiento “dual”, ademásde -o más que- la presentación del antí-geno propiamente dicha, es la expresióna nivel molecular del fenomeno de “res-tricción” inmunológica de las respuestasde células T por el que Rolf Zinkernagel yPaul Doherty obtuvieron recientemente elpremio Nobel de Fisiología y Medicina. Larestricción tiene múltiples consecuenciasmédicas de gran relevancia por lo que tra-taremos de resumir y fundamentar bre-vemente en los apartados siguientes al-gunas de las más importantes.En la figura 5 también se puede observarque la presencia de dos módulos de reco-nocimiento por complejo TCR/CD3 per-mite “concentrar” los complejos antígeno-MHC específicos al sitio de interacción
1278
ENFERMEDADES DEL SISTEMA INMUNE (I)

entre la APC y la célula T. En este senti-do conviene recordar que se estima quelas APC pueden presentar varios miles deantígenos distintos en las moléculas de MHC que expresan en su superficie, delas que existen hasta un millón de copias.Por ello se calcula que sólo una de cada500-1.000 moléculas de MHC evidencianel mismo antígeno específico (en la figu-ra en granate) para el complejo TCR/CD3de un clon T, mientras que el resto (porejemplo, antígenos amarillo, verde o fuc-sia) no son reconocidos por ese receptorsino por otras células T.El reconocimiento de antígeno-MHC porlas células T es muy sensible. Mark D. Da-vis y sus colaboradores han demostradoen modelos experimentales muy definidosque bastarían de tres a seis complejos an-tígeno-MHC específico para activar una cé-lula T, y que el reconocimiento del com-plejo antígeno-MHC por el αβTCR provocala agregación u oligomerización de recep-tores T, lo que desencadenaría la trans-ducción de señales. Para la mayoría de lasrespuestas inmunes se acepta que las cé-lulas T pueden responder de una forma ti-tulable a partir de un umbral inferior a 200copias de antígeno y aumentar de una for-ma cuantitativa proporcional su respues-ta (por ejemplo, producción de citocinas)en función de la dosis de antígeno-MHCsin alcanzar una saturación del efecto do-sis-respuesta hasta dosis 25-50 veces su-periores. Muchos investigadores defiendencon Davis la importancia de la agregacióncomo mecanismo de señalización por elcomplejo TCR/CD3. También defienden unmodelo de señalización “en serie”, comoel propuesto por Antonio Lanzavecchia y
sus colaboradores. En el modelo de seña-lización en serie, los complejos de tres aseis receptores que han reconocido el an-tígeno-MHC específico señalizan a la cé-lula T el reconocimiento específico; a con-tinuación, como parte del proceso deseñalización, se internalizarían por endo-citosis, permitiendo que otro grupo de receptores señalizara a continuación re-conociendo los mismos antígenos, segui-do de otro grupo, y luego otro y otro. Deesta forma se estima que se amplificaríala señal unas 200 veces, lo que explica-ría la enorme sensibilidad de las células Ta pequeñas cantidades de antígeno como
una sola copia de virus infectando una cé-lula. Aunque existen hipótesis de trabajoalternativas, pero todavía menos acepta-das para explicar el fenómeno de la sen-sibilidad, que entendemos está por su es-pecialización fuera de lugar discutir aquí,los estudios citados son de indudable in-terés para el lector de MEDICINE. Inde-pendientemente del resultado final de estadiscusión académica, la investigación ci-tada ya ha conducido a desarrollos expe-rimentales que tienen importantes aplica-ciones médicas como el uso de tetrámerosde moléculas de MHC y oligómeros de an-tígeno, como se explica más convenien-temente en las secciones siguientes trasrevisar el estado del conocimiento sobrelos ligandos del TCR.
Las moléculas de MHCcomo ligando del TCR y como receptorespresentadores de antígenoAunque la estructura de las moléculas deMHC y los mecanismos de presentaciónya fueron actualizados detalladamente enla 7ª serie de Medicine (N.o 50; pág. 2.187)la visualización a nivel molecular del com-plejo MHC antígeno puede ayudarnos aho-ra a comprender mejor este concepto ysus aplicaciones médicas. La figura 6
1279
EL RECEPTOR DE ANTÍGENO EN LOS LINFOCITOS T Y LAS MOLÉCULAS DE HISTOCOMPATIBILIDAD: PAREJAS INSEPARABLES
Fig. 5. Modelo en tres dimen-siones de la interacción entreun complejo TCR/CD3 bivalentey una pareja de moléculas de Ag-MHC que han sido selectiva-mente concentradas a la inter-fase T-APC entre un conjuntoheterogéneo de ligandos, en losque los agonistas son muy minoritarios. La ocupación delmódulo de reconocimientoαβTCR provoca la transducciónde señales a través de los nódu-los CD3 y ζ, que se indica porun destello.
Fig. 6. Imagen tomada de uncristal cuya celda contiene dosmoléculas de MHC de clase I hu-mano, del alelo HLA-A2.1 pre-sentando como antígeno un pép-tido de virus de la gripe. Lascoordenadas del cristal se hanrotado para poder observar si-multáneamente la estructura dela molécula con una cadena pe-sada (α) y la cadena asociada deβ2-microglobulina; y, un detallede la grieta de hélices alfa y sue-lo de hebras beta en la que sealoja el fragmento antigénico.

muestra dos moléculas de MHC de clase Ien dos orientaciones distintas –frontal ycoronal– dentro de la celda de cristaliza-ción de donde se obtuvo la imagen conuna resolución atómica (2Å). Es evidenteque el antígeno se aloja o encaja en una“grieta”. En dicha grieta, el antígeno (fi-deo curvado de color gris con extremos li-bres) está flanqueado a los lados por doshélices alfa (en fucsia en el diagrama re-presentativo de la estructura secundaria)y situado sobre un suelo de hebras betaantiparalelas (pintadas en amarillo).Es importante recalcar que en este dia-grama se representa la estructura real dela molécula de MHC y el antígeno, omi-tiendo las cadenas laterales de los ami-noácidos para ver mejor la estructura se-cundaria de plegamiento de los distintosdominios y la curvatura del antígeno. Sise incluyen dichos detalles -todos los áto-mos y no solo el esqueleto de la cadenapeptídica- (fig. 7) la interpretación pierdeesa información pero demuestra que nohay “huecos libres” y que el antígeno estácompletamente embebido en la moleculade MHC. Además, ayuda a identificar conclaridad los dominios globulares: α1, α2 yα3 de la cadena pesada de MHC de claseI, y la β2-microglobulina asociada no co-valentemente a ella. Recordemos que exis-ten dos grandes tipos de moléculas deMHC: clase I y clase II. La principal dife-
rencia estructural referente a la grieta depresentación es que los dos extremos dela grieta de las moléculas de MHC de cla-se I están cerrados, como el de la imagen,mientras que el de las de clase II estánabiertos. Consecuentemente, los péptidospresentados por moléculas de clase I tie-nen una longitud muy constante de ocho-diez aminoácidos, que es el tamaño delregistro “cerrado” en el que quedan em-bebidos, mientras que los péptidos pre-sentados por las moléculas de clase II notienen una longitud definida. En generalson más largos, de al menos trece ami-noácidos, y pueden llegar a casi duplicaresa longitud, al poder “colgar” en los ex-tremos de la grieta como los extremos deun perrito caliente en el bocadillo que losujeta.Un descubrimiento reciente muy impor-tante en este contexto médico ha sido quela interacción entre la molécula de MHC yel antígeno tiene especificidad. No se tra-ta de una grieta en la que se puede alojarcualquier fragmento de antígeno. El antí-geno tiene lugares de “anclaje” en la mo-lécula de MHC, dichas interacciones se re-alizan en “bolsillos” constituidos porresiduos de aminoácidos situados en losdos extremos de la grieta que posee la mo-lécula de MHC. Los “bolsillos” citados es-tán formados tanto por residuos de ami-noácidos situados en el fondo de hebras
beta como en las caras internas de las hé-lices alfa. Debe haber complementariedadentre los residuos de las moléculas de an-tígeno y la de los bolsillos del MHC, deuna forma análoga a una interación re-ceptor-ligando, lo que provoca que las mo-léculas de MHC “seleccionen” en ciertogrado los antígenos que presentan (o no).Como se puede observar en la figura 6, laregión central del antígeno está variable-mente curvada hacia arriba, lo que per-mite que esté mucho más expuesta paracontactar con los giros hipervariables 3 delTCR, que como hemos explicado son losmás diversos -únicos para cada clon- yocupan una posión central en el móduloαβTCR. De esta forma se logra explicar laespecificidad tan fina que exhiben las cé-lulas T en el reconocimiento de antígenosmuy similares.La unión de los péptidos al TCR ocurre in-tracelularmente, y es específica y con untiempo de disociación muy largo o pro-longado, que se estima en alrededor de undía. Los péptidos, una vez asociados, con-tribuyen a mantener la estructura nativade plegamiento de las moleculas de MHC,que es la mostrada en las figuras 6 y 7.Es importante recordar que sólo los pép-tidos con un umbral suficientemente altode afinidad se asocian de forma estable alMHC lo que es necesario para permitir unaexpresión en la superficie celular, comose detalla en el capítulo de células acce-sorias de Reyes y sus colaboradores queserá publicado en la Unidad Temática nº26 de esta 8ª serie de Medicine. Se acep-ta generalmente que la gran mayoría, sino todas, las moléculas de MHC expresa-das en la superficie contienen antígeno ensu grieta por ser éste necesario para man-tener la configuración nativa. Las molé-culas que se desprenden de la moléculade antígeno se desnaturalizan y son dia-na de degradación proteolítica, aunquepueden renaturalizarse tras su síntesis deforma forzada incubándolas con péptidoscomo veremos a continuación.El conocimiento tanto de la existencia desecuencias conservadas en los extremosde los fragmentos peptídicos de antígenopresentados por moléculas de MHC de cla-se I (y en posiciones con un espaciamientoanálogo a distancias variables de los ex-tremos “colgantes” en los péptidos pre-sentados por clase II) que son responsa-bles de la interacción de anclaje a lamolécula de MHC, como el conocimientode que la región principal presentada al
1280
ENFERMEDADES DEL SISTEMA INMUNE (I)
Fig. 7. Los antígenos están em-bebidos en la molécula de MHCofreciendo una superficie co-mún de contacto con el TCR elantígeno alojado y la moléculade MHC. A la representaciónde las moléculas de la figura 6se le ha introducido los datosde las coordenadas de los áto-mos de las cadenas lateralesde los aminoácidos, además delos del esqueleto peptídico re-presentado en la figura 6.

TCR es la central, ha llevado a distintosdesarrollos aplicados a la Medicina. Poruna parte, se ha descubierto que cambiosmínimos en la región central del péptido–entre los lugares de anclaje– pueden des-truir parcial o totalmente el papel agonis-ta del mismo para las respuestas T sinafectar a su capacidad de unión al MHC.Es interesante citar que se han descubiertoasí antígenos antagonistas, péptidos cuyapresencia simultánea en otras moléculasde MHC de la célula presentadora de an-tígeno inhibe de forma dominante nega-tiva la respuesta a los agonistas-MHC, quepor sí provocan respuesta cuando se pre-sentan “solos” a las mismas células T. Eluso de péptidos antagonistas está siendoobjeto de investigación muy activa comomecanismo potencial de inmunosupresiónespecífica que permitiría apagar una res-puesta inmune indeseable (por ejemplo,autoinmunidad, rechazo de trasplante,etc.); sin afectar colateralmente a otrasrespuestas específicas deseables comoocurre con los inmunosupresores al uso.Aunque hay éxitos en ensayos llevados acabo en modelos animales para enferme-dades como la esclerosis múltiple en pla-cas, su aplicabilibilidad en seres humanosrequerirá ensayos clínicos adicionales y re-solver problemas como la identificacióndel antígeno responsable y la naturalezapolimórfica del MHC en la población hu-mana que trataremos a continuación.Por otra parte, el descubrimiento de la im-portancia de la secuencias de anclaje y surelación con los “bolsillos” de los diferen-tes alelos del MHC está llevando a un es-tudio sistemático de las secuencias de an-claje que está permitiendo una selecciónracional de péptidos candidatos a provo-car una respuesta inmune eficiente encada alelo de MHC. Todo ello ayudará aun diseño más predictivo de las vacunasde síntesis y la localización de secuenciasadecuadas para su anclaje en los registrosde los bolsillos de las diferentes molécu-las de histocompatibilidad para lograr unefecto individualizado o universal del an-tígeno objeto de estudio en función de lasposibilidades. Este trabajo ha llevado ade-más a descubrir que deteminados pató-genos como el agente de la malaria con-tienen péptidos que tienen gran afinidadpor las moléculas de histocompatibilidaddel huésped. Gracias a ello compiten conantígenos protectores porque saturan condicha información las moléculas de MHCcon antígenos que no provocan respues-
tas protectoras, por lo que no sólo prote-gen al parásito sino que provocan una de-presión general de la respuesta inmune alimpedir una presentación más “variada”o diversa de antígenos.La teoría de agregación de complejosTCR/CD3 como mecanismo de activaciónde las células T, el conocimiento de la es-tructura de las moléculas presentadorasde antígeno y la posibilidad de predecir elespaciamiento entre los sitios de unión delantígeno al MHC, ha llevado a que JackStrominger y otros autores concibieran re-cientemente el diseño de multímeros depéptidos antigénicos, los denominados epí-topos T oligoméricos. Estos autores hansintetizado mediante técnicas de ingenie-ría génica péptidos repetitivos, en los quelas secuencias de los péptidos que se unenal MHC se repiten separaradas a distan-cias optimizadas por espaciadores quecontienen aminoácidos irrelevantes. Estostrabajos, realizados hasta la fecha en mo-delos experimentales in vitro con célulashumanas y en modelos animales, de-muestran que los epítopos oligoméricospueden tener una potencia farmacéuticamuy superior a los péptidos monoméricosusados hasta ahora, requiriendose con-centraciones molares del orden de 1.000a 10.000 veces menores para activar a lascélulas T in vitro. El efecto de aumento desensibilidad se observa tanto con agonis-tas como con antagonistas, y ya se ha de-mostrado su eficacia antagonista en la te-rapia de autoinmunidad en un modelo enratón para la esclerosis en placas. Las po-sibilidades de este diseño terapéutico envacunas antiinfecciosas convencionales yen la reprogramación de la respuesta in-mune que entendemos como clínicamen-te desviada o indeseable, como puede ocurrir en autoinmunidad y trasplante, requerirá más modelización y ensayos clí-nicos, pero ofrece un futuro más espe-ranzador al uso de péptidos como inmu-nomoduladores.
Las moléculas de MHCestán codificadas por un locus genéticoextenso, poligénico y muy polimórfico
La región del cromosoma 6 humano quecodifica las moléculas de histocompatibi-lidad (6p21.31) es una de las regiones de
mayor dimensión del genoma humano,con 3,67 millones de pares de bases (Mb)de ADN, cuya secuenciación se ha com-pletado ya, obteniéndose un mapa de suorganización génica de alta resolución (fig. 8). Cuando se descubrió el MHC hacemás de 50 años, los investigadores trata-ban de descubrir los genes clave en el re-chazo de órganos trasplantados. Así fuecomo descubrieron esta región genéti-ca como la más importante o “principal”(major) responsable del rechazo de tejidos,y consecuentemente le dieron ese nom-bre de MHC. La región de MHC humanose denomina HLA (de antígeno leucocita-rio humano). En otras especies en gene-ral se ha unificado la nomenclarura delMHC y se utiliza la sigla de la especie eninglés seguida de LA (por ejemplo, SLApara el cerdo, swine). Hay algunas espe-cies como el ratón o el pollo en las que seconserva la denominación original para elMHC: H2 en ratón y locus B en pollo.Como se ve en la figura 8, en el MHC sehan distinguido clásicamente tres regionesgénicas, una telomérica llamada de claseI, otra centromérica llamada de clase II, yuna tercera intermedia denominada claseIII. Esta última región de clase III contie-ne genes que codifican proteínas que notienen la estructura de las moléculas dehistocompatibilidad que hemos descritoen las secciones anteriores, aunque mu-chas de ellas sean muy relevantes a la res-puesta inmune. Entre ellos se encuentraun grupo de siete genes clave en el con-trol de la inflamación, que incluye losmiembros de la familia del factor de ne-crosis tumonoral/linfotoxina (TNFα, TNFβ/LTα y LTβ (genes LTa, TNFa, LTb), que al-gunos autores denominan clase IV y ge-nes codificantes de componentes del sis-tema de complementoEl locus HLA contiene 224 genes, de losque 128 se expresan y el resto son pseu-dogenes. La mayoría de los pseudogenesocurren en las regiones de MHC de claseI y II, y prácticamente no existen pseu-dogenes en la región de clase III, salvo enel caso de los haplotipos en los que la re-gión C4 se ha duplicado. Las regiones declase I y II se extienden mucho más alláde lo que originalmente se pensaba, y es-tas regiones son denominadas por ello “ex-tendidas” por algunos autores. Se piensaque los genes de clase I y II se han dupli-cado en numerosas ocasiones, a lo largode la evolución, generando nuevas fami-lias de genes que han hecho divergencia
1281
EL RECEPTOR DE ANTÍGENO EN LOS LINFOCITOS T Y LAS MOLÉCULAS DE HISTOCOMPATIBILIDAD: PAREJAS INSEPARABLES
1282
ENFERMEDADES DEL SISTEMA INMUNE (I)
HNRPA1 (c)TSBP830.2-L (c)BTL-IIHLA-DRAHLA-DRB9 (c)HLA-DRB3HLA-DRB2 (c)HLA-DRB1HLA-DDA1HLA-DOB1GLN-tRNA (c)
HLA-DOB3 (c)COX3-L (c)
HLA-DOA2 (c)
HLA-DOBTAP2LMP7TAP1RING9 (c)LMP2RING8 (c)IPP2 (c)RING14 (c)HLA-Z1 (c)HLA-DMBRING13 (c)
HLA-DOB2 (c)
HLA-DMARING3HLA-DOAHLA-DPA1RPL32-L (c)HLA-DPB1HLA-DPA2 (c)COL11A2 (c)HLA-DPB2 (c)HLA-DPA3 (c)Col11A2RXRBRING5RING2RING1ZFN-L (c)TAT-SF1-L (c)HSACM2LRPS18B3GALT4BING5 (c)BING4HKE2RGL2TAPBPBING1DAXXBING3 (c)rPL35A-L (c)rPL12-L (c)HSET
Cen
BAT1ATP8GNFKBIL1LTATNFaLTBLST11C7AIF1BAT2BAT3ApoM
BAT4G4
GSK2B
G5cBAT5G6fG6eG6dG6cG6bDDAHCLIC1MSH5MG23G7c
G5b
Val-TRSanRNPHSPA 1LHSPA 1AHSPA 1BG6NEUNG22G9aNG36G10G2BFRDSKI2WDOM3LSTK19C4BP450-C21BTNXBCREBL1NG7NG5PPT2NG3LPAATG16RAGEPBX2G18NOTCH4
ClaseIII
P5-15 (c)HCGIV-11 (c)HLA-FMICE (c)HCGIX-5 (c)
Tel
P5-14 (c)HCGIV-10 (c)HLA-75 (c)P5-13 (c)HCGIV-9 (c)HLA-90 (c)RPL79 (c)HCGII-B (c)P5-12 (c)HCGIV-B (c)P5-11 (c)HLA-GHCGVIII-2 (c)MICF (c)3.8-1.4 (c)P5-10 (c)HCGIV-7 (c)P5-9 (c)HLA-54 (c)P5-7 (c)HLA-16 (c)HCGII-73.8-1.3 (c)P5-6 (c)HCGIV-6P5-5 (c)HLA-70HLA-21 (c)HCGIV-5 (c)P5-4 (c)HLA-AP5-3 (c)HCGIV-4 (c)HLA-80 (c)HCGII-6 (c)MICD (c)HCGIX-43.8-1.2 (c)P5-2 (c)HCGIV-3 (c)HLA-59 (c)HCGVIII-1HCGVIIHTEX4HCGVHCGIRFB30ZNFB7ZNF173HLA-92 (c)CAT75XGT257HLA-30 (c)HCGII-5 (c)HCGII-4 (c)
3.8-1.5 (c)
MICG (c)HCGII-3 (c)TC4HLA-EHRS-1CAT-56ABC50FB19PROA-hom (c)DBP2RPL7A (c)KIAAD170TUBBFLOTILLINPRG1DDRTFHSPG8SC1OTF3NOB4 (c)HCGII-2 (c)HCGIX-3 (c)NOB-5 (c)HLA-CHCGIV-2 (c)KIAAOO55-hom (c)RPL3-hom (c)HCGII-1 (c)HLA-BHCGIV-1 (c)DHFRP (c)HLA-17 (c)P5-8 (c)NOB2 (c)NOB3 (c)NOB1 (c)HCGIX-2 (c)MICAHLA-X (c)P5-13.8-1.1HCGIX-1 (c)MICB
Clase IIclásico
Clase IIextendido
Clase Iclásico
C4A
OO
, C4B
1C
4A,C
4B,C
4B,C
4BC
4A,C
4B,C
4BC
4A,C
4B
4 5
87 6 6
DR
52D
R8
DR
1
DR
51
DR
53Fig. 8. Mapa genético completo del MHC humano. Los 224 ge-nes están ordenados del centromero al telómero del cromoso-ma 6 en la banda p21.31 sin guardar una proporción a escalarespecto a su tamaño y separación. Los genes que se han des-cubierto o localizado por primera vez como consecuencia dela secuenciación del genoma se indican como cajas negras, lamayoría de ellos son pseudogenes (ψ). Obsérvese que algunasregiones pueden tener inserciones y deleciones génicas, pre-sentando una serie de diferencias de contenido génico o ha-plotipos, distintas entre los individuos de la especie. En elcaso publicado se han secuenciado DR52 en la región de claseII clásico y C4AQO, C4B1 en la región de clase III. Debido a laexistencia de haplotipos y alelos el concepto de genoma hu-mano es una abstracción, se requerirá un trabajo adicional,ya en curso, secuenciación de todo el conjunto de dichos poli-morfismos para disponer del repertorio potencial de genomashumanos.

para ejecutar funciones independientes.Para explicar el mantenimiento de genesincapaces de generar mensaje o proteínafuncional (pseudogenes) se postula que setransmite un número tan alto de pseudo-genes para posibilitar la generación denuevos alelos por conversión génica. Laconversión génica es un mecanismo detransmisión de información entre dos ge-nes por recombinación homóloga duran-te la meiosis. En este caso, entre un pseu-dogen y un gen funcional el primerodonaría variabilidad de secuencia al se-gundo en un tramo o segmento de su es-tructura por oposición a la mutación pun-tual en el que aminoácidos puntualessufren cambios de una forma más dispersaen la estructura. Por este mecanismo, lasecuencia del pseudogén sería útil, aun-que no se pueda expresar por sí misma,por lo que se mantendría en el genoma.De los genes expresados, alrededor del40% ya tienen asignadas funciones im-portantes en el sistema inmune que hansido investigadas en los últimos 20 años.El HLA es la región génica con mayor den-sidad o concentración de genes estudiadahasta ahora en el genoma humano, fren-te a otras regiones poco densas o despo-bladas de genes como el cromosoma 21,recientemente secuenciado, que pese a sutamaño mucho mayor tiene un número degenes similar al MHC. Conviene hacer des-de un principio énfasis por una parte enque la región de MHC no solo contiene ge-nes que codifiquen moléculas de MHC pre-sentadoras de antígeno, como las que he-mos estudiado en las secciones anteriores.Esta afirmación es válida no solo para laregión III (y IV), sino también para las declase I y II clásicas. Por otra parte, comose observa en la figura 8, existen múlti-ples genes que codifican moléculas deMHC de clase I y clase II con capacidadpresentadora de antígeno. No solo hay unamolécula de MHC de clase I y otra de MHCde clase II. El primer concepto, no todaslas moléculas codificadas por el MHC tie-nen la misma estructura y función, inclu-so dentro de una región determinada, re-sulta evidente si se examina la figura 8.Por ejemplo, los genes TAP1 y TAP 2 –quecodifican las dos subunidades del trans-portador de péptidos que introduce en elretículo péptidos de origen citoplásmicopara su presentación por moléculas de cla-se I– están situados en la región de MHCII clásico, entre los genes HLA-DQ y HLA-DP. También, entre TAP1 y TAP2 se sitúa
el gen LMP7, uno de los componentes delinmunoproteasoma, una subunidad de uncomplejo de proteínas responsable de generar péptidos por degradación de pro-teínas citoplásmicas, que luego serán trans-portados a la luz del retículo endoplásmi-co por el transportador TAP1/TAP2 para serpresentados por las moléculas de MHC declase I (ver capítulo sobre procesamientode antígeno por células accesorias por Reyes y sus colaboradores en la Unidad Te-mática 26 de la 8.a serie de Medicine). Es-tos genes y otros como DM están implica-dos en el procesamiento intracelular deantígeno, pero no en su presentación en lasuperficie célular, que es el fenómeno queestudiamos en este capítulo.El polimorfismo de los componentes delsistema de procesamiento de antígeno esmuy relevante en el control de la respuestainmune y la susceptibilidad a la enferme-dad. No solo son polimórficas las mo-léculas que presentan antígeno sino tam-bién otras relacionadas funcionalmentecon ellas como subunidades del proteaso-ma, del transportador TAP1/2 o DM, res-ponsables de generar y facilitar la intro-ducción de péptidos en las moléculas declase I y II, respectivamente. Por ello, elrepertorio de fragmentos de antígeno va-ría no solo por la variabilidad de los “bol-sillos” de las moléculas de clase I y II delos diferentes alelos, sino también por loscambios alélicos de dichas moléculas fun-cionalmente relacionadas, implicadas enel procesamiento y carga del antígeno ytambién codificadas dentro del MHC.Otros genes no tienen una función obvia,por ahora, en inmunología como es el casoya discutido de los genes implicados enprocesamiento y presentación de antíge-no, regulación del sistema de comple-mento, síntesis de corticoides o inflama-ción por TNF, etc. citados en los párrafosprevios. Sin embargo, el descubrimientode esos genes con función desconocidapuede ayudar a definir causas de suscep-tibilidad a enfermedades tan frecuentescomo la psoriasis que se han asociado agenes vecinos a las moléculas de histo-compatibilidad de clase I clásica.En segundo lugar, se dice que el MHC esun sistema poligénico, al existir loci quecodifican productos relacionados pero di-vergentes como consecuencia del procesoantes descrito de duplicación génica. Ellohace que haya loci génicos íntimamenterelacionados dispuestos en tándem en elcromosoma. Como hemos dicho, este pro-
ceso es particularmente evidente en las re-giones de clase I y II. Existen múltiplesloci para codificar la cadena pesada de las moléculas de MHC de clase I, y no uno solo. En concreto se han identificadolos genes HLA-A, HLA-B, HLA-C, HLA-E,HLA-F, y HLA-G, cuyos productos son mo-léculas de histocompatibilidad con distri-bución celular y función distinta, pese aestar muy relacionadas molecularmente ytener una estructura de plegamiento si-milar a la descrita en las figuras 6 y 7. Laβ2-microglobulina, que se asocia con la ca-dena pesada de las moléculas de MHC declase I se codifica fuera del MHC. Las mo-léculas de HLA-A y HLA-B constituyen loque hoy se denomina MHC de clase I “clá-sico”, son muy polimórficas y se expresanen todas las células nucleadas del orga-nismo, aunque a niveles variables, siendoaltos en leucocitos y bajos en el sistemanervioso central, por ejemplo. En con-traste, las moléculas hoy denominadas declase I no clásico son muy poco polimór-ficas y pueden expresarse de forma espe-cífica de tejido. Es muy importante el ha-llazgo de que estas moléculas como HLA-C,-E, -F y -G, con función hasta ahora des-conocida son ligando para receptores decélulas Tγδ y receptores KIR y KAR de cé-lulas NK. Además son capaces de presen-tar moléculas distintas de péptidos, comolípidos, azúcares o ácidos nucléicos lo queposibilitaría el desarrollo de un sistemauniversal de reconocimiento, no restrin-gido a proteínas. Incluso en este últimocaso, las moléculas de MHC no clásico pu-dieran haber evolucionado para reconocerproductos proteicos de origen evolutiva-mente distante y por lo tanto con un “pa-trón” sistemáticamente extraño, como esel caso de péptidos con formil-metioninaun componente aminoacídico típico de mi-croorganismos bacterianos.Existen otras tres regiones genéticas fue-ra del MHC del cromosoma 6 que tienenuna organización genética similar y codi-fican proteínas estructuralmente y fun-cionalmente relacionadas, que se sospe-cha se originaron durante el proceso deduplicación génica pero no fueron rete-nidas en el complejo “principal” de histo-compatibilidad, sino que segregaron aotros cromosomas. Algunas moléculascomo las cinco codificadas en el locus CD1asemejan en su estructura, falta de poli-morfismo y restrición en la distribuciónde tejidos a moléculas de MHC de clase Ino clásico. Por ejemplo, CD1a que ha sido
1283
EL RECEPTOR DE ANTÍGENO EN LOS LINFOCITOS T Y LAS MOLÉCULAS DE HISTOCOMPATIBILIDAD: PAREJAS INSEPARABLES

considerado históricamente como un an-tígeno de diferenciación de células T esestructural y funcionalmente una “molé-cula de histocompatibilidad”. El estudio dela función de las moléculas de MHC ex-tendido/no clásico está en una fase muytemprana y su aplicación biomédica estásiendo objeto de investigación muy acti-va. La observación de que dichas mo-léculas son clave como ligandos de las células NK o presentando componentesglucolipídicos de micobacterias patógenasilustra el potencial de estos descubri-mientos recientes, y su importancia para“rellenar” las lagunas que dejan las mo-léculas de MHC “clasico” en el control dela respuesta inmune en situaciones de en-fermedad.Para las moléculas de MHC de clase II, he-terodímeros que tienen dos cadenas lla-madas α y β, se han identificado tres iso-tipos HLA-DP, HLA-DQ y HLA-DR quecodifican proteínas heterodiméricas dis-tintas capaces de presentar antígeno.Como ya hemos discutido, otros genes ho-mólogos que codifican para los productosDO y DM están situados entre HLA-DP yHLA-DQ, los loci HLA-DOA y HLA-DOB oHLA-DMA y HLA-DMB respectivamente,pero no tienen funciones de presentación.Están implicados en la regulación negati-va (DO) y positiva (DM) de la carga o in-troducción de antígenos en la grieta de lasmoléculas de clase II. Las razones exactaspor las que estos genes con funciones dis-tintas se han mantenido juntos en la evo-lución son desconocidas. Uno de los ar-gumentos habitualmente utilizados paraexplicar la sintenia de estos genes tieneinterés médico. Muchos de estos genescomo las moléculas de clase II, los com-ponentes del inmunoproteasoma, lostransportadores de péptidos o HLA-DM tie-nen que expresarse coordinadamente ensituaciones de inflamación. Para ello de-ben conservar las secuencias de los intro-nes implicados en la regulación de su ex-presión mucho más que las secuencias dela región exónica que codifica cada pro-teína expresada. Es interesante decir que,todos ellos responden aumentando sutranscripción en respuesta a interferóngamma (INFγ), pero no son estimuladospor INFα o INFβ, a través de la respuestaa un factor transcripcional (CIITA) que ac-tiva la expresión de todos ellos, aunqueoriginalmente fuese descubierto en molé-culas de clase II de donde tomó su nom-bre. Las deficiencias de CIITA provocan un
síndrome de inmunodeficiencia combi-nada severa (bare lymphocyte syndrome,BLS) que supone el 5% de los SCID hu-manos, cuyo principal mecanismo pato-génico son alteraciones de la expresiónde moléculas presentadoras y procesado-ras de antígeno, especialmente en la rutade clase II.Finalmente, es importante resaltar que laregión de HLA/MHC es muy polimórfica.Para algunos de los genes mostrados enel esquema hay más de 200 alelos, o va-riaciones de secuencia de un mismo locusgénico y su producto entre distintos indi-viduos de la misma especie. Por ejemplo,las variantes de HLA-B27 son uno de los207 alelos conocidos del locus B de la re-gión de MHC de clase I humano, sólo encaucásicos. Es decir, en cada uno de losdos cromosomas 6, que heredamos unode nuestra madre y otro del padre, hayuna posición o loci B que puede tener 1de entre al menos 207 secuencias distin-tas, con diferencias puntuales de unos po-cos aminoácidos entre sí. En caso de serheterozigoto, que es la situación más co-mún fuera de poblaciones con índices altos de endogamia, se expresan dos pro-teínas distintas por loci, por ser codomi-nante la expresión de los genes de estaregión MHC, lo que diversifica las posibi-lidades de presentación de antígenos en-tre los individuos y aumenta enorme-mente, o hace virtualmente completa, lade la especie en su conjunto. Esta situa-ción de polimorfismo no se limita a los ge-nes de clase I, otros como los que codifi-can la cadena HLA-DRβ de MHC de claseII tienen también un número elevado, (almenos 239 alelos), y también hay poli-morfismos menos frecuentes en genes declase III (y IV). Aunque el polimorfismo noes igual de extenso en todos los loci, pues-to que hay genes para los que sólo se haidentificado un único alelo como la cade-na HLA-DRα, el MHC es una región conun nivel de polimorfismo extraordinaria-mente alto para la media del genoma hu-mano. Habría pues genomas humanos yno un genoma humano, existiendo millo-nes de loci de MHC humanos por la di-versidad de combinaciones posibles quese han calculado. Esta diversidad hace queel tipaje del MHC sea útil con fines foren-ses de identificación de individuos (porejemplo, pruebas de paternidad, violacióny violencia), y antropológicos al ser dis-tinta la frecuencia de los diferentes alelosen las razas y poblaciones humanas.
Dicha variabilidad entre individuos no sedistribuye homogéneamente o al azar entoda la región HLA, sino que se concentraen determinadas regiones, fundamental-mente las de MHC de clase I y II clásico;y dentro de estos genes polimórficos se-lectivamente en determinados dominiosde la proteína como estudiaremos en la-siguiente sección.Es interesante que al tratarse la moléculade MHC II de un heterodímero, tanto lacadena α como la β contribuyen con susdominios α1 y β1 a la región implicada enla presentación de antígeno y reconoci-miento por el TCR, de forma análoga a losdominios α1 y α2 de las cadenas pesadasde MHC de clase I. En este sentido es im-portante decir que el ensamblaje de ca-dena de los distintos isotipos y alelos noes mutuamente excluyente generandosemoléculas híbridas entre ellos. Este con-cepto, aparentemente especializado y sinvalor práctico, tiene actualmente claro in-terés médico. Así, explicaría por qué endeterminadas enfermedades los hetero-zigotos (por ejemplo, HLA-DR3/DR4) tie-nen una modificación del riesgo genéticoque no se correlaciona con ninguno delos alelos por separado. Las posibilidades“combinatoriales” de los productos géni-cos de los loci HLA de clase II generan,pues, productos únicos que explicaríantambién riesgos distintos en haplotiposextendidos, como son las combinacionesde ciertos alelos de HLA-DP y –DR, porejemplo.
Asociación entre la regiónde MHC y enfermedad: de la inmunología a la patología médicaComo el sistema de MHC humano es muypolimórfico, las interacciones entre el an-tígeno y el MHC, y entre el MHC y el re-ceptor TCR/CD3 no son iguales para todaslas moléculas de MHC de la especie. Estoes especialmente cierto porque el poli-morfismo se concentra en los aminoáci-dos situados en los “bolsillos” en los quese ancla el antígeno, y en las regiones delas hélices α que entran en contacto conαβTCR, en vez de estar distribuido ho-mogéneamente por toda la molécula.Este hecho tiene múltiples consecuenciasde gran interés médico. Por ejemplo, hayantígenos que no pueden ser presentados
1284
ENFERMEDADES DEL SISTEMA INMUNE (I)

por los alelos de MHC que hereda un de-terminado individuo de la especie. Si elantígeno en cuestión puede desencadenaruna reacción autoinmune, se ha definidoque dicho alelo será “protector” (de la en-fermedad), frente a los alelos de “riesgo”que sí presentan eficientemente dicho an-tígeno y se “asocian” como genes de “mar-cadores de riesgo” de dicha enfermedad.Por lo tanto, el polimorfismo del MHC tie-ne consecuencias sobre el control de larespuesta inmune a nivel de presentación(y procesamiento) de antígeno, pero con-viene recordar que la no presentación noes un mecanismo universalmente benefi-cioso, y que el polimorfismo del MHC tie-ne consecuencias a otros niveles distintosde la presentación de antígeno.En primer lugar, como ocurre frecuente-mente en inmunología, el concepto deriesgo/protector es un arma de “doblefilo”. Si el antígeno que pretenden pre-sentar las moléculas de MHC procede deun patógeno infeccioso o un oncogén, lano presentación pasa a convertirse en un“riesgo”, mientras que la presentaciónpuede ser “protectora”. El conocimiento y“visualización” de los mecanismos gené-ticos y moleculares ayuda a escapar de lastrampas lógicas impuestas por la historiade la nomenclatura. Dado el carácter fuer-temente contextual de la activación de losmecanismos efectores del sistema inmu-ne, entendemos que es importante paralos lectores de MEDICINE comprender losmecanismos moleculares, que general-mente son neutrales. Conocer dichos me-canismos de una forma tan gráfica comoes hoy factible, permite entender de for-ma simple y fluida su valor patogénico yposibilidades de manipulación terapéuticade una forma racional, e independiente denomenclaturas.En segundo lugar, otras moléculas codifi-cadas por el MHC, distintas de las im-plicadas en presentación de antígeno, tam-bién exhiben polimorforfismos que cau-san diferencias funcionales entre los ale-los que justifican su asociación conenfermedades. Se olvida frecuentementeque estos genes también son polimórficosy responsables de/asociados a enferme-dad. Así, polimorfismos en genes de cla-se III que codifican proteínas como TNFα,complementos o proteínas de choque tér-mico (hsp), y otras implicadas en proce-samiento y presentación de antígenos yacitadas, también pueden afectar de formaevidente a la respuesta inmune.
Muchos otros genes no tienen una funcióninmune obvia sino funciones biológicas ge-nerales como control de crecimiento uotras. La importancia médica del MHC vie-ne subrayada por su asociación con másenfermedades que ninguna otra región delgenoma humano, incluyendo obviamentela mayoría de las enfermedades autoin-munes, pero también un amplio rango deotras enfermedades desde cáncer a trans-tornos del sueño y la lectura. Ello puedeser fruto, por una parte, de que se com-pletase la secuenciación de más de la mi-tad de los genes funcionales de esta re-gión en la primera mitad de los años 80–mucho antes de cualquier otra incursiónmulti-megabase aleatoria en el genoma hu-mano–-. Por otra parte, puede haber con-tribuido a la frecuencia de asociación conenfermedades la alta densidad de genesen la región de MHC, además de la im-portancia intrínseca del sistema inmune yla intensidad con la que se ha estudiadoen este fin de siglo. Desconocemos aún elsignificado funcional de la mitad de los ge-nes que contiene la región de MHC, perolo que ya conocemos supone para el lec-tor un ejemplo único del potencial apli-cado a la medicina del siglo XXI de la investigación sistemática del genoma hu-mano actualmente en curso.
Identificación ex vivo decélulas T específicas frentea complejos antígeno-MHC
Un problema fundamental en la historiadel estudio de las celulas T específicas deantígeno, implicadas en la defensa del or-ganismo o causantes de lesiones, ha sidoque reconocen dicho antígeno presenta-do en moléculas de MHC, y su frecuenciaes muy baja (por ejemplo, menos de unade cada diez mil a un millón de células Treconoce un antígeno dado, la mayoría re-conocen otros). Lás células T median suefecto en contacto íntimo con las célulasque expresan los complejos antígeno-MHCespecíficos (APC) y no secretan los recep-tores para mediar su efecto a distanciacomo las inmunoglobulinas. Este hecho,junto a la baja afinidad de los receptoresT por el MHC y la diversidad de antígenosirrelevantes a una respuesta específica quepueden presentar las APC, ha hecho muydifícil el estudio de las células T específi-cas ex vivo. Adicionalmente, el polimor-
fismo del MHC ha hecho que la identifi-cación de los ligandos antígeno-MHC res-ponsables de manifestaciones patológicaso defensivas sea una tarea especialmenteardua. En los últimos 20 años se han de-sarrollado técnicas de clonaje de células Tque han permitido obtener poblaciones decélulas homogéneas para sus receptoresde antígeno. Muy recientemente se ha logrado identificar células T especificas ex vivo, sin necesidad de activación y expansión in vitro, lo que ha revelado información de indudable interés biomé-dico.La afinidad del TCR por su ligando Ag-MHCes baja; se ha estimado experimental-mente una constante de disociación muyinferior a la de los anticuerpos (Kd = 10–4-10–6M) y tiempos de disociación muchomás rápidos (en torno a segundos). Davisy sus colaboradores razonaron que unaforma oligomérica del ligando antíge-no/MHC específico de un receptor T ten-dría una avidez mucho más alta, por elefecto cooperativo de la unión multiva-mente, permitiendo el uso de dichas mo-léculas para identificar las células T es-pecíficas de antígeno-MHC. Fieles a su hipótesis de trabajo construyeron mo-léculas de antígeno-MHC tetraméricas (fig. 9). Para ello sintetizaron por inge-niería de proteínas MHC recombinante so-luble, lo plegaron en presencia del pépti-do específico (u otros irrelevantes oantagonistas), y lo marcaron con biotinaen su cola terminal para que se pudiera
1285
EL RECEPTOR DE ANTÍGENO EN LOS LINFOCITOS T Y LAS MOLÉCULAS DE HISTOCOMPATIBILIDAD: PAREJAS INSEPARABLES
Fig. 9. El modelado in silico de tetrámeros de Ag-MHC de cla-se I realizado a partir de la estructura conocida del tetrámerode avidina (verde) y monómeros de la cadena α de MHC (añil)y β2m (celeste), anclando el péptido biotinado, añadido tras elextremo C-terminal de α3, a los sitios de unión a biotina exis-tentes en las cuatro moléculas de avidina. En morado, el Aginsertado en la grieta de presentación del MHC.

unir de una forma orientada a la molécu-la de streptavidina, que tiene cuatro sitiosindependientes de unión a biotina. A con-tinuación demostraron que dichas molé-culas pueden ser empleadas para identifi-car y purificar células T específicas, de unaforma similar al empleo generalizado deanticuerpos para cuantificar y separar sub-poblaciones celulares.Recientemente estos autores y otros handemostrado que esta metodología puedeser empleada para purificar, a partir debiopsias de pacientes, células T específi-cas de antígenos relevantes en medicinacomo oncogenes de melanoma o agentesinfecciosos como el virus de la inmuno-deficiencia humana (VIH). Más aún, estostrabajos han permitido demostrar que pue-den existir linfocitos anérgicos infiltrandolesiones en las que la respuesta inmuneprotectora es insuficiente y la patologíaprogresa. Estas células no son “clonables”por los métodos de cultivo tradicionalesdada la dificultad intrínseca de activar cé-lulas anérgicas. La posibilidad de emplearmoléculas de MHC/Ag multivalentes solu-bles abre pues una posibilidad inédita deestudiar las células T específicas ex vivo,en unas condiciones que pueden facilitarenormemente la compresión de mecanis-mos patogénicos crípticos hasta ahora.Además, la posibilidad de utilizar la for-ma restringida por el MHC de antígenosagonistas o antagonistas monoméricos ymultiméricos también ha abierto un áreanovedosa de posibles aplicaciones tera-péuticas.
Dos rutas de presentaciónpor moléculas de MHC para dos poblaciones decélulas T funcionalmentedistintas: papel de loscorreceptores CD4 y CD8Como dijimos al inicio de este trabajo,existen dos rutas independientes de pro-cesamiento de antígenos – extracelularese intracelulares- que tienen un profundoimpacto en la regulación del sistema in-mune al afectar a la actividad de dos sub-poblaciones de linfocitos funcionalmentedistintas, las células T CD4+, cooperado-ras (Th), y las células T CD8+, citóxico/su-presoras (Tc). Las moléculas de MHC declase I presentan péptidos de origen cito-sólico, que son generados por el protea-
soma y transportados al interior de la luzdel retículo endoplásmico rugoso, dondese sintetizan las moléculas de MHC de cla-se I con las que se asocian para permitirsu plegamiento correcto y transporte a lamembrana plasmática. Estos péptidos deorigen citosólico como virus, oncogenes oantígenos propios no se alojan en la grie-ta de presentación de las moléculas de cla-se II por impedirlo la cadena invariante,tal como se detalla en el capítulo escritopor Reyes y sus colaboradores y que serápublicado en la Unidad Temática número26 de esta serie de Medicine. Por el con-trario, los antígenos de origen extracelu-lar que introducidos al espacio vesiculardel interior de la célula por pinocitosis oendocitosis mediada por receptores seránpresentados por moléculas de MHC de cla-se II, y no se asocian con las moléculas deMHC de clase I. Esta segregación de lasrutas de procesamiento de antígeno a dis-tintas moléculas presentadoras es de granrelevancia funcional porque las moléculasde MHC de clase I y II son reconocidas pordistintas subpoblaciones de linfocitos. Lascélulas T cooperadoras reconocen antíge-nos presentados por moléculas de clase IImientras las citotóxicas lo hacen en el con-texto de restricción de clase I. Las molé-culas CD4 y CD8 son correceptores queexplican esta subdivisión funcional. Comose observa en la figura 10, el correceptorCD8 reconoce el complejo antígeno/MHCde clase I en un lugar distinto del módu-lo de reconocimiento αβTCR. CD8 tieneuna estructura dimérica, y cada una de lasdos cadenas tiene un dominio globular dela superfamilia de inmunoglobulinas en suextremo seguido de un segmento largo fle-
xible que se inserta en la membrana y secontinúa con una cola citoplásmica que noestán representadas en la figura. La figu-ra ilustra de forma muy clara cómo dichosdominios globulares CD8αβ reconocen ungiro expuesto en el dominio α3 de la ca-dena pesada de clase I, y por lo tanto seunen a una región distinta y alejada dellugar de contacto del complejo antíge-no/MHC con el módulo de reconocimien-to TCR. La interacción de CD4 ocurre enla región homóloga de las moléculas declase II, un giro expuesto en el dominioβ2, también separado del sitio de contac-to con el TCR (fig. 11). Ambos giros estánconservados en las moléculas de clase I yII, respectivamente, independientementede qué locus se trate y de la variación alé-
1286
ENFERMEDADES DEL SISTEMA INMUNE (I)
Fig. 11. Modelo de agregación del superdímero TCR/ CD3 con los correceptores (A) y formación de islas de complejos receptores(B) en la sinapsis inmunológica.
CD4
A
CD4
z
b
a z
e
a
CD4
B
CD4
z
b
a z
e
a
e d
e d
b
gCD4
CD4
z
b
a z
e
a
e g
CD4
CD4
z
b
a z
e
a
e d
b
d
b
g
b
g
Fig. 10. El heterodímero de moléculas CD8 (verde y ocre) reco-noce la molécula de Ag-MHC de clase I en un sitio separadodel lugar de unión del TCR. Arriba se entrevé en verde el antí-geno, insertado en una grieta de presentación del MHC. La ca-dena pesada de α de MHC (añil) y β2m (celeste). El sitio deunión de CD8αβ está situado en un giro del dominio α3. Lamayoría de las células T expresan heterodímeros CD8, unapequeña fracción de células Tαβ y bastantes de las célulasTγδy NK CD8+ expresan homodímeros CD8αα .

lica, con pocas excepciones, como HLA-A68 que es mal reconocido por las célu-las CD8.Estudios funcionales y bioquímicos handemostrado que la unión concurrente delos correceptores CD4 y CD8 a complejosTCR-Antígeno/MHC estabiliza dicha inte-racción aumentando la avidez y alargan-do notablemente los tiempos de disocia-ción, rebajando notablemente así elumbral de concentración de antígeno queactiva la célula T. El mecanismo de acciónde los correceptores no se limita a actuara nivel de reconocimiento. Se les dio di-cho nombre de correceptor porque tam-bién cooperan a nivel de la señalizaciónpor el complejo TCR/CD3. Se sabe que lascolas citoplásmicas de CD4 y CD8 tienenmotivos de asociación con la proteína-ti-rosina-quinasa p56lck. Según el modelomás aceptado, los correceptores CD4 oCD8 estarían distribuidos en la superficiede las células T independientemente delreceptor en células T en reposo (fig. 11A).Cuando dichas células reconocen un com-plejo antígeno-MHC específico con su re-ceptor TCR/CD3 se asocia en un comple-jo ternario el correceptor correspondiente;al asociarse la proteína p56lck que aportael correceptor puede actuar fosforilandolos ITAM de los módulos de transducciónζζ y CD3, lo que a su vez desencadena lacascada de señalización propia del com-plejo TCR/CD3, tal como se detalla en elcapítulo de Prieto y sus colaboradores queaparecerá publicado en la Unidad Temá-tica número 26 de esta serie de Medicine.Las moléculas CD4 pueden oligomerizar alas concentraciones a las que se encuen-tran en la grieta de interacción entre unacélula presentadora de antígeno y la célu-la T; nosotros y otros autores hemos pro-puesto que dicha dimerización CD4-CD4podría contribuir al proceso de agregaciónde los superdímeros de receptores T (fig.11B), permitiendo la formación de islas dereceptores en la membrana como se ex-plica en el apartado siguiente.Los timocitos adquieren el complejo re-ceptor αβTCR/CD3 en el estadío de célu-las “doble positivas”, CD4+CD8+. Sufrenun proceso de selección positiva a la quesólo sobreviven los que tienen afinidad porlos alelos de MHC que expresa el epiteliotímico (propios salvo el caso de trasplan-tes) y los que no los reconocen muerenpor apoptosis. En este proceso de selec-ción positiva, los correceptores CD4 favo-recen a los clones con receptores con es-
pecificidad para HLA-DR, -DP o DQ, mien-tras CD8 favorece el reconocimiento demoléculas de clase I. Cuando se producela señalización, las células seleccionadasmantienen la expresión del correceptorempleado (CD4 las células T cooperado-ras, y CD8 las células T citotóxicas), y pier-den la expresión del correceptor que noparticipó en el reconocimiento/señaliza-ción cuyo gen es metilado, y en condi-ciones generales deja de expresarse. Deesta forma, las células seleccionadas po-sitivamente adquieren el fenotipo “simplepositivo”, bien CD4+CD8– las cooperado-ras bien CD4–CD8+ las citotóxicas, propiode la mayoría de las células Tαβ maduras.Concurrentemente con el proceso de se-lección positiva ocurre la llamada selec-ción negativa. Aquellos clones cuyo TCRreconoce con alta afinidad los complejosantígeno-MHC presentes en el timo soneliminados por apoptosis. De esta formase “purga” el repertorio de αβTCR paraque tenga clones sólo con baja afinidadpor antígenos-MHC propio, pero sea capazde reconocer los alelos de MHC propio querealizan la función de restricción durantela presentación de antígeno. La presenciade un antígeno extraño que aumente laafinidad puede entonces desencadenar la respuesta inmune. En el caso de lostrasplantes de órganos en los que las mo-léculas de MHC del receptor, que son lasempleadas para “educar” su sistema in-mune, sean distintas de las expresadas porel órgano trasplantado ocurre que una fre-cuencia anormalmente alta de células T(más de 100 veces mayor que frente a an-tígenos convencionales) responde contraantígenos propios de la especie porque elelemento de restricción de otros alelos noempleados en la selección positiva/nega-tiva puede disparar el reconocimeinto deantígenos propios presentados por dichasmoléculas de MHC para las que no huboselección negativa.El conocimiento de la importancia de lafunción de los correceptores ha llevado aque se investigen como posibles dianas deacción terapéutica. Hay una línea de in-vestigación dirigida a interferir el recono-cimiento, la unión al giro conservado enlas moléculas de clase I o clase II con losque reaccionan CD8 y CD4, respectiva-mente empleando péptidos miméticos opequeñas moléculas para ese fin. Más re-cientemente, las compañias farmacéuticashan abordado la investigación de inhibi-dores específicos de PTK como p56lck, en
un intento de modular farmacológica-mente las rutas de señalización. Desgra-ciadamente esta tarea no es sencilla y seve adicionalmente complicada por la par-ticipación de p56lck como transductor deseñales en otros receptores, alguno deellos familiar a los lectores como el re-ceptor de interleucina-2 (IL-2).
La sinapsis inmunológica:organización topológica delos receptores de antígeno,correceptores y moléculascoestimuladoras
Las señales que conducen a la activaciónde las células T se producen en una es-tuctura de carácter sináptico entre la cé-lula T respondedora y la presentadora deantígeno (APC). Como muestra la figu-ra 12, el TCR y su ligando antígeno-MHCestán enriquecidos en una posición cen-tral entre la célula T y la APC, mientraslas moléculas de adhesión celular ICAM-1,integrinas y otras moléculas que estable-cen contactos intercelulares de forma an-tígeno-independiente forman una anilloque circunda el disco central donde se pro-duce la reacción inmunológicamente es-pecífica. Este modelo topológico de esti-mulación de las células T, propuesto porlos laboratorios de Kupfer, Dustin, Davisy otros, impone un orden estructural y fun-cional en el proceso casi inagotable de des-cubrimiento de nuevas moléculas impli-cadas.El proceso de activación sería un eventodinámico. En una etapa inicial, las célulasmigran y son atraídas hacia el lugar de le-sión por quimiocinas, como explican Me-llado y Frade en su revisión, que apareceen esta misma Unidad Temática. En unasegunda etapa, las células T se adhierende forma antígeno inespecífica a las célu-las APC empleando moléculas de adhesiónintercelular. En esta etapa, sin poder sa-ber si expresan el complejo antígeno-MHCespecífico de su TCR, “rastrean” la super-ficie célular para comprobar si se expre-sa dicho ligando. Las uniones celulares sonen esta fase muy dinámicas y plásticas,no firmes y estables; y si, no hay recoci-miento específico, la célula T se despegarápidamente de esa célula para “reciclar-se” y comenzar el rastreo de otra poten-cial célula diana, y otra secuencialmente.Esta función de adhesión reversible se sus-
1287
EL RECEPTOR DE ANTÍGENO EN LOS LINFOCITOS T Y LAS MOLÉCULAS DE HISTOCOMPATIBILIDAD: PAREJAS INSEPARABLES

pende si la célula encuentra un ligando es-pecífico para su TCR (a). Entonces la se-ñalización del TCR/CD3 provoca un cam-bio del patrón de adhesión de integrinascomo LFA-1, que aumentan su afinidadpor la célula diana en un proceso llamadoseñalización de dentro-hacia-afuera (in-outsignal) al modificar la señal intracelular delTCR/CD3 la conformación del dominio ex-tracelular de la integrina. Inicialmente lasmoleculas de adhesión ocupan un área ex-tensa, y central aunque dispersa o dis-continua, y en un punto de la periferia seproduce el reconocimiento de antígeno, lallamada señal 1 que se amplifica a travésde quinasas presentes en los correcepto-res CD4 y CD8 (b) y microdominios quecontienen LAT (c). Esta señal 1 provocauna reorganización de las moléculas de re-conocimiento en la superficie de la célulaT que conducen a la formación de la si-napsis mostrada en la figura 12, y de unaforma esquemática y más completa en lafigura 13. Las moléculas se redistribuiríande acuerdo con su tamaño, unas similaresen su altura al complejo TCR-antígeno-MHC se enriquecen en la región central,mientras que las más altas como CD45,CD43, o integrinas como LFA-1 y su li-
gando ICAM-1 son excluidas de esa regióny ocupan el anillo circundante “sellando”la región de unión con su poder de uniónintercelular. Además, al “apartar” a CD45(e) se aparta también la función fosfatasade su cola citoplásmica, que junto a otras fosfatasas como SHP-1 mantienen el proceso de activación del TCR/CD3 ba-jo regulación negativa en células T en re-poso (d).En este proceso de activación participanno solamente quimiocinas, moléculas deadhesión, y el complejo TCR/CD3 (y suscorreceptores que no se presentan, comolas moleculas CD3/ζ, para simplificar el es-quema). Además de los receptores que seactivan en serie (f) y se internalizan y de-gradan en los lisosomas tras transducir se-ñales (g), también participan de las molé-culas denominadas colectivamente comocoestimuladoras, y de las que el esquemarepresenta a CD28 como prototipo de ellas(h). En ausencia de coestimulación, el re-conocimiento de antígeno provoca esta-dos de anergia o apoptosis como revisanPrieto y sus colaboradores en otro capí-tulo que aparecerá publidado en la UnidadTemática número 26 de esta misma serie,al dominar el efecto de las fosfatasas ex-
cluidas cuando hay coestimulación (h). Lacoestimulación por CD28 no afecta direc-tamente a la extensión y a la cinética deactivación del TCR, pero amplifica el pro-ceso de activación en células vírgenes, yprotege a las células activadas/memoriade la muerte inducida por activación(AICD). Las células vírgenes tienen micro-dominios pequeños y niveles pobres dePTK por lo que la contribución de CD28es esencial para evitar la anergia. Aunqueen las células memoria CD28 no es esen-cial para la activación, sí es importantepara activar rutas de señalización antia-poptóticas dado que el incremento de sen-sibilidad a la activación se acompaña deun aumento paralelo de sensibilidad a lamuerte celular.La unión de CD28 a sus li-gandos en la APC conlleva el reclutamientode microdominios de la membrana cito-plásmica (Rafts) ricos en quinasas comoLck y adaptadores de transducción de se-ñales como LAT a la sinapsis inmunológi-ca, lo que potencia la señalización por elTCR/CD3 y la hace suficientemente soste-nida en el tiempo como para activar a lacélula T. En este mecanismo de activaciónse produce un proceso de reorganizacióndel esqueleto (j), al que contribuyen ade-más de CD28 otras moléculas de adhesióncon función coestimuladora como CD2,que, por una parte, favorece la formaciónde los patrones de complejos supramole-culares descritos en la sinapsis, y, por otra,la reorientación de la maquinaria de se-creción hacia la sinapsis. Este último me-canismo es importante porque permite redireccionar moléculas efectoras y regu-ladoras secretadas hacia la célula diana,bien sean estas perforinas, granzimas, fac-tor de necrosis tumoral o interferón se-cretadas por una célula T citotóxica fren-te a una APC infectada por virus o que seatumoral, bien sean citoquinas secretadaspor una célula T coperadora para “ayudar”a una célula B o Tc. De esta forma, el sis-tema inmune trata de delimitar daños co-laterales en sus respuestas efectoras y fo-calizar sus circuitos reguladores lograndocontactos intercelulares que combinan unadiversidad de poblaciones celulares, nú-mero de clones y plasticidad espacial ex-cepcional junto a la formacion de sinap-sis inmunitarias con contactos celularesíntimos. En los próximos años la aporta-ción del proyecto genoma humano va acompletar la imagen de los elementos queforman parte del “puzzle” o “mecano” in-munológico. Entonces, más que nunca
1288
ENFERMEDADES DEL SISTEMA INMUNE (I)
Fig. 12. La interacción eficiente entre una célula T y una célula presentadora de complejos antígeno-MHC específicos crea una si-napsis inmunológica en cuyo centro se encuentra una isla de complejos receptores TCR/CD3 (teñidos en verde). Éstos están rodea-dos de un anillo de moléculas de adhesión intercelular como ICAM-1 (teñido en rojo), en la zona de interfase entre ambos se sitúaun anillo de moléculas coestimuladoras como CD28 (no mostrado).

será necesario racionalizar y poner un or-den estructural y funcional a tanto CD y“marcador de diferenciación”, compren-diéndolos como moléculas con funcionesasociadas bien definidas. Jeraquizando sufunción y ordenándolos en caminos de re-conocimiento y señalización paralelos oen serie se producirán esquemas tan con-fortables como los que teníamos hace dosdecadas con las citoquinas, cuando sólo“existían” IL-1 e IL-2, con la diferencia no-table de que serán completos. Aunque pue-de estar próxima la fecha en que termine
la fase descriptiva de los componentes delsistema inmune, queda pues mucho tra-bajo por delante para ordenarlos de unaforma coherente y predictiva para la apli-cación biomédica del conocimiento. Sinembargo, hay razones de peso para dis-frutar de una perspectiva optimista. Hace25 años se describió el fenomeno de larestricción del reconocimiento T por elcomplejo de histocompatibilidad, cuya im-portancia mereció un Premio Nobel de Medicina aun cuando sus fundamentosmoleculares fueran completamente des-
conocidos, y sus aplicaciones una utopía.Hoy disponemos de la secuencia comple-ta de dicho complejo, y de la estructuradel TCR y sus ligandos, así como de múl-tiples componentes de la sinapsis immu-nológica y empezamos a comprender susfunciones. Hace pocos años era frecuentela pregunta ¿y vale para algo todo este co-nocimiento? Hoy nos encontramos estu-diando activamente las numerosas apli-caciones diagnósticas y terapéuticas deeste enorme avance del conocimiento enel área de la biología de las células T, quecontribuyen a mejorar la calidad de vidade nuestros pacientes, y haciéndonos estavez preguntas para investigar activamen-te otras nuevas aplicaciones antes insos-pechadas.
BIBLIOGRAFÍA RECOMENDADA
Benoist C, Mathis D. T-lymphocyte differentiation and biology.En: Paul WE, ed. Fundamental Immunology (4.a ed.). Filadel-fia: Lippincott-Raven Publishers, 1999; cap. 11: 367-409.Dave V, Cao Z-S, Browne C, Alarcón B, Fernández-MiguelG, Lafaille J, et al. CD3δ-deficiency arrests development ofthe αβ but not the γδ T cell lineage. EMBO J 1997; 6:1.360-1.370.Davis MM, Chien YH. T-cell antigen receptors. En: PaulWE, ed. Fundamental Immunology (4.a ed.). Filadelfia: Lip-pincott-Raven Publishers 1999; cap. 10: 341-346.Fernández-Miguel G, Alarcón B, Iglesias A, Bluethmann H,Álvarez-Mon M, Sanz E, Hera A de la. Multivalent structureof an αβT cell receptor. Proc Natl Acad Sci USA 1999;1.547-1.552.Germain R. Antigen Procesing and Presentation. En: PaulWE, ed. Fundamental Immunology (4.a ed.). Filadelfia: Lip-pincot-Raven Publishers, 1999; cap. 9: 287-340.Hera A de la, Muller U, Olsson C, Isaaz S, Tunnacliffe A.Structure of the T cell antigen receptor: two CD3ε subunitsin a functional TCR/CD3 complex. J Exp Med 1991; 173: 7-17.Margulies DH. The major histocompatibility complex: En:Paul WE, ed. Fundamental Immunology (4.a ed.). Filadelfia:Lippincott-Raven Publishers, 1999; cap. 8: 263-285.Reconocimiento de Antígeno por Linfocitos T. En: JanewayCh, Travers P, Walport M, Capra JD, eds. Immunobiología(1.a ed.). Barcelona: Editorial Masson, 2000; cap. 4.Rosen F, Geha R. Casos clínicos de Inmunología (1.a ed.).Barcelona: Editorial Masson, 2000.
1289
EL RECEPTOR DE ANTÍGENO EN LOS LINFOCITOS T Y LAS MOLÉCULAS DE HISTOCOMPATIBILIDAD: PAREJAS INSEPARABLES
APC
CD45(d)
SHP-1
(a)(f)
TCR
P(b)
LAT
P
LcK
Raft
PLC gras
Rac
Célula TLisosoma
ICAM-1(e)
CD43
LFA-1
ZAP-70
(g) (c)
APC
Microdominio
P Lck
Célula T
ZAP-70P P P P
CD2CD28
(h)
(i)
Fig. 13. Secuencia de eventos de señalización en una sinapsis inmunológica entre una célula T y la célula presentadora de antíge-no (APC) que reconoce en ausencia de coestimulación (arriba) o amplificada por la coestimulación (abajo).

Introducción
La estructura de los diferentes órganos ytejidos no es fruto de una organización alazar sino que responde a una necesidadde cumplir de forma óptima con una de-terminada función. Estas necesidades sehacen muy patentes en el caso del siste-ma inmunológico, ya que es un sistemaen continua renovación y, literalmente, encontinuo movimiento. El sistema inmunesirve como eficaz línea de defensa ba-sándose, fundamentalmente en un altogrado de diferenciación y especialización,tanto a nivel celular como tisular, y en lascomplejas interrelaciones que se estable-cen entre sus componentes. El funciona-miento inmunológico normal requiere delcontinuo tráfico linfocitario en labor de pa-trulla a través del torrente circulatorio yen los diferentes órganos y tejidos linfáti-cos y no linfáticos. Este proceso de recir-culación permite extender la respuesta in-munológica por todo el organismo ygarantiza la respuesta frente a agentes ex-traños e, incluso, frente a células del pro-pio organismo que han sufrido alguna al-teración. En respuesta a una de estascircunstancias, diversas poblaciones leu-cocitarias son reclutadas del torrente cir-culatorio hacia el foco inflamatorio, en un proceso denominado extravasación (fig. 1). Este proceso permite la salida deforma específica, ordenada y secuencialde las células más adecuadas para en-frentarse a la agresión. Para garantizar es-tas características, el proceso ocurre de-bido a un sumatorio de señales finamenteregulado de tal modo que definen un mo-delo secuencial; es decir con pasos suce-
sivos controlados por una gran variedadde moléculas, a veces incluso interrela-cionadas que justifican un alto número decombinaciones y finalmente condicionanla diversidad y especificidad de respues-ta. En este trabajo abordaremos el papelde algunas de ellas, selectinas, integrinasy quimiocinas, haciendo especial hincapiéen estas últimas dada la gran expansiónque han tenido en los últimos años.El primer paso en el proceso migratoriodepende de la detección de una serie deseñales por parte del leucocito circulante.Estas señales posibilitan su interacción conel endotelio y de hecho ocurre tambiénnormalmente, en ausencia de estímulo pa-tológico, y de él depende la migración ce-lular necesaria para construir y organizarlos órganos linfáticos. Así, en los órganos
linfáticos la adherencia de los linfocitos yla migración transendotelial ocurre en zonas postcapilares especializadas deno-minadas HEV (del inglés, High EndothelialVenules) que presentan de manera consti-tutiva una serie de proteínas en la super-ficie celular, más adelante referidas, quepermiten que la extravasación ocurra nor-malmente. Estas HEV son muy abundan-tes en las áreas de células T que rodeanlos folículos de células B y son la vía deentrada utilizada tanto por los linfocitos Tcomo por los B.Mientras las células no detectan las seña-les apropiadas, las interacciones con el en-dotelio se producen al azar y no tienenmayores consecuencias; la célula no aban-dona el torrente circulatorio. Sin embar-go, cuando una célula debe extravasarse,recibe un estímulo que hace que esas in-teracciones se conviertan en muy especí-ficas y de hecho provocan que el movi-miento del linfocito sea más lento y puedarodar por la superficie del endotelio vas-cular. Es éste el primer momento en elque se establece una cierta selección pues,del mismo modo que no todas las célulasse extravasan a la vez, tampoco comien-zan a rodar a un mismo tiempo. El fenó-meno del rodamiento por el endotelio escontrolado por unas moléculas denomi-nadas selectinas, nombre que hace refe-rencia precisamente a que son las mo-léculas encargadas de establecer una pri-mera selección.
1290
RECIRCULACIÓN Y LOCALIZACIÓNTISULAR DE CÉLULAS DEL SISTEMAINMUNE: PAPEL DE LASQUIMIOCINASM. Mellado García y J.M. Rodríguez FradeDepartamento de Inmunología y Oncología. Centro Nacional de Biotecnología (CSIC). Madrid.
Medicine 2000; 8(25): 1290-1298Fig. 1. Esquema de migración celular durante la extravasación. Se representan los pasos sucesivos y las moléculas implicadas enla extravasación leucocitaria desde el torrente circulatorio a los focos de inflamación.
Rodamiento
Extravasación
Proteoglicanos
Citocinas
Integrinas
Quimiocinas
Receptores de quimiocinas Selectinas
Adhesión firme
Foco inflamatorio

Selectinas
Las selectinas son una familia de recep-tores de adhesión estructuralmente ca-racterizadas por la presencia de varios do-minios (fig. 2). Uno N terminal, homólogoal de las lectinas dependientes de calcio,unido con otro de tipo del factor de cre-cimiento epidérmico (EGF) y un númerovariable de repeticiones consenso simila-res a las encontradas en proteínas queunen complemento; son las zonas SCR (delinglés Short Consensus Repeats). La prote-ína atraviesa la membrana celular sólo unavez y presenta también una región cito-plasmática, C-terminal, muy corta.Existen tres tipos diferentes de selectinascon características estructurales y funcio-nales específicas. Así, en seres humanos,la selectina-L contiene dos dominios SCR, la selectina-E seis y la selectina-P nue-ve. En cuanto a su expresión, la selectina-L está presente en la membrana de los leucocitos, la selectina-P se encuentra al-macenada en los denominados cuerpos deWeibel-Palade de las células endotelialesy en los gránulos α de las plaquetas des-de donde es rápidamente movilizada enrespuesta a mediadores de inflamacióncomo trombina o histamina. La selectina-E
se induce en el endotelio vascular en res-puesta a citocinas como interleucina 1 yfactor de necrosis tumoral (TNF) o lipo-polisacáridos bacterianos. La expresión deselectina-E, por lo tanto, es mucho máslenta pues requiere su síntesis de novo, loque indica que las respuestas inmediatasvendrán mediadas por la selectina-P.Las selectinas reconocen sialil-carbohi-dratos presentes en sus correspondientesligandos a través de sus dominios lectinacon ligeras variaciones de unas a otras (fig. 2). Los carbohidratos que reconocenlas selectinas L y P están unidos a molé-culas tipo mucina. De hecho, selectina-Lreconoce dos mucinas presentes en las es-tructuras antes referidas como HEV, y par-ticipa en la unión inicial de los linfocitosa esas estructuras pero también en la en-trada de linfocitos y neutrófilos en los fo-cos inflamatorios aunque en este últimocaso es la selectina-P la que parece iniciarla fase más temprana del reclutamientoleucocitario. Es destacable que la selecti-na-E puede también unir selectina-L y queel ligando más específico de la selectina-P es el PGSL-1, que, aunque se expresa enlas diferentes poblaciones leucocitarias,sólo presenta la glucosilación adecuadapara ser reconocido por la selectina en
neutrófilos, monocitos y células natural ki-ller (NK).Después de que los leucocitos hayan ini-ciado el rodamiento dirigido por el endo-telio vascular, como consecuencia de la in-teracción lábil entre las selectinas y susligandos, se produce la adhesión firme alendotelio. Este proceso es mediado porotras moléculas, las integrinas, que al in-teraccionar directamente con el citosque-leto celular transmiten rápidamente la señal al interior de la célula, de ahí preci-samente su nombre, ya que son integra-doras de señales al interior celular.
IntegrinasLas integrinas son proteínas heterotrimé-ricas formadas por asociación no covalentede subunidades α y β. Se conocen 16 su-bunidades α y 8 β y su combinación dalugar al menos a 24 integrinas diferentes.Seis integrinas del repertorio presente enleucocitos son importantes en la interac-ción de estas células con el endotelio y pa-rece ser la subunidad α quien aporta la es-pecificidad por el ligando. Los cationesdivalentes desempeñan un papel decisivoen la función adhesiva pues influyen en la
1291
RECIRCULACIÓN Y LOCALIZACIÓN TISULAR DE CÉLULAS DEL SISTEMA INMUNE: PAPEL DE LAS QUIMIOCINAS
Fig. 2. Selectinas. Representación de las selectinas, estructura y ligandos con atención a los tipos celulares que los expresan y las condiciones que condicionan su expresión.
Selectinas
LeucocitosEndotelio
Ligandos
GlyCAM-1MadCAM
CD34Sgp200
Selectina-E
MonocitosNeutrófilosEosinófilosLinfocitos
NeutrófilosMonocitosCélulas NK
NeutrófilosMonocitosEosinófilosBasófilosCélulas NKLinfocitos
Ligandos
PSGL-1CD24
Ligandos
ESL-1PSGL-1
NCASelectina-L
Selectina-P
Selectina-E
Endotelio(activado por citocinas)
Endotelio(gránulos de Weibel-Palade)
HEVEndotelio activado
Selectina-L
Dominios tipo EGF Dominios tipo lectinaDominio transmembrana+ región citoplasmática
Dominios SCR

afinidad y especificidad por el ligando. Elmecanismo para explicar la función de es-tas proteínas es su rápida transición des-de un estado de baja afinidad por su li-gando, estado inactivo, a otro transitorioactivo de alta afinidad. Basándose en elreconocimiento por anticuerpos de las di-ferentes subunidades se ha concluido queesos dos estados se caracterizan por uncambio conformacional real de las inte-grinas.Como resumen de lo anterior podríamosdecir que las integrinas regulan la paraday la fuerte adhesión de los leucocitos queruedan por el endotelio, pero la transiciónsucesiva entre la forma activa e inactivade estas moléculas es clave para que acontinuación se inicie el movimiento mi-gratorio.Los ligandos para las integrinas pertene-cen a la superfamilia de las inmunoglo-bulinas (tabla 1), entre ellos la subfami-lia de los ICAM (Cytokine-Induced Adhe-sion Molecule), VCAM (Vascular Cell Ad-hesion Molecule), MadCAM-1 (Mucosal addressin Cell Adhesion Molecule), PE-CAM-1, etc... Los ICAM son productos degenes homólogos pero diferentes y se di-ferencian entre sí en el número de domi-nios de inmunoglobulina que incluyen ensu estructura. ICAM-1 se expresa tanto encélulas hematopoyéticas como no hema-topoyéticas, expresión que es aumentadapor mediadores inflamatorios por lo quese le asigna un papel relevante en la in-teracción célula-célula y en la extravasa-ción linfocitaria a los focos inflamatorios.ICAM-2 se expresa en células endotelialesy algo en linfocitos en reposo; su expre-sión constitutiva le relaciona con el tráfi-co de linfocitos a tejidos no inflamados ysu recirculación. ICAM-3 se expresa ma-yoritariamente en los linfocitos en repo-
so, monocitos, neutrófilos y algunas célu-las endoteliales involucradas en procesostumorales.VCAM-1 se expresa principalmente en cé-lulas endoteliales y células dendríticas, yes inducible por citocinas. Su papel se re-laciona con la activación del linfocito porcélulas presentadoras de antígeno.MadCAM-1 contiene tres dominios de in-munoglobulina y una región de estructu-ra tipo mucina entre los dominios dos ytres. Además de unir integrinas puede, através del dominio mucina, unir selecti-na-L y de ese modo mediar no solo ad-hesión sino también el rodamiento del lin-focito.PECAM se expresa principalmente en cé-lulas endoteliales, monocitos, granulocitosy plaquetas, se localiza en las uniones cé-lula-célula, desempeñando por lo tanto unpapel relevante en las interacciones ho-motípicas aunque también ha sido impli-cado en la migración de monocitos y neu-trófilos.La célula firmemente adherida inicia en-tonces el movimiento de diapédesis usan-do como tracción las interacciones de lasintegrinas con sus ligandos y su paso deconformación activa a inactiva. El movi-miento no ocurre al azar sino que es diri-gido por un gradiente quimiotáctico pro-ducido por las quimiocinas.
QuimiocinasLas citocinas quimioatrayentes o quimio-cinas son una familia de proteínas de bajopeso molecular (7-12 kDa), con un alto gra-do de homología, originalmente identifi-cadas por su capacidad de atraer pobla-ciones leucocitarias.Estructuralmente son proteínas muy rela-cionadas entre sí, presentan cuatro resi-
duos de cisteína altamente conservados;aunque de la disposición de los dos máscercanos al extremo N-terminal surge pre-cisamente la clasificación actual de estasmoléculas (tabla 2), clasificación que, porcierto, coincide con una cierta especifici-dad de acción por algún tipo celular con-creto e incluso con la localización cromo-sómica de los genes que las codifican.Así, las quimiocinas de la familia CC pre-sentan las dos cisteínas situadas en el ex-tremo N-terminal adyacentes y actúanprincipalmente sobre distintas poblacio-nes de linfocitos T, eosinófilos, basófilos,células NK y células dendríticas. A esta fa-milia pertenece el grupo de las proteínasquimioatrayentes de monocitos, (MCP), lasproteínas inflamatorias que afectan a ma-crófagos, (MIP y RANTES). A éstas, mejorconocidas, se han añadido recientementeotras nuevas como TARC, I-309, 6Ckine oTECK, que son un grupo de quimiocinascon actividades relacionadas con procesoshomeostásicos en lugar de inflamatorios.Las quimiocinas pertenecientes a la fami-lia CXC presentan un residuo aminoacídi-co entre las dos primeras cisteínas, y ejer-cen su acción fundamentalmente sobreneutrófilos, monocitos y células T. Es ungrupo bastante uniforme y en algunos desus miembros destaca la presencia de lasecuencia Glu, Leu, Arg (ELR) que origi-nalmente diferenciaba las quimiocinas queactuan sobre neutrófilos de otras con actividad sobre otro tipo de poblacionescelulares. De hecho la presencia de esemotivo coincide con la descripción en estas quimiocinas de actividad angiogénica. Éste es el caso de la interleuquina 8 (IL-8), GRO-α o ENA-78 (Epithelial derived Neu-trophil Attractant-78). Entre las que no incluyen el motivo ERL cabe destacar IP-10 (IFN-γ-Inducible Protein-10) y MIG
1292
ENFERMEDADES DEL SISTEMA INMUNE (I)
TABLA 1Implicación de miembros de la superfamilia de las inmunoglobulinas en adhesión
y migración leucocitaria
Moléculas Integrinas Células Inductores Implicacionesde adhesión de la expresión
ICAM-1 LFA-1 Células hematopoyéticas y no hematopoyéticas Mediadores de inflamación Extravasación a foco inflamatorioMAC
ICAM-2 LFA-1 Células endoteliales Constitutiva Homeostasis
ICAM-3 LFA-1 Linfocitos, monocitos, neutrófilos Constitutiva Extravasación a focos inflamatorios y adhesión celular
VCAM-1 VLA-4 Células endoteliales y dendríticas Citocinas Activación de linfocitosα4/β7
PECAM αvβ3 Células endoteliales monocitos y neutrófilos Constitutiva Migración celular y adhesiones homotípicas
La tabla hace referencia a las integrinas que unen, las células que los expresan así como las condiciones de expresión y los fenómenos en los que ha sido descrita su participación.

(Monokine Induced by IFN-γ), ambas indu-cibles por interferón γ, que además pre-sentan actividad angiostática. También aeste segundo grupo pertenece el factor de-rivado de estroma, SDF-1 (Stroma derivedFactor), que es una quimiocina que actúafundamentalmente sobre células T, peroque además es el principal factor quimio-atrayente de progenitores hematopoyéti-cos CD34+.Además de las quimiocinas hasta ahorareferidas existen otras que sólo presentanuna cisteína en el extremo N-terminal,como las linfotactinas α y β que actúanprincipalmente sobre células T en reposoy constituyen la familia de quimiocinas C,y otras agrupadas en la familia CX3C for-mada por aquellas que poseen tres ami-noácidos entre las dos cisteínas iniciales.En realidad, esta última familia tiene úni-camente un miembro, la fractalquina queactúa principalmente sobre células NK ycuya característica fundamental es la deestar unida a la membrana célular me-diante un dominio rico en mucinas.Esta clasificación aceptada clásicamenteestá dejando paso en la actualidad a otraindependiente de condicionamientos es-tructurales que agrupa a las quimiocinassegún el tipo de proceso que regulan, he-cho que está directamente relacionado conque su expresión sea constitutiva o indu-
cible. Al primer grupo pertenecen las qui-miocinas que desempeñan un papel másrelacionado con la propia organización es-tructural del sistema inmune, es decir conlos procesos de homeostasis, entre ellasestán SDF-1, TECK o SLC. El segundo gru-po engloba a aquellas quimiocinas rela-cionadas con procesos inflamatorios, en-tre las que se encuentran algunas de las quimiocinas inicialmente conocidas,(IL-8, GRO-α , IP-10, MIG, MCP, MIP o RANTES).Las quimiocinas, independientemente dela familia a la que pertenezcan y del grado de homología en su secuencia, pre-sentan un plegamiento de la cadena pep-tídica muy parecido entre sí, organizan-dose en tres láminas β antiparalelas y unahélice α en el extremo C-terminal. Los da-tos sobre la estructura terciaria indicanque, salvo alguna excepción, todas pue-den formar dímeros, encontrándose lazona de unión en el extremo N-terminal.En la actualidad existe una amplia con-troversia sobre la relevancia fisiológica deestos dímeros ya que si bien parece queen algunos casos el monómero es funcio-nal, en otros casos se han descrito qui-miocinas modificadas químicamente que,siendo inactivas, mantienen la capacidadde dimerizar con la quimiocina nativa yde actuar como antagonistas bloquean-
do los efectos de la quimiocina original.Las quimiocinas ejercen sus acciones porunión a receptores presentes en la mem-brana de la célula diana (tabla 2). Estosreceptores pertenecen a la familia de re-ceptores de siete dominios transmem-brana acoplados a proteínas G (GPCR [GProtein-Coupled Receptor]). Constan deuna única cadena proteíca que cruza sie-te veces la membrana celular y activanvías de señalización entre las que estánlas mediadas por proteínas que unen nu-cleótidos de guanina. Estos receptores seorganizan de modo que en la unión delligando intervienen el dominio N-termi-nal y los tres bucles extracelulares, zonasque además presentan residuos suscepti-bles de ser glucosilados, mientras que elextremo C- terminal y los bucles intrace-lulares intervienen en el acoplamiento demoléculas transductoras de la señalización. Los receptores de quimiocinas tienen unacaracterística importante, la presencia dela secuencia DRYLAIV que integra el mo-tivo DRY, altamente conservado en todala familia de GPCR sean o no de quimio-cinas, y cuya presencia es crítica para laseñalización posterior. Por sus implica-ciones en los mecanismos de desensibi-lización e internalización del receptorcabe destacar también una región muyrica en residuos de serina y treonina enel extremo C-terminal de los receptores.Igual que ocurre con las quimiocinas, losreceptores también se clasifican en CCR,CXCR, CR y CX3CR según unan las qui-miocinas de alguna de las familias antesreferidas. Hasta la fecha se han descritocinco receptores para quimiocinas CXC,diez para las CC, uno para linfotactina, qui-miocina C, y otro para, fractalquina, quimiocina CX3C. Conviene hacer notaraquí el alto grado de promiscuidad exis-tente en la unión de los ligandos a estosreceptores; de hecho, distintos ligandospueden unir al mismo receptor y diferen-tes receptores pueden unir el mismo li-gando, aunque siempre se mantenga la es-pecificidad de familia. La expresión deestos receptores, al igual que de las qui-miocinas, está sujeta a un control muy finopor parte de citocinas y de las propias qui-miocinas, de ahí que la relativa promis-cuidad pueda entenderse como un pasomás en el control de la acción de estasmoléculas.Además de éstos, existen otros receptoresde quimiocinas que no se ajustan a estasclasificación. Éste es el caso del receptor
1293
RECIRCULACIÓN Y LOCALIZACIÓN TISULAR DE CÉLULAS DEL SISTEMA INMUNE: PAPEL DE LAS QUIMIOCINAS
TABLA 2Clasificación de quimiocinas. Familias de quimiocinas y receptores definidos en la bibliografía junto
con los tipos celulares sobre los que ejercen su acción
Familia Receptor Quimiocina Tipo celular
CXC CXCR1 IL-8, GCP-2 Ne
CXCR2 IL-8, GCP-2, ENA-78, NAP-2, GRAOα/β/γ Ne
CXCR3 IP-10, MIG, I-TAC NK, T
CXCR4 SDF-1α Mo, T, DC, B
CXCR5 BCA-1 B
CC CCR RANTES, MCP-2/3, MPIF-1, MIP-1α/δ Eos, Mo, DC, T
CCR2 MCP-1/2/3/4 Ba, DC, NK, B, Mo, T
CCR3 Eotaxina, MCP-2/3/4, RANTES, MIP-1δ Eos, Ba, DC
CCR4 TARC, MDC, RANTES DC, T
CCR5 MIP, 1α/β, RANTES Mo, DC, NK, T
CCR6 MIP-3α DC
CCR7 6CKINE, MIP-3β T
CCR8 I-309, TARC, MIP-1β Mo
CCR9 TECK *
CCR10 Skinquina **
CX3C CX3CR1 Fractalquina NK,T
C XCR1 Linfotactcina α y β T
Ne: neutrófilos; NK: natural killer; T: linfocitos T; Mo: monocitos; DC: células dendríticas; B: linfocitos B; Eos: eosinófilos; Ba: basófilos.Se ha detectado expresión de CCR9 en células en timo (*) y de CCR10 = en células de intestino (**).

DARC, que se corresponde con el antíge-no Duffy presente en los eritrocitos. Estereceptor es capaz de unir tanto quimioci-nas CC como CXC y tiene la particulari-dad de no señalizar en respuesta a estosligandos; con este receptor es con quieninteracciona Plasmodium vivax para infec-tar a las células. Otros receptores de qui-miocinas especiales son los codificadospor virus como los citomegalovirus (CMV)o Herpesvirus saimirii (HHV8). El signifi-cado biológico de estos receptores se des-conoce, igual que la razón por la que al-gunos virus como Moluscum contagiosumcodifican quimiocinas; sin embargo, se es-pecula con su participación durante la in-fección de células huésped secuestrandoquimiocinas.
Señalización por quimiocinas
La unión de la quimiocina a su receptorproduce la dimerización de éste, hechoque es un paso clave para desencadenarla cascada de señalización (fig. 3). Loscambios conformacionales que produce elligando y la propia dimerización promue-ven la asociación al receptor de una qui-nasa de la familia de las JANUS quinasas,implicadas en la señalización por citoci-nas y factores de crecimiento, y la rápidafosforilación en residuos de tirosina delpropio receptor. Debido a los cambios con-formacionales producidos en el receptorpor todos estos eventos se ponen de ma-nifiesto lugares que permiten la asociaciónde la proteína Gi, lo que desencadena laseñalización ligada a esta proteína: incre-mento de calcio intracelular, inhibición dela actividad adenilato ciclasa, liberaciónde las subunidades βγ de la propia proteí-na Gi y consiguiente activación de la fos-fatidilinositol-3-quinasa, activación de qui-nasas relacionadas con el citosqueletocelular como la quinasa de adhesión fo-cal, FAK, activación de MAP quinasas,etc... Por su parte, la JAK quinasa activatambién su propia vía de señalización loque conduce a la activación de factores detranscripción de la familia STAT, que de-bería, sin duda, y por analogía con lo quesucede en los factores de crecimiento, con-ducir al incremento de la expresión géni-ca. Sin embargo, esta vía está silenciadapor la activación de tirosin fosfatasas queimpiden la translocación de los factoresde transcripción al núcleo. La justificación
para este silenciamiento debe buscarse enque la mayor parte de los linfocitos pro-ducen quimiocinas que por unión a sus receptores mantendrían continuamenteactiva esta vía con la consiguiente des-regulación de la expresión de nuevos genes y la consiguiente pérdida de funciónde la célula.Una característica de los GPCR presentelógicamente en los receptores de quimio-cinas también es su susceptibilidad a serdesensibilizados, volviéndose insensiblesa un segundo estímulo de la misma qui-miocina. Este proceso conocido como desensibilización homóloga conduce fi-nalmente al desacoplamiento de la ma-quinaria transductora de la señalización ya la internalización del receptor lo que con-
lleva su desaparición de la superficie celular.En la señalización por quimiocinas pue-den distinguirse los acontecimientos que suceden rápidamente: dimerizaciónde los receptores, activación de la vía delas quinasas JANUS y activación de la pro-teína Gi que serían responsables de efec-tos observables a corto plazo como mo-vilización de calcio, y los eventos queocurren a largo plazo, muchas veces re-lacionados directamente con los prime-ros, como puede ser la activación de otrasquinasas relacionadas con el citoesquele-to celular que finalmente tiene su refle-jo en los efectos de polarización y mi-gración inducidos por estas moléculas (fig. 3).
1294
ENFERMEDADES DEL SISTEMA INMUNE (I)
Fig. 3. Principales vías de señalización activadas por las quimiocinas. Se representan algunas de las vías de señalización activadas por las quimiocinas relacionándolos con los efectos finales observados, detallando además algunas de las moléculas implicadas.
N
C
N
C
N
C
N
C
Tyr quinasasJAK
Factores detranscripción
(STAT)
Ser/Thr quinasasGRK
Internalización
b-arrestinaclatrina
dinamina
Proteína GQuinasas de
adhesiónfocal
Expresiónde genes
Reorganizacióndel
citoesqueleto
Expresiónde genes
Reorganizacióndel
citoesqueleto
PI3KRac
Cdc42Rho
MAPK

Efectos fisiológicosde las quimiocinas
Quimiocinas y polarización celular
Ya hemos indicado con anterioridad la im-portancia de la migración celular y la grancantidad de moléculas implicadas en es-tos fenómenos para garantizar su eficacia.El primer requerimiento para que un lin-focito inicie la migración es la adquisi-ción de un fenotipo polarizado, hecho quepuede ser inducido por las propias qui-miocinas o por otros estímulos como IL-2 ó IL-15. Este fenotipo polarizado (fig. 4) consiste en un cambio morfológi-co de la célula con la aparición de dos re-giones diferentes: el frente de avance encontacto con el sustrato y una prolonga-ción no observada en otras células euca-riotas migratorias con funciones rela-cionadas con la adhesión denominadaurópodo. La polarización conlleva cambiosen la distribución de una mólecula del ci-toesqueleto, la F-actina, lo que directa-mente induce la formación de extensio-nes celulares denominadas filopodio ylamelipodio.El movimiento se inicia dirigido por laorientación del gradiente quimiotáctico yusando como fuerza motriz los contactosdel cuerpo celular con el sustrato, que asu vez están promovidos por los cambiosconformacionales de las integrinas. La po-larización celular, resulta imprescindiblepara la migración celular, pero la adqui-sición de ese fenotipo desempeña tam-bién un papel importante en otros pro-cesos como por ejemplo, el contacto delos linfocitos con las células presentado-ras de antígeno o con las células dianaasí como en otras fases de la respuestainmune que requieren interacciones ce-lulares, por ejemplo los linfocitos T cito-líticos, y las células NK mantienen un fe-notipo polarizado cuando se unen a suscélulas diana.Como ya hemos indicado en el caso de laF-actina, cabría esperar que los cambiosque origina la polarización celular conlle-vasen la redistribución de otras proteínascelulares. De hecho, aunque hay datoscontrovertidos en la bibliografía, se sabeque los receptores de quimiocinas pasande localizarse de forma uniforme en lamembrana celular a localizarse en el fren-te de avance donde parecen ejercer unafunción de orientación del movimiento ce-lular; ya que durante la migración a favor
de gradiente quimiotáctico las células po-larizadas orientan su frente de avance ha-cia el foco de quimiocina, actuando comouna auténtica nariz celular. Además de losreceptores parece que también se trasla-dan al mismo lugar las moléculas necesa-rias para su señalización, entre ellas lasproteínas G.De acuerdo con la función que ejercen, lasintegrinas ocupan el cuerpo celular en con-tacto con el sustrato. La conformación ac-tiva se encuentra en las integrinas cerca-nas al frente de avance mientras laspresentes en la zona trasera, más próxi-mas al urópodo están en conformacióninactiva.En el urópodo se acumulan mientras tan-to moléculas de adhesión, ICAM, CD43,CD44, selectina-L, etc... lo que indica que el urópodo es una estructura con funciones adhesivas que permite unirotras células, y que en el caso de la mi-gración permitiría incrementar el reclu-tamiento celular y la transmigración en-dotelial.Además de redistribución de proteínas ce-lulares, el cambio fenotípico incluye unacompleta reorganización del citosqueletocelular que involucra a una gran cantidadde moléculas (fig. 4). Así, durante la mi-gración el centro organizador de microtú-bulos se localiza en el urópodo, lo cual in-crementa la capacidad de deformación de
las células, facilitando su paso por espa-cios constreñidos. La actina se reorienta yse localiza unida a los receptores de su-perficie interaccionando con el sustratopor lo que forma una red que actúa comomotor del movimiento al polimerizar inin-terrupidamente en el frente de avancemientras se produce la degradación de lospolímeros en la zona trasera del cuerpocelular.
Quimiocinas y respuesta Th1 y Th2
La activación de células T ocurre en lu-gares especializados del sistema linfáticoy requiere que la célula T se encuentrecon las células dendríticas que portan elantígeno. La combinación de citocinaspresente durante la presentación antigé-nica determina la polarización de la res-puesta y, por lo tanto, el patrón de cito-cinas que producirán las células Tefectoras. Así, las células Th1 CD4+ pro-ducen principalmente interferón γ, IL-12y activan macrófagos. Por otro lado, lascélulas Th2 CD4+ producen entre otrosmediadores interleucinas 4 y 5 y activanlinfocitos B. La respuesta inmunológicarequiere que además de las diferentes ca-pacidades efectoras, las células Th1 y Th2puedan migrar a los distintos tejidos. Des-de hace tiempo se sabía que las selecti-nas e integrinas expresadas en células T
1295
RECIRCULACIÓN Y LOCALIZACIÓN TISULAR DE CÉLULAS DEL SISTEMA INMUNE: PAPEL DE LAS QUIMIOCINAS
Fig. 4. Representación esquemática de la polarización celular. Se muestra el fenotipo polarizado que adoptan los leucocitos quemigran, así como la distribución que adoptan los receptores de quimiocinas, las integrinas y las moléculas de adhesión.
IntegrinasInactinas
Activas
Actina polimerizada
Molécula de adhesión
Selectinas
Receptor de quimiocina
Maquinaria de señalización
Quimiocina
Citocinas
Proteoglicano

vírgenes, efectoras o memoria eran dife-rentes, sin embargo recientemente se havisto que además expresan en su super-ficie distintos receptores de quimiocinas,hecho que condiciona su posicionamien-to en todo momento. En primer lugar, lacélula T virgen expresa CCR7, receptorpara 6Ckine y MIP-3β, y CXCR4, recep-tor para SDF-1, lo que la permite moversea los órganos linfáticos secundarios y con-diciona que se encuentre con la célulapresentadora. Tras la activación y comoconsecuencia del patrón de citocinas pre-sentes, se regulan positivamente los re-ceptores CCR1, 2, 5 y CXCR3 en las cé-lulas Th1 y el CCR2, 3 y 4 en las Th2, loque posibilita que las células se posicio-nen en los tejidos periféricos inflamadoso sean atrapadas en los órganos linfáti-cos secundarios donde derivan hacia cé-lulas de memoria o inician la expansiónclonal. Se especula con la posibilidad deque las células dendríticas en tejidos in-flamados produzcan quimiocinas de lasdenominadas constitutivas lo que sinduda atraería células T recién activadasprovocando reacciones inflamatorias cró-nicas.
Quimiocinas y homeostasis
Como antes se ha referido, las quimioci-nas desempeñan también un papel muyimportante en las funciones homeostási-cas del sistema inmunológico, de maneraparticular en la arquitectura de los órga-nos linfáticos secundarios, ganglios linfá-ticos, bazo y placas de Peyer. A modo deejemplo cabe indicar que las células den-dríticas inmaduras residen en tejidos nolinfáticos, son incapaces de estimular a lascélulas T pero están especializadas en cap-turar antígenos. Cuando lo hacen, madu-ran con rapidez y migran a los ganglioslinfáticos zonales donde activan a las cé-lulas T virgen; es precisamente la madu-ración lo que las confiere una alta capa-cidad migratoria. Junto a la migraciónocurre un profundo cambio del tipo de res-puesta quimiotáctica como consecuenciade la aparición y desaparición de recep-tores de quimiocinas de la superficie ce-lular. Las células, entonces, responden algradiente de quimiocinas constitutivas es-pecíficas de órganos linfáticos secundarioscomo son SLC y ELC. Ambas quimiocinasse expresan en las zonas de células T delos ganglios linfáticos y del bazo lo que ga-rantiza la llegada allí de las células den-
dríticas maduras. Otro ejemplo de la im-portancia de las quimiocinas en la ho-meostasis surge del estudio fenotípico deratones KO para el receptor quimiotácti-co CXCR5. La desaparición del gen paraeste receptor, cuyo ligando es BCL (qui-mioatrayente de células B) provoca fallosarquitectónicos en el bazo y en las pla-cas de Peyer e incluso desaparición deganglios linfáticos inguinales. De hecho,las células B entran en el ganglio linfáticopero se quedan en la zona que normalmente ocupan las células T y nollegan al folículo, impidiendo por lo tanto el normal funcionamiento del ór-gano.
Quimiocinas y angiogénesis
Se define angiogénesis como el procesode formación de nuevos vasos sanguíne-os a partir de otros preexistentes. Esteproceso es fundamental en muchos fe-nómenos biológicos como, por ejemplo,la embriogénesis, pero también tiene unagran importancia en el curso de muchosprocesos patológicos cuya evolución de-pende directamente de esa neovasculari-zación. Durante la angiogénesis, las células endoteliales sufren una reorgani-zación completa de su citoesqueleto, mo-difican el patrón de moléculas de adhe-sión que expresan en su superficie ycomienzan a segregar enzimas proteolí-ticas que modifican la matriz extracelu-lar adyacente. Aparecen entonces nuevasestructuras que finalmente forman nue-vos vasos. Hay factores autocrinos y pa-racrinos que favorecen este proceso, de-nominados factores angiogénicos, y otrosque interfieren en él, son los factores an-giostáticos. El control preciso del equili-brio entre estos factores gobierna todo elproceso angiogénico. La mayor parte delos factores angiogénicos son polipépti-dos que inducen migración, proliferacióny diferenciación en estructuras tubularesde las células endoteliales y actúan di-rectamente interaccionando con recep-tores presentes en la superficie de la cé-lula endotelial o atrayendo a la zona otrascélulas que a su vez producen los facto-res angiogénicos.Pues bien, las quimiocinas de la familiaCXC están en general involucradas en laangiogénesis; de hecho se ha demostra-do el papel angiogénico de IL-8, Gro-α yENA-78 mientras que otras como IP-10 y MIG son angiostáticas.
Quimiocinas y procesospatológicos
Se han detectado variaciones en la se-crección de quimiocinas en muchos pro-cesos patológicos (tabla 3), siempre rela-cionados con la activación y acumulaciónde leucocitos en los tejidos afectados. Sinembargo, no todas las quimiocinas pare-cen tener un papel relevante en las en-fermedades inflamatorias, de hecho ya seha mencionado anteriormente que son lasquimiocinas inducibles las implicadas enestos procesos. La composición celular delinfiltrado tisular dependerá del patrón dequimiocinas expresado por el tejido en-fermo. Por ejemplo, en muchos procesosagudos como las infecciones por Strepto-coccus pneumoniae hay una infiltración ma-siva de neutrófilos, que coincide con unaalta concentración de IL-8 en el fluidobroncoalveolar de los pacientes. En las me-ningitis víricas la infiltración de células mo-nonucleares coincide con la alta concen-tración de IP-10 o MCP-1 en el líquidocefalorraquídeo.En la mayor parte de las enfermedadescrónicas hay en mayor o menor medidainfiltración tisular de linfocitos y macró-fagos que en todos los casos se corres-ponde con una alta concentración local dealguna de las quimiocinas responsables.Por ejemplo, en asma, rinitis y dermatitisatópica, se produce un cúmulo selectivo yactivación de eosinófilos y mastocitos. Losmediadores derivados de estas células, porejemplo histamina, participan después enla patología de esas enfermedades alérgi-cas. Pues bien, la eotaxina y la familia deMCP desempeñan un papel fundamentalen esas enfermedades por ser los respon-sables de la atracción de los eosinófilos yser además factores liberadores de hista-mina.La colitis ulcerosa y la enfermedad deCrohn se caracterizan por la alternanciade fases de inflamación crónica con fa-ses de inflamación aguda. En la fase cró-nica son los macrófagos y los linfocitosquienes mayoritariamente infiltran el in-testino, consecuencia de la expresión deMCP-1, MIP, Rantes y eotaxina mientrasque en la fase aguda, y por efecto IP-10 yMCP-3 son los neutrófilos y los eosinófi-los quienes abandonan el torrente circu-latorio para internarse en la mucosa in-testinal.En los enfermos con psoriasis, las lesio-nes contienen neutrófilos y células T acti-
1296
ENFERMEDADES DEL SISTEMA INMUNE (I)
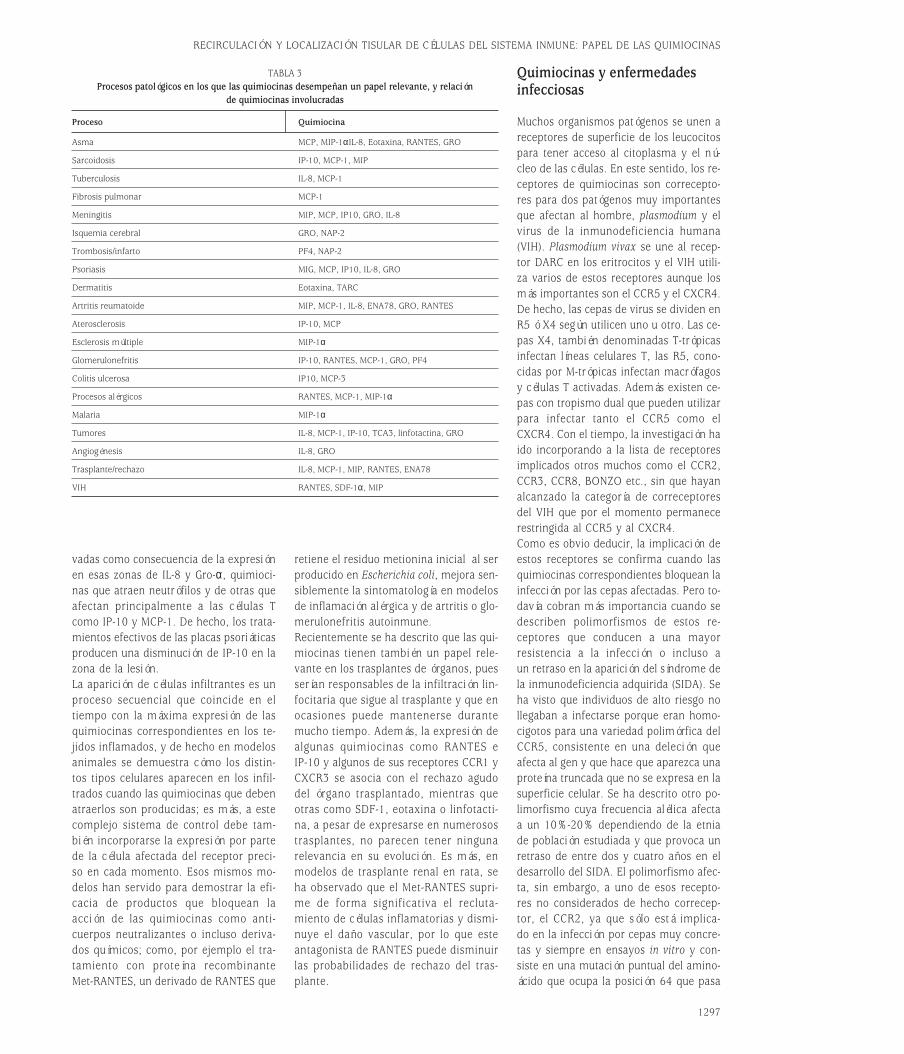
vadas como consecuencia de la expresiónen esas zonas de IL-8 y Gro-α, quimioci-nas que atraen neutrófilos y de otras queafectan principalmente a las células Tcomo IP-10 y MCP-1. De hecho, los trata-mientos efectivos de las placas psoriáticasproducen una disminución de IP-10 en lazona de la lesión.La aparición de células infiltrantes es unproceso secuencial que coincide en eltiempo con la máxima expresión de lasquimiocinas correspondientes en los te-jidos inflamados, y de hecho en modelosanimales se demuestra cómo los distin-tos tipos celulares aparecen en los infil-trados cuando las quimiocinas que debenatraerlos son producidas; es más, a estecomplejo sistema de control debe tam-bién incorporarse la expresión por partede la célula afectada del receptor preci-so en cada momento. Esos mismos mo-delos han servido para demostrar la efi-cacia de productos que bloquean laacción de las quimiocinas como anti-cuerpos neutralizantes o incluso deriva-dos químicos; como, por ejemplo el tra-tamiento con proteína recombinanteMet-RANTES, un derivado de RANTES que
retiene el residuo metionina inicial al serproducido en Escherichia coli, mejora sen-siblemente la sintomatología en modelosde inflamación alérgica y de artritis o glo-merulonefritis autoinmune.Recientemente se ha descrito que las qui-miocinas tienen también un papel rele-vante en los trasplantes de órganos, puesserían responsables de la infiltración lin-focitaria que sigue al trasplante y que enocasiones puede mantenerse durantemucho tiempo. Además, la expresión dealgunas quimiocinas como RANTES e IP-10 y algunos de sus receptores CCR1 y CXCR3 se asocia con el rechazo agudodel órgano trasplantado, mientras queotras como SDF-1, eotaxina o linfotacti-na, a pesar de expresarse en numerosostrasplantes, no parecen tener ningunarelevancia en su evolución. Es más, enmodelos de trasplante renal en rata, seha observado que el Met-RANTES supri-me de forma significativa el recluta-miento de células inflamatorias y dismi-nuye el daño vascular, por lo que esteantagonista de RANTES puede disminuirlas probabilidades de rechazo del tras-plante.
Quimiocinas y enfermedadesinfecciosas
Muchos organismos patógenos se unen areceptores de superficie de los leucocitospara tener acceso al citoplasma y el nú-cleo de las células. En este sentido, los re-ceptores de quimiocinas son correcepto-res para dos patógenos muy importantesque afectan al hombre, plasmodium y elvirus de la inmunodeficiencia humana(VIH). Plasmodium vivax se une al recep-tor DARC en los eritrocitos y el VIH utili-za varios de estos receptores aunque losmás importantes son el CCR5 y el CXCR4.De hecho, las cepas de virus se dividen enR5 ó X4 según utilicen uno u otro. Las ce-pas X4, también denominadas T-trópicasinfectan líneas celulares T, las R5, cono-cidas por M-trópicas infectan macrófagosy células T activadas. Además existen ce-pas con tropismo dual que pueden utilizarpara infectar tanto el CCR5 como elCXCR4. Con el tiempo, la investigación haido incorporando a la lista de receptoresimplicados otros muchos como el CCR2,CCR3, CCR8, BONZO etc., sin que hayanalcanzado la categoría de correceptoresdel VIH que por el momento permanecerestringida al CCR5 y al CXCR4.Como es obvio deducir, la implicación deestos receptores se confirma cuando lasquimiocinas correspondientes bloquean lainfección por las cepas afectadas. Pero to-davía cobran más importancia cuando sedescriben polimorfismos de estos re-ceptores que conducen a una mayor resistencia a la infección o incluso a un retraso en la aparición del síndrome dela inmunodeficiencia adquirida (SIDA). Seha visto que individuos de alto riesgo nollegaban a infectarse porque eran homo-cigotos para una variedad polimórfica delCCR5, consistente en una deleción queafecta al gen y que hace que aparezca unaproteína truncada que no se expresa en lasuperficie celular. Se ha descrito otro po-limorfismo cuya frecuencia alélica afectaa un 10%-20% dependiendo de la etniade población estudiada y que provoca unretraso de entre dos y cuatro años en eldesarrollo del SIDA. El polimorfismo afec-ta, sin embargo, a uno de esos recepto-res no considerados de hecho correcep-tor, el CCR2, ya que sólo está implica-do en la infección por cepas muy concre-tas y siempre en ensayos in vitro y con-siste en una mutación puntual del amino-ácido que ocupa la posición 64 que pasa
1297
RECIRCULACIÓN Y LOCALIZACIÓN TISULAR DE CÉLULAS DEL SISTEMA INMUNE: PAPEL DE LAS QUIMIOCINAS
TABLA 3Procesos patológicos en los que las quimiocinas desempeñan un papel relevante, y relación
de quimiocinas involucradas
Proceso Quimiocina
Asma MCP, MIP-1αIL-8, Eotaxina, RANTES, GRO
Sarcoidosis IP-10, MCP-1, MIP
Tuberculosis IL-8, MCP-1
Fibrosis pulmonar MCP-1
Meningitis MIP, MCP, IP10, GRO, IL-8
Isquemia cerebral GRO, NAP-2
Trombosis/infarto PF4, NAP-2
Psoriasis MIG, MCP, IP10, IL-8, GRO
Dermatitis Eotaxina, TARC
Artritis reumatoide MIP, MCP-1, IL-8, ENA78, GRO, RANTES
Aterosclerosis IP-10, MCP
Esclerosis múltiple MIP-1α
Glomerulonefritis IP-10, RANTES, MCP-1, GRO, PF4
Colitis ulcerosa IP10, MCP-3
Procesos alérgicos RANTES, MCP-1, MIP-1α
Malaria MIP-1α
Tumores IL-8, MCP-1, IP-10, TCA3, linfotactina, GRO
Angiogénesis IL-8, GRO
Trasplante/rechazo IL-8, MCP-1, MIP, RANTES, ENA78
VIH RANTES, SDF-1α, MIP

de ser una valina a una isoleucina,CCR2V64I.Existen varias teorías para explicar cómolas quimiocinas bloquean la infección porel VIH (fig. 5). Por un lado, está el impe-dimento estérico que tiene el virus paraunirse a receptores ocupados por las qui-miocinas, aunque no siempre el bloqueode la unión de la quimiocina a su recep-tor bloquea también la interacción del vi-rus indicando que ambos podrían coexis-tir sobre el receptor. Otra teoría implicaque la unión de la quimiocina a su re-ceptor dispara la cascada señalizadora queculmina con la internalización del recep-tor y, por lo tanto, con su desapariciónde la membrana celular. Una última teo-ría, a caballo entre las anteriores, se basaen el mecanismo íntimo de unión de laquimiocina al receptor de tal modo queel virus puede unirse al receptor mono-mérico pero no al dímero que se produ-ce en el momento que el ligando ocupasu lugar en el receptor. La causa proba-ble es que la dimerización provoca uncambio conformacional en el receptor queoculta el epítopo al que se unía el virus.Precisamente la dimerización de los re-ceptores de quimiocinas, que se produceincluso entre distintos receptores cuandocada uno de ellos está activado por su li-gando correspondiente (proceso que seconoce como heterodimerización) permi-te dar una explicación a cómo la muta-
ción puntual, antes referida, que afecta alCCR2 condiciona un retraso en la apari-ción del síndrome y una disminución delos receptores tipo del virus CCR5 yCXCR4. El receptor CCR2 mutante es ca-paz de dimerizar con cada uno de los co-rreceptores impidiendo entonces la unióndel virus y su entrada en el interior celu-lar.
ResumenEl funcionamiento normal del sistema in-munológico requiere la extravasación y mi-gración de las células que lo componentanto en situaciones de respuestas infla-matorias como para la organización y ar-quitectura del propio sistema. Quizá porla importancia de este proceso secuencial,numerosas moléculas y receptores estáninvolucrados y además sujetos a una re-gulación muy fina. Un grupo de esas mo-léculas, las quimiocinas tienen un papelrelevante en dirigir el movimiento que fi-nalmente posibilita el destino celular.Como consecuencia de esta función lasquimiocinas están implicadas en numero-sos procesos fisiopatológicos y su estudiopermite comprender mejor cómo se de-sarrolla la patología y sobre todo aumen-ta las posibilidades de intervención sobreella. Recientemente se ha descrito el pa-pel que algunos de sus receptores desem-
peñan en la infección por VIH lo que sinduda ha relanzado el interés por conocercómo actúan estas moléculas y cómo sepuede interferir en su actuación.
BIBLIOGRAFÍA RECOMENDADA
Berger EA, Murphy PM, Farber JM. Chemokines as HIV-1coreceptors: roles in viral entry, tropism and disease. AnnuRev Immunol 1999; 17: 657-700.Butcher EC. Leukocyte-endothelial cell recognition: three(or more) steps to specificity and diversity. Cell 1991; 67:1.033-1.036.De Melker AA, Sonnenberg A. Integrins:alternative splicingas a mechanism to regulate ligand binding and integrin sig-naling events. BioEssays 1999; 21:499-509.Kunkel SL. Through the looking glass: the diverse in vivoactivities of chemokines. J Clin Invest 1999; 104: 1.333-1.334.Luster AD. Chemokines- Chemotactic cytokines that me-diate inflammation. New Engl J Med 1998; 338: 436-445.Pelchen-Matthews A, Signoret N, Klasse PJ, Fraile-Ramos A,Marsh M. Chemokine receptor trafficking and viral replica-tion. Immunological Reviews 1997: 168: 33-49.Rollins B. Chemokines. Blood 1999; 90: 909-928.Sánchez-Madrid F, del Pozo MA. Leukocyte polarization incell migration and immune interactions. EMBO J 1999; 18:501-511.Springer T. Traffic signals for lymphocyte recirculation andleukocyte emigration:the multistep paradigm. Cell 1994;76: 301-314.Vestweber D, Blanks JE. Mechanisms that regulate thefunction of the selectins and their ligands. Physiological Re-views 1999; 79: 181-213.Ward SG, Bacon K, Westwick J. Chemokines and T lymp-hocytes: More than attraction. Immunity 1998; 9:1-11.Zlotnik A, Morales J, Hedrick JA. Recent advances in che-mokines and chemokine receptors. Critical Review in Im-munology 1999; 19: 1-47.
1298
ENFERMEDADES DEL SISTEMA INMUNE (I)
Infección
Impedimentoestérico
Internalización Dimerización
Bloqueo de la infección
Fig. 5. Representación esquemática de los diferentes modelos para explicar el bloqueo de la infección por VIH por parte de las quimiocinas: modelo de impedimento estérico, modelo de internalización y modelo de dimerización.

1351
Definición y detección de los anticuerposanticitoplasma de losneutrófilos
Los anticuerpos anticitoplasma de neu-trófilo (ANCA, del inglés antineutrophil cy-toplasmic antibodies) son inmunoglobuli-nas dirigidas contra proteínas que selocalizan en el citoplasma de los neutrófi-los. Su detección en la práctica clínica se realiza mediante: a) la técnica de in-munofluorescencia indirecta, que permitela visualización de diferentes patrones ci-toplasmáticos, y b) el método de ELISA,que posibilita la determinación de las en-tidades antigénicas reconocidas por dichosanticuerpos. Los ANCA representan unmarcador de considerable utilidad clínicaen el diagnóstico y seguimiento de un gru-po de vasculitis de pequeño vaso, consti-tuido por la enfermedad de Wegener, lapoliangeítis microscópica y el síndrome deChurg-Strauss. También aportan informa-ción valiosa para el manejo clínico de untipo de glomerulonefritis rápidamente pro-gresiva idiopática con escasos depósitosinmunes (pauciinmune), considerada pordiversos autores la forma limitada renalde la poliangeítis microscópica.Por inmunofluorescencia indirecta (IFI) sehan establecido cuatro patrones citomor-fológicos, con implicaciones clínicas dife-renciales y relevantes:1. C-ANCA o patrón citoplasmático, en elque la fluorescencia se expresa difusa-mente en el citoplasma con acentuacióncentral interlobar (entre los lóbulos del nú-cleo de los neutrófilos), asociado funda-mentalmente a la enfermedad de Wege-ner.
2. P-ANCA o patrón perinuclear, caracteri-zado por la visualización de la zona cito-plasmática inmediatamente adyacente alos lóbulos nucleares, con posible exten-sión de la fluorescencia al interior del nú-cleo. En realidad, el patrón perinuclear esel resultado de un artefacto técnico comoconsecuencia de la fijación de los neutró-filos por etanol, previo a su marcaje porIFI. La fijación con etanol permeabiliza lamembrana de los gránulos intracitoplas-máticos, lo cual permite que las proteínascargadas positivamente, que condicionaneste tipo de patrón de fluorescencia, mi-gren alrededor de la membrana nuclearcargada negativamente, e incluso puedanintroducirse en el interior del núcleo. Te-niendo en cuenta esta disposición cito-morfológica, con frecuencia es difícil con-firmar la presencia de anticuerpos P-ANCAen una muestra en la que coexisten anti-
cuerpos antinucleares (ANA). Para poderdiscriminar ambos anticuerpos con segu-ridad, la fijación de los granulocitos se realiza con otras sustancias como la for-
Medicine 2000; 8(26): 1351-1353
INDICACIONES E INTERPRETACIÓNCLÍNICA DE LOS ANTICUERPOS
ANTICITOPLASMA DE LOS NEUTRÓFILOSL. González Camacho, L. Aranzábal Orgaz y L. Manzano Espinosa
Servicio de Medicina Interna. Hospital Universitario Ramón y Cajal. Departamento de Medicina. Universidad de Alcalá.
TABLA 1Correlación entre los patrones de inmunofluorescencia indirecta (IFI) y las especificidades antigénicas de los ANCA y su asociación clínica
Patrón de IFI Antígenos Asociación clínica*
C-ANCA** PR3 Granulomatosis de Wegener (80%-90%)Poliangeítis microscópica (20%-40%)GNRPI «pauciinmune» (20%-40%)Síndrome de Churg-Strauss (15%)
P-ANCA*** MPO solo Poliangeítis microscópica (50%-80%)GNRPI «pauciinmune» (50%)Síndrome de Churg-Strauss (45%)Granulomatosis de Wegener (10%)
Múltiples especificidades: Enfermedad inflamatoria intestinalcoexistentes: catalasa, Artritis reumatoideenolasa, actina, lactoferrina, Vasculitis inducida por fármacoselastasa, catepsina... Hepatitis crónica autoinmune
C-ANCA atípico BPI solo Fibrosis quística (80%)
Múltiples especificidades Enfermedad inflamatoria intestinalcoexistentes: BPI, MPO... Colangitis esclerosante primaria
Artritis reumatoide
ANCA-atípico Múltiples especificidades Vasculitis inducida por drogascoexistentes: catalasa, Enfermedad inflamatoria intestinalenolasa, actina, lactoferrina... Artristis reumatoide
PR3: proteinasa 3; MPO: mieloperoxidasa; BPI: proteína incrementadora de la permeabilidad; GNRPI: glomerulonefritis rápidamente progresivaidiopática. *Porcentaje de casos positivos. **En muy pocos casos el patrón C-ANCA se corresponde con el antígeno MPO. ***Excepcionalmenteel patrón P-ANCA se corresponde con el antígeno PR3.
TABLA 2Indicaciones para la determinación de los ANCA
Glomerulonefritis, especialmente la forma rápidamente progresiva
Hemorragia pulmonar, sobre todo en el contextodel síndrome reno-pulmonar
Vasculitis cutánea con manifestaciones sistémicas
Nódulos pulmonares múltiples
Lesiones necrótico-inflamatorias crónicas de lasvías aéreas superiores
Sinusitis y otitis de larga duración
Estenosis traqueal subglótica
Mononeuritis múltiple u otras neuropatíasperiféricas
Masa retroorbitaria
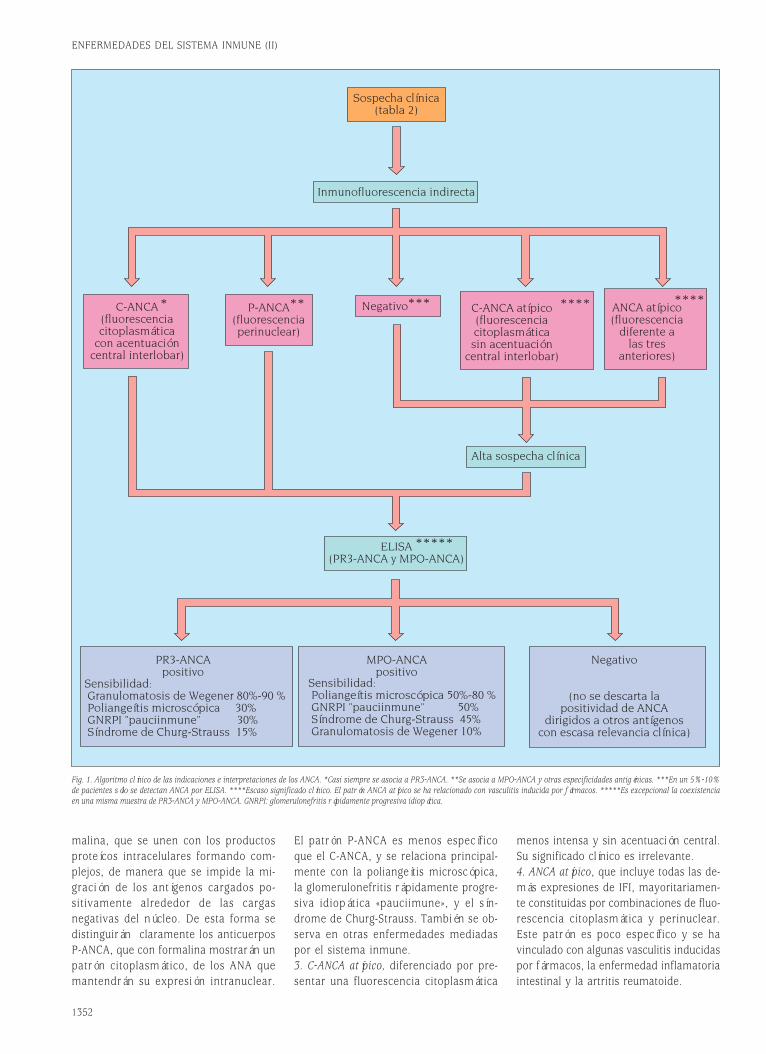
malina, que se unen con los productosproteícos intracelulares formando com-plejos, de manera que se impide la mi-gración de los antígenos cargados po-sitivamente alrededor de las cargas negativas del núcleo. De esta forma sedistinguirán claramente los anticuerposP-ANCA, que con formalina mostrarán unpatrón citoplasmático, de los ANA quemantendrán su expresión intranuclear.
El patrón P-ANCA es menos específicoque el C-ANCA, y se relaciona principal-mente con la poliangeítis microscópica,la glomerulonefritis rápidamente progre-siva idiopática «pauciimune», y el sín-drome de Churg-Strauss. También se ob-serva en otras enfermedades mediadaspor el sistema inmune.3. C-ANCA atípico, diferenciado por pre-sentar una fluorescencia citoplasmática
menos intensa y sin acentuación central.Su significado clínico es irrelevante.4. ANCA atípico, que incluye todas las de-más expresiones de IFI, mayoritariamen-te constituidas por combinaciones de fluo-rescencia citoplasmática y perinuclear.Este patrón es poco específico y se ha vinculado con algunas vasculitis inducidaspor fármacos, la enfermedad inflamatoriaintestinal y la artritis reumatoide.
1352
ENFERMEDADES DEL SISTEMA INMUNE (II)
Sensibilidad: Granulomatosis de Wegener 80%-90 % Poliangeítis microscópica 30% GNRPI "pauciinmune" 30% Síndrome de Churg-Strauss 15%
PR3-ANCApositivo
Sensibilidad: Poliangeítis microscópica 50%-80 % GNRPI "pauciinmune" 50% Síndrome de Churg-Strauss 45% Granulomatosis de Wegener 10%
MPO-ANCApositivo
Negativo
(no se descarta lapositividad de ANCA
dirigidos a otros antígenoscon escasa relevancia clínica)
Sospecha clínica(tabla 2)
Inmunofluorescencia indirecta
ELISA(PR3-ANCA y MPO-ANCA)
C-ANCA(fluorescenciacitoplasmática
con acentuacióncentral interlobar)
P-ANCA(fluorescenciaperinuclear)
C-ANCA atípico(fluorescenciacitoplasmáticasin acentuación
central interlobar)
ANCA atípico(fluorescencia
diferente alas tres
anteriores)
Negativo
Alta sospecha clínica
* * * * * * * * ** * *
* * * * *
* *
Fig. 1. Algoritmo clínico de las indicaciones e interpretaciones de los ANCA. *Casi siempre se asocia a PR3-ANCA. **Se asocia a MPO-ANCA y otras especificidades antigénicas. ***En un 5%-10%de pacientes sólo se detectan ANCA por ELISA. ****Escaso significado clínico. El patrón ANCA atípico se ha relacionado con vasculitis inducida por fármacos. *****Es excepcional la coexistenciaen una misma muestra de PR3-ANCA y MPO-ANCA. GNRPI: glomerulonefritis rápidamente progresiva idiopática.

La detección del antígeno específico queinteracciona con los ANCA se obtiene portécnicas de ELISA. Las proteínas identifi-cadas de mayor relevancia clínica son laproteinasa 3 (PR3) y la mieloperoxidasa(MPO); no obstante, el espectro de antí-genos frente a los que se dirigen los ANCAes muy amplio y engloba, entre otros, lalactoferrina, lisozima, elastasa o BPI (pro-teína incrementadora de la permeabili-dad). Existe una asociación estrecha, aun-que no absoluta, entre los anticuerposdirigidos frente a estas proteínas y el tipode patrón de fluorescencia. En la tabla 1se exponen la correlación de los patronesde IFI de los ANCA con las especifidadesantigénicas, y su significado clínico.Es importante subrayar que al menos enel 10% de las muestras de pacientes conenfermedad de Wegener o poliangeítis mi-croscópica sólo se detectan ANCA por unade las dos técnicas comentadas (IFI y ELI-SA). En consecuencia, ante una sospechaclínica sólida es aconsejable emplear am-bos métodos.
Utilidad diagnóstica de los ANCAAunque los ANCA representan un pará-metro de inestimable valor clínico, su determinación debe interpretarse, en elmomento actual, como un elemento com-plementario, junto con los datos clínicosy anatomopatológicos, en la valoracióndiagnóstica y seguimiento de la enferme-dad. Por tanto, la decisión terapéutica nose fundamentará exclusivamente en la po-sitividad de estos autoanticuerpos.1. En la granulomatosis de Wegener, los C-ANCA con especifidad PR3 tienen unaalta sensibilidad, aunque depende de la
extensión y actividad de la enfermedad.Se estima que en las formas generaliza-das activas con afectación renal, la fre-cuencia de estos autoanticuerpos es su-perior al 95%, mientras que en las formaslimitadas al tracto respiratorio es del 50%.Su especificidad en pacientes con clínicasugestiva de granulomatosis de Wegeneres alrededor del 90%. Conviene resaltarque la concentración sérica de los C-ANCApresenta una considerable relación con elcurso de la enfermedad, de manera que,tras la remisión de la actividad inflama-toria, se observa habitualmente un des-censo importante o desaparición de los tí-tulos de ANCA, elevándose en el 50% delos casos que posteriormente desarrollanuna recaída. Por tanto, la persistencia oelevación de los títulos de ANCA en pa-cientes en remisión traduce un notableriesgo de brote inflamatorio. Sin embar-go, en ocasiones el incremento sérico delos ANCA puede deberse a sobreinfeccio-nes como consecuencia del tratamientoinmunosupresor. En un pequeño porcen-taje de pacientes sólo se detectan P-ANCAque interaccionan con la MPO, cuyo sig-nificado clínico parece ser similar al ex-puesto para los C-ANCA.2. La poliangeítis microscópica y la glome-rulenofritis rápidamente progresiva idiopá-tica «pauciinmune» se asocian en el 90%de los casos con la presencia de ANCA. Elpatrón mayoritario es P-ANCA con espe-cificidad MPO (50%-80%). En el resto delos enfermos se observa anticuerpos tipoC-ANCA frente a la PR3 (20%-40%). Se hacomunicado que la existencia de PR3-ANCA se acompaña de un curso evolutivomás grave que los casos asociados a MPO-ANCA. Al igual que en las granulomatosisde Wegener, los títulos ANCA son de uti-lidad para la actividad de la enfermedad
y la monitorización de la respuesta tera-péutica.3. La detección de ANCA en el síndromede Churg-Strauss sucede en el 60% de loscasos, predominando claramente el patrónP-ANCA con especificidad MPO. Su posi-ble valor como marcador de actividad dela enfermedad aún no se ha podido es-clarecer.4. Se ha observado la presencia de ANCAen otras enfermedades de naturaleza in-mune o infecciosa, cuya determinaciónaporta escasa utilidad clínica, tanto diag-nóstica como pronóstica (tabla 1). En ge-neral, el patrón de fluorescencia observa-do es heterogéneo, en forma de P-ANCA,C-ANCA atípico o ANCA-atípico, con múl-tiples identidades antigénicas. Es impor-tante subrayar que en estos casos es ex-cepcional la existencia de PR3-ANCA ypoco frecuente la detección de MPO-ANCA.En la tabla 2 se describen las principalesindicaciones clínicas de la determinaciónde ANCA y en la figura 1 se presenta unalgoritmo sobre el empleo e interpretaciónde las distintas técnicas de detección deANCA.
BIBLIOGRAFÍA RECOMENDADA
Hoffman S, Specks U. Antineutrophil cytoplasmic antibo-dies. Arthritis Rheum 1998; 41: 1.521-1.537.Savage S, Harper L, Adu D. Primary systemic vasculitis.Lancet 1997; 349: 553-558.Savige J, Gillis D, Benson E, Davies D, Esnault V, Falk RJ, et al. International consensus statement on testing and re-porting of antineutrophil cytoplasmic antibodies (ANCA).Am J Clin Pathol 1999; 111: 507-513.Savige J, Davies D, Falk RJ, Jennette C, Wiik A. Antineu-trophil cytoplasmic antibodies and associated diseases: Areview of the clinical and laboratory features. Kidney Int2000; 57: 846-862
1353
INDICACIONES E INTERPRETACIÓN CLÍNICA DE LOS ANTICUERPOS ANTICITOPLASMA DE LOS NEUTRÓFILOS

1354
IntroducciónLos anticuerpos antinucleares (ANA) soninmunoglobulinas dirigidas contra antíge-nos nucleares. Estos anticuerpos se unena epítopes de moléculas de ADN, ARN oproteínas de localización nuclear o cito-plasmática. Pueden ser detectados enlaboratorio mediante técnicas de inmu-nofluorescencia indirecta (IFI) o inmuno-enzimáticas. Aunque es típica su presen-cia en el lupus eritematoso sistémico (LES)(constituye uno de los criterios diagnósti-cos) tambien se encuentra en otros pro-cesos, especialmente enfermedades au-toinmunes, si bien la relación de estosautoanticuerpos con la inmunopatogeniade la enfermedad no ha sido probada enmuchos casos. Los ANA pueden describirsesegún los patrones observados por IFI; es-tos patrones dependen de la unión del an-ticuerpo a estructuras intracelulares (ac-tualmente se utilizan preparaciones concultivos de líneas celulares humanas en lu-gar de roedor). Los patrones de IFI des-critos (homogéneo, moteado, periférico,nucleolar y citoplasmático) no aportan in-formación diagnóstica ni permiten identi-ficar el anticuerpo específico que producela positividad de la prueba, ya que existeun importante solapamiento entre losdiferentes patrones, los anticuerpos po-tencialmente responsables y las enferme-dades en que pueden hallarse. Una ex-cepción a esta afirmación es la presenciadel patrón centromérico, que se relacionacon la presencia de anticuerpos anticen-trómero, asociados al síndrome de CREST(Calcinosis, fenómeno de Raynaud, afec-tación Esofágica, Esclerodactilia y Telan-giectasias) una forma limitada de esclero-
dermia. Los títulos de ANA para cada pa-ciente se expresan como la máxima dilu-ción del suero que produce fluorescenciadetectable. Habitualmente se dan comopositivos títulos de 1/40 o superiores aun-que son los títulos a partir de 1/160 losque presentan un significado clínico másevidente. Los títulos de ANA dependen dela técnica utilizada; habitualmente no sonequivalentes los valores obtenidos por IFI,enzimoinmunoanálisis o radioinmunoa-nálisis ni intercambiables las determina-ciones de distintos laboratorios.
IndicacionesDebe determinarse la presencia de ANAen todo paciente con enfermedad multi-sistémica sugestiva de LES. También sonútiles en la evaluación de pacientes confotosensibilidad, poliartritis, nefritis, o ci-topenias no explicadas por otras causas.Los ANA están considerados como el mar-cador más importante para el diagnósticode pacientes con sospecha de conectivo-patía. Sin embargo, excepto para el LESla frecuencia y la especificidad diagnósti-ca de los ANA en las enfermedades au-toinmunes y conectivopatías son variables.Además, títulos bajos de ANA son fre-cuentes en otro tipo de enfermedadescomo infecciones crónicas o tumores (tabla 1). Algunos fármacos se asocian contítulos aumentados de ANA (procainami-da, hidralazina, isoniazida, clorpromazinao bloqueadores beta) habitualmente sinllegar a inducir la aparición de lupus eri-tematoso clínicamente significativo. Tam-bién aparecen ANA en ausencia de enfer-medad detectable y su frecuencia aumentacon la edad. Así, un 2% de jóvenes sanospresentan títulos significativos, cifra queaumenta hasta el 5% de los ancianos sa-nos. La presencia de ANA en situacionesclínicas tan diversas convierte su deter-
minación en una prueba poco específica.El mayor valor de los ANA reside en seruna prueba de detección muy sensible(98%) para el LES y con aceptable sensi-bilidad para otras conectivopatías. Prácti-camente todos los pacientes afectos deLES presentan títulos positivos de ANA, losfalsos negativos son muy raros y se sue-len ver en pacientes con otras conectivo-patías, enfermedad tiroidea autoinmune oLES inducido por fármacos. El valor pre-dictivo negativo es mayor del 99% (comocorresponde a una prueba muy sensible)por lo que un resultado negativo práctica-mente descarta la existencia de LES. Como
Medicine 2000; 8(26): 1354-1356
INDICACIONESE INTERPRETACIÓN CLÍNICA
DE LOS ANTICUERPOS ANTINUCLEARESO. Ordóñez Recio, J. Moya Moradas y J. Sabán Ruíz
Servicio de Medicina Interna. Hospital Universitario Ramón y Cajal. Departamento de Medicina. Universidad de Alcalá.
TABLA 1Enfermedades y situaciones asociadas a
la presencia de ANA
Enfermedades autoinmunes
Lupus eritematoso sistémico
Enfermedad mixta del tejido conectivo
Artritis reumatoide
Esclerodermia
Síndrome de Sjögren
Polimiositis-dermatomiositis
Hepatitis crónica autoinmune
Lupus eritematoso discoide
Enfermedades infecciosas
Endocarditis bacteriana
Infección por VIH
Malaria
Abscesos crónicos
Neoplasias
Linfomas
Otras neoplasias
Otras situaciones
Fenómeno de Raynaud
Mujeres sanas (especialmente en tratamientocon anticonceptivos orales)
Ancianos
VIH: virus de la inmunodeficiencia humana
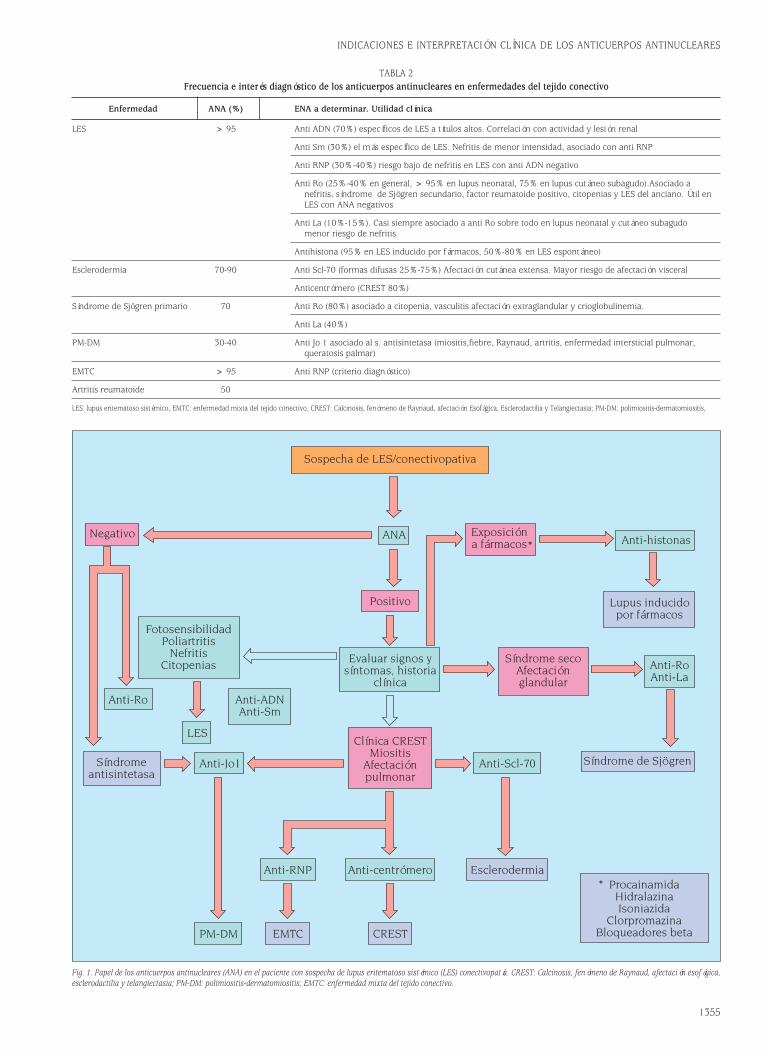
1355
INDICACIONES E INTERPRETACIÓN CLÍNICA DE LOS ANTICUERPOS ANTINUCLEARES
TABLA 2Frecuencia e interés diagnóstico de los anticuerpos antinucleares en enfermedades del tejido conectivo
Enfermedad ANA (%) ENA a determinar. Utilidad clínica
LES > 95 Anti ADN (70%) específicos de LES a títulos altos. Correlación con actividad y lesión renal
Anti Sm (30%) el más específico de LES. Nefritis de menor intensidad, asociado con anti RNP
Anti RNP (30%-40%) riesgo bajo de nefritis en LES con anti ADN negativo
Anti Ro (25%-40% en general, > 95% en lupus neonatal, 75% en lupus cutáneo subagudo).Asociado a nefritis, síndrome de Sjögren secundario, factor reumatoide positivo, citopenias y LES del anciano. Útil en LES con ANA negativos
Anti La (10%-15%). Casi siempre asociado a anti Ro sobre todo en lupus neonatal y cutáneo subagudo menor riesgo de nefritis
Antihistona (95% en LES inducido por fármacos, 50%-80% en LES espontáneo)
Esclerodermia 70-90 Anti Scl-70 (formas difusas 25%-75%) Afectación cutánea extensa. Mayor riesgo de afectación visceral
Anticentrómero (CREST 80%)
Síndrome de Sjögren primario 70 Anti Ro (80%) asociado a citopenia, vasculitis afectación extraglandular y crioglobulinemia.
Anti La (40%)
PM-DM 30-40 Anti Jo 1 asociado al s. antisintetasa (miositis,fiebre, Raynaud, artritis, enfermedad intersticial pulmonar, queratosis palmar)
EMTC > 95 Anti RNP (criterio diagnóstico)
Artritis reumatoide 50
LES: lupus eritematoso sistémico, EMTC: enfermedad mixta del tejido conectivo, CREST: Calcinosis, fenómeno de Raynaud, afectación Esofágica, Esclerodactilia y Telangiectasia; PM-DM: polimiositis-dermatomiositis,
Sospecha de LES/conectivopativa
ANANegativo Exposicióna fármacos Anti-histonas
Positivo
Evaluar signos ysíntomas, historia
clínica
FotosensibilidadPoliartritis
NefritisCitopenias
Lupus inducidopor fármacos
Síndrome secoAfectaciónglandular
Anti-RoAnti-La
Síndrome de Sjögren
Clínica CRESTMiositis
Afectaciónpulmonar
Anti-Scl-70
EsclerodermiaAnti-centrómero
CREST
Anti-Jo1
Anti-RNP
EMTCPM-DM
Síndromeantisintetasa
LES
Anti-Ro Anti-ADNAnti-Sm
*
ProcainamidaHidralazinaIsoniazida
ClorpromazinaBloqueadores beta
*
Fig. 1. Papel de los anticuerpos antinucleares (ANA) en el paciente con sospecha de lupus eritematoso sistémico (LES) conectivopatía. CREST: Calcinosis, fenómeno de Raynaud, afectación esofágica,esclerodactilia y telangiectasia; PM-DM: polimiositis-dermatomiositis; EMTC: enfermedad mixta del tejido conectivo.

1356
ENFERMEDADES DEL SISTEMA INMUNE (II)
consecuencia de la existencia de ANA enmúltiples enfermedades e incluso en po-blación sana, el valor predictivo positivopara la detección de LES en poblacionesno seleccionadas es bajo (30 %-40%); estosupone que dos tercios de los pacientescon ANA positivos no padecen LES. Es fre-cuente encontrar títulos altos de ANA enpacientes con otras conectivopatías; porejemplo, hasta en el 90% de pacientes conesclerodermia y 70% de pacientes con sín-drome de Sjögren (tabla 2). Debido a estasituación, los ANA suponen una de las pri-meras determinaciones que se deben realizar en los pacientes con sospecha deconectivopatía. Si los ANA son negativos,la probabilidad de padecer una enferme-dad autoinmune disminuye, aunque con-viene descartar mediante determinación
de anticuerpos específicos aquellos pro-cesos en los que la presencia de ANA noes habitual (polimiositis/dermatomiositis).En el caso de alta sospecha de LES y ANAnegativos es útil determinar el anticuerpoanti-Ro/SSA (en ocasiones único marcadordel proceso). La negatividad para ANA debeevitar el uso indiscriminado de baterías deautoanticuerpos, ya que en menos del 5%de estos casos se obtienen resultados po-sitivos. En los pacientes con ANA positi-vos las pruebas posteriores deben tratarde identificar los anticuerpos específicosresponsables del resultado positivo (espe-cialmente los anticuerpos con implicacio-nes en el LES), estas determinaciones de-ben ser guiadas por la sintomatología y lasospecha clínica (fig. 1). Solicitar los anti-cuerpos adecuados según el proceso que
se sospeche aumenta el valor predictivopositivo de las pruebas. Los títulos de ANAno son útiles para evaluar el curso de laenfermedad (LES u otra conectivopatía) yno reflejan los cambios en la actividad dela enfermedad ni en la situación clínicadel paciente.
BIBLIOGRAFÍA RECOMENDADA
Barland P, Lipstein J. Selection and use of Laboratory testsin the Rheumathic diseases. Am J Med 1996: 100 (sup2A)16-23.Homburger H. Cascade testin for autoantibodies in Con-nective tissue diseases. Mayo Clin Proc 1995; 70:183-184.Kavanaugh A. The role of the laboratory in the evaluation ofRheumatic Diseases. Clinical Cornerstone 1999; 2(2):11-21.Moder K. Use and interpretation of rheumatologic tests: Aguide for clinicians. Mayo Clin Proc 1996; 71:391-396.Wallace DJ. SLE and Sjögren Syndrome. Curr Opin Rheum1999; 11: 321-329.

1359
Definición
Se llama factor reumatoide (FR) a un sub-tipo de inmunoglobulinas con actividad au-toanticuerpo contra determinantes anti-génicos localizados en la porción Fc de lasIgG2 e IgG3 humanas. Dichas inmunoglo-bulinas pueden ser IgM (las más frecuen-tes), IgG e IgA.Si bien hasta la fecha el FR es el marca-dor serológico más sensible de la artritisreumatoide (AR), pues se halla presenteen el 70%-80% de los enfermos, dicho au-toanticuerpo puede estar presente en múl-tiples patologías (tabla 1) e, incluso, en su-jetos sanos.Esta baja especificidad diagnóstica conlle-va que aproximadamente sólo en un ter-cio de los casos en que se detectan FR esla AR la enfermedad implicada. En el res-to de las ocasiones aparecen otros proce-sos asociados, tales como síndromes lin-foproliferativos de células B (cerca del 90%en la crioglobulinemia mixta esencial), in-fecciones crónicas u otras enfermedadesautoinmunes.
Técnicas de detección de factor reumatoide
Existen varios métodos que permiten de-tectar y cuantificar la presencia de factorreumatoide.
Aglutinación de partículasrecubiertas de IgG
Consiste en poner en contacto el sueroproblema con una solución que contienepartículas (de látex, gelatinas, etc.) recu-biertas de IgG. Si existen inmunoglobuli-nas con actividad FR, y la proporción an-tígeno-anticuerpo es suficiente, se produce
aglutinación de las partículas, con el fac-tor reumatoide como nexo de unión entreellas, al unirse el FR con los determinan-tes antigénicos de las fracciones Fc de lasIgG que recubren las partículas. Esto pro-voca en la placa de análisis una aglutina-ción reconocible a simple vista. La con-centración de FR se expresa como ladilución más alta en la que se produce aglu-tinación, considerándose resultados posi-tivos títulos iguales o superiores a 1/80.
Es una técnica muy sensible para la de-tección de AR; sin embargo, su especifi-cidad es baja. Actualmente, ha sido des-plazada en la práctica clínica diaria por lanefelometría láser.
Prueba de Waaler-Rose
Es la técnica clásica mediante la cual Wa-aler descubrió en 1940, la presencia de FRen el suero de enfermos con AR.
Medicine 2000; 8(26): 1359-1360
INDICACIONESE INTERPRETACIÓN CLÍNICADEL FACTOR REUMATOIDEJ. Moya Moradas, O. Ordóñez Recio y J.L. Calleja López
Servicio de Medicina Interna. Hospital Universitario Ramón y Cajal. Departamento de Medicina. Universidad de Alcalá.
Historia clínica y exploración físicasugestivas de artritis reumatoide
(tabla 2)
Sí No
Determinación de FR Otras determinaciones analíticasy/o procesos diagnósticos
Positivo Negativo
Sospecha clínica Sospecha clínica
Alta Baja
Tratamiento Tratamiento sintomático
Alta
Tratamiento
Reevaluación diagnóstica Repetir determinaciónFR en 6 - 12 meses
Fig. 1. Algoritmo de interpretación clínica de la determinación de factor reumatoide (FR).

En esta técnica se utilizan hematíes de carnero recubiertos con IgG de conejo.Posteriormente se pone en contacto dichasolución con el suero problema, produ-ciéndose aglutinación visible si existen in-munoglobulinas con actividad FR.Esta prueba es más especifica de AR quelas técnicas de aglutinación con látex, perosu sensibilidad es menor.En la actualidad, la prueba de Waaler-Roseno se utiliza en la práctica clínica, por sumayor complejidad técnica y por necesi-tar más tiempo para su realización. Sinembargo, al ser mayor su especificidad,es útil en estudios epidemiológicos de pre-valencia de artritis reumatoide.
Nefelometría láser
Es la técnica más utilizada actualmente enla práctica clínica diaria, presenta una sensibilidad y especificidad similares a lastécnicas de aglutinación con látex, y per-mite procesar en serie y a bajo coste numerosas muestras.
Consiste en exponer una solución de par-tículas de IgG humanas al suero problema,midiendo en el nefelómetro el cambio enla intensidad de ondas láser antes y des-pués de la exposición.Los resultados se emiten en unidades in-ternacionales (UI), tras la calibración delaparato con un estándar internacional.
Interpretación clínica del factor reumatoide
La baja especificidad de la determinaciónde FR para la detección de AR se debe ala presencia de dicho autoanticuerpo enmúltiples enfermedades diferentes e, in-cluso, en sujetos sanos.En la AR aproximadamente el 70%-80%de los pacientes son seropositivos (positi-vidad para FR), el resto son seronegativos(títulos de FR inferiores al umbral de positividad de la técnica utilizada). Estosenfermos seronegativos pueden serocon-vertir a lo largo de la evolución de la en-fermedad. Por otro lado, los enfermos con
AR seropositivos para FR presentan unaexpresión clínica más agresiva, con ma-yor gravedad de las lesiones articulares,parámetros analíticos de actividad infla-matoria más elevados y mayor frecuenciade manifestaciones extraarticulares.Cabe señalar que aunque el FR constituyeel marcador serológico más característicode la AR, éste no es el pilar del diagnósti-co de la enfermedad, sino que el mismo esun ejercicio fundamentalmente clínico, enel que la determinación de FR forma par-te de uno de los criterios diagnósticos (ta-bla 2). Además, como vemos en la figura1, es la sospecha clínica la que marca la ac-titud terapéutica a seguir, independiente-mente de la positividad o no para FR.En resumen, podemos concluir que dadala escasa especificidad de la determina-ción de FR para artritis reumatoide, su uti-lidad clínica es escasa fuera de un con-texto clínico de sospecha. Por dichomotivo, su determinación inicial sin haberrealizado previamente una correcta histo-ria clínica y exploración física, es contra-producente para el paciente afecto de unaenfermedad diferente de una AR, puesprovocaría una actitud terapéutica inicialprobablemente inadecuada y un retrasoen el diagnóstico definitivo de la enfer-medad real de base.
BIBLIOGRAFÍA RECOMENDADA
Barland P, Lipstein E. Selection an use of laboratory test inthe rheumatic diseases. Am J Med 1996; 100: 1.987-1.988.Steffan G. Rheumatoid arthritis. Curr Op in Rheumatol1999; Vol 11.Tighe H, Carson D. Rheumatoid factors. Texbook of Rheuma-tology (5a ed). Philadelphia: WB Saunders, 1997; 241-249.
1360
ENFERMEDADES DEL SISTEMA INMUNE (II)
Neoplasias
Linfomas y leucemias B
Macroglobulinemia de Waldestrom
Carcinoma de colon
Tras tratamientos quimioterápicos y/oradioterápicos
Infecciones bacterianas
Tuberculosis
Endocarditis bacteriana subaguda
Lepra
Brucelosis
Enfermedad de Lyme
Enfermedades reumatológicas
Crioglobulinemia mixta esencial
Artritis reumatoide
Enfermedad mixta del tejido conectivo
Lupus eritematoso sistémico
Síndrome de Sjögren
Polimiositis-dermatomiositis
Esclerodermia
Vasculitis necrotizante
Vasculitis leucocitoclástica
Artritis crónica juvenil
Artritis psoriásica
Artritis por pirofosfato
Otras entidades con hiperactividad de linfocitos B
Sarcoidosis
Silicosis
Cirrosis biliar primaria
Cirrosis hepática etílica
Fibrosis pulmonar idiopática
Politransfundidos
Edad avanzada
Infecciones parasitarias y por protozoos
Esquistosomiasis
Tripanosomiasis
Enfermedad de Chagas
Hidatidosis
Leishmaniasis
Malaria
Toxoplasmosis
Infecciones víricas
Infección por VIH
Hepatitis vírica
Rubéola
Parotiditis
Mononucleosis infecciosa
TABLA 1Enfermedades en las que se ha descrito positividad para factor reumatoide
En negrita aparecen aquellas patologías en las que la frecuencia de positividad para factor reumatoide es mayor del 20%. VIH: virus de la inmu-nodeficiencia humana.
TABLA 2Criterios revisados en 1987 (ARA) para el diagnóstico de la artritis reumatoide
1. Rigidez matutina: rigidez en y alrededor de las articulaciones que dura al menos una horaantes de alcanzar la mejoría funcional máxima
2. Artritis de tres o más áreas articulares detectadasde forma simultánea
3. Artritis de las articulaciones de las manos
4. Afectación articular simétrica
5. Nódulos reumatoideos
6. Factor reumatoideo positivo en suero
7. Alteraciones radiológicas
– Se necesitan cuatro de los siete criterios paradiagnosticar a un paciente como afecto por artritisreumatoide
– Los criterios 1-4 deben estar presentes durante al menos seis semanas
– Los criterios 2-5 deben ser observados por unmédico

1357
Introducción
En este protocolo se comentan las indi-caciones de la determinación en suero delos autoanticuerpos órgano-específicos demayor interés en la práctica médica, se-leccionados en función de su relevanciapara el diagnóstico y/o seguimiento clí-nicos. También son considerados otros anticuerpos no propiamente órgano-espe-cíficos, pero característicos de enferme-dades autoinmunes con afectación anato-moclínica predominantemente localizada,como los antimitocondriales o antiendo-misio.Excluímos los anticuerpos célula-específi-cos como los antiplaquetarios, antieritro-citarios y anticitoplasma de neutrófilo queserán analizados en otros protocolos.
Indicaciones einterpretación de resultados
En la tabla 1 se relacionan las entidadesnosológicas en las que la detección de autoanticuerpos está especialmente indi-cada, reseñando el porcentaje medio depositividades de los diferentes anticuer-pos. Así mismo, en la tabla 2, se estable-ce una valoración global de la utilidad clínica de estos anticuerpos para el diag-nóstico y seguimiento de las correspon-dientes enfermedades autoinmunes.Para una adecuada interpretación de losresultados es preciso tener en cuenta lassiguientes consideraciones:1. Aunque los anticuerpos órgano-especí-ficos constituyen en muchos casos unmarcador de inestimable valor en la prác-tica médica, su determinación debe inter-
pretarse siempre como un elemento com-plementario, junto con los datos clínicosy anatomopatológicos, en la confirmacióndiagnóstica de la enfermedad. En conse-cuencia, el diagnóstico y la toma de deci-siones correspondientes no se fundamenta-rán exclusivamente en la positividad onegatividad de estos anticuerpos. Su detec-ción, a títulos bajos, puede reflejar un es-tado de autorreactividad fisiológica «con-trolada» no condicionante de lesión, porlo que ninguna medida terapéutica actualestá justificada en individuos sin eviden-cia de alteración morfofuncional orgánica.
No obstante, es previsible que el progre-so en el conocimiento de los mecanismospatogénicos implicados en el desarrollo delas enfermedades autoinmunes permita la introducción, en un futuro próximo, demedidas inmunoterapéuticas más eficacesy específicas en fases precoces subclí-nicas. Por otra parte, la ausencia de títu-los detectables de estos anticuerpos nodescarta la enfermedad autoinmune aso-ciada.2. Es frecuente la coincidencia en un mismopaciente de autoanticuerpos característicosde diferentes entidades nosológicas. El sig-
Medicine 2000; 8(26): 1357-1358
INDICACIONES E INTERPRETACIÓNCLÍNICA DE LOS AUTOANTICUERPOS
ÓRGANO-ESPECÍFICOSL. Aranzábal Orgaz, L. González Camacho y L. Manzano Espinosa
Servicio de Medicina Interna. Hospital Universitario Ramón y Cajal. Departamento de Medicina. Universidad de Alcalá..
TABLA 1Indicaciones de la determinación de autoanticuerpos órgano-específicos
Órgano o Entidad nosológica Autoanticuerpo Casos sistema afectado positivos
Sistema endocrino Enfermedad de Graves Antiperoxidasa tiroidea* 70%Antitiroglobulina 70%Antirreceptor de THS 95%
Hipotiroidismo primario Antiperoxidasa tiroidea* 90%Antitiroglobulina 70%
Tiroiditis de Hashimoto Antiperoxidasa tiroidea* 95%Antitiroglobulina 80%
Diabetes mellitus tipo 1 Anticélulas de los islotes pancreáticos 60%-90%
Enfermedad de Addison Anticélulas suprarrenales 80%
Hígado Cirrosis biliar primaria Antimitocondriales 90%
Hepatitis crónica autoinume Antimúsculo liso 95%Anti-LKM1 100%**
Hepatitis crónica vírica C Anti-LKM1 7%
Hepatitis crónica vírica D Anti-LKM3 12%
Tubo digestivo Enfermedad celíaca Antigliadina 90%Antiendomisio 90%Antirreticulina 60%
Anemia perniciosa Anticélulas parietales 90%Antifactor intrínseco 70%
Músculo estriado Miastenia gravis Antirreceptor de acetilcolina 90%Antimúsculo estriado 80%***
Síndrome de Eaton-Lambert Anticanales del calcio tipo P/Q 85%
Riñón Síndrome de Goodpasture Antimembrana basal glomerular 90%
Piel Pénfigo vulgar Antidesmogleína 3 >95%Pénfigo foliáceo Antidesmogleína 1 >95%Penfigoide ampollar Anti-BP230 90%
*Antimicrosomales. **Hepatitis crónica autoinmune tipo II. ***En pacientes con miastenia gravis asociada a timoma.

nificado clínico de esta superposición ocombinación de autoanticuerpos es doble.Por una parte, revela la existencia de unabase inmunopatológica común a determi-nadas enfermedades autoinmunes, queimplica la necesidad de investigar con-juntamente procesos autoinmunes rela-cionados. Por otra parte, la positividad de estos anticuerpos no sólo apoya el diagnóstico de su entidad específica, sinotambién el de otras enfermedades rela-cionadas. En este sentido, es clásica la aso-ciación de anemia perniciosa, tiroiditis deHashimoto, enfermedad de Graves, en-fermedad de Addison, miastenia gravis ydiabetes mellitus tipo I. Otras asociacio-nes usuales son la de la cirrosis biliar pri-maria con la tiroiditis de Hashimoto y de-terminadas conectivopatías (síndrome deSjögren, esclerodermia); la de la hepatitiscrónica autoinmune con la tiroiditis deHashimoto y la diabetes mellitus tipo 1; yla del pénfigo vulgar con la miastenia gra-vis y timoma.3. Además de la utilidad como criteriodiagnóstico, la detección de la concentra-ción sérica de autoanticuerpos puede ser deayuda para el seguimiento clínico. En algu-nos procesos patológicos existe correla-ción entre los títulos de anticuerpos y laactividad clínica inflamatoria (tabla 2), porlo que pueden ser empleados como pará-metros biológicos para la confirmación de
los brotes de la enfermedad y para la mo-nitorización de la respuesta terapéutica.Sin embargo, conviene destacar que unadeterminación aislada de cualquiera de es-tos anticuerpos carece de significado pre-dictivo de la actividad inflamatoria. Por ello,la valoración de una cifra concreta de an-ticuerpos debe realizarse de forma indivi-dual en cada paciente, comparándola conresultados previos obtenidos en diferen-tes fases clínicas de la enfermedad. Es in-teresante matizar que la persistencia deanticuerpos antimembrana basal glome-rular contraindica la realización de tras-plante renal por la alta incidencia de re-cidivas en el riñón trasplantado.
BIBLIOGRAFÍA RECOMENDADA
Bigazzi PE. Organ-localized autoimmune diseases. En:Rose NR, Conway de Macario E, Folds JD, Lane HC, Naka-mura RM, eds. Manual of clinical laboratory Inmunology(5.ª ed.). Washington: ASM press, 1997; 969-1.130.Bylund DJ, Nakamura RM. Organ-directed autoimmune di-seases. En: Henry JB, ed. Clinical diagnosis and mane-gement by laboratory methods. (19,ª ed.). Philadelphia:W.B. Saunders Company, 1996; 1.036-1.049.Peter JB, Shoenfeld Y, eds. Autoantibodies. Amsterdam: El-sevier, 1996.
1358
ENFERMEDADES DEL SISTEMA INMUNE (II)
TABLA 2Estimación del valor clínico de la determinación
de autoanticuerpos órgano-específicos
AutoanticuerpoUtilidad clínica
Diagnóstico Seguimiento
Antiperoxidasa tiroidea* +++ –
Antitiroglobulina ++ –
Antirreceptor de TSH ++ ++
Anticélulas de los islotespancreáticos + –
Anticélulas suprarrenales +/++ –
Antimitocondriales +++ –
Antimúsculo liso +++ –
Anti-LKM1 +++ –
Antigliadina ++ ++
Antiendomisio ++ ++
Antirreticulina ++ ++
Anticélulas parietales ++ –
Antifactor intrínseco ++ –
Antirreceptor de acetilcolina +++ ++
Antimúsculo estriado ++ –
Anticanales del calciotipo P/Q ++ –
Antimembrana basalglomerular +++ +/++
Antidesmogleína 3 +++ +
Antidesmogleína 1 +++ +
Anti-BP230 ++ –
*Antimicrosomales. – Nula. + Limitada. ++ Complementaria.+++ Relevante.

1299
Definición y basesinmunológicasLa función de los linfocitos T se puede ex-plorar in vivo de una forma sencilla me-diante las pruebas cutáneas de hipersen-sibilidad retardada.En la primera exposición a un antígeno,se generan células memoria T CD4+ es-pecíficas. Posteriormente, cuando el antí-geno es inoculado intradérmicamente, lascélulas presentadoras de antígeno lo pro-cesan y exponen, en pequeños fragmen-tos, en su superficie celular, unidos a mo-léculas de clase II del complejo principalde histocompatibilidad, para su reconoci-miento por las células T memoria antíge-no-específicas. Este reconocimiento indu-ce la activación celular, la secreción decitocinas tipo Th1 y el reclutamiento y lasubsecuente activación de monocitos y macrófagos, que producen, precozmen-te, en el lugar de la inoculación, eritemay edema, y posteriormente, a las 48-72horas, dan lugar a la aparición de indura-ción que disminuye gradualmente a par-tir de ese momento1. La presencia de in-duración es la única alteración local queindica la existencia de una reacción de hi-persensibilidad retardada.Una prueba cutánea de hipersensibilidadretardada positiva es un indicador sensi-ble de inmunidad celular intacta, pero unresultado negativo no siempre indica unaalteración de la inmunidad celular, por loque debe interpretarse con precaución.La anergia consiste en la pérdida de la res-puesta cutánea a un antígeno previamen-te expuesto. La anergia específica es la
pérdida de reactividad a un antígeno con-creto, mientras que la generalizada es laincapacidad para reaccionar con cualquierantígeno. Las situaciones de inmunocom-promiso, el síndrome de inmunodeficien-cia humana (SIDA) como prototipo, son elejemplo más conocido y estudiado deanergia generalizada, pero hay otros fac-tores etiológicos, como la malnutriciónproteica, que pueden conducir a esta si-tuación. Los cuadros clínicos asociados conuna producción excesiva de PGE2, comoson los traumatismos graves, han sido co-nocidos como causa de anergia temporal,que pueden ser revertidas con inhibidoresde la ciclooxigenasa. Existen otros me-diadores solubles que modulan esta res-puesta como la histamina o los bloquea-dores del calcio.En pacientes con una exposición remotao infrecuente al antígeno, las células me-moria pueden ser escasas y la reactivi-dad cutánea disminuye con el tiempo. Lareexposición al antígeno puede expandirla población especifica de células me-moria y producir una respuesta positivaen una exposición posterior. Éste es elmecanismo del llamado efecto booster quese pone de manifiesto con la repeticiónde la prueba en un plazo de una a dossemanas y que se utiliza ampliamentecon la prueba de la tuberculina. Sin em-bargo, la cantidad de antígeno inyectadono es suficiente para producir sensibili-zación a sujetos no expuestos y no danresultados falsos positivos en la pruebade la tuberculina.En la actualidad, las pruebas cutáneas dehipersensibilidad retardada tienen dosgrandes aplicaciones:1. El diagnóstico clínico de infección, paralo que se utilizan antígenos de diferentes
microorganismos. La prueba cutánea dela tuberculina o intradermorreacción de Mantoux es la que más se emplean enla clínica. Otras pruebas similares, que uti-lizan leishmanina (prueba cutánea de Mon-tenegro), la melitina o la lepronina, tienenuna aplicación clínica mucho menor o li-mitada a estudios epidemiológicos.2. La demostración de anergia cutáneaen el estudio de pacientes con alteraciónde la inmunidad celular, para lo que seutilizan preparados de múltiples antí-genos.
Prueba de la tuberculinaLa tuberculina fue preparada por primeravez por Roberto Koch en 1890 a partir decultivos de Mycobacterium tuberculosisinactivados por el calor. La prueba cutá-nea preferida es la intradermorreacción deMantoux, que se realiza mediante una in-yección intradérmica de 0,1 ml de PPD-S(derivado proteico purificado de Mycobac-terium tuberculosis) generalmente en lacara volar del antebrazo2. La dosis están-dar de PPD utilizada es de 5 unidades detuberculina (TU), que es denominada comode potencia intermedia; las pruebas conmúltiples punciones y las pruebas que uti-lizan PPD de 1 TU ó 250 TU no ofrecen lasuficiente precisión y no deben utilizarse3.La lectura debe realizarse 48-72 horas des-pués de la inyección. Se mide el diámetrode la induración, expresado en milímetros.El método de lectura más fiable es “el dela punta del bolígrafo” que consiste en tra-zar una línea hacia el área de induracióny notar un aumento en la resistencia almovimiento de la punta cuando se alcan-za el borde de la región indurada. Este pro-Medicine 2000; 8(25): 1299-1302
PROTOCOLO DE INDICACIONESE INTERPRETACIÓN CLÍNICA
DE LAS PRUEBAS DEINMUNIDAD RETARDADA
M. Rodríguez Zapata*, L. Sánchez Martínez**, E. Ruano Soriano** y M.L. Montes Ramírez*Departamento de Medicina. Universidad de Alcalá. Servicio de Medicina Interna. Hospital Universitario de Guadalajara.
**Servicio de Medicina Interna. Hospital Universitario de Guadalajara.

cedimiento se repite en el otro lado de lareacción cutánea, y se toma el diámetromayor. La inspección visual o la palpacióntienen menos sensibilidad4.La conversión de una prueba cutánea dela tuberculina negativa a positiva se pro-duce entre 4 y 10 semanas después de lainfección primaria; sin embargo, entre un10% a 25% de los pacientes diagnostica-dos de tuberculosis activa (enfermedad tu-berculosa) tienen una prueba cutánea ne-gativa. Así, más del 50% de los pacientescon tuberculosis diseminada, meningitistuberculosa o tuberculosis asociada a in-fección por virus de la inmunodeficienciahumana (VIH) tienen una prueba de la tu-berculina negativa. La sarcoidosis, la mal-nutrición proteica, algunas infecciones víricas intercurrentes, enfermedades retí-culoendoteliales, el tratamiento inmuno-supresor o las neoplasias pueden produ-cir falsas reacciones negativas. Por estosmotivos una reacción de Mantoux negativanunca puede excluir la existencia de una en-fermedad tuberculosa activa.La prueba cutánea de la tuberculina es elúnico método probado para identificar lainfección latente por Mycobacterium tu-berculosis y, aunque su sensibilidad y es-pecificidad son menores del 100%, en laactualidad no existe, un método diagnós-tico mejor. Para poder interpretar ade-cuadamente el resultado es necesario co-nocer su sensibilidad, especificidad y valorpredictivo positivo, que varían en funciónde distintos parámetros clínicos y epide-miológicos. En individuos con infecciónlatente por Mycobacterium tuberculosis yuna respuesta inmune normal, la sensi-bilidad se aproxima al 100%. Sin embar-go, en individuos infectados por mico-bacterias no tuberculosas o que han sidovacunados con bacilo de Calmette-Guèrin(BCG) pueden existir falsos positivos. Laespecificidad de la prueba depende delcriterio utilizado para definirla como po-sitiva y mejora según aumenta el tamañode la induración, aunque disminuye lasensibilidad. Por este motivo, las reco-mendaciones de los Center for DiseaseControl (CDC) varían en función del ries-go de cada individuo para desarrollar tu-berculosis5.La vacunación con el BCG produce una re-acción positiva indistinguible de la produ-cida por la infección natural por mico-bacterias que habitualmente disminuyecon el tiempo; sin embargo, puede utili-zarse para apoyar o excluir el diagnostico
de infección por Mycobacterium tuberculo-sis en aquellos casos en los que el pacientetenga un riesgo elevado de infección re-ciente o alguna situación clínica que au-mente el riesgo de enfermedad tubercu-losa. Una reacción mayor de 20 mm deinduración es muy improbable que se pro-duzca como consecuencia de la vacuna-ción por BCG.
Indicaciones
Aunque la prueba de la tuberculina puedeser útil en la evaluación de los pacientesclínicamente sospechosos de enfermedadtuberculosa, su indicación principal con-siste en la detección de individuos con in-fección latente por Mycobacterium tuber-culosis, con riesgo elevado de desarrollartuberculosis y determina la necesidad deinstaurar el tratamiento preventivo co-rrespondiente. Por lo tanto, la decisión derealizar la prueba de la tuberculina debeir acompañada de la decisión de tratar lainfección latente. Por este motivo, los CDCrecomiendan la prueba de la tuberculinaen grupos de riesgo de infección recientey aquellos que con independencia de laduración de la infección, tengan un ries-go elevado de progresión de la infecciónlatente a tuberculosis activa5.
Grupos y factores de riesgo parala infección con Mycobacteriumtuberculosis
Con la excepción de las pruebas realiza-das a aquellos individuos que tienen unriesgo pequeño, pero cuya actividad futu-ra va a suponer un riesgo elevado de exposición, el estudio sistemático de in-dividuos con un riesgo bajo está desa-consejado debido a que distrae recursosde otras actividades con una prioridad ma-yor. Además, una proporción sustancial delos individuos tuberculín-positivos perte-necientes a poblaciones de bajo riesgopueden ser falsos positivos.Los individuos con un alto riesgo de de-sarrollar una tuberculosis activa (riesgosustancialmente mayor que la poblacióngeneral) son aquellos que han adquiridouna infección reciente con Mycobacteriumtuberculosis o tienen alguna enfermedado situación clínica que se asocia a un ries-go aumentado de progresión de la infec-ción latente a tuberculosis activa, (tablas1 y 2)5.
Interpretación de los resultados
Los pacientes que tienen una reacciónpositiva a la tuberculina se denominanreactores. El problema estriba en deter-minar el tamaño de la induración a par-tir del cual una prueba de la tuberculinase considera positiva e indica infeccióntuberculosa. Por otra parte, el diámetrode una reacción positiva de la prueba dela tuberculina depende de múltiples fac-tores como el tipo de exposición a la tu-berculosis, la situación inmunológica dela población estudiada y la prevalenciade infección tuberculosa y por micobac-terias no tuberculosas. Por este motivola ATS y los CDC5 han recomendado diferentes criterios para determinar unareacción positiva basada en el riesgo de desarrollar enfermedad tuberculosa(fig. 1).En individuos con una reacción a la tu-berculina negativa, en los que se repita lamisma de forma periódica (personal sani-tario), un incremento del diámetro de lareacción igual o mayor de 10 mm en unperíodo de 2 años debe ser consideradouna conversión e indica infección recien-te por M. tuberculosis.El efecto booster consiste en una reac-ción positiva cuando se repite la prue-
1300
ENFERMEDADES DEL SISTEMA INMUNE (I)
TABLA 1Pacientes con riesgo elevado para desarrollar
tuberculosis activa
Infección tuberculosa reciente
Infección por VIH
Adictos a drogas por vía parenteral
Lesiones en la radiografía de tórax sugestivade tuberculosis anterior (no diagnosticada)
Pérdida de peso superior al 10% del peso ideal
TABLA 2Pacientes con riesgo relativo para desarrollar
tuberculosis activa
Silicosis
Diabetes mellitus
Insuficiencia renal crónica/hemodiálisis
Gastrectomía
By-pass yeyunoileal
Trasplante de órganos (renal, cardíaco)
Carcinoma de cabeza y cuello
Tratamiento con glucocorticoides o inmunosupresor

ba de la tuberculina 1-2 semanas después de una prueba negativa6. Ocurre en pa-cientes en los que la infección tubercu-losa se ha producido muchos años an-tes y en los que la intensidad de la reacción de hipersensibilidad retardadaha disminuido con el tiempo. Tambiénse observa en pacientes con historia devacunación por BCG, y en aquellos in-fectados con micobacterias no tubercu-losas. Son sujetos con un riesgo bajo ypueden ser manejados como no conver-tores.
Prueba cutánea de anergia(multitest)Como antes se ha señalado, las pruebascutáneas de hipersensibilidad retardadaexploran in vivo el estado funcional de lainmunidad mediada por células, por estemotivo son útiles para el estudio de un pa-ciente en el que se sospeche algún defec-to del sistema inmune1. Para poder in-terpretar los resultados es importante con-siderar su experiencia antigénica, comoinmunizaciones o infecciones previas y la
posible exposición a diferentes antígenos;así mismo es fundamental determinar lareactividad a varios antígenos de forma si-multanea.
Método
Para el estudio de la hipersensibilidad re-tardada se utiliza la inyección intradérmi-ca de 0,1 ml de distintos antígenos talescomo el toxoide tetánico, antígeno de pa-rotiditis, y antígeno de Candida. Existendispositivos que permiten realizar múlti-
1301
PROTOCOLO DE INDICACIONES E INTERPRETACIÓN CLÍNICA DE LAS PRUEBAS DE INMUNIDAD RETARDADA
Criterios de una prueba de tuberculina positiva en función del grupo de riesgo
Reacción > 10 mm de induración Reacción > 15 mm de induraciónReacción > 5 mm de induración
Emigrantes recientes (menos de 5 años)de países con alta prevalencia
Personas sin factores de riesgopara tuberculosis
Pacientes VIH positivos
Adictos a drogas por vía parenteral
Cambios fibróticos en laradiografía de tórax compatibles
con tuberculosis pasada
Pacientes con trasplante deórganos y otras causas de
inmunosupresión(tratamiento > 15 mg de
prednisona durantemás de 1 mes)
Prisiones, residencias geriátricas,hospitales,instituciones de acogidas benéficas,
o para pacientes con infección por VIH
Personal de laboratorio de micobacterias
Contactos recientes con casosde tuberculosis
Pacientes con patologías que conllevenun riesgo elevado
Silicosis, diabetes mellitus, insuficiencia renal crónicaEnfermedades hematológicas como leucemias o linfomas
Otras neoplasias (cabeza, cuello, pulmón)Pérdida del 10% del peso ideal, gastrectomía, bypass yeyunoileal
Niños menores de 4 añosNiños y adolescentes expuestos a adultos con riesgo elevado
Fig. 1. Criterios de una prueba de tuberculina positiva en función del grupo de riesgo. VIH: virus de la inmunodeficiencia humana.

ples pruebas percutáneas de forma si-multánea (Multitest CMI, Cognauht), quecontienen una batería de antígenos dis-tintos como toxoide tetánico y de difteria,tuberculina y antígenos derivados de es-treptococos, Candida, Trichofiton y Proteus,además de glicerina que se utiliza comocontrol negativo de la prueba. La fiabili-dad de este dispositivo es generalmenteadecuada.La respuesta se mide a las 48-72 horasdespués de la inyección. Una induraciónde más de 5 mm de diámetro, se consi-dera positiva en todos los casos. En niños,la induración mayor de 2 mm es algunasveces aceptada como positiva. El eritemano indica una reacción positiva.La principal ventaja de las pruebas de hi-persensibilidad retardada es su facilidad yeconomía. Es un filtro útil en muchas cir-cunstancias en las que se sospeche un dé-ficit de inmunidad celular. Una prueba po-sitiva indica una inmunidad celular intacta,confirma la presencia de linfocitos TCD4+ funcionantes y excluye la mayoríade los defectos congénitos de la inmuni-dad celular, pero un resultado negativo noindica necesariamente un defecto del sis-tema inmune y debe interpretarse con pre-caución. La respuesta es poco fiable enniños menores de 1 año (incluso en pre-
sencia de una exposición anterior al antí-geno) y está suprimida frecuentemente enpresencia de una infección vírica y/o bac-teriana. Por este motivo, ante un resulta-
do negativo se debe repetir la prueba cu-tánea (frecuentemente después de unainmunización). Es importante conocer quela respuesta de hipersensibilidad retarda-da puede conservarse hasta estadios rela-tivamente avanzados de la infección porVIH, por este motivo, la presencia de unaprueba positiva cutánea no debe conside-rarse como una evidencia contra la infec-ción por VIH per se.Por otra parte, las pruebas de anergia sehan utilizado para intentar mejorar el va-lor predictivo de una prueba de la tuber-culina negativa, en pacientes con infec-ción por VIH, como ayuda al diagnósticode tuberculosis latente7,8. Permiten explo-rar la capacidad para desarrollar una res-puesta adecuada a otros antígenos y de-terminar la existencia, o no, de anergiageneralizada. En 1991, los CDC recomen-daron realizar pruebas de anergia cutáneaconjuntamente con la de la tuberculina entodos los pacientes con infección por VIH.Sin embargo, su utilidad no ha sido com-probada y pueden aportar informaciónerrónea9. En este sentido, se ha observa-do que la capacidad para responder a otrosantígenos no aumenta, sustancialmente,el significado clínico de una prueba de latuberculina negativa, ya que un resultadonegativo no excluye una infección latenteo una enfermedad activa por M. Tubercu-losis10. Por otra parte, la reacción positivaa los antígenos de control, frecuentemen-te, promueve una confianza falsa en los
resultados negativos de la tuberculina. Enla actualidad, los CDC no recomiendan larealización sistemática de pruebas de aner-gia cutánea en los pacientes con infecciónpor VIH5.
BIBLIOGRAFÍA
1. Abbas AK, Lichtman AH, Pober JS. Reacciones de hiper-sensibilidad de tipo retardado. En: Abbas AK, LichtmanAH, Pober JS, eds, Inmunología celular y molecular. Ma-drid: Mc Graw Hill. Interamericana, 1998; 312-323.2. Huebner RE, Schein MF, Bass JB. The tuberculin skintest. Clin Infect Dis 1993; 17: 968-975.3. American Thoracic Society/CDC. Diagnostic standardsand clasification of tuberculosis. Am Rev Respir Dis 1990;142: 725.4. Pouchot J, Grasland A, Collet C, Coste J, Esdante JM, Vin-ceneux. Reliability of tuberculin skin test measurement.Ann Intern Med 1997; 126: 210-214.5. Targeted tuberculin testing and treatment of latent tu-berculosis infection. MMWR Morb Mortal Wkly Rep2000;49(RR-6): 1-54.6. Menzies D. Interpretation of repeated tuberculin test.Boosting, conversion and reversion. Am J Respir Crit CareMed 1999; 159: 15.7. Purified protein derivative (PPD) tuberculin anergy andHIV infection. MMWR Morb Mortal Wkly Rep 1991; 40(RR-5): 27-32.8. Moreno S, Baraia-Etxaburu J, Bouza E, Parras F, PérezTascón M, Miralles P, Vicente T, Alberdi JC, et al. Risk fordeveloping tuberculosis among anergic patients infectedwith HIV. Ann Intern Med 1993; 119: 194-198.9. Markowitz N, Hansen NI, Wilcosky TC, Hopewell PC,Glassroth J, Kvale PA et al, for the Pulmonary Complica-tions of HIV infection Study Group. Tuberculin and anergytesting in HIV-seropositive and HIV-seronegative persons.Ann Intern Med 1993; 119: 185-193.10. Slovis BS, Plitman JD, Haas DW. The case againstanergy testing as a routine adjunct to tuberculin skin test.JAMA 2000; 283: 2.003-2.007.
1302
ENFERMEDADES DEL SISTEMA INMUNE (I)

1303
Introducción
Las alteraciones cualitativas o cuantitati-vas de las inmunoglobulinas forman par-te del cuadro clínico de numerosas enfer-medades y constituyen una característicafundamental y crítica en algunas de ellas.Para su estudio es necesario combinar dis-tintas pruebas de laboratorio, ya que nin-guna de ellas aporta, de forma individual,una información completa que permita ca-racterizar las diferentes situaciones pato-lógicas. La electroforesis de alta resoluciónde las proteínas en gel de agarosa es elprocedimiento inicial y permite un análi-sis cualitativo y semicuantitativo de lasproteínas en general y, concretamente, delas inmunoglobulinas1. La inmunofijacióno la inmunoelectroforesis son técnicas fundamentales para diferenciar entre unaumento policlonal y monoclonal de lasinmunoglobulinas. Las técnicas de inmu-nodifusión radial, nefelometría y turbido-metría permiten el estudio cuantitativo delas inmunoglobulinas. Además, en muchasocasiones, es necesario completar el es-tudio con otros procedimientos como ladeterminación de la eliminación urinariade proteínas y la inmunodifusión en ori-na de 24 horas o la medición de la visco-sidad sérica en pacientes con signos o sín-tomas sugerentes de un síndrome dehiperviscosidad2. Dado el elevado núme-ro de las técnicas de laboratorio, es esen-cial su utilización ordenada y racional, asícomo establecer una estrecha relación en-tre el clínico y el laboratorio para optimi-
zar el estudio de cada paciente3. A conti-nuación describiremos las indicaciones yel significado clínico de las principalespruebas de laboratorio implicadas en elestudio de las inmunoglobulinas.
Electroforesis de lasproteínas. ProteinogramaEl fraccionamiento de las proteínas séri-cas por electroforesis en gel de agarosa ocelulosa es un procedimiento sencillo y ba-rato, mediante el cual las proteínas emi-gran bajo la acción de un campo eléctri-co y se localizan en cinco grandes regionesque se denominan albúmina, alfa-1-glo-bulina, alfa-2-globulina, beta-globulina ygamma-globulina. Recientemente se ha de-sarrollado la electroforesis de alta resolu-ción que permite una mayor precisión enla separación de las proteínas.Las inmunoglobulinas (Ig) IgG, IgA, IgM,IgD, IgE están contenidas en la fraccióngamma del proteinograma, pero puedenencontrarse también en la región beta,
beta-gamma y, ocasionalmente, en la re-gión alfa-23.
Alteraciones de la fraccióngamma del proteinograma
Existen distintos patrones patológicos enel proteinograma sérico cuya descripciónexcede el contenido de este capítulo, porlo que nos limitaremos a la de las altera-ciones de la fracción gamma (fig. 1).
Hipogammaglobulinemia
La presencia de una fracción gamma in-ferior a 0-6 g/dl se denomina hipogamma-globulinemia y puede ser producida bienpor un defecto congénito, como es el casode algunas inmunodeficiencias primarias4,bien de forma adquirida formando partedel cuadro clínico de enfermedades comoel mieloma múltiple, la amiloidosis pri-maria, la leucemia linfática crónica, los lin-fomas, el síndrome nefrótico o el trata-miento con glucocorticoides5 (tabla 1). La
Medicine 2000; 8(25): 1303-1306
PROTOCOLO DE INDICACIONESE INTERPRETACIÓN CLÍNICA
DE LA INMUNOELECTROFORESISDE LAS INMUNOGLOBULINAS
EN LA PRÁCTICA CLÍNICA
M. Rodríguez Zapata*, L. Sánchez Martínez*, E. Ruano Soriano** y M.L. Montes Ramírez*Departamento de Medicina. Universidad de Alcalá. Servicio de Medicina Interna. Hospital Universitario de Guadalajara.
**Servicico de Medicina Interna. Hospital Universitario de Guadalajara.
TABLA 1Causas de hipogammaglobulinemia
Congénitas Adquiridas
Inmunodeficiencia asociada al cromoxoma X Mieloma múltiple
Inmunodeficiencia común variable Macroglubulinemia de Waldeström
Amiloidosis primaria
Leucemia linfática crónica
Linfoma
Síndrome nefrótico
Tratamiento con glucocorticoides

existencia de una hipogammaglobulinemiadebe siempre documentarse mediante lacuantificación de los niveles séricos de IgG,IgA e IgM.
Hipergammaglobulinemia
El aumento de la fracción gamma en elproteinograma sérico se denomina hiper-gammaglobulinemia. Este aumento puedeser heterogéneo (policlonal) u homogéneo(monoclonal).La hipergammaglobulinemia policlonal apa-rece como una banda ancha y difusa en laregión de la gammaglobulina. Traduce laexpansión de distintos clones de células B,que producen diferentes tipos de inmuno-globulinas y se caracteriza por la presenciade una o más clases de cadenas pesadas(γ, α y µ) y ambas clases de cadenas lige-ras (λ y κ).Se produce, habitualmente, en el seno deenfermedades inflamatorias, infecciosas oreactivas. Ejemplos de hipergammaglo-bulinemia policlonal son la hepatopatíacrónica, las enfermedades del colágenovascular, las infecciones crónicas o lasenfermedades linfoproliferativas (tabla 2).Sin embargo, en ocasiones, la presen-cia de una hipergammaglobulinemia po-
liclonal no se acompaña de ningún es-tado patológico identificable clínicamen-te5.
Gammapatías monoclonales
El aumento homogéneo de la fracción gam-ma se denomina gammapatía monoclonaly se reconoce como una banda estrecha odensa en el gel de agarosa, que se localizaen la fracción gamma o beta, y excepcio-nalmente en la fracción alfa-2 del protei-nograma sérico. Las gammapatías mono-clonales constituyen un grupo deenfermedades (tabla 3) caracterizadas porla proliferación de un único clono de célu-las B. Este clono produce una inmunoglo-bulina homogénea formada por dos cade-nas pesadas de una misma clase (γ, α, µ, δo ε) y dos cadenas ligeras de una única cla-se (λ o κ) que recibe distintas denomina-ciones, como paraproteína, proteína mo-noclonal, componente M o proteína M.
Indicaciones de laelectroforesis de lasproteínas
La electroforesis de las proteínas está indi-cada siempre que se sospeche la presenciade mieloma múltiple, macroglobulinemiade Waldeström, amiloidosis primaria u otradiscrasia de células plasmáticas (tabla 4).
1304
ENFERMEDADES DEL SISTEMA INMUNE (I)
Electroforesis de las proteínas séricas
Mieloma, macroglobulinemia,Amiloidosis primariaSignificado incierto
Monoclonal
Tabla 3
Enfermedades inflamatoriasEnfermedades infecciosasEnfermedades reactivas
Enfermedades neoplásicas
Policlonal
Tabla 2
Trastorno adquiridoMieloma,
macroglobulinemiaAmiloidosis primaria,
linfomaLeucemia linfática crónica,
esteroides
Trastorno congénitoInmunodeficiencias
primarias
Hipogammablobulinemia
Tabla 1 Tabla 1
Disminución de la fracción gamma inferior a 0 - 6g/dl Aumento de la fracción gamma
Fig. 1. Alteraciones de la fracción gamma del proteinograma sérico.
TABLA 2Causas de hipergammaglobulinemia policlonal
Enfermedades del sistema inmune
Conectivopatías
Sarcoidosis
Vasculitis
Enfermedades hepáticas
Cirrosis hepática
Hepatitis crónica activa
Hepatopatía alcohólica
Cirrosis biliar
Neoplasias
Síndromes linfoproliferativos
Neoplasias avanzadas
Infecciones crónicas
Leishmaniasis
Lepra
Infección por VIH
Endocarditis bacteriana
Parasitosis
VIH: virus de la inmunodeficiencia humana.

Además, está indicada en cualquier pa-ciente con signos o síntomas, por otra par-te inexplicados, que sugieran la existenciade alguna de estas enfermedades6,7.En presencia de alguno de estos hechos, osi se detecta una banda monoclonal en elproteinograma sérico, el estudio se debecompletar mediante la realización de unainmunofijación o una inmunoelectroforesispara definir el tipo de proteína anormal.
Inmunoelectroforesis
La inmunoelectroforesis es una técnica quecombina la separación electroforética delas proteínas con la precipitación inmu-nológica frente a antisueros específicosanti-cadenas pesadas (γ, µ, α, δ, ε) y anti-cadenas ligeras (κ o λ). Aunque fue la téc-nica original empleada para identificar pro-teínas monoclonales, es menos sensible,más lenta y, frecuentemente, más difícilde interpretar que la inmunofijación o lainmunosustracción. Por ello, en el mo-mento actual, la mayoría de los laborato-rios no la utilizan para el estudio de pro-teínas monoclonales en sangre u orina.
Inmunofijación
La inmunofijación es el método de elec-ción para identificar componentes mo-noclonales por su mayor resolución y ra-
pidez6,7. Es una técnica similar a la inmu-noelectroforesis, en la que después de unaelectroforesis se realiza una incubacióncon antisueros específicos. La inmunofija-ción es crítica para la diferenciación entreun aumento monoclonal o policlonal delas inmunoglobulinas5.
Indicaciones de la inmunofijación
La inmunofijación debe realizarse siempreque se detecte una banda estrecha o unpico en la electroforesis en gel de agaro-sa o cuando se sospeche la presencia deun mieloma múltiple, macroglobulinemia,amiloidosis primaria o enfermedades re-lacionadas (tabla 5).Indicaciones adicionales de la inmunofi-jación son: la detección de una pequeñacantidad de proteína M en presencia deinmunoglobulinas normales o aumenta-das1,3,6,7; pacientes con mieloma múltipleo macroglobulinemia en los que el trata-miento haya producido la desaparición dela proteína M en la electroforesis de ruti-na; el reconocimiento y distinción de gam-mapatias biclonales o triclonales. Además,se debe estudiar la posibilidad de una IgDo IgE monoclonal en todos los pacientescon presencia de una cadena ligera mo-noclonal en el suero pero sin reactividadfrente a antisueros anti-G, anti-A o anti-M.
Cuantificación de lasinmunoglobulinas
La cuantificación de las inmunoglobulinasen suero, habitualmente por nefelometríao por inmunodifusión radial, es una téc-nica obligada ante la presencia de unagammapatía monoclonal, tanto para dosi-ficar la Ig monoclonal como para medir laconcentración de las Ig normales, lo queayuda al diagnóstico diferencial entre loscasos de gammapatía monoclonal de ori-gen incierto de aquellas que son expresiónde una discrasia de células plasmáticas. Esuna técnica obligada en el estudio de unahipogammaglobulinemia.
Electroforesis einmunofijación de la orinaconcentrada
La electroforesis de la orina concentradaes la prueba de elección para detectar laeliminación urinaria de cadenas ligeras li-bres (proteinuria de Bence-Jones). La me-dida de la eliminación urinaria de cade-nas ligeras libres aporta una informaciónimportante sobre el índice de masa tu-moral y permite la monitorización futurade su aumento o disminución. Por otraparte, es útil para diferenciar una gam-mapatía monoclonal de significado inciertode las enfermedades proliferativas malig-nas de células plasmáticas.
Indicaciones
En todo paciente con una gammapatía mo-noclonal debe cuantificarse la proteinuriade 24 horas y efectuar una electroforesise inmunoelectroforesis o inmunfijación deuna muestra de orina concentrada. Está
1305
PROTOCOLO DE INDICACIONES E INTERPRETACIÓN CLÍNICA DE LA INMUNOELECTROFORESIS DE LAS INMUNOGLOBULINAS EN LA PRÁCTICA CLÍNICA
TABLA 3Causas de hipergammaglobulinemia monoclonal
Mieloma
Plasmocitoma solitario
Óseo
Extramedular
Mieloma asintomático
Mieloma múltiple
Síndrome POEMS
Macroglobulinemia de Waldeström
Enfermedad de cadenas pesadas
Enfermedades por depósito o infiltración de proteínas
Amiloidosis primaria
Enfermedad por depósito de inmunoglobulinas
Gammapatía monoclonal de significado incierto
Crónica
Transitoria (trasplante renal, trasplante de médulaósea y tratamiento mieloablativo)
Modificada de Alexanian R et al5.POEMS: neuropatía periférica, organomegalia, insuficiencia endocri-na, gammapatía monoclonal y pigmentación cutánea (skin).
TABLA 4Indicaciones de la electroforesis de las proteínas
Datos clínicos aislados que pueden sugerir la presencia de mieloma o macroglobulinemia
Elevación de la velocidad de sedimentación globular
Dolor vertebral
Astenia
Lesiones osteolíticas o fracturas patológicas
Hipercalcemia
Insuficiencia renal
Proteinuria en un paciente mayor de 50 años
Infecciones recurrentes
Datos clínicos aislados que pueden sugerir la presencia de amiloidosis primaria
Síndrome nefrótico
Insuficiencia cardíaca congestiva
Neuropatía autoinmune
Neuropatía periférica
Malabsorción
Macroglosia
Púrpura cutánea
Síndrome del túnel carpiano
TABLA 5Indicaciones de la inmunofijación
Detección de una banda estrecha en la electroforesis
Sospecha de mieloma múltiple, macroglobulinemia,amiloidosis primaria, plasmocitoma solitarioo extramedular o enfermedades asociadas
Detección de proteína M en presencia de inmunoglobulinas normales o aumentadas
Mieloma o macroglobulinemia tratados con desaparición de la banda en la electroforesis
Gammapatías biclonales o triclonales
Gammapatía monoclonal IgD o IgE

indicada, inicialmente, en todo pacientecon mieloma múltiple, macroglobulinemiade Waldeström, amiloidosis primaria, en-fermedad de cadenas pesadas o sospechade estas entidades, y en todos los pa-cientes en los que se detecten cadenas li-geras monoclonales en suero (proteinemiade Bence-Jones).
Evaluación y seguimientode un paciente con unagammapatía monoclonal
Si el paciente no tiene otros datos de dis-crasia de células plasmáticas y la proteí-na M en suero es menor de 1,5 g/dl, laelectroforesis de las proteínas se debe re-petir a intervalos anuales6. La mayoría delas veces no es necesario hacer punción obiopsia de médula ósea, radiografías delesqueleto ni inmunofijación de la orina de24 horas (fig. 2).Si el paciente está asintomático y tieneuna proteína M de 1,5 a 2,5 g/dl, deben
realizarse estudios adicionales, como lacuantificación de las inmunoglobulinas yelectroforesis e inmunofijación de la ori-na de 24 horas. La electroforesis séricadebe repetirse en tres a seis meses y sipermanece estable anualmente, o tanpronto como se presente algún síntoma ocomplicaciones.Si el pico sérico de IgG o IgA es mayor de2,5 g/dl, la posibilidad de que el pacientepresente un mieloma múltiple es mayor.Se debe hacer un estudio óseo metastási-co, incluyendo la visualización del húme-ro y del fémur, electroforesis e inmunofi-jación en orina de 24 horas, aspirado demédula ósea. Si los estudios son norma-les, la electroforesis se debe repetir en doso tres meses, y si el paciente está estableen tres o cuatro meses, y sí continúa es-table al año.La presencia de una proteína M de tipoIgM de más de 2,5 g/dl sugiere la presen-cia de una macroglobulinemia de Wal-deström, y se debe realizar un aspirado obiopsia de médula ósea y una tomografíaaxial computarizada abdominal.
Estos comentarios sirven como una apro-ximación sugerida y el médico debe alte-rar sus estudios dependiendo del cuadroclínico global.
BIBLIOGRAFÍA
1. Kyle RA, Katzman JA. Immunological characterizacion ofinmunoglobulinas. En: Rose NR, Conway de Macario E,Folds JD, eds. Manual of Clinical Laboratory Immunology.(5.ª ed) Washington DC: American Society for Microbio-logy, 1997.2. Keren DF, Alexanian R, Goeken JA, Gorevic PD, Kyle RA,Tomar RH. Guidelines for clinical laboratory evaluation ofpatients with monoclonal gammapathies. Arch Pathol LabMed 1999; 123: 106-107.3. Gallart T. Principales pruebas inmunológicas de interésclínico. En: Farreras Rozman, eds. Medicina Interna (14ªed.) Harcourt, 2000; 3.179-3.185.4. Rosen FS, Cooper MD, Wedgwood RJ. The primary im-munodeficiencies. New Engl J Med 1995; 333: 431-440.5. Alexanian R, Weber D, Liu F. Differential diagnosis ofmonoclonal gammopathies. Arch Pathol Lab Med 1999;123: 108-113.6. Kyle RA. Sequence of testing for monoclonal gammapa-thies. Arch Pathol Lab Med 1999; 123: 114-118.7. Keren DF. Procedures for the evaluation of monoclonalinmunoglobulins. Arch Pathol Lab Med 1999; 123: 126-132.
1306
ENFERMEDADES DEL SISTEMA INMUNE (I)
Proteína monoclonal en el proteinograma
Componente M en suero< 1,5 mg/dl
Sin evidencia de discrasiade células plasmáticas
Componente M en suero1,5-2,5 mg/dl
Paciente asintomático
Componente M en suero> 2,5 mg/dl (IgA oIgG)Posibilidad elevada de
mieloma múltiple
Componente M en suero> 2,5 mg/dl (IgM)
Macroglobulinemiade Waldeström
Repetir electroforesisanualmente
Cuantificación de lasinmunoglobulinas
Electroforesis einmunofijación en orina
de 24 horas
Repetir electroforesis séricaen 3-6 meses
Si permanece estable repetiranualmente
Repetir si síntomas ocomplicación
Estudio óseo metastásicoAspirado de médula óseaBeta 2 microblobulinas
y proteína C reactiva
Aspirado y biopsia demédula ósea
TAC abdominal
Cuantificación de lasinmunologlobulinas
Electroforesis einmunofijación en orina
de 24 horas
Cuantificación de lasinmunologlobulinas
Electroforesis einmunofijación en orina
de 24 horas
Si los estudios son normalesrepetir electroforesis en 2-3 meses
Si permanece estable repetir en 3-4 mesesSi continúa estable repetir al año
Fig. 2. Evaluación y seguimiento de un paciente con gammapatía monoclonal. TAC: tomografía axial computarizada.

IntroducciónEn el anterior capítulo describimos los pro-cedimientos e indicaciones utilizadas parael estudio de las inmunoglobulinas (Ig), asícomo la significación clínica de las prin-cipales alteraciones de los mismos, ha-ciendo un énfasis especial en las altera-ciones cualitativas. En este capítulodescribiremos con más detalle las indica-ciones y significación clínica de la dosifi-cación de las inmunoglobulinas y sus iso-tipos.La determinación de la cantidad y espe-cificidad de los distintos isotipos de Ig ensuero y otros líquidos biológicos es una delas exploraciones complementarias analí-ticas frecuente en la práctica médica. Losmétodos empleados son la nefelometría yla inmunodifusión radial simple.La primera prueba que se utiliza comoprueba inicial es la electroforesis de lasproteínas séricas, descrita en el capítuloanterior. La fracción gamma del protei-nograma sérico aporta una informacióncualitativa y semicuantitativa, sobre la can-tidad y especificidad de las inmunoglobu-linas, y en concreto sobre la concentra-ción de IgG, ya que está integrada en sumayor parte por esta Ig, y a su vez, casitoda la IgG se acumula en esta fracción.
IndicacionesAnte toda disminución de la gammaglo-bulina en el proteinograma sérico, se debeproceder a la cuantificación de las inmu-noglobulinas. Las principales causas de hi-pogammaglobulinemia se encuentran re-
flejadas en la tabla 1 del protocolo titula-do “Protocolo de indicaciones e interpre-tación clínica de la inmunoelectroforesisde las inmunoglobulinas en la práctica clí-nica” que se publica en esta Unidad Te-mática.
Inmunodeficiencias
La cuantificación de las Ig en suero se deberealizar siempre que haya sospecha clíni-ca de una inmunodeficiencia primaria osecundaria1. Así, la presencia de infeccio-nes recurrentes, víricas o bacterianas, deltracto respiratorio o gastrointestinal debenalertar al clínico de la existencia de unadeficiencia de la función de las células By determinar los niveles séricos de IgG,IgA o IgM.Algunas de estas enfermedades, como laagammaglobulinemia ligada al sexo, la inmunodeficiencia común variable o la in-munodeficiencia combinada severa, cur-san con hipogammaglobulinemia2, perootras inmunodeficiencias primarias, como
el déficit selectivo de IgA o el déficit se-lectivo de subtipos de IgG, tienen valoresnormales o poco disminuidos de la frac-ción gamma. En estas circunstancias, y siexiste predisposición a infecciones, prin-cipalmente respiratorias y digestivas esobligada la cuantificación de las Ig y la do-sificación de subclases de IgG, aunque lafracción gamma del proteinograma seanormal1. En la tabla 1 se exponen las prin-cipales causas de inmunodeficiencias pri-marias asociadas a déficit en la produc-ción de anticuerpos.
Gammapatías monoclonales
La cuantificación de Ig está indicada enpresencia de una gammapatía monoclonalen la electroforesis de las proteínas séri-cas, con el doble objetivo de dosificar laIg monoclonal y medir la concentraciónde las Ig normales, lo que sirve de ayudaal diagnóstico diferencial entre los casosde gammapatías monoclonales de origenincierto de aquellas que se deben a una
1307
Medicine 2000; 8(25): 1307-1308
PROTOCOLO DE INDICACIONES EINTERPRETACIÓN DE LA CUANTIFICACIÓN
DE LAS INMUNOGLOBULINAS Y SUSISOTIPOS EN LA PRÁCTICA CLÍNICA
M. Rodríguez Zapata*, L. Sánchez Martínez*, M.L. Montes Ramírez** y E. Ruano Soriano***Departamento de Medicina. Universidad de Alcalá. Servicio de Medicina Interna. Hospital Universitario de Guadalajara.
**Servicio de Medicina Interna. Hospital Universitario de Guadalajara.
TABLA 1Síndromes asociados a la deficiencia de anticuerpos
Enfermedad Inmunoglobulinas séricas
Agammaglobulinemia asociada al cromosoma X Panhipogammaglobulinemia
Inmunodeficiencia común variable Panhipogammaglobulinemia
Déficit selectivo de IgA Déficit de IgA con o sin déficit de IgG2
Síndrome de hiper-IgM Déficit de IgG, IgA e IgE, aumento de IgM e IgD
Déficit de subclases de IgG IgG total normal con déficit selectivo de subclaseespecífica de IgG
Inmunodeficiencia combinada severa Panhipogammaglobulinemia
Síndrome de Wiskott-Aldrich Déficit de IgM, IgG normal, IgA e IgE elevadas
Ataxia telangiectasia Déficit de IgA (70%), déficit de IgE (80%), déficit de IgG2y IgG4

discrasia de células plasmáticas3. Hay quehacer notar que algunas inmunoglobulinasmonoclonales pueden pasar inadvertidasen el proteinograma sérico. Así ocurre conlas compuestas exclusivamente por cade-nas ligeras que se eliminan por la orina(10%-20% de mielomas), las producidasen las enfermedades de las cadenas pe-sadas y las gammapatías monoclonales deescasa concentración sérica, como ocurreen algunos casos de tipo IgG, IgA e IgM y,prácticamente siempre, con las de claseIgD e IgE4.Además de estos dos grandes grupos deindicaciones, la cuantificación de las in-munoglobulinas séricas es un procedi-miento útil en el proceso diagnóstico y te-rapéutico de numerosas enfermedadesinfecciosas e inflamatorias crónicas y neo-plasias hematológicas sobre todo en leish-maniasis, aspergilosis broncopulmonaralérgica, sospecha de infección congénita(determinación de IgM en sangre de cor-dón umbilical)), cirrosis hepática, vasculi-tis de Churg-Strauss, esclerosis múltiple yatopia4.
Estudio de las inmunoglobulinasen otros líquidos orgánicos
El estudio de las inmunoglobulinas enotros líquidos orgánicos es mucho menosfrecuente que el realizado en suero. Noobstante, puede tener utilidad, en pacien-tes con gammapatías monoclonales, la de-terminación de la presencia en líquidopleural o líquido cefalorraquídeo (LCR), delcomponente M.Se debe realizar electroforesis e inmunofi-jación en LCR si la concentración de proteí-nas está elevada. Esto es particularmenteimportante si se encuentran células plas-máticas o linfoides en el estudio citológicodel LCR. Se debe realizar electroforesis e in-munofijación del suero y si no encuentra uncomponente M, hay que considerar la po-sibilidad de un linfoma del sistema nervio-so central o con menor frecuencia un plas-mocitoma5. La presencia de una proteína Men líquido pleural acompaña casi siempre auna proteína M en el suero y se debe ge-neralmente a un tumor de células plasmá-ticas de localización pleural.
La indicación más frecuente para realizarun estudio de las Ig en LCR es la evalua-ción de algunas enfermedades neurológi-cas en las que existe síntesis intratecal deIgG. Así, en la esclerosis múltiple o en lapanencefalitis esclerosante subaguda3 seobserva aumento de la concentración deIgG en LCR y un incremento del índice co-ciente IgG LCR/IgG en suero (IgG/albúmi-na en LCR dividido por el cociente IgG/al-búmina en suero <0,7) así como lapresencia de bandas oligoclonales.
BIBLIOGRAFÍA
1. Callies QC. Hypogammaglobulinemia. En: Hurst JW, ed.Medicine for the practicing phisician. Appleton and Lange,1996; 200-204.2. Rosen FS, Cooper MD, Wedgwood RJ. The primary im-munodeficiencies. New Engl J Med 1995; 333: 431-440.3. Gallart T. Principales pruebas inmunológicas de interésclínico. En: Farreras Rozman, eds. Medicina Interna (14.a
ed.) Harcourt 2000; 3.179-3.185.4. Kyle RA. Sequence of testing for monoclonal gamma-pathies. Arch Pathol Lab Med 1999; 123: 114-118.5. Manzano Espinosa L, García-Suárez. Cuantificación ycaracterización de los isotipos de inmunoglobulinas. Indi-caciones y significación. Medicine 1997; 7: 2.246-2.248.
1308
ENFERMEDADES DEL SISTEMA INMUNE (I)

Base celular de lasrespuestas inmunesantígeno específicas
Las respuestas inmunes antígeno especí-ficas implican tanto a las células presen-tadoras de antígenos (APC), como a lasque los reconocen de modo específico, loslinfocitos T y B. El reconocimiento del an-tígeno por las células T y B desencadenaen éstas procesos de activación, prolife-ración, diferenciación y especializaciónfuncional en células efectoras. El conoci-miento de los mecanismos subyacentes adichos procesos es necesario para lograruna comprensión adecuada de las res-puestas inmunes antígeno-específicas.Las células efectoras antígeno especificasinician tras su activación procesos de es-pecialización funcional. Los linfocitos T alactivarse se diferencian hacia la produc-ción de determinadas citocinas como in-terferón gamma (IFNγ) o interleucina 4 (IL-4)1,2. Las células B se diferencian a cé-lulas plasmáticas productoras de determi-nados isotipos de inmunoglobulinas3,4 condistintas propiedades efectoras. Los pro-cesos de diferenciación de los linfocitos Ty B están íntimamente relacionados pueslos linfocitos T efectores productores deIL-4 favorecen en las células B cambioshacia la producción de anticuerpos de iso-tipos no fijadores de complemento (IgA,IgE, e IgG4), mientras que los linfocitos Tproductores de IFNγ (Th1) favorecen enlas células B cambios hacia la producciónde isotipos de IgG fijadores de comple-mento (IgG1, IgG2 e IgG3).
Reconocimiento antigénicopor los linfocitos TLas células que reconocen un determina-do antígeno son muy infrecuentes y las
células que lo presentan también. Una cé-lula T entre decenas de miles tiene queencontrar a una APC que presente aquelantígeno que reconoce su receptor clono-típico. La recirculación de linfocitos y APCasí como su concentración en los órganoslinfoides secundarios permiten que estosencuentros tan improbables se produz-can5. Los linfocitos T pasan de sangre alos órganos linfoides secundarios extrava-sándose en las vénulas endoteliales altaso en los senos venosos. Los linfocitos T seunen de modo transitorio a través de mo-léculas de adhesión no antígeno-específi-cas a las APC. De este modo, el linfocitoT explora con sus receptores para el antí-geno (TCR) los péptidos unidos a las mo-léculas de histocompatibilidad (MHC) dela APC. Si no encuentra el antígeno quereconoce su receptor, se libera de las APCy a través de los vasos linfáticos eferen-tes retorna a la circulación sanguínea, dedonde vuelve a extravasarse en otro ór-gano linfoide secundario.Las escasas APC que presentan antígenosdel patógeno se concentran en el ganglioque recoge las APC y los mediadoresproinflamatorios solubles procedentes dela zona infectada. La inflamación del gan-glio altera la expresión de moléculas deadhesión en sus vénulas endoteliales altascon lo que aumenta la infiltración de lin-focitos T. El linfocito T específico del pa-tógeno que entre en el ganglio inflamadoencontrará en éste una elevada proporciónde células que presentan antígenos del pa-tógeno.
Reconocimiento de uncomplejo MHC-antígeno pormúltiples TCR en cadena
Cada APC presenta miles de antígenos dis-tintos, en unos 20 tipos de moléculas dehistocompatibilidad diferentes. Esto im-plica que sólo puede presentar unas po-
cas moléculas de cada antígeno particulary éstas se van a encontrar en una baja fre-cuencia en las MHC. Sólo entre 5 y 10 mo-léculas de histocompatibilidad de entre elmillón que expresa una APC contienen elligando antigénico correcto reconocido porun determinado TCR. Por tanto, el reco-nocimiento MHC-antígeno-TCR necesitaser sensible a concentraciones muy bajasde antígeno.La afinidad funcional de los linfocitos T lespermite responder a antígenos a concen-traciones tan bajas como 10–8 M. Sin em-bargo, la afinidad del TCR por los com-plejos MHC-antígeno es baja, de 10–4 a 10–6
M. La explicación a esta paradoja resideen que cada molécula de MHC-antígenoestimula sucesivamente hasta 300 molé-culas de TCR6. El reconocimiento antigé-nico en cadena aumenta la sensibilidad delos linfocitos T a concentraciones bajas de su ligando. Una molécula de MHC-Agse une secuencialmente a unas 40 molé-culas de TCR por hora. Por tanto, 10 com-plejos MHC-péptido antigénico reconocidopor un TCR específico estimulan secuen-cialmente 2.000 TCR en 5 h. Las moléculas de adhesión del linfocito T,CD2 e integrinas tienen afinidades bajaspero el inicio de la reacción de reconoci-miento antigénico incrementa su afinidady fija al linfocito T a la APC. La activacióndel linfocito T se produce cuando en pre-sencia de señales coestimuladoras ade-cuadas el número de moléculas de TCRque reconocen el antígeno supera un um-bral de 1.5006. Tras el reconocimiento an-tigénico, el linfocito inicia una fase de pro-liferación y su progenie se diferenciará alinfocitos T efectores.
Coestimulación y activaciónlinfocitariaLas APC, además de presentar el antíge-no estimulador en sus MHC, aportan al lin-focito T las señales coestimuladoras queson necesarias para su activación y proli-feración. Las señales mejor caracterizadasson los ligandos de CD28, un correceptorde los linfocitos T7,8. Estos ligandos sonB7.1 (CD80) y B7.2 (CD86) y son expre-sados por las APC profesionales.El reconocimiento antigénico en ausenciade las señales coestimuladoras no induci-rá la diferenciación en linfocitos T efecto-res. Este mecanismo de seguridad contri-buye a evitar la activación de los linfocitos
1321
ACTIVACIÓN Y ESPECIALIZACIÓNFUNCIONAL DE LINFOCITOS T Y B A.Prieto Martín*, J. García de Tena*, L. Manzano Espinosa*,** y M. Álvarez-Mon Soto*,****Departamento de Medicina Universidad de Alcalá. **Servicio de Medicina Interna. Hospital Ramón y Cajal. Madrid.***Servicio de Enfermedades del Sistema Inmune y Oncología. Hospital Universitario Principe de Asturias. Alcalá.
Medicine 2000; 8(26): 1321-1330

con autoantígenos que podría provocarrespuestas autoinmunes. Existen tambiénseñales coestimuladoras solubles. La IL-12es secretada por las APC fagocíticas y, sinembargo, no es secretada por otras APCcomo las células B. Puesto que la IL-12 esnecesaria para la diferenciación en célu-las T productoras de IFNγ (Th1), la exis-tencia o no de esta señal coestimuladoradeterminará la especialización funcionaldel linfocito T que reconoce el antígeno9.
Transducción de señales de estimulación y coestimulación
En los procesos de estimulación del linfo-cito T por el antígeno son importantes lasseñales transducidas por tres receptores:los correceptores CD4/CD8, los receptorespara el antígeno y los receptores de se-ñales coestimuladoras (CD28) (fig. 1). Laseñalización del receptor para el antígenoes aumentada por correceptores (CD4 oCD8) que unen el mismo ligando (com-
plejo antígeno-MHC). La señalización delreceptor coestimulador CD28 sigue una víaparalela a la dependiente del TCR. Las tresvías cooperan en la promoción de la ex-presión de genes como el de la IL-2, queson imprescindibles para estimular el cre-cimiento autocrino de los linfocitos T quereconocen el antígeno. Las tirosina-quinasas de la familia Src aso-ciadas a los correceptores CD4 y CD8 fos-forilan los motivos activadores basados entirosina (ITAM) de las cadenas ε y ζ delCD310. La acción de los correceptores au-menta la sensibilidad del receptor para elantígeno en un factor de 100. Los ITAMfosforilados por las tirosina-quinasas unenproteína quinasas ZAP-70 y las activan.ZAP 70 propaga la señal a proteínas G Rasy a la fosfolipasa Cγ (PLC-γ).Las proteínas G activan una cascada deproteínas quinasas que fosforilan factoresde transcripción activándolos11. La prime-ra quinasa de la cascada se denominaMAPquinasa-quinasa-quinasa (MAPKKK).Esta enzima fosforila a las MAP quinasaquinasas y éstas a su vez fosforilan a lasMAP quinasas. Las MAP quinasas en su es-
tado inactivo no fosforilado residen en elcitoplasma, pero cuando son fosforiladasse traslocan al núcleo donde fosforilan enserinas y treoninas a los factores de trans-cripción. Los factores de transcripción ac-tivados promueven la transcripción de ge-nes responsables de la activación dellinfocito T. La estimulación por el antíge-no del TCR provoca la activación del fac-tor intercambiador de nucleótidos de gua-nina SOS que activa a la proteína G Rasque, a su vez, activa la cascada de MAPquinasas que conduce a la activación su-cesiva de Raf, Mek y Erk. Erk fosforila elfactor de transcripción Elk activándolo. Laforma activada de Elk provoca la trans-cripción génica de genes de activacióncomo Fos.Las cascadas de MAP quinasas iniciadaspor factores intercambiadores de nucleó-tidos de guanina no sólo participan en latransmisión de señales procedentes delTCR, sino que también participan en latransmisión de señales de coestimulaciónprocedentes de moléculas coestimulado-ras como CD28. La transmisión de seña-les coestimuladoras procedentes de CD28provoca la activación de otro factor inter-cambiador de nucleótidos de guanina, Vav,que activa a la proteína G Rac que a suvez activa otra cascada de MAP quinasasque conduce a la activación sucesiva deMekk, Jnkk, y Jnk. La quinasa Jnk fosfori-la el factor de transcripción Jun activán-dolo. La asociación de Jun con Fos formael factor de transcripción AP-1.La fosfolipasa Cγ hidroliza fosfatidilinosi-tol en diacil glicerol y fosfoinositol trifos-fato. El diacil glicerol activa a la proteínaquinasa C mientras que el fosfoinositol tri-fosfato provoca el aumento de los nivelesde calcio citoplásmico libre. El aumentode Ca2+ citoplásmico estimula la actividadserina/treonina fosfatasa de la calcineuri-na. La acción de la calcineurina sobre elfactor nuclear de las células T (NFAT) pro-duce una forma desfosforilada y activa deéste que se transloca al núcleo donde in-teracciona con AP-1 y promueve la ex-presión génica. Los fármacos inmunosupresores ciclospo-rina y FK506 (tacrolimus) son inhibidoresselectivos de NFAT pues inhiben la calci-neurina y antagonizan la formación deNFAT activo necesario para la producciónde IL-2. Estos fármacos inhiben la activa-ción de los linfocitos T y se emplean paraprevenir el rechazo de órganos trasplan-tados mediante la inhibición de la activa-
1322
ENFERMEDADES DEL SISTEMA INMUNE (II)
Fig. 1. Vías de transducción de señales del receptor para el antígeno, correceptores CD4/CD8 y moléculas coestimuladoras en lin-focitos T.
Receptoresde membrana
CD4/CD8(correceptor)
TCR/CD3(receptor de antígeno)
CD28(receptor coestimulación)
Transcripcióndel gen
de laIL-2
Src (PTK) PTK PTKFosfotirosina quinasas
Fosfolipasa C PLCSOS
Factores intercambiadoresde nucleótidos de guanina Vav
Ras Proteínas G Rac
Raf(MAPKKK)
Mekk(MAPKKK)
Mek(MAPKK)
Jnkk(MAPKK)
Erk(MAPK)
Jnk(MAPK)
Activaciónen cadena
de quinasas
DAG
PKC
Calcineurina
IP3
[Ca 2+]
Factores nuclearesreguladores
de la transcripcióngénica
Elk Fos Jun
NF - AT NFkB AP - 1
Región promotora de la transcripción del gen de la IL-2

ción de los linfocitos T alorreactivos. Lainteracción NFAT-AP-1 es un ejemplo decómo las proteínas reguladoras de la ex-presión génica integran señales proce-dentes de distintas vías de estimulación.
Expansión clonal de los linfocitos TLos linfocitos T noveles (sin contacto pre-vio con el antígeno) pueden vivir durantelargos períodos de tiempo sin dividirse,pero cuando sus receptores reconocen elantígeno en presencia de las señales co-estimuladoras cesa su migración, se divi-den rápida y repetidamente y, en una se-mana, producen una progenie numerosa(clon) que se diferenciará a linfocitos Tefectores capaces de actuar contra el pa-tógeno cuyos antígenos reconocen. Un lin-focito CD4, al activarse por el antígenopuede sufrir hasta diez rondas sucesivasde divisiones celulares y da lugar a unaprogenie de unos 1.000 linfocitos CD4 conidéntica especificidad antigénica. Un lin-focito CD8 sufre hasta doce rondas de di-visiones sucesivas dando lugar a unos4.000 linfocitos CD8.Las moléculas coestimuladoras solubles di-rigen la proliferación (IL-2 e IL-15)12-14 y ladiferenciación (IL-4 e IL-12)9 de los linfo-citos noveles tras el encuentro antigénico.Las APC producen IL-12 e IL-15, mientrasque los linfocitos T activados producen IL-2 e IL-4 y receptores de alta afinidad paraellas. La unión de IL-2 a su receptor esti-mula de modo autocrino la proliferaciónde los linfocitos durante cuatro o cincodías y posteriormente su diferenciación acélulas T efectoras capaces de respondera menores concentraciones del antígenosin necesidad de coestimulación. Estas cé-lulas efectoras pierden la capacidad de pro-ducir IL-2 por lo que se vuelven depen-dientes de la IL-2 producida por otroslinfocitos T y de otros factores como la IL-12. Algunas células efectoras (Th2) pro-ducen su propio factor de crecimiento, laIL-4. Las señales coestimuladoras depen-dientes de CD28 contribuyen al aumentode la transcripción del gen de IL-2 y tam-bién a la estabilización de su ARN.
Diferenciación a linfocitos TefectoresLos linfocitos activados por el antígeno sediferencian a linfocitos T efectores que son
capaces de luchar contra la fuente del an-tígeno extraño. Los hay de varios tipos:linfocitos T citotóxicos (CTL) CD8+, linfo-citos T cooperadores productores de IFNγ(cooperadores en respuestas dominadaspor IgG1 e IgG3, Th1) o de IL-4 (coopera-dores en respuestas dominadas porIgE,Th2).
Los CTL expresan el correceptor CD8 paramoléculas de histocompatibilidad de cla-se I. Estos linfocitos efectores reconocenel antígeno en células infectadas e indu-cen su muerte programada. Los linfocitosCD4 productores de IFNγ reconocen antí-geno en macrófagos y células B. Sus fun-ciones incluyen la cooperación con ma-crófagos y dirigen los cambios de isotipoen células B hacia isotipos fijadores decomplemento15. Los linfocitos CD4 coo-peradores en las respuestas dominadas porIgE reconocen antígeno en macrófagos ycélulas B. Sus funciones incluyen la coo-peración con células B y la supresión dela activación de los macrófagos.
Diferenciación a célulasefectoras de los linfocitosCD4. Papel de las citocinas
Las respuestas inmunes específicas re-quieren la activación y proliferación de loslinfocitos T que son estimuladas por la IL-2 y la IL-15. Estas citocinas son facto-res de crecimiento para los linfocitos re-cientemente activados. Sin embargo, loslinfocitos ya diferenciados hacia la pro-ducción de IL-4 o IFNγ necesitan de otrosfactores de crecimiento que son respecti-vamente la IL-4 y la IL-12. Estas citocinasreciben el nombre de factores polarizan-tes pues su presencia contribuye a la di-ferenciación del linfocito novato activadohacia la producción de IFNγ (Th1) o de IL-4 (Th2).El desarrollo y la expansión clonal de loslinfocitos Th1 depende de la IL-12 produ-cida por la APC que estimula al linfocitoT a polarizarse hacia Th1 y posteriormenteestimula su crecimiento9. Por tanto, el mo-delo de desarrollo de los linfocitos Th1 noes autocrino sino paracrino. Por el con-trario, el desarrollo y expansión clonal delos linfocitos Th2 depende de la IL-4 pro-ducida por ellos mismos y es, por tanto,autocrino. Esta diferencia entre ambos mo-delos de desarrollo y crecimiento de losdos tipos de linfocitos efectores tiene im-
plicaciones muy relevantes en el procesode elección del tipo de respuesta inmunemás adecuada a cada antígeno o patóge-no.La permanencia del estimulo antigénico,es decir, del antígeno presentado por laAPC apropiada, mantiene la proliferacióny la viabilidad de los linfocitos efectores.Cuando el antígeno presentado por la APCadecuada desaparece los linfocitos efec-tores mueren por apoptosis aunque unapequeña parte de ellos se diferencian a células memoria de larga vida, capaces desobrevivir en ausencia de reestimulaciónantigénica y con capacidad para volver aconvertirse en linfocitos efectores espe-cializados en la producción de una deter-minada citocina si el antígeno reaparece16.Estos linfocitos memoria pueden dar lugara una progenie de linfocitos efectores ocontribuir a la educación de linfocitos no-vatos si el antígeno reaparece. La presen-cia de linfocitos T memoria permite el de-sarrollo de respuestas inmunes másrápidas e intensas a los estímulos antigé-nicos a los que el individuo estuvo ex-puesto en el pasado. Aunque todavía hay controversia, pareceque la memoria T se debe a la supervi-vencia de células T memoria que no ne-cesitan la reestimulación antigénica parasobrevivir. Los linfocitos CD8 memoria so-breviven con interacciones de baja afini-dad con la molécula de MHC que recono-cen en ausencia del antígeno reconocido.Cuando reaparece el antígeno vuelven aproducirse interacciones de mayor afini-dad y la célula memoria se diferencia acélulas efectoras. El estudio de las célulasT memoria es difícil pues es muy compli-cado diferenciar a las células memoria delas células T efectoras. Las característicasfenotípicas de los linfocitos novatos, efec-tores y memoria son mostradas en la ta-bla 1.
Competencia por elantígeno y polarización de la respuesta
Entre los linfocitos Th1 y Th2 se desarro-lla una competencia para dirigir el antí-geno a las APC que los activan fagocitosy células B respectivamente. Los linfocitosTh1 productores de IFNγ necesitan IL-12como factor de crecimiento. La IL-12 esproducida por las APC fagocíticas y éstas
1323
ACTIVACIÓN Y ESPECIALIZACIÓN FUNCIONAL DE LINFOCITOS T Y B

producen IL-12 cuando fagocitan inmu-nocomplejos. Los linfocitos Th1 favorecenel cambio hacia isotipos fijadores de com-plemento que en presencia de antígenoforman inmunocomplejos que serán cap-tados por las APC fagocíticas productorasde IL-1217.Los linfocitos Th2 producen de modo au-tocrino su factor de crecimiento, la IL-4,y sus APC afines, las células B, son capa-ces de captar antígeno libre por medio desus inmunoglobulinas de membrana. Portanto, estos linfocitos Th2 son estimula-dos por APC capaces de capturar el antí-geno libre sin necesidad de que éste for-me inmunocomplejos.
Tipos de linfocitos TefectoresLinfocitos T citotóxicos
Los antígenos procedentes de patógenosque se multiplican en el citosol, como losvirus, son transportados y presentados enMHC de clase I donde son reconocidos porlinfocitos CD8 citotóxicos capaces de ma-tar las células infectadas. Los linfocitos Tcitotóxicos pueden inducir apoptosis enlas células diana por dos mecanismos: unobasado en la liberación del contenido degránulos y otro en la expresión de molé-culas de membrana18. Los CTL CD8+ con-tienen gránulos de secreción con proteí-nas: perforinas, serina proteasas yproteínas transportadoras de poliadenila-to. Tanto los linfocitos T citotóxicos CD8+
como los CD4 productores de IFNγ ex-presan en sus membranas la molécula Fas-
ligando (Fas-L), un miembro unido a mem-brana de la familia del factor de necrosistumoral (TNF)19. Las moléculas de Fas-Lde los CTL son capaces de inducir apop-tosis en las células diana que expresen Fas(CD95).Los linfocitos T citotóxicos liberan el con-tenido de sus gránulos orientando su apa-rato secretor hacia la célula diana, y deeste modo, pueden matar a células espe-cíficas sin dañar a otras adyacentes. Lasperforinas se polimerizan en la membra-na de la célula diana formando una es-tructura cilíndrica con un centro hueco deun diámetro de 160 Å. Estos poros per-miten la entrada en la célula diana de lasserina proteasas y las proteínas transpor-tadoras de poliadenilato que inducen lafragmentación del ADN y apoptosis.
Tipos de linfocitos Tcooperadores (Th): elparadigma Th1/Th2
Las respuestas inmunes específicas se handividido clásicamente en función de los me-canismos efectores implicados en respues-tas mediadas por células y en respuestashumorales basadas en anticuerpos. Desdehace años se sospechaba que las citocinasparticipan en la determinación del tipo derespuesta inmune. Las respuestas media-das por células requieren que los linfoci-tos Th activen a los macrófagos, y esti-mulen el crecimiento y la diferenciaciónde linfocitos T citotóxicos. En las res-puestas mediadas por anticuerpos, los lin-focitos Th estimulan la proliferación de los
linfocitos B y su diferenciación hacia cé-lulas productoras de Igs.Si los linfocitos Th promovían tanto lasrespuestas de anticuerpos como las celu-lares, entonces los patrones de producciónde citocinas por los linfocitos Th efectoresdeberían ser heterogéneos. Así, diferentestipos de linfocitos Th estimularían distin-tas respuestas inmunes. Mosmann et alencontraron que los clones de células Tcooperadoras inmortalizadas podían serdivididos en dos tipos con distintos pa-trones de producción de citocinas. Los lin-focitos Th de tipo1 (Th1) producirían IL-2,IFNγ y TNFβ y promoverían las respues-tas celulares e inflamatorias (respuestasinmunes de tipo 1). Los linfocitos Th detipo 2 (Th2) producirían IL-4, IL-5, IL-6 eIL-10 y cooperarían con las células B en las respuestas humorales (respuestas detipo 2)1,2. El paradigma Th1/Th2 establece que exis-ten dos tipos de linfocitos T cooperadoresantagónicos que producen citocinas queles polarizan, retroalimentan su creci-miento autocrino y antagonizan con el delos linfocitos del tipo contrario2. La dife-renciación de las células cooperadoras apartir de linfocitos T noveles no compro-metidos sería regulada por el balance en-tre la IL-4 y la IL-2 presentes durante laactivación del linfocito. Según este mode-lo, las células Th1 y Th2 producen los fac-tores que estimulan de modo autocrino sucrecimiento (IL-4 e IL-2) y suprimen el delotro subtipo. La IL-2 y el IFNγ producidospor los linfocitos Th1 antagonizan a las cé-lulas Th2, mientras que la IL-4 y la IL-10producidas por los linfocitos Th2 antago-nizan a las Th1 (fig. 2). La competenciaantagónica entre las citocinas producidasconducirá a que una subpoblación se im-ponga y domine en la respuesta inmune.
Desafíos al paradigma Th1/Th2
A continuación se enumeran observacio-nes experimentales que contradicen el sis-tema de regulación basado en un antago-nismo simétrico y competitivo entre dospoblaciones de linfocitos Th proinflama-torios y promotores de respuestas de an-ticuerpos.
La asociación entre la producción de las citocinas no es dicotómica
Los estudios de los patrones de secreciónde citocinas en clones primarios y células
1324
ENFERMEDADES DEL SISTEMA INMUNE (II)
TABLA 1Características inmunofenotípicas de linfocitos T noveles, efectores y memoria
Antígeno de diferenciación Células Células Células noveles efectoras memoria
Moléculas de adhesión
CD11b – ++ –
CD49d – ++ +
CD62L (LNHR) + – +
Experiencia antigénica
CD57 – ++ –
CD45RA + + –
CD45RO – – +
Regulación de la apoptosis
CD95 – + +
CD95L – + ±
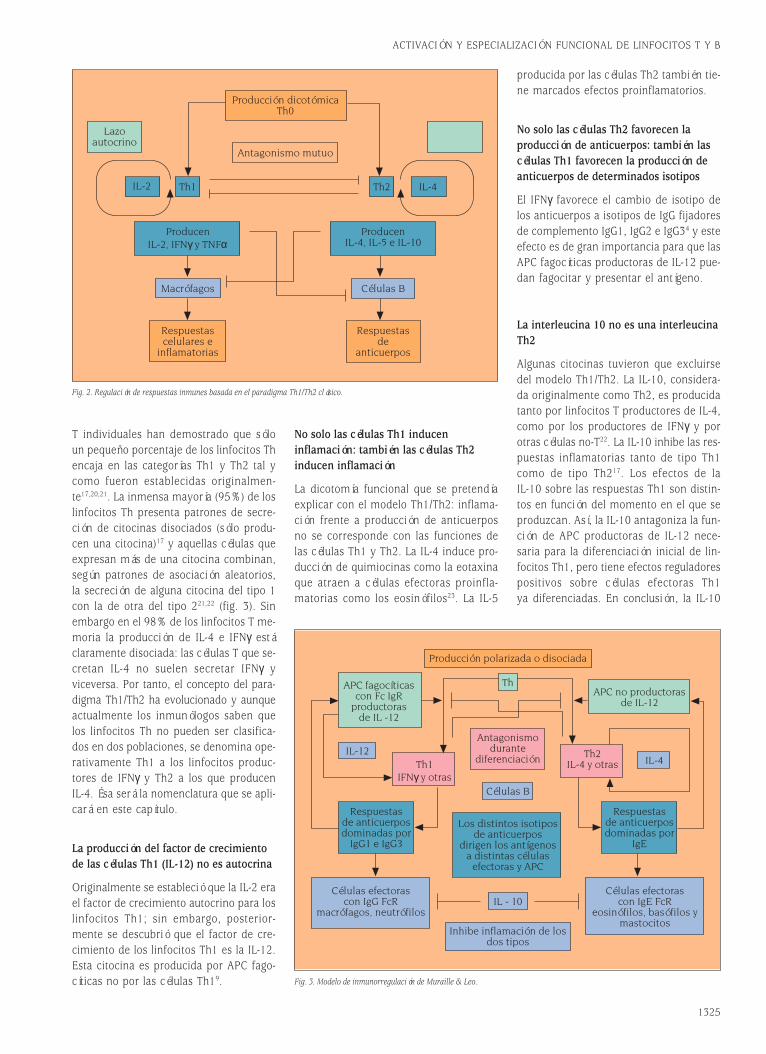
T individuales han demostrado que sóloun pequeño porcentaje de los linfocitos Thencaja en las categorías Th1 y Th2 tal ycomo fueron establecidas originalmen-te17,20,21. La inmensa mayoría (95%) de loslinfocitos Th presenta patrones de secre-ción de citocinas disociados (sólo produ-cen una citocina)17 y aquellas células queexpresan más de una citocina combinan,según patrones de asociación aleatorios,la secreción de alguna citocina del tipo 1con la de otra del tipo 221,22 (fig. 3). Sinembargo en el 98% de los linfocitos T me-moria la producción de IL-4 e IFNγ estáclaramente disociada: las células T que se-cretan IL-4 no suelen secretar IFNγ y viceversa. Por tanto, el concepto del para-digma Th1/Th2 ha evolucionado y aunque actualmente los inmunólogos saben quelos linfocitos Th no pueden ser clasifica-dos en dos poblaciones, se denomina ope-rativamente Th1 a los linfocitos produc-tores de IFNγ y Th2 a los que producenIL-4. Ésa será la nomenclatura que se apli-cará en este capítulo.
La producción del factor de crecimientode las células Th1 (IL-12) no es autocrina
Originalmente se estableció que la IL-2 erael factor de crecimiento autocrino para loslinfocitos Th1; sin embargo, posterior-mente se descubrió que el factor de cre-cimiento de los linfocitos Th1 es la IL-12.Esta citocina es producida por APC fago-cíticas no por las células Th19.
No solo las células Th1 induceninflamación: también las células Th2inducen inflamación
La dicotomía funcional que se pretendíaexplicar con el modelo Th1/Th2: inflama-ción frente a producción de anticuerposno se corresponde con las funciones delas células Th1 y Th2. La IL-4 induce pro-ducción de quimiocinas como la eotaxinaque atraen a células efectoras proinfla-matorias como los eosinófilos23. La IL-5
producida por las células Th2 también tie-ne marcados efectos proinflamatorios.
No solo las células Th2 favorecen laproducción de anticuerpos: también lascélulas Th1 favorecen la producción deanticuerpos de determinados isotipos
El IFNγ favorece el cambio de isotipo delos anticuerpos a isotipos de IgG fijadoresde complemento IgG1, IgG2 e IgG34 y esteefecto es de gran importancia para que lasAPC fagocíticas productoras de IL-12 pue-dan fagocitar y presentar el antígeno.
La interleucina 10 no es una interleucinaTh2
Algunas citocinas tuvieron que excluirsedel modelo Th1/Th2. La IL-10, considera-da originalmente como Th2, es producidatanto por linfocitos T productores de IL-4,como por los productores de IFNγ y porotras células no-T22. La IL-10 inhibe las res-puestas inflamatorias tanto de tipo Th1como de tipo Th217. Los efectos de la IL-10 sobre las respuestas Th1 son distin-tos en función del momento en el que seproduzcan. Así, la IL-10 antagoniza la fun-ción de APC productoras de IL-12 nece-saria para la diferenciación inicial de lin-focitos Th1, pero tiene efectos reguladorespositivos sobre células efectoras Th1 ya diferenciadas. En conclusión, la IL-10
1325
ACTIVACIÓN Y ESPECIALIZACIÓN FUNCIONAL DE LINFOCITOS T Y B
Lazoautocrino
Producción dicotómicaTh0
Antagonismo mutuo
Lazoautocrino
IL-2 Th1 Th2 IL-4
ProducenIL-2, IFNγ y TNFα
ProducenIL-4, IL-5 e IL-10
Macrófagos Células B
Respuestascelulares e
inflamatorias
Respuestasde
anticuerpos
Fig. 2. Regulación de respuestas inmunes basada en el paradigma Th1/Th2 clásico.
Fig. 3. Modelo de inmunorregulación de Muraille & Leo.
Producción polarizada o disociada
ThAPC fagocíticascon Fc IgR
productorasde IL -12
IL-12Th1
IFNγ y otras
Antagonismodurante
diferenciación
Respuestasde anticuerposdominadas por
IgG1 e IgG3
Células B
Los distintos isotiposde anticuerpos
dirigen los antígenosa distintas células
efectoras y APC
Células efectorascon IgG FcR
macrófagos, neutrófilosIL - 10
Inhibe inflamación de losdos tipos
Th2IL-4 y otras IL-4
APC no productorasde IL-12
Respuestasde anticuerposdominadas por
IgE
Células efectorascon IgE FcR
eosinófilos, basófilos ymastocitos

ni es producida en exclusiva por linfoci-tos Th2 ni sus efectos son los de una ci-tocina Th2.
La interleucina 12 no siempre antagonizalas respuestas Th2
Aunque la IL-12 tiene efectos negativos so-bre la iniciación de respuestas Th2 y so-bre la diferenciación de linfocitos novatoshacia células Th2, esta citocina tambiéntiene efectos positivos sobre respuestasTh2 preexistentes.
Tras una respuesta inmune coexistencélulas T memoria de los dos tipos
El paradigma Th1/Th2 predecía que lacompetencia antagónica de los dos tiposconduce a la selección de linfocitos coo-peradores memoria de un solo tipo. La co-existencia de células memoria Th1 y Th2se ha constatado en numerosas respues-tas inmunes17.
Modelo deinmunorregulación deMuraille &Leo
Este modelo17 que se expone a continua-ción pretende integrar los aspectos posi-tivos del paradigma Th1/Th2 en lo que aselección de respuesta inmune se refierecon otros aspectos de regulación de la to-xicidad que determinan la evolución tem-poral de las respuestas inmunes (fig. 3).Cuando el sistema inmune percibe algúnantígeno extraño y amenazador, respon-de utilizando células efectoras no antíge-no específicas (tóxicas e inflamatorias).Unas estimuladas por Th1 (monocito, cé-lulas dendríticas, macrófagos y células na-tural killer [NK]) y otras por Th2 (eosinó-filos, basófilos y mastocitos). Las célulasTh novatas no comprometidas proliferan.Los factores polarizantes IL-4 e IL-12 com-prometen células Th en una vía de dife-renciación a células efectoras. Si predo-mina la presentación antigénica por APCfagocíticas se estimulará una respuestaTh1, si por el contrario predomina la pre-sentación antigénica por células B se esti-mulará una respuesta Th2.La elección de efectores continúa duranteel desarrollo de la respuesta inmune. LasAPC compiten por el antígeno. Las Th1 ne-cesitan APC que fagocitan antígeno y pro-
duzcan IL-12; las Th2 sólo necesitan antí-geno presentado por APC que no produz-can IL-12. El tipo de linfocito cooperadorque recupere la mayoría del antígeno pre-dominará. A continuación, mediante el balance de IL-10 e IL-12 y la utilización de efectoresmás específicos como los CTL y los anti-cuerpos de alta afinidad se produce unafase de control de la inflamación que aumenta la especificidad de la respuestay reduce la toxicidad de la misma. El aumento de especificidad de las célulasefectoras suele conducir a la eliminacióndel patógeno y al desarrollo de inmunidadprotectora. En la respuesta Th1, las célu-las responsables de la inmunidad protec-tora son los propios linfocitos Th1, los CTLantígeno-específicos y las células plasmá-ticas productoras de anticuerpos de isoti-pos IgG1, IgG2, e IgG3 que dirigen la ac-tivación del complemento y actividadcitotóxica dependiente de anticuerpo eje-cutada por las células NK. En la respues-ta Th2, las células responsables de la in-munidad protectora son los linfocitos Th2y las células plasmáticas productoras deanticuerpos de los isotipos IgA, IgG4 e IgE.La IgE dirige la respuesta de mastocitos yeosinófilos. La IgG4 e IgA neutralizan elantígeno. El desarrollo de la memoria in-munológica vendría dado por la supervi-vencia de células memoria de varios tipos;en un caso, linfocitos Th1, CTL y célulasB memoria productoras de IgG1, IgG2 oIgG3 y, en otro, por linfocitos Th2 y célu-las B memoria productoras de IgA, IgG4 oIgE.
Regulación paracrina y autocrina de lapolarización de laspoblaciones cooperadoras
La polarización de la respuesta de las cé-lulas T cooperadoras hacia la producciónde IFNγ o IL-4 viene determinada por fac-tores que dependen de la naturaleza y vía de entrada del antígeno y del tras-fondo genético del huésped. En la ta-bla 2 se muestran los factores que influ-yen en la diferenciación de los linfoci-tos T cooperadores hacia la producciónde IL-4 o IFNγ. La presencia de IL-12 enel microambiente de la activación lin-focitaria es el factor más relevante parala polarización de la diferenciación haciael tipo Th1. Otros factores polarizantesconocidos se relacionan con la produc-ción de IL-12 por la APC y, por tanto, sumecanismo de acción puede relacionar-se con efectos indirectos mediados pordiferencias en la producción de IL-12. Lapresencia de APC adherentes o con ca-pacidad fagocítica favorecen la diferen-ciación a Th1, pero estas APC son pro-ductoras de IL-12. El antígeno forman-do inmunocomplejos y los patógenos intracelulares favorecen la diferencia-ción a Th1, pero estos antígenos son captados por APC fagocíticas producto-ras de IL-12. Entre las células produc-toras de IFNγ e IL-4 se producen rela-ciones de antagonismo recíproco, que in-hiben la polarización hacia el tipo con-trario.
1326
ENFERMEDADES DEL SISTEMA INMUNE (II)
TABLA 2Factores que promueven la diferenciación hacia Th1 y Th2
Polarización hacia producción Factores favorecedores IL-2 + IFNγ, IL-12de IFNγ Presencia de CTL CD8+ activados
APC adherentes (macrófagos)Antígeno que requiera fagocitosisPatógenos intracelularesDosis bajas de antígenosCélulas CD16+
Factores inhibidores IL-10 o IL-4Células productoras de IL-4
Polarización hacia la Factores favorecedores IL-4producción de IL-4 Células B como APC
Antígenos solublesBajas dosis de ciclosporina ADosis altas de antígenos
Factores inhibidores IFNγ, IL-12CTL CD8+ o células productoras de IFNγ
células CD16+
IL: interleucina; IFNγ: interferón gamma; CTL: linfocitos T citotóxicos; APC: células presentadoras de antígeno.

Patrones de expresión dereceptores de quimiocinas y polarización de larespuesta La eotaxina y su receptor CCR3 se impli-can en la atracción de basófilos y eosinó-filos y también en la de los linfocitos Tproductores de IL-4 que estimulan las res-puestas inmunes mediadas por basófilosy eosinófilos. El IFNα y el factor transfor-mante de crecimiento beta (TGFβ) inhibenpolarización a la producción de IL-4 y a laadquisición de CCR3 (tabla 3).
Funciones de los linfocitosT efectoresCooperación con células B:producción de anticuerpos
Los linfocitos B necesitan dos señales parasu activación. La primera es el antígenounido a sus moléculas de inmunoglobuli-nas de membrana. La segunda señal o se-ñal coestimuladora será diferente según laactivación de las células B sea timoinde-pendiente o timodependiente. En la acti-vación timoindependiente, la segunda se-ñal es transmitida por el receptor decomplemento CD21 que reconoce la mo-lécula C3b del complemento covalente-mente unida al antígeno reconocido porlas inmunoglobulinas de membrana24. Enla activación timodependiente, la segundaseñal proviene de los linfocitos coopera-dores que reconocen el antígeno presen-tado por el linfocito B. Los linfocitos CD4cooperadores expresan una molécula de-nominada CD40L que se une a CD40 unamolécula de membrana presente en célu-las B. La supervivencia de las células B quereconocen el antígeno depende de que susmoléculas de CD40 se unan a su ligandoexpresado por los linfocitos T cooperado-res25. Los linfocitos B estimulados por elantígeno y rescatados de la apoptosis porla señal transmitida a través de CD40, pro-liferan activamente y sufren mutaciones
somáticas que posibilitan la selección delos linfocitos B con mayor afinidad por elantígeno.
Cooperación con células B:regulación de su diferenciación
La diferenciación de los linfocitos B haciala producción de determinados isotipos deinmunoglobulinas está condicionada porlas citocinas producidas por los linfocitosCD4. El IFNγ actúa sobre los linfocitos Binhibiendo su proliferación y favorecien-do su diferenciación hacia células pro-ductoras de isotipos opsonizadores que fa-cilitan la captación de antígeno por lascélulas fagocíticas; esto es, los isotipos conmayor afinidad por los receptores Fc delas células fagocíticas y mayor capacidadpara fijar la molécula C1 del complemen-to. Estos isotipos son IgG1, IgG2 e IgG34.Por el contrario la IL-4 promueve la sín-tesis de IgM, la de IgE y la de isotipos deIgG no fijadores de complemento comoIgG43. La IgE se une a receptores Fc IgEde alta afinidad en eosinófilos, basófilos ymastocitos que liberaran el contenido desus gránulos cuando el antígeno se una ala IgE. La IL-5 tiene actividad como factorde diferenciación de células B promo-viendo la síntesis de inmunoglobulinas, es-pecialmente la de IgE e IgA. IL-4 e IL-5también son factores de crecimiento deeosinófilos. La diferenciación de estos lin-focitos B en células efectoras (células plas-máticas) o en células B memoria es de-pendiente de IL-10 y de las interaccionesa través de CD4026. Las células plasmáti-cas migran a la médula ósea, donde suproducción de anticuerpos es estimuladapor citocinas proinflamatorias.
Activación de macrófagos porlinfocitos productores de IFNγ
Los linfocitos Th1 secretan IFNγ, que es laprincipal citocina activadora de macrófa-gos, y linfotoxina que es una molécula ci-
totóxica para linfocitos T, fibroblastos ycélulas tumorales. Los macrófagos nece-sitan dos señales para su activación. Laprimera señal es el IFNγ15. La segunda estransmitida, bien por moléculas de mem-brana como una forma de membrana deTNFα de los linfocitos CD4 que reconocenel antígeno presentado por el macrófago,bien por pequeñas cantidades de lipopo-lisacárido bacteriano que se unen al re-ceptor de LPS del macrófago (CD14). Los macrófagos activados por los linfoci-tos proinflamatorios producen IL-12 (re-troalimentación Th1) y aumentan su ca-pacidad para destruir bacterias en elinterior de sus vesículas, sintetizando ra-dicales oxidantes, óxido nítrico y péptidosantibacterianos9. Estos agentes antibacte-rianos pueden causar daños en los tejidosdel huésped por lo que su secreción estáfinamente regulada. IL- 4 e IL-10 inhibenla mayoría de las funciones de los ma-crófagos, así como sus efectos estimula-dores de linfocitos Th1. Los linfocitos T CD4 proinflamatorios re-clutan las células fagocíticas en los sitiosde infección. Esta función la realizan dedos modos distintos: en primer lugar, pro-ducen IL-3 y GM-CSF que estimulan la producción de nuevas células fagocíticasen la médula ósea; en segundo lugar, pro-ducen TNFα y TNFβ que aumentan la ad-hesividad de las células endoteliales, demodo que los fagocitos se unen a las cé-lulas endoteliales de los vasos que atra-viesan lugares de infección.
CitocinasLas citocinas son mediadores solublesesenciales en la comunicación entre losleucocitos que participan en la regulaciónde la inflamación y de las respuestas in-munes. Su nomenclatura es mostrada enla tabla 4 y su clasificación en familias es-tructurales en la tabla 5. Una mejor com-prensión de los papeles de las citocinas enla regulación de la inflamación y de la res-puesta inmune permitirá desarrollar tera-pias basadas en citocinas para la mani-pulación de las respuestas inmunes confines terapéuticos.
Citocinas mediadoras de inmunidad natural
Son aquellas que protegen frente a la infec-ción vírica (interferones de tipo I, MIP 1-α,
1327
ACTIVACIÓN Y ESPECIALIZACIÓN FUNCIONAL DE LINFOCITOS T Y B
TABLA 3Expresión de receptores por subpoblaciones de linfocitos Tcooperadores
Subset Receptores de quimiocinas Citocinas
Th1 CXCR3, CCR1, CCR5 IL-2, IFNγ
Tnoveles CXCR4 (noveles>memoria), CCR7 noveles seminoveles IL-2
Th2 CCR3, CCR4 IL-4, IL-5

RANTES) y las que inician las reaccionesinflamatorias que defienden frente a lasinfecciones bacterianas (IL-1, IL-6, IL-8, IL-12, IL-15, IL-18 y TNFα,).Los interferones de tipo I (IFN) se sinteti-zan en respuesta a la infección vírica e in-cluyen dos grupos serológicamente dis-tintos de proteínas, denominados α y β.Los interferones α o leucocitarios son producidos preferentemente por fagocitosmononucleares; los interferones β o fibro-blastoideos preferentemente por fibro-blastos. Ambos interferones interaccionancon un mismo receptor. Los interferonesinhiben la replicación vírica mediante lainducción en las células diana de un esta-do de resistencia antivírica y la inhibición
de la proliferación celular. Los interfero-nes aumentan el potencial lítico de las cé-lulas NK y de los monocitos. También in-crementan la expresión de moléculas claseI e inhiben la expresión de moléculas declase II.
Citocinas proinflamatoriasproducidas por macrófagos
Estas citocinas incluyen el TNFα, y las in-terleucinas -1, -6, -8, -12 y -15. El TNFαse produce en grandes cantidades en lassepsis por bacterias gramnegativas. Lasconcentraciones extremadamente eleva-das de TNFα deprimen la contractilidad
del miocardio con la consiguiente reduc-ción de la perfusión tisular, relajan el tonodel músculo liso vascular, contribuyendoa la disminución de la presión sanguínea, yprovocan coagulación intravascular dise-minada. La IL-1 puede ejercer acciones en-docrinas y provoca, en el ámbito sistémi-co, algunos de los efectos que produce elTNFα: fiebre, síntesis de proteínas de faseaguda y caquexia. La IL-6, actúa como co-estimulador de la activación de las célulasT y de los timocitos. También actúa sobrelos hepatocitos, estimulando la síntesis dediferentes proteínas plasmáticas, como elfibrinógeno, que contribuyen a la res-puesta inflamatoria de fase aguda.La IL-12 tiene efectos antagónicos a los dela IL-109. La IL-12 es un factor de dife-renciación y crecimiento de linfocitos Th1.Los efectos antagónicos de la IL-12 sobrela función Th2 son seguramente indirec-tos y debidos a la estimulación de la pro-ducción de IFNγ y a la selección de célu-las no productoras de IL-4. La IL-15,estimula la generación y proliferación delinfocitos T citotóxicos. Sus efectos se so-lapan con los de la IL-2 con la que com-parte la cadena β de su receptor. Sin em-bargo, es mayoritariamente producida pormonocitos y fibroblastos. La IL-15 produ-cida en la sinovial participa como factorproinflamatorio en la sinovitis asociada ala artritis reumatoide. La IL-15 se detectaen los agregados de linfocitos de la mem-brana sinovial inflamada, su expresión secorrelaciona con los niveles de TNFα enliquido sinovial y disminuye con el trata-miento inmunosupresor. La IL-15 podríaser un factor primordial en la perpetua-ción de la inflamación sinovial. Por todolo anterior, los antagonistas del TNFα y dela IL-15 (anticuerpos, proteínas de fusióny receptores solubles) tienen un gran po-tencial terapéutico en el tratamiento de lassinovitis de etiología autoinmune14.
Citocinas que regulan laactivación linfocitaria T
Son producidas principalmente por linfo-citos activados por el antígeno. Promue-ven tanto las respuestas humorales comolas mediadas por contacto celular directo.La interleucina 2 (IL-2) es capaz de man-tener el crecimiento de los linfocitos T encultivo y es producida exclusivamente porcélulas T y NK. El receptor de alta afini-
1328
ENFERMEDADES DEL SISTEMA INMUNE (II)
TABLA 4Nomenclatura de las citocinas y sus receptores
Citocina Sinónimos Receptor
IL-1α Factor activador de los linfocitos (LAF) CD121a
IL-1β Pirógeno endógeno (EP) CD121b
IL-2 Factor de crecimiento de linfocitos T (T-CGF) CD25 - CD122
Factor cooperador de la citotoxicidad (KHF) IL-2Rγ
IL-3 Factor de crecimiento de mastocitos
IL-4 BSF-1 CD124
BCGF-1
IL-5 T cell replacing factor I CD125
Factor II de crecimiento de células B
Factor potenciador de la síntesis de IgA
Factor estimulador de colonias de eosinófilos
IL-6 Interferón b2 CD126
BSF-2 CD130
IL-7 Linfopoyetina I CD127
IL-8 Factor activador de los neutrófilos (NAF-1) CDw128
IL-10 Factor inhibidor de la síntesis de citocinas (CSIF). CD114
IL-12 Factor estimulador de las células NK IL-12R
IL-13 P-600
IL-14 Factor de crecimiento de células B de alto peso molecular
IL-15
GM-CSF CD116
TNFα Caquectina CD120a
CD120b
TNFβ Linfotoxina (LT) CD120a
CD120b
TGFβ Tipo I
Tipo II
Tipo III
IFNα Interferón leucocitario CD118
IFNβ Interferón fibroblastoideo CD118
IFNγ Interferón inmune, interferón tipo II CD119

dad está formado por tres cadenas (αβγ)y ejerce su acción por activación de unatirosina quinasa lck dependiente de las ca-denas β y γ. Los linfocitos T y B así comolos macrófagos en reposo expresan pe-queñas cantidades de receptores de afini-dad baja (α) y las células NK los presen-tan de afinidad intermedia (βγ). Las célulasT y B al activarse expresan un gran nú-mero de moléculas de la cadena α que setraduce en la formación de mayor núme-ro de receptores de alta (αβγ) y de bajaafinidad (α).La actividad biológica de la IL-2 es esencial en la regulación de la pro-liferación de los linfocitos T, en los que in-duce la síntesis de TNFβ e IFNγ entre otrascitocinas. Activa a los precursores citotó-xicos de las células NK; asimismo pro-mueve el crecimiento de las células NK eincrementa su potencial lítico. Tambiénpromueve el crecimiento de las células B,y estimula su diferenciación a células pro-ductoras y secretoras de Igs.
Citocinas promotoras de respuestas de anticuerpos
La IL-4, es un factor polarizante y de cre-cimiento para linfocitos Th2. Antagonizavarios de los efectos de la IL-2; en los lin-focitos B induce el cambio de isotipo deIg hacia la producción de IgE. Sobre losmacrófagos inhibe la expresión de los trestipos de receptores para la región Fc de laIgG y bloquea la producción de IL-1, IL-6,IL-8 y TNFα. Sobre los linfocitos T activa-dos inhibe la producción de IFNγ, aumentala capacidad lítica de los CTL y sobre lascélulas NK induce proliferación y activi-
dad LAK, pero antagoniza los efectos deIL-2. La IL-13 tiene una acción coestimu-ladora sobre las células B y de modo si-milar a la IL-4 induce el cambio de isoti-po hacia la producción de IgE e inhibe lasíntesis de citocinas en los monocitos.La interleucina 14 (IL-14) es producida porlas células foliculares dendríticas y linfo-citos T activados. Esta citocina parece de-sempeñar un importante papel en el de-sarrollo de las células B memoria, aumentala proliferación de las células B activadasy disminuye su síntesis de inmunoglobu-linas.
Citocinas proinflamatoriasproducidas por linfocitos T
Estas citocinas son producidas por linfo-citos T proinflamatorios activados por elantígeno y sirven para activar las funcio-nes de células efectoras que participan enla fase efectora de las respuestas inmunesmediadas por células. Algunas son produ-cidas por linfocitos Th1 (IFNγ y TNFβ) yotras por Th2 (IL-5).El IFNγ es una glucoproteína secretada porlos linfocitos T proinflamatorios y por lascélulas NK. Es el principal factor activadorde macrófagos en los que estimula la pro-ducción de IL-1 y TNFα, aumenta su ca-pacidad lítica e induce la expresión de mo-léculas de histocompatibilidad de clase Iy II del MHC con lo que aumenta la pre-sentación antigénica y la activación de lin-focitos CD4+ y CD8+15. El IFNγ tiene unefecto protector contra infecciones para-sitarias y patógenos intracelulares. Tam-bién regula positivamente la capacidad ci-
totóxica de los linfocitos T y de las célu-las NK, promoviendo su activación y ma-duración. Asimismo, actúa sobre los lin-focitos B inhibiendo su proliferación yfavoreciendo su diferenciación hacia cé-lulas productoras de IgG1, IgG2 o IgG34.El IFNγ activa los neutrófilos y las célulasendoteliales, promoviendo la adhesión einfiltración linfocitaria.El TNF β es producido por los linfocitos Tactivados27 y presenta un 30% de homo-logía en su secuencia de aminoácidos conel TNFα. El TNFα y el TNFβ comparten losmismos receptores y, en consecuencia, susactividades biológicas. La IL-5 es produci-da por linfocitos Th2 activados y tieneefectos proinflamatorios; estimula el cre-cimiento y la diferenciación de los eosi-nófilos y promueve su activación, que esnecesaria para la respuesta inmune fren-te a parásitos. Tiene actividad como fac-tor de diferenciación de células B promo-viendo la síntesis de inmunoglobulinas,especialmente la de IgA.
Citocinas con efecto supresor de las respuestas inmunes
El TGFβ es un polipéptido que induce encélulas no neoplásicas un crecimiento nosuprimible por contacto. El TGFβ es pro-ducido por casi todos los tipos celulares,los fagocitos mononucleares activados porLPS y los linfocitos B y T activados por an-tígenos. Inhibe el crecimiento de muchostipos celulares y estimula el crecimientode otros28. Aumenta la proliferación de losfibroblastos y la síntesis del colágeno. Así,en linfocitos T y B inhibe la proliferacióndependiente de IL-2 y en los linfocitos B,la secreción de Ig inducida por la IL-2 aun-que favorece su diferenciación hacia cé-lulas productoras de IgA. Inhibe la activa-ción de los macrófagos y de los linfocitoscitotóxicos. Su efecto como anticitocinadota de función supresora a los linfocitosT que lo sintetizan.Los receptores solubles de las citocinasson formas truncadas de los receptores demembrana a las que les faltan los domi-nios transmembrana e intracitoplásmico.Pueden liberarse por rotura proteolítica dereceptores transmembrana o por síntesisde ARN mensajero que han sido procesa-dos y sólo contienen la secuencia que co-difica la región extracelular del receptor.Pueden ejercer una función inmunorregu-ladora antagonista de la acción de las ci-
1329
ACTIVACIÓN Y ESPECIALIZACIÓN FUNCIONAL DE LINFOCITOS T Y B
TABLA 5Clasificación de las citocinas y sus receptores
Familia Estructura Clase del Cadena Citocinade la citocina receptor compartida
Hematopoyéticas 4 α hélice cadena corta Dominio CD132 (γ) IL-2, IL-4, IL-7, IL-9, hemopoyetina IL-10, IL-12, IL-13, IL-15
Hematopoyéticas 4 α hélice cadena corta Dominio CDw131 (β) IL-3, IL-5, GM-CSFhemopoyetina
Interferones 4 α hélice cadena corta IFNR IFNα, IFNβ, IFNγ
Hematopoyéticas 4 α hélice cadena larga Dominio CD130 IL-6, IL-11hemopoyetina (gp 130)
Del TNF Hoja plegada β TNFR/NGFR TNFα, TNFβ, CD27-L, CD30-L, CD40-L, NGF
Hoja plegada β Serina/treonina quinasa
Dominios Ig hoja plegada β Tipo IgR IL-1α, IL-1β

tocinas a las que se unen, y limitar su ra-dio de acción al entorno inmediato de loslugares de producción de las citocinas. Sinembargo, no sólo deben ser consideradoscomo agentes neutralizantes de las citoci-nas, ya que la presencia de receptores so-lubles en circulación puede estabilizar yprolongar la vida media de las citocinasen suero aumentando de este modo su ra-dio de acción sobre receptores distantesque posean mayor afinidad que el recep-tor soluble.El antagonista del receptor de IL-1 (IL-1Ra)es estructuralmente homólogo a IL-1β y seune a sus receptores, pero no transduceseñal por lo que actúa como inhibidorcompetitivo de la IL-1 y bloquea su acción.La IL-10 es producida por linfocitos T, B ymonocitos activados. Tiene efectos inmu-nosupresores y antiinflamatorios sobre res-puestas Th1 y Th217. Inhibe la acción co-estimuladora de los macrófagos sobre laproliferación y síntesis de citocinas en lin-focitos T al reducir la síntesis de citocinasproinflamatorias (IL-1, IL-6, IL-8, IL-12 y TNFα ) y aumentar la producción del IL-1Ra.La observación inicial de que la IL-10 blo-quea la producción de citocinas proinfla-matorias como IFNγ e IL-12 hizo pensarque la IL-10 sería una citocina Th2 conefectos antagonistas sobre la funciónproinflamatoria de los linfocitos Th1. Sinembargo, posteriormente se ha compro-bado que esta delimitación del papel de laIL-10 se basa en dos asunciones que sonfalsas. En primer lugar, la IL-10 no es unacitocina exclusivamente producida por cé-lulas Th1, también la producen los linfo-citos Th2 e incluso por muchos tipos ce-lulares distintos de los linfocitos T. Ensegundo lugar, la IL-10 no sólo antagoni-za respuestas inflamatorias agudas de tipoTh1 sino que también antagoniza las ac-ciones proinflamatorias de los linfocitosTh2 sobre eosinófilos, basófilos y masto-citos. La IL-10 también tiene propiedades regu-ladoras positivas: actúa como un factorquimiotrayente para CTL, estimula dife-renciación crecimiento y actividad citotó-xica de CTL y células NK. En células B, es-timula crecimiento, diferenciación, rescatede apoptosis y aumenta la expresión declase II. Finalmente, la IL-10 estimula res-puestas de memoria de anticuerpos y CTL.
En síntesis, la IL-10 es un inhibidor uni-versal de las respuestas inflamatorias y,sin embargo, tiene algunos efectos esti-muladores sobre respuestas antígeno es-pecíficas.
Regulación de lainflamación por elantagonismo entre IL-10-IL-12 La IL-10 es producida por casi cualquiertipo de célula inmune y es claro que tie-ne un gran potencial como factor regula-dor negativo de la inflamación17. La IL-12induce la síntesis de IL-10 iniciando unlazo de retroalimentación negativo que li-mita sus propiedades proinflamatorias.Muchos fármacos antinflamatorios que in-cluyen los que aumentan los niveles deAMPc como las prostaglandinas y los ago-nistas β-adrenérgicos, péptidos retrovíri-cos, inhibidores de las fosfodiesterasas es-timulan la síntesis de IL-1017. La radiaciónultravioleta y los glucocorticoides tambiénlo hacen además de ser citotóxicos paralas células productoras de IL-12.
BIBLIOGRAFÍA
1. Mosmann TR, Coffman RL. Th1 and Th2 cells: Differentpatterns of lymphokine secrection lead to different functio-nal properties. Annu Rev Immunol 1989; 7: 145-173. 2. Mosmann TR, Cherwinski H, Bond MW, Giedlin MA,Coffman RL. Two types of murine helper T cell clone. I. De-finition according to profiles of lymphokine activities andsecreted proteins. J Immunol 1986; 136: 2.348-2.357. 3. Blaser K Allergen dose dependent cytokine productionregulates specific IgE and IgG antibody production. AdvExp Med Biol 1996; 409: 295-303.4. Widhe M, Ekerfelt C Forsberg P, Bergtrom S, ErnerudhJ. IgG subclasses in Lyme borreliosis: a study of specific IgGsubclass distribution in an interferon-gamma-predomina-ted disease Scand J Inmunol 1998; 47: 575-581.5. Macatonia SE, Taylor PM, Knight SC, Asconas BA. Pri-mary stimulation by dendritic cells induces antiviral proli-feration and cytotoxic T cell responses in vitro. J Exp Med1988; 169: 1.255-1.264.6. Viola A, Lanzavecchia A. T cell activation determined byT cell receptor number and tunable thresolds. Science1996; 273: 104-106.7. Kohno K, Shibata Y, Matsua Y, Minowada J: CD28 as areceptor like function accesory signals in cell mediatedaugmentation of IL-2 production. Cell Immunol. 1990; 131:1-10.8. Linsley PS, Clark EA, Ledbetter JA. T-cell antigen CD28mediates adhesion with B cells by interacting with activa-tion antigen B7/BB1. Proc Natl Acad Sci USA 1990; 87:5.031-5.035.
9. Manetti R, Parronchi P, Giudizi MG, Piccinhi MP, MaggiE, Trinchieri G, Romagnan: S. Natural killer stimulatory fac-tor (NKSF/IL-12) induces Th-1 type specific immune res-ponses and inhibits the development of IL-4 producing Thcells J Exp Med 1993; 177: 1.199-1.204.10. Bolen JB, Brugge JS. Lymphocyte protein tyrosine kina-ses: potential targets for drug discovery Annu Rev Immu-nol 1997, 15: 371-404.11. Cantrell D. T cell antigen receptor signal transductionpathways Annu Rev Immunol 1996; 14: 259-274.12. Larson EL, Iscove N., Coutinho A. Two distinct factorsare required for induction of T-cell growth. Nature 1980;283: 664-666.13. Meuer SC, Hussey RE, Cantrel DA, Hodgdon JC, Sch-lossman Smith KA, Reinherz EL. Triggering of the T3-Ti an-tigen-receptor complex results in clonal T-cell proliferationthrough an interleukin 2- dependent autocrine pathway.Proc Natl Acad Sci USA 1984; 81: 1.509-1.513.14. Mc Innes, Liew FY. Interleukin 15: a proinflammatoryrole in rheumathoid arthritis synovitis. Immunol Today1998; 19: 75-79.15. Stout R Antigen specific activation of effector macropha-gues by interferon -gamma producing (TH1) T-cell clones: fai-lure of IL-4 producing (TH2) T-cell clones to activate effectorfunctions in macrophagues J Immunol 1989; 142: 760.16. Karulin AY, Hesse MD, Tary-Lechmann M, LehmannPV. Single-cytokine producing CD4 memory cells predomi-nate in type 1 and type 2 immunity. J Immunol 2000 164:1.862-1.87217. Muraille E, Leo O. Revisiting the Th1/Th2 paradigmScand J Immunol 1998; 47:1-9.18. Helgason CD, Shi L, Greenberg AH, Shi Y, Bromley P,Cotter TG, et al. DNA fragmentation induced by cytotoxic Tlymphocytes can result in target cell death. Exp Cell Res1993; 206: 302-310.19. Ju ST, Cui H, Panka DJ, Ettinger R, Marshak-RothsteinA. Participation of target Fas protein in apoptosis pathwayinduced by CD4+ Th1 and CD8+ cytotoxic T cells. ProcNatl Acad Sci USA 1994; 91: 4.185-4.189.20. Kelso A, Trout AB, Maraskovsky E, Gough NM, MorrisL, Pech MH, Thomsom JA. Heterogeneity in lymphokineprofiles of CD4+ and CD8+ T cells and clones activated invivo and in vitro Immunol rev 1991; 123: 85-114.21. Kelso A. Th1 and Th2 subsets: paradigms lost? Immu-nology Today 1995; 16: 374-379. 22. Assenmacher M, Schmitz J, Radbruch A: Flow cytome-tric determination of cytokines in activated murine T hel-per lymphocytes: expression of interleukin-10 in IFN-gam-ma and in interleukin-4-expressing cells. Eur J Immunol1994; 24: 1.097-1.101.23. Martin LB, Kita H, Leiferman KM, Gleich GJ. Eosinop-hils in allergy: role in disease, degranulation and cytokines.Int Arch Allergy Immunol 1996; 109: 207–215.24. Bonnefoy JY, Henchoz S, Hardie D, Holder MJ, GordonJ. A subset of anti-CD21 antibodies promote the rescue ofgerminal center B cells from apoptosis. Eur J Immunol1993; 23: 969-972.25. Tsubata T, Wu J, Honjo T. B cell apoptosis induced byantigen receptor croslinking is blocked by a T cell signaltrough CD40. Nature 1993; 364: 645-648.26. Arpin C, Déchanet J, Van Kooten C, Merville P,Grouard G, Brière F, et al. Generation of memory B cellsand plasma cells in vitro. Science 1995; 268: 720-722.27. Kerl JH, Álvarez-Mon M, Delsing GA, Fauci AS: Lymp-hotoxin is an important T cell-derived growth factor for hu-man B cells. Science 1987; 238: 1.144-1.146.28. Kehrl JH, Álvarez-Mon M, Fauci AS. Type B TGF sup-presses the growth and differentiation of human B and Tlymphocytes. Clin Res 1985; 33: 610.
1330
ENFERMEDADES DEL SISTEMA INMUNE (II)

Introducción
Se conoce como tolerancia inmunológica lacapacidad de bloquear la respuesta frentea un antígeno (Ag) concreto con el cual elsistema inmunitario ha interaccionado pre-viamente en unas condiciones particula-res.El sistema inmunitario posee dos estruc-turas únicas, la inmunoglobulina de su-perficie (sIg) de los linfocitos B y el re-ceptor antigénico de los linfocitos T (TCR),que le permiten activarse ante un Ag de-terminado de una forma muy selectiva (es-pecificidad). La recombinación aleatoria delos genes que codifican estas dos estruc-turas genera un repertorio tan amplio (di-versidad) que asegura la existencia de ele-mentos específicos frente a cualquieragente extraño y frente a las variacionesantigénicas que experimentan muchos mi-croorganismos. Como contrapartida a laproducción de diversidad, se generan ele-mentos capaces de reconocer cualquier Agdel propio organismo. Por ello, el sistemainmunitario debe desarrollar estrategiasde autotolerancia que neutralicen física ofuncionalmente los elementos reactivoscon Ag propios. Esencialmente, la consi-deración de un Ag como “propio” va a de-pender del momento y del lugar en quelos linfocitos T y B se encuentren con eseAg. En otras palabras, para que el sistemainmunitario “aprenda” a no responderfrente a un determinado Ag son decisivosel grado de maduración de los linfocitos ylos factores del microentorno donde seproduce el encuentro con el Ag. Así, lostres mecanismos básicos de instauraciónde “tolerancia hacia lo propio” son: a) laeliminación clonal de los linfocitos T y Bque reconocen Ag cuando no han alcan-zado aún la maduración funcional com-
pleta; b) la inactivación, funcional (aner-gia) o física (apoptosis), de los linfocitosmaduros que reconocen un Ag en ausen-cia de señales coestimuladoras; y c) la ig-norancia inmunológica de muchos Ag cuyaconcentración es tan baja que no desen-cadenan respuestas ni efectoras ni tolero-génicas. Existe un cuarto mecanismo, lageneración de células con actividad su-presora de la respuesta sobre un Ag con-creto, cuya relevancia es mayor en la to-lerancia de ciertos Ag externos, como losAg alimentarios. Además de prevenir la autorreactividad,el sistema inmunitario debe regular su res-puesta frente a los Ag externos para evi-tar el daño de las estructuras propias. Así,en la medida de lo posible, la reacciónfrente a un agente microbiano debe limi-tarse al propio agente y no generarelementos autorreactivos. Todos estos me-canismos de regulación y de autotoleran-cia consumen la mayor parte de la activi-dad del sistema inmunitario, aunque sufracaso puede conducir al desarrollo demanifestaciones autoinmunes. En el pre-sente artículo se revisan los mecanismosesenciales de tolerancia y regulación de larespuesta inmunitaria y se comentan po-sibles puntos débiles de estos sistemasdesde la óptica de la predisposición al de-sencadenamiento de reacciones autoin-munes. Con un objetivo puramente di-dáctico se exponen por separado losmecanismos de tolerancia que incumbena los linfocitos T y a los linfocitos B.
Tolerancia en loslinfocitos TDado el papel central que las células T de-sempeñan en la respuesta inmunitaria, pa-rece lógico que los mecanismos de tole-rancia se centren fundamentalmente eneste tipo celular. Existen tres mecanismosbásicos de tolerancia en las células T: to-
lerancia central, tolerancia periférica e ig-norancia inmunológica. La primera fasedel desarrollo de las células T, hasta ad-quirir una plena capacidad funcional, secompleta íntegramente en el timo. Lo pri-mero que ocurre a este nivel es el reor-denamiento de los genes de las regionesvariables del TCR, generándose un amplí-simo repertorio de receptores antigénicos.Seguidamente tienen lugar procesos deselección en los que sólo sobreviven loslinfocitos T funcionales (aquellos que re-conocen el complejo principal de histo-compatibilidad –MHC- propio) y que ade-más no reconocen autoAg presentes en eltimo a una alta concentración (autorreac-tivos). Este proceso no previene com-pletamente la salida del timo de célulaspotencialmente autorreactivas que reco-nozcan autoAg no expresados en el timo.Por ello, se requiere la existencia de me-canismos de tolerancia periférica que com-pleten el efecto de los anteriores.
Desarrollo y tolerancia de loslinfocitos T en el timo
Las células de linaje T más inmaduras, pro-cedentes de la médula ósea (MO), entranen el timo y comienzan su diferenciaciónhacia células T maduras en respuesta afactores del microambiente tímico. La ex-presión del complejo TCR-CD3 y de los co-rreceptores CD4 y CD8 en la superficie delos timocitos permite seguir groseramen-te sus diferentes estadios del desarrollo(fig. 1). Los precursores más tempranostienen un fenotipo CD4–CD8– doble nega-tivo (DN) y suponen alrededor del 3% delnúmero total de timocitos. Éstos proliferany se convierten en timocitos CD4+CD8+
doble positivos (DP), aún inmaduros, quesufrirán los procesos de selección depen-dientes del reconocimiento del receptorantigénico de la célula T (TCR). Para com-pletar su maduración hasta células TCD4+CD8– o CD4–CD8+, el repertorio ini-cial se selecciona negativa y positivamen-te a través de sus interacciones con lasMHC en el timo, de forma que sólo el 5%de los timocitos DP migran del timo a losórganos linfoides periféricos. El resto detimocitos defectuosos o peligrosos por supotencial autorreactivo son físicamente eli-minados mediante apoptosis (deleción).Algunos timocitos autorreactivos escapana la periferia, pero son funcionalmenteinactivos (anérgicos)1.
1331
DE LOS MECANISMOS DETOLERANCIA A LA AUTOINMUNIDADJ. Merino Pérez y M. López HoyosDepartamento de Biología Molecular de la Universidad de Cantabria. *Servicio de Inmunología, HospitalUniversitario Marqués de Valdecilla, Santander.
Medicine 2000; 8(26): 1331-1341

Los timocitos inmaduros DP necesitan unaseñal que evite una muerte celular pre-matura (selección positiva). Esta señal con-siste en la interacción de baja afinidad en-tre el TCR del timocito DP y el complejoMHC-péptido presentado por células ac-cesorias (fundamentalmente de tipo epi-telioide)2. Así, los timocitos DP cuyo TCRno interacciona con el complejo MHC-pép-tido no reciben señales de supervivenciay mueren por apoptosis. Este proceso tie-ne como objetivo abortar la salida a la pe-riferia de linfocitos T incapaces de reco-nocer Ag junto a moléculas MHC propiasy por lo tanto, no funcionales3. Además,el éxito de esta señal de supervivencia pa-rece depender del grado de afinidad en-tre el TCR del timocito y el complejo MHC-
péptido, de forma que sólo se van a con-servar los timocitos DP con afinidad bajao intermedia por alguno de estos comple-jos2. El proceso de selección positiva de-pendiente de las interacciones de baja afi-nidad implica que los timocitos DP sonmás sensibles a ligandos de baja afinidadque sus descendientes maduros, a pesarde expresar en su membrana unos nive-les de TCR diez veces menores.Por el contrario, los timocitos con alta afi-nidad por el complejo MHC-péptido serántratados como autorreactivos y, en con-secuencia, eliminados por apoptosis en unproceso conocido como “selección nega-tiva”4. Este fenómeno se considera el me-canismo principal de tolerancia frente aAg propios, ya que va a asegurar la eli-
minación de células T que reconocen Agexpresados de forma ubicua a altas con-centraciones, como las moléculas MHCpropias. La reactividad con MHC es espe-cialmente relevante, ya que entre el 1% yel 10% de los linfocitos T de un individuopueden llegar a ser reactivos con un ha-plotipo MHC no propio. Simplemente hayque considerar que en el caso de que nose produjera tolerancia frente al MHC pro-pio ocurriría una reacción similar a la ob-servada en la enfermedad injerto contrahuésped. La tolerancia frente a este tipode Ag es muy difícil de instaurar fuera deltimo una vez que se ha generado un re-pertorio funcional. Muestra de ello es lanecesidad de mantener indefinidamentela inmunosupresión en pacientes someti-dos a trasplante de órganos alogénicos.La eliminación de una parte importantedel repertorio de células T en los procesosde selección negativa del timo no afectasustancialmente a la capacidad del siste-ma inmunitario para reaccionar frente aAg externos. En efecto, la denominada ba-rrera hemato-tímica hace prácticamenteimposible el acceso de los Ag no propiosa este órgano, por lo que el repertorio delinfocitos T específicos frente a Ag exter-nos no se verá mermado, salvo en los ca-sos de péptidos exógenos que compartanidentidad antigénica con los Ag propios(mimetismo molecular)5.
Tolerancia y homeostasis de células T en la periferia
Resulta bastante obvio que la selección ne-gativa en el timo no es capaz de asegurarla tolerancia frente a todos los autoAg. Ellose debe fundamentalmente a que muchosde estos últimos no se expresan en el timoo lo hacen a concentraciones tan bajas queno alcanzan una representación suficien-te en la membrana de las células que realizan la selección de repertorios. A ellose une además el que la expresión de re-ceptores antigénicos en los timocitos in-maduros es mucho más baja que en loslinfocitos T maduros. Por lo tanto, el timova a dejar escapar un porcentaje signifi-cativo de linfocitos T autorreactivos, porlo que se hace necesaria la existencia demecanismos de control periféricos queprevengan el desarrollo de manifestacio-nes autoinmunes. Los mecanismos de tolerancia en la peri-feria no pueden sustentarse en los mis-
1332
ENFERMEDADES DEL SISTEMA INMUNE (II)
Selección Ag - independiente Selección Ag - dependiente
MÉDULA ÓSEA
ReordenamientoIgH
ReordenamientoIgL
Inmadura Madura
Defectuosos
Deleción clonal
APOPTOSIS
CÓRTEX TÍMICO
MÉDULATÍMICA
Bcl-2
Bcl-2
Bcl-X
Bcl-X
ReordenamientoTCRβ
ReordenamientoTCRα
Defectuosos
Deleción clonal
APOPTOSIS
Madura
Madura
L
-
-
-- +
L
pro-B pre-B pre-B IgMIgD
IgMIgD
TCRβCD44
CD4CD8
CD4CD8
CD4CD8 CD4
CD8CD4CD8
+-
+
++
+
+
TCRCD44 ++
+lo +
+
Fig. 1. Expresión de las proteínas antiapoptóticas Bcl-2 y Bcl-XL durante el desarrollo de los linfocitos B (arriba) y linfocitos T(abajo). Se señalan los dos puntos críticos de entrada en apoptosis de ambos subtipos celulares.

mos principios que la tolerancia central enel timo, ya que las células T han alcanza-do una maduración funcional completa yvan a responder frente a cualquier Ag quese les presente de forma adecuada. En estecaso, la prevención de la respuesta frentea un Ag determinado se fundamenta enmúltiples mecanismos basados en: a) laconcentración y forma de presentación delpropio Ag; b) la actividad supresora deotros linfocitos; c) el control de la apop-tosis de los linfocitos T activados; y d) laexistencia de correceptores con actividadinhibidora, como CTLA-4 (se expone másadelante, con las moléculas de función si-milar en las células B).
Papel del Ag en la inducción de tolerancia T periférica
Como se ha discutido en el capítulo pre-cedente, la activación de un linfocito T enla periferia va a depender de al menos dosseñales: la primera proporcionada por launión del TCR al complejo Ag-MHC y lasegunda por diferentes interacciones mo-leculares, especialmente entre el CD28 delos linfocitos T y sus ligandos B7-1 y B7-2, que sólo se expresan en las células pre-sentadoras de Ag (APC) profesionales (vercapítulo previo)6. La ausencia de esta se-gunda señal es el mecanismo principal detolerancia periférica. Así, cuando un lin-focito T virgen (naive) reconoce un Ag pre-sentado por una célula parenquimatosa,carente de moléculas coestimuladoras, nose activa, aunque tampoco entra en apop-tosis. En estas células se produce un blo-queo en la producción de varias citocinas,fundamentalmente interleucina 2 (IL-2),que deja a la célula en un estado refrac-tario para la activación, aunque interac-cione seguidamente con una APC profe-sional. Este estado, denominado anergia6,se mantiene durante varias semanas in vi-tro, aunque in vivo el estado de anergiaparece condicionar un acortamiento de lavida media del linfocito T. Además de noactivarse, las células anérgicas contribu-yen de forma específica al mantenimien-to de la tolerancia, ya que compiten conotras células virgen tanto por el Ag comopor citocinas proactivadoras que puedanhaber sido liberadas al medio extrace-lular. Este mecanismo también puede actuar enlos tejidos sobre células T autorreactivasque hayan sido activadas por APC profe-sionales en los órganos linfoides. La
ausencia de coestimulación en los tejidoslimita normalmente la capacidad agresorade las células T activadas, cuya respuestaes leve y corta en el tiempo y mueren porun mecanismo aún desconocido. La inter-ferencia en las señales coestimuladoras delos linfocitos T ha comenzado a ser ex-plorada en modelos experimentales de au-toinmunidad como estrategia terapéutica7.Así, se ha comprobado la eficacia del blo-queo de los ligandos de CD28 en la pre-vención de la glomerulonefritis fatal quesufren los ratones NZB/W que espontáne-amente desarrollan una enfermedad simi-lar al lupus eritematoso sistémico.Otra cualidad de algunos autoAg, su bajaconcentración, puede ayudar también aprevenir la activación de linfocitos T au-torreactivos específicos que simplementelos ignoran. La ignorancia clonal define unaforma de tolerancia “pasiva” hacia estosAg que no llegan a alcanzar concentra-ciones suficientes para inducir toleranciatímica ni anergia clonal, pero tampoco ac-tivan a los linfocitos T reactivos contraellos, que normalmente existen en los ór-ganos linfoides8. Este mecanismo no de-bería ser considerado una forma de tole-rancia, dado que no se trata de un procesoactivamente aprendido. Sin embargo, esposible que la mayoría de las reaccionesautoinmunes reflejen la activación de es-tos linfocitos “ignorantes”.Una forma especial de tolerancia ocurreen ciertas localizaciones anatómicas co-nocidas desde hace tiempo por tolerar me-jor que otras los aloinjertos que allí se ubi-quen, a pesar de que existan diferenciasen aloantígenos. Estos “santuarios”, co-nocidos como sitios inmunológicamente pri-vilegiados, son entre otros el cerebro, losovarios, los testículos, la placenta y la cá-mara anterior ocular. El mejor ejemplo de este privilegio inmunológico es lasupervivencia de injertos de córnea en au-sencia de compatibilidad HLA o de trata-miento inmunosupresor9. La justificaciónde este fenómeno es compleja. Al princi-pio se pensó que los autoAg de estos si-tios no salían de allí y, por lo tanto, no in-ducían respuestas inmunitarias, de formasimilar a la ignorancia inmunológica. Sinembargo, se ha comprobado que estos au-toAg salen e inducen respuestas, bien to-lerogénicas bien activadoras pero no con-dicionan daño al tejido. Estos hallazgos seatribuyen a la salida de citocinas inhibi-doras, como factor de crecimiento tumo-ral beta (TGFβ), junto con el Ag. Un factor
anatómico adicional en este fenómeno esque el líquido intersticial de estos tejidosno circula por los vasos linfáticos con-vencionales, con lo que sus componentesno son presentados a los linfocitos T deforma convencional. Recientemente se haobservado que algunas células parenqui-matosas de estos sitios inmunoprivilegia-dos expresan concentraciones elevadas delligando de la molécula proapoptótica Fas(Fas-L). Como se expone más adelante,Fas-L es una molécula que expresan al ac-tivarse los linfocitos T (CD4+ o CD8+) concapacidad citolítica. La interacción de Fas-L con la molécula Fas, expresada consti-tutivamente en la mayoría de las células,induce la activación del programa de apop-tosis en la célula diana y es en gran par-te responsable del poder citotóxico de es-tos linfocitos. Por lo tanto, la inhibición dela respuesta inmunitaria en los tejidos pri-vilegiados podría deberse, al menos enparte, a que Fas-L presente en estos teji-dos induciría la apoptosis de los linfocitosque los infiltran, los cuales, si están acti-vados, expresan niveles elevados de Fas9.
Actividad supresora de las células T
La idea de una célula T con actividad su-presora específica de una respuesta in-munitaria ha sido y es objeto de contro-versia desde su descripción. El conceptode célula T supresora nace con la demos-tración de la transferencia de toleranciafrente a un Ag concreto con células T deun animal con tolerancia a ese Ag. El fe-nómeno se aplica inmediatamente a si-tuaciones inmunopatológicas y, en la dé-cada de los 70 y principios de los 80, másde 5.000 publicaciones científicas impli-can a los linfocitos T-supresores en la pa-togenia de todas las enfermedades au-toinmunes. Sin embargo, en aquellos añosla función supresora se evaluaba funcio-nalmente de forma global (no específica)y la función supresora de las células T seatribuía a un único subtipo celular, los linfocitos T-CD8+, en contraposición alsubtipo T-CD4+ al que claramente se asig-naba un papel “helper” o colaborador. Enaños posteriores, la mayor parte de la co-munidad inmunológica adopta una opiniónescéptica sobre las células T supresoras,dada la falta de evidencia de que las cé-lulas T-CD8+ supriman específicamenterespuestas inmunitarias y ante la comple-jidad de un número creciente de factoressolubles (citocinas).
1333
DE LOS MECANISMOS DE TOLERANCIA A LA AUTOINMUNIDAD

La idea que se tiene actualmente sobreeste tema es que, efectivamente, las cé-lulas T son capaces de suprimir específi-camente respuestas frente a un determi-nado Ag. Sin embargo, no existe unsubtipo celular, fenotípicamente identifi-cable, con función exclusivamente supre-sora, sino que una misma célula T puedesuprimir o estimular una determinada res-puesta inmunitaria en función de múlti-ples factores (célula que presenta el Ag,citocinas presentes en el entorno, etc). In-cluso, una misma célula puede cambiar suactividad supresora en activadora, o vice-versa, si se modifican las condiciones am-bientales. Por lo tanto, es más correcto hablar de funciones inmunosupresoras de-sarrolladas por las células T que de célu-las T supresoras. El mecanismo de supresión mejor esta-blecido en las células T es el balanceTh1/Th2 (fig. 2). Como se expuso con de-talle en un capítulo previo, las células T-CD4+ tienen básicamente dos patronesde activación: el patrón Th1, que consis-te en la producción de las citocinas IL-2,interferón gamma (IFNγ) y factor de cre-cimiento tumoral beta (TNFβ), y el patrónTh2, con citocinas del tipo de IL-4, IL-5,IL-10 e IL-1310. En ambos casos se produ-cen una serie de citocinas comunes (IL-3,GM-CSF, TNFα). Algunas de las citocinassecretadas por cada uno de estos subtiposcelulares tienen un efecto claramente su-presor del otro subtipo: IFNγ inhibe la res-puesta Th2, mientras que IL-4 e IL-10 in-
hiben la respuesta de tipo Th1. Este con-cepto viene confirmado por la demostra-ción de una modulación de la respuestainmunitaria mediante la administraciónexógena de estas citocinas. En líneas ge-nerales, podemos decir que, desde el pun-to de vista de los mecanismos de tolerancia,las células T-CD4+ con un fenotipo TH2 seconsideran las principales células supreso-ras de las respuestas inmunitarias en don-de predominan los fenómenos de citotoxici-dad mediada por células T-CD8+, célulasnatural killer NK e incluso linfocitos TH110.Recientemente se ha descrito que un por-centaje significativo de linfocitos T-CD4+
del tejido linfoide asociado a las mucosassecretan TGFβ, citocina con función su-presora de la respuesta de otros linfocitosT-CD4+, tanto Th1 como Th210. Se ha pro-puesto para estas células la denominaciónde linfocitos Th3, y podrían constituir unnuevo tipo de linfocito T CD4+ con fun-ción eminentemente supresora11. A nivelfisiológico, estas células secretoras deTGFβ desempeñan un papel relevante enla inducción de tolerancia a los Ag ali-mentarios (ver más adelante). Además, elinterés de este mecanismo regulador me-diado por el TGFβ queda evidenciado porel síndrome lúpico observado en los rato-nes deficientes en TGFβ12.
Control de la apoptosis en los linfocitos T
Entre los mecanismos de tolerancia de cé-lulas T en la periferia están adquiriendouna especial relevancia los fenómenos quecontrolan la apoptosis de las células T ma-duras, sobre todo tras el encuentro de és-tas con el Ag. Básicamente, existen dos
vías de apoptosis linfocitaria, muy simila-res en lo que respecta a los mecanismosefectores, pero diferentes tanto en su pa-pel fisiológico como en sus mecanismosreguladores (tabla 1).La primera, conocida como muerte celularpasiva o por negligencia, es consecuenciade la falta de factores de crecimiento y deotras señales de supervivencia (citocinas,señales coestimuladoras…)13. Este tipo deapoptosis se produce tanto en la selecciónpositiva de las células T en el timo (verarriba), como en el control homeostáticoen la periferia de los linfocitos T activadospor el Ag (fig. 3). Así, tras un estímulo an-tigénico, la frecuencia de linfocitos T es-pecíficos para un Ag aumenta mil veces,de 10-6 a 10-3. Al cabo de 4-12 semanas,si el Ag se ha eliminado y disminuyen lasseñales de supervivencia, esta frecuenciaregresa a sus valores iniciales por apop-tosis pasiva de la mayoría de linfocitos Tespecíficos efectores. Sólo queda una po-blación de células T memoria capaces desobrevivir mucho más tiempo que las cé-lulas virgen sin recibir estímulo alguno. Secree que el aumento en la supervivenciade las células memoria es debido al estí-mulo continuado por una fracción persis-tente del Ag primitivo o por Ag ambien-tales o autoAg que tienen reacción cruzadacon el Ag primitivo13 (fig. 3) La apoptosispasiva se ha atribuido a una pérdida deexpresión de moléculas anti-apoptóticasde la familia de Bcl-2, fundamentalmenteBcl-2 y Bcl-XL, las cuales mantienen nive-les más sostenidos de expresión en las cé-lulas memoria14.La segunda vía de apoptosis linfocitaria seinduce por estimulación antigénica cróni-ca y se denomina muerte celular inducida
1334
ENFERMEDADES DEL SISTEMA INMUNE (II)
Fig. 2. Subtipos funcionales de linfocitos T-CD4+ en funciónde las citocinas secretadas. Se indican las citocinas que ejer-cen un papel regulador sobre el subtipo funcional opuesto.
DTH
IL-2
TNFβ
IFNγ
GM-CSFIL-3
TNFαIL-12
Th1
Th2IL-4
IL-10
IL-5
IL-6
Ac
TABLA 1Tipos de apoptosis linfocitaria
Muerte pasiva Muerte inducida tras activación
Inducción Ausencia de estímulos: antígeno, citocinas, Estímulo antigénico repetidofactores de crecimiento, etc.
Células sensibles Virgen y activada Sólo activadas
Aumento permeabilidad mitocondria Entrecruzamiento Fas o TNFR 1Mecanismos bioquímicos Liberación citocromo c al citoplasma Unión de FADD y TRADD
Interviene caspasa 9 Interviene caspasa 8
Papel de IL-2 Impide Potencia
Papel de Fas Ninguno Imprescindible
Papel de Bcl-2/Bcl-x Potencia Ninguno
Elimina linfocitos inmadurosdefectuosos o autorreactivos Eliminación de linfocitos
Papel fisiológico Elimina células maduras una vez autorreactivos madurosactivadas por el antígeno ¿Homeostasis linfocitaria?(homeostasis linfocitaria)

tras activación (AICD). Los linfocitos T queson estimulados de forma repetida por elAg expresan en su superficie receptoresde la familia del receptor de TNF (TNFR)y sus ligandos, fundamentalmente Fas yFas-L11, cuya unión dispara la apoptosis delos linfocitos. Actualmente se cree que estaAICD de los linfocitos T desempeña un pa-pel esencial en la eliminación de aquellascélulas que se encuentran repetidamentecon Ag persistentes, como los autoAg, demanera que la interferencia en la AICDprovoca reacciones autoinmunes, como secomenta más adelante. Curiosamente, eneste tipo de muerte celular interviene IL-2, una citocina a la que hasta el mo-mento se le había concedido una misiónproliferativa pero que también potencia laAICD15. La hipótesis que se baraja es que,en las fases iniciales de la activación de lacélula T, cuando la concentración de IL-2es baja, funciona como un factor de su-pervivencia y crecimiento y se necesitapara la expansión clonal en respuesta alAg. En cambio, tras un estímulo conti-nuado la IL-2 se acumula facilitando laAICD mediante dos mecanismos: aumen-tando la expresión de Fas-L e inhibiendola transcripción FLIP, molécula antagonis-ta de la señal apoptótica a través de Fas13
(fig. 3).
Tolerancia en loslinfocitos BEl repertorio antigénico de los linfocitos Btambién se genera mediante recombina-ción genética al azar en los genes de lasregiones variables de las cadenas pesadasy ligeras de las Ig. El hallazgo de linfocitosB autorreactivos en la sangre de sujetossanos hizo pensar, en un principio, que losprocesos de selección clonal, anterior-mente descritos en las células T, no se ex-tendían al compartimento B. Este hechono debería implicar un riesgo de autoin-munidad, habida cuenta de que las célu-las B precisan la colaboración de las cé-lulas T para generar respuestas eficaces.Se ha sugerido incluso que la existencia decélulas B autorreactivas es beneficiosa parael sistema inmunitario, ya que incremen-ta el repertorio frente a agentes externos.No obstante, y sin que ello reste validez aestos argumentos, se han demostrado fe-nómenos de selección clonal de linfocitosB tanto en la médula ósea como en los ór-ganos linfoides secundarios (OLS).
Desarrollo y tolerancia de loslinfocitos B en la médula ósea
También los linfocitos B sufren deleciónclonal cuando interaccionan con Ag du-rante su maduración en la MO (fig. 1). Esteproceso se produce fundamentalmente en
el estadio de células B inmaduras IgM+
IgD–, cuando estas células se unen con afi-nidad media o alta a Ag multivalentes queabundan en esta localización. El estado delAg condiciona dos fenómenos: a) cuandoel Ag está unido a membranas celulares,como las moléculas MHC o los Ag del gru-
1335
DE LOS MECANISMOS DE TOLERANCIA A LA AUTOINMUNIDAD
Célula T virgen
CélulaT naive
TCR CélulaT naive Muerte celular
inducida trasactivación (AICD)
APCCélula Tactivada
IL-2
IL-2
Ausenciade unión
al TCR
Muertecelularpasiva
BcL-2Bcl-XL
Supervivencia
Bélula Tactivada
Célula Tactivada
FLIP
FasL
Fas
Sensibilidad a AICD
AICD
Proliferación
Mantenimientode señales
de supervivencia
Célula T memoria
Diferenciación
Célula T efectora
Muertecelularpasiva
Eliminacióndel Ag y otras
señales desupervivencia
Célulapresentadora
de Ag
Fig. 3. Homeostasis de los linfocitos T en la periferia.

po sanguíneo AB0, el linfocito B entra rá-pidamente en apoptosis; b) si el Ag estáen fase soluble la célula no muere inme-diatamente, pero entra en un estadoarreactivo de anergia que suele seguirsede un acortamiento marcado de su vidamedia. En un bajo porcentaje de célulasB autorreactivas se ha descrito un meca-nismo de rescate de la apoptosis deno-minado edición del receptor16. Consiste en la reactivación de las recombinasas RAG-1 y RAG-2 las cuales llevan a caboun nuevo reordenamiento de los genesde las regiones variables de las Ig en unintento de modificar el carácter autorre-activo del receptor antigénico. Si el nue-vo receptor resultante no reconoce auto-Ag, el linfocito B podría alcanzar elestadio de célula B madura inmunocom-petente.
Tolerancia y homeostasis decélulas B en la periferia
Como se comentó anteriormente, muchosAg propios sólo se expresan en determi-nados órganos, precisándose también me-canismos que impidan la reacción de lascélulas B contra ellos. Experimentos re-cientes indican que, a diferencia de lascélulas T, la mayor parte de los fenóme-nos de eliminación clonal de las célulasB tienen lugar en el mismo lugar en queéstas inician la respuesta inmunitaria fren-te a los Ag externos, las zonas T de losórganos linfoides secundarios. Estas zo-nas presentan una frecuencia relativa-mente elevada de células B HSA+, un mar-cador de linfocitos T y B inmaduros cuyaexpresión en células T se restringe altimo. La mayor parte de estas células tie-ne una vida media corta (menos de 48horas), responden muy débilmente a mi-tógenos y no llegan a pasar a los folícu-los linfoides, caracterizados por la pre-sencia fundamental de células B. Se hademostrado que estas células B anérgicasde vida media corta son linfocitos B in-maduros que reconocen Ag pero no reci-ben señales coestimuladoras de las célu-las T. Por el contrario, sólo aquellos queno reconocen Ag entran en los folículosy completan su maduración. Estas últi-mas van a componer la población de cé-lulas B periféricas maduras, de vida me-dia alrededor de cuatro semanas, querecirculan colonizando otros OLS. Estamaduración de las células B en un entor-
no público, a diferencia del entorno ce-rrado del timo, tiene implicaciones im-portantes en el mantenimiento de la auto-tolerancia y puede contribuir al desarrollode enfermedades autoinmunes17. Así, úl-timamente está adquiriendo una mayorimportancia el fracaso de la tolerancia delas células B en la periferia como meca-nismo de producción de autoanticuerposy enfermedades autoinmunes.
Control de las células B en los centros germinales de los OLS
El destino de un linfocito B maduro queencuentra su Ag en la periferia va a ve-nir determinado por señales coestimula-doras dependientes del entorno y del pro-pio Ag. En general, las células B madurasque reconocen Ag proteicos son elimina-das o entran en un estado de anergia sino reciben señales coestimuladoras decélulas T-CD4+. Una excepción es el re-conocimiento de Ag con múltiples epíto-pos de repetición que entrecruzan grannúmero de Ig en la membrana de las cé-
lulas B, como los Ag polisacarídicos (Ag T-independientes). La respuesta fren-te a estos Ag no precisa la colaboraciónde las células T, pero sólo suscitan res-puestas humorales de tipo IgM de bajaafinidad y no establecen memoria inmu-nológica.En la mayoría de los casos, y sobre todofrente a Ag proteicos, la activación de unlinfocito B sólo se produce si el reconoci-miento del Ag se acompaña de la coope-ración con la célula T. Esta ayuda consis-te en la liberación de citocinas activadoras(IL-2, IL-4, etc) y en el establecimiento deinteracciones moleculares, entre las quedestaca la interacción CD40/CD40L18.Como resultado de todas estas señales, ellinfocito B se expande clonalmente y se di-ferencia, formando una estructura carac-terística denominada folículo linfoide se-cundario o centro germinal (CG)19. Durantela fase de proliferación algunos linfocitosB experimentan mutaciones puntuales so-máticas en las regiones variables de su Igque pueden dar lugar a cambios estructu-rales en el sitio de reconocimiento del Ag(fig. 4). El resultado es una disminución oun aumento de afinidad por el Ag primiti-
1336
ENFERMEDADES DEL SISTEMA INMUNE (II)
Fig. 4. Organización arquitectónica de un folículo linfoide secundario (centro germinal) en los órganos linfoides secundarios.
APOPTOSIS
Pérdida de especificidad
Baja afinidad
AUTORREACTIVA
Célula Bmemoria
Célulaplasmática
Y
Y
Y
Centrocito sIg+
M
CDF
Blastos Bsecundarios
Centroblasto sIg -Ag

vo o la aparición de una nueva especifici-dad antigénica. El significado fisiológico deeste proceso es doble: a) algunos linfoci-tos B mutados habrán aumentado su afi-nidad por el Ag primitivo, aumentando lacalidad de sus anticuerpos (maduración dela afinidad); b) éste podría ser un intentode ampliación de un repertorio antigénicomuy mermado tras los procesos de selec-ción negativa de linfocitos B inmaduros,que habrán creado auténticos “agujeros”en el repertorio de células B frente a de-terminados Ag externos17. El riesgo que conlleva el fenómeno de hi-permutación somática es la conversión enautorreactivos de algunos linfocitos B, conel consiguiente peligro de producción dereacciones autoinmunes. Además, se ge-nerarán células B cuya afinidad por el Agprimitivo disminuya hasta el punto de ha-cerlas irrelevantes en la respuesta inmu-nitaria que motivó su activación. Para pa-liar estos inconvenientes, en los CG selleva a cabo una selección positiva de lascélulas B, función en la que desempeñanun papel esencial las células dendríticasfoliculares. Estas células portan el Ag pri-mitivo en sus prolongaciones dendríticas,para que lo puedan volver a reconocer lascélulas B del CG. Las células que reco-nozcan de nuevo el Ag con alta afinidadrecibirán señales de supervivencia y com-pletarán su diferenciación a célula plas-mática o a célula B memoria. Por el con-trario, aquellas cuya afinidad por el Agprimitivo haya disminuido o desaparecidoentrarán en apoptosis19. Al igual que se describió en las células T,los mecanismos de control de la apopto-sis van a desempeñar un papel relevan-te en la función de los CG y en particu-lar en la prevención de reaccionesautoinmunes. Así, se ha demostrado quealgunos genes con función antiapoptóti-ca, como bcl-2, disminuyen su expresiónen la fase en que las células B son so-metidas a los procesos de selección enlos CG, muy probablemente con objetode facilitar la eliminación de las célulasautorreactivas o inútiles (fig. 4)20. La po-sible relevancia de este fenómeno vienerefrendada en experimentos recientesque demuestran cómo la inhibición de losprocesos de apoptosis en las células B delos CG, mediante hiperexpresión de Bcl-2 en ratones transgénicos, provoca unsíndrome autoinmune en el que destacala producción de autoanticuerpos pato-génicos que originan una glomerulone-
fritis fatal (M. López Hoyos et al, envia-do a publicación).
Receptores de membranacon capacidad inhibitoriaComo se ha expuesto en artículos previos,la activación de los linfocitos T y B es laconsecuencia directa de las interaccionesde secuencias activadoras existentes enlos segmentos intracitoplasmáticos de losreceptores antigénicos y de las moléculascorreceptoras. Estas secuencias, conoci-das como dominios ITAM (motivos de ac-tivación basados en tirosina de los inmu-norreceptores), disparan la activación defactores de transcripción para la prolife-ración celular y la síntesis de citocinas pro-activadoras. En la membrana de las células linfoidesse han descrito otras moléculas capacesde contrarrestar estas señales de activa-ción, pudiendo incluso bloquearlas com-pletamente. Su efecto inhibidor se rela-ciona principalmente con secuenciasespecíficas en sus segmentos intracito-plasmáticos denominados dominios ITIM(motivos de inhibición basados en tirosi-na de los inmunorreceptores)21. La com-posición característica de estos dominioses (I/V)XYXXL: un residuo hidrofóbico (va-lina o isoleucina), seguido de un aminoá-cido cualquiera, una tirosina, otros dosaminoácidos y una leucina. Este dominiorecluta alguna de las fosfatasas inhibido-ras SHP-1 o SHIP. SHP-1 es una protein ti-rosina fosfatasa que elimina grupos fosfa-tos añadidos por las protein cinasas queinician los procesos de activación en loslinfocitos. SHIP es una inositol fosfatasaque elimina el fosfato de la posición 5’ delfosfatidil inositol trifosfato (PI-3,4,5-P). Sedesconoce el mecanismo exacto por el queSHIP regula la activación celular, aunquese cree que lo hace inhibiendo la activa-ción de PLC-γ y, por lo tanto, la produc-ción de DAG e IP3
21.Las células B disponen de alguna de estasmoléculas inhibidoras. La más conocidaes el receptor de tipo II para el fragmen-to Fc de IgG (FcγRII). Desde hace tiempose sabía que el entrecruzamiento de la Igde superficie de la célula B con el FcγRIIinhibía la activación de las células B vír-genes, siendo la base de estrategias tera-péuticas como la inyección de Ig en algu-nos procesos autoinmunes o el empleo deanticuerpos anti-Rh para prevenir la in-
munización de madres Rh(-) cuyos fetosportan el Ag Rh. Recientemente, se ha ca-racterizado el efecto inhibidor de otras dosmoléculas, CD22 y el PIR-B (paired immu-noglobulin-like receptor), sobre la activa-ción de las células B22.A nivel de las células T, una molécula confunción inhibidora asociada a motivos ITIMes CTLA-4 (CD152). Esta molécula tieneuna homología del 30% con la moléculacoestimuladora CD28 y utiliza sus ligan-dos B7-1 y B7-2, aunque su avidez por losmismos es 100 veces mayor que la deCD28. La expresión de CTLA-4 suele mar-car el final de la activación de la célula T23.Finalmente, en las células NK se han des-crito receptores inhibidores de muerte(KIR) con especificidad por moléculas MHCde clase I, cuya confluencia inhibe la li-beración de los factores citotóxicos de es-tas células cuando interaccionan con cé-lulas sanas24. Estos receptores, provistosde motivos ITIM en sus colas intracito-plasmáticas, podrían desempeñar un pa-pel relevante en el mantenimiento de latolerancia materno fetal en la decidua placentaria, lugar donde abundan las cé-lulas NK.
Tolerancia oral a losantígenos alimentariosEn las primeras 24-48 horas que siguena la administración de un Ag proteico porvía de mucosas se pueden detectar célu-las reguladoras de la respuesta frente aese Ag, tanto localmente como en losganglios linfáticos. A los 5-7 días pode-mos hablar ya de un estado de toleran-cia, que persiste al menos un año y me-dio después de la administración del Ag25.La naturaleza y la dosis del Ag influyenen el establecimiento de tolerancia oral:así, los Ag T-dependientes solubles songeneralmente tolerógenos, un hecho queha dificultado el éxito en el desarrollo devacunas orales. Por el contrario, el con-tacto del tejido linfoide de las mucosascon patógenos vivos, capaces de multi-plicarse, activa la respuesta inmunitarialocal y sistémica, probablemente por sucaptación preferencial por las células M,eficaces presentadoras de Ag (fig. 5). Porotro lado, los Ag administrados a dosisaltas o repetidas suelen ser tolerogénicos,mientras que los administrados a dosismuy bajas sensibilizan respuestas localesy sistémicas25.
1337
DE LOS MECANISMOS DE TOLERANCIA A LA AUTOINMUNIDAD

La inducción de tolerancia oral parecefundamentarse en la inducción de aner-gia en las células T, posiblemente comoconsecuencia de una presentación de-fectuosa del Ag por APC, como los ente-rocitos, que expresan niveles bajos demoléculas MHC de clase II pero carecende moléculas coestimuladoras como B7-1/B7-2, ICAM-1 u otras. Además, los en-terocitos expresan un ligando para célu-las CD8 similar al CD1, sugiriendo quelos linfocitos CD8+, probablemente me-diante producción de TGFβ, están tam-bién implicados en el establecimiento detolerancia oral25. También parecen parti-cipar en este proceso las APC conven-cionales presentes en los epitelios, MALTy ganglios linfáticos de drenaje. En estecontexto, las células dendríticas podríandesempeñar un papel destacado, ya quein situ expresan niveles bajos de B7-1 yCD40, y cuando se estimula su expansiónlocal con el factor de crecimiento FIt3L(ligando de la tirosina-cinasa fms-like) seincrementa la susceptibilidad para in-ducción de tolerancia oral26. El efecto to-lerogénico de estas células dendríticas po-dría revertirse cuando se activan conpatógenos vivos, y es posible también es-
pecular que algunos fármacos inmunoes-timuladores pudieran tener un efecto si-milar. Experimentos de transferencia celularapoyan la activación de mecanismos su-presores en el tejido linfoide asociado alintestino (GALT), mediados fundamental-mente por linfocitos T-CD4+ y linfocitosTγδ+, los cuales serían claves en el man-tenimiento de la tolerancia oral. Así, seha propuesto que la tolerancia oral refle-jaría una activación preferencial de célu-las TH2, que inhibirían respuestas TH1(fig. 5)10,26. Sin embargo, por un lado, estetipo de tolerancia puede inducirse en ra-tones deficientes en IL-4 o en ratones tra-tados con anti-IL-10 y, por otro, la pro-ducción de IgE Ag-específica puedesuprimirse también tras administracióndel Ag26. Un subtipo particular de célulasT-CD4+ parece tener un papel central eneste fenómeno. Estas células, que algu-nos autores han comenzado a denominarTH3, secretan la citocina TGF-β que ejer-ce un potente efecto supresor sobre di-versos tipos de células inmunocompe-tentes10. La activación Ag-específica deestas células podrían inducir la anulaciónfuncional de otras células de su entorno
por un mecanismo bystander (mirón ino-cente).
Fracaso de la tolerancia e inducción deautoinmunidad
La mayor frecuencia de enfermedades au-toinmunes entre gemelos monocigóticosque entre gemelos dicigóticos de pacien-tes con enfermedades autoinmunes de-muestra la intervención de factores ge-néticos en estos procesos27. Al mismotiempo, el que un porcentaje significativode gemelos monocigóticos no padezcan laenfermedad autoinmune que desarrolla elotro gemelo pone claramente de mani-fiesto la intervención de factores ambien-tales en la ruptura de la tolerancia hacialo propio que tiene lugar en estos pacien-tes.
Factores genéticos quepredisponen a autoinmunidad
Genes del sistema principal dehistocompatibilidad
Entre los factores que predisponen a au-toinmunidad destaca la asociación de di-versas enfermedades autoinmunes conciertos haplotipos MHC. Estas asociacio-nes se comentan con detalle en otros apar-tados de esta monografía. En la mayoríade los casos los procesos autoinmunes seasocian a moléculas MHC de clase II con-cretas, aunque también hay claras aso-ciaciones con moléculas MHC de clase I.Dos razones justifican esencialmente laasociación MHC-enfermedad autoinmune: 1. Las moléculas MHC son cruciales en lapresentación de Ag a las células T y, porlo tanto, en su activación. La capacidad depresentación de un determinado Ag esta-rá supeditada a la formación de un com-plejo estable entre la molécula MHC y eseAg. Por lo tanto, es evidente que algunasmoléculas MHC tendrán más capacidadque otras para unirse y presentar un de-terminado autoAg27. Uno de los ejemplosmás evidentes es la asociación de diabe-tes mellitus con determinadas moléculasMHC, tanto de clase I como de clase II (versección correspondiente). Por el contrario,se ha identificado un haplotipo DQβ cuyaexpresión protege del desarrollo de dia-betes, probablemente por incapacidad de
1338
ENFERMEDADES DEL SISTEMA INMUNE (II)
Administración oral de un Ag
Tejido linfoideasociado al intestino
Dosis bajas:presentación del Ag
en el intestino
Dosis altas:presentación del Ag
a nivel sistémico
Factores que influyen en la tolerancia oral: Requerimiento de coestimulación (presentación por enterocitos) Citocinas en el ambiente
Inducción de células reguladoras:Cel. TH2 (IL-4 / IL-10)Cel. productoras de TGFb (Th3)
Inhibición de las células Th1 (IL-2, IFNg) y Th2 (IL-4 / IL-10)
Órgano diana:supresión activa
Órgano linfoide:inhibe la generación
de cel. efectoras
Anergia Deleción
Tolerancia periférica
Fig. 5. Mecanismos implicados en la instauración de tolerancia a los antígenos que penetran por vía de mucosas

esta molécula DQ para presentar un Agdiana fundamental en el desarrollo de estaenfermedad. 2. La selección de repertorios de células Tviene guiada por las moléculas MHC y porlos Ag que éstas presenten1. Así, puedeconsiderarse que ciertas moléculas MHCpueden no ser capaces de llevar a cabouna eliminación eficaz de los timocitos re-activos con ciertos autoAg que una vez enla periferia, y con la confluencia de fac-tores ambientales, pueden desencadenarla agresión de estos linfocitos T autorre-activos frente a tejidos que expresen losautoAg correspondientes.
Genes reguladores de la apoptosis
Siguiendo el mismo esquema que se ex-puso en los mecanismos que inducen to-
lerancia, se puede considerar que los ge-nes que controlan la apoptosis pueden in-tervenir en el desarrollo de manifestacio-nes autoinmunes a dos niveles, la AICD yla apoptosis pasiva.La importancia de la AICD en el manteni-miento de la tolerancia a autoAg se pusode manifiesto por primera vez con los ra-tones portadores de las mutaciones lpr ygld. La mutación lpr consiste en una mu-tación por inserción de un retrotranspo-són del gen Fas, mientras que la mutacióngld implica una mutación puntual en el genFas-L que condiciona la expresión de unamolécula no funcional11. Los ratones por-tadores de ambos tipos de mutaciones de-sarrollan una enfermedad sistémica muysimilar al lupus eritematoso sistémico(LES), caracterizado por linfadenopatía,producción de múltiples autoanticuerpos
y desarrollo de nefritis. Las mutacionesdescritas provocan una supervivencia pro-longada de las células T y B autorreacti-vas que acaban provocando el cortejo sin-tomático descrito. En los seres humanosse ha descrito un síndrome similar al delos ratones con las mutaciones lpr y gldque se ha denominado síndrome autoin-mune linfoproliferativo (ALPS). Se han des-crito tres tipos de ALPS según la molécu-la reguladora de apoptosis que estégenéticamente alterada: ALPS Ia (mutaciónde Fas), ALPS Ib (mutación de Fas-L) yALPS II (mutación en la ruta intracelularde Fas)28. Sin embargo, es importante des-tacar que, al igual que en los modelos mu-rinos, la alteración aislada de los meca-nismos de control de la apoptosis no escapaz de desencadenar un síndrome au-toinmune, sino que necesita de la pre-
1339
DE LOS MECANISMOS DE TOLERANCIA A LA AUTOINMUNIDAD
Fig. 6. Efectos de la falta de función de la vía de apoptosis a través de la molécula Fas en el desarrollo de apoptosis, tomando como ejemplo las mutaciones lpr y gld consistentes, respectivamente, enel defecto de las moléculas Fas y Fas-ligando.
Normal Ipr/Ipr o gld/gld
FasAuto Ag
APCprofesional
Célula Tautorreactiva
FasL
Fas Activación
AICD
Y YY
Activaciónde cortaduración
APCprofesional
Célula Tautorreactiva
Activación
Supervivenciaincrementada
Ipr
gld
YY
YYYY YYYY
YYYY YYYY
Activación delarga duración
Célula Bautorreactiva

sencia de otras alteraciones genéticas y/oambientales que complementen a la alte-ración de la vía Fas/Fas-L (fig. 6).También se ha descrito el desarrollo delinfadenopatía y autoinmunidad en rato-nes que no expresan IL-2 ni las cadenasα y β del receptor de IL-2. Aunque en unprincipio este hallazgo fue sorprendente,la capacidad de IL-2 de potenciar la AICDde células T autorreactivas, como se haexplicado en la primera parte de este ar-tículo, explica al menos de forma parcialla enfermedad observada en esos rato-nes14,15.Respecto a la apoptosis pasiva, los rato-nes transgénicos que hiperexpresan losmiembros anti-apoptóticos de la familiade Bcl-2 o que se han hecho deficientes(gene knockout) en estos genes, ponen demanifiesto, al igual que se ha explicado enlos mecanismos de tolerancia, que estetipo de muerte celular no interviene en losmecanismos de tolerancia periférica de lascélulas T. En concreto, se ha comprobadoque los ratones transgénicos que hiperex-presan Bcl-2 en células T, y que en con-secuencia tienen una supervivencia pro-longada, no desarrollan enfermedadautoinmune alguna. En cambio, la muer-te celular por negligencia sí interviene enel control de la respuesta de las células Ta Ag extraños y el mantenimiento de lahomeostasis periférica, además de en losmecanismos de tolerancia tímica14.
Genes que controlan fosforilación de los autoAg
La fosforilación de determinados epítopospor serina/treonina cinasas puede desem-peñar un papel importante en la produc-ción de enfermedades autoinmunes, es-pecialmente en aquellas enfermedades enlas que sólo el autoAg humano, y no elequivalente de otras especies, es capaz deprovocar enfermedad autoinmune29. Porlo tanto, se puede suponer que la activa-ción patológica de estas proteina cinasascondicione una fosforilación anormal deun Ag, modificando su inmunogenicidad.Ejemplos de este mecanismo mediador deautoinmunidad pueden ser el síndrome deGoodpasture o la esclerosis múltiple, loscuales se asocian también a un determi-nado haplotipo MHC (HLA-DR15). Es po-sible que los autoAg implicados de esasenfermedades se presentan mejor por esamolécula de histocompatibilidad cuandose encuentran fosforilados.
En los tejidos afectos de los pacientes consíndrome de Goodpasture se ha demos-trado la expresión de una proteína cinasanueva encargada de fosforilar el autoAgresponsable de la enfermedad (cadena α3del colágeno de tipo IV). Este hallazgo abreuna nueva ruta de estudio de genes im-plicados en el desencadenamiento de au-toinmunidad.Estudios recientes han demostrado que va-rios autoAc en pacientes con LES se diri-gen frente a autoAg específicamente fos-forilados por una familia de cinasasactivadas por estrés y que desempeñan unpapel fundamental en la señalización dela apoptosis inducida por estrés. De estamanera, la fosforilación que ocurre du-rante el fenómeno de la apoptosis de loscomponentes proteicos de diversos com-plejos macromoleculares, como los RNP,puede producir neo-epítopos que escapana los mecanismos normales de toleranciaperiférica. Otra posibilidad es que la fos-forilación de las proteínas las predispon-ga a la proteólisis y/o a la traslocación alas “blebs” apoptóticas, facilitando la pre-sentación de esos antígenos30. En la ac-tualidad, diversos trabajos se centran endefinir mejor los mecanismos de fosfori-lación proteica durante la apoptosis y ensaber si la fosforilación de los autoAg con-fiere una mayor patogenicidad a los au-toanticuerpos.
Factores ambientales en laproducción de enfermedadautoinmune
Diversas evidencias indican que los facto-res ambientales tienen un papel destaca-do en el desarrollo de enfermedades au-toinmunes: a) individuos normales, sinevidencia de enfermedad autoinmune, pre-sentan linfocitos con capacidad autorre-activa; b) alrededor del 75% de los ge-melos monocigóticos de pacientes conenfermedades autoinmunes son normales;c) las enfermedades autoinmunes suelencursar con brotes de reagudización entrelos cuales el paciente puede estar inclusoasintomático. De entre todos los posibles factores am-bientales capaces de facilitar reaccionesautoinmunes destacan los agentes infec-ciosos. En la mayoría de las enfermeda-des autoinmunes, un episodio infecciososuele preceder al comienzo de la enfer-medad o a un episodio de reagudización
de la misma. Además, a nivel experimen-tal se sabe que el adyuvante completo deFreund (que contiene Mycobacterium tu-berculosis muerta) posibilita la respuestainmunitaria frente a muchos antígenos queprovocan enfermedades autoinmunes se-mejantes a enfermedades humanas, comola artritis reumatoide o la esclerosis múl-tiple. Los microorganismos pueden de-sencadenar fenómenos autoinmunes almenos por cinco mecanismos31:1. Activación de las células presentadorasde autoAg por factores producidos por losmicroorganismos, los cuales pueden in-ducir moléculas coestimuladoras que faci-litan la activación de células T autorreac-tivas. Uno de los más estudiados es laendotoxina bacteriana, que además de es-timular a los macrófagos induce activaciónpoliclonal de los linfocitos B. 2. Presentación de antígenos que normal-mente están ocultos al sistema inmunita-rio, en sujetos predispuestos genética-mente. Un ejemplo de producción deautoinmunidad por este mecanismo es laoftalmía simpática.3. La respuesta frente a un Ag microbia-no puede comprometer a autoAg (mime-tismo molecular). Este mecanismo podríaexplicar la activación de linfocitos T auto-rreactivos con autoAg presentes en dosissubóptimas. Una vez activados, puedenperpetuar una reacción autoinmune con-tra los autoAg contra los que antes eranincapaces de reaccionar.4.La unión de agentes infecciosos a auto-Ag puede modificar el estado de toleran-cia hacia los mismos. En este caso, elagente infeccioso actuando de carrier fa-cilitaría la respuesta autoinmune. 5. Algunos agentes infecciosos son capa-ces de inducir la activación policlonal delos linfocitos T. Se denominan superantí-genos y, a diferencia de los péptidos con-vencionales que estimulan menos del0,01% de los linfocitos virgen, un únicosuperAg puede estimular más del 5% delreservorio de linfocitos virgen. Los supe-rAg son proteínas que se unen a las mo-léculas MHC de clase II en las APC y queinteraccionan con regiones Vβ del TCR me-nos variables que el sitio habitual de re-conocimiento del Ag. De este modo, unsuperAg es capaz de estimular todas aque-llas células que porten ese segmento deVβ del TCR específico, habiéndoseles im-plicado en la producción de enfermedadesautoinmunes y en los brotes que suelenocurrir en estas enfermedades.
1340
ENFERMEDADES DEL SISTEMA INMUNE (II)

Finalmente, en la patogenia de algunosprocesos autoinmunes se han visto impli-cados diversos agentes químicos. Entreotros se encuentran: el mercurio, el síliceo fármacos como la procainamida, la hi-dralazina o las sales de oro. Estas sustan-cias pueden reaccionar con diversas mo-léculas existentes en la membrana, elcitoplasma o el núcleo celular y modificarla fisiología de las células inmunocompe-tentes32. Se han propuesto tres mecanis-mos, no excluyentes entre sí, para expli-car la asociación de los agentes químicoscon la producción de reacciones autoin-munes:1. Modificación de la estructura de las mo-léculas MHC o de los autoAg que son pre-sentados, de modo que las células T re-accionan frente a ellos como si fuesenextraños.2. Alteración de la molécula del TCR bienpor unión directa o por modificación a ni-vel genético.3. Amplificación de las interacciones en-tre la célula T y la APC al unirse a molé-culas de la membrana celular de un modosimilar a los superAg.
BIBLIOGRAFÍA
1. Goldrath AW, Bevan MJ. Selecting and maintaining a di-verse T-cell repertoire. Nature 1999; 402: 255-262.2. Ashton-Rickardt PG, Bandeira A, Delaney JR, Van KaerL, Pircher HP, Zinkernagel RM, Tonegawa S. Evidence for adifferential avidity model of T-cell selection in the thymus.Cell 1994; 76: 651-663.3. Von Boehmer H. Positive selection of lymphocytes. Cell1994; 76: 219-228.
4. Nossal GJ. Negative selection of lymphocytes. Cell 1994;76: 229-239.5. Wucherpfennig KW, Strominger JL. Molecular mimicryin T cell-mediated autoimmunity: viral peptides activatehuman T cell clones specific for myelin basic protein. Cell1995; 80: 695-705.6. Harding FA, McArthur JG, Gross JA, Raulet DH, AllisonJP. CD28-mediated signaling co-stimulates murine T cellsand prevents induction of anergy in T-cell clones. Nature1992; 356: 607-609.7. Finck BK, Linsley PS, Wofsy D. Treatment of murine lu-pus with CTLA4Ig. Science 1994; 265: 1.225-1.227.8. Theofilopoulos AN. The basis of autoimmunity: Part I.Mechanisms of aberrant self-recognition. Immunol Today1995; 16: 90-98.9. Griffith TS, Brunner T, Fletcher S, Green DR, FergusonTA. Fas ligand-induced apoptosis as a mechanism of im-mune privilege. Science 1995; 270: 1.189-1.192.10. Abbas AK, Murphy DM, Sher A. Functional diversity ofhelper T lymphocytes. Nature 1996; 383: 787-793.11. Nagata S. Apoptosis by death factor. Cell 1997; 88:355-365.12. Letterio JJ, Geiser AG, Kulkarni AB, Dang H, Kong L,Nakabayashi T, et al. Autoimmunity associated with TGF-beta1-deficiency in mice is dependent on MHC class II anti-gen expression. J Clin Invest 1996; 98: 2.109-2.119.13. Van Parijs L, Abbas AK. Homeostasis and self-tolerancein the immune system: turning lymphocytes off. Science1998; 280: 243-248.14. Van Parijs L, Peterson DA, Abbas AK. The Fas/Fas li-gand pathway and Bcl-2 regulate T cell responses to modelself and foreign antigens. Immunity 1998; 8: 265-274.15. Lenardo MJ. Interleukin-2 programs mouse αβ T lymp-hocytes for apoptosis. Nature 1991; 353: 858-861.16. Nemazee D. Receptor editing in B cells. Adv Immunol2000; 74: 89-126.17. Townsend SE, Weintraub BC, Goodnow CC. Growingup on the streets: why B-cell development differs from T-cell development. Immunol Today 1999; 20: 217-220.18. Randall TD, Heath AW, Santos-Argumedo, L, HowardMC, Weissman IL, Lund, FE. Arrest of B lymphocyte termi-nal differentiation by CD40 signaling: mechanisms for lackof antibody-secreting in germinal centers. Immunity 1998;8: 733-742.19. Liu Y-J, Arpin C. Germinal center development. Immu-nol Rev 1997; 156: 111-126.
20. Martínez-Valdez H, Guret C, de Bouteiller O, Fugier I,Banchereau J, Lui YJ. Human germinal center B cells ex-press the apoptosis inducing genes Fas, c-myc, P53 andBax but not the survival gene bcl-2. J Exp Med 1996; 183:971-977.21. Bolland S, Ravetch JV. Inhibitory pathways triggered byITIM-containing receptors. Adv Immunol 1999; 72: 149-177.22. Maeda A, Kurosaki M, Ono M, Takai T, Kurosaki T. Re-quirement of SH2-containing protein tyrosine phosphata-ses SHP-1 and SHP-2 for paired immunoglobulin-like re-ceptor B (PIR-B)-mediated inhibitory signal. J Exp Med1998; 187: 1.355-1.360.23. McAdam AJ, Schweitzer AN, Sharpe AH. The role of B7co-stimulation in activation and differentiation of CD4+and CD8+ T cells. Immunol Rev 1998; 165: 231-247.24. López-Botet M, Bellón T. Natural killer cell activationand inhibition by receptors for MHC class I. Curr Opin Im-munol 1999;11:301-307.25. Strobel S, Mowat AM. Immune responses to dietary an-tigens: oral tolerance. Immunol Today 1998; 19: 173-181.26. Viney JL, Mowat AM, O’Malley JM, Williamson E, Fan-ger NA. Expanding dendritic cells in vivo enhances the in-duction of oral tolerance. J Immunol 1998; 160: 5.815-5.825.27. Wakeland EK, Wandstrat AE, Liu K, Morel L. Geneticdissection of systemic lupus erythematosus. Curr Opin Im-munol 1999; 11: 701-707.28. Straus SE, Sneller M, Lenardo MJ, Puck JM, Strober W.An inherited disorder of lymphocyte apoptosis: the autoim-mune lymphoproliferative syndrome. Ann Intern Med1999; 130: 591-601.29. Raya A, Revert F, Navarro S, Saus J. Characterization ofa novel type of serine/threonine kinase that specificallyphosphorylates the human Goodpasture antigen. J BiolChem 1999; 274: 12.642-12.649.30. Utz PJ, Hottelet M, Schur PH, Anderson P. Proteinsphosphorylated during stress-induced apoptosis are commontargets for autoantibody production in patients with systemiclupus erythematosus. J Exp Med 1997; 185: 843-854.31. Horwitz MC, Sarvetnick N. Viruses, host responses, andautoimmunity. Immunol Rev 1999; 169: 241-253.32. Goldman M, Druet P, Gleichmann E. Th2 cells in syste-mic autoimmunity: insights from allogeneic diseases andchemically-induced autoimmunity. Immunol Today 1991;12: 223-227.
1341
DE LOS MECANISMOS DE TOLERANCIA A LA AUTOINMUNIDAD

Introducción
Los enormes avances realizados en las úl-timas décadas en el conocimiento de lasbases celulares y moleculares que regulanel sistema inmunitario junto con el des-cubrimiento de potentes fármacos inmu-nosupresores y el desarrollo y perfeccio-namiento de nuevas técnicas quirúrgicas,han convertido al trasplante alogénico deórganos (trasplante de órganos entre in-dividuos de la misma especie) en el tra-tamiento de elección de múltiples proce-sos patológicos. En la actualidad, elnúmero y el tipo de órganos que puedenser trasplantados continúa aumentando yel éxito de este procedimiento es incues-tionable. Por ello, entre los países desa-rrollados un gran número de hospitales haincorporado a su actividad clínica habitualla realización de este tipo de terapia.En España se ha asistido año tras año aun aumento progresivo del número detrasplantes realizados (tabla 1). En lo querespecta al trasplante renal, en el año 1999se realizaron un total de 2.023 trasplan-tes, mayoritariamente a partir de donan-te de cadáver, lo que representa una me-dia de 51,87 trasplantes por millón depoblación (PMP). Lo mismo podemos decir con relación al trasplante cardíaco yhepático, asistiendo a un progresivo au-mento del número de trasplantes realiza-dos hasta llegar a una tasa en 1999 de 8,1PMP y 24,6 PMP, respectivamente. El pro-grama de trasplante pulmonar es el másjoven de todos los programas de tras-plante, pero se halla ya plenamente esta-blecido tanto en su modalidad de tras-plante unipulmonar como bipulmonarhabiéndose realizado en 1999 un total de135 trasplantes pulmonares lo que repre-senta una tasa de trasplante de 2,7 PMP.Por último, cabe destacar que debido alelevado número de donaciones, funda-mentalmente procedentes de cadáveres,y al excelente sistema organizativo pues-to en marcha, las cifras de enfermos tras-
plantados en España son claramente su-periores a las existentes en otros paísesde la Unión Europea y Estados Unidos (ta-bla 1).A pesar de los importantes avances reali-zados en el campo del trasplante alogéni-co, la universalización de este tipo de te-rapia se ve obstaculizada por dos grandesproblemas aún sin resolver en su totali-dad. En primer lugar, el éxito del tras-plante alogénico en el tratamiento de unnúmero cada vez mayor de enfermedadesha provocado un aumento aún mayor enel número de pacientes que espera reci-bir un órgano. Sin embargo, este aumen-to en la demanda de órganos no se acom-paña de un incremento paralelo delnúmero de donantes. En segundo lugar,aunque los tratamientos inmunosupreso-res utilizados son eficaces en la preven-ción y tratamiento de las formas más agu-das de rechazo, en la práctica totalidad delos enfermos trasplantados el órgano in-jertado es finalmente destruido en un pla-zo más o menos largo sin que hasta el momento se disponga de tratamientos eficaces que lo eviten. Por ello, el interés
prioritario en trasplante se centra, por unlado, en el descubrimiento de nuevas te-rapias inmunosupresoras y/o inmunomo-duladoras que induzcan en el receptor unestado de tolerancia al injerto completo y permanente; y, por otro, en el desarro-llo de nuevas fuentes, en principio inago-tables, de órganos. En esta Unidad Temá-tica se analizarán en primer lugar los mecanismos inmunopatogénicos respon-sables del desarrollo alogénico de órganossólidos así como el desarrollo de nuevasestrategias terapéuticas experimentales di-rigidas a la prevención total del mismo.En segundo lugar, se abordará el tema delxenotrasplante (trasplante de órganos en-tre individuos de especies diferentes).
Inmunobiología del rechazoalogénicoTipos de rechazo
Desde el punto de vista inmunológico ytambién cronológico se pueden distinguirvarias fases en el fenómeno de rechazo aun aloinjerto vascularizado1. Una primeraforma de rechazo es el denominado re-chazo hiperagudo (RHA) el cual puede ocurrir minutos u horas después de pro-ducirse la revascularización del injerto. Posteriormente, el aloinjerto puede serdestruido mediante el denominado re-chazo agudo (RA), el cual puede apareceren los pocos días o semanas después de
1342
RECHAZO DE TRASPLANTESR. Merino Pérez, G. Fernández Fresnedo* y M. Arias Rodríguez*Laboratorio de Inmunología del Trasplante. Unidad de Investigación y *Servicio de Nefrología.. Hospital Universitario Marqués de Valdecilla. Santander.
Medicine 2000; 8(26): 1342-1349
TABLA 1Actividad de trasplante en España y en el mundo
Años Trasplante Trasplante Trasplante Trasplante renal hepático cardíaco pulmonar
1981-1985 3.264 19 32 0
1986-1990 5.465 700 435 23
1991-1995 7.784 2.687 1.073 114
1996-1999 7.586 3.349 1.285 447
Número de trasplantes por millón de población (año 1998)
País Trasplante Trasplante Trasplante Trasplante renal hepático cardíaco pulmonar
España 47,5 8,1 20,1 2,7
Alemania 27,4 6,8 9 1,5
Bélgica 41,7 11,3 13,7 3,4
Francia 29,1 7,2 10,7 1,5
Holanda 33 3,4 5,7 0,7
Reino Unido 29,1 5,2 11,1 ND
EE.UU. 36,5 8,9 15,5 3,7
ND: no disponible.

la realización del trasplante. Por último,existe una tercera forma de rechazo, el re-chazo crónico (RC), mediante el cual el te-jido trasplantado es destruido de una for-ma larvada y a largo plazo.
Rechazo hiperagudo
Hace más de 25 años se observó que pa-cientes sensibilizados sometidos a alo-trasplantes renales rechazaban el órganotrasplantado de una forma violenta en lospocos minutos u horas siguientes a la re-vascularización del injerto. Este tipo deRHA, el cual como se comentará más ade-lante depende de la existencia de anti-cuerpos alorreactivos preformados, es hoyen día fácilmente predecible, y por lo tan-to evitable, mediante la determinación delgrupo sanguíneo del donante y del recep-tor (compatibilidad ABO y Rh), el tipajeHLA y mediante el análisis de la presen-cia en el receptor del trasplante de anti-cuerpos circulantes reactivos contra el do-nante (prueba cruzada)2. Sin embargo,existe un grupo significativo de pacientes,fundamentalmente enfermos en lista deespera para trasplante renal, que poseenniveles circulantes elevados de estos anti-cuerpos preformados presentando un altoriesgo de desarrollar RHA al ser trasplan-tados. Las causas más comunes de inmu-nización en estos pacientes hipersensibi-lizados son la recepción previa detransfusiones sanguíneas, comunes en pa-cientes con enfermedad renal crónica, em-barazos o alotrasplantes anteriores. El usoen los últimos años de la eritropoyetinarecombinante en el tratamiento de la ane-mia asociada a la insuficiencia renal cró-nica ha conducido a una disminución enel numero de transfusiones sanguíneas queestos pacientes recibían y, por lo tanto, auna disminución de los niveles de anti-cuerpos alorreactivos preformados en es-tos individuos.El hallazgo anatomopatológico más rele-vante en el RHA es la presencia de lesio-nes vasculares a nivel de los vasos de pequeño calibre con fenómenos de trom-bosis intravascular, edema y hemorragiaintersticial. Otros hallazgos típicos en losórganos rechazados son la presencia defocos necróticos e infiltrados de polimor-fonucleares2.Desde el punto de vista fisiopatogénico, elRHA está mediado por la fijación al injer-to de anticuerpos alorreactivos preforma-dos y la activación de la cascada del com-
plemento, aunque elementos celularescomo plaquetas y polimorfonucleares pue-den también desempeñar un papel pato-génico importante. La diana principal delos anticuerpos alorreactivos preformadoses el endotelio vascular. Se ha observadoque los primeros signos de RHA precedenincluso la aparición de fenómenos demuerte celular y conllevan la activaciónde la célula endotelial. Como consecuen-cia de estos fenómenos de activación, lascélulas endoteliales tienden a separarse,permitiendo la extravasación de fluido ycélulas al intersticio. Al mismo tiempo, seproduce una pérdida en la célula endote-lial de heparán sulfato lo cual provocacambios en la actividad procoagulante dela superficie endotelial2,3.
Rechazo agudo
El RA ha sido clásicamente consideradocomo una reacción mediada por linfocitosque puede provocar la destrucción delaloinjerto en los pocos días o semanas des-pués de realizarse el trasplante. Una pro-porción elevada de pacientes que recibenun trasplante, y con independencia del ór-gano injertado, suelen sufrir uno o variosepisodios de RA que en general son biencontrolados con un tratamiento inmuno-supresor adecuado (tabla 2). Histológica-mente el hallazgo más característico delRA es la presencia de un infiltrado de cé-lulas mononucleares en un principio loca-lizados en las áreas perivasculares y quecon el tiempo se extienden por el restodel injerto. Este infiltrado celular está com-puesto por linfocitos T y B, macrófagos ycélulas natural killer (NK). Junto con los in-filtrados celulares, se observan fenómenosde trombosis intravascular y necrosis ce-lular. Al igual que en el RHA, la célula en-dotelial parece ser la primera diana en estetipo de rechazo produciéndose un fenó-meno de activación endotelial que a dife-rencia del caso anterior, provoca un au-mento en la transcripción génica y en lasíntesis de proteínas4. Por consideracionesdidácticas, los mecanismos inmunopato-génicos implicados en este tipo de recha-zo serán discutidos conjuntamente con losresponsables del RC.
Rechazo crónico
El desarrollo en estas últimas décadas denuevos tratamientos inmunosupresores, lamejora en las condiciones de extracción y
preservación de los órganos, y el trata-miento efectivo de los cuadros infeccio-sos que aparecen como complicacionesde la inmunosupresión, ha provocado unaumento significativo tanto en la viabili-dad inmediata de los aloinjertos como ensupervivencia de los pacientes trasplan-tados. Sin embargo, a pesar de estosavances a corto plazo, la incidencia dealteraciones funcionales en los órganostrasplantados después del primer año noha mejorado sustancialmente. La causaprincipal de la pérdida del injerto en es-tos pacientes es el RC. La evolución clí-nica característica del RC es un deterio-ro lento, progresivo y gradual de lafunción del injerto, cuyo inicio suele serasintomático en la mayor parte de los ca-sos. Desde el punto de vista histológico,e independientemente del órgano que seanalice, la característica principal en elRC es la fibrosis obliterante de las es-tructuras huecas del injerto (vasos san-guíneos, bronquiolos, conductos biliares),la presencia de infiltrados celulares mo-derados constituidos fundamentalmentepor linfocitos, macrófagos y células NK,la fibrosis intersticial y la aparición de ar-terioesclerosis5.El desarrollo de RC se ve influido por unaserie de factores de riesgo que van a con-dicionar la severidad y cronología del mis-mo. Entre ellos, el más importante es lapresencia de episodios de RA previos.Otros factores de riesgo son la hiperlipi-demia, lesiones secundarias al síndromeisquemia-reperfusión, infecciones pos-trasplante, edad del donante, hipertensióny obesidad del receptor6.
Conceptos básicos de larespuesta inmune frente a aloantígenos
Los mecanismos inmunológicos a nivel ce-lular y molecular responsables tanto delRA como del RC son en general mal co-nocidos y son la consecuencia de la acti-vación de los linfocitos T del receptor através del reconocimiento de aloantígenosdel donante. Dentro del grupo de aloantí-genos, los más importantes son las molé-culas del sistema principal de histocom-patibilidad o MHC (clase I: HLA –A, –B y–C, clase II: HLA-DR, –DQ, –DP). Otrosaloantígenos, mucho peor caracterizadospero que también pueden provocar fenó-menos de rechazo aunque con menor in-
1343
RECHAZO DE TRASPLANTES

tensidad, son los denominados antígenosmenores de histocompatibilidad5.Existen dos formas de reconocimiento delos aloantígenos MHC del donante por par-te de los linfocitos T del receptor (fig.1 ).Un primer tipo de reconocimiento es eldenominado directo, mediante el cual lin-focitos T CD4+ y/o CD8+ del receptor re-conocen determinantes peptídicos direc-tamente en la molécula MHC intacta deldonante expresada en la superficie de lascélulas del injerto; MHC de clase I en elcaso de los linfocitos CD8+ y MHC de cla-se II en el caso de las células CD4+. La fre-cuencia de este tipo de linfocitos T alo-rreactivos que reconocen aloantígenos porla vía directa es muy elevada y la diversi-dad de sus receptores antigénicos es tam-bién alta7. En paralelo con esta vía de re-conocimiento, las moléculas MHC deldonante son procesadas y presentadascomo péptidos por parte de las células pre-sentadoras de antígeno del receptor. Estetipo de respuesta inmune es, por lo tan-to, una respuesta restringida al MHC propio. A diferencia del reconocimientodirecto, la frecuencia de células T alorre-activas de la vía indirecta es mucho me-
nor y el repertorio de TCR utilizado mucho más restringido7. La contribuciónde cada una de estas vías de reconoci-miento en la regulación de la respuestaalogénica es en gran medida desconocida.Algunos autores han postulado que una in-teracción directa entre linfocitos T del re-ceptor y aloantígenos MHC en células pre-sentadoras de antígeno del donante,capaces de emigrar tras el trasplante a losórganos linfoides secundarios del recep-tor, podría ser el estímulo iniciador de larespuesta alogénica dado que en deter-minados modelos experimentales la eli-minación en el injerto, previamente a larealización del trasplante, de estas pobla-ciones de células presentadoras de antí-geno, inhibe o retrasa considerableme-mente el desarrollo de rechazo7,8. Sinembargo, la relevancia de esta hipótesisen los fenómenos de rechazo de órganosvascularizados puede quedar en entredi-cho, ya que como veremos más adelanteel endotelio vascular del donante expresaconstitutivamente moléculas MHC tantode clase I como de clase II y es capaz desuministrar señales coactivadoras a los lin-focitos T alorreactivos.
Además de la interacción entre el recep-tor antigénico en los linfocitos T del hués-ped y el aloantígeno, se requiere la pre-sencia de otras señales accesorias para laactivación efectiva de los linfocitos alo-rreactivos (figs. 1 y 2)9. Estas señales co-activadoras son mediadas por la interac-ción entre receptores no polimórficosexpresados en la superficie de las célulasT alorreactivas y sus respectivos ligandosen la superficie de las células presentado-ras de antígeno del donante (vía directa)o del receptor (vía indirecta). La ausenciade estas señales coactivadoras puede pro-vocar la tolerización en vez de activaciónde los linfocitos T alorreactivos. De entrelas posibles interacciones celulares que seestablecen entre ambas poblaciones celu-lares, resaltamos por su importancia la in-teracción entre las moléculas CD28 y CTTLA-4 y sus ligandos B7.1 y B7.2 y en-tre las moléculas CD40L y CD409. CD28es la principal molécula coestimuladora enlos linfocitos T. Sus ligandos en las célu-las presentadoras de antígeno son las mo-léculas B7.1 (CD80) y B7.2 (CD86). La in-teracción de CD28, presente tanto en lascélulas T vírgenes como activadas, con
1344
ENFERMEDADES DEL SISTEMA INMUNE (II)
TABLA 2Tratamiento inmunosupresor en el trasplante renal
Tratamiento de inducción
Anticalcineurínicos Ciclosporina A Uno de los dos Tacrólimus o FK 506
Antimetabolitos Azatioprina Uno de los dos Ácido micofenólico
Corticosteroides Bolus inicial intravenoso y luego oral
Anticuerpos ALG o ATC: antilinfocitarios Sólo en pacientes de alto riesgo: hiperinmunizados con > 50% anticuerpos HLA o anti-timocíticos o injerto previos perdidos por rechazo agudo en menos de 1 mes. Se usan en
OKT3: Ac monoclonales terapia secuencial con ciclosporina, es decir la ciclosporina se inicia un poco más tarde
Anti-CD25 humanizado En pacientes de alto riesgo pero todavía no claros los resultados
El tratamiento habitual suele ser la triple terapia consistente en ciclosporina A, azatioprina o ácido micofenólico y esteroides
Tratamiento de mantenimiento
Anticalcineurínicos Ciclosporina A Uno u otro Tacrólimus
Corticosteroides
Antimetabolitos Azatioprina Uno u otro Ácido micofenólico
Sirolimus Su uso en España no está aprobado todavía Se suele usar en combinación con ciclosporina A por su diferente mecanismo de acción
Tratamiento del rechazo
Corticosteroides vía intravenosa Tratamiento básico
Ac monoclonales: OKT3 RA córtico-resistente
Ac anti-timocíticos
Tacrólimus Estudios recientes sugieren su utilización en el tratamiento de pacientes con RA recurrente
A título de ejemplo se describen los protocolos de tratamiento inmunosupresor en los enfermos con trasplante renal en el Hospital Universitario Marqués de Valdecilla.

las moléculas B7 promueve la proliferaciónde los linfocitos T. La molécula CTLA-4, unhomólogo de CD28 que se induce tras laactivación celular, interactúa con los mis-mos ligandos que CD28 aunque con unaafinidad unas 20 veces superior. Sin em-bargo, a diferencia de CD28, CTLA-4 trans-duce señales inhibidoras a la célula T10. Porsu parte, la molécula CD40L (CD154) esuna proteína de membrana presente en lasuperficie de las células T perteneciente ala familia del factor de necrosis tumoral(TNF). Su ligando en la célula presentado-ra de antígeno es la molécula CD40, unmiembro de la familia del receptor de TNF.La interacción entre CD40L y CD40 es unestímulo coactivador en la célula T y entreotros efectos promueve la expresión de lasmoléculas B7 en la célula presentadora deantígeno facilitando por lo tanto la activa-ción de los linfocitos T vía la moléculaCD2811. La relevancia de las interaccionesmoleculares mencionadas anteriormenteen la activación alogénica y en los fenó-
menos de rechazo viene avalada por el he-cho de que en diversos modelos experi-mentales el bloqueo de tales interaccionespromueve la inducción de tolerancia de loslinfocitos T alorreactivos y la persistenciade los injertos12.Una vez activados los linfocitos T alorre-activos del receptor, éstos pueden provo-car la destrucción del injerto por múltiplesmecanismos; mediante un efecto citotó-xico directo en el caso de los linfocitosCD8+, mediante la secreción de citocinasproinflamatorias, o mediante la coopera-ción con linfocitos B en la producción deanticuerpos alorreactivos (fig. 1).
Relevancia del endoteliovascular en la activación decélulas alorreactivas
El endotelio vascular de los aloinjertosconstituye la barrera donde se estableceel primer contacto entre las células inmu-
nocompetentes del huésped y las célulasdel donante. Uno de los primeros dañosque se produce durante los fenómenos derechazo en el injerto, ya sea durante elRHA como el RA o el RC, es la activaciónendotelial. En el caso del RA y del RC, estaactivación endotelial se acompaña de unaumento en la expresión de numerosasmoléculas de superficie y en la secreciónde citocinas y quimiocinas que van a re-gular directamente el proceso inflamato-rio. Básicamente, el endotelio vascular delos aloinjertos vascularizados puede in-fluenciar el rechazo mediado por linfoci-tos T alorreactivos del huésped por dosmecanismos principales. En primer lugar,el endotelio va a desempeñar un papelcentral regulando la extravasación de lin-focitos efectores al parénquima del injer-to. En segundo lugar, las células endote-liales van a participar directamente en laactivación de las células T alorreactivas víala presentación de aloantígenos y me-diante el suministro de señales coestimu-ladoras (fig. 2)13.Tras la activación de las células T vírge-nes en el curso de una respuesta inmu-nitaria éstas se diferencian bien a célu-las efectoras o a células T memoria. Loslinfocitos diferenciados son atraídos pos-teriormente a los focos inflamatorios enun proceso de migración celular media-do por la interacción entre moléculas deadhesión expresadas en la superficie delas células endoteliales activadas, y susligandos en la superficie de los linfocitosT diferenciados. En el caso del alotras-plante, se ha observado que poco tiem-po después de efectuarse el mismo, e in-cluso antes de que aparezcan signos deRA, se produce un aumento en la expre-sión de selectina-E en el endotelio del in-jerto. Así mismo, se ha observado un au-mento en la expresión endotelial de lamolécula VCAM-1 si bien este incremen-to se asocia con la presencia de infiltra-dos celulares y con el fenómeno de RA.Así mismo, durante el RA, y como con-secuencia de la acción de diferentes ci-tocinas, interferón gamma (IFNγ) y TNFαfundamentalmente, se produce un au-mento en la expresión endotelial deICAM-1. La presencia de niveles elevadosde estas moléculas de adhesión tiene unpapel importante en la inducción tantode RA como de RC, ya que éstos puedenprevenirse en ratones mediante el trata-miento con anticuerpos monoclonales dirigidos contra las moléculas LFA-1 o
1345
RECHAZO DE TRASPLANTES
Célulaplasmática
Anticuerposalorreactivos
YY
Y
YY
Y Y
YY
Y
Y
Y
B
Alorreconocimientoindirecto
Alorreconocimientodirecto
CD8
Aloantígenos
Citotoxicidad
MHC clase Idonante
MHC clase IIdonante
CPAhuésped
CPAdonante
Señ
alco
esti
mu
lad
ora
Señ
alco
esti
mu
lad
ora
MHC clase IIhuésped
CD4 CD4
Fig. 1. Mecanismos de activación de las células T y B alorreactivas. Los linfocitos T pueden reconocer los aloantígenos presenta-dos por las células presentadoras de antígeno (ACP) mediante la vía directa (derecha) e indirecta (izquierda) en presencia de estí-mulos coestimuladores. Una vez activadas, las células T alorreactivas pueden destruir el injerto mediante la cooperación con loslinfocitos B alorreactivos en la producción de aloanticuerpos (parte superior) o mediante citotoxicidad directa en el caso de los lin-focitos CD8 (parte inferior).

VLA-4, los ligandos naturales de ICAM-1y VCAM-1 respectivamente13.Otros factores que van a regular el tráfi-co de linfocitos alorreactivos al interiordel injerto durante los procesos de re-chazo son las quimiocinas o factores qui-miotácticos. Múltiples quimiocinas se ex-presan en los aloinjertos tras daños talescomo el síndrome isquemia-reperfusión,episodios de RA y RC y en asociación coninfecciones víricas. Aparte de su funciónquimiotáctica, las quimiocinas pueden fa-cilitar la migración al injerto de leucoci-tos mediante el aumento en la capacidadadhesiva de determinadas integrinas quese expresan en la superficie de las célu-las T y sus ligandos en la superficie en-dotelial. Éste es, por ejemplo, el caso dela quimiocina SDF-1 (stromal-cell-derivedfactor 1) que uniéndose a la célula endo-telial promueve la adhesión de los leu-cocitos a ICAM-1. Por otro lado, la ex-presión de quimiocinas en el endoteliodel aloinjerto puede estar implicada en elreclutamiento preferencial al interior delaloinjerto de determinados subtipos fun-cionales de linfocitos CD4+ con distintopotencial patogénico. En este sentido, re-cientemente se ha demostrado que loslinfocitos CD4+ del tipo TH1, productores
de IFNγ, y TH2, productores de IL-4, ex-presan diferentes receptores de quimio-cinas13.El papel de las diferentes citocinas y delos subtipos funcionales de linfocitosCD4+ en el rechazo, tanto en el RA comoen el RC, es confuso y controvertido. Elanálisis de la producción de citocinas enaloinjertos llevó en un principio a postu-lar que los linfocitos TH1 podrían consti-tuir la subpoblación de células CD4+ im-plicada en el rechazo. Sin embargo,estudios recientes utilizando animales de-ficientes en citocinas tipo TH1 o TH2 hanlevantado serias dudas sobre la veracidadde esta hipótesis e indican que, depen-diendo del modelo de alotrasplante utili-zado, tanto citocinas tipo TH1, IFNγ e IL-2, o TH2, IL-4, pueden ser factores fa-vorecedores o protectores del RA yRC13,14.Otra posible participación del endoteliovascular del injerto en el control de la ac-tivación alogénica de los linfocitos T delreceptor durante el rechazo es actuandocomo células presentadoras de antígeno.Esto es posible dado que en primer lugarel endotelio vascular expresa moléculasMHC tanto de clase I como de clase II pu-diendo activar por la vía directa a linfo-
citos T CD4 y/o CD8 alorreactivos. Porotra parte, el endotelio activado expresauna serie de moléculas que pueden apor-tar las señales coestimuladoras necesa-rias para la activación adecuada de di-chas células alorreactivas. Aparte de laanteriormente mencionada ICAM-1, el en-dotelio vascular expresa CD58 (LFA-3) yfundamentalmente CD40 y CD40L. La ex-presión de CD40 y CD58 puede suminis-trar señales coestimuladoras a los linfo-citos T necesarias para su activación ypara la síntesis y secreción de citocinasproinflamatorias. Por su parte, la inte-racción entre CD40L en la superficie en-dotelial del aloinjerto y CD40 en célulaspresentadoras de antígeno del receptorpuede promover en estas últimas la ex-presión de moléculas de la familia B7, po-tenciando de esta forma la activación delinfocitos T alorreactivos mediante la víaindirecta (fig. 2)13.Múltiples estudios han demostrado queuno de los mecanismos principales porlos que se regula la duración de la res-puesta inmune es mediante la elimina-ción por apoptosis de los linfocitos acti-vados. Este proceso de muerte celularprogramada linfocitario es dependientede la molécula Fas, un miembro de la fa-milia del receptor de TNF que se expre-sa entre otros en la superficie de los lin-focitos y que transduce señales demuerte. La interacción entre Fas y su li-gando, Fas-L, activa una cascada de pro-teasas intracelulares que inducen el pro-ceso apoptótico15. Recientemente, se haobservado que la expresión de Fas-L seasocia a la capacidad que presentan al-gunos tejidos, tales como las células deSertoli en los testículos y la córnea, a noser rechazados tras ser trasplantados. Enestos tejidos inmunoprivilegiados, la ex-presión de Fas-L podría inducir la apari-ción de apoptosis de los linfocitos T alo-rreactivos que intentan destruirlos. Elendotelio vascular es otro tejido que ex-presa Fas-L. Sin embargo, citocinas proin-flamatorias como TNFα, el cual se liberatras la activación alogénica, regulan ne-gativamente la expresión de Fas-L en elendotelio vascular13. La importancia deeste fenómeno en la patogenia del re-chazo todavía no se conoce, aunque ma-niobras encaminadas a impedir la regu-lación negativa de Fas-L en el endoteliovascular durante la activación alogénicapodrían tener consecuencias beneficiosasen el control del rechazo alogénico.
1346
ENFERMEDADES DEL SISTEMA INMUNE (II)
Endoteliovascular
Activación
Citocinasproinflamatorias
Migración
ICAM-1/LFA-1
VLA-4/VCAM-1
Endoteliovascular
CPAhuésped
CD4
MHC
TCRCD58
CD2
B7
CD28
CD40L
CD40
B7
CD28
CD40L CD40
TCR
MHC
CD4
Fig. 2. Interacciones entre el endotelio vascular del aloinjerto y los linfocitos T alorreactivos del huésped durante el rechazo. Estasinteracciones son de una gran complejidad y en ellas participan moléculas de adhesión intercelular, moléculas coestimuladoras,citocinas pro-inflamatorias y quimiocinas (ver texto).

Papel de la inmunidad humoraly de las células NK en el rechazo
Además del papel de los anticuerpos alo-rreactivos en el desarrollo de RHA, la in-munidad humoral también parece inter-venir en la patogenia del RA y del RC.Rápidamente tras el trasplante se obser-va un aumento en los niveles circulantesde anticuerpos anti-MHC del donante, pri-mero de tipo IgM y posteriormente detipo IgG. La producción de anticuerpos de tipo IgG indica que este fenómeno estámediado por linfocitos T CD4+ alorreac-tivos. Asimismo, en los infiltrados celu-lares de los tejidos con RA se observa lapresencia de linfocitos B y células plas-máticas. La participación de los anti-cuerpos antidonante en el desarrollo deRA podría ser múltiple; mediante la acti-vación de la cascada del complemento,induciendo la activación del endoteliovascular del aloinjerto y promoviendo fe-nómenos de citotoxicidad celular media-da por anticuerpos (ADCC). Asimismo, seobservan infiltrados celulares de linfoci-tos B y depósitos de inmunoglobulinas enlas lesiones de RC. Estas inmunoglobuli-nas pueden ser tanto específicas comoinespecíficas, ya que durante el RC, ycomo consecuencia de la alteración en-dotelial, se produce un aumento en la permeabilidad capilar. Por último, cabedestacar que en determinados modelosexperimentales de trasplante cardíaco, la administración continuada de anti-cuerpos antidonante es capaz de provo-car lesiones de arterioesclerosis en el injerto4,5.A pesar de su capacidad de distinguir en-tre lo propio y lo extraño, y de su pre-sencia en los infiltrados celulares típicosdel RA y RC, las células NK no parecen es-tar involucradas, al menos de una formarelevante, en el rechazo de aloinjertos só-lidos. Sin embargo, las células NK sí cons-tituyen una barrera importante durante eltrasplante de células hematopoyéticas ysobre todo, como se comentará poste-riormente, en el xenotrasplante16.
Mecanismos patogénicos de la arterioesclerosis asociadaal trasplante
La limitación mayor para la supervivenciaa largo plazo de los alotrasplantes vascu-larizados es, sin duda alguna, la aparición
de lesiones de arterioesclerosis en losmismos. Aunque los mecanismos etiopa-togénicos precisos de esta lesión no sonbien conocidos, en ellos intervienen tan-to factores inmunológicos como no in-munológicos. Desde el punto de vista in-munológico se ha implicado a diversascitocinas en el desarrollo de esta lesióntales como la IL-1, IL-6, factor de creci-miento derivado de las plaquetas (PDGF)y fundamentalmente el factor transfor-mador del crecimiento beta o TGFβ. LosTGFβ son unas citocinas fundamentalesdurante la fibrinogénesis y son produci-das por casi todos los tejidos incluidoslos linfocitos y macrófagos presentes en los infiltrados celulares durante el RC.Así, durante los fenómenos inflamatoriosagudos (crisis de RA) o larvados en elaloinjerto y en respuesta al daño tisularproducido se puede poner en marcha lamaquinaria de reparación tisular provo-cando un aumento en la producción y se-creción de las formas activas de TGFβ.Estos factores de crecimiento podríansubsiguientemente promover lesiones dearterioesclerosis mediante varios meca-nismos; la inducción de la proliferacióncelular, la síntesis de proteínas de la ma-triz extracelular (diversos colágenos, fi-bronectina, osteopondina, etc.) y la delinhibidor tisular de metaloproteinasas loque disminuiría la degradación ulteriorde la matriz neoformada17.
Nuevas estrategiasterapéuticas en laprevención del rechazoalogénicoComo hemos comentado previamente,los tratamientos inmunosupresores utili-zados en la actualidad en los enfermostrasplantados han mostrado una eficaciaaceptable en la prevención y control delRA, pero son ineficaces en la prevencióndel RC y por lo tanto en la supervivenciaa largo término del implante. Por ello, ungran número de laboratorios en todo elmundo están investigando nuevas estra-tegias terapéuticas que tienen como fi-nalidad la inducción de un estado de to-lerancia completo y persistente hacia losaloantígenos del donante. Aunque múlti-ples protocolos han resultado ser efica-ces en la prevención de RC en modelosde alotrasplante en el ratón, dichos tra-
tamientos no han dado los mismos re-sultados en modelos con animales gran-des como el cerdo o primates, probable-mente debido a que el endotelio vascularde estos últimos, y no de los primeros,expresa moléculas MHC de clase II. Acontinuación comentaremos muy breve-mente algunas de las estrategias tera-péuticas más prometedoras, todavía enfase experimental, que están desarro-llándose para el control del rechazo alo-génico en animales grandes.
Utilización de células demédula ósea del donante parala inducción de tolerancia
En estos casos se intenta promover en elhuésped un estado de quimerismo utili-zando células de médula ósea del donante(CMOD). Estas células portadoras de losaloantígenos serían capaces de inducir enel huésped un estado de tolerancia. Parainducir quimerismo es preciso someter alhuésped a tratamientos de acondiciona-miento cuya finalidad es evitar el recha-zo prematuro de las CMOD y/o el desa-rrollo de manifestaciones de enfermedadinjerto contra huésped. En estos trata-mientos de acondicionamiento previos ala administración de las CMOD se ha uti-lizado la irradiación linfoide total, la irra-diación total y la administración de sue-ros antilinfocitarios. Dependiendo delgrado de quimerismo inducido, el trata-miento de acondicionamiento previo ydel momento pre- o postrasplante en elque se administran las CMOD, estos pro-tocolos son capaces de inducir un esta-do de tolerancia T en el huésped bienmediante la eliminación clonal de célu-las alorreactivas en el timo, bien me-diante la inducción de células inmuno-rreguladoras-supresoras en la periferia.Los resultados obtenidos con los dife-rentes protocolos utilizados son variados.En todos los casos, se ha observado unaumento en la supervivencia a corto pla-zo de los aloinjertos aunque solamenteaquellos protocolos que inducen un es-tado de macro-quimerismo prolongadoson capaces de inhibir la producción deanticuerpos alorreactivos y el desarrollode RC. Sin embargo, los tratamientos deacondicionaimiento aplicados en estoscasos son altamente agresivos provo-cando numerosas complicaciones gra-ves18.
1347
RECHAZO DE TRASPLANTES

Tratamientos encaminados a bloquear la coestimulación de linfocitos alorreactivos
La activación de las células T alorreactivasrequiere al menos dos señales, una señalprocedente de la interacción del receptorantigénico con el aloantígeno y otra me-diada por la interacción entre moléculasde superficie coestimuladoras en la célulaT alorreactiva con sus ligandos en la cé-lula presentadora de antígeno. El bloqueode estas señales coestimuladoras puedeprovocar la inactivación funcional de loslinfocitos T antígeno-específicos. Basán-dose en este principio, se está evaluandola eficacia del bloqueo de las interaccio-nes entre CD28 y B7.1 o B7.2 y entreCD40L y CD40 en la prevención del re-chazo alogénico en animales grandes. Es-tos tratamientos han demostrado ser muyefectivos en modelos de alotrasplante enmodelos murinos. Para la realización deestos tratamientos, se han desarrollado an-ticuerpos monoclonales humanizados anti-CD40L (hu5C8) y moléculas solubles CTLA-4-Ig. Los primeros resultados obtenidosson sumamente esperanzadores, ya queestos tratamientos prolongan la supervi-vencia de los aloinjertos aunque no soncapaces de bloquear la producción de an-ticuerpos alorreactivos14,18.En definitiva, la utilización de estas nue-vas estrategias terapéuticas abre una nue-va vía para el control del rechazo alogé-nico. Aunque han demostrado ser eficacesa corto plazo todavía no se conoce concerteza si son capaces de prevenir la apa-rición de RC, en la actualidad la mayor li-mitación en alotrasplante.
XenotrasplanteUna posible solución al problema que plan-tea la escasez de órganos en alotrasplan-te es la utilización de órganos proceden-tes de otras especies animales. Existe unconsenso generalizado de que el cerdo,entre las posibles especies animales es laespecie ideal en xenotrasplante. Sin em-bargo, el xenotrasplante está aún lejos deconvertirse en la panacea terapéutica queresuelva el problema de la limitación dedonantes. Dos son los problemas funda-mentales que hacen inviable en la actua-lidad este abordaje terapéutico. En primerlugar, la experiencia que se dispone sobrexenotrasplante, bien en los estudios reali-
zados en modelos animales experimenta-les bien en los ensayos clínicos realizadosen seres humanos, indican que los fenó-menos de rechazo hacia los xenoinjertos,aunque respondan a mecanismos inmu-nopatogénicos similares, son mucho másintensos que en el caso de aloinjertos, ydifícilmente controlables con los trata-mientos inmunosupresores actuales. Ensegundo lugar, existe la posibilidad de quela utilización de tejidos de otras especiessea la puerta de entrada de nuevas infec-ciones, fundamentalmente víricas, en laespecie humana.Al igual que con el trasplante alogénico,los xenoinjertos pueden ser destruidos me-diante los tres tipos de rechazo mencio-nados anteriormente, RHA, RA y RC. Enla actualidad, la mayor barrera en xeno-trasplante de órganos de cerdo es el de-sarrollo de RHA. Este tipo de rechazo estáprovocado por la interacción de anticuer-pos naturales anti-Galα1,3Gal del receptorcon el antígeno Galα1,3Gal del cerdo y laactivación del sistema del complemento.El determinante antigénico Galα1,3Gal,presente en el cerdo y en la mayor par-te de las especies animales con la excep-ción de los seres humanos y los primatessuperiores, se forma tras la glucosilaciónde la N-acetil-lactosamina por el enzima α-galactosil transferasa. En seres humanosy en los primates superiores, el gen codi-ficante para el enzima α-galactosil trans-ferasa ha desaparecido usándose en su lu-gar el enzima α1-2-fucosil transferasa paraproducir, a partir del mismo sustrato, lasustancia H, la cual dependiendo del gru-po sanguíneo del individuo sufre procesosde glucosilación diferencial. En esencia,esto significa que el cerdo posee un siste-ma de grupos sanguíneos antigénicamen-te diferente al humano. Debido a que eldeterminante antigénico Galα1,3Gal estátambién presente en bacterias y virus, lapráctica totalidad de los individuos pre-sentan constitutivamente anticuerpos cir-culantes anti-Galα1,3Gal, fundamental-mente del tipo IgM e IgG3, que puedenestar desempeñando una función fisioló-gica de defensa contra patógenos bacte-rianos o víricos. Se están aplicando di-versas estrategias para intentar superar labarrera del RHA en xenotrasplantes cer-do→humano. Diversos laboratorios handesarrollado cepas de cerdos transgénicosque expresan a nivel del endotelio vascu-lar moléculas reguladoras del comple-mento humanas tales como CD55, CD46
y CD59. Estas moléculas son capaces deinhibir la activación del complemento y,por lo tanto, conferir una protección con-tra el RHA. Esta inhibición es específicade especie de tal forma que la moléculaCD59 humana, por ejemplo, modulará laactividad de la cascada del complementode su propia especie. Ensayos de xeno-trasplante cerdo→primate utilizando ór-ganos de estos animales transgénicos handemostrado que este tipo de estrategiapuede ser de gran utilidad en el control deesta forma de rechazo. Otras posibilida-des todavía en fase experimental están encaminadas a inactivar la actividad de la enzima α-galactosil transferasa y, porlo tanto, impedir la generación de Galα1,3Gal19,20.Con respecto a los otros tipos de rechazode xenotrasplantes, el RA y el RC, existenuna serie de diferencias fisiopatogénicasque los distinguen del rechazo alogénicoy que van a influir de una forma muy im-portante en la severidad de los mismos.Al igual que en rechazo alogénico, los xe-noantígenos de cerdo pueden ser recono-cidos por las células alorreactivas huma-nas tanto mediante la vía directa comopor la indirecta. Sin embargo, la intensi-dad de las respuestas T es más intensa enel caso de las reacciones xenogénicas. Es-tas diferencias posiblemente estén rela-cionadas con el hecho de que mientras enlas reacciones alogénicas las respuestasiban dirigidas fundamentalmente hacia losantígenos MHC del aloinjerto, el númerode antígenos potenciales involucrados enla respuesta xenogénica humano→cerdosea muy superior19.La segunda diferencia importante entre elRA y RC de aloinjertos y de xenoinjertoshace referencia al papel de las células NK.Como se comentó en apartados anterio-res, las células NK no parecen desempe-ñar un papel relevante en la patogenia delRA y RC de aloinjertos. Sin embargo, di-ferentes estudios in vitro sugieren que lascélulas NK pueden inducir, bien directa-mente bien mediante un mecanismo deADCC, una activación del endotelio vas-cular del xenoinjerto. Por otro lado, se haobservado que las células NK pueden in-ducir la lisis del injerto en modelos in vi-tro de xenotrasplante cerdo→humano. Es-tas diferencias parecen estar relacionadascon la ausencia de interacciones entre losreceptores KIR de las células NK, los cua-les modulan negativamente la actividad deestas células, y las moléculas MHC de
1348
ENFERMEDADES DEL SISTEMA INMUNE (II)

clase I xenogénicas permitiendo de estaforma su activación. De esta manera, tra-tamientos encaminados a regular negati-vamente la actividad de las células NK pue-den ser de gran utilidad en el control delrechazo de xenoinjertos16,19.Un problema añadido que plantea la utili-zación de tejidos de otras especies ani-males para trasplantes en seres humanosradica en la posibilidad de que microor-ganismos presentes en esas especies ani-males puedan cruzar la barrera interes-pecie e infectar el huésped, en este casoal hombre. Durante estos últimos años,este problema ha sido motivo de gran po-lémica sobre todo a partir de varios estu-dios en los que se han caracterizado di-versos retrovirus endógenos de cerdocapaces de infectar células humanas encultivo. Hasta qué punto este tipo de zo-onosis representa un peligro grave de apa-rición de nuevas pandemias es algo a loque actualmente nadie puede respondercon certeza. La experiencia que se tieneen xenotrasplante, bien de órganos o te-jidos como la piel, bien de células, es muylimitada y no permite establecer conclu-siones sólidas a este respecto. Sin em-bargo, en los pocos pacientes estudiadosque han estado en contacto con células
porcinas, durante períodos en algunos ca-sos prolongados, no se ha observado has-ta el momento la aparición de nuevas en-fermedades infecciosas21,22.
BIBLIOGRAFÍA
1. VanBuskirk AM, Pidwell DJ, Adams PW, Orosz CG.Transplantation immunology. JAMA 1997; 278: 1.993-1.999.2. Kupiec-Wegliski JW, Hancock W. Hyperactive and acce-lerated allograft rejection. En: Tilney NL, Strom TB, PaulLC, eds. Transplantation biology: cellular and molecular as-pects. Philadelphia: Lippincott-Raven, 1996; 541-556.3. Platt JL, Vercellotti GM, Lindman BJ, Oegena TR Jr,Bach. FH, Dalmasso AP. Release of heparan sulfate fromendothelial cells: implications for patogenesis of hyperacu-te rejection. J Exp Med 1990; 171: 1.363-1.368.4. Tilney NL, Strom TB. Acute rejection. En: Tilney NL,Strom TB, Paul LC, eds. Transplantation biology: cellularand molecular aspects. Philadelphia: Lippincott-Raven,1996; 557-565.5. Paul LC, Tilney NL. Alloantigen-dependent events in chronic rejection. En: Tilney NL, Strom TB, Paul LC eds. Transplantation biology: cellular and molecular as-pects. Philadelphia: Lippincott-Raven, 1996; 565-575.6. Almond PS, Matas A, Gillingham KJ, Dunn DL, PayneWD, Gores P, et al. Risk factors for chronic rejection in re-nal allograft recipient. Transplantation 1993; 55: 752-757.7. Benichou G. Direct and indirect antigen recognition: thepathways to allograft immune rejection. Front Biosci 1999;4: 476-480.8. Lacy PE, Davie JM, Finke EH. Prolongation of islet allo-grafth survival following in vitro culture (24 °C) and a sin-gle injection of ALS. Science 1979; 204: 312.
9. Jenkins MK, Johnson JG. Molecules involved in T-cellcostimulation. Curr Opin Immunol 1993; 5: 361-367.10. Lee KM, Chuang E, Griffin M, Krattri R, Hong DK,Zhang W, et al. Molecular basis of T cell inactivation byCTLA-4. Science 1998; 282: 2.263-2.266.11. Grewal IS, Flavell RA. CD40 and CD154 in cell-media-ted immunity. Annu Rev Immunol 1998; 16: 111-135.12. Kirk AD, Harlan DM, Armstrong NN, Davis TA, DongY, Gray GS, et al. CTLA4-Ig and anti-CD40 ligand preventrenal allograft rejection in primates. Proc Natl Acad SciUSA 1997; 94: 8.789-8.794.13. Briscoe DM, Alexander SI, Lichtman AH. Interactionsbetween T lymphocytes and endothelial cells in allograftrejection. Curr Opin Immunol 1998; 10: 525-531.14. Dai Z, Lakkis FG. The role of cytokines, CTLA-4 andcostimulation in trasplant tolerance and rejection. CurrOpin Immunol 1999; 11: 504-508.15. Peter ME, Krammer PH. Mechanism of CD95 (APO-1/Fas)-mediated cell death. Curr Opin Immunol 1998; 10:545-551.16. Manilay JO, Sykes M. Natural killer cells and their rolein graft rejection. Curr Opin Immunol 1998; 10: 523-538.17. Libby P. Transplantation-associated arteriosclerosis:potential mechanisms. En: Tilney NL, Strom TB, Paul LC,eds. Transplantation biology: cellular and molecular as-pects. Philadelphia: Lippincott-Raven, 1996; 577-586.18. Kawai T, Sachs DH, Cosimi AB. Tolerance to vasculari-zed organ allografts in large-animal models. Curr Opin Im-munol 1999; 11: 516-520.19. Auchincloss H, Sachs DH. Xenogeneic transplantation.Annu Rev Immunol 1998; 16: 433-470.20. Sandrin MS, McKenzie IFC. Recent advances in xeno-transplantation. Curr Opin Immunol 1999; 11: 527-531.21. Patiente C, Takeuchi Y, Weiss RA. Zoonosis in xeno-trasplantation. Curr Opin Immunol 1998; 10: 539-542.22. Isacson O, Breakefield XO. Benefits and risks of hos-ting animal cells in the human. Nat Med 1997; 3: 964-969.
1349
RECHAZO DE TRASPLANTES

Conexiones entre la inmunidad natural y las respuestas inmunes adquiridas
E. Reyes Martína J. Monserrat Sanzb E. San Antonio Sánchez E. San Antonio Sánchezb A. Prieto Martínb aDepartamento de I+D. Industrial Farmacéutica Cantabria. bDepartamento de Medicina. Universidad de Alcalá.
Introducción
El sistema inmune está involucrado en la sofisticada tarea biológica de distinguir lo propio de lo no propio. Esta discriminación, necesaria para que el huésped controle la invasión, colonización y daño tisular causados por microorganismos patógenos, está mediada por una gran variedad de mecanismos. Existen mecanismos no clonales o innatos, y clonales o adaptativos (fig. 1). Los primeros, no clonales, constituyen una resistencia entendida como no antígeno específica. Es la primera que se desarrolla, inmediatamente o en pocas horas, y está formada por varias líneas de defensa. La primera línea es la formada por barreras físicas (epitelios, ácidos grasos), factores solubles (sistema de complemento) y citoquinas (incluidas las quimioquinas)1. La segunda línea de defensa está constituida por las características de los microorganismos invasores que permiten diferenciarlos de los distintos componentes propios del huésped (la presencia de formil-metionil-leucil-fenilalanina [fMLP], ARN de cadena doble, lipopoliacárido [LPS] y otros componentes de paredes bacterianas) (fig. 2).
Fig. 1. Mecanismos no clonales o innatos, y clonales o adaptativos.

Fig. 2. Dependencia en el desencadenamiento y regulación de los mecanismos de defensa clonales y no clonales. NK: naturalkiller; DC: células dendríticas.
Los mecanismos clonales de defensa, en los que están involucradas las células T y B, no se pueden comprender sin los conceptos de resistencia específica a la infección y la inmunidad adaptativa. La iniciación de una respuesta inmune adaptativa, comienza con el procesamiento y presentación del antígeno, necesario para el reconocimiento del mismo por los linfocitos T. La expansión clonal de linfocitos que reconocen específicamente el antígeno conduce a la adquisición de funciones efectoras1.
Los mecanismos de defensa, clonales y no clonales, son dependientes entre sí, en su desencadenamiento y en su regulación, existiendo por lo tanto, múltiples conexiones entre la inmunidad natural y las respuestas inmunes adquiridas (fig. 2). Así, mecanismos innatos como la activación del complemento contribuyen a la discriminación entre lo propio y lo extraño, mientras que los anticuerpos, resultado de la respuesta clonal de células B, desencadenan la fijación de complemento y la fagocitosis que son mecanismos de defensa no adaptativos.
Fagocitosis y presentación antigénica
Cuando un patógeno supera las barreras físicas de defensa del huésped y elude las respuestas inmunes no clonales (fig. 2), el huésped puede desarrollar estrategias de actuación contra el patógeno. Estas estrategias pueden estar dirigidas contra el patógeno en su forma extracelular y sus productos de secreción, o, contra el patógeno que permanece dentro de las células del huésped.
Existe una clara desventaja numérica entre la cantidad de linfocitos con capacidad de reconocer el antígeno y responder de forma clonal y los patógenos que deben eliminar o aclarar, que poseen, además, una gran capacidad de replicación. De ahí, que el control de la homeostasis en el número de linfocitos, independiente en células T y B, es de vital importancia para el huésped2.

Los linfocitos B son capaces de suavizar esta desventaja mediante la secreción de formas solubles de inmunoglobulinas generadas por mutación somática o selección durante su diferenciación en los centros germinales. Sin embargo, esta estrategia es efectiva en su inicio a nivel extracelular (crecimiento del patógeno o desplazamiento del mismo por el huésped).
En la mayoría de los casos, el sistema permanece ignorante ante la presencia de patógenos hasta que moléculas del patógeno son expuestas al medio y reconocidas por inmunoglobulinas solubles y de superficie. Las inmunoglobulinas tienen por lo tanto una gran eficiencia inhibiendo nuevas infecciones; sin embargo, esta estrategia fracasa cuando la replicación vírica está muy avanzada en el momento en que es localizada una molécula de su superficie por las inmunoglobulinas o cuan do va dirigido contra parásitos intracelulares.
Dadas estas limitaciones en la actuación por parte de las células B, deben coexistir formas de identificación de patógenos presentes en el interior celular (citosol o endosomas) preferiblemente antes de que se produzca la replicación de estos patógenos3. El sistema inmune corta las moléculas de los patógenos invasores en pequeñas piezas que son mostradas en la superficie celular unidas a moléculas de histocompatibilidad (MHC) de clase I o II4. El reconocimiento por parte de las células T de péptidos en el contexto de MHC5, conlleva a la solución de este problema.
Todos los organismos contienen y producen una o varias proteínas que son únicas de especie. Pequeños fragmentos de estas proteínas son generados por proteolisis, capturados por MHC y expresados en la superficie celular mostrando capacidad de interacción con células T, participando en la homeostasis en el número de células T novatas y memoria2. En este contexto, la presencia de moléculas de histocompatibilidad vacías (sin péptido) supondría una distracción para el sistema, pudiendo inhibir un reconocimiento eficiente por parte de las células T. Por lo tanto, las células necesitan mecanismos que limiten la presencia de moléculas de histocompatibilidad vacías en la superficie de células presentadoras de antígeno (APC)6.
El reconocimiento de péptidos tiene una ventaja adicional como estrategia en la detección de patógenos, ya que las proteínas pueden variar evolutivamente en secuencias no imprescindibles para llevar a cabo su función. Sin embargo, el núcleo de estas proteínas (core) se mantiene con relativa constancia en el tiempo.
El complemento
El sistema de complemento es vital en la inmunidad no clonal, pero también desempeña una importante función en la discriminación de lo propio y extraño así como en la regulación positiva de mecanismos clonales7. Este sistema, distribuido entre el plasma sanguíneo y la superficie celular, consiste en un complejo conjunto de proteínas bioactivas (enzimas proteolíticas, proteínas inflamatorias, receptores de superficie y proteínas con capacidad de provocar lisis celular) que se caracteriza por una estrecha regulación8 (fig. 3). Este sistema puede reconocer y unirse directamente a la superficie de las bacterias y los hongos. El sistema de complemento también puede interactuar con la inmunidad clonal, complementando literalmente procesos de muerte celular y retirada de microorganismos reconocidos por anticuerpos. Las anafilotoxinas (C3a y C5a), fragmentos ligeros resultantes de la hidrólisis de C3 y C5, tienen función proinflamatoria y quimioatrayente (fig. 4). Estos mediadores están relacionados con importantes procesos que regulan el delicado balance entre los efectos beneficiosos y adversos de la inflamación9 que aparecen en numerosas patologías (artritis reumatoide, lupus eritematoso sistémico, psoriasis, glomerulonefritis y vasculitis entre otras)10 (fig. 4). C3a se une a receptores presentes en basófilos y mastocitos, dando como consecuencia la generación de histamina y otros mediadores anafilácticos. C5a interviene como factor quimioatrayente para neutrófilos y monocitos, induciendo la secreción de mediadores inflamatorios como el factor de necrosis tumoral alfa (TNF*) por estas células y su receptor (CD88), modulado negativamente por la presencia de TNF*, y media en la defensa a las infecciones vía mucosas11.
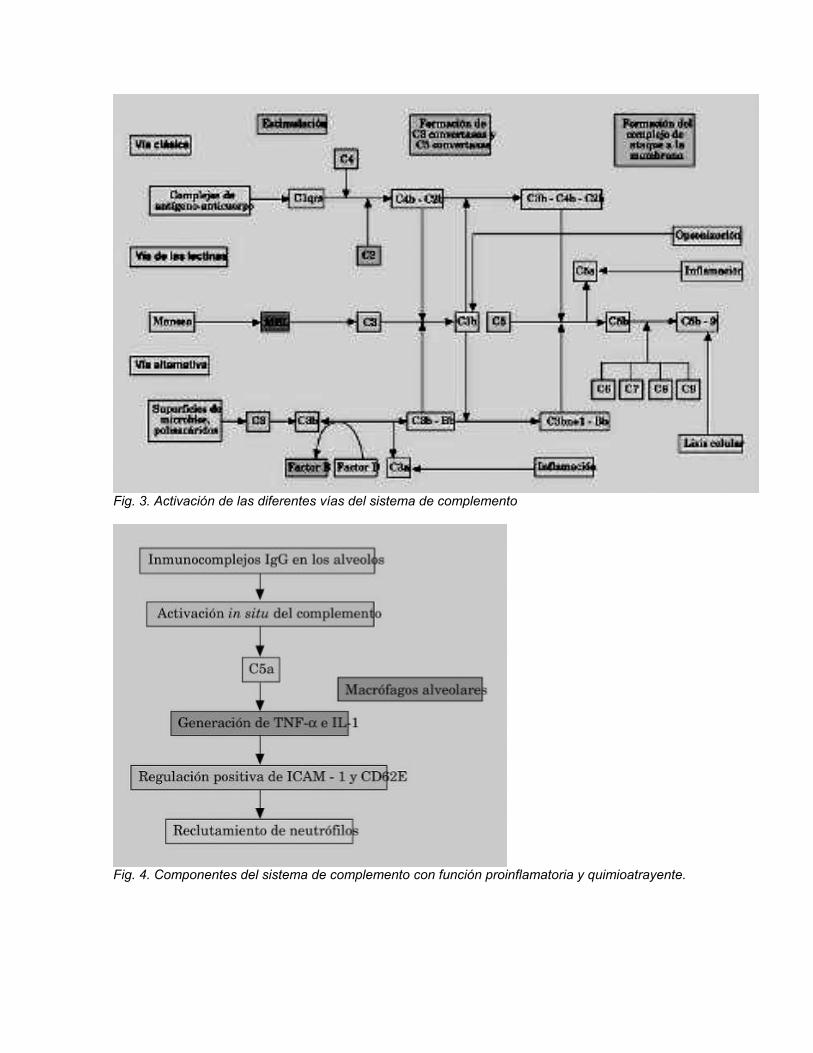
Fig. 3. Activación de las diferentes vías del sistema de complemento
Fig. 4. Componentes del sistema de complemento con función proinflamatoria y quimioatrayente.

Las principales funciones del sistema de complemento son tres: a) recubrir los patógenos para promover su fagocitosis (opsonización), b) iniciar respuestas inflamatorias mediante la interacción con el músculo liso (contracción), vasodilatación y atracción de leucocitos, y c) lisar patógenos y células infectadas mediante la formación de poros en sus membranas.
Existen tres vías de activación diferentes que culminan en la activación del C3: la vía clásica, la vía de las lectinas y la vía alternativa. La vía clásica se inicia mediante la formación de complejos de antígeno-anticuerpo con IgM o IgG. La vía de las lectinas se inicia por mecanismos anticuerpo independientes mediados por lectinas que unen manosa (MBL) (fig. 3). La vía alternativa, evolutivamente más antigua, puede ser activada por estímulos no específicos (lipopolisacáridos bacterianos) o por complejos antígeno-anticuerpo. Las vías clásica y alternativa no son independientes en su regulación, ya que fragmentos provenientes de la vía clásica (C4b2a) pueden activar de forma potente en presencia de factor D, la formación de convertasa (C3bBb).
Como consecuencia de la activación de las diferentes vías del sistema de complemento, C3 da lugar a C3b por mediación de C3 convertasas. C3b es una molécula importante, ya que es reconocido por receptores presentes en diferentes tipos celulares incluidos los macrófagos (esencial para desencadenar una actividad fagocítica) y las células B. C3 también es necesario para la formación de C5 convertasas que desencadenan la polimerización de complejos de ataque a la membrana celular que causará la lisis celular.
El proceso de activación de la cascada de complemento está estrechamente regulado (tabla 1). Alteraciones en el sistema de complemento o en su regulación, se pueden manifestar con un amplio espectro de patologías (angioedema hereditario o hemoglobinuria paroxística nocturna).
Rutas de procesamiento antigénico
La localización de los patógenos deriva en diferentes estrategias para su aclaramie nto definitivo (tabla 2). Todos los virus y ciertas bacterias se replican en el citosol.

Sus antígenos son presentados en moléculas de histocompatibilidad de clase I a los linfocitos T CD8+. Algunas bacterias y algunos parásitos, son fagocitados y localizados en endosomas. Sus antígenos son presentados en el contexto de moléculas de histocompatibilidad de clase II a los linfocitos CD4+. Sin embargo, las proteínas derivadas de patógenos extracelulares pueden localizarse en vesículas endocíticas tras su unión a receptores de la superficie celular.
En el sistema principal de histocompatibilidad radica la base molecular de la discriminación por parte de los linfocitos T de lo propio de lo no propio1. Existen dos clases de moléculas de histocompatibilidad generadas por los genes HLA (HLA-A, -B, -C, -DR, -DQ y -DP), que tienen una estructura tridimensional similar. Las moléculas de clase I son heterodímeros, formados por una glucoproteína tipo I, muy polimórfica, con tres dominios de inmunoglobulina (*1, *2 y *3) de 43 Kda, codificada en el cromosoma 6, en la región de MHC y una cadena monomórfica no anclada en la membrana citoplasmática (ß2 microglobulina) de 12 Kda unida a la anterior de forma no covalente y codificada en el cromosoma 15. Las moléculas de histocompatibilidad de clase II están constituidas por dos proteínas tipo I unidas de forma no covalente (* y ß) de 34 y 29 KDa respectivamente. Las moléculas de clase I se expresan en todas las células nucleadas del organismo. Sin embargo, las MHC clase II se expresan en células T, B, macrófagos, otras células presentadoras de antígeno y células epiteliales del timo.
La degradación de las proteínas presentes en el citosol se realiza mediante un complejo sistema denominado proteosoma. Este sistema, evolutivamente muy conservado, consta de 28 unidades organizadas en cuatro anillos de siete unidades cada uno. Los péptidos que unen las moléculas de histocompatibilidad de clase I, algunos de ellos inducidos por la presencia de interferón gamma (IFN*) secretado en respuestas no clonales12, son transportados activamente al retículo endoplásmico mediante un sistema de transportadores asociados con el procesamiento antigénico (TAP) constituido por un heterodímero (TAP-1 y TAP-2)6,13.
El ensamblaje de moléculas de histocompatibilidad de clase I es secuencial y dependiente de péptidos con actividad transportadora y plegadora de proteínas (chaperonas). La cadena * de las moléculas de histocompatibilidad de clase I, antes de unirse definitivamente a la ß2 microglobulina, se une en primer lugar a una proteína chaperona denominada calnexina. Tras el ensamblaje entre la cadena * y ß2 microglobulina, se forma un complejo en el que intervienen otras chaperonas denominadas calreticulina y tapasina. La primera estabiliza las MHC de clase I, mientras que la segunda media en su interacción con TAP-1 del sistema TAP. Los péptidos generados en el citosol por el proteosoma atraviesan la membrana del retículo endoplásmico mediante el sistema TAP y se sitúan en el complejo formado por la cadena * y la ß2 microglobulina.

Las moléculas de histocompatibilidad de clase I no abandonan el retículo endoplásmico hacia la superficie celular, hasta que no tienen unido un péptido antigénico (fig. 5A)6,13.
Fig. 5. Vías de procesamiento antigénico.
Los péptidos presentados en moléculas de MHC de clase II son generados en vesículas endocíticas (fig. 5B). La presencia de la cadena invariante evita que el sitio de unión para los péptidos antigénicos sea ocupado por péptidos presentes en el retículo endoplásmico. La cadena invariante es degradada cuando las MHC de clase II se incorporan a vesículas de exocitosis, manteniéndose tan sólo una pequeña región (CLIP) que ocupa el lugar del fragmento peptídico que se presentará. La fusión entre las vesículas endocíticas y las vesículas de transporte da lugar al desalojo de la región CLIP con la ayuda de moléculas HLA-DM, fomentando la unión irreversible de péptidos presentes en las vesículas endocíticas14.
Existe una presentación antigénica no dependiente del sistema MHC. Existen algunas moléculas que son capaces de presentar sustancias antigénicas no peptídicas al 5% 10% de los linfocitos T circulantes: linfocitos CD3+CD4CD8. Son las moléculas CD115,16, que presentan ácido micólico y lipoarabinomananos17. Existen cinco isotipos diferentes que están presentes en la mayoría de las células presentadoras de antígeno pero a diferencia de las MHC de clase I y II, no son polimórficas. Ha sido propuesto como un mecanismo para potenciar la capacidad del sistema inmune en el reconocimiento de antígenos bacterianos de naturaleza lipídica.
Como ya hemos mencionado, la localización del patógeno deriva en diferentes estrategias para su aclaramiento definitivo. De forma paralela, muchos virus son capaces de eludir estas estrategias. Así, existen virus con capacidad de modular de forma negativa la expresión de MHC de clase I a nivel génico o afectando al sistema TAP o a la formación del proteosoma12.
Células presentadoras de antígeno
Las APC profesionales son células accesorias (caracterizadas por la presencia de receptores Fc) (tabla 3), que están especializadas en la captación de antígenos, así como en su procesamiento y su presentación a los linfocitos T. Las APC profesionales están concentradas en los órganos linfoides periféricos, lugares donde se produce la interacción inicial entre los linfocitos novatos y los antígenos. Los patógenos en los tejidos son recogidos y atrapados en los ganglios que los drenan. Los que entran en la sangre son atrapados en el bazo, los que infectan la superficie de las mucosas se atrapan en las placas de Peyer o en las amígdalas.
Los tres tipos principales de APC profesionales en los órganos linfoides periféricos son los macrófagos, las células dendríticas y las células B (tabla 3). Cada tipo de APC está especializado en procesar y presentar antígenos de distinta procedencia. Así, los macrófagos se encuentran en todas las áreas del ganglio e ingieren microbios y antígenos particulados. Las células dendríticas están presentes en las áreas T y son especialmente importantes en la presentación de antígenos víricos. Y por último, las células B se encuentran en los folículos linfoides y son especialmente eficientes atrapando antígenos solubles, como las toxinas bacterianas, a través de sus inmunoglobulinas de superficie. Los macrófagos y las células B, además de ser APC, también son las células efectoras sobre las que ejercerán su acción cooperadora los dos tipos de linfocitos CD4 efectores: los linfocitos T proinflamatorios y los linfocitos cooperadores.
La alarma de un proceso lesivo sea infeccioso o no para un tejido es dada por células parenquimatosas del tejido que producen quimiocinas que atraen a las células inflamatorias. Primero, leucocitos polimorfonucleares y después mononucleares. Los monocitos que se extravasan expresan receptores para complemento, inmunoglobulinas, receptores de lectinas con los que captan antígenos. Los monocitos extravasados, en 24 horas en presencia de GM-CSF se transforman en células dendríticas inmaduras. Si están infectadas aumentan la expresión de MHC

de clase I (reconocidas por linfocitos CD8) pero no son capaces todavía de estimular a los linfocitos T CD4. Si no están infectadas pierden la capacidad de captar antígeno y adquieren la de presentarlo en MHC de clase II. Expresan hasta un millón de moléculas de clase II y moléculas coestimuladoras. La vida media de las moléculas de clase II se alarga a cientos de horas.
Las células dentríticas expresan diferentes receptores de quimiocinas secuencialmente durante la activación de modo que primero son atraídas por los focos inflamatorios y después se liberan para ir a los ganglios. El número de receptores CCR1, CCR2a y CCR2b (de tropismo a tejidos inflamados) baja y además pierden capacidad de respuesta, mientras que aumenta la expresión de CCR7 (de tropismo hacia ganglios); de este modo, las células dendríticas pueden abandonar el tejido para ir al ganglio.
Coestimulación y activación linfocitaria
Las APC dan a los linfocitos T las dos señales necesarias para su activación: la primera el antígeno en el contexto de MHC, y la segunda, señales coestimuladoras de membrana. De entre estas señales coestimuladoras en células APC destacan el CD80 y CD86, ligandos de los receptores de coestimulación CD28 y CD152 en linfocitos T. Las señales coestimuladoras dependientes de CD28 contribuyen al aumento de la transcripción del gen de IL-2 y también a la estabilización de su ARN. Los fármacos inmunosupresores como ciclosporina A y FK506 inhiben la producción de IL-2 al interferir las señales de activación provenientes del receptor del antígeno.
El reconocimiento antigénico en ausencia de las señales coestimuladoras no inducirá la diferenciación a linfocitos T efectores, de este modo se produce la tolerancia a los antígenos propios. Este mecanismo de seguridad previene la activación de los linfocitos con autoantígenos que podría provocar respuestas inmunes que dañarían los tejidos propios.
La proliferación y la diferenciación de los linfocitos novatos tras el encuentro antigénico también es dirigida por moléculas coestimuladoras solubles como la IL-2 e IL-15. Los linfocitos T tras la activación producen IL-2 y receptores de alta afinidad formados por tres cadenas (*, ß y *). La unión de IL-2 a su receptor estimula de modo autocrino la proliferación de los linfocitos durante varios días y posteriormente su diferenciación a células T efectoras capaces de responder a menores concentraciones del antígeno.
El reconocimiento antigénico tanto en determinadas condiciones de coestimulación (predominio de unión de CD152) como en ausencia de coestimulación puede producir alternativamente apoptosis o anergia. Estos procesos (anergia y apoptosis), inducidos por el reconocimiento antigénico, son de crucial importancia en el mantenimiento de la tolerancia a los antígenos propios, ya que no todos los linfocitos potencialmente autorreactivos son eliminados en el timo durante la selección tímica.
A diferencia de los procesos de anergia que son potencialmente reversibles, la apoptosis implica la muerte celular de los linfocitos que reconocen el antígeno por lo que es irreversible.
Los procesos de apoptosis eliminan los linfocitos innecesarios mientras los procesos de anergia mantienen linfocitos en un estado de no reactividad que requiere una inversión de nutrientes en células que no van a responder a la estimulación antigénica. Al ser la apoptosis un proceso mucho mas económico para el organismo. la permanencia de los linfocitos T en estado anérgico es un hecho que requiere una explicación funcional. Es posible que estas células anérgicas eviten respuestas inmunes a antígenos propios mimetizados por los patógenos.
Las células anérgicas se unirían a los complejos péptido imitador-MHC en la superficie de células presentadoras de antígeno sin responder a ellos, compitiendo con los linfocitos T potencialmente autorreactivos de la misma especificidad.

Los linfocitos T anérgicos podrían prevenir la activación accidental de otros linfocitos T autorreactivos por agentes infecciosos contribuyendo de un modo activo al mantenimiento de la tolerancia a los antígenos propios.
Células dendríticas
Las células dendríticas (DC) también denominadas células de Langerhans (LC) cuando se localizan en la piel18 son como ya hemos mencionado APC y desempeñan una función de gran importancia en el control de la inmunidad (tabla 4). Dada su escasez (0,1% al 1% de las células en sangre periférica y médula ósea) y su heterogeneidad, han sido poco estudiadas aunque su presencia es conocida desde 186819. Sin embargo, las DC son de gran importancia en la presentación antigénica a linfocitos T novatos18. Estas células expresan grandes cantidades de moléculas de clase II (HLA-DR, -DQ, -DP), de coestimulación (CD80, CD86) y de comunicación celular (CD40, TRANCE/RANK). Las DC inmaduras son incapaces de interactuar con los linfocitos T, pero tras la captación de antígenos, su activación y migración hacia órganos linfoides (bazo y ganglios) son capaces de inducir una fuerte activación de los linfocitos T19 (tabla 5).

Origen y linaje
Se definen inmunofenotípicamente como células CD3 CD14 CD19 CD56 pero HLADR+ pudiendo expresar o no el CD16. Las DC más frecuentes expresan CD16. Las DC CD16 (CD3 CD14 CD19 CD56 HLADR+ CD16) se pueden clasificar en dos tipos según sea su expresión de CD123 (una de las cadenas del receptor para IL-3) y CD33. Así, unas expresan un elevado número de moléculas de CD123 y bajo de CD33, mientras que otras expresan pocas moléculas de CD123 y un elevados número de moléculas de CD33 (fig. 6).
Fig. 6. Las DC pueden ser identificadas mediante marcadores de superficie NK: natural killer.

Para la identificación de estos tipos celulares, se plantean protocolos de citometría de flujo de adquisición de células raras. En primer lugar, se adquieren todas las células, en segundo lugar todas las células HLADR+, dentro de las cuales se identifican las células CD123 y CD33. Por último, se identifican las células CD16+.
Los diferentes tipos de DC se caracterizan por mostrar una diferente capacidad de producir citocinas, diferente actividad fagocítica y oxidativa. Así, las células CD123 no producen citocinas y poseen una escasa capacidad fagocítica y oxidativa. Las DC CD33+ producen TNF, y poseen también una escasa capacidad fagocítica y oxidativa. Gran parte de las células CD16+ producen gran cantidad de TNF, son las que mayor capacidad fagocítica muestran aunque su capacidad de generar radicales oxidantes es inferior a la de los monocitos/macrófagos.
Potencial de las DC en la inmunoterapia contra el cáncer
Dado el papel inmunoestimulador de las DC sobre los linfocitos T, es lógico pensar utilizar este tipo celular para modular respuestas inmunes en las que los linfocitos T están involucrados: trasplantes, alergias, enfermedades autoinmunes, resistencia a la infección y tumores, inmunodeficiencias y vacunas18,19. La idea de poder expandir las DC in vitro es realmente estimulante dado el gran número de aplicaciones clínicas que hemos mencionado. Se han planteado estrategias contra linfomas humanos o el virus del papiloma humano (HPV-60) en modelos murinos. Las estrategias antitumorales basadas en las DC se plantean en cuatro pasos secuenciales: aislamiento de precusores inmaduros, crecimiento y diferenciación de DC inmaduras, pulsación con antígenos in vitro y reinfusión.
Parece haber numerosas vías para generar DC si existe la apropiada combinación de citocinas19. Para llevar a cabo el aislamiento de precursores inmaduros, las células CD34+ de sangre periférica se movilizan mediante el uso de citocinas (GM-CSF, G-CSF, IL-3) o combinaciones de ellas (G-CSF + IL-3). Recordemos que las células CD34+ pueden dar lugar a dos poblaciones diferentes de DC: las LC (DC epidérmicas) y las dérmicas o DC intersticiales que inducen a las células B a producir anticuerpos20. También ha sido descrito que las DC pueden derivar directamente desde los monocitos18. Estas células pueden seleccionarse mediante técnicas de separación inmunomagnéticas. La diferenciación in vitro se realiza con 50.000 células /ml en cultivo de siete a catorce días en presencia de GM-CSF, TNF-* o IL-4. La tercera fase o pulsación con antígenos se puede realizar con antígenos (purificados o de síntesis), con chaperonas (hsp70), con células apoptóticas o con lisados tumorales. Por último, la reinfusión se puede realizar por vía intravenosa, vía subcutánea (desde donde migrarán a los ganglios), por punción directa en ganglios accesibles por ecografia o por punción intratumoral.
Ciencia básica para la medicina clínica
En general, el sistema inmune debe ser considerado como un conjunto de me canismos cuya finalidad es la defensa del huésped ante un universo de potenciales patógenos. Estos mecanismos están basados en un delicado equilibrio adquirido evolutivamente entre la protección y la propia destrucción. Defectos en la regulación del sistema inmune producen desórdenes clínicos que en ocasiones contribuyen a esclarecer estos mecanismos. Los avances en la etiopatogenia y el diagnóstico de estos desórdenes repercutirán en la medicina clínica del futuro.

Referencias Bibliográficas: 1. Huston DP The biology of the immune system. JAMA 1997, 278: 1.804-1.814. 2. Goldrath AW, Bevan MJ Selecting and maintaining a diverse T-cell repertoire. Nature 1999, 402: 255-262. 3. Jones SL, Lindberg FP, Brown EJ Phagocytosis En: Paul WE, ed. Fundamental Immunology (4th ed). Philadelphia: Lippincott-Raven, 1999. 4. Wilson IA Another Twist to MHC-Peptide recognition. Science 1996; 272: 973-974. 5. Zinkernagel RM, Doherty PC Restriction of in vitro T cellmediated cytotoxicity in lymphocytic choriomeningitis within a syngeneic or semiallogeneic system. Nature 1974, 248: 701-710. 6. Germain RN Antigen Processing and Presentation. En: Paul WE, ed. Fundamental Immunology (4th ed). Philadelphia: Lippincott-Raven, 1999. 7. Peter H Immunology: A complement to host defense. Nature 1996; 383:25-26. 8. Frank MM, Fries LF The role of complement in inflammation and phagocytosis. Immunol Today 1991; 12: 322-326. 9. Colten HR, Krause JE Pulmonary inflammation A balancing act. N Engl J Med 1997; 336: 1.094-1.097. 10. Ward PA Recruitment of inflammatory cells into lung: Rolesof cytokines, adhesion molecules, and complement. J of Laboratory and Clinical Medicine 1997; 192: 400- 404. 11. Hopken UE, Lu B, Gerard NP, Gerard C The C5a chemoattractant receptor mediates mucosal defense to infection. Nature 1996, 383: 86-89. 12. Ploegh HL Viral strategies of immune evasion. Science 1998; 280: 248-253. 13. Janeway CA Antigen recognition by T lymphocytes in Immuno biology. The immune system in health and disease. (4th ed) Current Biology publications, New York: Garland publishing: 1999. 14. Lanzavecchia A, Reid PA, Watts C Irreversible association of peptides with class II MHC molecules in living cells. Nature 1992; 357: 249-252. 15. Zeng ZH, Castano AR, Segelke B, Stura EA, Peterson PA, Wilson IA The crystal structure of murine CD1: and MHC-like fold with a large hydrophobic binding groove. Science 1997; 277: 339-345. 16. Wilson IA, Bjorkman PJ Unusual MHC-like molecules: CD1, Fc receptor, the hemochromatosis gene product, and viral homologs. Curr Opin Immunol 1998; 10: 67-73. 17. Sieling PA, Chatterjee D, Porcelli SA, Prigozy TI, Mazzaccaro RJ, Soriano T, et al CD1-restricted T cell recognition of microbial lypoglycan antigens. Science 1995; 269: 227-230. 18. Reid CDL The dendritics cell lineage in haemopoiesis. British J Haematol 1997; 96: 217-223. 19. Banchereau J, Steinman RM Dendritics cells and the control of immunity. Nature 1998; 392: 245-252. 20. Claux C CD34 sup + hematopoietic progenitors from human cord blood diferénciate along two independent dendritic cell pathway in response to GM-CSF + TNFalpha: II Functional analysis. Blood 1997; 90: 1.458-1.470

1361
IntroducciónLas crioglobulinas son inmunoglobulinasque precipitan de manera reversible a tem-peraturas bajas (inferiores a 37º C, redi-solviéndose con el calor). Su presencia enla sangre se denomina crioglobulinemia ypuede provocar fenómenos de hipervis-cosidad sanguínea, vasculíticos y por de-pósitos de inmunocomplejos.
ClasificaciónEn 1974, Brouet et al establecieron unaclasificación de las crioglobulinemias queaún sigue vigente, en función del tipo deinmunoglobulina (tabla 1).
Crioglobulinemia de tipo I
Está constituida por una única inmuno-globulina monoclonal, generalmente IgGo IgM (sin actividad factor reumatoide).Son más frecuentes las crioglobulinas IgGque las IgM.Las crioglobulinemias de tipo I se asociana discrasias de células plasmáticas, comoel mieloma múltiple y enfermedad de Wäl-denstrom, así como a síndromes linfo-proliferativos.Aparecen manifestaciones clínicas apro-ximadamente en la mitad de los casos, se-cundarias a hiperviscosidad y al depósitode agregados de inmuglobulinas, lo queda lugar a fenómenos vasooclusivos. Nosuelen presentar enfermedad por inmu-nocomplejos (con depósito de comple-mento y vasculitis) ni muestran lesioneshistológicas características en la biopsia.El hallazgo histológico más frecuente es ladetección de precipitados de crioglobuli-nas en el interior de los vasos dérmicos,
pulmonares, cerebrales y glomérulos re-nales. Las manifestaciones clínicas másfrecuentes consisten en fenómeno de Ray-naud, urticaria inducida por frío, úlcerasisquémicas y gangrena distal por oclusiónde pequeños vasos, hemorragias retinia-nas, trastornos visuales, cefalea y encefa-lopatía por afectación de la microcircula-ción del sistema nervioso central.Las crioglobulinas de tipo I suelen estarpresentes en grandes cantidades en sue-ro (1-5 gr/dl), por lo que se detectan confacilidad como componentes monoclona-les en la electroforesis, bien en suero com-pleto o en el crioprecipitado aislado.
Crioglobulinemia mixta (tipos II y III)
La crioglobulinemia tipo II está constitui-da por IgG policlonal e IgM monoclonalcon actividad factor reumatoide, la crio-globulinemia tipo III por IgG policlonal eIgM policlonal con actividad factor reu-matoide. Las crioglobulinemias de tipos IIy III, al estar formadas por dos clases di-ferentes de inmunoglobulinas, se englo-
ban bajo el término común de crioglobu-linemia mixta (CM). Las crioglobulinas ti-pos II y III son a menudo difíciles de de-tectar (a diferencia de las tipo I), ya queprecipitan muy lentamente y suelen estarpresentes en pequeñas cantidades en sue-ro (50-500 mg/dl). La mayor parte de lasCM se asocian a diversas patologías, prin-cipalmente enfermedades autoinmunes,infecciones y procesos linfoproliferativos.
Medicine 2000; 8(26): 1361-1362
INDICACIONESE INTERPRETACIÓN CLÍNICA
DE LAS CRIOGLOBULINASS. Diz Fariña, A. Ruedas López y J.L. Patier de la Peña
Sevicio de Medicina Interna. Hospital Universitario Ramón y Cajal. Departamento de Medicina. Universidad de Alcalá.
TABLA 1Características diferenciales entre crioglobulinemia tipo I y crioglobulinemias mixtas (tipos II y III)
Tipo I Tipos II y III
Composición Ig Única (IgG o IgM) Mixta (IgG+IgM)
Principal etiología Neoplasia hematológica Infección VHC
Mecanismo etiopatogénico Hiperviscosidad. Depósito Depósito de Inmunocomplejosagregados Ig y complemento
Histología Inespecífica Específica
Principales órganos afectados Piel SNC PielArticulacionesRiñónSNP/SNCHígado
Detección en suero Grandes cantidades (1-5g/dl) Pequeñas cantidades (0,5-0,05 g/dl)
Actividad factor reumatoide No Sí
VHC: virus de la hepatitis C; SNP: sistema nervioso periférico; SNC: sistema nervioso central
Tabla 2Principales enfermedades asociadas a la
crioglobulinemia mixta
InfeccionesVíricas (VHC, VHB, VHA, CMV, VEB)Bacterianas (sífilis, lepra, glomerulonefritispostestreptocócica)Parasitarias (esquistosomiasis, paludismo, kala-azar)
Enfermedades autoinmunesLES, AR, PAN, Esclerosis múltiple, Síndrome de.Sjögren
Trastornos linfoproliferativosMacroglobulinemia, LLC, Linfoma
VHC: virus hepatitis C; VHB: virus de la hepatitis B; VHA: virus de lahepatitis A; CMV: citomegalovirus; VEB: virus de Epstein Barr. LES:lupus eritematoso sistémico, AR: artritis reumatoide; PAN: panarteri-tis nodosa. LLC: leucemia linfática crónica.

No obstante, en un 30% de los casos nose encuentra patología asociada, por lo quese utiliza el término de crioglobulinemiamixta esencial (tabla 2).Cada vez son más numerosos los estudiosque establecen como principal causa deCM la infección crónica por el virus de lahepatitis C (VHC). Se sabe que la CM estápresente en aproximadamente la mitad delos casos de hepatopatía por VHC y se hademostrado la presencia de anticuerposanti-VHC y del ARN del virus en más del80% de los casos de CM, así como la deestos anticuerpos frente al virus forman-
do parte de los complejos inmunes pre-sentes en el crioprecipitado.Las manifestaciones clínicas más fre-cuentes que aparecen en la CM son ar-tralgias, debilidad muscular, neuropatíaperiférica, púrpura palpable (síntoma másfrecuente) y afectación renal. Se han des-crito también hemorragia alveolar, fiebre,hepatoesplenomegalia e isquemia intesti-nal y encefalopatía difusa. Estas manifes-taciones clínicas son debidas al depósitode inmunocomplejos circulantes en la pa-red de los vasos de pequeño y medianotamaño, iniciando una respuesta inflama-
toria que origina una vasculitis sistémica,afectándose vasos pequeños y medianosde múltiples órganos, con mayor frecuen-cia los de piel y riñón. La lesión cutáneatípica es la vasculitis leucocitoclástica.
ConclusionesLa crioglobulinemia es una enfermedadsistémica, cuya afectación multiorgánicaobliga, en numerosas ocasiones, a un tra-tamiento específico.La presencia de crioglobulinas en el sue-ro de un paciente no se asocia necesaria-mente con la presencia de manifestacio-nes clínicas, por lo que la determinaciónde crioglobulinas y la posterior interven-ción terapéutica debe realizarse en fun-ción de la clínica.Así, la investigación de crioglobulinas esobligada en todo paciente que presentemanifestaciones clínicas de vasculitis depequeño y mediano vaso (especialmenteen pacientes afectados de enfermedadeshematológicas o con infección crónica porVHC), así como en todas aquellas gam-mapatías monoclonales asociadas a ma-nifestaciones de síndrome de hipervisco-sidad plasmática, acrocianosis, livedoreticularis o fenómeno de Raynaud, pro-vocados o no por exposición al frío (fig. 1).La infección por VHC debe investigarse entoda CM (mediante ELISA de 3ª genera-ción y/o detección de ARN vírico)
BIBLIOGRAFÍA RECOMENDADA
Guillevin R, Lhote F, Gherardi R. The spectrum and treat-ment of virus-associated vasculitides. Curr Op in Rheuma-tol 1997;9:31-36Miescher P A, Huang Y-P and Izui S. Type II cryoglobuline-mia. Sem Hematol 1995; 32: 80-85. Wener M H, Johnson R J and Sasso E H. Hepatitis C virusand rheumatic disease. J Rheumatol 1996;23: 6.953-6.958Willson R A. Extrahepatic manifestations of Chronic viralhepatitis. Am J Gastroenterol 1997; 92: 4-15.
1362
ENFERMEDADES DEL SISTEMA INMUNE (II)
Manifestacionesclínicas de vasculitis
de pequeño ymediano vaso
Gammapatías monoclonales asociadas a: Síndrome de hiperviscosidad plasmática Acrocianosis Livedo reticularis Fenómeno de Raynaud
Cuantificación yanálisis de
componentes
Ig monoclonal única Crioglobulinemia tipo I
Determinación decrioglobulinas
Positivo Negativo
Ig policlonales Crioglobulinemia tipo III
IgM monoclonalIgG policlonal Crioglobulinemia tipo II
Fig. 1. Algoritmo diagnóstico de crioglobulinemias.

1363
PROTOCOLO DE ACTUACIÓN EN LA DIARREA DEL VIAJERO

Concepto
La sarcoidosis es una enfermedad granu-lomatosa sistémica de causa desconocidaque se presenta típicamente en adultos jó-venes y que afecta sobre todo al pulmón.Con frecuencia cursa con adenopatías hiliares bilaterales, infiltrado pulmonar, lesiones oculares y lesiones cutáneas, aunque también puede comprometer aotros ganglios linfáticos, hígado, bazo y otros órganos. Se establece el diagnós-tico cuando los hallazgos clínico-radiográ-ficos se apoyan en una evidencia histoló-gica de granulomas de células epitelioidesno caseificantes presentes en más de unórgano o sistema1,2.Aunque habitualmente existe depresión delas pruebas de hipersensibilidad cutánearetardada, en las lesiones de la enferme-dad hay un aumento de la respuesta delos linfocitos T cooperadores y tambiénpueden encontrarse datos de hiperactivi-dad de las células B.
EpidemiologíaLa sarcoidosis es más frecuente entre los20 y los 40 años, con un ligero predomi-nio del sexo femenino, aunque puede pre-sentarse a cualquier edad. Las estima-ciones sobre su prevalencia son muy variables y oscilan entre 0,2 y 64 ca-sos/100.000 habitantes, con máximos enlas poblaciones escandinava y afroameri-cana. En EE.UU. se ha estimado una inci-dencia anual ajustada a edad de 10,9 por100.000 para personas de raza blanca y3 veces superior para la raza negra3. Laincidencia de la enfermedad en España esdesconocida, un estudio mediante en-cuestas realizado en 1979 y referido a laprovincia de Barcelona obtuvo una tasa deincidencia anual de 1,2 casos por cada100.000 habitantes4.
Etiopatogenia
Aunque la causa de la enfermedad es des-conocida, se piensa que la sarcoidosis esel resultado de la exposición de un hués-ped genéticamente predispuesto a agen-tes ambientales específicos que se com-portan como antígenos.
Factores genéticos
Se han descrito familias con varios miem-bros afectados y concordancia en gemelosmonocigotos y parece ser que la herencia espoligénica. Los genes más investigados sonlos del complejo de histocompatibilidad prin-cipal (HLA) y los hallazgos varían según lospaíses, pero es posible que, dentro de cadaetnia, condicionen la forma clínica y pro-nóstico del paciente1,2.
Factores ambientales
Se considera que actúan como antígenospersistentes que son presentados a lin-focitos T específicos por células presen-tadoras. Se han propuesto agentes in-fecciosos (virus, micobacterias, borrelias,micoplasmas), agentes inorgánicos (alu-minio, zirconio, talco) y agentes orgáni-cos (polen de pino, tiza). Las principalesinvestigaciones se han centrado en la hipótesis infecciosa, en particular por micobacterias y se ha encontrado unamayor frecuencia de anticuerpos a mi-cobacterias en el suero de pacientes con sarcoidosis. La interpretación de es-tos hallazgos debe ser cautelosa, pues en la sarcoidosis existe una estimulaciónpoliclonal que puede dar lugar a anti-cuerpos contra antígenos comunes. Otros hallazgos más recientes que se refierena la presencia de ADN y ARN ribosomalde micobacterias en células obteni-das mediante lavado bronquial requie-ren confirmación5,6. En cualquier caso,debe recordarse que la sarcoidosis si-gue siendo un diagnóstico de exclusión,una vez que se han descartado otras causas de enfermedad granulomatosa (tabla 1)2.
1715
SARCOIDOSISJ.M. Martín SantosSección de Reumatología. Hospital Universitario del Río Hortega. Valladolid.
Medicine 2001; 8(33): 1715-1722
TABLA 1Clasificación etiológica de las enfermedades granulomatosas
Causa Enfermedad resultante
Agentes infecciosos
Micobacterias TuberculosisInfección por micobacterias atípicas
Hongos Histoplasmosis
Bacterias BrucelosisFiebre QInfección por ChlamydiaeTularemia
Espiroquetas Sífilis
Parásitos LeishmaniasisToxoplasmosis
Agentes ambientales u ocupacionales
Agentes orgánicos o inorgánicos Neumonitis por hipersensibilidad (bacterias, hongos, proteínas, isocianatos, etc...)
Enfermedad crónica por berilioEnfermedades crónicas por otros metales (titanio, aluminio, zirconio)TalcoNeumonitis por metotrexato
Otras condiciones
Neoplasia LinfomaGranulomas relacionados con tumor
Enfermedades autoinmunes Granulomatosis de WegenerCirrosis biliar primariaEnfermedad de Churg-Strauss
Otros Sarcoidosis
Adaptada de Newman LS, et a2.

La respuesta inflamatoria de la sarcoidosis
La mayoría de los estudios se han hechoen pulmón (fig. 1). En fases iniciales seproduce una acumulación de linfocitos Tcooperadores CD4+ y de monocito-ma-crófagos, en ambos casos procedentes dela circulación y derivados de proliferaciónlocal7. La formación de granulomas de-pende de la acción de citocinas derivadasde linfocitos T cooperadores con fenotipoTh1 (interleucina 2 [IL-2] e interferón[IFN]γ principalmente) y de citocinas de-rivadas de monocitos (IL-1, IL-6, factor de necrosis tumoral alfa [TNF-α], IL-15, IP-10, RANTES). En los casos en que seproduce progresión a fibrosis, ésta se de-be a factores secretados por macrófagos(TGF-β, PDGF y IGF-1) y también se ha re-lacionado con la transición de un fenoti-po linfocitario Th1 a un tipo Th2 (secretorde IL-4, una citocina con propiedades fi-brosantes conocidas, IL-5 e IL-10)8.
Anatomía patológicaLa lesión característica es el granulomasarcoide (fig. 2). En su forma típica, se tra-ta de granulomas de células epitelioidesno caseificantes de aspecto compacto ybien definido y de presentación múltiple
y en diferentes órganos. Constan de uncore integrado por células derivadas demacrófagos muy diferenciadas (células epi-telioides y células gigantes multinucleadasde tipo Langhans) y de linfocitos T CD4+.El citoplasma de las células gigantes pue-de contener inclusiones como los cuerposasteroideos y los cuerpos de Schaumann.En la periferia existe un anillo compuestopor células T CD4+, células T CD8+ y enfases más evolucionadas, también fibro-blastos y fibras de colágeno indicativas defibrosis.Los granulomas sarcoides son frecuentesen ganglios linfáticos (especialmente in-tratorácicos), pulmones, hígado, bazo ypiel. En el pulmón son más frecuentes enel intersticio, lo que los hace accesibles ala biopsia transbronquial.Los granulomas aislados en localizacionesatípicas pueden inducir a error y exigenconsiderar diagnósticos alternativos9. Des-de un punto de vista histológico tiene in-terés recordar que en presencia de ne-crosis o localización en espacios aéreosdebe sospecharse infección (tuberculosis,hongos) y descartarse mediante tincionesadecuadas. La presencia de pequeñas áre-as de necrosis fibrinoide en ausencia deinfiltrado polimorfonuclear no debe excluirel diagnóstico de sarcoidosis si el contex-to clínico lo sugiere9. Por otra parte, debeconsiderarse la posibilidad de una reac-
ción sarcoidal local secundaria a una ne-oplasia. Es el caso de los granulomas epi-telioides no caseificantes presentes en gan-glios linfáticos regionales de carcinomas,en hígado o bazo de pacientes con Hodg-kin y linfomas no Hodgkin y en el senode algunos tumores primarios como se-minomas y disgerminomas.
ClasificaciónEn la práctica resulta útil distinguir entreuna forma aguda o subaguda de sarcoi-dosis, que se desarrolla bruscamente enel curso de unas pocas semanas y que ha-bitualmente cursa con síntomas genera-les como fiebre, astenia, anorexia y pér-dida de peso; y una forma crónica, quese desarrolla de forma insidiosa en el cur-so de meses y que cursa principalmentecon síntomas respiratorios, en ausenciade fiebre. Esta última forma crónica es laforma más frecuente de sarcoidosis enEE.UU., donde la mayoría de los pacien-tes son afroamericanos. Dentro de las for-mas agudas se incluyen dos síndromes ca-racterísticos: el síndrome de Löfgren,caracterizado por la presencia de eritemanudoso, adenopatías hiliares bilaterales yartralgias, que es común en Europa y laforma más frecuente de presentación desarcoidosis en España10,11; y el síndromede Heerfordt, consistente en fiebre, tu-mefacción parotídea, uveítis anterior y parálisis facial. La diferenciación entre for-mas agudas y formas crónicas de sarcoi-dosis es útil porque las primeras suelenafectar a sujetos más jóvenes y por lo ge-neral tienen un carácter benigno con re-solución sin necesidad de tratamiento.Otras características diferenciales se re-cogen en la tabla 212.Mención aparte merecen las formas asin-tomáticas de sarcoidosis, habitualmentedetectadas en exámenes radiográficos detórax rutinarios y de frecuencia muy va-
1716
ENFERMEDADES DEL SISTEMA INMUNE (IX)
Macrófago
Linfocito TCD4 +
AcúmuloCD8 +
AcúmuloCD4 +
Fibrosis
IL-4
Granuloma
IL-2 IFN-γ
Th1 Th2
IL-12 +
IL-12
Fig. 1. Hipótesis patogénica dela sarcoidosis.
Fig. 2. Microfotografía de ganglio superficial que muestra elcaracterístico granuloma sarcoide.
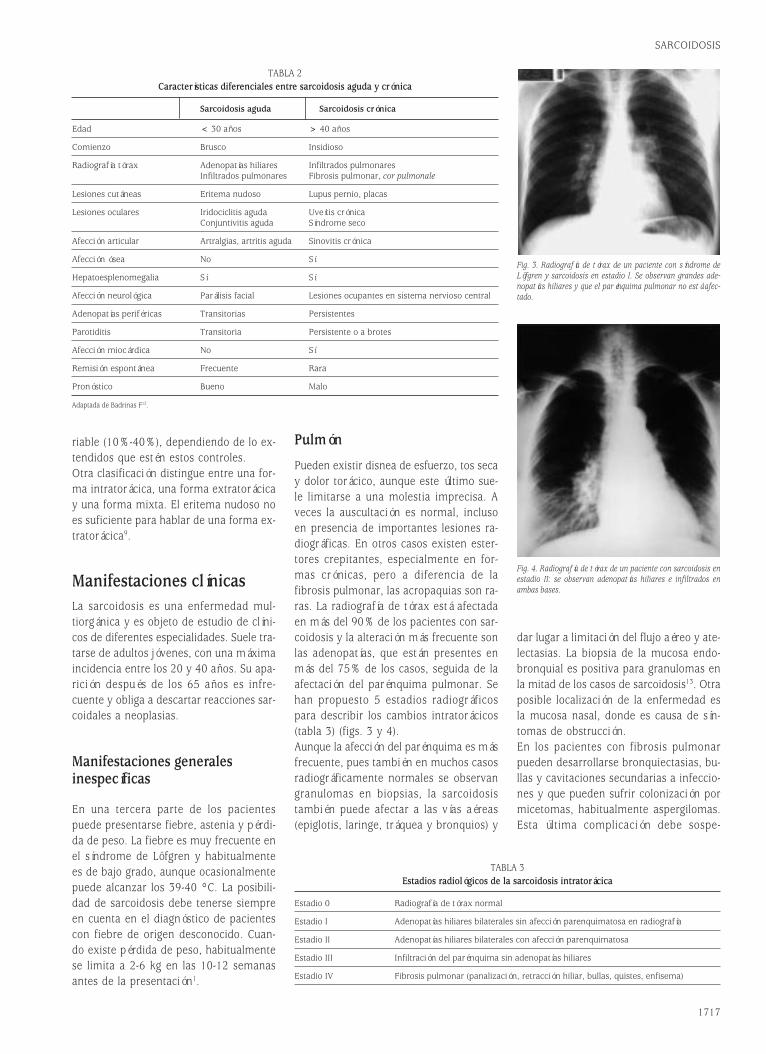
riable (10%-40%), dependiendo de lo ex-tendidos que estén estos controles.Otra clasificación distingue entre una for-ma intratorácica, una forma extratorácicay una forma mixta. El eritema nudoso noes suficiente para hablar de una forma ex-tratorácica9.
Manifestaciones clínicasLa sarcoidosis es una enfermedad mul-tiorgánica y es objeto de estudio de clíni-cos de diferentes especialidades. Suele tra-tarse de adultos jóvenes, con una máximaincidencia entre los 20 y 40 años. Su apa-rición después de los 65 años es infre-cuente y obliga a descartar reacciones sar-coidales a neoplasias.
Manifestaciones generalesinespecíficas
En una tercera parte de los pacientes puede presentarse fiebre, astenia y pérdi-da de peso. La fiebre es muy frecuente enel síndrome de Löfgren y habitualmentees de bajo grado, aunque ocasionalmentepuede alcanzar los 39-40 °C. La posibili-dad de sarcoidosis debe tenerse siempreen cuenta en el diagnóstico de pacientescon fiebre de origen desconocido. Cuan-do existe pérdida de peso, habitualmentese limita a 2-6 kg en las 10-12 semanasantes de la presentación1.
Pulmón
Pueden existir disnea de esfuerzo, tos secay dolor torácico, aunque este último sue-le limitarse a una molestia imprecisa. Aveces la auscultación es normal, inclusoen presencia de importantes lesiones ra-diográficas. En otros casos existen ester-tores crepitantes, especialmente en for-mas crónicas, pero a diferencia de lafibrosis pulmonar, las acropaquias son ra-ras. La radiografía de tórax está afectadaen más del 90% de los pacientes con sar-coidosis y la alteración más frecuente sonlas adenopatías, que están presentes enmás del 75% de los casos, seguida de laafectación del parénquima pulmonar. Sehan propuesto 5 estadios radiográficospara describir los cambios intratorácicos(tabla 3) (figs. 3 y 4).Aunque la afección del parénquima es másfrecuente, pues también en muchos casosradiográficamente normales se observangranulomas en biopsias, la sarcoidosistambién puede afectar a las vías aéreas(epiglotis, laringe, tráquea y bronquios) y
dar lugar a limitación del flujo aéreo y ate-lectasias. La biopsia de la mucosa endo-bronquial es positiva para granulomas enla mitad de los casos de sarcoidosis13. Otraposible localización de la enfermedad esla mucosa nasal, donde es causa de sín-tomas de obstrucción.En los pacientes con fibrosis pulmonarpueden desarrollarse bronquiectasias, bu-llas y cavitaciones secundarias a infeccio-nes y que pueden sufrir colonización pormicetomas, habitualmente aspergilomas.Esta última complicación debe sospe-
1717
SARCOIDOSIS
TABLA 2Características diferenciales entre sarcoidosis aguda y crónica
Sarcoidosis aguda Sarcoidosis crónica
Edad < 30 años > 40 años
Comienzo Brusco Insidioso
Radiografía tórax Adenopatías hiliares Infiltrados pulmonaresInfiltrados pulmonares Fibrosis pulmonar, cor pulmonale
Lesiones cutáneas Eritema nudoso Lupus pernio, placas
Lesiones oculares Iridociclitis aguda Uveítis crónicaConjuntivitis aguda Síndrome seco
Afección articular Artralgias, artritis aguda Sinovitis crónica
Afección ósea No Sí
Hepatoesplenomegalia Sí Sí
Afección neurológica Parálisis facial Lesiones ocupantes en sistema nervioso central
Adenopatías periféricas Transitorias Persistentes
Parotiditis Transitoria Persistente o a brotes
Afección miocárdica No Sí
Remisión espontánea Frecuente Rara
Pronóstico Bueno Malo
Adaptada de Badrinas F12.
TABLA 3Estadios radiológicos de la sarcoidosis intratorácica
Estadio 0 Radiografía de tórax normal
Estadio I Adenopatías hiliares bilaterales sin afección parenquimatosa en radiografía
Estadio II Adenopatías hiliares bilaterales con afección parenquimatosa
Estadio III Infiltración del parénquima sin adenopatías hiliares
Estadio IV Fibrosis pulmonar (panalización, retracción hiliar, bullas, quistes, enfisema)
Fig. 3. Radiografía de tórax de un paciente con síndrome deLöfgren y sarcoidosis en estadio I. Se observan grandes ade-nopatías hiliares y que el parénquima pulmonar no está afec-tado.
Fig. 4. Radiografía de tórax de un paciente con sarcoidosis enestadio II: se observan adenopatías hiliares e infiltrados enambas bases.

charse ante situaciones como hemoptisis.El neumotórax espontáneo es otra com-plicación infrecuente de la fibrosis. El de-rrame pleural es raro y obliga a descartarotras causas.
Sistema linfático
En promedio, se observa agrandamientode ganglios linfáticos intratorácicos en un 80% de los pacientes con sarcoidosis.Como se ha dicho, los más frecuente-mente afectados son los ganglios hiliaresde ambos lados. Incluso cuando tienengran tamaño (“ganglios patata”), suelenrespetar la ventana aireada normal queexiste entre el hilio, la arteria pulmonardescendente y el borde derecho de la silueta cardíaca (espacio de Chaperon), a diferencia de los linfomas14. En la ra-diografía convencional son también frecuentes las adenopatías paratraquea-les derechas y de la ventana aortopul-monar. Mediante tomografía axial com-putarizada (TAC) se aprecia que tambiénse afectan las paratraqueales izquier-das y en menor grado también las sub-carinales y las mediastínicas anteriores,estas últimas tradicionalmente conside-radas altamente sugestivas de linfoma(figs. 5 y 6).Un tercio de los pacientes con sarcoidosistienen adenopatías periféricas palpables.Son más frecuentes en las formas agudasque en las formas crónicas de la enfer-medad y suelen localizarse en el cuello (enparticular en los triángulos posteriores),
axilas y regiones epitroclear e inguinal. Ha-bitualmente se trata de ganglios agranda-dos que son móviles y no dolorosos y queno se ulceran. La esplenomegalia es in-frecuente, aunque puede dar lugar a sín-tomas por compresión local e incluso apancitopenia. Las adenopatías abdomina-les son peor conocidas y aparentementecon poca correlación con las endotoráci-cas15 (fig. 7).
Corazón
Clínicamente está afectado en un 5% delos pacientes con sarcoidosis, si bien elporcentaje en autopsias es mayor. Puedenpresentarse arritmias, bloqueos y sínco-pes. Además del electrocardiograma (ECG),cuando existe sospecha de compromisomiocárdico debe hacerse ecocardiogramapara descartar disfunción diastólica y gam-magrafía con 201Tl para detectar anorma-lidades de la contracción16. Se desconoceel significado de estas pruebas en pacien-tes asintomáticos. La biopsia endomio-
cárdica puede proporcionar granulomas ono hacerlo.
Hígado
Aunque la hepatomegalia sólo se apreciaen un 20% de los casos, son muy fre-cuentes leves aumentos de fosfatasa alca-lina y transaminasas, y se detectan gra-nulomas en las biopsias en más de lamitad de los pacientes, por lo que la biop-sia hepática tiene utilidad para el diag-nóstico en un contexto clínico adecuado.Ocasionalmente pueden presentarse sig-nos de obstrucción biliar intrahepática oictericia o signos de hipertensión portal,que son indicativos de mal pronóstico ypobre respuesta a corticoides.
Piel
La piel está afectada en un 25% de los pa-cientes. Son típicos el eritema nudoso y ellupus pernio. El eritema nudoso es unamanifestación de sarcoidosis aguda (sín-drome de Löfgren) muy frecuente en Eu-ropa y consiste en nódulos elevados, ro-jos y dolorosos en la cara anterior de laspiernas y con edema local (fig. 8). Histo-lógicamente se trata de una paniculitis sep-tal y no son típicos los granulomas. Lasarticulaciones adyacentes habitualmenteestán inflamadas y dolorosas. El eritemanudoso suele durar 6-8 semanas y es ra-ro que recidive después de 12 meses, algoque exigiría una biopsia de confirmaciónsi no se había hecho antes. El lupus per-nio es una forma de sarcoidosis crónicaque es más frecuente en mujeres afroa-mericanas y que consiste en placas indu-radas e hipopigmentadas que aparecen ennariz, mejillas, labios y orejas (partesacras). El lupus pernio se asocia con fre-cuencia a quistes óseos y fibrosis pulmo-nar. Existen también otras lesiones de sar-
1718
ENFERMEDADES DEL SISTEMA INMUNE (IX)
Fig. 5. Esquema que muestra la localización más frecuente de las adenopatías mediastínicas en la sarcoidosis modificada de Mo-rera Prat et al14.
Mediastino anterior 16%
Paratraquealderecha 71%
Hiliar97%
Subcarinal21% Mediastino
posterior 2%
Hiliar97%
Ventanaaorto-
pulmonar76%
Fig. 6. Tomografía axial computarizada de tórax que muestralas características adenopatías hiliares.
Fig. 7. Adenopatías retroperitoneales en una paciente con sar-coidosis crónica (biopsiadas).

coidosis crónica como nódulos, pápulas yplacas hipo o hiperpigmentadas (fig. 9).Característicamente las lesiones cutáneasde sarcoidosis crónica no son dolorosas nipruriginosas y no se ulceran. Histológica-mente muestran granulomas sarcoides tí-picos.
Lesiones oculares
La manifestación más frecuente es la uve-ítis anterior, que puede ser aguda o cró-nica. Las formas agudas pueden respon-der a corticoide tópico, pero las crónicasy los casos de uveítis posterior requierencorticoide por vía general para evitar elglaucoma. Con menos frecuencia la sar-coidosis crónica puede afectar también ala conjuntiva en forma de pequeños nó-dulos amarillentos o a las glándulas lacri-males y dar lugar a queratoconjuntivitisseca.
Neurosarcoidosis
El sistema nervioso está afectado en un5% de los pacientes, y en casi la mitad delos casos es una manifestación de co-mienzo. La manifestación más frecuentees la parálisis facial periférica. En otros casos se producen trastornos del hipotá-lamo o de la hipófisis o meningitis asép-tica. Su estudio exige TAC o, mejor, reso-nancia magnética (RM) con gadolinio,aunque las imágenes son inespecíficas. Elestudio de líquido cefaleorraquídeo (LCR)es importante, pues muy a menudo exis-te linfocitosis y aumento de proteínas ypermite descartar infecciones.
Sistema musculoesquelético
Las artralgias son comunes en la sarcoi-dosis. La artritis es menos frecuente, aun-que están descritas formas agudas y for-mas crónicas17. La (poli)artritis aguda enla mayoría de los casos se asocia a erite-ma nudoso y forma parte del síndromede Löfgren. En ausencia de eritema nu-doso se presenta en menos del 5% de lospacientes. En general, afecta a grandesarticulaciones (rodillas y tobillos), aunquepuede afectar a articulaciones de miem-bros superiores e incluso a las manos y alos talones. El grado de inflamación es va-riable, desde no existir ningún signo ob-jetivo a presentar importante tumefacciónperiarticular e incluso derrame sinovial.La biopsia sinovial sólo muestra infiltra-do mononuclear. El ataque suele durar po-cas semanas y rara vez recidiva. La(poli)artritis crónica es casi exclusiva dela raza negra y afecta tanto a grandescomo a pequeñas articulaciones y vainastendinosas. El líquido sinovial es de pre-dominio mononuclear y poco inflamato-rio, y el diagnóstico por lo general re-quiere confirmación de granulomas en eltejido sinovial. En la sarcoidosis crónica también puedehaber cambios óseos por infiltración gra-nulomatosa. Suele tratarse de pacientescon lupus pernio u otras lesiones cutáne-as granulomatosas, mucho más frecuen-tes en la raza negra. La manifestación másfrecuente es la dactilitis sarcoide, que cur-sa con tumefacción y dolor del dedo y dis-trofia ungueal. En las radiografías se apre-cian quistes óseos en las falanges sinreacción perióstica. Otras lesiones óseas,en general raras, pueden afectar a huesos
de cráneo, huesos nasales, vértebras, cos-tillas o pelvis. Se piensa que existe compromiso muscu-lar en dos tercios de los pacientes con sar-coidosis y tanto en el síndrome de Löfgrencomo en formas crónicas, pero se trata deformas asintomáticas en las que sólo oca-sionalmente la biopsia ha permitido eldiagnóstico de la enfermedad. La miopa-tía sarcoide sintomática es rara (menos del0,5%) y en la mayoría de los casos es cró-nica, con pérdida de fuerza lentamenteprogresiva. Las formas agudas o subagu-das con dolor y aumento de enzimas mus-culares son excepcionales. Más rara vezse trata de formas nodulares que cursancomo masas localizadas dolorosas que cap-tan en la gammagrafía con 67Ga y que sevisualizan bien mediante resonancia mag-nética. El diagnóstico requiere biopsia18.
Glándulas salivares
Puede producirse engrosamiento parotí-deo hasta en un 6% de los pacientes ycasi siempre es bilateral. La combinaciónde fiebre, tumefacción parotídea, parálisisfacial y uveítis anterior constituye el sín-drome de Heerfordt. Las glándulas saliva-res afectadas pueden captar el citrato de67Ga y dar un aspecto típico de oso pan-da en la gammagrafía que, aunque ines-pecífico, es muy sugestivo de sarcoidosis(fig. 10).
LaboratorioExiste hipercalcemia en un 2%-10% delos pacientes con sarcoidosis. La hiper-calciuria es 2-3 veces más frecuente. Se
1719
SARCOIDOSIS
Fig. 8. Eritema nodoso en el curso de un síndrome de Löfgren.
Fig. 9. Sarcoidosis cutánea (confirmada histológicamente)(cortesía Dr. T. Pozo).
Fig. 10. Sarcoidosis que afecta a las glándulas salivares. Gam-magrafía con 67Ga que muestra el característico patrón de osopanda (cortesía del Dr. G. Iglesias).

debe a un aumento de la producción de1,25(OH)2 vitamina D3 (calcitriol) por losmacrófagos y granulomas. Si no se con-trola puede conducir a cálculos renales,nefrocalcinosis e insuficiencia renal.La enzima conversora de la angiotensina(ECA) es producida por las células epite-lioides de los granulomas y está elevadaen suero en dos tercios de los pacientescon sarcoidosis. Es útil como apoyo, másque como prueba diagnóstica, pues pue-de elevarse en condiciones como diabe-tes, silicosis, asbestosis, beriliosis, tuber-culosis, neumonitis por hipersensibilidad,hepatitis granulomatosas, cirrosis, hiper-tiroidismo, linfoma y enfermedad de Gau-cher. Aunque disminuye tras el trata-miento con corticoides, se correlacionapoco con las manifestaciones clínicas y esde poca ayuda en el seguimiento9.Otras alteraciones de laboratorio que pue-den presentarse, como anemia crónica,linfopenia, aumento de fosfatasa alcalina,aumento de velocidad de sedimentaciónglobular (VSG) o proteína C reactiva (PCR)o hipergammaglobulinemia policlonal, soninespecíficas. Con frecuencia también pue-de detectarse factor reumatoide o anti-cuerpos antinucleares.
Formas de comienzo y evoluciónEn la raza blanca comienzan como erite-ma nudoso, formando parte del síndromede Löfgren un 20%-50% de los pacientes(44% de una serie de 425 pacientes des-crita en nuestro país por Badrinas10). Enotros casos se diagnostica tras detectarseadenopatías hiliares bilaterales en pacien-tes asintomáticos en radiografías de tóraxrutinarias. Muchos estudios sugieren queen la raza blanca la enfermedad puede serleve y con evolución espontánea favora-ble y así la mayoría de los casos de fiebrey/o eritema nudoso se resuelven sin ne-cesidad de corticoides en 6 semanas, aun-que las adenopatías puedan persistir unoo más años. Las formas crónicas de sarcoidosis co-mienzan principalmente por síntomas res-piratorios (tos y disnea de esfuerzo) y lasmanifestaciones extratorácicas, cuandoexisten, suelen ser posteriores. En pre-sencia de cambios radiográficos progresi-vos o de pruebas de función pulmonar in-dicativas de compromiso del parénquimapulmonar hay que considerar el trata-
miento con corticoides para evitar la evo-lución a la fibrosis9. La indicación de tra-tamiento viene dada por la progresión dela enfermedad, pues incluso un 40%-70%de los pacientes en estadio II y un 10%-20% de los que están en estadio III pue-den experimentar remisión espontánea sinrecidiva posterior1. Por el contrario, cuan-do se administran corticoides, en la ma-yoría de los casos la capacidad vital for-zada mejora con tratamiento y la mejoríaes máxima en el primer año en un 59%de los casos, aunque es frecuente un de-terioro posterior. La mortalidad de la sar-coidosis es difícil de establecer, en gene-ral, es inferior al 5%, aunque depende delas poblaciones objeto de estudio. Las cau-sas más frecuentes son la insuficiencia res-piratoria y la hemoptisis masiva.
DiagnósticoEl diagnóstico de la sarcoidosis requiere 3condiciones: un cuadro clínico-radiográfi-co compatible, demostración histológicade granulomas no caseificantes y exclu-sión de otras enfermedades. La actituddiagnóstica puede dividirse en 3 etapas:1. Obtención de la biopsia.2. Valoración de la extensión y gravedadde la enfermedad.3. Determinación de si la enfermedad estáestable o tiende a progresar.
Obtención de la biopsia
La existencia de adenopatías hiliares bila-terales en ausencia de otras manifesta-ciones no debe usarse como único crite-rio para el diagnóstico de sarcoidosis, pueseste patrón ocasionalmente se observa enlinfomas, tuberculosis, brucelosis, infec-ciones micóticas y carcinoma broncogé-nico y debe hacerse un estudio exhausti-vo para descartar estas enfermedades2.Algunos autores han propuesto que el sín-drome de Löfgren que se resuelve total-mente en pocas semanas no requiere con-firmación histológica, pero algunas de lascondiciones anteriores, como tuberculosiso linfomas también son causas de erite-ma nodoso y además, es frecuente que lasadenopatías persistan durante más de unaño, con la consiguiente incertidumbretanto para el paciente como para el mé-dico.En la mayoría de los casos el procedi-miento de elección es la biopsia trans-
bronquial, que tiene una rentabilidad del40%-90%. Pueden hacerse en el mismoacto una biopsia de mucosa bronquial yun lavado bronquial. Cuando éste mues-tra un cociente de linfocitos CD4/CD8 su-perior a 3,5 la especificidad para sarcoi-dosis es del 94%, aunque con unasensibilidad baja. Otros posibles lugaresde biopsia vienen determinados por la clí-nica (piel, reacciones granulomatosas sobre cicatrices antiguas, ganglios, con-juntiva, etc.). Rara vez la biopsia trans-bronquial no es diagnóstica y no son ac-cesibles otras localizaciones; en estoscasos la TAC de tórax puede definir la con-ducta a tomar: las lesiones de parénqui-ma se abordan por toracoscopia o biopsiaabierta, pero si existen adenopatías me-diastínicas, la biopsia por mediastinosco-pia tiene menos complicaciones1.Cuando no puedan hacerse biopsias ocuando las radiografías y TAC de tóraxsean normales, en los centros en que estédisponible puede ser útil la prueba deKveim-Siltzbach. Es el caso de los pacien-tes con uveítis de causa desconocida, hi-percalciurias, enfermedad granulomatosahepática, sospecha de neurosarcoidosis oeritema nudoso recidivante. Consiste enla intradermorreacción con extracto debazo o ganglio afectados de sarcoidosis yobtenidos de otro paciente. La piel inyec-tada se biopsia a las 4-6 semanas bus-cando granulomas. La prueba tiene unaespecificidad superior al 97% para el diag-nóstico de sarcoidosis, por lo que es muyútil en los casos en que es positiva, lo queocurre en un 84% de las sarcoidosis agu-das/subagudas y en un 61% de las formascrónicas19. La falta de accesibilidad y laposibilidad de infección, incluidos retro-virus, limitan mucho la aplicación de estatécnica.
Extensión de la enfermedad
A todos los pacientes debe hacerse radio-grafía de tórax, espirometría y difusión deCO, electrocardiograma, exploración of-talmológica y prueba de tuberculina1. Laanergia tuberculínica es típica de la sar-coidosis, aunque existen casos de positi-vidad9. También deben hacerse estudiosde laboratorio que incluyan hemograma,perfil hepático y renal y calcio en sangrey en orina. También se ha propuesto quedeben hacerse de rutina pruebas para de-tectar infección por el virus de la inmu-
1720
ENFERMEDADES DEL SISTEMA INMUNE (IX)

nodeficiencia humana (VIH), una condi-ción en la que frecuentemente existe lin-fopenia, anomalías radiográficas pulmo-nares y granulomas pulmonares. Laprueba está especialmente indicada cuan-do el lavado broncoalveolar muestre uncociente de linfocitos CD4+/CD8+ infe-rior a 120.La TAC de tórax está indicada cuando haysospecha de sarcoidosis y las radiografíasson normales, cuando las radiografías sonatípicas o cuando se desee descartar com-plicaciones como bronquiectasias, aspergi-loma, fibrosis pulmonar, infección añadidao cáncer. En los casos en que se sospechesarcoidosis extratorácica las exploracionesvendrán dadas por la clínica: Holter, RM decorazón, cráneo o músculo, etc.
Valoración de la actividad o estabilización
La actividad se valora según datos clínicosy de laboratorio y exploraciones comple-mentarias, sobre todo radiografía de tóraxy espirometría. La aparición de nuevas ma-nifestaciones o la persistencia o progre-sión de las previas se considera índice deactividad. La radiografía de tórax no per-mite distinguir entre lesiones inflamato-rias y fibrosis, y la TAC de tórax, inclusode alta resolución, rara vez modifica lasdecisiones terapéuticas. Con esta últimatécnica se ha propuesto que los patronesde vidrio deslustrado, nodular, de opaci-dades lineales irregulares y de engrosa-miento septal interlobular representancondiciones potencialmente reversibles,mientras que los hallazgos de espacios aéreos quísticos y la distorsión de la ar-quitectura sugieren enfermedad pulmonarirreversible21. La gammagrafía con citratode 67Ga, el lavado broncoalveolar y la de-terminación de la enzima conversora dela angiotensina, como se ha comentado,tienen un interés diagnóstico relativo ypoca utilidad para el seguimiento de la en-fermedad2.
Tratamiento
Indicaciones del tratamiento con corticoides
Los síntomas o los hallazgos que necesi-tan tratamiento con corticoides por vía
oral son controvertidos, al no existir en-sayos controlados que demuestren su efi-cacia en el control a largo plazo. En ge-neral, los pacientes que sólo tienenlesiones cutáneas, uveítis anterior o tosúnicamente precisan corticoides tópicos,al menos de entrada. Los pacientes consíndrome de Löfgren con sarcoidosis gra-do I tampoco necesitan corticoides, puesel reposo y los antiinflamatorios son sufi-cientes en la mayoría de los casos. En ge-neral, se admiten las siguientes indicacio-nes de tratamiento con corticoides por víageneral1,2:1. Hipercalcemia significativa.2.Neurosarcoidosis o afectación miocár-dica.3.Uveítis que no responde a tratamientotópico.4. Pacientes con sarcoidosis pulmonar enestadio II que tengan síntomas respirato-rios progresivos o que tengan infiltradospersistentes o limitación progresiva de laspruebas de función respiratoria, inclusosin síntomas.5. Pacientes con sarcoidosis pulmonar enestadio III.6. Síndrome de Löfgren con deficiente evo-lución de las manifestaciones sistémicasdespués de 6 semanas (las adenopatíaspueden persistir más de 1 año).
Pautas de tratamiento con corticoides
Las dosis y duración del tratamiento de-ben ser individualizadas. En general, lasarcoidosis pulmonar se trata con 30-40
mg/día de prednisona o equivalente endosis única diaria con el desayuno. Se eva-lúa la respuesta en 2-3 meses y si el pa-ciente está mejor se reduce gradualmen-te la dosis diaria hasta un mantenimientode 5-10 mg/día. Esta dosis o su equiva-lente se mantienen un mínimo de 12 me-ses. La tabla 4 recoge la pauta de trata-miento con prednisona recomendada porJohns y Michele, especialistas con una di-latada experiencia en sarcoidosis9. Segúneste protocolo, deben hacerse valoraciónclínica, espirometría y difusión de CO a2 meses y a 8 meses de tratamiento. Enel primer caso para confirmar la eficacia,pues si no se ha producido respuesta, espoco probable que se consiga prolongán-dolo y debe valorarse si es por dosis in-suficiente o porque existe fibrosis1. La va-loración a 8 meses se hace para confirmarque se ha alcanzado la mejoría estable an-tes de iniciar el descenso. Si en ese mo-mento se ha producido deterioro, es pocoprobable que se deba a tratamiento ina-decuado y hay que asegurarse de que elpaciente lo hace correctamente y de queno existan causas intercurrentes como in-fección o insuficiencia cardíaca. Al ter-minar el primer año de tratamiento debehacerse nueva valoración y en caso de re-cidiva, volver a tratar comenzando por 20 mg/día.Existen series de pacientes con sarcoido-sis pulmonar o bronquial tratados con cor-ticoides tópicos inhalados (budesonida),cuya eficacia no ha sido confirmada22.Los casos de sarcoidosis con afectaciónneurológica central o periférica y la sar-coidosis miocárdica requieren dosis de
1721
SARCOIDOSIS
TABLA 4Pauta de tratamiento con prednisona para la sarcoidosis pulmonar
Dosis Duración
40 mg/día 2 semanas
30 mg/día 2 semanas
Revisión clínica, radiografía de tórax, espirometría y difusión de CO
25 mg/día 2 semanas
20 mg/día 2 semanas
15 mg/día 6,5 meses (10 mg/día puede ser suficiente en la última parte de este período)
Revisión clínica, radiografía de tórax, espirometría y difusión de CO
7,5 mg/día 1 mes
5 mg/día 1 mes
2,5 mg/día 1 mes
2,5mg/48 h 1 mes
Revisión clínica, radiografía de tórax, espirometría y difusión de CO
Tomada de Johns CJ, Michele TM9.

40-80 mg/día, aunque existe poca expe-riencia23. Las dosis de corticoides a ad-ministrar en la hipercalcemia clínica-mente significativa son del orden de 10-20 mg/día2.
Otras alternativas de tratamiento
Citotóxicos
Se han empleado metotrexato y azatio-prina, en monoterapia o asociados a pred-nisona, en formas crónicas de sarcoidosispulmonar o cutánea con la intención dereducir o evitar los efectos del corticoidea largo plazo o en pacientes que se nie-gan a tomarlos. Existe poca experienciade estos fármacos en sarcoidosis, aunqueen general son bien tolerados y relativa-mente seguros cuando se adoptan las pre-cauciones de monitorización bioquímica yhematológica y prevención de embarazo.Otros citotóxicos como ciclofosfamida oclorambucilo muy rara vez se emplean porlos graves riesgos que conllevan.
Antipalúdicos de síntesis
La cloroquina (250 mg/día) y la hidroxi-cloroquina (400 mg/día) por períodos de6 meses se han mostrado eficaces en for-mas de sarcoidosis cutánea, sarcoidosispulmonar e hipercalcemia. El riesgo másimportante de su administración a largoplazo es la toxicidad retiniana, que es me-nor con la hidroxicloroquina y que debevigilarse mediante controles oftalmológi-cos cada 3 meses que incluyan fondo deojo y test de los colores.
Ketoconazol
Es un antifúngico que se ha empleado paratratar la hipercalcemia de la sarcoidosispor su capacidad para reducir los nivelesaumentados de 1,25(OH)2 vitamina D3.
Control clínico
Todos los pacientes con sarcoidosis debenser seguidos durante al menos 3 años, enparticular los grados II, III y IV de sarcoi-dosis pulmonar, pues son muy frecuenteslas recidivas tras la supresión del corti-coide, algo infrecuente si la remisión esespontánea. Los casos de recidiva tras tra-tamiento pueden requerir el manteni-miento de dosis bajas de corticoides a lar-go plazo.
Prevención de las complicacionesOtras precauciones a tener en cuenta son:a) profilaxis de la osteoporosis inducidapor corticoides con medidas similares alas de otras indicaciones de corticotera-pia, si nos aseguramos de que los nivelesde calcitriol, calcemia y calciuria se man-tengan normales (precaución con la ad-ministración de vitamina D); y b) quimio-profilaxis tuberculosa secundaria conisoniacida en casos de prueba de tuber-culina positiva, pues lo propio de la sar-coidosis es la anergia.
BIBLIOGRAFÍA
1. Hunninghake GW, Costabel U, Ando M, Baughman R,Cordier JF, du Bois R, et al. ATS/ERS/WASOG Statement onSarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 1999; 16:149-173.2. Newman LS, Rose CS, Maier LA. Sarcoidosis. N Eng JMed 1997; 336: 1.224-1.234.3. Rybicki BA, Major M, Popovich J, Maliaric MJ, IannuziMC. Racial differences in sarcoidosis incidence: a 5-yearstudy in a health maintenance organization. Am J Epide-miol 1997; 145: 234-241.4. Morera J, Aranda A, Badrinas F. Sarcoidosis in Spain.En: Chretien J, Marsac J, Saltier JC (eds) Proc IX Internatio-nal Conference on Sarcoidosis, 625. París: PergamonPress. 1983.5. Saboor SA, Johnson NM, McFadden J. Detection of my-cobacterial DNA in sarcoidosis and tuberculosis with poly-merase chain reaction. Lancet 1992; 339: 1.012-1.015.
6. Mitchell IC, Tmk JL, Mitchell DN. Detection of mycobac-terial rRNA in sarcoidosis with liquid-phase. Lancet 1992;339: 1.015-1.017.7. Agostini C, Adami F, Semenzato G. New pathogeneticinsights into the sarcoid granuloma. Curr Opin Rheumatol2000; 12: 71-76.8. Kunkel SL, Lukacs NW, Strieter RM, Chensue SW. Th1and Th2 responses regulate experimental lung granulomadevelopment. Sarcoidosis Vasc Diffuse Lung Dis 1996; 13:120-138.9. Johns CJ, Michele TM. The clinical management of sar-coidosis. Medicine (Baltimore) 1999; 78: 65-111.10. Badrinas F, Morera J, Fité E, Mañá J, Vidal R, RuizManzano J, et al. Sarcoidosis en Cataluña: análisis de 425casos. Med Clin (Barc) 1989; 93: 81-87.11. Atanes A, de Toro J, Gómez N, Aspe B, Graña J, BursónJM, Galdo F. Estudio de 94 casos de sarcoidosis con espe-cial referencia al eritema nudoso. Rev Clin Esp 1992; 191;65-70.12. Badrinas F. Definición. Manifestaciones clínicas: gene-ralidades. En: Badrinas F. Definición. Manifestaciones clíni-cas: generalidades. En Badrinas F, Morera J, eds. Sarcoido-sis. Barcelona: Doyma, 1989; 10-20.13. Bjermer L, Thunell M, Rosenhall L, Stjernberg N. Endo-bronchial biopsy positive sarcoidosis: relation to bronchoal-veolar lavage and course of disease. Respir Med 1991; 85:229-234.14. Morera J, Domingo C, Fité E, Ruiz J. Sarcoidosis endo-torácica: manifestaciones clínicas y radiológicas. En: Badri-nas F, Morera J, eds. Sarcoidosis. Barcelona: Doyma, 1989;79-115.15. Folz SJ. Abdominal manifestations of sarcoidosis in CTstudies. J Comput Assist Tomography 1995; 19: 573-579.16. Fahy GJ, Marwick T, McCreery CJ, Quingley PJ, MaurerBJ. Doppler echocardiographic detection of left ventriculardiastolic dysfunction in patients with pulmonary sarcoido-sis. Chest 1996; 109: 62-66.17. Mitchell DN. Sarcoidosis with skeletal involvement.En: Klippel JH, Dieppe PA, eds. Rheumatology, St. Louis:Mosby, 1994; 3.37.1-3.37.8.18. Zisman DA, Biermann JS, Martínez FJ, Devaney KO,Lynch JP. Sarcoidosis presenting as tumorlike muscular le-sion. Medicine (Baltimore) 1999; 78: 112-122.19. Mañá J, Pujol R, Salazar A, Morera J, Fité E, BadrinasF. La prueba de Kvein-Stilzbach en la sarcoidosis. Med Clin(Barc) 1995; 104: 645-647.20. Juega J, Pedreira JD, Verea H. Sarcoidosis e infecciónpor VIH. Med Clin (Barc) 1996; 107: 115-116.21. Murdoch J, Müller NL. Pulmonary sarcoidosis: changeson follow-up CT examination. Am J Roentgenol 1992; 159:473-477. 22. Alberts C, van der Mark TW, Jansen HM, Dutch StudyGroup on Pulmonary Sarcoidosis. Inhaled budesonide inpulmonary sarcoidosis: a double-blind, placebo-controlledstudy. Eur Respir J 1995; 8: 682-688.23. Sharma OP, Sharma AM. Sarcoidosis of the nervoussystem: a clinical approach. Arch Intern Med 1991; 151:1.317-1.321.
1722
ENFERMEDADES DEL SISTEMA INMUNE (IX)

ConceptoLa amiloidosis es un síndrome que se ca-racteriza por el depósito extracelular deproteínas fibrilares, que después de tin-ción con rojo Congo presentan una birre-fringencia verde al microscopio óptico deluz polarizada. Las fibrillas de amiloide es-tán formadas por diversas proteínas o frag-mentos de éstas que tienen en común lacapacidad de adoptar una estructura mo-lecular terciaria en disposición β-plegada,que es propia de la sustancia amiloide yes la responsable de su insolubilidad y altaresistencia a la digestión proteolítica. Eldepósito mantenido de sustancia amiloi-de ocasiona la sustitución y destruccióndel parénquima en los órganos afectos,con la posterior pérdida de función de éstos1.El termino “amiloide” (equivalente a al-midón o celulosa) fue introducido por Virchow en 1854 al observar que los depósitos amiloides tenían el mismo comportamiento que la celulosa en la tin-ción con yodo y ácido sulfúrico. Friede-rich y Kekulé descubrieron su naturalezaproteica unos años más tarde y no fuehasta 1922 cuando se observó su afini-dad a la tinción por el rojo Congo (con-gofilia).En estos momentos se ha descrito la se-cuencia de aminoácidos de hasta 17 tiposdistintos de proteínas o fragmentos de pro-teínas fibrilares presentes en los depósi-tos de sustancia amiloide. Estas proteínasson las que definen cada variedad de ami-loidosis. En cambio, en todos los tipos deamiloidosis se encuentra el denominadocomponente P de la amiloide, común a to-das las variedades; su precursor es unaglucoproteína del plasma, que pertenecea la familia de las pentraxinas (como laproteína C reactiva) y es sintetizada en elhígado1-3.
ClasificaciónLa amiloidosis se puede clasificar en fun-ción de dos criterios: a) por la distribu-ción de los depósitos amiloides: formaslocalizadas y sistémicas; b) por la proteí-na fibrilar específica de cada variedad. Laterminología utilizada incluye dos letras,la primera es la A de Amiloide, y la se-gunda define la proteína fibrilar especí-fica o características clínicas relevantes(tabla 1)4.
Formas sistémicas
Amiloidosis AL
La proteína fibrilar está constituida por cadenas ligeras de inmunoglobulina (másfrecuentemente lambda que kappa en una relación 2:1). En más del 90% de casos se observa componente M en el suero. Es el tipo de amiloidosis que se ob-serva en las discrasias de células plasmá-ticas (primaria o idiopática y la asociadaa mieloma múltiple y macroglobulinemia).
Amiloidosis AA
Denominada amiloidosis secundaria o re-activa. Es la que se relaciona con proce-
sos infecciosos e inflamatorios crónicos5.Es también el tipo de amiloidosis queacompaña a la fiebre mediterránea fami-liar y al síndrome de Muckle Wells. Laamiloidosis AA está formada por la pro-teína fibrilar AA (proteína amiloide A), conuna estructura constituida por 76 amino-ácidos y con un peso molecular de 8,5 ki-lodaltons. La proteína AA deriva de la porción N terminal de un precursor plas-mático denominado proteína sérica A de la amiloide (SAA) que circula en plas-ma unido a una lipoproteína de alta densidad (HDL3). La SAA es un reactantede fase aguda, de síntesis hepática (aun-que en fechas recientes ha podido de-mostrarse su expresión también en ma-crófagos y endotelio, así como síntesislocal en tejidos inflamados como la si-novial reumatoide) con incrementos marcados, de hasta 1.000 veces sus va-lores basales, en caso de estímulos infla-matorios adecuados. Existen diferentesisoformas de SAA productos de dos genesSAA1 y SAA2 con distintas variedades alé-licas. La proteína SAA1 es la que predo-mina en el plasma y los depósitos ami-loides6.
Amiloidosis AH
Se observa en pacientes afectos de insu-ficiencia renal crónica terminal en pro-grama de diálisis. La proteína fibrilar deeste tipo de amiloidosis es la β2-micro-globulina; se trata de una proteína plas-mática, que forma parte de la moléculaHLA de clase I, y que se acumula cuandoexiste un déficit importante de la funciónrenal.
1709
AMILOIDOSISR. Sanmartí Sala y J. Muñoz GómezServicio de Reumatología. Institut Clínic de l’Aparell Locomotor. Hospital Clínic de Barcelona.
Medicine 2001; 8(33): 1709-1714
TABLA 1Clasificación de la amiloidosis
Tipo de amiloidosis Síndrome clínico Proteína amiloide
AL Primaria o asociada a mieloma/ Cadenas ligeras de macroglobulinemia inmunoglobulinas
AA Secundaria o reactiva Proteína AFiebre mediterránea familiar
Aβ2-M Asociada a diálisis β2-microglobulina
AF Formas familiares Transtirretina, apolipoproteína(polineuropatía, cardiopatía) A1, gelsolina
AS Amiloidosis senil Transtirretina
AE Amiloidosis endocrina Calcitonina, amilina
AD Amiloidosis cutánea Queratina
AB Amiloidosis cerebral β-proteína o A4(síndrome de Down,enfermedad de Alzheimer)

Amiloidosis AF
Define diversas formas de amiloidosis he-reditarias (polineuropatía amiloidea fami-liar [PAF], la variedad cardiomiopática tipodanés, y otras). La PAF es la forma másfrecuente de amiloidosis familiar, con he-rencia autosómica dominante y formadapor el depósito de la proteína fibrilar transtirretina (anteriormente denominada prealbúmina), habiéndose identificadomúltiples variedades distintas como con-secuencia de distintas mutaciones. Otrasproteínas responsables de amiloidosis fa-miliar son la apolipoproteína A1, el fibri-nógeno y la gelsolina.
Formas localizadas
Son variedades de amiloidosis con depó-sitos localizados en determinados tejidosu órganos.
Amiloidosis senil
Corresponde a una variedad de amiloido-sis localizada, que se caracteriza por pe-queños depósitos en cerebro, corazón,páncreas y bazo; se considera el resulta-do de la desnaturalización de las proteí-nas por el propio envejecimiento tisular.Aunque en general es asintomática, pue-de ser responsable de trastornos del rit-mo cardíaco. La proteína fibrilar amiloidees la transtirretina; en el corazón se pue-den detectar depósitos de sustancia ami-loide formados por el factor natriuréticoatrial.
Amiloidosis endocrina
Se relaciona con tumores endocrinos, es-pecialmente del sistema APUD. Existe re-lación entre la proteína fibrilar amiloidecaracterística de cada variedad y la hor-mona sintetizada por el tumor. La más fre-cuente es la asociada al carcinoma me-dular de tiroides secretor de calcitonina.En los islotes pancreáticos de pacientesdiabéticos se ha encontrado sustancia ami-loide cuya proteína fibrilar es la amilina(polipéptido amiloide de los islotes); sedesconoce su significado patogenético.
Amiloidosis cerebral
En este grupo se incluyen las formas deamiloidosis asociada a la demencia de Alz-heimer y al síndrome de Down. La proteí-
na fibrilar responsable es la denominadaß-proteína o proteína A4. Existen otros ti-pos de amiloidosis exclusivamente cere-bral como la amiloidosis cerebral hemo-rrágica tipo islandés y holandés, cuyasproteínas fibrilares son la cistatina C y unamutuación de la β-proteína respectiva-mente. En fechas recientes se ha demostrado lapresencia de depósitos amiloides en en-fermedades neurodegenerativas transmi-sibles como el kuru, enfermedad deCreutzfeldt-Jacob y la encefalopatía es-pongiforme bovina. La proteína fibrilar deeste tipo de amiloidosis es la proteínaprion (PrP) modificada, que constituye unagente infeccioso distinto de los virus7.
Amiloidosis cutánea
Es una variedad de amiloidosis localizadaen la piel, con un significado patogenéti-co similar a la amiloidosis senil. La prote-ína constituyente es la propia queratina.
PatogeniaNo se conoce con exactitud la etiopato-genia1,6,8 de la amiloidosis. Las distintasvariedades de sustancia amiloide derivande un precursor plasmático específico, quees finalmente degradado a proteína fibri-lar. Así pues, la amiloidogénesis se pro-duciría por la coexistencia de dos facto-res: por una parte, la presencia dealteraciones en la concentración o estruc-tura primaria del precursor plasmático delas fibrillas amiloides y, por otra, la exis-tencia de un metabolismo alterado del pre-cursor de la proteína fibrilar. En las ami-loidosis AA o AL, existe una síntesisaumentada del precursor plasmático y unproceso posterior de proteólisis a proteí-na fibrilar amiloide. En la amiloidosis AHy AF, las proteínas fibrilares, β2-micro-globulina y transtirretina tienen la mismaestructura molecular que sus precursores,por lo que no es preciso un proceso pre-vio de proteólisis o degradación.La amiloidosis AA es la variedad mejor es-tudiada. Como se ha comentado previa-mente, el primer requisito es la presenciade concentraciones elevadas de la SAA deforma persistente en el tiempo como con-secuencia de un estímulo inflamatorio ade-cuado. La síntesis de SAA está inducidapor diferentes citocinas, especialmente in-terleucinas 1 y 6 y factor de necrosis tu-
moral. La SAA incrementa la afinidad porlas lipoproteínas de alta densidad en lu-gares donde existe acumulación de ma-crófagos y es en dichas regiones dondepredominantemente se acumula la sus-tancia amiloide A, como la zona perifoli-cular del bazo, los sinusoides hepáticos yel mesangio glomerular. La SAA tambiénmodula la respuesta inflamatoria, ya quese ha demostrado que tiene poder qui-miotáctico para monocitos y polimorfo-nucleares. Se han sugerido dos mecanis-mos para el desarrollo de amiloidosis AA;el proceso inicial sería o bien la degrada-ción enzimática de la SAA con su poste-rior depósito en tejidos, o bien existiríauna precipitación inicial de fibrillas SAA,con lo que la conversión a proteína AA se-ría un fenómeno postfibrilogénico. Las in-vestigaciones realizadas en amiloidosis ex-perimental parecen apoyar el segundomecanismo. La catepsina B participaría di-rectamente en la génesis de proteína AAa partir de su precursor SAA.Existen también otros factores que pare-cen desempeñar un papel decisivo en eldesarrollo de amiloidosis. El llamado fac-tor favorecedor (o acelerador) de la ami-loidosis (FVA) es una glucoproteína queestá presente en los depósitos de amiloi-de, incluso horas previas al desarrollo deldepósito, en modelos de amiloidosis ex-perimental en animales, en los que se hademostrado su capacidad de acelerar eldepósito de sustancia amiloide. Por otraparte, existe una presencia constante deproteinoglicanos o glucosaminoglicanos entodas las variedades de amiloidosis; asípues, es probable que diversas interac-ciones con algunos proteinoglicanos comoel perlecan u otras proteínas de la mem-brana basal desempeñen un papel pri-mordial en el proceso de amiloidogénesis.Se desconoce exactamente el papel delcomponente P, presente en todas las va-riedades de amiloidosis, tanto en su fun-ción fisiológica como en el proceso de ami-loidogénesis.
EpidemiologíaEs difícil obtener cifras fiables de la pre-valencia e incidencia de la amiloidosis sis-témica. Los casos familiares predominanen ciertas áreas como la forma familiar conpolineuropatía en Portugal, Japón o Sueciay la amiloidosis asociada a fiebre medite-rránea familiar en Oriente próximo. La in-
1710
ENFERMEDADES DEL SISTEMA INMUNE (IX)

cidencia anual de amiloidosis AL en un es-tudio americano se cifró en 8,9 por millónde habitantes. La relación entre formas ALy AA es extremadamente variable; desdeuna relación AL:AA de 17:1 en Estados Uni-dos a una de 1:2 en Europa9.En nuestra área geográfica, la forma másfrecuente de amiloidosis es la variedad AA. En la actualidad, la enfermedad cró-nica que con mayor frecuencia es causade amiloidosis AA ya no es una enferme-dad infecciosa sino inflamatoria y, másconcretamente, la artritis reumatoide (ta-bla 2)9-11. La prevalencia de amiloidosis AAclínica en los distintos reumatismos infla-matorios es variable según las distintasáreas geográficas y metodología utilizadaen los diferentes estudios. Se cifra apro-ximadamente en el 5%-10% de los casosde artritis reumatoide evolucionada y ar-tritis crónica juvenil, siendo la forma sis-témica (enfermedad de Still) la más ami-loidogénica. En general, los pacientes conartritis reumatoide y amiloidosis AA sue-len tener varios años de evolución de unaenfermedad activa, con marcada destruc-ción articular y resistente a la terapia an-tirreumática. Se desconoce, no obstante,el porqué en enfermos con un nivel simi-lar de actividad inflamatoria unos desa-rrollen amiloidosis y otros no; es posibleque existan factores del huésped, quizasrelacionados con la presencia de distintosalelos del gen SAA1, que predispongan ala amiloidogénesis12.
Manifestaciones clínicasLas manifestaciones clínicas2,3,9,13,14 de laamiloidosis pueden ser muy diversas, enfunción de la magnitud del depósito, delórgano afecto y del tipo de amiloidosis. Laamiloidosis AL y AA son las formas más
frecuentes de amiloidosis sistémica; la pri-mera representa la forma más grave, afec-tando el riñón, aparato digestivo, hígado,suprarrenales, corazón, articulaciones ysistema nervioso central y periférico. Enla amiloidosis AA se afecta fundamental-mente el riñón, suprarrenales, hígado,bazo y tubo digestivo. El cuadro clínico dela amiloidosis AF varía en función del tipode proteína fibrilar amiloide. La variedadmás frecuente es una polineuropatía mix-ta de predominio sensitivo, con afecta-ción cardíaca en forma de trastornos de conducción y/o insuficiencia cardíaca con-gestiva. Las manifestaciones clínicas de la amiloidosis asociada a la diálisis por β2-microglobulina incluyen el síndromedel túnel carpiano, artropatía y fracturaspatológicas.
Riñón
Constituye la localización más frecuentede la enfermedad; en las amiloidosis sis-témicas tipo AL y AA la participación delriñón es prácticamente constante (un 90%de casos). La afección glomerular es la másfrecuente, y es responsable de la protei-nuria de grado variable, pero que puedeprogresar a un verdadero síndrome ne-frótico. La afección vascular es tambiénfrecuente y suele acompañar a la afecciónglomerular. El proceso generalmente evo-luciona hacia la insuficiencia renal pro-gresiva y finalmente insuficiencia renal terminal. Cuando el depósito es primor-dialmente vascular, los pacientes puedentener deterioro de la función renal con pro-teinuria mínima; es raro que la hematuriaaislada sea debida a amiloidosis renal ysuele aparecer acompañando a la protei-nuria. La amiloidosis puede infiltrar tam-bién las vías urinarias y manifestarse como
hematuria macroscópica. En algunos ca-sos los depósitos de sustancia amiloidepredominan a nivel del túbulo renal, oca-sionando síndromes tubulares tipo Fan-coni o acidosis tubular renal. La hiperten-sión arterial se presenta aproximadamenteen el 20%-30% de pacientes con nefro-patía amiloidea. Aunque se ha descrito quelos pacientes muestran riñones de mayortamaño en el estudio ecográfico, esta cir-cunstancia no se observa en muchos pa-cientes con amiloidosis renal, especial-mente en fases más avanzadas de laenfermedad.
Corazón
La amiloidosis cardíaca es una entidad gra-ve y de mal pronóstico. Es una localiza-ción frecuente en la amiloidosis AL y mu-cho menos común en la amiloidosis AA.La infiltración miocárdica por amiloideprovoca típicamente una míocardiopatíarestrictiva resistente al tratamiento médi-co, que cursa con insuficiencia cardíacacongestiva, y trastornos del ritmo cardía-co. La participación de otras estructurascardíacas, como válvulas, endocardio y pe-ricardio es posible en el curso de la ami-loidosis sistémica provocando diversas al-teraciones funcionales. Es típico un bajovoltaje del complejo QRS en el elec-trocardiograma, con alteraciones en laconducción aurículo-ventricular e intra-ventricular, y presencia de arritmias. Elecocardiograma bidimensional permite ob-servar engrosamiento simétrico de las pa-redes del ventrículo izquierdo y del tabi-que interventricular, e hipoquinesia de lascavidades izquierdas. La infiltración mio-cárdica por amiloide ocasiona una imagenecocardiográfica casi patognomónica conun granulado brillante en la pared del ven-trículo izquierdo y tabique interventricu-lar (fig. 1).
1711
AMILOIDOSIS
TABLA 2Prevalencia de distintas enfermedades en tres series de amiloidosis secundaria (AA)
Hazenberg9 Gertz, Kyle10 De la Sierra11
Año 1994 Año 1991 Año 1985(n = 91) (n = 64) (n = 38)
Artritis reumatoide 56% 48% 57%
Otras artropatías 5% 17% –(espondiloartropatías, artritis juvenil)
Infecciones (tuberculosis, 16% 17% 42%bronquiectasias, osteomielitis, etc.)
Otras (enfermedad inflamatoria 23% 17% –intestinal, Fiebre mediterráneafamiliar, etc.)
Fig. 1. Eocardiografía en un paciente con cardiopatía amiloi-dea. Se observa el típico granulado brillante en el septo inter-ventricular (flechas).

Aparato digestivo
La afección anatómica del tracto gas-trointestinal es frecuente, aunque las ma-nifestaciones clínicas son algo menos co-munes. Algunas manifestaciones como lamacroglosia son exclusivas de la amiloi-dosis AL. Las manifestaciones más fre-cuentes son la diarrea crónica, muchas veces acompañada de un verdadero sín-drome de malabsorción intestinal, debidoa infiltración de vellosidades intestinalesy más probablemente a disfunción auto-nómica. La afección hepática suele estarpresente en forma de hepatomegalia y al-teración biológica (colestasis anictérica)siendo rara la insuficiencia hepática; esmás frecuente en la amiloidosis AL que enla amiloidosis AA. La amiloidosis intesti-nal puede ser también responsable del desarrollo de cuadros de hemorragia di-gestiva, pseudoobstrucción, perforación in-testinal y acalasia
Sistema nervioso
La polineuropatía mixta se da sobre todoen las variedades de amiloidosis heredi-tarias (PAF) y en la amiloidosis AL, y apa-rece pocas veces en la amiloidosis AA. Esfrecuente la disfunción autonómica que semanifiesta por hipotensión ortostática, in-continencia de esfínteres y alteraciones enla sudación. En la amiloidosis AL y sobretodo en la amiloidosis asociada a la diáli-sis es frecuente la presencia de síndromedel túnel carpiano.
Piel
La afección cutánea es frecuente en la ami-loidosis AL. Suele tratarse de pápulas so-breelevadas y confluyentes que se locali-zan en pliegues como la axila, ingle ycuello. Son muy características las equi-mosis en la región periorbitaria, en rela-ción con la diátesis hemorrágica que pue-de observarse en la amiloidosis AL.
Sistema endocrino
Se pueden afectar tanto las glándulas exo-crinas como endocrinas. Suelen cursar demanera asintomática, aunque la infiltra-ción de tiroides y glándulas suprarrenalespuede conllevar a hipotiroidismo e insu-ficiencia suprarrenal. En el tipo AL, el
depósito de amiloide en las glándulas sub-mandibulares puede conllevar a un sín-drome seco.
Aparato locomotor
Sólo ocasionalmente la amiloidosis AApuede asociarse a pequeños depósitos deamiloide AA en la membrana sinovial delas articulaciones, cursando de maneraasintomática. En cambio, la presencia deartropatía es muy característica en la ami-loidosis AL y por depósito de β2 micro-globulina. La denominada artropatía ami-loidea puede ser la manifestación inicialde la amiloidosis AL y simular una artritisreumatoide. Uno de los hallazgos más ca-racterísticos son los hombros acolchadoso de jugador de rugby que pueden obser-varse en el 10%-20% de casos de amiloi-dosis AL a lo largo de la evolución de laenfermedad (fig. 2). Un cuadro clínico si-milar se observa en la amiloidosis β2M delpaciente en diálisis, con evidencia de pro-liferación sinovial, tenosinovitis y presen-cia de imágenes quísticas radiolúcidas yerosiones a la radiología (figs. 3, 4, 5). Eneste tipo de amiloidosis es característicatambién la existencia de fracturas espon-táneas y síndrome del túnel carpiano. Esprobable que los depósitos de β2-micro-globulina sean también responsables dealgunas formas de espondiloartropatía des-tructiva que se observan en pacientes conuremia terminal.
Aparato respiratorio
La amiloidosis AL puede ocasionar infil-tración de laringe, traquea y bronquiosprincipales. Se han descrito también ca-sos de depósitos localizados en el tractorespiratorio.
DiagnósticoEl diagnóstico de amiloidosis se realizadespués de la demostración histológica delos depósitos de sustancia amiloide porbiopsia o aspiración. Dichos depósitos seidentifican por sus características tinto-riales: afinidad por el rojo Congo (fig. 6)y birrefringencia verde-manzana en el exa-
1712
ENFERMEDADES DEL SISTEMA INMUNE (IX)
Fig. 3. Amiloidosis por β2-microglobulina. Radiografía de ar-ticulación coxofemoral donde se aprecian importantes geodasacetabulares.
Fig. 2. Artropatía amiloidea en una paciente con amiloidosisAL asociada a mieloma múltiple. Imagen típica de hombrosacolchados o de jugador de rugby.
Fig. 4. Resonancia magnética nuclear del hombro de un pa-ciente con amiloidosis por β2 microglobulina, donde se apre-cia el depósito amiloideo en cabeza humeral.
Fig. 5. Amiloidosis por β2-microglobulina. Imagen necrópsicadonde se observan los depósitos de amiloide en cuello femo-ral.

men con luz polarizada15. La técnica de ladecoloración con permanganato potásico,permite diferenciar la amiloidosis AL deotras formas de amiloidosis, pero su bajasensibilidad y especificidad, limitan su uti-lización. El estudio inmunohistoquímicocon anticuerpos monoclonales (contra pro-teína AA, cadenas ligeras, transtirretina,etc) del depósito de amiloide permite confirmar el diagnóstico específico de lavariedad de amiloidosis. El estudio con mi-croscopía electrónica demuestra la es-tructura fibrilar característica de la sus-tancia amiloide. Diversas técnicas depurificación de proteínas y de biología mo-lecular con la secuenciación del ADN, per-miten determinar la variedad específica yla mutación concreta en los casos de ami-loidosis hereditaria.La demostración anatomopatológica de losdepósitos amiloides se realiza en los ór-ganos con sospecha clínica y/o analíticade infiltración: riñón, hígado, nervio peri-férico, corazón, sinovial o mediante la re-alización de biopsias en zonas con altorendimiento diagnóstico (grasa subcutá-nea, recto y glándulas salivares). Proba-blemente, la aspiración de la grasa sub-cutánea abdominal representa la técnicade elección en el diagnóstico de la ami-loidosis sistémica, por su alto rendimien-to y escasa agresividad16 (fig. 7). En la
tabla 3 se exponen las diferentes sensibi-lidades diagnósticas de diversos órganospara el diagnóstico de amiloidosis sisté-mica. La gammagrafía con componente P de laamiloide marcado con 123I es una técnicanovedosa de imagen con una sensibilidadpara el diagnóstico de amiloidosis sisté-mica AL y AA cercana al 100% y que permite además realizar el seguimiento ycontrol del paciente; no obstante, su dis-ponibilidad está limitada por dificultadestécnicas y no está al alcance de la mayo-ría de hospitales17.
PronósticoEl pronóstico de la amiloidosis depende-rá del tipo de amiloidosis y de la reper-cusión en los órganos afectos en el mo-mento de su diagnóstico.La amiloidosis AL es la que tiene un peorpronóstico con una supervivencia media-na de 14 meses una vez realizado el diagnóstico; no obstante, ocasionalmente pueden observarse pacientes con una su-pervivencia de varios años. Las causas másfrecuentes de muerte son la míocardiopa-tía y la afección renal. Se han identifi-cado los siguientes factores de mal pro-nóstico: insuficiencia cardíaca, niveles ele-vados de β2-microglobulina, componentemonoclonal de cadenas ligeras en plasmay orina, hepatomegalia y coexistencia demieloma múltiple18. La amiloidosis AA tiene mejor pronósticoque la amiloidosis AL y dependerá en granmanera de la posibilidad de minimizar laactividad inflamatoria de la enfermedadde base. La supervivencia mediana en unestudio americano se cifró en 2 años10,aunque existen pacientes en que tras con-seguir un buen control de su enfermedadde base, los signos y síntomas de amiloi-
dosis mejoran notablemente o incluso de-saparecen, con remisiones prolongadas.En algunos casos se ha demostrado unaregresión de los depósitos de amiloide conla gammagrafía con componente P en pa-cientes con artritis crónica juvenil y ar-tritis reumatoide. La causa más frecuen-te de muerte es la nefropatía y se haidentificado la presencia de insuficienciarenal (cifras de creatinina sérica superiora 2 mg/dl) y la hipoalbuminemia como losprincipales factores de mal pronóstico10,19.Otras causas de muerte son la hemorra-gia digestiva o los procesos infecciosos.En la artritis reumatoide del adulto, laamiloidosis representa la causa de muer-te del 2%-10% de casos20 y en la artritiscrónica juvenil se constituye en la causamás frecuente de fallecimiento despuésde los accidentes. Un aspecto a destacaren este tipo de amiloidosis es la existen-cia de depósitos subclínicos en un por-centaje importante de casos si se realizauna búsqueda sistemática de los mismos.En nuestro Servicio de Reumatología un16% y 7% de pacientes con artritis reu-matoide y espondilitis anquilosante res-pectivamente, de más de cinco años deevolución, presentan depósitos de ami-loide en el aspirado de la grasa subcutá-nea abdominal; en la mayoría de casosestos depósitos permanecen subclínicos,incluso después de varios años de segui-miento21,22. En la amiloidosis del paciente en diálisis,el cuadro clínico es progresivo y sólo me-jora con el trasplante renal. En las formasde amiloidosis hereditaria el pronósticoes malo, a excepción de algunas varieda-des en que el trasplante hepático ha dadoresultados esperanzadores.
TratamientoTratamiento de la amiloidosis AL
No existe un tratamiento curativo para estetipo de amiloidosis, que como se ha co-mentado es de muy mal pronóstico. Laquimioterapia con melfalan y prednisonaaumenta muy discretamente la supervi-vencia de estos enfermos. El trasplante demédula ósea o de precursores hematopo-yéticos puede ser eficaz en aquellos casosen que no existe una gran pérdida de fun-ción de los órganos afectos, aunque noestá exento de riesgos importantes y mor-talidad postoperatoria23.
1713
AMILOIDOSIS
Fig. 6. Biopsia rectal en un paciente con amiloidosis AA. Pre-sencia de sustancia amiloide que se tiñe con rojo Congo en lasuperficie de la mucosa rectal.
Fig. 7. Biopsia-aspiración de grasa subcutánea abdominal enun paciente con artritis reumatoide. Presencia de sustanciaamiloide en la grasa que se tiñe con rojo Congo y presenta bi-rrefringencia verde-manzana al microscopio óptico de luz po-larizada.
TABLA 3Sensibilidad diagnóstica aproximada de losdistintos tipos de biopsia en la amiloidosis
sistémica
Renal 90%-95%
Hepática 60%-90%
Rectal 70%-85%
Grasa subcutánea abdominal 60%-85%
Gingival 50%-65%
Gastroduodenal 70%-90%
Esplénica 100%

Tratamiento de la amiloidosis AA
El tratamiento básico consiste en dismi-nuir al máximo la actividad de la enfer-medad en los procesos inflamatorios cró-nicos y curar las infecciones crónicas conlos antibióticos adecuados, con lo que seintenta disminuir la síntesis de SAA. En elcaso de los reumatismos inflamatorios, di-versos citostáticos han demostrado su uti-lidad, como la azatioprina, metotrexato,ciclofosfamida y clorambucil; no obstan-te, los dos últimos y en especial el clo-rambucil son hoy en día los fármacos máseficaces, sobre todo en la artritis reuma-toide del adulto y la artritis crónica juve-nil24,25. En estos pacientes los episodios in-fecciosos deben tratarse de forma rápidapara prevenir nuevos aumentos de SAA.En la fiebre mediterránea familiar, la col-chicina a dosis superiores a 1,5 mg/díaademás de controlar los episodios agudosy recidivantes de artritis, serositis y fie-bre, logra prevenir el desarrollo de ami-loidosis26. La eficacia de la colchicina enla amiloidosis AA no debida a fiebre me-diterránea familiar está por demostrar.
Tratamiento de otras formas de amiloidosis
No existe un tratamiento específico paralas otras variedades de amiloidosis. Comose ha comentado, el trasplante hepáticoha sido eficaz en formas concretas de ami-loidosis heredofamiliar, como la debida aldepósito de transtirretina por la mutaciónde la metionina en la posición 3027. En laamiloidosis por β2-microglobulina, el tras-plante renal mejora la sintomatología clí-nica de los pacientes, pero probablemen-te no reduce los depósitos de amiloide yaestablecidos28. Finalmente, es preciso reseñar la impor-tancia de las medidas de soporte en el tratamiento sintomático de la amiloidosis;dichas medidas dependerán de la distri-bución y repercusión orgánica de los de-pósitos. La diálisis constituye un métodoefectivo que incrementa la supervivenciade pacientes con amiloidosis AA e insufi-ciencia renal terminal. Existe poca ex-
periencia con el trasplante renal en la nefropatía amiloidea AA, aunque los re-sultados hasta la fecha parecen aceptables. Por otra parte, se están ensayando nue-vas modalidades terapéuticas dirigidas adisminuir el proceso de formación de pro-teína fibrilar29. En este sentido, la terbu-talina y aminofilina, a través de una ele-vación del AMP cíclico han demostrado sueficacia en inhibir la amiloidosis AA en unmodelo experimental murino30. Es proba-ble que en un futuro próximo se den a co-nocer los resultados de distintas aproxi-maciones terapéuticas en seres humanos,destinadas a disminuir el proceso de ami-loidogénesis a través de fármacos com-petidores en la interacción entre la prote-ína fibrilar y otras sustancias presentes enel depósito de amiloide como los protei-noglicanos.
BIBLIOGRAFÍA
1. Falk RH, Comenzo RL, Skinner M. The systemic amyloi-dosis. N Engl J Med, 1997; 337: 898-909.2. Campistol JM. Amiloidosis. En: Farreras Rozman. Medi-cina Interna (14ed). Barcelona: Harcourt ed. 2000; I59:I305-I309.3. Roselló R. Amiloidosis primaria y secundaria. En: Pas-cual E, Rodríguez V, Carbonell J, Gómez-Reino JJ. Tratadode Reumatología. Madrid: Aran ed. 1998; 1.189-1.199.4. Husby G, Araki S, Benditt E. The 1990 Guidelines fornomenclature and classification of amyloid and amyloido-sis. En: Natvig J, ed. Amyloid and amyloidosis 1990, Dor-drecht: Kluwer Academic Publisher, 1991; 7-11.5. Husby G. Amyloidosis. Semin Arthritis Rheum 1992;22: 67-82.6. Friman C, Pettersson T. Amyloidosis. Cur Op Rheuma-tol 1996; 8: 62-71.7. Prusiner SB, Scott MR, DeArmond SJ, Cohen FE. PrionProtein Biology. Cell 1998; 93: 337-348.8. Pepys MB. Amyloidosis: some recent developments. Q JMed 1988; 252: 283-298.9. Hazenberg BPC, van Rijswijk MH. Clinical and therapeu-tic aspects of AA amyloidosis. Baill Clin Rheumatol 1994;8: 661-690.10. Gertz MA. Kyle RA. Secondary systemic amyloidosis:response and survival in 64 patients. Medicine (Baltimore)1991; 70: 246-256.11. De la Sierra A, Cardellach F, Ingelmo M, Roselló R, Bal-cells A. Amiloidosis: aspectos clínicos y biológicos en 60casos. Med Clin (Barc) 1985; 84: 297-301.12. Moriguchi M, Terai C, Koseki Y, Uesato M, NakajimaA, Inada S, et al. Influence of genotypes at SAA 1 and SAA2 loci on development and the lenght of latent period of se-condary AA-amyloidosis in patients with rheumatoid arthri-tis. Hum Genet 1999; 105: 360-366.
13. Muñoz Gómez J. Amiloidosis secundaria en las enfer-medades reumáticas. Seminarios de la Fundación Españolade Reumatología 2000; 4: 225-231.14. Muñoz Gómez J, Bergadà-Barado E, Gómez-Pérez R,Llopart-Buisan E, Subias-Sobrevia E, Roter-Querol J, Sole-Arques M. Amyloid arthropathy in patients undergoing pe-riodical hemodiálisis for chronic renal failure: a new com-plication. Ann Rheum Dis 1985; 44: 729-733. 15. Muñoz Gómez J. Diagnóstico y seguimiento de la ami-loidosis. Rev Esp Reumatol 1996; 23: 21-25.16. Rosselló R, Solé M. La biopsia de la grasa subcutáneaabdominal en el diagnóstico de amiloidosis. Med Clin(Barc) 1985; 85: 691-694.17. Hawkins PN, Lavender JP, Pepys MB. Evaluation ofsystemic amyloidosis by scintigraphy with 123 I-labeled serum amyloid P component. N Engl J Med 1990; 323:508- 513.18. Kyle RA, Gertz MA. Primary systemic amyloidosis: cli-nical and laboratory features in 474 cases. Semin Haema-tol 1995; 32: 45-49.19. Fiter Areste J, Nolla Sole JM, Gómez Vaquero J, Valver-de García J, Roig Escofet D. Amiloidosis secundaria a la ar-tritis reumatoide. Estudio clínico de una serie de 29 casos.An Med Interna 1999; 16: 615-619.20. Mitchell DM, Spitz PW, Young DY, Bloch DA, McShaneDJ, Fries JF. Survival, prognosis and causes of death inrheumatoid arthritis. Arthritis Rheum 1986; 29: 706-714.21. Gratacós J, Orellana C, Sanmartí R, Sole M, Collado A,Gómez-Casanovas E, et al. Secondary amyloidosis in anky-losing spondylitis. A systematic survey of 137 patientsusing abdominal fat aspiration. J Rheumatol 1997; 24: 912-915.22. Gómez-Casanovas E, Sanmartí R, Solé M, Cañete JD,Muñoz-Gómez J. The clinical significance of amyloid fat de-posits in rheumatoid artritis: a systematic long-term follow-up study using subcutaneous abdominal fat aspitration.Arthritis Rheum 2001; 44: 66-72.23. Sezer O, Eucker J, Schmid P, Possinger K. New thera-peutic approaches in primary systemical amiloidosis. AnnHematol 2000; 79: 1-6.24. Berglund K, Thysell H, Keller C. Results, principles andpitfalls in the management of renal AA amyloidosis: 10-21year follow-up of 16 patients with rheumatic disease trea-ted with alkylating cytostatics. J Rheumatol, 1993; 20:2.051-2.057.25. Woo P. Amyloidosis in pediatric rheumatic diseases. JRheumatol 1992; 19: 10-16.26. Samuels J, Aksentijevich I, Torosyan Y, Centola M,Deng Z, Sood R, Kastner DL. Familial Mediterranean feverat the millennium. Clinical spectrum, ancient mutations,and a survey of 100 American referrals to the National Institutes of Health. Medicine (Baltimore), 1998; 77: 268-297.27. Suhr OB, Herlenius G, Friman S, Ericzon BG. Livertransplantation for hereditary transthtretin amyloidosis. Li-ver Transpl 2000; 6: 263-276.28. Campistol JM, Muñoz-Gómez J, Oppenheimer F, Ri-card HJ, Villardell J, Andreu J. Renal transplantation fordyalisis arthropathy. Lancet 1988; I: 900.29. Gilmore JD, Hawkins PN, Pepys M. Amyloidosis: A re-view of recent diagnostic and therapeutic developments.Br J Haematol, 1997; 99: 245-256.30. Brandwein S, Sipe JD, Cohen AS. Combined treatmentwith terbutaline and aminophyline inhibits experimentalamyloidosis in mice. Arthritis Rheum 1994; 37: 1757-1760.
1714
ENFERMEDADES DEL SISTEMA INMUNE (IX)

Anatomía y fisiología de lahipodermis
La hipodermis o tejido celular subcutáneoes la parte profunda de las tres estructu-ras de la piel, localizándose entre la dermisprofunda o reticular y la fascia muscularsuperficial. Está formada por lobulillos detejido adiposo compuestos por adipocitosmaduros, células encargadas de fabricar yacumular lípidos en su interior, cuyo nú-cleo está comprimido contra la membra-na citoplasmática adoptando el aspecto decélulas en anillo de sello. Estos lobulillosestán separados por tabiques conectivosfinos que constituyen los septos interlo-bulillares. Los septos son una continuacióndel tejido conectivo de la dermis reticularsuprayacente y por ellos discurren las es-tructuras vasculares y nerviosas. Están for-mados por fibras colágenas y elásticas.La hipodermis tiene un grosor variable de-pendiendo de la región anatómica, la edad,el sexo, estado nutricional, factores gené-ticos, endocrinológicos y metabólicos. Esdos a tres veces más ancha que la dermis,excepto en los párpados, algunas partesde la cara, los genitales y los dedos.El tejido subcutáneo actúa como aislantetérmico y protector mecánico frente atraumatismos, y como reservorio energé-tico. Su vascularización es terminal a di-ferencia del resto de la piel, teniendo unplexo propio sin anastomosis con la der-mis. También en la hipodermis hay ane-jos cutáneos, los bulbos del folículo pilo-so y la porción secretora de las glándulassudoríparas, que se disponen entre los lo-bulillos.
Clasificación de laspaniculitisLas paniculitis son el grupo de procesosen los que existe una inflamación prima-
ria del tejido subcutáneo o hipodermis. Lamayoría de estas enfermedades se carac-terizan clínicamente por la aparición denódulos eritematosos de consistencia fir-me, múltiples y dolorosos, en las piernas.Para su diagnóstico es fundamental el es-tudio histopatológico, siendo necesario ha-cer una biopsia grande y profunda de unalesión reciente no evolucionada en la ma-yoría de los casos. No es aconsejable lautilización del punch o sacabocados.En la actualidad, la mejor forma de clasi-ficar las paniculitis es desde el punto devista histopatólogico (tabla 1), dependien-do de si la afectación es principalmenteen el lobulillo o en el septo y si se asociaa vasculitis o no. También es importantela composición celular del infiltrado infla-matorio. En realidad no existen las pani-culitis septales o lobulillares puras, todas
son mixtas, pero sí son predominante-mente septales o predominantemente lo-bulillares.Desde el punto de vista del médico clíni-co, pueden clasificarse según su etiología(tabla 2), que es la clasificación que va-mos a utilizar en el desarrollo de estetema.
Paniculitis reactivas
Eritema nudoso
Epidemiología y etiopatogenia
El eritema nudoso es un patrón reactivocutáneo relativamente frecuente que pue-de ser causado por múltiples procesos (ta-bla 3). Bastantes casos son idiopáticos1.En nuestro medio la causa más frecuenteen los niños es la infección estreptocóci-ca, en las mujeres jóvenes los anticon-ceptivos orales y en los adultos la infec-ción por micobacterias y la sarcoidosis.Aparece con más frecuencia a los 20-30años, y predomina en el sexo femenino(3:1). Existe una variación estacional, sien-do más frecuente en primavera y menosen verano.
1723
PANICULITISG. Roustan GullónServicio de Dermatología. Hospital Universitario. Clínica Puerta de Hierro. Madrid.
Medicine 2001; 8(33): 1723-1730
TABLA 1Clasificación de las paniculitis desde el punto
de vista histopatológico
Lobulillares
Con vasculitisEritema indurado de BazinEritema nudoso leprosoFenómeno de LucioEnfermedad de Crohn
Sin vasculitisPaniculitis lúpicaPaniculitis asociada a dermatomiositisPaniculitis asociada a artritis reumatoidePanicultis pancreáticaNecrosis grasa del recién nacidoEscleredema neonatorumPaniculitis postesteroideaPaniculitis facticia y traumáticaInfecciones por micobacterias atípicasMicosis subcutáneasLipodistrofia
Septales
Con vasculitisPeriarteritis nodosaVasculitis leucocitoclásticaTromboflebitis migratoria superficialCalcifilaxis
Sin vasculitisEritema nudosoMorfea profundaNecrobiosis lipoidícaGranuloma anularNódulo reumatoideXantogranuloma necrobiótico
TABLA 2Clasificación de las paniculitis desde el punto de
vista etiopatogénico
Paniculitis reactivas
Eritema nudosoVasculitis nodular
Paniculitis en enfermedades inmunológicas o autoinmunes
Lupus paniculitisPaniculitis en esclerodermiaPaniculitis en dermatomiositisPaniculitis en artritis reumatoidePeriarteritis nodosa cutánea
Paniculitis metabólica
Necrosis grasa pancreáticaNecrosis grasa del recién nacidoEscleredema neonatorumPaniculitis post-esteroideaPaniculitis por deficiencia de α-1 antitripsinaCalcifilaxis
Paniculitis físicas
Paniculitis traumáticaPaniculitis facticia
Paniculitis infecciosa
Paniculitis neoplásica
Paniculitis histiocítica citofágica
Paniculitis paraneoplásica
Tromboflebitis migratoria superficial

El mecanismo patogénico es poco cono-cido. Se produciría una reacción de hi-persensibilidad retardada desencadenadapor diversos estímulos antigénicos queafectaría principalmente a los elementosvasculares de los septos.
Clínica
Se caracteriza clínicamente por la apari-ción aguda de nódulos redondeados eri-tematosos (fig. 1), calientes y dolorosos,ligeramente sobreelevados, mal delimita-dos, de superficie brillante, con un tama-ño de 2-6 centímetros de diámetro, quese localizan en la cara anterior de losmiembros inferiores, sobre todo en laspiernas, alrededor de rodillas y tobillos.Pueden aparecer también en brazos y másraramente en cara y cuello. Las lesionessuelen ser múltiples, con una distribuciónbilateral y con cierta simetría (fig. 2). Aveces los nódulos confluyen y forman pla-cas grandes. Evolucionan por brotes du-
rante una semana con una tendencia pos-terior a la remisión espontánea en un mes,cambiando de color como un hematoma(cambios contusiformes) sin dejar cicatrizresidual. No suelen ulcerarse.El cuadro puede estar precedido por unepisodio de faringitis o infección respira-toria previa, y se suele acompañar de afec-tación del estado general, con fiebre alta,cefalea, astenia, diarrea, y pérdida depeso. Aparecen artralgias en al menos lamitad de los casos, sobre todo en la rodi-lla. La sinovitis periférica es también fre-cuente.Existe una variante rara, el eritema nu-doso crónico, que se caracteriza por le-siones solitarias unilaterales migratoriaslocalizadas en una pierna y de duraciónmás prolongada, con una morfología ar-ciforme y un centro violáceo, que afectasobre todo a mujeres adultas, y que no seacompaña de sintomatología general.
Las exploraciones complementarias mues-tran una elevación de la velocidad de se-dimentación globular (VSG) y la β-2 glo-bulina. En estos pacientes es obligado unestudio general minucioso y completo (he-mograma, exudado faríngeo, ASLO, ra-diografía de tórax, Mantoux) para descar-tar las diferentes causas que puedendesencadenar el eritema nudoso.
Histopatología
Los hallazgos histopatológicos son, de-pendiendo del tiempo de evolución, unapaniculitis septal sin vasculitis (fig. 3) conla presencia de un infiltrado inflamatoriocompuesto por neutrófilos en fases ini-ciales e histiocitos y linfocitos en fases más avanzadas. Un hallazgo muy carac-terístico es el granuloma radial de Mies-cher2, histiocitos pequeños agrupados circularmente alrededor de una grieta ohendidura. En los casos asociados al sín-drome de Sweet puede observarse un in-filtrado neutrofílico abundante. En lesio-nes evolucionadas, la fibrosis septal esprominente.
Tratamiento y evolución
El tratamiento consiste principalmente eneliminar la causa desencadenante. Al evo-lucionar la mayor parte de los casos a laremisión espontánea suele ser suficientecon reposo y antiinflamatorios no este-roideos (AINE).El yoduro potásico es un fármaco bastan-te eficaz en ésta y otras formas de pa-
1724
ENFERMEDADES DEL SISTEMA INMUNE (IX)
TABLA 3Enfermedades asociadas a eritema nudoso
Infecciones bacterianas
Estreptococo β hemolítico*Infección respiratoria de vías altas**Yersinia enterocolítica*Tuberculosis*LeptospirosisClamidiasSalmonelosis
Infecciones víricas
Infección respiratoria de vías altas**Mononucleosis infecciosaHepatitis BNódulo de los ordeñadores
Infecciones por hongos
DermatofitosisHistoplasmosisBlastomicosisCoccidiodomicosisEsporotricosis
Protozoos
ToxoplasmosisGiardiasis
Fármacos
Anticonceptivos orales*SulfonamidasCompuestos halogenadosBromurosAntibióticos orales
Otras enfermedades
Enfermedad inflamatoria intestinal*Sarcoidosis*Enfermedad de BehçetSíndrome de SweetLinfomas y Leucemias
*Más frecuentes; **agente causal no identificado.
Fig. 1. Nódulos con superficie eritematosa característicos del eritemanudoso.
Fig. 2. Eritema nudoso: nódulos múltiples, bilaterales, en cara anteriorde extremidades inferiores. Fig. 3. Eritema nudoso: paniculitis de predominio septal.

niculitis. Se utiliza a una dosis de 300mgcada 8 horas. Su mecanismo de acción se desconoce. No debe ser usado en el embarazo por el riesgo de bocio e hipoti-roidismo en el feto. Puede producir reac-ciones adversas como iododerma, hiper-secreción salivar y lacrimal, trastornosgastrointestinales, ansiedad, depresión, ohipotiroidismo. El uso de prednisona oralen casos extensos y con sintomatologíageneral importante debe ser valorado con cuidado, sobre todo si existe la posi-bilidad de un proceso infeccioso subya-cente.
Vasculitis nodular/eritemaindurado de Bazin
Epidemiología y etiopatogenia
La vasculitis nodular es un patrón reacti-vo cutáneo de patogenia diferente de unasáreas geográficas a otras. Se utiliza el tér-mino general de vasculitis nodular si es decausa desconocida, y si está en relacióncon la tuberculosis se denomina eritemaindurado de Bazin. Es un proceso frecuente que afecta sobretodo a mujeres en la edad media de lavida. La insuficiencia venosa, el frío, o la hu-medad actuarían como factores predispo-nentes en algunos casos.El mecanismo etiopatogénico exacto de lavasculitis nodular es desconocido. Podríaser multifactorial. Por un lado existiría unproceso vasculítico mediado por inmuno-complejos que llevaría a la aparición defenómenos tromboisquémicos con la con-siguiente necrosis de adipocitos y apari-ción de inflamación granulomatosa. Porotro existiría en algunos casos una reac-ción de hipersensibiliadad retardada fren-te al bacilo tuberculoso u otros antígenosbacterianos. Con las nuevas técnicas dereacción en cadena de la polimerasa (PCR)se ha conseguido identificar el ADN delbacilo Mycobacterium tuberculosis en le-siones de eritema indurado en bastantescasos3. Sin embargo, nunca se ha podidocultivar, por lo que no tendría capacidadinfectiva. No existe aún acuerdo sobre siel daño vascular primario ocurre en venaso arterias.
Clínica
Se caracteriza por la aparición progresivade nódulos hipodérmicos en la cara pos-
terior de las piernas con descamaciónsuperficial (fig. 4). Las lesiones son múlti-ples, dolorosas a la presión, de consis-tencia firme, con una evolución crónica yrecidivante. En el transcurso de la enfer-medad pueden ulcerarse (fig. 5) y al curardejar una cicatriz atrófica residual. La dis-tribución de los nódulos es bilateral, pu-diendo aparecer también en muslos y bra-zos, e incluso diseminarse. En ocasionesasientan sobre piel eritemato-cianótica yes frecuente que empeoren en inviernopor el frío.
Histopatología
Se observa una paniculitis lobulillar (fig. 6),con necrosis eosinofílica de adipocitos,asociada a inflamación de la pared y ne-crosis fibrinoide en las grandes venas delos septos4. La necrosis del lobulillo grasorecuerda al caseum, que se encuentra ro-deada de granulomas tuberculoides for-mados por células epitelioides y célulasgigantes multinucleadas. No existen dife-rencias histológicas significativas entre loscasos en que la PCR para el bacilo tu-
berculoso es positiva de los casos en quees negativa.
Tratamiento y evolución
Es importante el reposo y evitar el frío. Seutilizan los AINE y en casos graves los es-teroides orales (descartando previamenteun proceso tuberculoso). También es efi-caz el yoduro potásico. En algunos casosel tratamiento con tuberculostáticos con-sigue una desaparición de las lesiones cu-táneas. El proceso es crónico en la mayo-ría de los pacientes y persiste durantemucho tiempo.
Paniculitis en enfermedadesinmunológicas o autoinmunes
Paniculitis lúpica
Epidemiología y patogenia
La paniculitis lúpica o lupus profundo esuna variedad infrecuente de lupus erite-matoso5. Es la única manifestación clíni-ca en más de la mitad de los casos, aso-ciándose a lupus eritematoso cutáneocrónico en un 20%-50% de los casos y al lupus eritematoso sistémico en un 10%-35% de los casos, aunque en pocasocasiones con afectación renal o grave.Excepcionalmente se asocia a otras en-fermedades autoinmunes como el síndro-me de Sjögren, artritis reumatoide y en-fermedad mixta del tejido conjuntivo.Es más frecuente en pacientes de sexo fe-menino (4:1), y suele aparecer entre los30-60 años de edad. Existe algún caso aso-ciado a lupus neonatal. Su etiopatogeniaes similar al lupus eritematoso cutáneocrónico o sistémico.
1725
PANICULITIS
Fig. 5. Vasculitis nodular: lesiones ulceradas.
Fig. 4. Vasculitis nodular: nódulos en cara posterior de piernas con fe-nómenos de atrofia e hiperpigmentación en su superficie.
Fig. 6. Vasculitis nodular: paniculitis de predominio lobulillar.
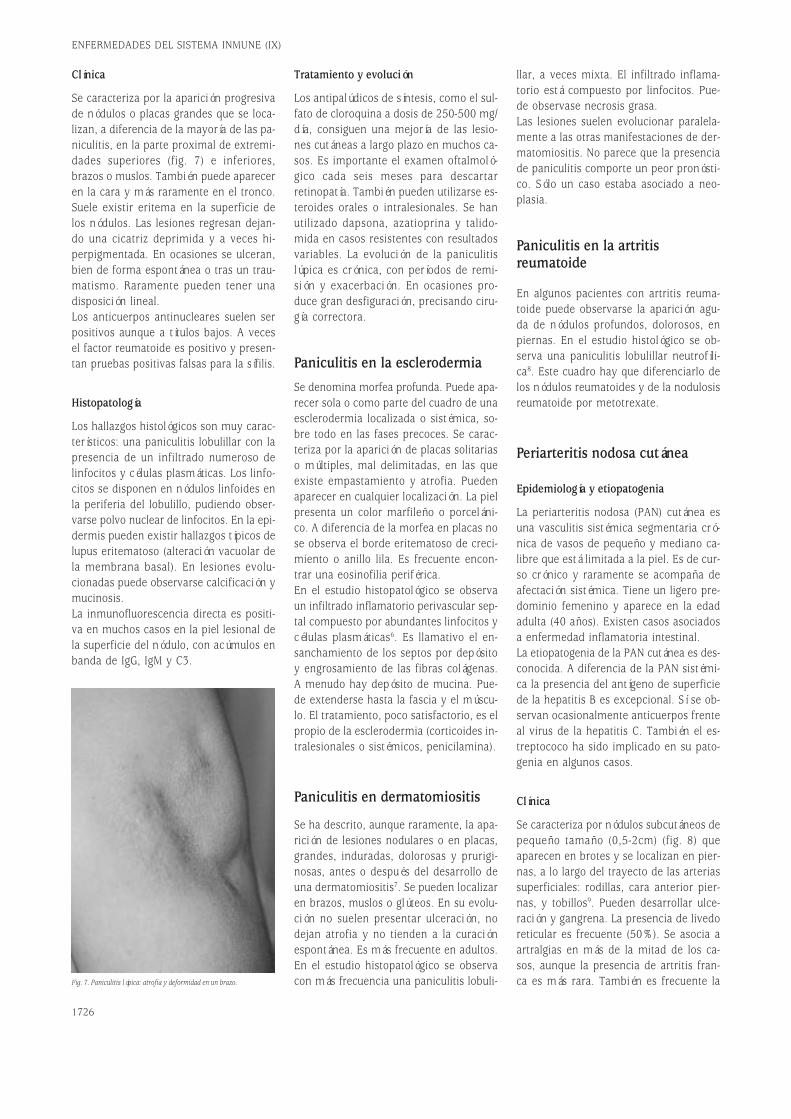
Clínica
Se caracteriza por la aparición progresivade nódulos o placas grandes que se loca-lizan, a diferencia de la mayoría de las pa-niculitis, en la parte proximal de extremi-dades superiores (fig. 7) e inferiores,brazos o muslos. También puede apareceren la cara y más raramente en el tronco.Suele existir eritema en la superficie delos nódulos. Las lesiones regresan dejan-do una cicatriz deprimida y a veces hi-perpigmentada. En ocasiones se ulceran,bien de forma espontánea o tras un trau-matismo. Raramente pueden tener unadisposición lineal.Los anticuerpos antinucleares suelen serpositivos aunque a títulos bajos. A vecesel factor reumatoide es positivo y presen-tan pruebas positivas falsas para la sífilis.
Histopatología
Los hallazgos histológicos son muy carac-terísticos: una paniculitis lobulillar con lapresencia de un infiltrado numeroso delinfocitos y células plasmáticas. Los linfo-citos se disponen en nódulos linfoides enla periferia del lobulillo, pudiendo obser-varse polvo nuclear de linfocitos. En la epi-dermis pueden existir hallazgos típicos delupus eritematoso (alteración vacuolar dela membrana basal). En lesiones evolu-cionadas puede observarse calcificación ymucinosis.La inmunofluorescencia directa es positi-va en muchos casos en la piel lesional dela superficie del nódulo, con acúmulos enbanda de IgG, IgM y C3.
Tratamiento y evolución
Los antipalúdicos de síntesis, como el sul-fato de cloroquina a dosis de 250-500 mg/día, consiguen una mejoría de las lesio-nes cutáneas a largo plazo en muchos ca-sos. Es importante el examen oftalmoló-gico cada seis meses para descartarretinopatía. También pueden utilizarse es-teroides orales o intralesionales. Se hanutilizado dapsona, azatioprina y talido-mida en casos resistentes con resultadosvariables. La evolución de la paniculitislúpica es crónica, con períodos de remi-sión y exacerbación. En ocasiones pro-duce gran desfiguración, precisando ciru-gía correctora.
Paniculitis en la esclerodermia
Se denomina morfea profunda. Puede apa-recer sola o como parte del cuadro de unaesclerodermia localizada o sistémica, so-bre todo en las fases precoces. Se carac-teriza por la aparición de placas solitariaso múltiples, mal delimitadas, en las queexiste empastamiento y atrofia. Puedenaparecer en cualquier localización. La pielpresenta un color marfileño o porceláni-co. A diferencia de la morfea en placas nose observa el borde eritematoso de creci-miento o anillo lila. Es frecuente encon-trar una eosinofilia periférica.En el estudio histopatológico se observaun infiltrado inflamatorio perivascular sep-tal compuesto por abundantes linfocitos ycélulas plasmáticas6. Es llamativo el en-sanchamiento de los septos por depósitoy engrosamiento de las fibras colágenas.A menudo hay depósito de mucina. Pue-de extenderse hasta la fascia y el múscu-lo. El tratamiento, poco satisfactorio, es elpropio de la esclerodermia (corticoides in-tralesionales o sistémicos, penicilamina).
Paniculitis en dermatomiositis
Se ha descrito, aunque raramente, la apa-rición de lesiones nodulares o en placas,grandes, induradas, dolorosas y prurigi-nosas, antes o después del desarrollo deuna dermatomiositis7. Se pueden localizaren brazos, muslos o glúteos. En su evolu-ción no suelen presentar ulceración, nodejan atrofia y no tienden a la curaciónespontánea. Es más frecuente en adultos.En el estudio histopatológico se observacon más frecuencia una paniculitis lobuli-
llar, a veces mixta. El infiltrado inflama-torio está compuesto por linfocitos. Pue-de observase necrosis grasa.Las lesiones suelen evolucionar paralela-mente a las otras manifestaciones de der-matomiositis. No parece que la presenciade paniculitis comporte un peor pronósti-co. Sólo un caso estaba asociado a neo-plasia.
Paniculitis en la artritisreumatoide
En algunos pacientes con artritis reuma-toide puede observarse la aparición agu-da de nódulos profundos, dolorosos, enpiernas. En el estudio histológico se ob-serva una paniculitis lobulillar neutrofíli-ca8. Este cuadro hay que diferenciarlo delos nódulos reumatoides y de la nodulosisreumatoide por metotrexate.
Periarteritis nodosa cutánea
Epidemiología y etiopatogenia
La periarteritis nodosa (PAN) cutánea esuna vasculitis sistémica segmentaria cró-nica de vasos de pequeño y mediano ca-libre que está limitada a la piel. Es de cur-so crónico y raramente se acompaña deafectación sistémica. Tiene un ligero pre-dominio femenino y aparece en la edadadulta (40 años). Existen casos asociadosa enfermedad inflamatoria intestinal.La etiopatogenia de la PAN cutánea es des-conocida. A diferencia de la PAN sistémi-ca la presencia del antígeno de superficiede la hepatitis B es excepcional. Sí se ob-servan ocasionalmente anticuerpos frenteal virus de la hepatitis C. También el es-treptococo ha sido implicado en su pato-genia en algunos casos.
Clínica
Se caracteriza por nódulos subcutáneos depequeño tamaño (0,5-2cm) (fig. 8) queaparecen en brotes y se localizan en pier-nas, a lo largo del trayecto de las arteriassuperficiales: rodillas, cara anterior pier-nas, y tobillos9. Pueden desarrollar ulce-ración y gangrena. La presencia de livedoreticular es frecuente (50%). Se asocia aartralgias en más de la mitad de los ca-sos, aunque la presencia de artritis fran-ca es más rara. También es frecuente la
1726
ENFERMEDADES DEL SISTEMA INMUNE (IX)
Fig. 7. Paniculitis lúpica: atrofia y deformidad en un brazo.

mononeuritis periférica múltiple. Otros da-tos acompañantes son fiebre, anemia yelevación de la VSG. La ausencia de anor-malidades inmunológicas es la norma (an-ticuerpos antinucleares [ANA], factor reu-matoide [FR], crioglobulinas, disminucióndel complemento).En la PAN sistémica también es frecuen-te la presencia de lesiones cutáneas, peroa diferencia de la PAN cutánea lo carac-terístico es la aparición de lesiones de vas-culitis leucocitoclásica clásica o púrpurapalpable acompañando a todo el cortejosintomático general (hipertensión, protei-nuria…).Algunos autores sugieren que en la PANexistiría, al igual que en el lupus eritema-toso, un espectro de enfermedad que fluc-tuaría desde una afectación cutánea loca-lizada en los casos más leves hasta unaafectación multiorgánica generalizada enlos casos más graves.
Histopatología
Se observa una paniculitis septal y vascu-litis con depósitos fibrinoides, trombosisde arteriolas y arterias en dermis profun-da e hipodermis. Al ser característica laafectación segmentaria a veces hay quehacer varios cortes. La inmunofluorescen-cia directa es positiva en un 60% de loscasos.
Tratamiento y evolución
Los corticoides orales consiguen una re-misión satisfactoria de los brotes agudos.En casos leves pueden utilizarse los AINE,solos o asociados a prednisona a dosis ba-jas (20 mg/día).La evolución del proceso es crónica, conbrotes de reagudización. Es importante elseguimiento de estos pacientes ya que al-gunos casos, sobre todo los ulcerados, pue-den evolucionar a PAN sistémica.
Paniculitis metabólicas
Paniculitis pancreáticas
Epidemiología y etiopatogenia
La necrosis grasa asociada a enfermedadpancreática es una forma rara de pa-niculitis. Se observa en menos del 2% delos enfermos pancreáticos. El factor etio-lógico más frecuente asociado es la pancreatitis aguda secundaria a alcoho-lismo crónico y a colelitiasis10. Otro fac-tor etiológico importante es el carcinomade páncreas (20%-40%), sobre todo decélulas acinares, debido a que este tipode neoplasia conserva su capacidad secretora. Más raramente se observa enrelación con medicamentos (corticoides,sulfasalazina, tiazidas, anticonceptivosorales, AINE), seudoquistes, pancreatitiscrónica, traumatismos, o malformacio-nes. Aparece entre los 40-60 años conpredomino de un sexo u otro según laetiología del proceso pancreático.El desarrollo de la paniculitis pancreáticase debería a enzimas pancreáticas comola lipasa (preferentemente), la amilasa, latripsina, la quimiotripsina, y la elastasa.Al ser liberadas pasarían al torrente cir-culatorio linfático y sanguíneo para depo-sitarse secundariamente en la grasa de lahipodermis y de otros sitios, produciendonecrosis de los adipocitos.
Clínica
Aparecen nódulos subcutáneos dolorososeritematovioláceos de tamaño variable. Selocalizan en piernas, con frecuencia en región maleolar, muslos o nalgas. A ve-ces pueden localizarse en tronco y extre-midades superiores. Los casos más gra-ves se ulceran drenando un materialcremoso. Se resuelven dejando atrofia yuna hiperpigmentación residual. Es el signo de presentación de la enfermedadpancreática en un 40% de los casos, conmayor frecuencia aún cuando se asocia a carcinoma, siendo un factor de mal pronóstico, con una supervivencia menorde un año en la mayoría de los pacien-tes. El estudio de las enzimas pancreáticas re-vela un aumento de los niveles de amila-sa (sobre todo) y/o lipasa en sangre peri-férica, y amilasa en orina. En muchoscasos sólo aumenta el valor de una de
ellas. También se acompaña de leucocito-sis. La eosinofilia se observa más en loscasos asociados a carcinoma.No es infrecuente que la paniculitis pan-creática se acompañe de afectación deotras estructuras. Artralgias y artritis es-tán presentes en un 50%-80% de los ca-sos, afectando sobre todo a las articula-ciones de miembros inferiores. El líquidoobtenido por punción presenta un aspec-to cremoso debido a la necrosis grasa pe-riarticular. Cuando afecta a las membra-nas serosas se puede observar derramepleural, derrame pericárdico y ascitis. Ladestrucción del tejido graso intramedulardel hueso origina lesiones osteolíticas muydolorosas.
Histopatología
Los hallazgos histopatológicos son tan característicos que constituyen la clavedel diagnóstico. Se observa una paniculi-tis lobulillar con necrosis basofílica ex-tensa de adipocitos con infiltrados neu-trofílicos abundantes y presencia deadipocitos fantasmas ( sin núcleo y conuna gruesa membrana) (fig. 9). La causade esta imagen tan típica es la hidroliza-ción de las grasas neutras de los adipo-citos en glicerol y ácidos grasos libres por la lipasa y la amilasa, mientras quelas membranas celulares permanecen in-tactas.
1727
PANICULITIS
Fig. 8. Periarteritis nodosa cutánea: múltiples nódulos de pequeño tama-ño en extremidades inferiores.
Fig. 9. Paniculitis pancreática: necrosis de adipocitos y presencia de cé-lulas fantasma.

Tratamiento y evolución
La evolución de las lesiones cutáneas estáen relación con la pancreatopatía por loque lo fundamental es tratar la enferme-dad subyacente.
Paniculitis del recién nacido
La grasa del recién nacido tiene una ma-yor fragilidad que la del adulto al estaraumentada la proporción superficie cor-poral/peso y tener una proporción mayorde ácidos grasos saturados. También tie-ne un punto de solidificación y fundicióno melting más elevado. Se distinguen tresformas de paniculitis en el recién nacido:necrosis grasa subcutánea, escleredemaneonatorum y paniculitis post-esteroidea.Todas ellas son poco frecuentes. Los fac-tores precipitantes de las paniculitis enel recién nacido son: la hipotermia, la hipoxia, y el traumatismo mecánico delparto.
Necrosis grasa subcutánea del recién nacido
Se observa en recién nacidos a término opostmaduros con algún problema perina-tal. También ocurre tras cirugía cardíacahipotérmica en lactantes. Aparecen en lasprimeras semanas de vida uno o variosnódulos o placas circunscritas profundasen la hipodermis que se distribuyen de forma simétrica. Se localizan con frecuen-cia en tronco (hombros y espalda), y también en muslo, brazos o mejilla11. Tie-nen una consistencia firme, son despla-zables, no están adheridos y están cu-biertos por piel normal o violácea. Seasocia con frecuencia a hipercalcemia,una complicación que puede ser grave yque precisa una monitorización cuidado-sa. Más raramente se acompaña de trom-bopenia.Los hallazgos histopatológicos muestranuna paniculitis lobulillar con necrosis deadipocitos y sin vasculitis, con un infiltra-do granulomatoso con células gigantesmultinucleadas a cuerpo extraño. Las cé-lulas (histiocitos, lipocitos, células gigan-tes) tienen hendiduras como agujas deno-minadas aciculares en su interior, que soncristales de lípidos. Puede haber depósi-tos de calcio.El cuadro persiste durante semanas evo-lucionando a la curación espontánea sindejar cicatriz residual o una ligera atrofia,
teniendo muy buen pronóstico. Excepcio-nalmente se ulceran drenando un mate-rial aceitoso y dejando cicatriz.
Escleredema neonatorum
Se observa en niños prematuros con pro-blemas graves en las primeras semanasde vida. Suele aparecer en los primerosdías de vida un empastamiento rápido dela hipodermis, comenzando de forma si-métrica en glúteos y extendiéndose pro-gresivamente hasta afectar a toda la su-perficie corporal12. No afecta palmas,plantas ni genitales. La piel está edema-tosa, cérea y dura al tacto. En algún casose afecta también la grasa visceral.En el estudio histopatológico la necrosisgrasa no es muy llamativa y el infiltradoinflamatorio es muy escaso. Los lipocitosestán alargados y presentan hendidurasaciculares. La fibrosis es muy abundante.Al ser una enfermedad con tendencia a laprogresión, el escleredema neonatorum tie-ne muy mal pronóstico.
Paniculitis lobulillar post-esteroidea
Es un proceso muy raro que ocurre sóloen niños. Se caracteriza por la apariciónde nódulos subcutáneos de 1-4 centíme-tros, asintomáticos o dolorosos, localiza-dos en mejillas, brazo, parte superior deltronco; 1-13 días después de suspenderun tratamiento con esteroides por vía oral(por una leucemia, un síndrome nefróti-co…). Es debida a una rápida movilizaciónde lípidos. La histopatología es similar ala necrosis grasa subcutánea del recién nacido13.El tratamiento es la reinstauración de losesteroides y su supresión gradual poste-rior. La resolución espontánea a las pocassemanas es la norma, aunque existe algúncaso fatal.
Paniculitis por déficit de α1-antitripsina
La α1-antitripsina es una glucoproteínaque se sintetiza en el hígado y que regu-la la tripsina y otras enzimas proteolíti-cas. Cuando hay niveles insuficientes deα1-antitripsina la activación de las enzi-mas proteolíticas puede llevar a daño ti-sular grave. La deficiencia de α1-antitrip-sina es un defecto hereditario común encaucásicos. El locus es pleomórfico y se
han identificado 75 alelos. Es más graveen homocigóticos (fenotipos SS y ZZ). Pue-de aparecer a cualquier edad y a veces hayuna historia previa de un traumatismocomo factor desencadenante. En ocasio-nes hay varios miembros de la familiaafectados. Esta enfermedad afecta a mu-chos órganos, sobre todo al pulmón (en-fisema) y al hígado (hepatitis y cirrosis).La afectación cutánea se caracteriza pornódulos profundos, dolorosos, múltiplescon tendencia a la abscesificación y ul-ceración con drenaje de un fluido sero-sanguinolento14. Las lesiones se localizansobre todo en parte proximal de extremi-dades y tronco y se acompañan de afec-tación del estado general (fiebre, náuseas,vómitos…). También puede aparecer urti-caria y angioedema.En el estudio histológico se observa en es-tadios precoces una paniculitis septal sinalteraciones vasculares, con destrucciónde las fibras colágenas y elásticas; y uninfiltrado inflamatorio compuesto porabundantes neutrófilos. Posteriormente seafecta parte del lobulillo, entremezclán-dose áreas de necrosis grasa con áreas dehipodermis normal con la presencia de histiocitos y macrófagos rellenos degrasa.La evolución es crónica y recidivante, conuna elevada mortalidad. Para su diagnós-tico se precisa una determinación en san-gre de los niveles de α1-antitripsina. Eltratamiento consiste en la infusión de con-centrados de inhibidor de α1-proteinasa.A veces los pacientes responden a dap-sona.
Calcifilaxis
La paniculitis calcificante es un procesoraro y grave que se presenta en pacientesen diálisis o trasplantados renales15. Suetiopatogenia es desconocida, probable-mente multifactorial.Se caracteriza por nódulos subcutáneosmuy dolorosos que aparecen en extremi-dades inferiores y placas equimóticas ynecróticas con un patrón livedoide.En el estudio histopatológico se observauna calcificación y trombosis de los vasosde mediano calibre de la hipodermis,acompañado de necrosis cutánea. La in-flamación suele ser escasa.El cuadro evoluciona rápidamente y su cur-so suele ser fatal, falleciendo por sepsisen muchos casos.
1728
ENFERMEDADES DEL SISTEMA INMUNE (IX)

Paniculitis físicas
Paniculitis traumática
Se caracteriza por nódulos o placas indu-radas en zonas que han sufrido un trau-ma o una presión. Las lesiones son dolo-rosas, eritematosas. Posteriormente sevuelven violáceas, persistiendo la zona in-durada durante semanas y remitiendo es-pontáneamente a los varios meses sin de-jar rastro. Se localizan sobre todo enmiembros inferiores y en la mama, mássusceptibles a golpes. En ocasiones el trau-ma pone en marcha otros tipos de pani-culitis: deficiencia de α1-antitripsina, pa-niculitis pancreática.En el estudio histológico es característicoencontrar unas formaciones quísticas detamaño variable, observándose en su pared una formación membranosa fes-toneada con proyecciones papilares se-mejantes a la cutícula de algunos pará-sitos y que está consituído por un mate-rial PAS positivo y diastasa resistente16.Se pueden ver histiocitos espumosos a su alrededor. Existen también tractos fi-brosos.No precisa tratamiento ya que remite es-pontáneamente. Es importante pensar enella a la hora del diagnóstico diferencialcon otros tipos de paniculitis.
Paniculitis facticia
Se produce por la inyección de diversassustancias (povidona, pentazocina, me-peridina, morfina, heces, leche, saliva…).Suelen ser pacientes con trastornos gra-ves de la personalidad, por lo que debenser evaluados y tratados por psiquiatría.También se ha observado asociada al usode parafina o silicona con fines estéticos.Puede ocurrir en niños y adultos. La clí-nica es muy variable, dependiendo de lanaturaleza de la sustancia inyectada y elmodo de introducción17. Suele apareceren sitios accesibles para inyectarse por elpropio paciente, con fenómenos de infla-mación intensa. A veces se forman abs-cesos y fístulas, con ulceración de las le-siones. La histopatología es similar a lanecrosis grasa del recién nacido pero nopresentan hendiduras aciculares y el in-filtrado está compuesto por neutrófilos.En los casos por silicona o parafina loslinfocitos e histiocitos son las células pre-dominantes.
Paniculitis infecciosa
La presencia de nódulos o placas profun-das con eritema e inflamación en extre-midades, que se vuelven fluctuantes ul-cerándose y supurando un material purulento, son habituales en la práctica clínica. Pueden observarse en sujetos sanos aunque es más frecuente en in-munodeprimidos. Los agentes causales pueden ser bacterias (estreptococo β-he-molítico, pseudomona, xanthomona), mi-cosis (candida, fusarium) o micobacterias(fig. 10). También se han descrito por elvirus de Epstein-Barr y el citomegalovirus.En el estudio histológico se observa unapaniculitis lobulillar abscesificada, con uninfiltrado muy abundante de neutrófilos.En ocasiones el infiltrado es granuloma-toso, sobre todo si el agente causal es unamicobacteria. Existen fenómenos de ne-crosis focal y la dermis profunda suele es-tar también afectada18. Hay que hacer tin-ciones especiales para los diferentesmicroorganismos (Gram, Zhiel, PAS).Para su diagnóstico es muy importante elcultivo del material supurado y de la biop-sia. No es raro que sean necesarios varioscultivos. El tratamiento se instaurará unavez identificado el agente causal.
Paniculitis neoplásica:paniculitis histiocíticacitofágica
La paniculitis histiocítica citofágica es unproceso heterogéneo que se asocia con fre-cuencia al síndrome hemofagocítico. Lamayoría de los casos son verdaderos lin-fomas T (sobre todo) muy agresivos, conmal pronóstico, detectándose en ellos elvirus de Epstein-Barr (por hibridación insitu o PCR). En cambio, en los casos noasociados a linfoma no hay evidencia delvirus de Epstein-Barr.
La patogénesis de la hemofagocitosis esaún desconocida, se cree que es debida alefecto de algunas citoquinas circulantesdel suero. Existiría una disregulación in-mune de linfocitos T.Es una paniculitis poco frecuente, tiene unpredominio por el sexo femenino, y apa-rece en la edad adulta.Se caracteriza por nódulos eritematosos,dolorosos, de tamaño variable, múltiplesy extensamente distribuidos. Se asocian aepisodios febriles, leucopenia, hepatoes-plenomegalia, úlceras mucosas y derramepleural19. Suele haber un deterioro pro-gresivo, con alteración de la función hepática, equimosis, hemorragias con pan-hemocitopenia, fracaso hepático y coagu-lación intravascular diseminada.En el estudio histológico se observa unapaniculitis lobulillar sin vasculitis. Hay una marcada necrosis del lobulillo, acom-pañada de un infiltrado denso de linfoci-tos, histiocitos y plasmáticas. Los macró-fagos tienen un citoplasma espumoso ymuchos contienen restos de neutrófilos y eritrocitos que les da el aspecto de unsaco de judías (citohemofagocitosis). Loslinfocitos se disponen en anillo alrededorde los adipocitos necrosados. Se encuen-tran también acúmulos de histiocitos enmédula, ganglios, hígado, y bazo.El tratamiento es poco efectivo. A vecesmejora con ciclosporina y prednisona. Loscasos asociados a linfoma tienen un pro-nóstico fatal a pesar de la quimioterapiao la radioterapia. Casi todos los pacientesmueren de esta enfermedad. Evolucionacomo un proceso maligno en todos los casos.
Paniculitis paraneoplásica:tromboflebitis migratoriasuperficial
Se considera una dermatosis paraneoplá-sica, ya que suele aparecer asociada a ade-nocarcinoma visceral, sobre todo de ori-gen pancreático. También se ha observadoen pacientes con enfermedad de Behçet yanticoagulante lúpico.El mecanismo patogénico no está com-pletamente aclarado. Se produciría un es-tado de hipercoagulabilidad sanguíneaasociado a un proceso maligno.Se presenta más en pacientes de sexomasculino como brotes de nódulos erite-matosos profundos que suelen localizarse
1729
PANICULITIS
Fig. 10. Paniculitis infecciosa por Mycobacterium fortuitum.

en las piernas. Al evolucionar se distribu-yen de forma lineal o cordonal.En el estudio histopatológico se encuentrauna trombosis de venas superficiales conprácticamente ningún infiltrado inflama-torio o necrosis grasa, por lo que no esuna panicultis en sentido estricto. Para su tratamiento se aconseja el uso deheparina. Suele mejorar con el tratamien-to de la enfermedad de base.
Paniculitis de Weber-ChristianEste síndrome fue descrito originalmentecomo la aparición de brotes de nódulosrecurrentes subcutáneos acompañados defiebre y síntomas generales en mujeres de 20-60 años. Se ha descrito tambiénuna variante infantil, apirética, con afec-tación exclusiva cutánea, con pocos nó-dulos, denominada síndrome de Roth-man-Makai.En la actualidad se considera que la ma-yoría de estos pacientes presentaban al-gún tipo de las paniculitis anteriormentedescritas que en su momento no fue po-sible diagnosticar o una paniculitis a la queno puede llegarse todavía a un diagnósti-co definitivo (inespecíficas), por lo que mu-
chos autores creen que este término de-bería ser abandonado20.
AGRADECIMIENTOS
Al Doctor Luis Requena Caballero, del Servicio de Dermato-logía de la Fundación Jiménez Díaz, por su valiosa contri-bución en la elaboración de este manuscrito.
BIBLIOGRAFÍA
1. García Porrúa C, Gonzáles Gay MA, Vázquez CarunchoM, López Lázaro L, Lueiro M, Fernández ML. Erythema no-dosum: etiologic and predictive factors in a defined popula-tion. Arthritis Rheum 2000; 43: 584-592.2. Sánchez Yus E, Sanz Vico MD, De Diego V. Miescher´sradial granuloma: a characteristic marker of erythema no-dosum. Am J Dermatopathol 1989; 11: 432-442.3. Baselga E, Margall N, Barnadas MA, Coll P, Moragas JM.Detection of mycobacterium tuberculosis DNA in lobulargranulomatous panniculitis (erythema induratum-nodularvasculitis). Arch Dermatol 1997; 133:457-462.4. Sanz Vico MD, De Diego V, Sánchez Yus E. Erythemanodosum versus nodular vasculitis. Int J Dermatol 1993;32: 108-112.5. Martens PB, Moder KG, Ahmed I. Lupus panniculitis: cli-nical perspectives from a case series. J Rheumatol 1999;26: 68-72.6. Su WP, Person JR. Morphea profunda. A new conceptand a histopathologic study of 23 cases. Am J Dermatopat-hol 1981; 3: 251-260.7. Lorenzo Alemán JA, Hernández Santana J, Báez MarreroO, Castro Piñeiro I, Torrado González RM, Rodríguez LópezJ, et al. Paniculitis lobulillar como forma de presentación deuna dermatomiositis: presentación de un caso y revisión dela literatura. Actas Dermosifiliogr 1998; 89: 400-404.8. Tran JA, Du Pree M, Carlson JA. Neutrophilic lobular(pustular) panniculitis associated with rheumatoid arthritis:
case report and review of the literature. Am J Dermatopat-hol 1999; 21: 247-252.9. Daoud MS, Hutton KP, Gibson LE. Cutaneous periarteri-tis nodosa: a clinicopathological study of 79 cases. Br J Der-matol 1997; 136: 706-713.10. Segurado Rodríguez A, Guerra Tapia A, Jaén Olasolo P,Cuevas Santos J. Paniculitis pancreática: estudio de 12 ca-sos y valoración comparativa de sus caracteres epidemioló-gicos, clínicos, histopatológicos y terapeúticos. Actas Der-mosifiliogr 1999; 90: 227-234.11. Burden AD, Krafchik BR. Subcutaneous fat necrosis ofthe newborn: a review. Pediatr Dermatol 1999; 16: 384-387.12. Fretzin DF, Arias AM. Sclerema neonatorum and sub-cutaneous fat necrosis of the newborn. Pediatr Dermatol1987; 4: 112-122.13. Saxena AK, Nigam PK. Panniculitis following steroidtherapy. Cutis 1988; 42:156-157.14. Su WP, Smith KC, Pittelkow MR, Winikelman RK. Alp-ha-1 antitrypsin deficiency panniculitis. Am J Dermatopat-hol 1988; 18: 684-692.15. Fariña MC, De Sequera P, Soriano ML. Calcifilaxis. Ac-tas Dermosifiliogr 1997; 88: 333-336.16. Dorado J, Daudén E, Gil R, Vanaclocha F, Iglesias L. Pa-niculitis traumática. Estudio clínico e histológico de ochocasos. Actas Dermosifiliogr 1988; 74: 498-502.17. Forstrom L, Winkelman RK. Factitial panniculitis. Archdermatol 1974; 11: 747-750.18. Patterson JW, Brown PC, Broecker AH. Infection-indu-ced panniculitis. J Cutan Pathol 1989; 16: 183-193.19. Craig AJ, Cualing H, Thomas G, Lamerson C, Smith R.Cytophagic histiocytic panniculitis- a syndrome associatedwith benign and malignant panniculitis: case comparisonand review of the literature. J Am Acad Dermatol 1998; 39:721-736.20. White JW, Winkelman RK. Weber-Christian panniculi-tis: a review of 30 cases with this diagnosis. J Am AcadDermatol 1998; 39: 56-62.
1730
ENFERMEDADES DEL SISTEMA INMUNE (IX)